

SUMMARY
Precise targeting of activation-induced cytidine deaminase (AID) to immunoglobulin (Ig) loci promotes antibody class switch recombination (CSR) and somatic hypermutation (SHM), whereas AID targeting of non-Ig loci can generate oncogenic DNA lesions. Here, we examined the contribution of G-quadruplex (G4) nucleic acid structures to AID targeting in vivo. Mice bearing a mutation in Aicda (AIDG133V) that disrupts AID-G4 binding modeled the pathology of hyper-IgM syndrome patients with an orthologous mutation, lacked CSR and SHM, and had broad defects in genome-wide AIDG133V chromatin localization. Genome-wide analyses also revealed that wild type AID localized to MHCII genes, and AID expression correlated with decreased MHCII expression in germinal center B cells and diffuse large B-cell lymphoma. Our findings indicate a crucial role for G4-binding in AID targeting, and suggest that AID activity may extend beyond Ig loci to regulate the expression of genes relevant to the physiology and pathology of activated B cells.
Keywords: Activation-induced cytidine deaminase, class switch recombination, somatic hypermutation, G-quadruplex, hyper-IgM syndrome, MHC class II (MHCII), germinal center B cells, diffuse large B-cell lymphoma (DLBCL)
Graphical Abstract
INTRODUCTION
The vertebrate humoral immune system possesses the remarkable ability to recognize a vast array of antigens, activate antigen-specific B cells, and select cells with high-affinity B cell receptors (BCRs) to produce antibodies (abs) in a process of accelerated Darwinian natural selection. Activation-induced cytidine deaminase (AID) plays a critical role in humoral immunity by orchestrating the secondary diversification of BCRs. Targeted AID deamination of the immunoglobulin (Ig) locus converts deoxycytidines into deoxyuridines, supercharging the frequency of somatic mutations that fuel class switch recombination (CSR) and somatic hypermutation (SHM) (Muramatsu et al., 2000; Muramatsu et al., 1999; Petersen-Mahrt et al., 2002; Revy et al., 2000). Mutations in AICDA can result in hyper-immunoglobulin M syndrome type 2 (HIGM2), a primary immunodeficiency disease characterized by a severe defect in CSR that is frequently coupled with a deficiency in SHM (Durandy et al., 2006; Revy et al., 2000).
CSR is a molecular rearrangement that deletes and recombines segments of the immunoglobulin (Ig) heavy chain (Igh) locus, exchanging the default IgM and IgD constant region gene segments for a set of downstream exons. During CSR, AID is targeted to repetitive DNA elements called switch (S) regions, located upstream of each constant region gene segment. AID deamination within S regions initiates a cascade of DNA processing events leading to the formation of DNA double-strand breaks (DSBs). Ligation of donor and acceptor S region DSBs, primarily by non-homologous end-joining, juxtaposes the upstream V(D)J region with a new downstream constant region gene segment, switching activated B cells from expressing IgM and IgD to a secondary heavy chain isotype like IgG, IgE, IgA or in rare cases, expressing solely IgD (Gilliam et al., 1984; Matthews et al., 2014; Xu et al., 2012). Pathogens and responding immune cells provide molecular cues that direct CSR to particular antibody isotypes, equipping the adaptive immune system with tailored antibody-mediated effector functions to optimally combat a variety of pathogens.
In addition to S regions, AID targets the variable region of the Igh, Igk, and Igl loci to perform SHM, where deamination and subsequent processing by error prone DNA repair generate somatic mutations (Di Noia and Neuberger, 2007). SHM occurs in microanatomical structures called germinal centers (GC) located within secondary lymphoid organs. Here, competition between B cells for T cell help leads to the selection of B cells with higher affinity BCRs (Mesin et al., 2016). While AID targeting to Ig loci is critical for CSR and SHM, AID can localize to hundreds of non-Ig genes, many of which have been shown to be mutated (Alvarez-Prado et al., 2018; Chiarle et al., 2011; Klein et al., 2011; Liu et al., 2008; Pavri et al., 2010; Yamane et al., 2011). Understanding AID specificity is paramount, since AID targeting of non-Ig loci can generate oncogenic mutations and translocations, such as with Myc or Bcl6, that characterize many B cell lymphomas (Lieber, 2016; Lu et al., 2013; Pasqualucci et al., 2008; Ramiro et al., 2004).
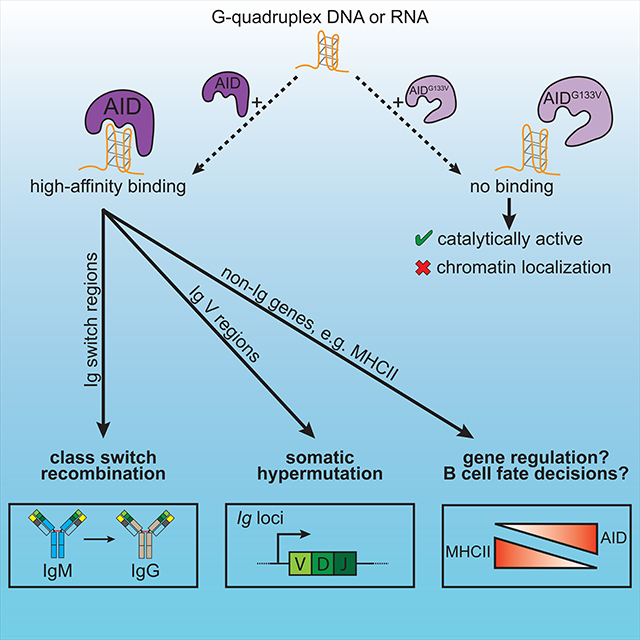
Myriad molecular factors affect AID targeting, including R-loops, the transcriptional and splicing machineries, the RNA exosome, and G-quadruplex structures (G4s) (Casellas et al., 2016). G4s are non-canonical nucleic acid structures that can form in guanine-rich sequences via Hoogsteen base-pairing of planar guanine tetrads, generating a parallel four-stranded structure (Gellert et al., 1962; Sen and Gilbert, 1988). DNA G4s have been observed following in vitro transcription of S regions (Carrasco-Salas et al., 2019; Duquette et al., 2004; Neaves et al., 2009), which are composed of G-rich tandem repeats (Dunnick et al., 1993). AID binding to transcription-induced G4s in vitro suggests that these structures may recruit AID in cis (Duquette et al., 2005). AID also binds to S region RNA G4s (Qiao et al., 2017; Zheng et al., 2015), and G4s bind to AID with 10-fold higher affinity than linear forms of the same sequence, perhaps accounting for the preferential deamination of deoxycytidine residues adjacent to G4 structures (Qiao et al., 2017).
There is mounting functional evidence for a critical role of AID-G4 binding in CSR. Cooperative AID-G4 binding seeds the formation of large AID-G4 oligomers, and mutations that disrupt cooperativity and oligomer formation impair CSR without altering deamination activity (Qiao et al., 2017). Further, a Gly-to-Val mutation at residue 133 of AID (AIDG133V) disrupts AID-G4 RNA binding, abolishing AID targeting to S regions and CSR in ex vivo activated B cells (Zheng et al., 2015). Intriguingly, expression of S region transcripts that generate G4s, but not their antisense counterparts, rescues CSR in B cells with defective RNA splicing, suggesting that S region G4 RNAs “guide” AID to the Igh locus, analogous to guide RNAs targeting Cas9 (Zheng et al., 2015). Lastly, the identification of HIGM2 patients with homozygous G133V mutations in AICDA (Mahdaviani et al., 2012) suggests a conserved role for AID-G4 binding in CSR, and underscores the importance of studying AID-G4 binding in vivo, for which experimental models are currently lacking.
To address this gap in knowledge, we generated mice bearing an engineered allele of Aicda encoding a Gly-to-Val mutation (AIDG133V). Whereas AIDG133V and wild type AID (AIDWT) had comparable DNA deaminase activity in vitro and in ex vivo activated B cells, AicdaG133V/G133V (AicdaGV/GV) mice lacked CSR and SHM. AIDG133V failed to localize to Igh S regions, and ChIP-sequencing (ChIP-seq) uncovered a broad defect in genome-wide AIDG133V chromatin localization. These genome-wide analyses also revealed that AIDWT localized to MHCII genes, and AID expression correlated with decreased MHCII expression in GC B cells. Additionally, human diffuse large B-cell lymphoma (DLBCL) tumors with the highest AID levels exhibited decreased expression of multiple MHCII presentation pathway genes. Our findings point to a central role for G4 binding in AID targeting, and suggest that AID-dependent gene regulation of non-Ig loci may play a critical role in regulating GC B cell fates, as well as affect the genesis and prognosis of DLBCL.
RESULTS
AicdaGV/GV mice model HIGM2 syndrome
Using CRISPR-Cas9 targeted mutagenesis, we generated a mouse strain with a Gly-to-Val mutation at residue 133 (G133V) of the Aicda gene (Figure S1A) that has been shown to disrupt AID-G4 binding (Zheng et al., 2015). The modified allele was expressed, properly spliced and contained only the specified G133V encoding mutation (Figure S1B). We next compared the development of B cells in AicdaGV/GV mice to WT and AicdaGV/+ littermate controls, and found no overt differences in the absolute number or frequency of developing or mature B cell subsets within the bone marrow or spleen, or within T cell subsets (Figure S1).
To test if AicdaGV/GV mice model human HIGM2, we used ELISAs to measure serum Ig isotype composition. AicdaGV/GV mice had a nearly 4-fold higher IgM concentration compared to WT controls (Figure 1A), similar to the fold change in HIGM2 patients (Mahdaviani et al., 2012; Revy et al., 2000). Compared to WT and Aicda+/− mice, the levels of all class-switched abs in AicdaGV/GV and Aicda−/− mice were reduced 10- to 1,000-fold (Figure 1A), indicating a major CSR defect.
Figure 1. AicdaGV/GV mice model hyper-IgM type 2 syndrome.

(A) Quantification of homeostatic serum immunoglobulin concentrations by ELISA. (B-D) Homeostatic germinal center (GC) hyperplasia in AicdaGV/GV secondary lymphoid organs. (B) Representative GC B cell gates (GL7+Fas+ of B220+ live cells) from mesenteric lymph node (LN). Quantification of GC B cell frequency within the mesenteric LN (C) or Peyer’s patches (D). (E-J) Influenza A virus GC hyperplasia. Mice were intranasally infected with 50 TCID50 PR8 and analyzed at d21 post-infection. (E) Representative GC B cell gates (GL7+Fas+ of B220+ live cells) from mediastinal LN. Quantification of GC B cell frequency within the mediastinal LN (F) or spleen (G). (H) Representative immunofluorescent images from splenic sections stained with PNA (red), anti-IgD (green), anti-B220 (grey), and DAPI (blue). Scale bar represents 50 μm. Quantification of GC size (I) and density (J). Data in (A-D) are from 2 independent experiments with 1–6 mice per genotype, (E-G) are from 5 independent experiments with 2–8 mice per genotype, (H-J) are from 1 experiment with 3–4 mice per genotype. AicdaGV/GV, AicdaG133V/G133V. Error bars represent the mean ± std. dev. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; p-values calculated using a one-way ANOVA with Tukey’s multiple comparisons test without pairing.
Aicda−/− mice and HIGM2 patients exhibit lymph node (LN) hyperplasia resulting from enlarged GCs (Muramatsu et al., 2000; Revy et al., 2000). To test if AicdaGV/GV mice develop GC B cell hyperplasia, we analyzed homeostatic GCs in secondary lymphoid organs. We found elevated frequencies of GC B cells in the mesenteric LNs (Figure 1B,C), Peyer’s patches (Figure 1D), and spleen (Figure S2A) of AicdaGV/GV mice, and the absolute number of GC B cells in Peyer’s patches was approximately 6.5-fold higher on average, and 5-fold higher per individual Peyer’s patch in AicdaGV/GV mice versus WT controls (Figure S2B–D).
To determine if AIDG133V-induced hyperplasia occurs during acute immune responses, we infected mice intranasally with 50 TCID50 of PR8, a highly mouse-adapted influenza A virus (IAV) strain. We harvested spleen and mediastinal LNs 21 days post-infection (dpi) and quantified the frequency of GC B cells, and the number and size of GCs in splenic sections. AicdaGV/GV and Aicda−/− mice had significantly elevated frequencies of mediastinal (draining) LN (Figure 1E,F) and splenic (Figure 1G) GC B cells. This can be attributed to the larger size of individual GCs in AicdaGV/GV and Aicda−/− mice (Figure 1H,I), as all genotypes had similar numbers of GCs (Figure 1J). Additionally, we found that AicdaGV/GV GC B cells predominately occupied the light zone (LZ) (Figure S2E,F), as previously shown for Aicda−/− GC B cells (Boulianne et al., 2013; Hogenbirk et al., 2013), whereas WT GC B cells predominantly populate the dark zone (DZ). Overall, the lack of class-switched antibodies, elevated serum IgM and lymphoid GC hyperplasia indicate that AicdaGV/GV mice model HIGM2 pathology.
AicdaGV/GV B cells do not class switch in response to viral infection
To determine if the greatly reduced serum levels of class-switched abs in AicdaGV/GV mice result from defective CSR, we analyzed GC B cell CSR by flow cytometry. WT and Aicda+/− mice had substantial frequencies of IgG1 and IgA class-switched B cells in chronic homeostatic GCs within mesenteric LNs and Peyer’s patches, whereas AicdaGV/GV mice had no detectable levels of class-switched B cells in either lymphoid organ, similar to Aicda−/− controls (Figure 2A–D).
Figure 2. AicdaGV/GV B cells do not undergo CSR at homeostasis or during acute viral infection.

(A-D) Homeostatic class switch recombination (CSR) in secondary lymphoid organs. Representative gates of IgG1 (A) or IgA (B) class-switched B cells in mesenteric lymph node (LN) (A) or Peyer’s patch (B) germinal centers (GC) (IgG1+ or IgA+ of GL7+Fas+ B220+ live cells). Quantification of class-switched B cells within the mesenteric LN (C) or Peyer’s patch GCs (D). (E-J) CSR in response to influenza A virus infection. Mice were intranasally infected with 50 TCID50 PR8 or J1 and analyzed at d21 post-infection. (E) Representative gates showing hemagglutinin-specific (HA+) GC B cells (HA+ of GL7+Fas+B220+ live cells) in the mediastinal LN. Quantification of HA+ frequency within GC B cells in the mediastinal LN (F) or spleen (G). Representative gates showing IgG1+ and IgG2c+ (H) or IgD−IgM− and IgM+ (I) B cell populations in mediastinal LN GC HA+ cells (IgG1+, IgG2c+, IgD−IgM−, or IgM+ of HA+GL7+Fas+B220+ live cells). (J) Quantification of HA+ class-switched B cells within mediastinal LN GCs. Data in (A-D) are from 2 independent experiments with 3–6 mice per genotype, (E-J) are from 5 independent experiments with 2–8 mice per genotype. AicdaGV/GV, AicdaG133V/G133V. Error bars represent the mean ± std. dev. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; ns, not significant, p ≥ 0.05. (C,D) all comparisons p < 0.0001 unless noted; (J) all comparisons p < 0.05 unless noted. p-values calculated using a one-way ANOVA with Tukey’s multiple comparisons test without pairing.
Extending these findings to IAV infection, we harvested the spleen and mediastinal LN 21dpi during the peak of the GC B cell response (Frank et al., 2015; Rothaeusler and Baumgarth, 2010). We measured CSR in total GC B cells, as well as hemagglutinin (HA) -specific GC B cells (Figure 2E); HA is the immunodominant antigen following IAV infection (Altman et al., 2018). We identified HA-specific cells by staining with fluorescent HA containing a mutation that abrogates HA binding to terminal sialic acid residues, which are abundant on B cells (Whittle et al., 2014). As a negative control for HA staining, Aicda+/− mice were infected with the J1 strain of IAV (Figure 2E), a reassortant virus that is identical to PR8 except for an H3 HA gene, which does not cross-react serologically with PR8 HA (Palese, 1977). Both AicdaGV/GV and Aicda−/− mice had higher frequencies of HA-specific B cells within mediastinal LN and splenic GCs, although this difference was only statistically significant in the spleen (Figure 2E–G). While approximately 80% of HA-specific GC B cells class-switched to IgG1, IgG2c, or IgG2b in WT mediastinal LNs, AicdaGV/GV HA-specific GC B cells were virtually 100% IgM+, as were Aicda−/− GC B cells (Figure 2H–J). This held true for total GC B cells in the mediastinal LN (Figure S3A–C), and total (Figure S3D) and HA-specific (Figure S3E) splenic GC B cells.
We next immunized AicdaGV/GV mice with the model antigen 4-Hydroxy-3-nitrophenylacetyl (NP) conjugated to chicken gamma globulin (NP-CGG) and measured NP-specific IgM and IgG1 ab responses over 28 days. Both WT and AicdaGV/GV mice had significant increases in NP-specific IgM abs following immunization (Figure S3F), however, class-switched NP-specific IgG1 abs were undetectable in AicdaGV/GV and Aicda−/− mice (Figure S3G). Taken together, these data demonstrate that AicdaGV/GV B cells have a complete CSR defect during a primary immune response.
AicdaGV/GV B cells do not undergo CSR ex vivo
For mechanistic insight underlying the CSR deficiency in AicdaGV/GV mice, we used a tractable ex vivo system wherein purified naïve splenic B cells are stimulated with various cytokine plus mitogen cocktails to induce CSR. AicdaGV/GV B cells failed to undergo detectable CSR to IgG1 (Figure 3A,B), IgG3 (Figure 3C,D), or IgA (Figure 3E,F). AicdaGV/+ B cells underwent approximately 80% the levels of WT IgG1 CSR following 96hrs LPS plus IL-4 stimulation (Figure 3B). Because Aicda+/− B cells also undergo 80% of WT IgG1 CSR under similar stimulation conditions (McBride et al., 2008), the partial CSR defect in AicdaGV/+ B cells is likely due to decreased AIDWT protein levels, versus a dominant negative interaction between AIDWT and AIDG133V proteins. Retroviral expression of AIDWT, but not catalytically dead AIDH56R/E58Q (AIDCD) (Papavasiliou and Schatz, 2002), rescued the CSR defect in AicdaGV/GV B cells, ruling out off-target mutations introduced during CRISPR-Cas9 targeting causing the CSR defect (Figure 3G).
