

Abstract
This review provides the feasible literature on drug discovery through ML tools and techniques that are enforced in every phase of drug development to accelerate the research process and deduce the risk and expenditure in clinical trials. Machine learning techniques improve the decision-making in pharmaceutical data across various applications like QSAR analysis, hit discoveries, de novo drug architectures to retrieve accurate outcomes. Target validation, prognostic biomarkers, digital pathology are considered under problem statements in this review. ML challenges must be applicable for the main cause of inadequacy in interpretability outcomes that may restrict the applications in drug discovery. In clinical trials, absolute and methodological data must be generated to tackle many puzzles in validating ML techniques, improving decision-making, promoting awareness in ML approaches, and deducing risk failures in drug discovery.
Keywords: Artificial intelligence, Drug discovery, Machine learning, Target validation, Prognostic biomarkers, Digital pathology
Introduction
In computer science, Artificial intelligence (AI) additionally attributed as machine intelligence because machines are trained or customized to perform activities like a human brain (Poole et al. 1998; Vinod and Anand 2021; Gopal 2018). Artificial Intelligence (AI) can be categorized here as the field is dealing with a wide range of utilization and layouts of numerous algorithms for interpreting and attaining knowledge from data. And the AI concept is firmly related to many fields like pattern recognition, probability theory, statistics, machine learning, and numerous procedures like fuzzy models, neural networks which are collectively known as “Computational Intelligence” Vinod and Anand (2021), Engelbrecht (2007), Konar (2006), Duda et al. (2012), Webb (2003), Friedman et al. (2001). Multiple complicated usages engaged with AI strategies like classification, regression, predictions and also optimization techniques. Machine learning needs to be modified well in the utilization of any kind of information i.e., initially, a particular model must be characterized along with parameters. So, machines can be gain proficiency in the model with accessible parameters through the utilization of trained data. Furthermore, the model can predict the data in the future for recovering information from data (Alpaydin 2020).
In this review, we are primarily focusing on qualities of AI approaches that are appropriate for drug development and discovery (Duch et al. 2007). Recently various factors were developed due to greater enthusiasm for utilizing machine learning approaches in the pharmaceutical industry. Figure 1 shows that the various fields of Drug Discovery and advancements utilized through machine learning. Every phase was performed like a pipeline to represent therapeutic concepts. The respective phases represent unique iterations in time and cost expenditure. Here each phase is carried out to prove the effectiveness of the remedial treatment. The medical information was being mined and estimated accurately by using some ‘omics’ and ‘smart automation tools’. Enlarging these techniques into the biological field gives more opportunities as well as challenges in the pharmaceutical industry. Since numerous pharmaceutical enterprises’ objective is to distinguish the persuasive clinical hypothesis. With the obtained results, practitioners or clinicians can develop the medications. For establishing any type of drug in pharmaceutical industries, the usage of machine learning approaches has checked out the performance. At this point, if included with unlimited storage, improvement appeared in datasets like size, types can provide premises to machine learning. In this way, it can access enormous data from pharmaceutical industries. Data types can have different configurations like textual data, images, assay information, biometrics, and furthermore high dimensional omics data (Mamoshina et al. 2018).
Fig. 1.
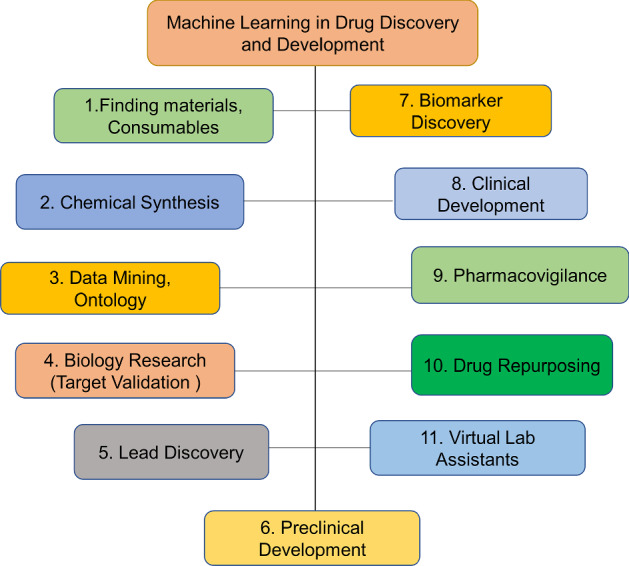
Various fields in Drug discovery by using Machine Learning
Thus, the AI field has developed from theoretical knowledge to real-world data. Information was widely improved for utilizing in PC hardware, for example, Graphical Processing Units (GPU), which makes faster in processing (i.e., in computational techniques). Recently, the deep learning model is one of the machine learning algorithms (LeCun et al. 2015), it develops the models for making more accomplishment in broad daylight challenges (Chen et al. 2018; Hinton 2018). For the past 2 years, the usage of ML algorithms has a great extension within pharmaceutical enterprises.
