

Abstract
Olivocochlear neurons make temporary cholinergic synapses on inner hair cells of the rodent cochlea in the first 2 to 3 wk after birth. Repetitive stimulation of these efferent neurons causes facilitation of evoked release and increased spontaneous release that continues for seconds to minutes. Presynaptic nicotinic acetylcholine receptors (nAChRs) are known to modulate neurotransmitter release from brain neurons. The present study explores the hypothesis that presynaptic nAChRs help to increase spontaneous release from efferent terminals on cochlear hair cells. Direct application of nicotine (which does not activate the hair cells’ α9α10-containing nAChRs) produces sustained efferent transmitter release, implicating presynaptic nAChRs in this response. The effect of nicotine was reduced by application of ryanodine that reduces release of calcium from intraterminal stores.
NEW & NOTEWORTHY Sensory organs exhibit spontaneous activity before the onset of response to external stimuli. Such activity in the cochlea is subject to modulation by cholinergic efferent neurons that directly inhibit sensory hair cells (inner hair cells). Those efferent neurons are themselves subject to various modulatory mechanisms. One such mechanism is positive feedback by released acetylcholine onto presynaptic nicotinic acetylcholine receptors causing further release of acetylcholine.
Keywords: cochlea, efferent, hair cell, nicotinic, presynaptic
INTRODUCTION
Efferent cholinergic neurons project from periolivary nuclei in the brainstem to innervate the mammalian cochlea. Medial olivocochlear (MOC) efferent neurons form synapses onto mature outer hair cells (OHCs) and temporarily onto inner hair cells (IHCs) before the onset of hearing. The MOC efferent terminals release acetylcholine (ACh) to hyperpolarize and shunt hair cells through the action of α9α10-containing nicotinic ACh receptors (nAChRs) coupled to calcium-dependent potassium channels (SK2 and/or BK) (Rohmann et al. 2015; Wersinger et al. 2010). These actions reduce OHC electromotility, the mechanical amplification that confers cochlear sensitivity and frequency selectivity (Guinan 2010). Olivocochlear feedback also plays a role during development. Transient efferent inhibition of IHCs before the onset of hearing influences afferent synaptic maturation of the IHC (Johnson et al. 2013) and shapes spontaneous activity that instructs the connectivity of central auditory nuclei (Clause et al. 2017). Efferent synapses on both IHCs and OHCs show short-term facilitation (Ballestero et al. 2011; Boero et al. 2018; Goutman et al. 2005) and longer-term modulation through a variety of mechanisms (Kong et al. 2013; Wedemeyer et al. 2013; Ye et al. 2017; Zorrilla de San Martín et al. 2010). The probability of release from these efferent neurons in the resting condition is quite low (fewer than one vesicle released per action potential), so they must facilitate during repetitive firing to inhibit hair cells effectively. Recordings in adult animals showed that efferent neurons were sensitive and sharply tuned to acoustic stimulation that could cause firing at rates exceeding 100 Hz (Brown et al. 1998), which rates can cause maximal facilitation of ACh release onto OHCs ex vivo (Ballestero et al. 2011; Goutman et al. 2005). Prior to the onset of hearing, spontaneous activity arising in the cochlea also can include higher frequency components (Jones et al. 2007; Tritsch et al. 2010). Through central connections, afferent neurons may in turn drive facilitated release of ACh from efferent terminals on IHCs. Prolonged efferent activation not only facilitates ongoing transmitter release but also can cause prolonged delayed release (Goutman et al. 2005) and long-lasting potentiation of spontaneous transmitter release. One possibility is that activation of presynaptic nAChRs by spillover of ACh from the synaptic cleft helps to drive delayed and increased spontaneous release from cochlear efferent terminals.
Presynaptic nAChRs have long been implicated in the modulation of synaptic function and connectivity at the neuromuscular junction, and in the central nervous system (McGehee et al. 1995) (reviewed in Picciotto et al. 2012; Role and Berg 1996), including within the auditory thalamus-medial geniculate body (Sottile et al. 2017). Their effects can range from facilitation to longer-term potentiation of evoked transmitter release to modification of innervation during development. Activation of presynaptic nAChRs can enhance evoked and/or spontaneous transmitter release via mechanisms that include voltage- or ligand-gated calcium entry, calcium release from internal stores, and modification of synapsins by second-messenger-dependent kinases (CAMKII and PKA) (Cheng and Yakel 2015). These longer-lasting effects of presynaptic nAChR activation may be particularly relevant for developmental and pathogenic effects of nicotine exposure in general and for the auditory system in particular (Morley 2005; Paquette et al. 2018; Sottile et al. 2017). This possibility extends to developmental plasticity of cochlear innervation. MOC efferent neurons provide functional input to rodent IHCs for 2 to 3 wk after birth (Glowatzki and Fuchs 2000; Katz et al. 2004; Roux et al. 2011), during which time cochlear hair cells and afferents exhibit patterned electrical activity (Marcotti et al. 2003; Tritsch et al. 2007) but the animal does not yet respond to sound. Intriguingly, functional efferent synapses return to IHCs of deaf adult mice (Corns et al. 2018; Lauer et al. 2012; Zachary and Fuchs 2015), further motivating a study of modulatory mechanisms.
The present work demonstrates that nicotine, which itself does not activate α9α10-containing nAChRs in hair cells (Elgoyhen et al. 1994), instead evokes stochastic ACh release from cochlear efferent neurons. This will be referred to throughout as “nicotine-evoked” release as distinct from transmitter release evoked by efferent action potentials (“shock-evoked”). Mouse pups of either sex, ranging in age from 8 to 11 days postnatal were studied. Both immature IHCs and OHCs were examined, with the majority of experiments carried out on IHCs.
METHODS
Mice.
All procedures involving animals were approved by the Johns Hopkins University Animal Care and Use Committee. In brief, mice were deeply anaesthetized (isoflurane or CO2 inhalation) till unresponsive to toe or tail pinch, then decapitated. C57BL/6J mice were obtained from The Jackson Laboratory (no. 000664). Mice lacking functional α7 nAChRs (B6.129S7-Chrna7tm1Bay/J) were also purchased from the Jackson Laboratory (no. 003232). Mice of either sex (sexually immature) were studied from postnatal day 8 (P8) to P11. The day of birth was counted as P0. Mice from at least three litters were used for most experiments.
Tissue preparation.
The apical cochlear turn (average length of 3 exemplar dissections was 1.5 ± 0.1 mm, ∼20% of the total length) was dissected from the otic capsule and transferred to a saline-filled chamber within a few minutes of death. Tissue was secured to a coverslip using fine pins as spring clips. The coverslip with attached tissue was placed in a holding chamber on the stage of a Zeiss Axioskop 2 and superfused with external solution (mM): 5.8 KCl, 144 NaCl, 1.3 CaCl2, 0.9 MgCl2, 0.7 NaH2PO4, 5.6 d-glucose, and 10 HEPES [300 mosmol/kgH2O, pH 7.4 (adjusted with NaOH)]. For some experiments, the external calcium was increased to 5.0 mM. “High potassium” (40 mM) saline substituted sodium with potassium. Experimental solutions (e.g., containing blocking agents) were delivered by switching between reservoirs attached to the bath perfusion array. All recordings were carried out at room temperature (∼22°C).
Intracellular recording from hair cells.