Figure 3. AicdaGV/GV B cells do not undergo CSR ex vivo.

(A-F) Naïve splenic B cells from mice of the indicated genotypes were stimulated for 72 or 96 hrs with LPS plus IL-4 (A,B), LPS (C,D), or B cell activating factor (BAFF), retinoic acid (RA), IL-4, TGFβ, IL-5, and LPS (E,F). Class switch recombination (CSR) to IgG1 (B), IgG3 (D), or IgA (F) was quantified at 72 and 96 hrs post-stimulation. Representative CSR gates shown in (A), (C), and (E) (gated on live cells). (G) Retroviral rescue of CSR in AicdaGV/GV B cells. AicdaGV/GV B cells were infected with retroviruses expressing GFP only (empty vector, EV), wild type AID (AIDWT) or catalytically dead AIDH56R/E58Q (AIDCD), stimulated with LPS plus IL-4, and IgG1 CSR was quantified as a frequency of live infected cells (GFP+) at 72 and 96 hrs following stimulation. (H) Expression of AIDG133V ex vivo. Whole cell extracts were prepared from WT and AicdaGV/GV purified naïve splenic B cells stimulated for 96 hrs with LPS plus IL-4, and immunoblotted with anti-AID antibodies; loading control, HSP90; quantification shown in Figure S4C. (I) Expression of AIDG133V in vivo. Following immunization with SRBCs, splenic GC B cells were sorted at d7, whole cell extracts were prepared and immunoblotted with anti-AID antibodies; loading control, α-Tubulin. Data in (A-F, H) are from, or representative of, 4 independent experiments with 1–6 mice per genotype, (G) are from 2 independent experiments with 3 mice, (I) are from one experiment with 2–3 mice per genotype. AicdaGV/GV, AicdaG133V/G133V. Error bars represent the mean ± std. dev. ****p < 0.0001; ns, not significant, p ≥ 0.05. All comparisons p < 0.001 in (B) unless noted; all comparisons p < 0.0001 in (D,F) unless noted. p-values calculated using a one-way ANOVA with Tukey’s multiple comparisons test with (G), or without pairing (B,D,F).
Compromised CSR can result from multiple defects, including cell proliferation, transcription and splicing of switch regions, as well as AID expression and targeting to the Igh locus (Matthews et al., 2014; Yewdell and Chaudhuri, 2017). AicdaGV/GV B cells proliferate comparably to WT (Figure S4A) and have no significant differences in the levels of Sμ or Sγ1 germline transcripts (Figure S4B). To analyze AID expression, we prepared B cell lysates 96hrs post-stimulation and quantified AID protein levels by immunoblotting (Figure 3H, S4C) and intracellular flow cytometry (Figure S4D). AIDG133V was expressed at 70% of AIDWT levels on average, despite AicdaGV/GV B cells having slightly more abundant Aicda mRNA (Figure S4E). Extending these findings, sorted GC B cells from mice immunized with sheep red blood cells (SRBCs) expressed AIDG133V at slightly lower levels compared to WT controls (Figure 3I).
Intriguingly, AIDG133V extracted from ex vivo activated B cells and GC B cells migrated faster than AIDWT under denaturing SDS-PAGE conditions (Figure 3H,I). Recombinant N-terminal maltose binding protein (MBP) tagged protein purified from E. coli, MBP-AIDG133V, and AIDG133V proteolytically cleaved from MBP, also migrated faster than AIDWT, whereas AIDCD did not (Figure S4F–J). Thus, the accelerated migration is not intrinsic to eukaryotic cell expression or dependent on AID catalytic activity. Because the MBP fusion protects the AIDG133V N-terminus, an N-terminal truncation is not likely to cause the accelerated migration. Additionally, mass spectrometry failed to reveal alterations to C-terminal peptides (Figure S4K). It is possible that AIDG133V and AIDWT differ in a post-translational modification common to both mouse and bacterial cells; however, we did not detect any such modifications in our mass spectrometry analysis. It is more likely that the mobility of AID in SDS-PAGE experiments is highly sensitive to the primary amino acid sequence, as AIDCD also migrates differently than AIDWT (Figure S4I,J), and the altered migration of other AID point mutants is observable in previous reports (Methot et al., 2018; Mondal et al., 2016).
AIDG133V retains DNA deamination activity
The indistinguishable phenotypes of AicdaGV/GV and Aicda−/− mice suggest that AIDG133V may be catalytically impaired. However, in vitro deamination assays (Figure 4A) using purified proteins revealed that MBP-AIDWT and MBP-AIDG133V have comparable deamination activity across 3 different protein preparations, whereas the negative control MBP-AIDCD has no detectable activity (Figure 4B, S5A). Additionally, we expressed and purified human AID (hAID) MBP fusion proteins from E. coli (Figure S5B), and found that MBP-hAIDWT and MBP-hAIDG133V also had comparable deamination activity, whereas MBP-hAIDCD had no detectable activity (Figure S5C,D). To more quantitatively compare AIDWT and AIDG133V deamination activity, we performed deamination assays across a wide concentration of purified proteins. We found no differences between AIDG133V and AIDWT deamination activity at all concentrations tested (Figure 4C, S5E). Thus, these data indicate that AIDWT and AIDG133V have comparable in vitro deamination activity.
Figure 4. AIDG133V retains DNA deamination activity.

(A) Schematic of AID in vitro deamination assay. FAM, 6-carboxyfluorescein. TGU, positive control representing 100% cytidine deamination; TGC, AID deamination substrate. (B) Representative TBE-Urea gel showing the products of in vitro deamination reactions with purified MBP-mouse AID (MBP-mAID) proteins, quantified in Figure S5A. Purified MBP-mAID proteins shown in Figure S4F–J. CD, catalytically dead AIDH56R/E58Q. (C) Quantification of deamination assays with varying amounts of purified MBP-mAID proteins, representative gels shown in Figure S5E. (D) G4 substrate derived from Igh-Sμ locus (Sμ4G) and mutant sequence unable to form G4 (Sμ4Gmut). Substrate characterization in Figure S5F. (E,F) Representative TBE-Urea gels showing the products of in vitro deamination reactions with purified mouse MBP-mAID proteins, performed with or without G4 DNA or RNA, quantified in (G,H). Mut, Sμ4Gmut; Li+, G4 folded in presence of Li+ (destabilizes G4s); K+, G4 folded in presence of K+ (stabilizes G4s). Data in (B) are representative of 2 independent experiments, (C, E-H) are from, or representative of, 4 or 5 independent experiments using 2 (C) or 3 (E-H) independent protein preparations. (B, E-F) “s” denotes substrate, “p” denotes product. Error bars represent the mean ± std. dev. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001; p-values calculated using a one-way ANOVA with Tukey’s multiple comparisons test with (G,H) or without (C) pairing.
Because AID binds to G4s cooperatively, creating large AID-G4 oligomers (Qiao et al., 2017), we tested whether the addition of G4 substrates could alter MBP-AID’s deamination activity on the linear substrate. Deamination assays performed in the presence of DNA or RNA G4 substrates composed of 4 tandem Sμ repeats (Sμ4G) (Figure 4D, S5F) previously shown to bind AID (Zheng et al., 2015) inhibited deamination of the linear substrate by MBP-AIDWT (Figure 4E,G,H, S5G). This is consistent with the previous observations that AID binds to G4 substrates with > 10-fold affinity than it binds to linear substrates, and that AID binds to DNA and RNA G4 substrates with similar affinities (Qiao et al., 2017). Disrupting the G4 substrate by mutating the sequence (Sμ4Gmut) or folding Sμ4G in the presence of Li+, which destabilizes G4s (Bardin and Leroy, 2008; Bhattacharyya et al., 2016) (Figure S5F), abrogates G4-dependent inhibition (Figure 4E,G,H S5G). Consistent with a defect in G4 nucleic acid binding, the addition of DNA or RNA Sμ4G did not inhibit MBP-AIDG133V deamination of the linear substrate (Figure 4F–H, S5H), despite a 5-fold molar excess of Sμ4G versus linear substrate. Thus, these data indicate that AIDWT binding to a G4 structure can inhibit its deamination of a linear substrate, and reinforce the notion that AIDG133V does not bind to G4 nucleic acids.
AIDG133V has impaired Igh targeting
We next tested the ability of AIDG133V to localize to the Sμ region of the Igh locus by performing ChIP-qPCR experiments. We first confirmed that the anti-AID ab immunoprecipitated both AIDWT and AIDG133V proteins during the ChIP protocol (Figure 5A). The amount of AIDG133V immunoprecipitated was approximately 70% of the AIDWT levels (Figure 5B), mirroring the relative protein levels of AIDWT and AIDG133V that we detected in B cell lysates (Figure 3H, S4C). As an additional control, we performed nuclear-cytoplasmic fractionation experiments followed by immunoblotting to test if AIDG133V localizes to the nucleus. Because upwards of 90% of AID is localized in the cytoplasm (Methot et al., 2015; Pasqualucci et al., 2004; Rada et al., 2002), detecting endogenous AID in nuclear extracts is challenging. As expected, both AIDWT and AIDG133V were readily detected in the cytoplasmic fraction, and sparse in the nuclear fraction (Figure 5C). qPCR revealed a significant enrichment of Sμ DNA in AID ChIP samples from WT B cells versus Aicda−/− B cells. However, AIDG133V ChIP samples were not significantly enriched (Figure 5D), indicating a defect in AIDG133V localization to Sμ.
Figure 5. AIDG133V has impaired Igh targeting.

(A) Representative immunoblot showing AID immunoprecipitation during ChIP protocol, fraction of total lysate loaded is indicated. Exp, exposure. (B) Quantification of data shown in (A). (C) Representative immunoblots from nuclear-cytoplasmic fractionation experiments. Cytoplasmic loading control, HSP 90; nuclear loading control, Lamin-B1. (D) ChIP qPCR quantifying the amount of Sμ DNA immunoprecipitated with indicated antibodies (ab) from various genotypes. (E-G) Analysis of somatic hypermutation within the Jh4 intron. Germinal center (GC) B cells were sorted from Peyer’s patches at homeostasis (E), or the spleen at d7 post-SRBC immunization (F), and Jh4 intron sequences were cloned and sequenced. Total number of clones and total base pairs (bp) analyzed indicated inside circles, average mutation frequency indicated below circles, fraction of clones with the indicated number of mutations represented as section of circle. (G) Quantification of data in (E,F). (H) Representative gates from retroviral rescue of CSR in Aicda−/− B cells. Aicda−/− B cells were infected with retroviruses expressing GFP only (empty vector, EV), wild type AID (AIDWT), AIDG133V, catalytically dead AIDH56R/E58Q (AIDCD), or AIDWT-AIDCD, AIDG133V-AIDCD, AIDCD-AIDCD, and AIDG133V-mCherry fusions. Cells were stimulated with LPS plus IL-4, and IgG1 CSR was quantified as a frequency of live infected cells (GFP+) at 96 hrs post-stimulation. (I) Quantification of data shown in (H), raw CSR values shown in Table S1. Data in (A,B) are representative of 3 independent experiments with 1 mouse per genotype, (C) are representative of 2 independent experiments with 2 mice per genotype, (D) are from 4 independent experiments with 1 mouse per genotype, (E,F) are from 1 experiment with 3–4 mice per genotype, (H,I) are from 2 or 3 independent experiments with 1 or 2 mice per experiment. AicdaGV/GV, AicdaG133V/G133V. Error bars represent the mean ± std. dev. *p < 0.05; **p < 0.01; ***p < 0.001; ****p < 0.0001. p-values calculated using a paired (B,I) or unpaired (G) t-test.
To assess AIDG133V localization to the V(D)J segments, we sorted GC B cells from Peyer’s patches at homeostasis, or spleens following SRBC immunization, and PCR amplified and sequenced the 5’ region of the Jh4 intron. Sequencing of the Jh4 intron is commonly used as a proxy to measure variable region SHM, as hypermutation continues past the rearranged V(D)J and into the downstream intron (Jolly et al., 1997). WT and Aicda+/− GC B cells had mutation rates ranging from 2 × 10−3 - 6 ×10−3/bp (Figure 5E–G), consistent with previous Jh4 mutation rates within these lymphoid organs (Wei et al., 2011). AicdaGV/GV mutation rates (1 × 10−5 - 5 × 10−5/bp) were two orders of magnitude lower than WT, and indistinguishable from Aicda−/−, consistent with the complete absence of SHM in AicdaGV/GV GC B cells. Taken together, the above data suggest that the lack of CSR and SHM in AicdaGV/GV B cells results from a defect in AIDG133V Igh localization.
Forced targeting of AIDG133V to S regions can restore CSR
To determine the extent that defective AIDG133V Igh localization limits CSR, and reinforce the notion that AIDG133V is catalytically active, we sought to forcibly target AIDG133V to S regions to rescue the CSR defect. We fused AIDG133V to the N terminus of AIDCD, which localizes to S regions but lacks deamination activity (Methot et al., 2018; Vuong et al., 2013). We expressed the AIDG133V-AIDCD fusion protein retrovirally, along with AIDWT-AIDCD and AIDCD-AIDCD fusions as positive and negative controls, respectively, and assessed their ability to restore CSR in Aicda−/− B cells. The AIDWT-AIDCD fusion reduced IgG1 CSR efficiency to ~71% of the levels of AIDWT, whereas the negative control AIDCD-AIDCD and the empty vector (EV) control resulted in no detectable CSR. Remarkably, while AIDG133V and AIDCD induced no detectable CSR on their own, the AIDG133V-AIDCD fusion restored IgG1 CSR in Aicda−/− B cells to ~66% of the AIDWT-AIDCD levels (Figure 5,H,I). The CSR activity of AIDG133V-AIDCD is likely not due to simple protein stabilization, as an AIDG133V-mCherry fusion did not support detectable CSR.
The inability of the AIDG133V-AIDCD fusion to fully restore CSR to control levels could be due to the AIDWT-AIDCD fusion containing two moieties capable of targeting S regions, versus only one in the AIDG133V-AIDCD fusion. Additionally, because AIDWT binds to G4s with 10-fold higher affinity than a linear substrate of the same sequence, and preferentially deaminates deoxycytidine residues adjacent to G4s (Qiao et al., 2017), the AIDG133V-AIDCD fusion may be less efficient at deaminating S regions than AIDWT-AIDCD. Overall, these data demonstrate that first, AIDG133V can be catalytically active in activated B cells, and second, AIDG133V can deaminate S regions if properly targeted.
AIDG133V has impaired genome-wide chromatin localization
To uncover the contribution of G4 binding to AID chromatin localization outside the Igh locus, we performed ChIP-seq experiments in WT and AicdaGV/GV B cells. In order to allay reproducibility concerns regarding previous AID ChIP-seq analyses (Hogenbirk et al., 2012), we analyzed sequencing libraries from 4 biological replicates of WT and AicdaGV/GV ChIP experiments, and 3 biological replicates plus one technical replicate of Aicda−/−. After filtering low quality and duplicate reads, putative sites of binding were identified and analyzed for differentially bound regions across genotype comparisons, accounting for differences between biological replicates (see methods for details). We identified 37 peaks significantly enriched in WT versus either Aicda−/− or AicdaGV/GV, of which 21 were significantly enriched in both comparisons, 14 in comparison to AicdaGV/GV only, and 2 in comparison to Aicda−/− only (Figure 6A, table S2,3,4). There were no significantly enriched peaks in the AicdaGV/GV versus Aicda−/− comparison, indicating a major genome-wide defect in AIDG133V chromatin localization (Table S4).
Figure 6. AIDG133V has impaired genome-wide chromatin localization.

(A) Heat map displaying ChIP-seq peaks that were significantly enriched in WT versus either AicdaGV/GV, Aicda−/−, or both. Each row represents a peak region, and each tile within the row represents normalized read counts averaged from 100bp bins spanning +/− 2.5kb from the peak center. Significance of peak enrichment indicated by green shading, * p < 0.05. Gene names shown with corresponding peak number, purple text indicates a previously identified AID target verified by either deep-sequencing of AID-dependent somatic mutations (Alvarez-Prado et al., 2018; Klein et al., 2011; Liu et al., 2008; Pavri et al., 2010; Yamane et al., 2011), or as an AID-dependent translocation partner (Chiarle et al., 2011; Klein et al., 2011). (B) Normalized read count tracks for selected genes. Dashed lines denote peak regions; closed rectangles denote exons; arrows indicate direction of transcription. (C) Mean signal of all significantly enriched regions described in (A) relative to the closest transcriptional start site (TSS). Data in (A-C) are from 4 independent experiments with 1 mouse per genotype. AicdaGV/GV, AicdaG133V/G133V. Complete ChIP-seq data shown in Table S2–4.
As expected and previously reported, we detected AIDWT enriched at two Igh S regions (Ighm-Sμ and Ighg1-Sγ1) that are targeted by AID as sites of IgG1 CSR following LPS plus IL-4 stimulation (Figure 6A,B) (Yamane et al., 2011). We also detected peaks at the Igl and Igk light chain genes; the higher fold-change and significance of the Igk peak versus the Igl peak is consistent with the observation that over 90% of mouse B cells express κ light chains (Eisen and Reilly, 1985; LeJeune et al., 1982; Woloschak and Krco, 1987) (Table S5). Genome-wide AID occupancy is highly correlated with RNA polymerase II, peaking around transcriptional start sites (TSS) (Pavri et al., 2010; Yamane et al., 2011). Similarly, we found that the mean AID signal at significantly enriched regions dramatically increased immediately 3’ of TSSs (Figure 6C).