In the clinical field, developing a new drug for persistent disease primarily relied on new medications. As of late, various drugs are improvised for recognizing dynamic components from traditional treatments such as penicillin. In chemical laboratories, it consists of natural substances, small molecules that aid in therapeutic medicine to detect substances such as cells or intact organisms. This procedure is called old-style pharmacology.
High throughput screening with multiple libraries has normally expanded because of the human genome has permitted cloning strategies and furthermore improving refining of proteins in huge quantities. Screening activity for large compounds through biological targets can be used to achieve a change in a disease called reverse pharmacology. Multiple hits can be generated from screening activity to provide cells and furthermore tests have been conducted in creatures for adequacy. In modern days, drug discovery has engaged with the performance of identifications on screening hits, optimization techniques can build the drug effectiveness, affinity, stability of metabolic. If all requirements are satisfied by the compound, a particular drug will be developed in clinical trials if the drug is successful. In process of drug development and discovery, it requires lead optimization, target identification and validation, hit discovery, clinical trials (Vohora and Singh 2018). In novel drug development, the cost expenditure can approximately 2.558 billion USD (DiMasi et al. 2016) and it is a tedious procedure in light of the fact that about 10–15 years have taken for selling in the market (Turner 2010). To accomplish a small number of molecules in drug development, many investors are putting a lot of cash in developing exact progress in clinical trials. And still, 13% precision rate is lagging with disappointment. So as to conquer this issue, clinicians have utilized the Computer-assisted Drug Design CADD technique (Hassan Baig et al. 2016). By utilizing this strategy in drug discovery, the artificial techniques not just provide the molecular properties (i.e., selectivity, distribution, absorption, bioactivity, metabolism, side effects, and excretion in the theoretical levels) but also provides the lead compounds such as ideal attributes in silico. Also, attrition cost in the preclinical state can be decreased through the utilization of multi-objective optimization techniques.
In drug discovery, computational intelligence provides various techniques for analyzing, learning and furthermore clarifies how such pharmaceutical was identified with AI for finding numerous medications in a programmed and integrated format (Duch et al. 2007). Therefore, many pharmaceutical industries have shown greater enthusiasm for contributing to technologies, resources for retrieving accurate results in drug discovery. At last, this survey proposes AI techniques in the drug discovery area for targeting multiple applications in drug discovery and development by utilizing deep learning techniques. Along these lines, the AI field provides expected outcomes in concern of computational intelligence in drug development and discovery (Table 1).
Table 1.