IHCs and OHCs were targeted by visual inspection using a ×40 water immersion objective and differential interference contrast optics. “Patch” pipettes were advanced to contact a targeted hair cell using remotely controlled piezo-electric manipulators (Sutter Instruments ROE 200 and MP 285). Recording pipettes were pulled from 1 mm borosilicate class capillaries (World Precision Instrument Kwik-Fil no. 1B100F-4) and fire-polished to attain resistances of 3–6 MΩ. Sylgard (184-Dow Corning) was applied to reduce electrode capacitance for some recordings. Pipettes were filled with internal solution containing (in mM) 135 KCl, 3.5 MgCl2, 0.1 CaCl2, 5 EGTA, 5 HEPES, and 2.5 Na2-ATP, pH 7.2 (290−310 mosmol/kgH2O, adjusted with KOH). Electrode series resistance ranged from 7 to 20 MΩ and was not compensated for recording the small membrane currents described here. Hair cell membrane currents were recorded either with an Axopatch 200B or a Multiclamp 700B amplifier (Axon Instruments) and analyzed using pClamp (Axon, RRID:SCR_011323), MiniAnalysis (Synaptosoft, RRID:SCR_002184), GraphPad Prism 4 (GraphPad Software, RRID:SCR_002798), and Excel (Microsoft) software. Voltage command potentials are reported as membrane potential, adding −4 mV for the junction potential in these solutions.
Rapid perfusion of nicotinic agonists was achieved using a “puffer” pipette positioned near the targeted hair cell. Fluid was ejected by a pressure pulse from a Picospritzer (General Valve Corp.) controlled by the data acquisition software (Clampex 10, Axon Instruments). Some experiments used a Smart Squirt Micro-Perfusion System (250-μm internal diameter) positioned ∼50 μm from the recorded IHC, connected to a ValveLink8.2 Controller (Automate Scientific). Antagonists, channel blockers and ryanodine were added to the perfusion reservoirs for gradual bath replacement. Efferent axons were stimulated by current pulses delivered between a silver wire inserted into a glass micropipette, filled with external saline (diameter of 20–30 μm), and a return wire inserted in the bath. The shock pipette was positioned ∼20 μm modiolar to the base of a targeted hair cell. An electrically isolated constant current source (Digitimer Limited, model DS3) was triggered within the acquisition software to deliver pulses of ∼50–100 μA, 30–300 μs in duration.
Drugs.
Nicotine solutions (nicotine tartrate salt, MP Biomedicals SKU 0215355491) were prepared daily from powder and adjusted for pH with NaOH. Acetylcholine chloride (no. A6625), (±)-epibatidine dihydrochloride hydrate (no. E1145) and strychnine hydrochloride (no. S8753) were obtained from Sigma-Aldrich. Tetrodotoxin (citrate salt, Tocris no. 1069), ryanodine (Tocris no. 1329), and dihydro-β-erythroidine hydrobromide (Tocris no. 2349) were dissolved in external solution to their final concentrations from frozen stocks daily. Calcium channel blockers were obtained from Alomone Laboratories: ω-conotoxin GVIA (no. C-300), ω-agatoxin IVA (no. STA-500), and SNX-482 (no. RTS-500).
Data analysis.
Spontaneous release after facilitated trains of synaptic currents, or evoked by application of nAChR agonist, occurred sporadically for periods of seconds to minutes. The effect of an experimental manipulation was assessed by summing spontaneous synaptic currents for 40 s after nicotine application. When individual synaptic events could be distinguished this was accomplished by marking each event (semiautomated using a fixed amplitude and area threshold in MiniAnalysis), then calculating waveform area and summing the area of all events within the designated time interval. In those cases where the response to nicotine was more complex and individual events could not be distinguished, the entire recording was integrated for 40 s (in Clampfit after setting the current baseline to zero) following nicotine application and that integral compared between control and experimental conditions.
Trains of electrical shocks produced facilitated and summated synaptic currents, followed by “tail” currents that decayed for several hundred milliseconds (“delayed release”) and an increase in ongoing spontaneous synaptic events. To quantify delayed release, shock trains were repeated 5 to 10 times and the average current and its standard deviation determined. The standard deviation of the tail current was integrated for 1 to 2 s after the final shock and compared between control and experimental conditions. A second method made direct measurement of individual synaptic events after the last efferent shock. Event area was summed and averaged for multiple recordings in each condition. Where group data are compared, statistical significance was determined by two-tailed paired or unpaired t test, for within cell or between group comparisons, respectively.
RESULTS
Cholinergic activation evokes delayed transient currents in cochlear hair cells.
Olivocochlear efferent neurons form cholinergic synapses on outer hair cells (OHCs) and transiently on inner hair cells (IHCs) before the onset of hearing. Electrical stimulation in excised cochlear tissue has been used to show that both types of efferent contacts have initially low probabilities of transmitter release but facilitate markedly with repetitive activation (Ballestero et al. 2011; Goutman et al. 2005; Katz and Elgoyhen 2014). Sustained efferent release of ACh can be followed by randomly timed synaptic events, seen here as delayed release (Fig. 1, A1 and A2). Direct application of ACh produces large membrane currents in hair cells, which also can give rise to seconds-long flurries of transient currents similar to synaptic currents resulting from quantized release by the efferent terminal (Fig. 1, B1 and B2). Transmitter release from olivocochlear efferent terminals has been shown to be modulated by a number of different mechanisms (Kong et al. 2013; Wedemeyer et al. 2013; Ye et al. 2017; Zorrilla de San Martín et al. 2010). The present study aims to learn whether presynaptic nicotinic ACh receptors (nAChRs), presumably activated by ACh spillover during sustained efferent release, participate in modulation of these synapses and to test whether they could influence evoked or spontaneous transmitter release. This effort was carried out using nicotine, which itself does not activate the α9α10-containing nAChRs expressed in hair cells (Elgoyhen et al. 1994; McNiven et al. 1996), nor does it otherwise activate membrane current directly in hair cells (Roux et al. 2016), arguing against the presence of functional nicotine-sensitive nAChRs in hair cells.
Fig. 1.

Sustained cholinergic activation evokes delayed transient postsynaptic currents. A1: a train of 10 shocks at 40 Hz elicits a facilitated and summated postsynaptic response in a young [postnatal day (P) 9] inner hair cell (inward going at a holding potential of −94 mV). Delayed release is seen as randomly timed transient membrane currents that continue long after the last shock. A2: delayed transient currents expanded and enlarged, beginning near the asterisk in A1. B1: application of 1 mM ACh evoked a large inward current at −94 mV followed by numerous small transient currents. B2: transient currents expanded and enlarged beginning near the time marked by asterisk in B1. The membrane current (Im) of the cells is presented as a function of time.
Nicotine evokes transient currents in hair cells.
A nearby puffer pipette applied 1 mM nicotine briefly (500 ms) during intracellular recordings from IHCs in apical cochlear turns excised from young (P8–P11) mice of either sex. This stimulus reliably and reproducibly evoked a prolonged flurry of transient membrane currents (in more than 90% of cells tested). Transient synaptic currents could continue for more than one minute after nicotine application. In some cases after repeated exposures, ongoing transmitter release frequency remained elevated for 10 min or longer. The magnitude of effect varied from cell to cell, in many cells with overlapping events that summed for a sustained inward current at −94 mV (Fig. 2, A1 and A2). At −34 mV, the nicotine-evoked currents were outward-going (Fig. 2, B1 and B2). The waveforms of well-resolved transient currents could be analyzed in a minority of IHCs with lesser response to 1 mM nicotine (Fig. 2C1). The waveforms of nicotine-evoked events occurring after the nicotine puff (Fig. 2C2) were similar to efferent postsynaptic currents for IHCs reported previously (Glowatzki and Fuchs 2000; Katz et al. 2004; Roux et al. 2011). Average amplitude was −27.5 pA (±12.3 SD) at −94 mV (n = 484; 4 IHCs, 3 mice P9–10). The average decay time constant (at 22°C) from single exponential fits of these events was 50.3 ms (±19.7 SD).
Fig. 2.

“Puffer” application of 1 mM nicotine (at shaded vertical bars) reliably evoked long-lasting bursts of transient currents in young (postnatal day 9) mouse inner hair cells. A1: nicotine evoked inward currents at a holding potential of −94 mV. A2: expanded time base. B1: the currents evoked by nicotine were outward at−34 mV (same cell as A). B2: expanded time base. C1: the lesser effect of nicotine on a different inner hair cell. C2 expanded time base for one event marked by the asterisk in C1, single exponential fit to decay (red) with time constant of 52 ms.