Many of the non-Ig peaks identified, such as Gas5, mir142, and Il4rα, were among the top AID targets in a previous ChIP-seq analysis (Yamane et al., 2011). Additionally, 11/37 peaks were previously identified as AID-dependent c-myc or Igh translocation partners (Chiarle et al., 2011; Klein et al., 2011), 18/37 have been validated as AID hypermutation targets via sequencing analysis of ex vivo activated B cells (Klein et al., 2011; Pavri et al., 2010; Yamane et al., 2011) or sorted GC B cells (Alvarez-Prado et al., 2018; Liu et al., 2008), and 17/37 were identified as AID-dependent sites of RPA recruitment (Qian et al., 2014) (Table S5). The identification of numerous AID targets that have been verified across multiple experimental approaches indicates that novel targets uncovered by our ChIP-seq analysis may reveal overlooked aspects of AID biology.
AID localizes to MHCII genes
ChIP-seq revealed that 3 of the top 10 AIDWT targets are the MHCII genes H2-Aa, H2-Eb1 and the invariant chain Cd74. Additionally, H2-Dma was significantly enriched and H2-Ab1 was near the significance cut-off (Table S5). Revisiting a previous AID ChIP analysis, we found that H2-Aa, H2-Eb1, and H2-Ab1 ranked within the top 4%, and H2-Dma within the top 14%, of AID targets (Yamane et al., 2011). To determine if MHCII genes are hypermutated by AID, we cross-referenced two previous studies that performed DNA deep-sequencing to identify AID targets in GC B cells isolated from either Ung−/−Msh2−/− or Aicda−/− mice (Alvarez-Prado et al., 2018; Liu et al., 2008). Due to their inability to remove dG:dU mismatches, Ung−/−Msh2−/− B cells are commonly used to trap AID deamination events. Both studies found Cd74 mutation rates that were 10-fold higher in Ung−/−Msh2−/− B cells versus Aicda−/− controls (Table S6) (Alvarez-Prado et al., 2018; Liu et al., 2008). Additionally, CD74 is thought to be a target of recurrent AID SHM in DLBCL samples (Khodabakhshi et al., 2012), and was identified as a site of AID-dependent RPA recruitment (Qian et al., 2014). Thus, while the data from two independent AID ChIP-seq analyses suggest AID localizes to multiple MHCII genes in ex vivo activated B cells (Figure 6, (Yamane et al., 2011)), we conclude that Cd74 is the only MHCII AID target currently known to be mutated in GC B cells.
AID expression correlates with decreased MHCII expression in mouse GC B cells
Given that GC B cells primarily express AID within the DZ (Greiner et al., 2005; Moldenhauer et al., 2006; Victora et al., 2012), and MHCII expression is downregulated in DZ versus LZ B cells (Bannard et al., 2016; Victora et al., 2012), we hypothesized that AID targeting could negatively regulate MHCII expression. To test this hypothesis, we immunized WT, Aicda+/−, and Aicda−/− mice intraperitoneally with UV inactivated IAV, and assessed MHCII surface levels in LZ and DZ GC B cells 12 days post-immunization. Similar to homeostatic and IAV infection-induced GCs, Aicda−/− mice were characterized by GC hyperplasia and an increase in the frequency of LZ B cells (Figure 7A, S6A,B), while all genotypes had a similar frequency of HA+ GC B cells (Figure S6C,D).
Figure 7. AID expression correlates with decreased MHCII expression in mouse GC B cells and human DLBCL.

(A-D) MHCII expression in germinal center (GC) B cells. Mice were immunized intraperitoneally with UV-inactivated influenza A virus and analyzed at d12 post-immunization. (A) Representative GC B cell gates (GL7+Fas+ of B220+ live cells) (top), and GC dark zone (DZ) (CXCR4hiCD86lo of GL7+Fas+B220+ live cells) and light zone (LZ) (CXCR4loCD86hi of GL7+Fas+B220+ live cells) gates (bottom) in the spleen (Quantified in Figure S6A,B). (B) Representative histograms depicting MHCII surface staining in LZ and DZ GC B cells, and representative MHCIIlo gate (%MHCIIlo of DZ or LZ GC B cells). (C) Quantification of MHCIIlo frequency within DZ and LZ GC B cells. (D) Quantification of MHCII MFI ratio, calculated as DZ MHCII MFI/LZ MHCII MFI (MFI values quantified in Figure S6F). (E) Violin plots depicting AICDA expression values within activated B cell-like (ABC), germinal center B cell-like (GCB), or unclassified human diffuse large B-cell lymphoma (DLBCL) tumors. Box and whiskers plot displayed inside; TPM, transcripts per million. (F) Volcano plots depicting RNA-seq data, analyzed as TPM, from ABC, GCB or unclassified human DLBCL tumors. Log2 fold change (top 10% of AID expressing samples (AIDtop10%) versus the bottom 90% (AIDbot90%)) shown on the x-axis and –log10 (p-value) on the y-axis. Circles represent genes plotted within boundaries; triangles represent genes that are off the axis, plotted at the nearest point within the boundaries. Green, MHCII genes; red, control genes; purple, AICDA and CIITA. Dotted lines mark p-value = 0.05 and log2(fold change) = ± 0.5. Scatter plots correlating AICDA and MHCII expression, analyzed as transcripts per million (TPM), shown in Figure S7. Data in (A-D) are from 3 independent experiments with 2–6 mice per genotype. Error bars represent the mean ± std. dev. **p < 0.01; ***p < 0.001; ****p < 0.0001. p-values calculated using a one-way ANOVA with Tukey’s multiple comparisons test (C-E), or from DEseq analysis (F) (see methods for details).
As previously reported in WT mice (Bannard et al., 2016; Victora et al., 2012), we observed a distinct population of DZ B cells expressing low levels of MHCII (MHCIIlo) in comparison to their LZ counterparts. This MHCIIlo population was clearly diminished in Aicda−/− mice (Figure 7B). Indeed, the frequency of MHCIIlo DZ B cells was nearly 4-fold greater in WT vs. Aicda−/− mice, and 2-fold greater in Aicda+/− vs. Aicda−/− mice, whereas the frequency of MHCIIlo LZ B cells was close to zero for all genotypes (Figure 7C). To control for considerable fluctuations in MHCII MFIs across different mice and experiments (Figure S6E,F), we calculated the ratio of DZ/LZ MFIs per mouse. While the DZ MHCII MFI was ~65% of the matched LZ MFI in WT mice, this ratio increased significantly to ~80% in Aicda−/− GCs (Figure 7D). Similar to the frequency of MHCIIlo DZ B cells, the DZ/LZ MHCII MFI ratio scaled with the increasing amounts of AID present within Aicda−/−, Aicda+/−, and WT GC B cells. Thus, it appears that MHCII expression is inversely correlated with AID levels in GC B cells.
Human DLBCLs with the highest AID levels have decreased MHCII expression
DLBCL, the most common human lymphoma, is fatal in approximately 30% of patients (Pasqualucci and Dalla-Favera, 2018). DLBCLs can be classified into three subgroups based on gene expression profiling: activated B cell-like (ABC), germinal center B cell-like (GCB), or unclassified (Alizadeh et al., 2000; Rosenwald et al., 2002). DLBCLs frequently express AID (Arima et al., 2018; Greeve et al., 2003; Kawamura et al., 2016; Lossos et al., 2004; Pasqualucci et al., 2004), and ABC tumors generally express the highest levels of AID versus the other subgroups (Lossos et al., 2004; Pasqualucci et al., 2004). Given that DLBCLs also have varying levels of MHCII expression (Miller et al., 1988; Momburg et al., 1987; Rimsza et al., 2004; Rosenwald et al., 2002), we hypothesized that the inverse correlation between AID expression and MHCII observed in GC B cells might also occur in DLBCLs. To test this, we analyzed RNA-sequencing (RNA-seq) data generated from 562 DLBCL biopsy samples, consisting of 286 ABC, 162 GCB, and 114 unclassified tumors (Schmitz et al., 2018).
As expected, the ABC tumors had ~3-fold higher AICDA expression versus GCB tumors, and ~2.5 fold higher expression versus unclassified tumors (Figure 7E). We correlated AICDA expression with MHCII expression amongst each DLBCL subgroup, and observed that DLBCLs with the highest levels of AID expression appeared to be enriched for low expression of several MHCII presentation pathway genes, particularly amongst ABC tumors (Figure S7A–C). To quantify this, we performed differential expression analyses comparing MHCII genes amongst the top 10% of AID expressing samples (AIDtop10%) versus the bottom 90% (AIDbot90%) within each subgroup.
11 MHCII genes were significantly decreased in the AIDtop10% within ABC tumors, including CD74, HLA-DQA2 (H2-Aa), HLA-DMA (H2-Dma), whereas the housekeeping genes ACTB and GAPDH were similarly expressed in the AIDtop10% and AIDbot90% tumors (Figure 7F). Interestingly, the MHCII transactivator (CIITA) was also significantly decreased in the AIDtop10% within the ABC tumors, consistent with AID targeting of CIITA previously reported in primary mediastinal large B-cell lymphoma (Mottok et al., 2015) and a previous AID ChIP analysis (Yamane et al., 2011). Additionally, Ciita has also been identified as a site of AID-dependent RPA recruitment (Qian et al., 2014). In contrast, GCB tumors had only 4 MHCII genes that were significantly decreased, and the unclassified tumors had none. Thus, these data indicate that ABC DLBCLs with the highest AID levels exhibit decreased expression of multiple MHCII presentation pathway genes.
DISCUSSION
Nearly 25 years after the structural basis for G4 nucleic acids was first proposed (Gellert et al., 1962), Igh S regions were amongst the first DNA sequences shown to self-associate into G4 structures (Sen and Gilbert, 1988). Over the last decade, G4 biology has become an exciting area of investigation in myriad biological processes (Hansel-Hertsch et al., 2017), including recent work implicating G4s as critical effectors of class switch recombination and AID biology (Qiao et al., 2017; Zheng et al., 2015). Given the lack of models currently available to study AID-G4 binding function in vivo, we generated AicdaGV/GV mice.
We found that AicdaGV/GV mice completely lack CSR and SHM due to a broad, drastic defect in genome-wide AIDG133V chromatin localization. Crucially, while AIDG133V has impaired G4-binding, it is catalytically active both in vitro and in ex vivo activated B cells. Importantly, overexpression of AIDG133V does not restore any detectable CSR, suggesting that the 30% decrease in AIDG133V expression is not limiting. While the lower AIDG133V expression may reflect an intrinsic difference in protein stability compared to AIDWT, the ability of the AIDG133V-AIDCD fusion to restore significant levels of CSR indicate that AIDG133V is sufficiently stable to deaminate S regions if properly targeted. Further, the inability of the AIDG133V-mCherry fusion to restore CSR indicates that a C-terminal protein fusion cannot simply stabilize an otherwise unstable AIDG133V. Thus, to the extent of our current knowledge, AIDG133V largely separates DNA deamination from G4 binding, and can be used to parse the importance of G4 binding to AID function.
Given this separation of function, the failure of AIDG133V to localize to Sμ, coupled with the observations that 1) transcription of S regions triggers the formation of DNA G4s (Carrasco-Salas et al., 2019; Duquette et al., 2004; Neaves et al., 2009), and 2) G4s can bind purified AID with high affinity (Duquette et al., 2005; Qiao et al., 2017), indicate that S region G4s may target AID in cis, or trans via an RNA-guided mechanism analogous to CRISPR-Cas9 targeting (Zheng et al., 2015). Importantly, G4-mediated targeting in cis or trans are not mutually exclusive, and could also involve an S region RNA/DNA hybrid G4. Notably, AID also targets to the G4-forming telomeric repeats (Cortizas et al., 2016).
Replacement of the endogenous mouse Sγ1 region with 4kb of the AT-rich Xenopus Sμ region is sufficient to promote CSR (Zarrin et al., 2004), implying that AID targeting to S regions does not absolutely require AID binding to G4 sequences at the targeted S region. Until recently, this observation was clearly inconsistent with a G4-mediated AID targeting model for S regions, and the complete absence of CSR in AicdaGV/GV B cells. However, a recent study possibly reconciles these inconsistencies by proposing Igh chromatin loop extrusion as a mechanism that juxtaposes downstream S regions with Sμ, creating a class switch recombination center (CSRC) (Zhang et al., 2019). In this light, efficient AID targeting to Sμ and the formation of the CSRC could endow targeting to the downstream G4-deficient Xenopus Sγ1 region. Such a mechanism could also account for the lack of SHM within the Jh4 intron, as V(D)J segments are not predisposed to form G4s, and the Sμ region is merely 3–4 kb away from the rearranged V(D)J segments (depending on the utilized J segment). Thus, SHM at the Igh locus may rely upon efficient AID localization to Sμ, which is defective in AicdaGV/GV B cells. Therefore, G-rich proximal sequences, as well as the 3-dimensional chromatin architecture exemplified by the CSRC, may affect the efficiency of AID targeting to the Igh locus.
Outside of the Igh locus, the computationally predicted propensity of a sequence to form G4s has been suggested to correlate with genome-wide AID targeting (Duquette et al., 2007; Zheng et al., 2015). However, the mutation frequency of over 100 genes expressed in GC B cells does not correlate with G-richness (Liu et al., 2008). It is possible that chromatin looping interactions, such as those that establish the CSRC, play a role in genome-wide AID chromatin localization. Alternatively, G4 binding could precipitate the formation of large AID-G4 oligomers (Qiao et al., 2017), and AIDWT, but not AIDG133V, forms high molecular weight, RNA-dependent complexes containing multiple RNA-binding proteins (Mondal et al., 2016). The formation of a G4-dependent high-avidity AID complex could thus function to increase AID binding affinity to target loci. An intriguing alternative possibility is that AID targeting depends on a G4-dependent phase separation of AID and other binding partners. While AID is predicted to have only a small disordered region at its N-terminus, its various binding partners could potentially provide larger disordered regions that associate into phase-separated complexes. Further investigation is required to elucidate the molecular requirements for G4 binding in localizing AID to S regions, as well as other prominent non-Ig loci.
A central question in AID biology is the extent to which SHM extends to non-Ig genes, and how this affects B cell function. AID ChIP revealed localization to multiple MHCII genes, and two previous studies found that Cd74 is mutated by AID in GC B cells (Alvarez-Prado et al., 2018; Liu et al., 2008). This lead to the hypothesis that AID directly regulates MHCII gene expression in GC B cells, as DNA DSBs and deamination can regulate transcription (Kim et al., 2016; Pankotai et al., 2012; Periyasamy et al., 2015; Shanbhag et al., 2010). Consistent with this, we found that first, AID expression correlates with decreased MHCII surface staining within DZ GC B cells, and second, human ABC DLBCLs with the highest AID levels exhibit decreased expression of multiple MHCII presentation pathway genes. Notably, only DLBCLs with the highest AID expression approach physiological AID levels found within GC B cells (Lossos et al., 2004). Taken together, these data suggest there may be a threshold for AID expression that is required for AID-dependent gene regulation, and while this threshold is routinely achieved in GC B cells, it is reached only in a fraction of ABC tumors. It is also important to note that DLBCLs have frequent genetic alterations (Wright et al., 2020) that could lead to AID-independent regulation of MHCII genes.
AID-dependent regulation of MHCII expression has critical pathological implications, particularly for DLBCLs, where loss of MHCII expression is highly correlated with decreased patient survival (Rimsza et al., 2004). MHCII molecules enable CD4+ T cell immunosurveillance of B cell lymphomas, and DLBCLs with low MHCII have severely diminished numbers of tumor infiltrating lymphocytes (Rimsza et al., 2004). MHCII molecule transcription is regulated by CIITA, which is frequently translocated or mutated in DLBCLs, and has been proposed to be a target of aberrant AID SHM (Khodabakhshi et al., 2012; Mottok et al., 2015; Steidl et al., 2011). In addition to our finding decreased CIITA expression in the AIDtop10% ABC tumors, Ciita ranked within the top 1% of AID targets in a previous ChIP analysis (Yamane et al., 2011), and was identified as a site of AID-dependent RPA recruitment (Qian et al., 2014). Thus, in addition to directly blocking MHCII gene transcription, AID may also regulate MHCII transcription by targeting CIITA.
While non-Ig AID targets are generally considered to be collateral damage incurred during BCR secondary diversification, we venture that AID-dependent regulation of gene expression is a physiological adaption in B cell responses. Alterations in MHCII expression would be expected to modulate T cell help within the GC, a critical factor in B cell expansion and affinity maturation. Intriguingly, another critical regulator of GC B cell biology, Bcl6, is not only the most highly mutated non-Ig AID target, but is also thought to be selectively permissive to DNA damage (Liu et al., 2008; Shen et al., 1998; Ye et al., 1993). Furthermore, the levels of Bcl6 mRNA are increased 10-fold in Aicda−/− vs. WT DLBCL cell lines (Jiao et al., 2020), hinting at a role for AID-dependent regulation.
AID-dependent gene regulation of MHCII and Bcl6 could affect the probability of a B cell to leave the GC, acting as rheostat to prevent B cells from staying in an environment that is dangerously permissive to DNA damage. The observations that 1) Aicda−/− GC B cells do not efficiently differentiate into plasma cells (Boulianne et al., 2013), and 2) LZ GC B cells expressing low levels of BCL6 are thought to favor GC exit and differentiation into plasma cells (Ise et al., 2018), suggest that AID may play a critical role regulating GC B cell fates. Furthermore, the absence of this regulation could contribute to the GC B cell hyperplasia observed in Aicda−/− mice. Importantly, AID-deficient mice and CRISPR-Cas9 technology will allow many of these ideas to transition into testable hypotheses, making AID-dependent regulation of gene expression an exciting avenue for future investigation in physiological and pathological B cells.