List of Major Abbreviations
| ADMET | Absorption, Distribution, Metabolism And ExcretionToxicology |
|---|---|
| AE | AutoEncoder |
| AI | Artificial Intellingence |
| ANN | Artificial Neural Networks |
| AUC | Area under the ROC Curve |
| CNN | Convolution Neural Networks |
| CT | Computed Tomography |
| DL | Deep Learning |
| DNN | Deep Neural Networks |
| DPI | Drug Protein Interaction |
| GPCR | G-Protein coupled receptors |
| GPU | Graphical Processing Unit |
| HARF | Heterogeneity Aware Random Forest |
| HTVS | High-Throughput Virtual Screening |
| LDA | Linear Discriminant Analysis |
| LR | Logistic Regression |
| MAE | Mean Absolute Error |
| ML | Machine Learning |
| MLP | Multi Layer Perceptron |
| MLR | Multiple Linear Regression |
| MRI | Magnetic Resonance Imaging |
| NBC | Naive Bayesian Classification |
| NCE | New Chemical Entities |
| PNN | Probabilistic Neural Networks |
| PPI | Protein to Protein Interaction |
| QSAR | Quantitative Structure-Activity Relationship |
| RBN | Radial Basis function Network |
| RF | Random Forest |
| RMSE | Root Mean Square Error |
| RNA | Ribonucleic Acid |
| RNN | Recurrent Neural Networks |
| ROC | Receiver Operating Characteristic curve |
| SARS-CoV-2 | Severe Acute Respiratory Syndrome CoronaVirus 2 |
| SMILES | Simplified Molecular Input Line Entry Specifications |
| SVM | Support Vector Machines |
| VAE | Variational AutoEncoders |
Roadmap
The rest of the article is arranged in the following way: Sect. 2 describes the application of AI in Drug design. Then, the various machine learning methods towards Drug discovery are discussed in Sect. 3. Various Drug design applications are discussed in Sect. 4. In Sect. 5, different Drug design problems have discussed. Finally, Sect. 6 presents the research challenges with few possible suggestions in Drug discovery using Machine learning, and Sect. 7 concludes the article and provided some future directions.
Application of AI in drug design
This section discusses a few applications in AI which relate to drug study. The activity of protein structure is considered as the application in drug design. Many impurities have appeared in the human body due to protein dysfunctions. Structural drug design strategies are used to differentiate small molecules in protein targets. Protein structure in 3D format requires more money and time for predicting the 3D structure. And still, it faces the problem i.e., in making more exactness over de-novo prediction in 3D structure. By using deep learning and feature extraction tools, it is mandatory to predict the secondary structure (Spencer et al. 2014) and residing the protein contacts (Li et al. 2017). It precisely gains the information on the connection among structure and sequence from feature extraction. The further goal is to predict the 3D- protein structure by utilizing deep learning techniques for improving the accuracy. To retrieve information from drug design of protein-protein computer structure, then it is mandatory to conduct investigations on PPI interface (Xue et al. 2015).
Artificial Intelligence has been used in various applications like a prediction on drug–protein interactions, the discovery of drug efficacy, ensuring the safety biomarkers. The detailed discussion is given as follows
Prediction on drug–protein interactions
The crucial step of drug development in silico is consisting of multiple biological sources for predicting drug–protein interactions. Here complications can be seen in large predictions, which relied on the countless unknown interactions. Therefore, semi-supervised training techniques should be used to address these unlabelled and labeled date complications. Usually, only labeled data will produce better results. In addition, the semi-supervised technology integrates chemical structure, drug–protein interaction network data, and genome sequence data. Finally, in this article, drug–protein interactions of various data sets such as ions, enzymes, and nuclear receptors provided well predictable results (Xia et al. 2010).
Drugs have an important priority in therapeutic activity, which is regulated by protein interactions. The drug–protein interaction database (DPI) focuses primarily on therapeutic protein targets, while knowledge of non-targets has been limited and resolved. Thus, computational techniques can fill the knowledge gap for predicting protein targets for distributed drug molecules. In that study, the pool of 35 predictors had a major impact on the similarity between protein and drug targets. Drug structure, target sequence, and drug profile are three types of similarity developed from the results of 35 predictors. Finally, the significant content, relationships, and implications between database sources are of great importance for therapeutic activity (Wang and Kurgan 2020).
In drug repurposing, the unexpected detection of drug–protein interactions is essential. Thus, the dominant drug may be useful for repurposing, while drug side effects are unavoidable and about 1,000 human proteins can cause critical side effects. The proteomic scale method was used to predict side effects and protein goals. FINDSITEcomb is used to predict drug–protein interactions. The estimates showed greater disruption with a mean of 329 human targets for each drug (Zhou et al. 2015).