While 1 mM nicotine reliably evoked transmitter release onto young IHCs, there was little effect on OHCs at these ages. This may reflect the later arrival of synaptic contacts on OHCs (Roux et al. 2011) or the smaller number of efferent fibers contacting each OHC compared with developing IHCs. Synaptic activity in control conditions was observed only rarely in OHCs in the excised apical turns of young cochleas, even in elevated external potassium to increase the normally very low rate of spontaneous release from efferent terminals. For the minority of OHCs with potassium-accelerated transmitter release (8 OHCs from 5 mice), the average frequency of transient currents for 100 s in 40 mM external K+ before nicotine exposure was 0.47 Hz ± 0.25 SD and was not increased after adding 1 mM nicotine (40 mM K+ continuing); 0.29 Hz ± 0.21 SD (control versus nicotine: P = 0.18, t = 1.42, df = 14, two-tailed paired t test). Transient current amplitudes were 304 pA ± 113 SD in control (at −94 mV in elevated external potassium) and 294 pA ± 124 SD with nicotine in the same conditions (control versus nicotine: P = 0.868, t = 0.169, df = 14, two-tailed paired t test). Decay time constants (at 22°C) from single exponential fits were 65 ms (±23 SD) in control, 55 ms ± 20 SD) after nicotine (control versus nicotine: P = 0.369, t = 0.928, df = 14). Under these recording conditions, the inward current is a combination of flux through the nAChRs and through associated calcium-activated (SK2) potassium channels.
Neuronal nicotinic AChRs modulate presynaptic transmitter release in central nervous system neurons. Homomeric α7 nAChRs are commonly found in this role (McGehee et al. 1995). This possibility was examined by recording from inner hair cells in cochlear tissue excised from prehearing mice lacking the α7 nAChR. Application of ACh or nicotine evoked transient currents in 4 of 5 IHCs from four α7 knockout mice (all were offspring of homozygous parents, genotype confirmed in three pups by PCR) (Fig. 3A). These observations rule out homomeric α7 nAChRs as necessary presynaptic modulators in cochlear efferent neurons. Another candidate neuronal nAChR is composed of α4β2 subunits, which are particularly sensitive to the nicotine analog epibatidine and 20- to 200-fold less sensitive to nicotine itself (Traynor 1998). Application of 10 μM epibatidine evoked transient currents in 6 of 6 IHCs tested (Fig. 3B). In contrast, at 10 µM, nicotine slightly increased the occurrence of synaptic currents in only 2 of 8 cells tested (Fig. 3C), while at 100 µM, nicotine increased synaptic currents in only 4 of 11 cells tested (Fig. 3D). These results, while preliminary, nonetheless suggest that transmitter release from cochlear efferent terminals is modulated by presynaptic nAChRs with an α4β2-like agonist profile.
Fig. 3.

Preliminary classification of presynaptic nicotinic ACh receptors. A: synaptic release elicited by nicotine (1 mM) on an inner hair cell (IHC) from an α7 knockout mouse [representative of 4 of 5 IHCs at postnatal day 9 (P9)]. Membrane potential of −94 mV. Nicotine application indicated by vertical bar. B: the nicotinic agonist epibatidine (10 µM) evoked transient currents in 6 of 6 young IHCs tested (P8, −94 mV). C: nicotine (10 µM) evoked transient currents (at −94 mV) in only 2 of 8 IHCs (P8). D: nicotine (100 µM) evoked currents in an IHC (P9) at −94 mV, representative of response observed in 4 of 11 IHCs tested.
In some IHCs, repeated presentations of micromolar nicotine led to a gradual increase in the frequency of spontaneous synaptic currents lasting many minutes. Overall however, brief application of nicotine at micromolar concentration was of limited utility for these studies to probe underlying mechanisms of action. Therefore, nicotine was applied at 1 mM in experiments to investigate its mechanism of action within the usual time frame available for intracellular recordings in excised tissue.
Nicotine-evoked transient currents result from activation of hair cell nAChRs.
Transient currents evoked by nicotine were inward at −94 mV and outward at −34 mV (Fig. 2). At an intermediate membrane potential (−64 mV), biphasic currents were evident (Fig. 4A). These effects of membrane potential on response waveform are the hallmark of efferent synaptic currents that result from the combined activation of hair cell nAChRs providing cation influx, followed by activation of calcium-dependent potassium channels (Fuchs and Murrow 1992; Glowatzki and Fuchs 2000). In support of that conclusion, bath application of strychnine (10 µM), a potent antagonist of α9α10-containing nAChRs expressed in hair cells (Gómez-Casati et al. 2005) completely eliminated the effect of nicotine in three IHCs (Fig. 4B) and was reversible. This concentration of strychnine also blocked completely the synaptic response evoked by electrical stimulation of the efferents (averaged responses during 5 Hz trains of 50 shocks) in three IHCs (not shown).
Fig. 4.

Nicotine-evoked postsynaptic currents in young inner hair cells. A: the effect of hair cell membrane potential (indicated in mV above each recording) on nicotine-evoked currents. Average waveforms aligned by rise time. The majority of current flows through calcium-activated potassium channels and so reverses around the potassium equilibrium potential of −80 mV. At −64 mV, the current is biphasic due to sequential flux through the hair cell’s nAChR and calcium-activated potassium channels. B: nicotine-evoked currents before (black) and in the presence of strychnine (red; 10 µM applied in the bath), a potent antagonist of α9-containing nAChRs. C: nicotine-evoked currents before (black) and in the presence of tetrodotoxin (red; 1 µM applied in the bath). Vertical gray-shaded bars indicate 500-ms puff of 1 mM nicotine.
Thus, the effect of nicotine depends on activation of α9α10-containing nAChRs in the hair cell. That is, nicotine does not evoke membrane current on its own but rather acts presynaptically to cause ACh release from efferent terminals to activate the hair cell’s cholinergic response. However, nicotine-evoked transmitter release does not require presynaptic action potentials. While bath perfusion with 1 µM tetrodotoxin could completely block shock-evoked release of ACh within several minutes (in 3 of 3 IHCs from 2 mice) (not shown), the response to nicotine was unaffected by exposure to 1 µM tetrodotoxin for 15 min or more (Fig. 4C). Synaptic activity evoked by nicotine was quantified by summation of the areas of individual waveforms for 40 s after nicotine application or by integration of total current over that same time interval when the membrane current fluctuations were too complex to be measured individually. Either method provided a single value that was compared between experimental and control conditions. The ratio of total nicotine-evoked current in tetrodotoxin to that in the preceding control was 0.92 (±0.36 SD, n = 4 cells from 3 mice). For equivalently timed pairs of recordings of the effect of nicotine without exposure to tetrodotoxin, this ratio was 1.11 (±0.32 SD, n = 5 cells from 5 mice, P = 0.42, t = 0.86, df = 7 for comparison to tetrodotoxin exposed, unpaired two-tailed t test).
Thus, the effect of nicotine does not require action potentials in the efferent terminal. However, nicotine might act by depolarizing the efferent terminal to open voltage-gated calcium channels directly and thereby drive transmitter release. This possibility was examined using blockers of voltage-gated calcium channels (VGCCs). Following the lead of earlier studies (Kearney et al. 2019; Zorrilla de San Martín et al. 2010), a combination of three antagonists was used to block the complex of VGCCs that can operate in efferent terminals: the P/Q channel blocker ω-agatoxin IVA (400 nM), the N-type channel blocker ω-conotoxin GVIA (1 µM), and the R-type channel blocker SNX-482 (500 nM). Evoked release was assayed by trains of 50 shocks at 1 to 5 Hz or by paired-pulse stimulation with intervals ranging from 100 to 20 ms in different recordings. Bath perfusion of toxins reduced the shock-evoked response (probability of release) to 16 ± 7% (SD) of control within 10–20 min of perfusion (initial release probability ranged from 0.5 to 0.9 in 3 IHCs from 3 mice) (Fig. 5A). Channel blocking peptides required prolonged perfusion time to achieve this level of inhibition. The effect of these calcium channel blockers on the response to nicotine was assessed by summing the area of individual synaptic currents for 40 s after nicotine application, or by integration of the total current over this same period. The response to nicotine was 13 ± 10% that of the control response in six IHCs from four mice ages P8–P10 (Fig. 5B). Thus, over a similar time course, this trio of channel blockers reduced the effect of nicotine to an equivalent extent compared with their effect on evoked release (P = 0.56, t = 0.59 df = 8 for comparison of blocking effect on shock-evoked versus nicotine-evoked transient currents, unpaired two-tailed t test).