LIMITATIONS OF STUDY
There are a few key limitations to the interpretations of the data presented herein. Firstly, the inherent difficulties in purifying active full-length mouse AID impaired our ability to thoroughly assess AID catalytic activity. Mutations and truncations are commonly made to stabilize purified AID proteins in order to produce maximally active preps, and mouse AID in particular is notoriously finicky. We prioritized controlling for as few perturbations as possible when comparing AIDWT and AIDG133V. Thus, it is possible that our biochemical analysis was not sensitive enough to detect subtle differences between AIDWT and AIDG133V catalytic activity in vitro. Secondly, while there is a reasonable amount of evidence demonstrating that S region sequences can form G4 structures, this is not the case for the Ig variable region genes or the non-Ig loci identified in our AID ChIP-seq analysis. While G4s generated by S region DNA or RNA may function to target AID to S regions, further investigation is required to determine the G4-dependent mechanisms of AID targeting to loci whose underlying sequences may not support G4 formation. Thirdly, while we focused on disrupting AID-G4 binding in vivo via the AIDG133V mutation, mutating genomic G4 sequences and assessing AIDWT targeting in vivo would strengthen the conclusions of this study. Additionally, sufficiency experiments in which the ability of G4 structures to recruit AID to an ectopic locus is tested would further bolster the conclusions of this study. Lastly, further studies are required to test the proposed mechanisms underlying the model for AID-dependent regulation of MHCII gene expression.
STAR METHODS
RESOURCE AVAILIABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Jayanta Chaudhuri, (chaudhuj@mskcc.org)
Materials Availability
All materials generated in this study are available upon request with a completed Materials Transfer Agreement.
Data and Code Availability
ChIP-seq datasets generated during this study are available at the Gene Omnibus Repository under accession code GSE136959.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Mice
AicdaGV/GV mice were generated at the MSKCC transgenic core facility using CRISPR-Cas9 targeted mutagenesis and the following oligos: G133V gRNA, G133V donor oligo. Two independent founders were identified by DNA sequencing of the Aicda locus, and backcrossed to the C57BL/6J background. All mice used in subsequent experiments were derived from these 2 founder lines. AicdaGV/GV mice were genotyped using sG133V and asG133V primers. PCR products were digested with SfcI and AvaII restriction enzymes to differentiate between WT and AicdaGV/GV alleles. Aicda−/− mice were a kind gift from Dr. T. Honjo, and were generated from homozygous breeders. Experiments were conducted using 3–5 month old littermate controls, or age matched controls when littermates were not available. Mice were housed under specific pathogen-free conditions and handled in accordance with the guidelines for animal care of MSKCC Research Animal Resource Center and the Institutional Animal Care and Use Committee (IACUC).
Primary B cell ex vivo CSR assays
Splenic single cell suspensions were prepared from ~12 week-old mice by mashing through a 70μm cell strainer (352350, Corning). Cells were collected by centrifugation and resuspended in 2mLs of red blood cell lysis buffer for 5 min at 25°C, followed by neutralization with B cell media. Naive B cells were purified by negative selection using anti-CD43 magnetic beads (130–049-801, Miltenyi Biotec) according to the manufacturer’s protocol. 2 × 106 B cells were plated at a density of 1 × 106 cells/mL in a 6-well dish and cultured in B cell media. B cells were immediately stimulated with one of the following conditions: LPS plus IL-4, 30 μg/mL LPS (L4130, Sigma), 25 ng/mL IL-4 (404-ML-010, R&D systems); LPS, 30 μg/mL LPS (L4130, Sigma); BAFF plus retinoic acid (RA) plus IL-4 plus TGFβ plus IL-5 plus LPS, 20 ng/mL BAFF (AG-40B-0022, Adipogen), 10 nM RA (R2625, Sigma),10 ng/mL IL-4 (404-ML-010, R&D systems), 10 ng/mL TGFβ (240-B-010, R&D systems), 10 ng/mL IL-5 (215–15, Peprotech), 10 μg/mL LPS (L4130, Sigma). Cell cultures were split 1:2 and 1:3 at 48 hrs and 72 hrs post-stimulation, respectively. B cells were harvested at 48 hrs to purify total RNA for quantitative PCR (qPCR) analysis, or at 72 hrs and 96 hrs for flow cytometry and immunoblotting analysis.
Cell lines
HEK293T cells were cultured in DMEM (11965118, Gibco) supplemented with 10% FBS and 1% Pen-Strep (400–109, Gemini), and were used for the production of retroviruses. See “B cell retroviral infections” below.
METHOD DETAILS
Infections and immunizations
Influenza A/Puerto Rico/8/34 (PR8) and J1 were grown in 10 day-old embryonated chicken eggs. For intranasal infections, mice were anesthetized with 3% isofluorane and nasally inoculated with 50 TCID50 of PR8 or J1 diluted in PBS plus 0.1% BSA.
For NP-CGG immunizations, NP(33)-CGG (N-5055D-5, Biosearch Technologies) was precipitated with Imject alum adjuvant (77161, Thermo Scientific). Briefly, NP(33)-CGG was resuspended to a final concentration of 1 mg/mL in PBS. 2 mLs of Imject alum was added dropwise to 2 mLs of NP(33)-CGG, and mixed by vortexing for 30 min at 25°C. Mice were immunized intraperitoneally with 200 μL (100 μg NP(33)-CGG) of this solution. Blood was collected at day 0,7,14,21, and 28 post-immunization by submandibular bleeding, and serum was isolated using BD microtainer SST blood tubes (365967, BD Biosciences) and stored at −80°C.
For SRBC immunizations, packed 10% SRBCs (ISHRBC10P15ML, Innovative Research) were diluted to 1×109 cells/mL in PBS, mice were immunized intraperitoneally with 200 uL (2×108 cells), and analyzed at day 7 post-immunization.
For IAV immunizations, PR8 was inactivated by exposing to 254nm UV-C light using an 8 watt MRL-58 multiple-ray lamp (95–0313-01, Analytikjena) with a shortwave, 254nm, 8 watt bulb (34–0007-01, Analytikjena). Briefly, 1mL of PR8 allantoic fluid (with a titer of ~1.2 × 109 TCID50/mL) was placed into 1 well of a 6-well dish on ice, and exposed to UV-C light for 10 min, at a distance of 6 inches from the bulb, with shaking every 2 min. To assess infectivity post-inactivation, TCID50 values were calculated using the Reed and Muench method. To ensure UV-inactivation did not overtly damage the PR8 HA, hemagglutination assays were performed and compared to non-inactivated samples. Mice were immunized intraperitoneally with 180uL of UV-inactivated PR8, and the spleens were analyzed at day 12 post-immunization.
Flow cytometry
Single cell suspensions were prepared from mouse lymph nodes, spleen, Peyer’s patches, or thymus by mashing through a 70μm cell strainer (352350, Corning), and bone marrow was flushed from the tibia bone using a syringe. Single cell suspensions from the spleen and bone marrow were resuspended in 2mLs of red blood cell lysis buffer (150 mM NH4Cl, 10 mM KHCO3, 0.1 mM EDTA) for 5 min at 25°C, followed by neutralization with B cell media (RPMI 1640 plus L-Glutamine (11875, Gibco), supplemented with 15% FBS, 1% Pen-Strep (400–109, Gemini), 55 μM β-Mercaptoethanol (21985023, Gibco), and 2 mM L-Glutamine). Approximately 5–10 × 106 cells were washed once with PBS, stained with Zombie Red fixable viability dye (423109, BioLegend, 1:500) for 15 min at 25°C, washed with once with FACS buffer (PBS + 2.5% FBS), stained with rat anti-mouse CD16/CD32 Fc Block (553142, BD Biosciences, 1:50) for 5 min at 25°C, and washed once with FACS buffer before staining with the following antibody cocktails at 4°C for 30 min. Splenic B cell development stain: B220-BV786 (563894, BD Biosciences, 1:750), IgM-BUV395 (743329, BD Biosciences, 1:750), IgD-AF700 (405729, BioLegend, 1:750), CD43-FITC (11–0431-82, eBioscience, 1:300), CD21-eFluor450 (48–0212-82, eBioscience, 1:750), CD23-PE-Cy7 (25–0232-81, eBioscience, 1:750), IgG1-BV510 (742476, BD Biosciences, 1:750), IgA-PE (1040–09, Southern Biotech, 1:750), CD38-PerCP-eFluor710 (46–0381-80, eBioscience, 1:750), CD19-APC-H7 (560143, BD Biosciences, 1:400). Bone marrow B cell development stain: B220-BV786 (563894, BD Biosciences, 1:750), IgM-BUV395 (743329, BD Biosciences, 1:750), CD25-APC-eFluor780 (47–0251-82, eBioscience, 1:500), IgD-BV510 (405723, BioLegend, 1:750), CD138 -PE-Cy7 (142514, BioLegend, 1:500), CD21-eFluor450 (48–0212-82, eBioscience, 1:750), CD19-AF700 (115528, BioLegend, 1:750), CD43-FITC (11–0431-82, eBioscience, 1:300), cKit-APC (17–1171-81, eBioscience, 1:100). Thymus T cell stain: B220-BV786 (563894, BD Biosciences, 1;750), CD3ε-eFluor450 (48–0032-80, eBioscience, 1:750), CD8-FITC (35–0081, Tonbo Biosciences, 1:500), TCRβ-AF700 (109224, BioLegend, 1:750), CD4-PerCP-Cy5.5 (45–0042-80, eBioscience, 1:400), CD62L-PE-Cy7 (25–0621-82, eBioscience, 1:400), CD69-APC (17–0691-82, eBioscience, 1:500), CD44-PE (12–0441-83, eBioscience, 1:400). Homeostatic GC B cell stain: B220-BV786 (563894, BD Biosciences, 1:750), IgM-BUV395 (743329, BD Biosciences, 1:750), IgD-APC-eFluor780 (47–5993-80, eBioscience, 1:750), IgG1-BV510 (742476, BD Biosciences, 1:750), Fas-PE-Cy7 (557653, BD Biosciences, 1:750), GL7-eFluor450 (48–5902-80, eBioscience, 1:750), CD38-AF700 (56–0381-82, eBioscience, 1:750), IgG2c-FITC (1079–02, Southern Biotech, 1:200), CD19-PerCP-Cy5.5 (45–0193-80, eBioscience, 1:400), IgA-PE (1040–09, Southern Biotech, 1:750). Influenza GC B cell stain (infections): B220-BV786 (563894, BD Biosciences, 1:750), IgM-BUV395 (743329, BD Biosciences, 1:750), IgG2b-APC-Cy7 (1090–19, Southern Biotech, 1:150), IgG1-BV510 (742476, BD Biosciences, 1:750), Fas-PE-Cy7 (557653, BD Biosciences, 1:750), GL7-eFluor450 (48–5902-80, eBioscience, 1:750), CD38-AF700 (56–0381-82, eBioscience, 1:750), IgG2c-FITC (1079–02, Southern Biotech, 1:200), CD86-PE (12–0862-82, eBioscience, 1:5,000), IgD-BV711 (405731, BioLegend, 1:750), CXCR4-PerCP-eFluor710 (46–9991-82, eBioscience, 1:250), biotinylated HA trimer (recombinant protein from A.B.M., final concentration = 2.3nM), streptavidin-APC (405207, BioLegend, 1:1700). Influenza GC B cell stain (immunizations): IgM-BV786 (743328, BD Biosciences, 1:750), CD86-BUV395 (564199, BD Biosciences, 1:4000), IgG2b-APC-Cy7 (1090–19, Southern Biotech, 1:150), IgG1-BV510 (742476, BD Biosciences, 1:750), Fas-PE-Cy7 (557653, BD Biosciences, 1:750), GL7-eFluor450 (48–5902, eBioscience, 1:750), CD38-AF700 (56–0381, eBioscience, 1:750), IgG2c-FITC (1079–02, Southern Biotech, 1:200), MHC Class II (I-A/I-E)-APC (17–5321-81, eBioscience, 1:1000), Rat IgG2b-APC, k isotype control (400612, BioLegend, 1:1000), IgD-BV711 (405731, BioLegend, 1:750), CXCR4-PerCP-eFluor710 (46–9991-82, eBioscience, 1:250), B220-BUV737 (612838, BD Biosciences, 1:750), biotinylated HA trimer (recombinant protein from A.B.M., final concentration = 2.3nM), streptavidin-PE (405204, BioLegend, 1:1700). Ex vivo CSR stain: B220-BV786 (563894, BD Biosciences, 1:750), IgM-BUV395 (743329, BD Biosciences, 1:750), IgD-APC-eFluor780 (47–5993, eBioscience, 1:750), IgG1-BV510 (742476, BD Biosciences, 1:750), IgG3-FITC (553403, BD Biosciences, 1:300), IgA-PE (1040–09, Southern Biotech, 1:750).
Data was acquired using an LSR II flow cytometer (BD Biosciences), and analyzed using FlowJo software (version 9.9).
B cell retroviral infections
Retroviruses were prepared by co-transfecting 30 μgs of various pMIG vectors with 20 μgs of the packaging vector pCL-Eco per 10 cm dish of 50–70% confluent HEK293T cells by the calcium phosphate method. 10 mLs of retroviral supernatant were collected from per 10 cm dish of HEK293T cells at 48 hrs and 72 hrs post-transfection, and polybrene was added to a final concentration of 8 μg/mL. 2 × 106 purified naïve splenic B cells were plated at a density of 1 × 106 cells per mL in B cell media and were immediately stimulated with 30 μg/mL LPS (L4130, Sigma) plus 25 ng/mL IL-4 (404-ML-010, R&D systems). After stimulation for either 24 hrs or 48 hrs, media were aspirated, leaving approximately 1 mL of media per well of a six-well dish, and 3 mLs of retroviral supernatant (out of 10 mLs total per 10cm dish of transfected HEK239T cells) were added to each well of a six-well dish. Six-well dishes were spun at 2,000 g for 90 min at 32°C, after which viral supernatants were aspirated and fresh B-cell media plus LPS and IL-4 was added.
B cell lysates
Lysates were prepared from B cell pellets stored at −80°C or from B cell cultures collected by centrifugation. Approximately 5 × 106 B cells were resuspended in 200 μL B cell lysis buffer (0.5% NP-40, 50 mM Tris pH 7.4, 200 mM NaCl, 1 mM EDTA, 1 mM Benzamidine HCl (B6506, Sigma), 1 mM PMSF (36978, Thermo Scientific), 5 μM β-mercaptoethanol, and 1 cOmplete, Mini, EDTA-free Protease Inhibitor Cocktail tablet per 10 mL buffer (11836170001,Sigma)), and incubated on ice for 30 min. Lysates were sonicated using a Branson digital sonifier with a microtip until no longer viscous, and protein concentrations were measured by Bradford assay (5000001, Bio-Rad). 4x protein sample buffer (10% SDS, 50% glycerol, 0.05% Bromophenol blue, 200 mM Tris pH 6.8, 40 mM EDTA, 20% β-mercaptoethanol) was added to a final concentration of 1x, samples were boiled at 98°C 10 min and stored at −20°C.
Nuclear-cytoplasmic fractionation
Nuclear and cytoplasmic lysates were prepared at 72 hrs following stimulation of purified naïve splenic B cells with LPS plus IL-4 using the NE-PER Nuclear and Cytoplasmic Extraction Reagents (78835, Thermo Scientific), and according to the manufacturers protocol. Briefly, approximately 5 × 106 B cells were resuspended in 200 μL of CER I buffer, and incubated on ice for 10 min. 11 μL of CER II buffer was added, samples were vortexed at max speed for 5 sec, incubated on ice for 1 min, vortexed at max speed for 5 sec, and centrifuged for 5 min at 16,000 g at 4°C. The cytoplasmic fraction contained in the supernatant was transferred to a clean 1.5 mL tube, 4x protein sample buffer was added to a 1x final concentration, samples were boiled at 98°C for 10 min and stored at −20°C. The remaining pellet was resuspended in 100 μL NER and incubated at 4°C for 40 min, with 15 sec of max speed vortexing every 10 min. Samples were centrifuged at 16,000 g for 10 min at 4°C, and the nuclear fraction contained in the supernatant was transferred to a clean 1.5 mL tube. 4x protein sample buffer was added to a 1x final concentration, samples were boiled at 98°C for 10 min and stored at −20°C.
Immunoblotting
SDS-polyacrylamide gels were transferred to a PVDF membrane (IPVH00010, Millipore) using a Trans-Blot SD Semi-Dry Transfer Cell (1703940, Bio-Rad) for 1 hr at 19 V. Immunoblots were blocked with PBST (PBS, 0.1% Tween 20) plus 4% non-fat dry milk (NC9121673, Fisher) for 30 min at 25°C, and all subsequent incubations were carried out in this solution. Rabbit anti-AID polyclonal antibodies were generated by Covance. The following antibodies were used for immunoblotting: rabbit anti-AID (Chaudhuri Lab, (Chaudhuri et al., 2003), 1:500), mouse anti-HSP90 (MAB3286, R&D systems, 0.25 μg/mL), rabbit anti-Lamin-B1 (PA5–19468, ThermoFisher Scientific, 0.2 μg/mL), mouse anti-α-Tubulin (T9026, Sigma, 1:2,000), mouse anti-MBP (E8032, NEB, 1:10,000). Primary antibodies were incubated for ~12 hrs at 4°C, with the exception of anti-HSP90, which was incubated for 1 hr at 25°C.
qPCR
RNA was isolated from purified naïve splenic B cells stimulated with LPS plus IL-4 for 48 hrs using the TRIzol reagent (15596026, Invitrogen) according to the manufacturers protocol. RNA concentration and purity was assessed using a NanoDrop spectrophotometer. cDNA was synthesized from 1 μg of purified total RNA using the SuperScript First-Strand Synthesis System (18080051, Invitrogen) according to the manufacturers protocol, and using random hexamers for priming. All qPCR experiments were performed in a 384-well format using an Applied Biosystems QuantStudio 6 Flex instrument. Briefly, 1 μL of cDNA (5% of total) was used with the PowerUp SYBR Green Master Mix (A25741, Applied Biosystems) according to the manufacturers protocol, and using the following primer pairs: Imu F and Cmu R for μGLTs, Ig1 F and Cg1 R for γ1GLTs, sAID and asAID for Aicda mRNA, sAct and asAct for Actb mRNA. Data was normalized to Actb mRNA expression, and to the average of WT samples for each experiment.