Discover of drug efficacy
Usually, a drug effect assessment looks at its biochemical activity. The effectiveness of the therapeutic activity has posed a challenge to be properly coupled with the biochemical activity. The collection of a large amount of data on the effects of cellular drugs was undertaken to fill a gap that has been explored in the extensive content of cellular estimations and while this estimation is classified as a psychotropic drug. Here, the microarray data can be analyzed by applying random trees to the forest and classifying them, providing a profile for the efficiency of biomarker gene expression. Accuracy of 88.9% of the classification tree and 83.3% of the random forest model used this efficacy profile for a drug treatment analysis. Therefore, at the cellular assessment level, general genomic data are acceptable to reconcile the effects of new physiological drugs with clinical applications. Finally, in vitro signatures of gene expression data can identify the effectiveness of therapeutic activities that can help validate targeting and drug development (Gunther et al. 2003).
In drug development, increasing profitability by validating new drugs requires predicting effectiveness and identifying targets. The proximity of medical illnesses helps to reduce the effectiveness of the treatment and also releases drugs that are effective in therapeutic activity. The study treated 78 diseases with 238 drugs to demonstrate the drug’s effectiveness in therapeutic activity, as well as problems with gene efficacy and various disorders. Here the network-based system is used to develop a drug-disease proximate measure that assesses the interactions between the disease and the drug target. Therefore, the proximity of network-based systems makes it possible to predict associations for novel drug diseases, offering a wide range of possibilities for conflict detection and drug repurposing (Guney et al. 2016).
Ensuring the safety biomarkers
In drug development, the use of biomarkers supports the provision of safety measures that critically determine the biological and analytical indicators of a particular biomarker. In this way, stakeholders can assess and manage whether claims are defended for a particular purpose and whether the desired standards are being met. For shareholders in the implementation of evaluating the experiment agreement, a stakeholder evaluation process is needed to adjust the unique characteristics of the biomarkers, as well as to determine how these innovations are analyzed, integrated, and interpreted, and how improved biomarkers and conventional comparators are measured (Sistare et al. 2010).
In the survey, we found that modern medicines are no safer than older drugs, even though with longer medical trial programs. These trails are placed on the market and impractical inspections are carried out which are not sufficient to be carried out systematically to ensure safety. Previous drug-related signals can help in improving drug safety as well as identify underlying biomarkers, making them more toxic. However, the safety markers can be different for different target systems. However, no other approach can provide assurance that medicines are very safe, but we can develop a common understanding of benefit and risk assessment by communicating with the public (Rolan et al. 2007).
Various deep learning techniques are carried out here to predict the PPI interface and show fantastic results when contrasted with the SVM technique (Du et al. 2016). Thus, the PPI’s became more complex to utilize in biological techniques (Falchi et al. 2014; Scott et al. 2016). Each PPI can be a mixture of various residues (Cukuroglu et al. 2014). New PPI can act as a modern class for pharmaceutical targets where disparate for different targets i.e., ion channels, GPCRs (G-Protein coupled receptors), kinases (Higueruelo et al. 2013; Santos et al. 2017). iFitDock is a docking tool used for investigating a few hotspots in PPIs. Further, AI techniques have been utilized for distinguishing structures and hotspots in PPI interface (Fig. 2).
Fig. 2.

Applications of AI in Drug discovery depicts the Machine learning mechanisms
Machine learning methods to drug discovery
AI innovation has a high priority in drug design through the enhancement of ML approaches and the collection of pharmacological data. AI does not rely upon any hypothetical improvements, but it has more essence in transforming medical information into studies like reusable methods. In general, there are different approaches such as Random Forest, Naive Bayesian Classification (NBC), Multiple Linear Regression (MLR), Logistic Regression (LR), Linear Discriminant Analysis (LDA), Probabilistic Neural Networks (PNN), Multi-Layer Perceptron (MLP), Support Vector Machine (SVM), etc are considered in the context of ML (Lavecchia and Di Giovanni 2013). In order to gain capability in feature extraction and feature generalization, AI advancements are specifically used as a deep learning technique towards drug design. Also, Fig. 3 shows respective applications which illustrate an outline of AI procedures utilized to respond to drug discovery queries in the review. A scope of classifier and regression strategies i.e. supervised learning techniques utilized to respond addresses desire expectations in continuous or categorical data factors, also unsupervised methods utilized in creating a model which empowers the clustering data.