Fig. 5.

Voltage-gated calcium channels and nicotine. A: a cocktail of voltage-gated calcium channel antagonists (400 nM ω-agatoxin IVA, 1 µM ω-conotoxin GVIA, and 500 nM SNX-482) blocked shock-evoked postsynaptic currents. Shock artifacts precede the postsynaptic currents (inward at the holding potential of −94 mV). B: the same cocktail of antagonists reduced nicotine-evoked transient membrane currents. Toxin block required long application time (>15 min). Nicotine (1 mM) was applied during time of shaded vertical bar.
Inhibition of the nicotine effect with ryanodine.
The effect of calcium channel blockers suggests that nicotine simply depolarizes the efferent terminal, opening voltage-gated calcium channels to promote transmitter release. Other observations however argue that the effect of nicotine is more complex. Nicotine produces a long-lasting increase in transmitter release from the efferent neurons innervating IHCs. The long time course of the response and the sustained increase of spontaneous synaptic currents suggests that nicotine might act through second-messenger mediated pathways, rather than directly by voltage-gated calcium influx to trigger transmitter release. This has been found in other studies where presynaptic facilitation by nicotine occurs through release of calcium from ryanodine-sensitive cytoplasmic stores (Dickinson et al. 2008; Sharma et al. 2008). To test whether a similar mechanism is involved here, the response to nicotine was compared in control conditions and in presence of 100 µM ryanodine (by bath perfusion) to block release from stores. Ryanodine severely curtailed nicotine’s ability to evoke postsynaptic currents (Fig. 6, A and B). The effect of ryanodine was quantified by integrating the membrane current or by summation of the waveform area of transient currents for 40 s following a 500-ms-long puff of 1 mM nicotine. The average ratio of integrated or summated current to control (ryanodine/control) for four IHCs from three mice was 0.06 ± 0.03 (SD), whereas repeated applications of 1 mM nicotine produced similar responses in control conditions (or in presence of tetrodotoxin, for example).
Fig. 6.

Ryanodine (100 µM) prevents nicotine-evoked efferent postsynaptic currents and reduces delayed release onto inner hair cells. A: nicotine (1 mM for 500 ms) was applied at time marked by vertical gray bar in control conditions (black) and after bath application of ryanodine (red). B: integral of nicotine-evoked membrane currents from A (after end of puff) in control conditions (black) and after exposure to ryanodine (red). C: 10 shocks at 40 Hz produced facilitated and summated efferent synaptic currents (black). Bath application of 100 µM ryanodine did not change the evoked response significantly but smoothed the decay phase (red). D: integrated standard deviation of averaged currents following shock train (integral begins after last shock). Exposure to ryanodine reduced delayed release seen as its contribution to current variance (red trace).
In contrast, 100 µM ryanodine had less effect on release evoked by trains of 10 shocks at 40 Hz (Fig. 6, C and D). For seven IHCs from five mice, the ratio of peak current amplitude in the presence of 100 µM ryanodine to that in control was 0.78 ± 0.21 (SD). However, ryanodine did reduce the transient currents appearing on the decay phase (tail current) of the synaptic response (delayed release) (Fig. 6C). Delayed release was quantified by averaging several responses, then integrating the standard deviation of the decaying tail current for 1 to 2 s after the last shock-evoked response (Fig. 6D). The integrated standard deviation of the tail current was then compared between control responses and responses obtained after exposure to 100 µM ryanodine applied for 15 min or longer. The ratio of delayed release in 100 µM ryanodine to that in control, evaluated at 1 s post-shock train, averaged 0.54 (±0.25 SD, n = 7 IHCs from 5 mice). Although a two-tailed t test of the comparison of ryanodine’s effect on evoked and delayed responses just failed significance (P = 0.07, t = 1.94, df = 12), exposure to ryanodine could substantially reduce delayed release without an appreciable change in the evoked response (Fig. 6, C and D). The implication is that delayed release is partially dependent on calcium release from internal stores, but evoked release less so.
Another test of this hypothesis would be to quantify delayed release during specific pharmacological blockade of presynaptic nAChRs. This is somewhat problematic since most nicotinic antagonists also affect the α9-containing nAChR of the hair cell to some extent (Verbitsky et al. 2000). A potential strategy is to employ a competitive antagonist with greater affinity for presynaptic nAChRs than for those on the hair cells. The nAChR antagonist dihydro beta erythroidine (DHβE) blocks alpha9-containing receptors (IC50 24 µM) (Verbitsky et al. 2000), but neuronal nAChRs such as α4β2 more potently (IC50 80 nM) (Buisson et al. 1996). As for the test of ryanodine, trains of shocks were used to produce facilitated postsynaptic currents that were followed by delayed release as in Fig. 1A. Delayed release was quantified by evaluating the integrated standard deviation of the averaged tail current at 1 s after a facilitated train of synaptic currents (Fig. 7). Application of 100 nM DHβE reduced delayed release in three of four IHCs. In a fourth IHC, delayed and spontaneous release were initially absent then increased gradually throughout the recording, possibly a result of calcium loading in damaged efferent terminals. On average for three cells delayed release fell to 40% (±27%, SD) that of control (Fig. 7B). In those same cells the evoked response (peak) was 79% (±31%) of control (P = 0.176, t = 1.643, df = 4, comparing percent change of delayed release vs percent change of the evoked response peak). Although the antagonist did not block postsynaptic and presynaptic nAChRs to a significantly different extent, delayed release was substantially reduced. Of note, a reduction of the evoked response does not mean that less ACh is released, only that it has less postsynaptic effect. It is still available to act on presynaptic nAChRs.
Fig. 7.

Evoked and delayed release in the presence of the nicotinic ACh receptor antagonist, dihydro beta erythroidine (DHβE). A: average and standard deviation of membrane current evoked by a train of 10 shocks at 40 Hz. The evoked response facilitates and summates throughout the train and is followed by a slow decay (“tail current”) that includes delayed synaptic events (less visible in this averaged record). B: the standard deviation of the tail current was integrated from the end of the shock train for the next 1.5 s to quantify delayed release. Delayed release was reduced by exposure to 100 nM DHβE (n = 4) and recovered partially after washout.
Nicotine and the probability of evoked release.
Nicotine at 1 mM reliably evoked transmitter release without activating presynaptic action potentials, presumably through mechanisms of spontaneous rather than evoked transmitter release. Could nicotine also alter action-potential-dependent evoked transmitter release? This question was approached by quantifying the probability of evoked release in the presence of micromolar concentrations of nicotine applied by bath perfusion. These lower concentrations avoid the robust release caused by 1 mM nicotine that obscured the shock-evoked response [and incidentally, approach nicotine levels in heavy smokers (Matta et al. 2007; Russell et al. 1975)]. A variety of stimulation protocols was employed, ranging from long trains of shocks (100) at frequencies of 1 Hz or lower, to brief (3-shock) trains at higher frequencies that facilitate the probability of transmitter release (Goutman et al. 2005). These protocols were repeated several times to obtain control values, then repeated several times again in the presence of nicotine at concentrations ranging from 300 nM to 100 µM. The average number of evoked responses was recorded for 100 shocks at 1 Hz, as well as the number of spontaneous events (not timed to the shock). For 16 IHCs from 11 cochleas the grand average response fraction in control was 0.49 (±0.27 SD). After exposure to any nicotine, the grand average response fraction was 0.51 (±0.27 SD). Pairwise comparisons for shock-evoked and spontaneous event counts (1 Hz, 100 s) in each nicotine concentration are shown in Fig. 8 with P values greater than 0.07 in all cases (see Fig. 8 legend). Thus, under these experimental conditions, exposure to nicotine over a range of micromolar concentrations had no significant effect on the probability of evoked release at 1 Hz.