Immunofluorescent imaging and quantification
Spleen samples were embedded in optimal cutting tissue reagent (4583, Sakura). Tissue sections of 6 μm thickness were fixed in 4% paraformaldehyde and stained with primary antibodies, followed by appropriate secondary reagents. Primary antibodies: anti-IgD (1120–01, Southern Biotech), PNA (B-1075, Vector Laboratories), anti-B220 (ab64100; Abcam). Secondary antibodies: donkey anti-rat AlexaFluor 488 (A21208, Invitrogen), donkey anti-rat 594 (A21209, Invitrogen), Streptavidin-Cy5 (016–170-084, Jackson ImmunoResearch). Primary antibodies with irrelevant binding activity and the appropriate secondary reagents were used to validate the specificity of tissue staining. In all cases, nuclear DNA was stained with DAPI (4’, 6-diamidine-2’-phenylindole dihydrochloride; D1306, Invitrogen). Coverslips were applied with FluorSave (345789–20, Calbiochem). Slides were scanned with Panoramic Flash (3DHistech) using 203/0.8NA objective, and regions of interest were drawn manually using CaseViewer (3DHistech) and exported into TIFF files. Raw unedited images were then analyzed using ImageJ/FIJI, by which the area of the interest was measured. In all cases, DAPI channel of the scan was used as a mask to exclude artifacts outside the tissue. Signals of interest were thresholded, and the area and count were measured. Scoring of desired area was done manually with randomly shuffled pictures to reduce bias.
MBP-AID expression
WT, AIDG133V and AIDCD sequences were cloned into the pMAL-c5X vector with a modified MCS using AscI and PacI restriction sites. BL21(DE3) competent E. coli cells (C2527, NEB) were transformed with MBP-AID plasmids and the trigger factor chaperone plasmid (courtesy of Kohli Lab, via LiChung Ma and Gaetano T. Montelione) (Kohli et al., 2009). Colonies were picked and cultured at 37°C in 25mL LB media containing 100 μg/mL carbenicillin and 33 μg/mL chloramphenicol overnight with shaking. 2L of 2X YT media containing 100 μg/mL carbenicillin and 33 μg/mL chloramphenicol was inoculated with the overnight cultures and grown at 37°C with shaking until the OD600 reached ~0.6–0.8, after which cultures were cooled to 16°C. IPTG was added to a final concentration of 0.3 mM and the cells were cultured at 16°C for 18 hours with shaking. Bacterial cells were harvested by centrifugation at 6,000g at 4°C for 15 min, washed with PBS, frozen with liquid nitrogen and stored at −80°C.
MBP-AID purification
Bacterial pellets were thawed at 37°C, resuspended with 50 mL lysis buffer (50mM Tris-Cl pH 80, 15mM NaCl, 10μM ZnCl2, 10% glycerol, 0.1% Tween-20, 5 mM β-mercaptoethanol; 1 mM PMSF (36978, Thermo Scientific), 1 mM benzamidine hydrochloride (B6506, Sigma), 0.5 mg/mL lysozyme from hen egg-white (10837059001, Sigma), 1 tablet cOmplete Mini EDTA-free Protease Inhibitor Cocktail per 10mL buffer, (11836170001,Sigma)), and incubated at 4°C for 30 min with magnetic stirring. Lysates were sonicated using a Branson 450 digital sonifier and a microtip until no longer viscous, while kept on ice. Samples were centrifugated at 27,000 g for 1 hour at 4°C. 6 mL amylose resin (E8021, NEB) was equilibrated with 30 mL binding buffer (50mM Tris-Cl pH 8.0, 15mM NaCl, 10μM ZnCl2, 10% glycerol, 0.1% Tween-20, 5mM β-mercaptoethanol), loaded with the lysate supernatant using a peristaltic pump, washed with 100 mL binding buffer, and eluted with 25 mL elution buffer (binding buffer + 10mM maltose) over twelve 2 mL fractions. The protein concentration for each fraction was measured using Bradford assay, and fractions were analyzed by SDS-PAGE and Coomassie staining. Fractions were frozen in liquid nitrogen and stored at −80°C. Fractions with significant full-length MBP-AID band (67kDa) on Coomassie gels were pooled and concentrated with Amicon Ultra-15 centrifugal 30kDa filter units (C7715, Millipore) and incubated with 20 μg Ambion RNase A (AM2271, Invitrogen) on ice for 2 hours. RNA-digested samples were loaded onto a Superdex 200 10/300 GL gel filtration column (17517501, GE) with gel filtration elution buffer (50mM Tris-Cl pH 8.0, 150mM KCl, 10μM ZnCl2, 5mM β-mercaptoethanol, 10% glycerol, 0.1% Tween-20). Each fraction was frozen in liquid nitrogen and stored at −80°C. Fractions with significant full-length MBP-AID band (67kDa) on Coomassie gel were thawed, pooled, concentrated and buffer-exchanged to deamination reaction buffer (20mM Tris-Cl pH 8.0, 20mM NaCl, 20μM ZnCl2, 5mM β-mercaptoethanol, 5% glycerol) with Amicon Ultra-15 centrifugal 30kDa filter units (C7715, Millipore). Samples were diluted to 4 mg/mL, aliquoted, frozen with liquid nitrogen, and stored at −80°C. For linker digestion, ~80 μg MBP-AID was incubated with 2U PreScission protease (27–0843-01, GE Healthcare) at 4°C for 4 hours.
In vitro deamination assay
MBP-AID aliquots were thawed on ice. S30-TGC, S30-TGU, or 15mer oligos were used as substrates (Integrated DNA Technologies). Deamination reactions were performed at 30°C for 16 hrs in a final volume of 10 or 20 μL, and contained the following: 20 mM Tris-Cl pH 8.0, 20 mM NaCl, 20 μM ZnCl2, 5 mM β-mercaptoethanol, 5% glycerol, 0.5 μM substrate, and 1 mg/mL MBP-AID, aside from Figure 4C in which μg of AID used is indicated. For G4 inhibition experiments, 2.5 μM of folded DNA or RNA Sμ4G or Sμ4Gmut oligos and 2U/μl of Ambion RNase Inhibitor (AM2682, Invitrogen) were added to the deamination reaction. 2U of Uracil-DNA glycosylase (UDG) (M0280, NEB) was added and incubated at 37°C for 1 hr. Sample were boiled at 95°C in 1x RNA loading buffer (B0363, NEB) supplemented with 150 mM NaOH for 30 min. Samples were run on a 13.3% TBE-Urea gel and imaged with a Typhoon gel imager (GE). Images were analyzed with ImageJ software. Deamination activity (%) is defined as [(sample product ratio) - (Negative control product ratio)] / [(positive control product ratio) - (negative control product ratio)] x 100. Product ratio is defined as (product signal pixel count) / (total signal pixel count) for each lane.
Mass spectrometry
Samples were excised, washed, reduced with DTT, alkylated with IAA, and digested overnight with either Asp-N/Lys-C or Glu-C at 37°C. Peptides were then desalted using C18 zip tips, and dried by vacuum centrifugation. Each sample was reconstituted in 0.1% (vol/vol) formic acid and analyzed by microcapillary liquid chromatography with tandem mass spectrometry using the NanoAcquity (Waters) with a 100-μm inner-diameter × 10-cm- length C18 column (1.7 um BEH130, Waters) configured with a 180-μm × 2-cm trap column coupled to a Q-Exactive Plus mass spectrometer (Thermo Fisher Scientific). Peptides were eluted with a linear gradient of 0–30% acetonitrile (0.1% formic acid) in water (0.1% formic acid) over 50 mins with a flow rate of 300 nL/min. The QE+ was operated in automatic, data-dependent MS/MS acquisition mode with one MS full scan (380–1600 m/z) at 70,000 mass resolution and up to ten concurrent MS/MS scans for the ten most intense peaks selected from each survey scan. Survey scans were acquired in profile mode and MS/MS scans were acquired in centroid mode at 17,500 resolution and isolation window of 1.5 amu and normalized collision energy of 27. AGC was set to 1 × 106 for MS1 and 5 × 104 and 50 ms IT for MS2. Charge exclusion of unassigned, +1, and greater than +6 enabled with a dynamic exclusion of 15 s. Mass spectrometry data files were converted to mascot generic files, searched using MASCOT (v. 2.3.02) against a custom database, and imported into Scaffold (v. 4.8.4).
G4 folding
For DNA and RNA G4 folding, 10 μM Sμ4G or Sμ4Gmut oligos (Integrated DNA Technologies) were boiled in folding buffer (20 mM Tris-Cl pH 7.6, 100 mM KCl or LiCl, 1 mM EDTA) at 95°C for 10 min and cooled at 25°C for 2 hours. Folded DNA and RNAs (4 μM) were incubated with 12 μM hemin (H-5533, Sigma) resuspended in DMSO, in 1x NEBuffer 2 (NEB) at 37°C for 1 hr. Substrate solution (2 mM ABTS (A9941, Sigma), 2 mM hydrogen peroxide (H1009, Sigma), 25 mM HEPES pH 7.4, 0.2 M NaCl, 10 mM KCl, 0.05% Triton X-100, 1% DMSO) was added. Color development was allowed for 15 min and absorbance was measured from 400 nm to 500 nm using Synergy HT Biotek plate reader (513148) with Gen5 software.
Jh4 intron DNA sequencing
Peyer’s patches were isolated from 5–7 month-old mice, or spleens at day 7 post-immunization with SRBCs, processed into single cell suspensions, stained with surface antibodies, and GC B cells were sorted into B cell media. For SRBC immunizations, B cells were enriched by positive selection using anti-mouse CD19 MicroBeads (130–121-301, Miltenyi Biotec) and LS columns (130–042-401, Miltenyi Biotec) prior to surface staining. Sorted cells were collected by centrifugation and resuspended in 500 μL of genomic DNA cell lysis buffer (100 mM Tris 8.8, 200 mM NaCl, 5 mM EDTA, 0.2% SDS) plus 80 ug of protease K, and incubated at 56°C for 12hrs. Genomic DNA was precipitated with isopropanol and resuspended in 25 μL of H2O. Jh4 intron sequences were amplified by PCR using Jh4 forward and Jh4 reverse primers. The following PCR program was used: 1) 98°C 3 min, 2) 98°C 30 sec, 3) 72°C 1 min, 4) go to step 2 34 times, 5) 72°C 10 min 6) 4°C hold. PCR amplicons that were approximately 1200 bp were gel purified, ligated into the pCR-Blunt II-TOPO vector (K2800J10, Invitrogen) and transformed into One Shot TOP10 chemically competent E. coli (K280020, Thermo Fisher Scientific) according to the manufacturers protocol. Clones were sequenced using the bacterial colony Sanger sequencing service provided by Genewiz (South Plainfield, NJ), and the Jh4 seq primer. Sequences were aligned using SeqMan Pro (DNASTAR) and analyzed for mutations.
ChIP
ChIP was performed according to the protocol of the ChIP Assay Kit (Millipore, 17–295). Briefly, purified naïve splenic B cells were stimulated with LPS (30 μg/mL, L4130, Sigma) plus mouse IL-4 (404-ML-010, R&D systems) for 72 hours. 10 × 106 activated B cells were crosslinked by adding fresh 16% formaldehyde (28908, Thermo Scientific) to a final concentration of 1% and incubating at 25°C for 10 min with shaking. The reaction was neutralized by adding 1:20 volumes of 2.5 M glycine pH 8 with shaking for 5 min. Crosslinked cells were washed with PBS Cell Wash Buffer (PBS, 0.1% FBS, 2 mM EDTA). Cells were lysed according to the protocol of truChIP Chromatin Shearing Kit with Formaldehyde (520154, Covaris,). Cell lysates were sonicated in the M220 Focused-ultrasonicator (5002295, Covaris) to shear the chromatin, and debris was removed by centrifugation at 18,000g at 4°C for 10 min. 50 μL (5%) of the lysate was removed for the input fraction. The remaining sonicated lysates were diluted to 1.5 mL in ChIP dilution buffer (0.01% SDS, 1.1% Triton X100, 1.2 mM EDTA, 16.7 mM Tris-HCl pH 8.1, 167 mM NaCl) (20–153, Millipore) and precleared according to protocol with Salmon Sperm DNA/Protein A Agarose Slurry for 1 hour at 4°C. Precleared lysates were divided into three parts of 500 μL each and incubated with either 1 μg of anti-H3 antibody (ab1791, Abcam), 2 μg of anti-AID antibody (Chaudhuri Lab) or 1 μg control IgG (011–000-003, Jackson Immunoresearch Labs) at 4°C overnight. Immunocomplexes were recovered by incubation with Salmon Sperm DNA/Protein A Agarose Slurry (16–157C, Millipore) and washed with 1 mL of the following sequence of buffers,: Low Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 150 mM NaCl) (20–154, Millipore), High Salt Buffer (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris-HCl, pH 8.1, 500 mM NaCl) (20–155, Millipore), LiCl Immune Complex Wash Buffer (0.25 M LiCl, 1% IGEPAL-CA630, 1% deoxycholic acid (sodium salt), 1 mM EDTA, 10 mM Tris, pH 8.1) (20–156, Millipore), and TE Buffer (10 mM Tris-HCl, 1 mM EDTA, pH 8.0) (20–157, Millipore). Immunocomplexes were eluted from the beads using 500 μL of fresh elution buffer (1% SDS and 0.1M NaHCO3). Cross-linking for immunoprecipitated and input samples was reversed by incubation in 0.3M NaCl and RNase A (30 μg /mL) (19101, Qiagen) at 65°C for 6 hours. Samples were treated with 20 μL of 1 M Tris pH 6.5, 10 μL of 0.5 M EDTA and 2 μL proteinase K (10mg/ml), (97062–242, VWR) for 1 hour at 45°C. DNA was recovered by phenol-chloroform extraction and ethanol precipitation with 20 μg carrier glycogen. Pellets were resuspended in 35 μL of water for use as ChIP DNA or 50 μL of water for use as input DNA. Sμ DNA was analyzed by qPCR using PowerUp SYBR™ Green Master Mix (Applied Biosystems) and sSμ and asSμ primers.
ELISAs
4 month-old mice were used to generate all ELISA data. Assays were done in MaxiSorp clear, flat-bottom, 96- well plates (439454, Thermo Fisher Scientific). Coating abs for binding IgM, IgG1, IgG2b, IgG2c, IgG3, and IgA (1020–01, 1070–01, 1090–01, 1079–01, 1100–01, 1040–01, respectively, Southern Biotech) were used at 3 μg/mL in PBS pH 8. Each plate was coated with ab overnight at 4°C (100 μl per well). Plates were washed 4x with 0.05% PBS plus 0.1% Tween-20 (PBST) and blocked with 250 μl of ELISA diluent per well (00–4202-56, eBioscience) for 3 hours at 25°C or overnight at 4°C. Plates were washed 3x, loaded with 100 μl of serum samples and standards per well, and incubated for 2.5 hrs at 25°C or overnight at 4°C. The following isotype standards were used to calculate absolute concentration values: IgM (14–4752-81; eBioscience), IgG1 (0102–01; Southern Biotech), IgG2b (14–4732-81; eBioscience), IgG2c (0122–01; Southern Biotech), IgG3 (553486; BD Pharmingen), and IgA (553476; BD Pharmingen). Plates were washed 6x and secondary abs for detecting IgM, IgG1, IgG2b, IgG2c, IgG3, and IgA (1020–05, 1070–05, 1090–05, 1079–05, 1100–05, 1040–05, respectively, Southern Biotech) were used at 1:2,000 for 1.5 hrs at RT (100 μl per well). After 7 washes, 100 μl of TMB substrate (00–4201-56, eBioscience) per well was used to develop for 30–60 sec, and 1 M phosphoric acid was used to stop development. Plates were read at 450 nm on a BioTek Synergy HT detector. Absolute concentrations of serum abs were determined by interpolation from the standard curve, while keeping within standard and sample linear ranges. All samples were done in triplicate over a six-step dilution series. An eleven-step standard curve was generated for each plate. For NP-specific assays, plates were coated with 3 μg/ml NP(8)-BSA (N-5050L-10; Biosearch Technologies) or NP(30)-BSA (N-5050H-10; Biosearch Technologies) resuspended in PBS. Relative titers were determined by interpolation of the plate reference curve generated for each plate using a constant sample, with attention paid to keeping within plate reference and sample linear ranges. All samples were done in triplicate over a six-step dilution series.
Cell proliferation assays
Naïve splenic B cells were resuspended in PBS at a concentration of 10 × 106 cells/mL. CellTrace Violet (C34571, Invitrogen) stock solution in DMSO was added to a final working solution of 4.25 μM. Cells were vortexed briefly and incubated for 10 min at RT in the dark. B cell media was added at 5x the original staining volume and cells were incubated for 5 min to remove the free dye. The cells were then pelleted, resuspended in warm B cell media and incubated for 30 min at 25°C before cell stimulation or analysis.
Library preparation for ChIP-Seq
Immunoprecipitated DNA was quantified by PicoGreen and the size was evaluated by Agilent BioAnalyzer. Illumina libraries were prepared using the KAPA HTP Library Preparation Kit (Kapa Biosystems KK8234) according to the manufacturer’s instructions with 0 (undetectable)-4.6 ng input DNA and 8–12 cycles of PCR. Barcoded libraries were run on the Hiseq 2500 in Rapid mode and the HiSeq 4000 in 50bp/50bp paired end runs, using the HiSeq Rapid SBS Kit v2 and HiSeq 3000/4000 SBS Kit, respectively (Illumina). An average of 35 million paired reads were generated per sample.
ChIP-Seq analysis
Paired-end reads were trimmed for adaptors and removal of low quality reads using Trimmomatic (v.0.36) (Bolger et al., 2014). Trimmed reads were mapped to the Mus musculus genome (mm10 assembly) using Bowtie2 (v2.2.9)(Langmead and Salzberg, 2012). Concordantly aligned paired mates pooled from all samples were used for peak calling by MACS2 (v2.1.1.20160309)(Zhang et al., 2008) with the arguments “-f BAMPE --broad”. Peak assignment to genes was performed using ChipPeakAnno (Zhu et al., 2010) and the UCSC Known Gene transcript models (v10) (Speir et al., 2016). For some peaks (e.g. those around the Igh locus), version 11 of the UCSC Known Gene transcript model was used to manually reference and reassign peaks. Overlapping blacklist regions downloaded from the ENCODE portal (Davis et al., 2018) (accession ENCFF547MET) and regions found in non-standard or mitochondrial chromosomes were removed. This yielded an atlas of 15,052 quantifiable regions used for statistical comparisons.