Fig. 3.
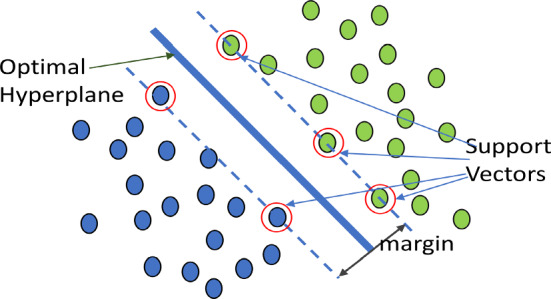
Maximum-margin hyperplane and margins for an SVM trained with samples from two classes. Samples on the margin are called the support vectors
Many designed features in traditional ML models are performed manually, but deep learning approaches will accelerate various features through available initialized data automatically because multi-layer feature extraction techniques are used to convert straightforward features into complex features. One advantage of using deep learning approaches was, presence of low quantity generalization blunders, so it recovers more exact results. CNN, RNN, Auto Encoder, DNN, and RBN are considered as different deep learning techniques. Summary of deep learning algorithms can be identified (LeCun et al. 2015; Angermueller et al. 2016; Schmidhuber 2015) and provides detailed information about deep learning techniques which are available in Deep Learning literature (Goodfellow et al. 2016).
In drug discovery and development, many AI calculations are associated to analyse and predict the data. Here, few popular models like SVM, RF, and MLP discuss their effective use in drug discovery.
Support vector machines
SVM model is a supervised learning algorithm basically utilized in predicting the class labelled data i.e., binary data. In SVM, x is considered as feature vector i.e., input to SVM model. At that point, where n is a dimension feature vector. Y acts as a class i.e., output for svm. . Here, Binary values are considered as classification task. Parameters in SVM u and b have considered for learning data in training set. In dataset, are considered as sample. Y can be represented as follows:
A class Y can be written as . Finally, SVM algorithm goal is to satisfy:
In SVM, seperation between any two boundaries ought to be augmented i.e., the distance between two hyperplane and should be maximized. In this way, . Finally, it have to solve
Complete samples need to classify effectively in the SVM i.e.,
Then, it produces quadratic optimization problem i.e. . So that, .
The above equation was a hard-margin SVM, and we can avoid this problem through applying linearly separable method. Using the slack variable as constraints. In training data, each sample has its own slack variable. Then,
| 1 |
Now, it’s a soft margin SVM, where ‘C’ is considered as a penalty of the error term. Involving function to allow more flexibility in mapping. So, it maps multiple features like original space to high dimensional space (Noble 2006). Then, the quadratic optimization problem updates Eq. 1 as the following:
The SVM widely used in drug discovery using its various kernels (Smola and Schölkopf 2004). Various problems like Screen radiation protection and Gene Interaction using SVM-RBF(Radial Basis Function) (Matsumoto et al. 2016; Guo et al. 2008), Assess target-ligand interactions using Regression-SVM (Li et al. 2011), Identify drug target interaction by Biased SVM (Wang et al. 2017), Predicting drug sensitivity prediction by Ensemble SVM, and the Linear SVM used in Identify novel drug targets (Volkamer et al. 2012), Anti/non-anticancer molecule classification (Kapoorb et al. 2020), Kinase mutaion activation (Patil et al. 2021).
The SVM approach (Huang et al. 2018) was used to quantify anti-cancer drugs based on cancer cell properties. To understand the relationship between cancer cell properties and drug resistance, 24 drugs were tested on cancer cell lines (Gupta et al. 2016). In the treatment of oral cancer, the SVM-RBF (Radial Basis Function) approach has been used to find therapeutic compounds from a large collection of public databases (Bundela et al. 2015), the RBF is the popular kernel function used in various learning algorithms. The RBF kernel takes two samples S1 and S2, represented as feature vectors in some input space where is used to recognized as the squared Euclidean distance between two vectors and is a free parameter. Here the RBF is used and hybridized as many variations with different parameter values.