Fig. 8.
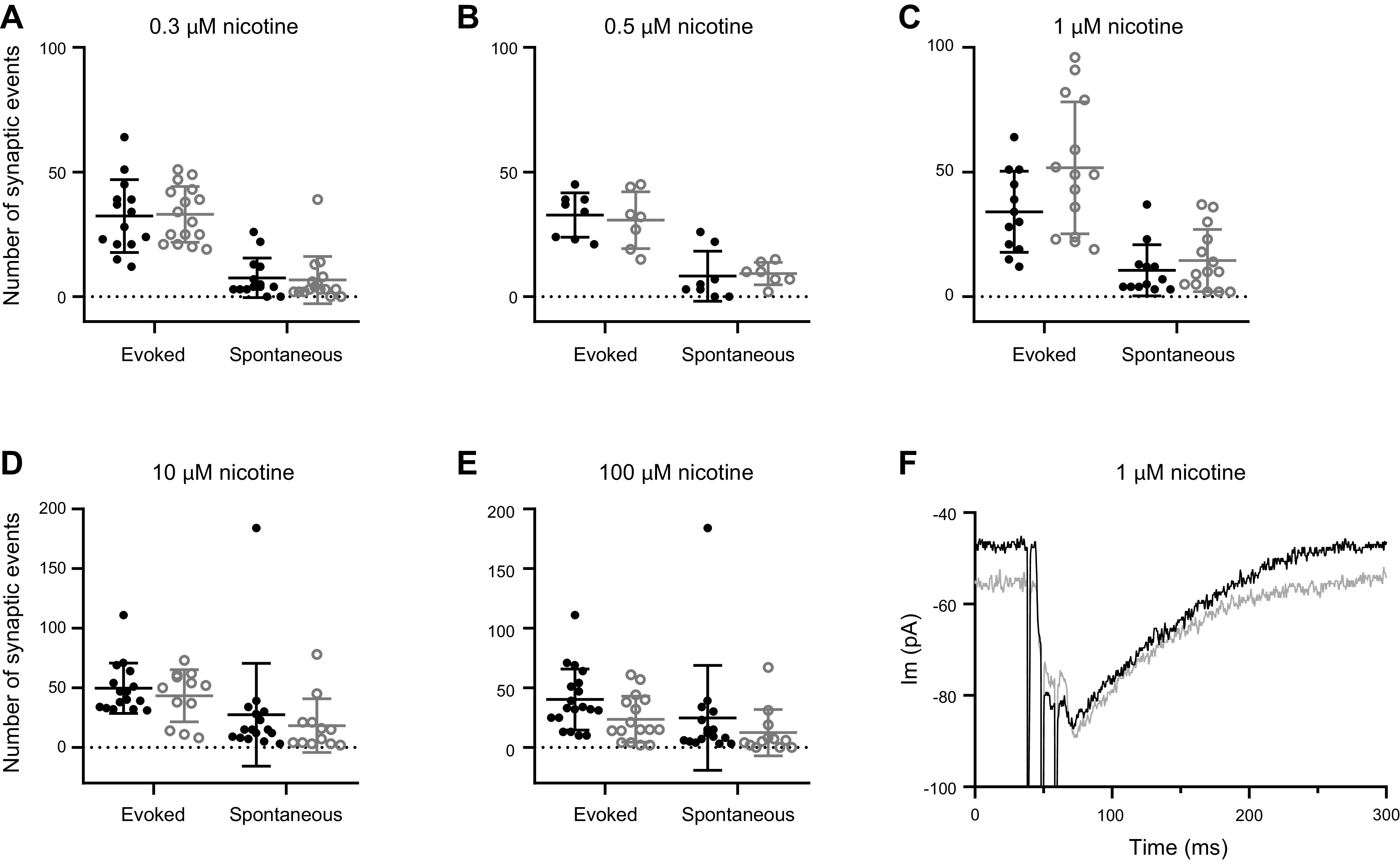
Micromolar nicotine did not increase shocked-evoked efferent transmitter release. A–E: number of evoked and spontaneous synaptic responses during 100 shocks at 1 Hz, repeated before and during perfusion with nicotine. F: exemplar recordings of postsynaptic currents evoked by short, facilitating shock trains in control and 1 µM nicotine. Circles correspond to values for individual recordings. Mean and standard deviation are presented for each condition. Control values are in black, nicotine values in gray. Paired two-tailed t tests were used to compare control versus experimental data A through F. A: effect of 0.3 µM nicotine (for n = 4 cells, no difference for control evoked versus nicotine evoked, P = 0.723, t = 0.337, df = 3, control spontaneous versus nicotine spontaneous P = 0.07, t = 2.74, df = 3). B: effect of 0.5 µM nicotine (for n = 2 cells, no difference for control evoked versus nicotine evoked, P = 0.685, t = 0.0538, df = 1, control spontaneous versus nicotine spontaneous P = 0.118, t = 5.32, df = 1). C: effect of 1 µM nicotine (for n = 3 cells, no difference for control evoked versus nicotine evoked, P = 0.245, t = 1.626, df = 2, control spontaneous versus nicotine spontaneous P = 0.593, t = 0.630, df = 2). D: effect of 10 µM nicotine (for n = 3 cells, no difference for control evoked versus nicotine evoked, P = 0.323, t = 1.302, df = 2, control spontaneous versus nicotine spontaneous P = 0.671, t = 0.493, df = 2). E: effect of 100 µM nicotine (for n = 4 cells, no difference for control evoked versus nicotine evoked, P = 0.076, t = 2.664, df = 3, control spontaneous versus nicotine spontaneous P = 0.589, t = 0.603, df = 3). F: example from one cell of the effect of 1 µM nicotine on a short (3-shock) train at 100 Hz to cause facilitated release from the efferents, recorded at −94 mV. Black is the average of 5 control responses from the exemplar cell, gray is the average of 5 responses in nicotine from that same cell. For n = 6 cells the average peak amplitude in control (50.8 pA, ± 40.5 SD) versus nicotine (36.1 pA, ± 24.9 SD) was not different (P = 0.1205, t = 1.869, df = 5).
To learn whether nicotine might alter higher probability transmitter release, trains of three shocks at 100 Hz were employed to produce facilitated and summated postsynaptic responses (the product of release probability and response amplitude) (Fig. 8F). To increase release probability further, the external calcium concentration was raised from 1.3 to 5.0 mM. For six IHCs from four cochleas the average peak synaptic response to three shocks at 100 Hz in 1 µM nicotine was 36.1 pA (±24.9 SD) compared with controls in the same cells (50.8 pA ± 40.5 SD; P = 0.33, t = 1.01, df = 14). In two additional cells, the average ratio of peak amplitude in 10 µM nicotine to that in control was 1.01 (±0.58 SD). Therefore, even under these conditions of facilitated transmitter release, exposure to micromolar nicotine did not increase the efficacy of evoked synaptic transmission.
DISCUSSION
Acoustic stimuli activate medial olivocochlear (MOC) efferent neurons via second order neurons in the brainstem. MOC neurons inhibit cochlear hair cell activity by the release of ACh. MOC neurons have an initially low probability of release, but facilitate markedly for firing frequencies greater than 5 Hz. Facilitation is accompanied by delayed release lasting several seconds and by elevated rates of spontaneous release that can last minutes. Evidence presented here demonstrates that a component of that enhanced activity may be driven by presynaptic nicotinic ACh receptors (nAChRs) presumably responding to spillover of ACh from the synaptic cleft. Presynaptic nAChRs are known to facilitate transmitter release from cholinergic neurons (McGehee et al. 1995) by calcium entry through ionotropic nAChRs, through activation of voltage-gated calcium channels, or by release of calcium from internal stores (McGehee and Role 1996; Role and Berg 1996; Sharma et al. 2008). The present work suggests that voltage-gated calcium influx supplies intracellular calcium stores whose release supports the presynaptic effect of nicotine on cochlear efferents.