For differential binding, duplicate reads were removed from concordantly aligned paired mates using the default settings of MarkDuplicates from Picard (http://broadinstitute.github.io/picard/) (MACS2 peak calling ignores duplicates, thus deduplication was not required at an earlier stage). These filtered reads were counted within all atlas regions using the summarizeOverlaps function from the GenomicAlignments package (v1.18.1)(Lawrence et al., 2013) using arguments “mode=‘IntersectionNotEmpty’, ignore.strand=TRUE, singleEnd=FALSE”. Technical replicates were first collapsed and differential analyses were executed with the DESeq2 software (v1.22.2)(Love et al., 2014), accounting for differences between biological replicates and using the “normal” shrinkage estimator. Low count regions were removed by first calculating a common threshold for all samples. This threshold was determined by taking the geometric mean of independent filtering thresholds calculated by DESeq2 when performing pairwise contrasts between all conditions (WT, Aicda−/−, and AicdaGV/GV). Regions showing a mean count (using all samples) higher than this threshold were retained for final differential binding comparisons, with independent filtering disabled. Ultimately, 5,242 regions were tested and those that reported an FDR-adjusted p-value < 0.05 were considered differentially bound. AID-dependent regions were defined as differentially bound regions showing a negative log2 fold change when comparing Aicda−/− or AicdaGV/GV (numerator) to WT (denominator).
To generate gene tracks, paired-end reads from deduplicated BAM files were extended to represent fragment length using the bamCoverage function from deepTools (v3.2.1) (Ramirez et al., 2016). The resulting bigWig files were normalized for sequencing depth using size factors calculated by DESeq2 and visualized using the Gviz R package (v1.26.5) (Hahne and Ivanek, 2016).
To generate heatmaps and signal density plots, signal for each sample was quantified from bigWigs described above. For heatmaps, scores were generated using the computeMatrix function from deepTools, with arguments “--referencePoint center --missingDataAsZero -b 2500 -a 2500 -bs 100”. For each condition, scores from each bin position were summarized across condition replicates and plotted as a heatmap using the ComplexHeatmap R package (v1.99.7)(Gu et al., 2016). For signal density plots, individual scores at each base pair were computed +/− 2.5kb the nearest transcriptional start site per AID-dependent peak and then summarized as a mean score per position. The mean of these summarized scores across all replicates were plotted as a line across the 5 kb window for each condition.
Notably, this analysis identified 37 peaks that were significantly enriched in WT versus either Aicda−/− or AicdaGV/GV B cells, a stark contrast to a previous AID-ChIP study which identified thousands of genes as potential AID targets (Yamane et al., 2011). It is important to highlight key differences between these two analyses that can account for such a large discrepancy, such as the peak calling software and algorithms, as well as experimental design. Whereas the previous study pooled multiple anti-AID immunoprecipitations (which can serve to increase read depth and sensitivity to detect AID binding), this analysis focused on attempting to detect the most reproducible and robust AID-bound regions. Thus, 4 biological replicates for WT and AicdaGV/GV, or 3 biological replicates and one technical replicate for Aicda−/−, were incorporated in order to account for variation and to rigorously gauge statistical confidence. While the replicate variability undoubtedly decreased the overall number of AID binding events detected, it potentially yields relatively higher confidence that those that passed statistical thresholds are likely true AID binding events.
RNA-sequencing analysis
HTSeq counts files from the following projects were downloaded from the Genomic Data Commons Data Portal: NCICCR-DLBCL (dbGaP Study Accession phs001444), CTSP-DLBCL1 (dbGaP Study Accession phs001175), and TCGA-DLBC (dbGaP Study Accession phs000178). These were melted into a single count matrix and TPMs for each gene were determined using custom scripts (available upon request). The ggPlot2 package v3.3.2 (Wickham, 2009) was used to generate scatter plots for each gene, as well as violin plots for AICDA expression amongst DLBCL subgroups. In parallel, the counts files were imported into DESeq2 v1.24.0 (Love et al., 2014) using Tximport v1.12.3 (Soneson et al., 2015). Separately for each of the three classifications, DESeq2 was used to quantify differentially expressed genes between AIDtop10% (defined as the samples with the highest 10% AID TPM) and AIDbot90% (the remainder of the samples in that subgroup) tumor samples. Genes with fewer than a single count per sample were removed from consideration. The data was further visualized using distinct volcano plots for each tumor subgroup, with the following modification: data points with p values less than 10−3 or fold changes greater than 8 between the groups were mapped to the closest point on the plot. These modified points were depicted as triangles for clarity, while the unmodified data points were plotted as circles. MHC class II-associated genes, AID and two housekeeping genes were highlighted.
QUANTIFICATION AND STATISTICAL ANALYSIS
All statistical analyses were performed with GraphPad Prism 7 (GraphPad Software, Inc.), with the exception of ChIP-seq and RNA-seq analyses, which were performed using DESeq2 (Love et al., 2014). Flow cytometry data was analyzed using FlowJo software (version 9.9). Graphs were created using GraphPad Prism 7 or the ggPlot2 package (v3.3.2) (Wickham, 2009). Graph titles and axes labels were edited using Adobe Illustrator CS6. All graphs depict data collected from all performed experiments, and all error bars represent the mean plus or minus the standard deviation.
Supplementary Material
Table S2-4. Differentially bound regions from AID ChIP-seq analysis, Related toFigure 6. Listed from left to right are peak identifiers (pk.id), the averages of the normalized counts taken over all samples (baseMean), log2-transformed differences for indicated genotype contrasts (log2FC) and their standard errors (lfcSE), Wald statistics (stat) and Wald test p-values (pvalue), FDR-corrected p-values (padj), chromosome location (peak.chr), genomic start coordinates (peak.start), genomic end coordinates (peak.end), widths of peak regions (peak.width), associated transcript IDs (tx.id), associated Entrez Gene IDs (gene.id), descriptions of genes (description), -log10 q-values of peak calculated by the peak-calling software (qValue), and normalized counts for relevant samples (NormCounts_sample name). Peaks are listed by increasing p adj value. Gene IDs with no annotated identifier according to the gene model are labeled as “noID” followed by self-assigned numbers.
Table S2. Aicda−/− versus WT ChIP-seq analysis, Related to Figure 6.
Table S3. AicdaGV/GV versus WT ChIP-seq analysis, Related to Figure 6.
Table S4. AicdaGV/GV versus Aicda−/− ChIP-seq analysis, Related to Figure 6.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| BV786 Rat Anti-Mouse CD45R/B220 | BD Biosciences | Cat#: 563894; RRID: AB_2738472 |
| BUV395 Rat Anti-Mouse IgM | BD Biosciences | Cat#: 743329; RRID: AB_2741430 |
| Alexa Fluor® 700 anti-mouse IgD | BioLegend | Cat#: 405729, RRID: AB_2563340 |
| CD43 Monoclonal Antibody (eBioR2/60), FITC, eBioscience™ | Thermo Fisher Scientific | Cat#: 11-0431-82; RRID: AB_465040 |
| CD21/CD35 Monoclonal Antibody (eBio4E3 (4E3)), eFluor 450, eBioscience™ | Thermo Fisher Scientific | Cat#: 48-0212-82: RRID: AB_2016703) |
| CD23 Monoclonal Antibody (B3B4), PE-Cyanine7, eBioscience™ | Thermo Fisher Scientific | Cat#: 25-0232-81; RRID: AB_469603 |
| BV510 Rat Anti-Mouse IgG1 | BD Biosciences | Cat# 742476; RRID: AB_2740810 |
| Goat Anti-Mouse IgA-PE | Southern Biotech | Cat#: 1040-09; RRID: AB_2794375 |
| CD38 Monoclonal Antibody (90), PerCP-eFluor 710, eBioscience™ | Thermo Fisher Scientific | Cat#: 46-0381-80; RRID: AB_10852870 |
| APC-H7 Rat anti-Mouse CD19 | BD Biosciences | Cat#: 560143; RRID: AB_1645234 |
| CD25 Monoclonal Antibody (PC61.5). APC-eFluor 780, eBioscience™ | Thermo Fisher Scientific | Cat#: 47-0251-82; RRID: AB_1272179 |
| Brilliant Violet 510™ anti-mouse IgD | BioLegend | Cat#: 405723; RRID: AB_2562742 |
| PE/Cy7 anti-mouse CD138 (Syndecan-1) | BioLegend | Cat#: 142514; RRID: AB_2562198 |
| Aiexa Fluor® 700 anti-mouse CD19 | BioLegend | Cat#: 115528; RRID: AB_493735 |
| CD117 (c-Kit) Monoclonal Antibody (2B8), APC, eBioscience™ | Thermo Fisher Scientific | Cat#: 17-1171-81; RRID: AB_469429 |
| CD3 Monoclonal Antibody (17A2), eFIuor 450, eBioscience™ | Thermo Fisher Scientific | Cat#: 48-0032-80; RRID: AB_1272229 |
| FITC Anti-Mouse CD8a (53-6.7) | Tonbo Biosciences | Cat#: 35-0081; RRID: AB_2621671 |
| Aiexa Fluor® 700 anti-mouse TCR β chain | BioLegend | Cat#: 109224; RRID: AB_1027648 |
| CD4 Monoclonal Antibody (RM4-5), PerCP-Cyanine5.5, eBioscience™ | Thermo Fisher Scientific | Cat#: 45-0042-80: RRID: AB_906231 |
| CD62L (L-Selectin) Monoclonal Antibody (MEL-14), PE-Cyanine7, eBioscience™ | Thermo Fisher Scientific | Cat#: 25-0621-82; RRID: AB_469633 |
| CD69 Monoclonal Antibody (H1.2F3), APC, eBioscience™ | Thermo Fisher Scientific | Cat#: 17-0691-82; RRID: AB_1210795 |
| CD44 Monoclonal Antibody (IM7), PE, eBioscience™ | Thermo Fisher Scientific | Cat#: 12-0441-83: RRID: AB_465665 |
| IgD Monoclonal Antibody (11-26c(11-26)), APC-eFluor 780, eBioscience™ | Thermo Fisher Scientific | Cat#: 47-5993-80; RRID: AB_2573993 |
| PE-Cy™7 Hamster Anti-Mouse CD95 | BD Biosciences | Cat#: 557653; RRID: AB_396768 |
| GL7 Monoclonal Antibody (GL-7 (GL7)), eFluor 450, eBioscience™ | Thermo Fisher Scientific | Cat#: 48-5902-80; RRID: AB_10854881 |
| CD38 Monoclonal Antibody (90), Alexa Fluor 700, eBioscience™ | Thermo Fisher Scientific | Cat#: 56-0381-82; RRID:AB_657740 |
| Goat Anti-Mouse IgG2c, Human ads-FITC | Southern Biotech | Cat#: 1079-02; RRID: AB_2794465 |
| CD19 Monoclonal Antibody (eBio1D3 (1D3)), PerCP-Cyanine5.5, eBioscience™ | Thermo Fisher Scientific | Cat#: 45-0193-80; RRID: AB_906215 |
| Goat Anti-Mouse IgG2b, Human ads-APC/CY7 | Southern Biotech | Cat#: 1090-19; RRID: AB_2794530 |
| CD86 (B7-2) Monoclonal Antibody (GL1), PE, eBioscience™ | Thermo Fisher Scientific | Cat#: 12-0862-82; RRID: AB_465768 |
| Brilliant Violet 711™ anti-mouse IgD | BioLegend | Cat#: 405731; RRID: AB_2563342 |
| CD184 (CXCR4) Monoclonal Antibody (2B11), PerCP-eFluor 710, eBioscience™ | Thermo Fisher Scientific | Cat#: 46-9991-82; RRID: AB_10670489 |
| APC Streptavidin | BioLegend | Cat#: 405207 |
| BV786 Rat Anti-Mouse IgM | BD Biosciences | Cat#: 743328; RRID: AB_2741429 |
| BUV395 Rat Anti-Mouse CD86 | BD Biosciences | Cat#: 564199; RRID: AB_2738664 |
| MHC Class II (I-A/I-E) Monoclonal Antibody (M5/114.15.2), APC, eBioscience™ | Thermo Fisher Scientific | Cat#: 17-5321-81; RRID: AB_469454 |
| APC Rat IgG2b, kappa Isotype Ctrl | BioLegend | Cat#: 400612; RRID: AB_326556 |
| BUV737 Rat Anti-Mouse CD45R/B220 | BD Biosciences | Cat#: 612838 |
| PE Streptavidin | BioLegend | Cat#: 405204 |
| FITC Rat Anti-Mouse IgG3 | BD Biosciences | Cat#: 553403; RRID: AB_394840 |
| Rabbit anti-AID | Chaudhuri et al., 2003 | N/A |
| Human/Mouse/Rat HSP90 Antibody | R&D Systems | Cat#: MAB3286; RRID: AB_2121072 |
| Lamin B1 Polyclonal Antibody | Thermo Fisher Scientific | Cat#: PA5-19468; RRID: AB_10985414 |
| Monoclonal Anti-α-Tubulin antibody produced in mouse | Sigma-Aldrich | Cat#: T9026; RRID: AB_477593 |
| Anti-MBP Monoclonal Antibody | New England Biolabs | Cat#: E8032; RRID: AB_1559730 |
| Rat Anti-Mouse IgD-UNLB | Southern Biotech | Cat#: 1120-01; RRID: AB_2794607 |
| Peanut Agglutinin (PNA), Biotinylated | Vectors Laboratories | Cat#: B-1075; RRID: AB_2313597 |
| Anti-CD45R antibody [RA3-6B2] | Abcam | Cat#: ab64100; RRID: AB_1140036 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 488 | Thermo Fisher Scientific | Cat#: A-21208; RRID: AB_141709 |
| Donkey anti-Rat IgG (H+L) Highly Cross-Adsorbed Secondary Antibody, Alexa Fluor 594 | Thermo Fisher Scientific | Cat#: A-21209; RRID: AB_2535795 |
| Cy™5 Streptavidin | Jackson ImmunoResearch Labs | Cat#: 016-170-084; RRID: AB_2337245 |
| Anti-Histone H3 antibody - Nuclear Marker and ChIP Grade | Abcam | Cat#: ab1791; RRID: AB_302613 |
| ChromPure Rabbit IgG, whole molecule | Jackson ImmunoResearch Labs | Cat#: 011-000-003; RRID: AB_2337118 |
| Goat Anti-Mouse IgM, Human ads-UNLB | Southern Biotech | Cat#: 1020-01; RRID: AB_2794197 |
| Goat Anti-Mouse IgG1, Human ads-UNLB antibody | Southern Biotech | Cat#: 1070-01; RRID: AB_2794408 |
| Goat Anti-Mouse IgG2b, Human ads-UNLB antibody | Southern Biotech | Cat#: 1090-01; RRID: AB_2794517 |
| Goat Anti-Mouse IgG2c, Human ads-UNLB antibody | Southern Biotech | Cat#: 1079-01; RRID: AB_2794464 |
| Goat Anti-Mouse IgG3, Human ads-UNLB antibody | Southern Biotech | Cat#: 1100-01; RRID: AB_2794567 |
| Goat Anti-Mouse IgA-UNLB | Southern Biotech | Cat#: 1040-01; RRID: AB_2314669 |
| Mouse IgM Isotype Control (11E10), eBioscience™ | Thermo Fisher Scientific | Cat#: 14-4752-81; RRID: AB_470122 |
| Mouse IgG1-UNLB antibody | Southern Biotech | Cat#: 0102-01; RRID: AB_2793845 |
| Mouse IgG2b kappa Isotype Control (eBMG2b), eBioscience™ | Thermo Fisher Scientific | Cat#: 14-4732-81; RRID: AB_470116 |
| Mouse IgG2c-UNLB antibody | Southern Biotech | Cat#: 0122-01; RRID: AB_2794064 |
| Purified Mouse IgG3, κ Isotype Control | BD Biosciences | Cat#: 553486; RRID: AB_10054920 |
| Purified Mouse IgA, κ Isotype Control | BD Biosciences | Cat#: 553476; RRID: AB_479590 |
| Goat Anti-Mouse IgM, Human ads-HRP | Southern Biotech | Cat#: 1020-05; RRID: AB_2794201 |
| Goat Anti-Mouse IgG1, Human ads-HRP | Southern Biotech | Cat#: 1070-05; RRID: AB_2650509 |
| Goat Anti-Mouse IgG2b, Human ads-HRP | Southern Biotech | Cat#: 1090-05; RRID: AB_2794521 |
| Goat Anti-Mouse IgG2c, Human ads-HRP | Southern Biotech | Cat#: 1079-05; RRID: AB_2794466 |
| Goat Anti-Mouse IgG3, Human ads-HRP | Southern Biotech | Cat#: 1100-05; RRID: AB_2794573 |
| Goat Anti-Mouse IgA-HRP antibody | Southern Biotech | Cat#: 1040-05; RRID: AB_2714213 |
| Purified Rat Anti-Mouse CD16/CD32 (Mouse BD Fc Block™) | BD Biosciences | Cat#: 553142; RRID: AB_394657 |
| Bacterial and Virus Strains | ||
| Influenza A/Puerto Rico/8/34 (PR8) mouse adapted influenza strain, grown in 10 day-old embryonated chicken eggs | Yewdell laboratory, (NIAID, NIH) | N/A |
| J1, PR8 reassortant virus, mouse adapted influenza strain, grown in 10 day-old embryonated chicken eggs | Yewdell laboratory, (NIAID, NIH) | N/A |
| BL21(DE3) Competent E. coli | New England Biolabs | Cat#: C2527 |
| One Shot™ TOP10 Chemically Competent E. coli | Invitrogen | Cat#: C404010 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| NP(33)-CGG (Chicken Gamma Globulin), Ratio 30–39 | Biosearch Technologies | Cat#: N-5055D-5 |
| NP(8)-BSA | Biosearch Technologies | N-5050L-10 |
| NP(30)-BSA | Biosearch Technologies | N-5050H-10 |
| Imject™ Alum Adjuvant | Thermo Scientific | Cat#: 77161 |
| Sheep Red Blood Cells Packed 10% | Innovative Research | Cat#: ISHRBC10P15ML |
| Lipopolysaccharides from Escherichia coli O111:B4 | Sigma | L4130 |
| Recombinant Mouse IL-4 Protein | R&D Systems | Cat#: 404-ML-010 |
| BAFF, Soluble (mouse) (rec.) | Adipogen | Cat#: AG-40B-0022 |
| Retinoic acid, ≥98% (HPLC), powder | Sigma | Cat#: R2625 |
| Recombinant Human TGF-beta 1 Protein | R&D Systems | Cat#: 240-B-010 |
| Recombinant Murine IL-5 | Peprotech | Cat#: 215-15 |
| Benzamidine hydrochloride hydrate | Sigma | Cat#: B6506 |
| PMSF Protease Inhibitor | Thermo Scientific | Cat#: 36978 |
| cOmplete™, Mini, EDTA-free Protease Inhibitor Cocktail | Sigma | Cat#: 11836170001 |
| TRIzol™ Reagent | Invitrogen | Cat#: 15596026 |
| Tissue-Tek® O.C.T. Compound, Sakura® Finetek | Sakura | Cat#: 4583 |
| Ambion™ RNase A, affinity purified, 1 mg/mL | Invitrogen | Cat#: AM2270 |