An alternative explanation is that the transient currents observed here are not due to transmitter release, but rather are spontaneous transient outward currents (STOCs) triggered by calcium-induced calcium release from endoplasmic stores in the hair cell, as found in other cell types (Benham and Bolton 1986; Bolton and Imaizumi 1996; Merriam et al. 1999; Satin and Adams 1987). Perhaps nicotine causes such events via α1-containing nAChRs, whose mRNA has been detected in IHCs (Cai et al. 2015; Scheffer et al. 2007; Sinkkonen et al. 2011). There are several arguments against this alternative hypothesis. First, direct testing found no α1-dependent currents in hair cells (Roux et al. 2016). Second, the transient currents evoked by nicotine in hair cells were blocked by application of strychnine, a potent antagonist of α9α10-containing nAChRs in hair cells, but not other nAChRs, strongly implicating synaptic activation. Third, the effect of nicotine was significantly reduced by antagonists to voltage-gated calcium channels that support transmitter release from the efferent terminal but do not function in hair cells.
The effect of nicotine depends on calcium. Both evoked release and the effect of nicotine were suppressed by voltage-gated calcium channel blockade. However, nicotine does not cause transmitter release directly by triggering voltage-gated calcium influx. This is evident from the strong suppression of nicotine’s effect when calcium release from cytoplasmic stores was blocked by 100 µM ryanodine. This suggests that nicotine causes transmitter release via calcium release from ryanodine-sensitive calcium stores as found in other systems (Sharma et al. 2008). The reduction of nicotine’s effect by voltage-gated calcium channel blockers can be explained by the fact that cytoplasmic stores reflect a balance between influx, buffering, and active extrusion. Block of voltage-gated calcium influx shifts that balance, lowering the endoplasmic calcium levels (Verkhratsky and Petersen 1998; Zachary et al. 2018).
In this respect, the action of nicotine observed here differs in various respects from shock-evoked, and to some extent, delayed release. Electrical shocks drive tetrodotoxin-sensitive action potentials that open P/Q, N, and R-type voltage-gated calcium channels to trigger vesicle fusion directly, without immediate reliance on calcium stores. Similarly, delayed release after a facilitated train of evoked responses presumably depends on voltage-gated calcium influx that accumulates during the train, as well as on the action of presynaptic nAChRs acting through endoplasmic calcium stores, as observed here. Thus, while ryanodine receptor block reduced delayed release, the response to nicotine itself was reduced to a much greater extent.
MOC efferent neurons inhibit OHCs to suppress sensitivity of the mature cochlea. Prior to the onset of hearing, medial olivocochlear efferent neurons temporarily inhibit IHCs to modulate the pattern of spontaneous activity in afferent neurons. It is not known what drives the establishment and then departure of these temporary efferent contacts on IHCs. However it has been found that the expression of functional nAChRs precedes synapse formation (Roux et al. 2011); efferent innervation of OHCs varies in mice with altered expression of hair cell nAChRs (Murthy et al. 2009b) and efferent contacts are eliminated when the associated SK2 channel is absent from hair cells (Kong et al. 2008; Murthy et al. 2009a). Activity-dependent modulation of efferent innervation can occur through a number of mechanisms (Katz and Elgoyhen 2014; Wersinger and Fuchs 2011). The present work adds presynaptic nAChRs to this cohort. However, at least under the present experimental conditions, there was no consistent effect of nicotine on efferent synapses of OHCs. This may reflect the relative immaturity of these contacts on hair cells recorded here. Efferent synapses form on OHCs later than those on IHCs (Roux et al. 2011). Alternatively, this type of plasticity, and perhaps others, may be stronger for the transient efferent synapses on IHCs that form and then depart in a matter of weeks, while those made on OHCs are permanent.
The majority of experiments reported here examined the effect of 1 mM nicotine. This raises the question of whether activation of presynaptic nAChRs by released ACh can occur during normal operation of these synapses. Delayed release followed brief trains of shocks at 40 Hz, well within the range of firing recorded from MOC neurons in vivo [up to 100 Hz during maximal acoustic stimulation (Brown et al. 1998)]. Activity patterns of efferent neurons before the onset of hearing are unknown, but prehearing cochlear afferents have spontaneous activity that occurs in a bursting pattern (driven by IHC transmitter release). The instantaneous spike frequency within the burst can be more than 100 Hz (Jones et al. 2007). Furthermore, that pattern of activity is transmitted to neurons of the medial nucleus of the trapezoid body and inferior colliculus (Tritsch et al. 2010), suggesting that downstream neurons such as MOC efferents also could fire in patterns that would facilitate transmitter release and potentially lead to activation of presynaptic nAChRs. Thus, the present and previous studies (Goutman et al. 2005) have shown strong facilitation of efferent transmission to immature IHCs that includes a sustained increase in spontaneous release like that found after repeated nicotine application. The present studies on excised tissue examined acute, perhaps extreme effects to explore underlying mechanisms. Left unexplored is whether more modest activation of presynaptic nAChRs over a longer period of time could influence synaptic efficacy and perhaps play a role in activity-dependent synaptic rearrangements in the postnatal rodent cochlea (Johnson et al. 2013). In this context, it will be of interest to investigate the impact of sustained nicotine exposure on cochlear innervation in pre- and postnatal mice.
In conclusion, nicotine provoked sustained bursts of transmitter release from efferent terminals on “prehearing” IHCs by causing calcium release from ryanodine-sensitive stores. Cytoplasmic calcium stores also contribute to the delayed release of neurotransmitter that occurs after sustained activation of cholinergic efferent terminals on hair cells. Presynaptic nicotinic facilitation is one among several mechanisms that contribute to plasticity and modulation of efferent transmission onto young IHCs (Katz and Elgoyhen 2014). However, nicotine at micromolar concentration had no significant effect on evoked release over the time course of these experiments, thus pointing to a role for these presynaptic nAChRs in modulation of spontaneous release, and leaving open the possibility of a role in developmental regulation.
GRANTS
This research was supported by NIDCD R01 DC001508 to PF, NIDCD R03 DC013374 and the NIH intramural research program fund Z01-DC000060 to IR, NIDCD R01DC006476 and the John Mitchell, Jr. Trust to EG, and the David M. Rubenstein Professorship and Fund for Hearing Research.
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
I.R., Y.A., and P.F. conceived and designed research; Y.Z. and I.R. performed experiments; Y.Z., E.G., I.R. and P.F. analyzed data; Y.Z., E.G., I.R. and P.F. interpreted results of experiments; Y.Z., I.R. and P.F. prepared figures; P.F. drafted manuscript; Y.Z., E.G., I.R. and P.F. edited and revised manuscript; Y.Z., E.G., I.R. and P.F. approved final version of manuscript.
ACKNOWLEDGMENTS
We thank Dr. J. J. S. Wu for assistance with initial experiments and subsequent discussion.
Permanent address for Y. Zhang: Otolaryngology-Head and Neck Surgery, Renmin Hospital of Wuhan University, Wuhan, 430060, Hubei, China.
REFERENCES
- Ballestero J, Zorrilla de San Martín J, Goutman J, Elgoyhen AB, Fuchs PA, Katz E. Short-term synaptic plasticity regulates the level of olivocochlear inhibition to auditory hair cells. J Neurosci 31: 14763–14774, 2011. doi: 10.1523/JNEUROSCI.6788-10.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benham CD, Bolton TB. Spontaneous transient outward currents in single visceral and vascular smooth muscle cells of the rabbit. J Physiol 381: 385–406, 1986. doi: 10.1113/jphysiol.1986.sp016333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boero LE, Castagna VC, Di Guilmi MN, Goutman JD, Elgoyhen AB, Gómez-Casati ME. Enhancement of the medial olivocochlear system prevents hidden hearing loss. J Neurosci 38: 7440–7451, 2018. doi: 10.1523/JNEUROSCI.0363-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolton TB, Imaizumi Y. Spontaneous transient outward currents in smooth muscle cells. Cell Calcium 20: 141–152, 1996. doi: 10.1016/S0143-4160(96)90103-7. [DOI] [PubMed] [Google Scholar]
- Brown MC, Kujawa SG, Duca ML. Single olivocochlear neurons in the guinea pig. I. Binaural facilitation of responses to high-level noise. J Neurophysiol 79: 3077–3087, 1998. doi: 10.1152/jn.1998.79.6.3077. [DOI] [PubMed] [Google Scholar]
- Buisson B, Gopalakrishnan M, Arneric SP, Sullivan JP, Bertrand D. Human alpha4beta2 neuronal nicotinic acetylcholine receptor in HEK 293 cells: a patch-clamp study. J Neurosci 16: 7880–7891, 1996. doi: 10.1523/JNEUROSCI.16-24-07880.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cai T, Jen HI, Kang H, Klisch TJ, Zoghbi HY, Groves AK. Characterization of the transcriptome of nascent hair cells and identification of direct targets of the Atoh1 transcription factor. J Neurosci 35: 5870–5883, 2015. doi: 10.1523/JNEUROSCI.5083-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng Q, Yakel JL. The effect of α7 nicotinic receptor activation on glutamatergic transmission in the hippocampus. Biochem Pharmacol 97: 439–444, 2015. doi: 10.1016/j.bcp.2015.07.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clause A, Lauer AM, Kandler K. Mice lacking the alpha9 subunit of the nicotinic acetylcholine receptor exhibit deficits in frequency difference limens and sound localization. Front Cell Neurosci 11: 167, 2017. doi: 10.3389/fncel.2017.00167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Corns LF, Johnson SL, Roberts T, Ranatunga KM, Hendry A, Ceriani F, Safieddine S, Steel KP, Forge A, Petit C, Furness DN, Kros CJ, Marcotti W. Mechanotransduction is required for establishing and maintaining mature inner hair cells and regulating efferent innervation. Nat Commun 9: 4015, 2018. doi: 10.1038/s41467-018-06307-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dickinson JA, Kew JN, Wonnacott S. Presynaptic alpha 7- and beta 2-containing nicotinic acetylcholine receptors modulate excitatory amino acid release from rat prefrontal cortex nerve terminals via distinct cellular mechanisms. Mol Pharmacol 74: 348–359, 2008. doi: 10.1124/mol.108.046623. [DOI] [PubMed] [Google Scholar]
- Elgoyhen AB, Johnson DS, Boulter J, Vetter DE, Heinemann S. Alpha 9: an acetylcholine receptor with novel pharmacological properties expressed in rat cochlear hair cells. Cell 79: 705–715, 1994. doi: 10.1016/0092-8674(94)90555-X. [DOI] [PubMed] [Google Scholar]
- Fuchs PA, Murrow BW. Cholinergic inhibition of short (outer) hair cells of the chick’s cochlea. J Neurosci 12: 800–809, 1992. doi: 10.1523/JNEUROSCI.12-03-00800.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Glowatzki E, Fuchs PA. Cholinergic synaptic inhibition of inner hair cells in the neonatal mammalian cochlea. Science 288: 2366–2368, 2000. doi: 10.1126/science.288.5475.2366. [DOI] [PubMed] [Google Scholar]
- Gómez-Casati ME, Fuchs PA, Elgoyhen AB, Katz E. Biophysical and pharmacological characterization of nicotinic cholinergic receptors in rat cochlear inner hair cells. J Physiol 566: 103–118, 2005. doi: 10.1113/jphysiol.2005.087155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goutman JD, Fuchs PA, Glowatzki E. Facilitating efferent inhibition of inner hair cells in the cochlea of the neonatal rat. J Physiol 566: 49–59, 2005. doi: 10.1113/jphysiol.2005.087460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinan JJ Jr. Cochlear efferent innervation and function. Curr Opin Otolaryngol Head Neck Surg 18: 447–453, 2010. doi: 10.1097/MOO.0b013e32833e05d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson SL, Wedemeyer C, Vetter DE, Adachi R, Holley MC, Elgoyhen AB, Marcotti W. Cholinergic efferent synaptic transmission regulates the maturation of auditory hair cell ribbon synapses. Open Biol 3: 130163, 2013. doi: 10.1098/rsob.130163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones TA, Leake PA, Snyder RL, Stakhovskaya O, Bonham B. Spontaneous discharge patterns in cochlear spiral ganglion cells before the onset of hearing in cats. J Neurophysiol 98: 1898–1908, 2007. doi: 10.1152/jn.00472.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Elgoyhen AB. Short-term plasticity and modulation of synaptic transmission at mammalian inhibitory cholinergic olivocochlear synapses. Front Syst Neurosci 8: 224, 2014. doi: 10.3389/fnsys.2014.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katz E, Elgoyhen AB, Gómez-Casati ME, Knipper M, Vetter DE, Fuchs PA, Glowatzki E. Developmental regulation of nicotinic synapses on cochlear inner hair cells. J Neurosci 24: 7814–7820, 2004. doi: 10.1523/JNEUROSCI.2102-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kearney G, Zorrilla de San Martín J, Vattino LG, Elgoyhen AB, Wedemeyer C, Katz E. Developmental synaptic changes at the transient olivocochlear-inner hair cell synapse. J Neurosci 39: 3360–3375, 2019. doi: 10.1523/jneurosci.2746-18.2019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong JH, Adelman JP, Fuchs PA. Expression of the SK2 calcium-activated potassium channel is required for cholinergic function in mouse cochlear hair cells. J Physiol 586: 5471–5485, 2008. doi: 10.1113/jphysiol.2008.160077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kong JH, Zachary S, Rohmann KN, Fuchs PA. Retrograde facilitation of efferent synapses on cochlear hair cells. J Assoc Res Otolaryngol 14: 17–27, 2013. doi: 10.1007/s10162-012-0361-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauer AM, Fuchs PA, Ryugo DK, Francis HW. Efferent synapses return to inner hair cells in the aging cochlea. Neurobiol Aging 33: 2892–2902, 2012. doi: 10.1016/j.neurobiolaging.2012.02.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcotti W, Johnson SL, Rusch A, Kros CJ. Sodium and calcium currents shape action potentials in immature mouse inner hair cells. J Physiol 552: 743–761, 2003. doi: 10.1113/jphysiol.2003.043612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matta SG, Balfour DJ, Benowitz NL, Boyd RT, Buccafusco JJ, Caggiula AR, Craig CR, Collins AC, Damaj MI, Donny EC, Gardiner PS, Grady SR, Heberlein U, Leonard SS, Levin ED, Lukas RJ, Markou A, Marks MJ, McCallum SE, Parameswaran N, Perkins KA, Picciotto MR, Quik M, Rose JE, Rothenfluh A, Schafer WR, Stolerman IP, Tyndale RF, Wehner JM, Zirger JM. Guidelines on nicotine dose selection for in vivo research. Psychopharmacology (Berl) 190: 269–319, 2007. doi: 10.1007/s00213-006-0441-0. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Heath MJ, Gelber S, Devay P, Role LW. Nicotine enhancement of fast excitatory synaptic transmission in CNS by presynaptic receptors. Science 269: 1692–1696, 1995. doi: 10.1126/science.7569895. [DOI] [PubMed] [Google Scholar]