| PreScission protease | GE | Cat#: 27-0843-01 |
| Ambion RNase Inhibitor | Invitrogen | Cat#: AM2682 |
| Uracil-DNA glycosylase | New England Biolabs | Cat#: M0280 |
| Hemin, from bovine, ≥80% | Sigma | Cat#: H-5533 |
| 2,2′-Azino-bis(3-ethylbenzothiazoline-6-sulfonic acid) diammonium salt (ABTS) | Sigma | Cat#: A9941 |
| Pierce™ 16% Formaldehyde (w/v), Methanol-free | Thermo Scientific | Cat#: 28908 |
| RNase A | Qiagen | Cat#: 19101 |
| Proteinase K, Biotechnology Grade | VWR | Cat#: 97062-242 |
| eBioscience™ ELISA/ELISPOT Diluent (5X) | Invitrogen | Cat#: 00-4202-56 |
| eBioscience™ TMB Solution (1X) | Invitrogen | Cat#: 00-4201-56 |
| Recombinant PR8 HA-biotin | A. McDermott (VRC, NIAID,NIH) | NA |
| Critical Commercial Assays | ||
| RPMI 1640 Medium | Gibco | Cat#: 11875085 |
| DMEM, high glucose | Gibco | Cat#: 11965118 |
| Zombie Red™ Fixable Viability Kit | BioLegend | Cat#: 423110 |
| CD43 (Ly-48) MicroBeads, mouse | Miltenyi Biotec | Cat#: 130-049-801; RRID: AB_2861373 |
| CD19 MicroBeads, mouse | Miltenyi Biotec | Cat#: 130-121-301; RRID: AB_2827612 |
| NE-PER™ Nuclear and Cytoplasmic Extraction Reagents | Thermo Scientific | Cat#: 78835 |
| Superscript™ III First-Strand Synthesis System | Invitrogen | Cat#: 18080051 |
| PowerUp™ SYBR™ Green Master Mix | Applied Biosystems™ | Cat#: A25741 |
| Amylose Resin | New England Biolabs | Cat#: E8021 |
| Superdex 200 10/300 GL gel filtration column | GE | Cat#: 17517501 |
| Amicon Ultra-15 centrifugal 30kDa filter | Millipore | Cat#: C7715 |
| LS Columns | Miltenyi Bictec | Cat#: 130-042-401 |
| Zero Blunt™ TOPO™ PCR Cloning Kit, with One Shot™ TOP10 Chemically Competent E. coli cells | Invitrogen | Cat#: K2800J10 |
| Chromatin Immunoprecipitation (ChIP) Assay Kit | Millipore | Cat#: 17-295 |
| truChIP Chromatin Shearing Kit with Formaldehyde | Covaris | Cat#: 520154 |
| CellTrace™ Violet Cell Proliferation Kit, for flow cytometry | Invitrogen | Cat#: C34571 |
| Deposited Data | ||
| AID ChIP-seq raw and processed data | This paper | GEO: GSE136959, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE136959 |
| DLBCL RNA-Seq processed data | Schmitz et al., 2018 | dbGaP Study Accessions phs001444, phs001175, phs000178. |
| Experimental Models: Cell Lines | ||
| HEK293T cells | ATCC | ATCC® CRL-3216™ |
| Experimental Models: Organisms/Strains | ||
| Mouse: AicdaG133V | This paper | N/A |
| Mouse: Aicda−/− | T. Honjo, (Muramatsu et al., 2000) | N/A |
| Mouse: C57BL6/J | The Jackson Laboratory | JAX: 000664 |
| Oligonucleotides | ||
| See Table S7 | IDT | N/A |
| Recombinant DNA | ||
| pCL-Eco, retroviral packaging vector | Chaudhuri Lab | JCP 28 |
| pMIG, retroviral expression vector | Chaudhuri Lab | JCP 1 |
| Trigger factor plasmid | L. Ma. and G. Montelione, Rutgers. (Kohli et al., 2009) | JCP 128 |
| pMALc5x-MBP-AID (mouse) | This paper | JCP 20 |
| pMALc5x-MBP-AID G133V (mouse) | This paper | JCP 23 |
| pMALc5x-MBP-AID H56R E58Q (CD) (mouse) | This paper | JCP 22 |
| pMAL-MBP-AID (human), codon optimized | R. Kohli Laboratory | JCP 113 (RK016) |
| pMAL-MBP-AID E58 (CD) (human), codon optimized | R. Kohli Laboratory | JCP 114 (RK019) |
| pMAL-MBP-AID G133V (human), codon optimized | This paper | JCP 115 |
| pMIG-AID | Zheng et al., 2015 | JCP 24 |
| pMIG-AID G133V | Zheng et al., 2015 | JCP 27 |
| pMIG-AID H56R ES8Q (pMIG-AID CD) | Vuong et al., 2013 | JCP 22 |
| pMIG-AID-AID CD (fusion) | This paper | JCP 197 |
| pMIG-AID G133V-AID CD (fusion) | This paper | JCP 201 |
| pMIG-AID CD-AID CD (fusion) | This paper | JCP 221 |
| pMIG-AID G133V-mCherry (fusion) | This paper | JCP 220 |
| Software and Algorithms | ||
| Trimmomatic (v.0.36) | Bolger et al., 2014 | http://www.usadellab.org/cms/?page=trimmomatic |
| Bowtie2 (v2.2.9) | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2/index.shtml |
| MACS2 (v2.1.1.20160309) | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| ChipPeakAnno (v3.16.1) | Zhu et al., 2010 | https://bioconductor.org/packages/release/bioc/html/ChIPpeakAnno.html |
| UCSC mm10 Known Gene Annotation Package (v3.4.4) | Speir et al., 2016 | https://bioconductor.org/packages/release/data/annotation/html/TxDb.Mmusculus.UCSC.mm10.knownGene.html |
| GenomicAlignments (v.1.18.1) | Lawrence et al., 2013 | https://bioconductor.org/packages/release/bioc/html/GenomicAlignments.html |
| DESeq2 (v.1.22.2) | Love et al., 2014 | http://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| deeptools2 (v3.21.1) | Ramírez et al, 2016 | https://deeptools.readthedocs.io/en/develop/index.html |
| Gviz (v.1.18.2) | Hahne and Ivanek, 2016 | http://bioconductor.org/packages/release/bioc/html/Gviz.html |
| ComplexHeatmap (v1.99.7) | Gu et al, 2016 | https://bioconductor.org/packages/release/bioc/html/ComplexHeatmap.html |
| R (v.3.5.3) | https://www.r-project.org/ | https://www.r-project.org/ |
| Tximport (v1.12.3) | Soneson et al., 2015 | https://bioconductor.org/packages/release/bioc/html/tximport.html |
| ggPlot2 | Wickham, 2009 | https://ggplot2.tidyverse.org/ |
| Prism | Graphpad | RRID: SCR_002798 |
| FlowJo | TreeStar | RRID: SCR_008520 |
| Excel | Microsoft | RRID: SCR_016137 |
| Illustrator | Adobe | RRID: SCR_010279 |
| Photoshop | Adobe | RRID: SCR_014199 |
| FIJI | https://imagej.net/Fiji | RRID: SCR_002285 |
ACKNOWLEDGMENTS
W.T.Y. was supported by a Special Fellow award from the Leukemia & Lymphoma Society and an NIH T32 training grant (CA009149). C.M.L. is a Cancer Research Institute-Carson Family Fellow and was supported by a T32 award from the NIH (CA009149). J.W.Y. and D.A. were supported by the DIR, NIAID, Bethesda, MD. J.C.S. was supported by grants from the NIH (AI100874, AI130043, and P30CA008748), the Ludwig Center for Cancer Immunotherapy, the Burroughs Wellcome Fund, and the American Cancer Society. J.C. was supported by grants from the National Institutes of Health (R01AI072194, R01AI124186, R56AI072194, U54CA137788 and P30CA008748), the Starr Cancer Research Foundation, the Ludwig Center for Cancer Immunotherapy, MSKCC Functional Genomics, and the Geoffrey Beene Cancer Center. We acknowledge the use of the Integrated Genomics Operation Core, funded by the NCI Cancer Center Support Grant (CCSG, P30 CA08748), Cycle for Survival, and the Marie-Josée and Henry R. Kravis Center for Molecular Oncology. This work was supported by NCI Cancer Center support grant P30 CA08748 to Microchemistry and Proteomics Core Laboratory, Memorial Sloan-Kettering Cancer Center. We are grateful to the Molecular Cytology Core Facility at Memorial Sloan-Kettering Cancer Center funded by Core Grant (P30 CA008748) for immunofluorescence technical assistance. We thank A. Bravo for help with maintenance of the mouse colony. We thank Dr. Ashutosh Chaudhry for assistance with sorting experiments, Dr. Rahul Kohli for plasmids and advice regarding the MBP-AID purification strategy, and Dr. Louis Staudt for advice regarding the DLBCL analysis.
Footnotes
DECLARATION OF INTERESTS
The authors have no conflicts of interest to disclose.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Alizadeh AA, Eisen MB, Davis RE, Ma C, Lossos IS, Rosenwald A, Boldrick JC, Sabet H, Tran T, Yu X, et al. (2000). Distinct types of diffuse large B-cell lymphoma identified by gene expression profiling. Nature 403, 503–511. [DOI] [PubMed] [Google Scholar]
- Altman MO, Angeletti D, and Yewdell JW (2018). Antibody Immunodominance: The Key to Understanding Influenza Virus Antigenic Drift. Viral Immunol 31, 142–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alvarez-Prado AF, Perez-Duran P, Perez-Garcia A, Benguria A, Torroja C, de Yebenes VG, and Ramiro AR (2018). A broad atlas of somatic hypermutation allows prediction of activation-induced deaminase targets. The Journal of experimental medicine 215, 761–771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arima H, Fujimoto M, Nishikori M, Kitano T, Kishimoto W, Hishizawa M, Kondo T, Yamashita K, Hirata M, Haga H, et al. (2018). Prognostic impact of activation-induced cytidine deaminase expression for patients with diffuse large B-cell lymphoma. Leuk Lymphoma 59, 2085–2095. [DOI] [PubMed] [Google Scholar]
- Bannard O, McGowan SJ, Ersching J, Ishido S, Victora GD, Shin JS, and Cyster JG (2016). Ubiquitin-mediated fluctuations in MHC class II facilitate efficient germinal center B cell responses. The Journal of experimental medicine 213, 993–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardin C, and Leroy JL (2008). The formation pathway of tetramolecular G-quadruplexes. Nucleic acids research 36, 477–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhattacharyya D, Mirihana Arachchilage G, and Basu S (2016). Metal Cations in G-Quadruplex Folding and Stability. Front Chem 4, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boulianne B, Rojas OL, Haddad D, Zaheen A, Kapelnikov A, Nguyen T, Li C, Hakem R, Gommerman JL, and Martin A (2013). AID and caspase 8 shape the germinal center response through apoptosis. Journal of immunology 191, 5840–5847. [DOI] [PubMed] [Google Scholar]
- Carrasco-Salas Y, Malapert A, Sulthana S, Molcrette B, Chazot-Franguiadakis L, Bernard P, Chedin F, Faivre-Moskalenko C, and Vanoosthuyse V (2019). The extruded non-template strand determines the architecture of R-loops. Nucleic acids research. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casellas R, Basu U, Yewdell WT, Chaudhuri J, Robbiani DF, and Di Noia JM (2016). Mutations, kataegis and translocations in B cells: understanding AID promiscuous activity. Nature reviews Immunology 16, 164–176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhuri J, Tian M, Khuong C, Chua K, Pinaud E, and Alt FW (2003). Transcription-targeted DNA deamination by the AID antibody diversification enzyme. Nature 422, 726–730. [DOI] [PubMed] [Google Scholar]
- Chiarle R, Zhang Y, Frock RL, Lewis SM, Molinie B, Ho YJ, Myers DR, Choi VW, Compagno M, Malkin DJ, et al. (2011). Genome-wide translocation sequencing reveals mechanisms of chromosome breaks and rearrangements in B cells. Cell 147, 107–119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortizas EM, Zahn A, Safavi S, Reed JA, Vega F, Di Noia JM, and Verdun RE (2016). UNG protects B cells from AID-induced telomere loss. The Journal of experimental medicine 213, 2459–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis CA, Hitz BC, Sloan CA, Chan ET, Davidson JM, Gabdank I, Hilton JA, Jain K, Baymuradov UK, Narayanan AK, et al. (2018). The Encyclopedia of DNA elements (ENCODE): data portal update. Nucleic acids research 46, D794–D801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Noia JM, and Neuberger MS (2007). Molecular mechanisms of antibody somatic hypermutation. Annu Rev Biochem 76, 1–22. [DOI] [PubMed] [Google Scholar]
- Dunnick W, Hertz GZ, Scappino L, and Gritzmacher C (1993). DNA sequences at immunoglobulin switch region recombination sites. Nucleic acids research 21, 365–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette ML, Handa P, Vincent JA, Taylor AF, and Maizels N (2004). Intracellular transcription of G-rich DNAs induces formation of G-loops, novel structures containing G4 DNA. Genes & development 18, 1618–1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duquette ML, Huber MD, and Maizels N (2007). G-rich proto-oncogenes are targeted for genomic instability in B-cell lymphomas. Cancer research 67, 2586–2594. [DOI] [PubMed] [Google Scholar]
- Duquette ML, Pham P, Goodman MF, and Maizels N (2005). AID binds to transcription-induced structures in c-MYC that map to regions associated with translocation and hypermutation. Oncogene 24, 5791–5798. [DOI] [PubMed] [Google Scholar]
- Durandy A, Peron S, Taubenheim N, and Fischer A (2006). Activation-induced cytidine deaminase: structure-function relationship as based on the study of mutants. Hum Mutat 27, 1185–1191. [DOI] [PubMed] [Google Scholar]
- Eisen HN, and Reilly EB (1985). Lambda chains and genes in inbred mice. Annual review of immunology 3, 337–365. [DOI] [PubMed] [Google Scholar]
- Frank GM, Angeletti D, Ince WL, Gibbs JS, Khurana S, Wheatley AK, Max EE, McDermott AB, Golding H, Stevens J, et al. (2015). A Simple Flow-Cytometric Method Measuring B Cell Surface Immunoglobulin Avidity Enables Characterization of Affinity Maturation to Influenza A Virus. MBio 6, e01156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gellert M, Lipsett MN, and Davies DR (1962). Helix formation by guanylic acid. Proceedings of the National Academy of Sciences of the United States of America 48, 2013–2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilliam AC, Shen A, Richards JE, Blattner FR, Mushinski JF, and Tucker PW (1984). Illegitimate recombination generates a class switch from C mu to C delta in an IgD-secreting plasmacytoma. Proceedings of the National Academy of Sciences of the United States of America 81, 4164–4168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greeve J, Philipsen A, Krause K, Klapper W, Heidorn K, Castle BE, Janda J, Marcu KB, and Parwaresch R (2003). Expression of activation-induced cytidine deaminase in human B-cell non-Hodgkin lymphomas. Blood 101, 3574–3580. [DOI] [PubMed] [Google Scholar]
- Greiner A, Tobollik S, Buettner M, Jungnickel B, Herrmann K, Kremmer E, and Niedobitek G (2005). Differential expression of activation-induced cytidine deaminase (AID) in nodular lymphocyte-predominant and classical Hodgkin lymphoma. J Pathol 205, 541–547. [DOI] [PubMed] [Google Scholar]
- Gu Z, Eils R, and Schlesner M (2016). Complex heatmaps reveal patterns and correlations in multidimensional genomic data. Bioinformatics 32, 2847–2849. [DOI] [PubMed] [Google Scholar]
- Hahne F, and Ivanek R (2016). Visualizing Genomic Data Using Gviz and Bioconductor. Statistical Genomics: Methods and Protocols 1418, 335–351. [DOI] [PubMed] [Google Scholar]
- Hansel-Hertsch R, Di Antonio M, and Balasubramanian S (2017). DNA G-quadruplexes in the human genome: detection, functions and therapeutic potential. Nat Rev Mol Cell Biol 18, 279–284. [DOI] [PubMed] [Google Scholar]
- Hogenbirk MA, Heideman MR, Velds A, van den Berk PC, Kerkhoven RM, van Steensel B, and Jacobs H (2013). Differential programming of B cells in AID deficient mice. PLoS One 8, e69815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogenbirk MA, Velds A, Kerkhoven RM, and Jacobs H (2012). Reassessing genomic targeting of AID. Nature immunology 13, 797–798; author reply 798–800. [DOI] [PubMed] [Google Scholar]
- Ise W, Fujii K, Shiroguchi K, Ito A, Kometani K, Takeda K, Kawakami E, Yamashita K, Suzuki K, Okada T, et al. (2018). T Follicular Helper Cell-Germinal Center B Cell Interaction Strength Regulates Entry into Plasma Cell or Recycling Germinal Center Cell Fate. Immunity 48, 702–715 e704. [DOI] [PubMed] [Google Scholar]
- Jiao J, Lv Z, Zhang P, Wang Y, Yuan M, Yu X, Otieno Odhiambo W, Zheng M, Zhang H, Ma Y, et al. (2020). AID assists DNMT1 to attenuate BCL6 expression through DNA methylation in diffuse large B-cell lymphoma cell lines. Neoplasia 22, 142–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jolly CJ, Klix N, and Neuberger MS (1997). Rapid methods for the analysis of immunoglobulin gene hypermutation: application to transgenic and gene targeted mice. Nucleic acids research 25, 1913–1919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kawamura K, Wada A, Wang JY, Li Q, Ishii A, Tsujimura H, Takagi T, Itami M, Tada Y, Tatsumi K, et al. (2016). Expression of activation-induced cytidine deaminase is associated with a poor prognosis of diffuse large B cell lymphoma patients treated with CHOP-based chemotherapy. J Cancer Res Clin Oncol 142, 27–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khodabakhshi AH, Morin RD, Fejes AP, Mungall AJ, Mungall KL, Bolger-Munro M, Johnson NA, Connors JM, Gascoyne RD, Marra MA, et al. (2012). Recurrent targets of aberrant somatic hypermutation in lymphoma. Oncotarget 3, 1308–1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Sturgill D, Tran AD, Sinclair DA, and Oberdoerffer P (2016). Controlled DNA double-strand break induction in mice reveals post-damage transcriptome stability. Nucleic acids research 44, e64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klein IA, Resch W, Jankovic M, Oliveira T, Yamane A, Nakahashi H, Di Virgilio M, Bothmer A, Nussenzweig A, Robbiani DF, et al. (2011). Translocation-capture sequencing reveals the extent and nature of chromosomal rearrangements in B lymphocytes. Cell 147, 95–106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohli RM, Abrams SR, Gajula KS, Maul RW, Gearhart PJ, and Stivers JT (2009). A portable hot spot recognition loop transfers sequence preferences from APOBEC family members to activation-induced cytidine deaminase. The Journal of biological chemistry 284, 22898–22904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nature methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence M, Huber W, Pages H, Aboyoun P, Carlson M, Gentleman R, Morgan MT, and Carey VJ (2013). Software for computing and annotating genomic ranges. PLoS computational biology 9, e1003118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeJeune JM, Briles DE, Lawton AR, and Kearney JF (1982). Estimate of the light chain repertoire size of fetal and adult BALB/cJ and CBA/J mice. Journal of immunology 129, 673–677. [PubMed] [Google Scholar]
- Lieber MR (2016). Mechanisms of human lymphoid chromosomal translocations. Nat Rev Cancer 16, 387–398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu M, Duke JL, Richter DJ, Vinuesa CG, Goodnow CC, Kleinstein SH, and Schatz DG (2008). Two levels of protection for the B cell genome during somatic hypermutation. Nature 451, 841–845. [DOI] [PubMed] [Google Scholar]
- Lossos IS, Levy R, and Alizadeh AA (2004). AID is expressed in germinal center B-cell-like and activated B-cell-like diffuse large-cell lymphomas and is not correlated with intraclonal heterogeneity. Leukemia 18, 1775–1779. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Z, Tsai AG, Akasaka T, Ohno H, Jiang Y, Melnick AM, Greisman HA, and Lieber MR (2013). BCL6 breaks occur at different AID sequence motifs in Ig-BCL6 and non-Ig-BCL6 rearrangements. Blood 121, 4551–4554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mahdaviani SA, Hirbod-Mobarakeh A, Wang N, Aghamohammadi A, Hammarstrom L, Masjedi MR, Pan-Hammarstrom Q, and Rezaei N (2012). Novel mutation of the activation-induced cytidine deaminase gene in a Tajik family: special review on hyper-immunoglobulin M syndrome. Expert review of clinical immunology 8, 539–546. [DOI] [PubMed] [Google Scholar]
- Matthews AJ, Zheng S, DiMenna LJ, and Chaudhuri J (2014). Regulation of immunoglobulin class-switch recombination: choreography of noncoding transcription, targeted DNA deamination, and long-range DNA repair. Advances in immunology 122, 1–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McBride KM, Gazumyan A, Woo EM, Schwickert TA, Chait BT, and Nussenzweig MC (2008). Regulation of class switch recombination and somatic mutation by AID phosphorylation. The Journal of experimental medicine 205, 2585–2594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mesin L, Ersching J, and Victora GD (2016). Germinal Center B Cell Dynamics. Immunity 45, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot SP, Litzler LC, Subramani PG, Eranki AK, Fifield H, Patenaude AM, Gilmore JC, Santiago GE, Bagci H, Cote JF, et al. (2018). A licensing step links AID to transcription elongation for mutagenesis in B cells. Nat Commun 9, 1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Methot SP, Litzler LC, Trajtenberg F, Zahn A, Robert F, Pelletier J, Buschiazzo A, Magor BG, and Di Noia JM (2015). Consecutive interactions with HSP90 and eEF1A underlie a functional maturation and storage pathway of AID in the cytoplasm. The Journal of experimental medicine 212, 581–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller TP, Lippman SM, Spier CM, Slymen DJ, and Grogan TM (1988). HLA-DR (Ia) immune phenotype predicts outcome for patients with diffuse large cell lymphoma. The Journal of clinical investigation 82, 370–372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moldenhauer G, Popov SW, Wotschke B, Bruderlein S, Riedl P, Fissolo N, Schirmbeck R, Ritz O, Moller P, and Leithauser F (2006). AID expression identifies interfollicular large B cells as putative precursors of mature B-cell malignancies. Blood 107, 2470–2473. [DOI] [PubMed] [Google Scholar]
- Momburg F, Herrmann B, Moldenhauer G, and Moller P (1987). B-cell lymphomas of high-grade malignancy frequently lack HLA-DR, -DP and -DQ antigens and associated invariant chain. Int J Cancer 40, 598–603. [DOI] [PubMed] [Google Scholar]
- Mondal S, Begum NA, Hu W, and Honjo T (2016). Functional requirements of AID’s higher order structures and their interaction with RNA-binding proteins. Proceedings of the National Academy of Sciences of the United States of America 113, E1545–1554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mottok A, Woolcock B, Chan FC, Tong KM, Chong L, Farinha P, Telenius A, Chavez E, Ramchandani S, Drake M, et al. (2015). Genomic Alterations in CIITA Are Frequent in Primary Mediastinal Large B Cell Lymphoma and Are Associated with Diminished MHC Class II Expression. Cell Rep 13, 1418–1431. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Kinoshita K, Fagarasan S, Yamada S, Shinkai Y, and Honjo T (2000). Class switch recombination and hypermutation require activation-induced cytidine deaminase (AID), a potential RNA editing enzyme. Cell 102, 553–563. [DOI] [PubMed] [Google Scholar]
- Muramatsu M, Sankaranand VS, Anant S, Sugai M, Kinoshita K, Davidson NO, and Honjo T (1999). Specific expression of activation-induced cytidine deaminase (AID), a novel member of the RNA-editing deaminase family in germinal center B cells. The Journal of biological chemistry 274, 18470–18476. [DOI] [PubMed] [Google Scholar]
- Neaves KJ, Huppert JL, Henderson RM, and Edwardson JM (2009). Direct visualization of G-quadruplexes in DNA using atomic force microscopy. Nucleic acids research 37, 6269–6275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palese P (1977). The genes of influenza virus. Cell 10, 1–10. [DOI] [PubMed] [Google Scholar]
- Pankotai T., Bonhomme C., Chen D., and Soutoglou E. (2012). DNAPKcs-dependent arrest of RNA polymerase II transcription in the presence of DNA breaks. Nat Struct Mol Biol 19, 276–282. [DOI] [PubMed] [Google Scholar]
- Papavasiliou FN, and Schatz DG (2002). The activation-induced deaminase functions in a postcleavage step of the somatic hypermutation process. The Journal of experimental medicine 195, 1193–1198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Bhagat G, Jankovic M, Compagno M, Smith P, Muramatsu M, Honjo T, Morse HC 3rd, Nussenzweig MC, and Dalla-Favera R (2008). AID is required for germinal center-derived lymphomagenesis. Nat Genet 40, 108–112. [DOI] [PubMed] [Google Scholar]
- Pasqualucci L, and Dalla-Favera R (2018). Genetics of diffuse large B-cell lymphoma. Blood 131, 2307–2319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasqualucci L, Guglielmino R, Houldsworth J, Mohr J, Aoufouchi S, Polakiewicz R, Chaganti RS, and Dalla-Favera R (2004). Expression of the AID protein in normal and neoplastic B cells. Blood 104, 3318–3325. [DOI] [PubMed] [Google Scholar]
- Pavri R, Gazumyan A, Jankovic M, Di Virgilio M, Klein I, Ansarah-Sobrinho C, Resch W, Yamane A, Reina San-Martin B, Barreto V, et al. (2010). Activation-induced cytidine deaminase targets DNA at sites of RNA polymerase II stalling by interaction with Spt5. Cell 143, 122–133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Periyasamy M, Patel H, Lai CF, Nguyen VTM, Nevedomskaya E, Harrod A, Russell R, Remenyi J, Ochocka AM, Thomas RS, et al. (2015). APOBEC3B-Mediated Cytidine Deamination Is Required for Estrogen Receptor Action in Breast Cancer. Cell Rep 13, 108–121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen-Mahrt SK, Harris RS, and Neuberger MS (2002). AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 418, 99–103. [DOI] [PubMed] [Google Scholar]
- Qian J, Wang Q, Dose M, Pruett N, Kieffer-Kwon KR, Resch W, Liang G, Tang Z, Mathe E, Benner C, et al. (2014). B cell super-enhancers and regulatory clusters recruit AID tumorigenic activity. Cell 159, 1524–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiao Q, Wang L, Meng FL, Hwang JK, Alt FW, and Wu H (2017). AID Recognizes Structured DNA for Class Switch Recombination. Molecular cell 67, 361–373 e364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rada C, Jarvis JM, and Milstein C (2002). AID-GFP chimeric protein increases hypermutation of Ig genes with no evidence of nuclear localization. Proceedings of the National Academy of Sciences of the United States of America 99, 7003–7008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramirez F, Ryan DP, Gruning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dundar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic acids research 44, W160–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramiro AR, Jankovic M, Eisenreich T, Difilippantonio S, Chen-Kiang S, Muramatsu M, Honjo T, Nussenzweig A, and Nussenzweig MC (2004). AID is required for c-myc/IgH chromosome translocations in vivo. Cell 118, 431–438. [DOI] [PubMed] [Google Scholar]
- Revy P, Muto T, Levy Y, Geissmann F, Plebani A, Sanal O, Catalan N, Forveille M, Dufourcq-Labelouse R, Gennery A, et al. (2000). Activation-induced cytidine deaminase (AID) deficiency causes the autosomal recessive form of the Hyper-IgM syndrome (HIGM2). Cell 102, 565–575. [DOI] [PubMed] [Google Scholar]
- Rimsza LM, Roberts RA, Miller TP, Unger JM, LeBlanc M, Braziel RM, Weisenberger DD, Chan WC, Muller-Hermelink HK, Jaffe ES, et al. (2004). Loss of MHC class II gene and protein expression in diffuse large B-cell lymphoma is related to decreased tumor immunosurveillance and poor patient survival regardless of other prognostic factors: a follow-up study from the Leukemia and Lymphoma Molecular Profiling Project. Blood 103, 4251–4258. [DOI] [PubMed] [Google Scholar]
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al. (2002). The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 346, 1937–1947. [DOI] [PubMed] [Google Scholar]
- Rothaeusler K, and Baumgarth N (2010). B-cell fate decisions following influenza virus infection. European journal of immunology 40, 366–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmitz R, Wright GW, Huang DW, Johnson CA, Phelan JD, Wang JQ, Roulland S, Kasbekar M, Young RM, Shaffer AL, et al. (2018). Genetics and Pathogenesis of Diffuse Large B-Cell Lymphoma. N Engl J Med 378, 1396–1407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sen D, and Gilbert W (1988). Formation of parallel four-stranded complexes by guanine-rich motifs in DNA and its implications for meiosis. Nature 334, 364–366. [DOI] [PubMed] [Google Scholar]
- Shanbhag NM., Rafalska-Metcalf IU., Balane-Bolivar C., Janicki SM., and Greenberg RA. (2010). ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 141, 970–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen HM, Peters A, Baron B, Zhu X, and Storb U (1998). Mutation of BCL-6 gene in normal B cells by the process of somatic hypermutation of Ig genes. Science 280, 1750–1752. [DOI] [PubMed] [Google Scholar]
- Soneson C, Love MI, and Robinson MD (2015). Differential analyses for RNA-seq: transcript-level estimates improve gene-level inferences. F1000Res 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Speir ML, Zweig AS, Rosenbloom KR, Raney BJ, Paten B, Nejad P, Lee BT, Learned K, Karolchik D, Hinrichs AS, et al. (2016). The UCSC Genome Browser database: 2016 update. Nucleic acids research 44, D717–725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steidl C, Shah SP, Woolcock BW, Rui L, Kawahara M, Farinha P, Johnson NA, Zhao Y, Telenius A, Neriah SB, et al. (2011). MHC class II transactivator CIITA is a recurrent gene fusion partner in lymphoid cancers. Nature 471, 377–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Victora GD, Dominguez-Sola D, Holmes AB, Deroubaix S, Dalla-Favera R, and Nussenzweig MC (2012). Identification of human germinal center light and dark zone cells and their relationship to human B-cell lymphomas. Blood 120, 2240–2248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vuong BQ, Herrick-Reynolds K, Vaidyanathan B, Pucella JN, Ucher AJ, Donghia NM, Gu X, Nicolas L, Nowak U, Rahman N, et al. (2013). A DNA break- and phosphorylation-dependent positive feedback loop promotes immunoglobulin classs-witch recombination. Nature immunology 14, 1183–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei M, Shinkura R, Doi Y, Maruya M, Fagarasan S, and Honjo T (2011). Mice carrying a knock-in mutation of Aicda resulting in a defect in somatic hypermutation have impaired gut homeostasis and compromised mucosal defense. Nature immunology 12, 264–270. [DOI] [PubMed] [Google Scholar]
- Whittle JR, Wheatley AK, Wu L, Lingwood D, Kanekiyo M, Ma SS, Narpala SR, Yassine HM, Frank GM, Yewdell JW, et al. (2014). Flow cytometry reveals that H5N1 vaccination elicits cross-reactive stem-directed antibodies from multiple Ig heavy-chain lineages. J Virol 88, 4047–4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wickham H (2009). Ggplot2: Elegant Graphics for Data Analysis, 2nd edn (Springer Publishing Company Incorporated; ). [Google Scholar]
- Woloschak GE, and Krco CJ (1987). Regulation of kappa/lambda immunoglobulin light chain expression in normal murine lymphocytes. Molecular immunology 24, 751–757. [DOI] [PubMed] [Google Scholar]
- Wright GW, Huang DW, Phelan JD, Coulibaly ZA, Roulland S, Young RM, Wang JQ, Schmitz R, Morin RD, Tang J, et al. (2020). A Probabilistic Classification Tool for Genetic Subtypes of Diffuse Large B Cell Lymphoma with Therapeutic Implications. Cancer Cell 37, 551–568 e514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu Z, Zan H, Pone EJ, Mai T, and Casali P (2012). Immunoglobulin class-switch DNA recombination: induction, targeting and beyond. Nature reviews Immunology 12, 517–531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamane A, Resch W, Kuo N, Kuchen S, Li Z, Sun HW, Robbiani DF, McBride K, Nussenzweig MC, and Casellas R (2011). Deep-sequencing identification of the genomic targets of the cytidine deaminase AID and its cofactor RPA in B lymphocytes. Nature immunology 12, 62–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye BH, Lista F, Lo Coco F, Knowles DM, Offit K, Chaganti RS, and Dalla-Favera R (1993). Alterations of a zinc finger-encoding gene, BCL-6, in diffuse large-cell lymphoma. Science 262, 747–750. [DOI] [PubMed] [Google Scholar]
- Yewdell WT, and Chaudhuri J (2017). A transcriptional serenAID: the role of noncoding RNAs in class switch recombination. International immunology 29, 183–196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarrin AA, Alt FW, Chaudhuri J, Stokes N, Kaushal D, Du Pasquier L, and Tian M (2004). An evolutionarily conserved target motif for immunoglobulin class-switch recombination. Nature immunology 5, 1275–1281. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang Y, Ba Z, Kyritsis N, Casellas R, and Alt FW (2019). Fundamental roles of chromatin loop extrusion in antibody class switching. Nature 575, 385–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng S, Vuong BQ, Vaidyanathan B, Lin JY, Huang FT, and Chaudhuri J (2015). Non-coding RNA Generated following Lariat Debranching Mediates Targeting of AID to DNA. Cell 161, 762–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu LJ, Gazin C, Lawson ND, Pages H, Lin SM, Lapointe DS, and Green MR (2010). ChIPpeakAnno: a Bioconductor package to annotate ChIP-seq and ChIP-chip data. BMC Bioinformatics 11, 237. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S2. Aicda−/− versus WT ChIP-seq analysis, Related to Figure 6.
Table S3. AicdaGV/GV versus WT ChIP-seq analysis, Related to Figure 6.
Table S4. AicdaGV/GV versus Aicda−/− ChIP-seq analysis, Related to Figure 6.
Data Availability Statement
ChIP-seq datasets generated during this study are available at the Gene Omnibus Repository under accession code GSE136959.