- McGehee DS, Role LW. Presynaptic ionotropic receptors. Curr Opin Neurobiol 6: 342–349, 1996. doi: 10.1016/S0959-4388(96)80118-8. [DOI] [PubMed] [Google Scholar]
- McNiven AI, Yuhas WA, Fuchs PA. Ionic dependence and agonist preference of an acetylcholine receptor in hair cells. Aud Neurosci 2: 63–77, 1996. [Google Scholar]
- Merriam LA, Scornik FS, Parsons RL. Ca2+-induced Ca2+ release activates spontaneous miniature outward currents (SMOCs) in parasympathetic cardiac neurons. J Neurophysiol 82: 540–550, 1999. doi: 10.1152/jn.1999.82.2.540. [DOI] [PubMed] [Google Scholar]
- Morley BJ. Nicotinic cholinergic intercellular communication: implications for the developing auditory system. Hear Res 206: 74–88, 2005. doi: 10.1016/j.heares.2005.02.012. [DOI] [PubMed] [Google Scholar]
- Murthy V, Maison SF, Taranda J, Haque N, Bond CT, Elgoyhen AB, Adelman JP, Liberman MC, Vetter DE. SK2 channels are required for function and long-term survival of efferent synapses on mammalian outer hair cells. Mol Cell Neurosci 40: 39–49, 2009a. doi: 10.1016/j.mcn.2008.08.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murthy V, Taranda J, Elgoyhen AB, Vetter DE. Activity of nAChRs containing alpha9 subunits modulates synapse stabilization via bidirectional signaling programs. Dev Neurobiol 69: 931–949, 2009b. doi: 10.1002/dneu.20753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paquette ST, Dawes RP, Sundar IK, Rahman I, Brown EB, White PM. Chronic cigarette smoke exposure drives spiral ganglion neuron loss in mice. Sci Rep 8: 5746, 2018. doi: 10.1038/s41598-018-24166-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Picciotto MR, Higley MJ, Mineur YS. Acetylcholine as a neuromodulator: cholinergic signaling shapes nervous system function and behavior. Neuron 76: 116–129, 2012. doi: 10.1016/j.neuron.2012.08.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohmann KN, Wersinger E, Braude JP, Pyott SJ, Fuchs PA. Activation of BK and SK channels by efferent synapses on outer hair cells in high-frequency regions of the rodent cochlea. J Neurosci 35: 1821–1830, 2015. doi: 10.1523/JNEUROSCI.2790-14.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Role LW, Berg DK. Nicotinic receptors in the development and modulation of CNS synapses. Neuron 16: 1077–1085, 1996. doi: 10.1016/S0896-6273(00)80134-8. [DOI] [PubMed] [Google Scholar]
- Roux I, Wersinger E, McIntosh JM, Fuchs PA, Glowatzki E. Onset of cholinergic efferent synaptic function in sensory hair cells of the rat cochlea. J Neurosci 31: 15092–15101, 2011. doi: 10.1523/JNEUROSCI.2743-11.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roux I, Wu JS, McIntosh JM, Glowatzki E. Assessment of the expression and role of the α1-nAChR subunit in efferent cholinergic function during the development of the mammalian cochlea. J Neurophysiol 116: 479–492, 2016. doi: 10.1152/jn.01038.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Russell MA, Wilson C, Patel UA, Feyerabend C, Cole PV. Plasma nicotine levels after smoking cigarettes with high, medium, and low nicotine yields. BMJ 2: 414–416, 1975. doi: 10.1136/bmj.2.5968.414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satin LS, Adams PR. Spontaneous miniature outward currents in cultured bullfrog neurons. Brain Res 401: 331–339, 1987. doi: 10.1016/0006-8993(87)91417-X. [DOI] [PubMed] [Google Scholar]
- Scheffer D, Sage C, Plazas PV, Huang M, Wedemeyer C, Zhang DS, Chen ZY, Elgoyhen AB, Corey DP, Pingault V. The α1 subunit of nicotinic acetylcholine receptors in the inner ear: transcriptional regulation by ATOH1 and co-expression with the γ subunit in hair cells. J Neurochem 103: 2651–2664, 2007. doi: 10.1111/j.1471-4159.2007.04980.x. [DOI] [PubMed] [Google Scholar]
- Sharma G, Grybko M, Vijayaraghavan S. Action potential-independent and nicotinic receptor-mediated concerted release of multiple quanta at hippocampal CA3-mossy fiber synapses. J Neurosci 28: 2563–2575, 2008. doi: 10.1523/JNEUROSCI.5407-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinkkonen ST, Chai R, Jan TA, Hartman BH, Laske RD, Gahlen F, Sinkkonen W, Cheng AG, Oshima K, Heller S. Intrinsic regenerative potential of murine cochlear supporting cells. Sci Rep 1: 26, 2011. doi: 10.1038/srep00026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sottile SY, Hackett TA, Cai R, Ling L, Llano DA, Caspary DM. Presynaptic neuronal nicotinic receptors differentially shape select inputs to auditory thalamus and are negatively impacted by aging. J Neurosci 37: 11377–11389, 2017. doi: 10.1523/JNEUROSCI.1795-17.2017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Traynor JR. Epibatidine and pain. Br J Anaesth 81: 69–76, 1998. doi: 10.1093/bja/81.1.69. [DOI] [PubMed] [Google Scholar]
- Tritsch NX, Rodríguez-Contreras A, Crins TT, Wang HC, Borst JG, Bergles DE. Calcium action potentials in hair cells pattern auditory neuron activity before hearing onset. Nat Neurosci 13: 1050–1052, 2010. doi: 10.1038/nn.2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch NX, Yi E, Gale JE, Glowatzki E, Bergles DE. The origin of spontaneous activity in the developing auditory system. Nature 450: 50–55, 2007. doi: 10.1038/nature06233. [DOI] [PubMed] [Google Scholar]
- Verbitsky M, Rothlin CV, Katz E, Elgoyhen AB. Mixed nicotinic-muscarinic properties of the alpha9 nicotinic cholinergic receptor. Neuropharmacology 39: 2515–2524, 2000. doi: 10.1016/S0028-3908(00)00124-6. [DOI] [PubMed] [Google Scholar]
- Verkhratsky AJ, Petersen OH. Neuronal calcium stores. Cell Calcium 24: 333–343, 1998. doi: 10.1016/S0143-4160(98)90057-4. [DOI] [PubMed] [Google Scholar]
- Wedemeyer C, Zorrilla de San Martín J, Ballestero J, Gómez-Casati ME, Torbidoni AV, Fuchs PA, Bettler B, Elgoyhen AB, Katz E. Activation of presynaptic GABAB(1a,2) receptors inhibits synaptic transmission at mammalian inhibitory cholinergic olivocochlear-hair cell synapses. J Neurosci 33: 15477–15487, 2013. doi: 10.1523/JNEUROSCI.2554-13.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wersinger E, Fuchs PA. Modulation of hair cell efferents. Hear Res 279: 1–12, 2011. doi: 10.1016/j.heares.2010.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wersinger E, McLean WJ, Fuchs PA, Pyott SJ. BK channels mediate cholinergic inhibition of high frequency cochlear hair cells. PLoS One 5: e13836, 2010. doi: 10.1371/journal.pone.0013836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye Z, Goutman JD, Pyott SJ, Glowatzki E. mGluR1 enhances efferent inhibition of inner hair cells in the developing rat cochlea. J Physiol 595: 3483–3495, 2017. doi: 10.1113/JP272604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary S, Nowak N, Vyas P, Bonanni L, Fuchs PA. Voltage-gated calcium influx modifies cholinergic inhibition of inner hair cells in the immature rat cochlea. J Neurosci 38: 5677–5687, 2018. doi: 10.1523/JNEUROSCI.0230-18.2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zachary SP, Fuchs PA. Re-emergent inhibition of cochlear inner hair cells in a mouse model of hearing loss. J Neurosci 35: 9701–9706, 2015. doi: 10.1523/JNEUROSCI.0879-15.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zorrilla de San Martín J, Pyott S, Ballestero J, Katz E. Ca2+ and Ca2+-activated K+ channels that support and modulate transmitter release at the olivocochlear efferent-inner hair cell synapse. J Neurosci 30: 12157–12167, 2010. doi: 10.1523/JNEUROSCI.2541-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]