In general, radiation therapy techniques help to protect against cancer. Therefore, the SVM method is used in virtual screening (Matsumoto et al. 2016) to protect the radiation function. Radiation therapy also has side effects on normal cells and tissues (Morita et al. 2014). In this study, we found that the SVM approach worked better than other techniques. When the target protein is known, we can find a suitable compound for the target protein. However, the SVM technique is mainly used to predict the outcome of targeted drugs. SVM has used sites to link global descriptors, taking into account various properties such as compactness and size. These descriptors can determine drug scores for novel targets (Volkamer et al. 2012; Li et al. 2011).
In therapeutic activities, the use of SVM helps to find the active ingredient at various stages of the drug development process. In general, the active component of the connection is taken into account in the number of turns of the design process. The main goal is to find different lead series in the active compound to improve them in parallel in therapeutic activity (Warmuth et al. 2003). In contrast to other artificial neural networks, SVM demonstrated the ability to test drug similarity predictions of a wide variety of compounds. Because of this set of descriptors, the SVM outperformed the task and also reported that the SVM model predicted better enzyme inhibitor quality for conventional QSAR (Zernov et al. 2003).
Right now, the SVM model is the best methodology for predicting organic and compound properties. Recently, the SVM model has been utilized in the drug discovery region and turned out to be more famous in drug discovery applications like a prediction on properties, compound classification (Maltarollo et al. 2019). In designing new structures, the SVM approach was utilized for retrieving higher predicted results where depend on ligands (Hartenfeller and Schneider 2010). In the Activity process, to improve scoring capacity execution, the SVM approach was utilized for clarifying non-linear relationships of energy terms from eHiTS and binding data which shows a lot of improvement in scoring power and screening power (Kinnings et al. 2011; Zsoldos et al. 2007). SVM model was frequently utilized in virtual screening (Leelananda and Lindert 2016; Liew et al. 2009; Melville et al. 2009) and demonstrated best results (in the predicted ratio called hits) and furthermore false-hit rates are decreased concurrently (counterfeit hit rates in the predicted hits) (Ma et al. 2009). Creating meta-classifiers with SVM-based methodology can coordinate different methods for exploiting each complementarity and individual strengths (Maltarollo et al. 2019).
Random forest
The Random Forest algorithm was a supervised algorithm. The name itself says, ”This is a way of creating a forest from various perspectives to make it random”. The significant advantage of the Random Forest algorithm was, it can relevant for both regression and classification issues. In the procedure of regression and classification tasks, overfitting can happen normally, so the outcome will be in a worse state. We can defeat the overfitting issue through the usage of random forests algorithm with the availability of multiple trees in the forest. Random forests can apply trained algorithmic techniques i.e., bagging. Training set comprises,
, . Then, random samples can alternately selected from training data for fitting random forest tree.
Alternate samples with n trained examples from X, Y then .
Classification tree must be trained on data. Here, .
After training the data, invisible samples need to be predicted by averaging all individual trees on :
In classification trees, majority voting can be considered. Finally, random forest model produces better results due to the absence of increment in bias, it reduces variance in the model. The equation for individual regression tree on can be represented in standard deviation form i.e.,
where ‘A’ is a free parameter. In view of the size, nature of the trained data, a large number of trees can be used (Ho 1995). Also, the random forest can be appropriate in medication for deciding the right segments of grouping in therapy, and; investigating patient records can be supportive in recognizing the infections (Polamuri 2017). In ligand-protein binding affinity, using random forests can improve the scoring function performance (Kinnings et al. 2011; Zsoldos et al. 2007). Representation of scientific models and chemical structures are the fundamental issues in QSAR model (Dudek et al. 2006). At that point when descriptors are chosen, it is necessary to establish the best mathematical model for correct fitting in structure-activity correlation. So as to improve fitting standards in mathematical model (A Dobchev et al. 2014; Ning and Karypis 2011), a random forest algorithm was utilized (Fig. 4).
Fig. 4.
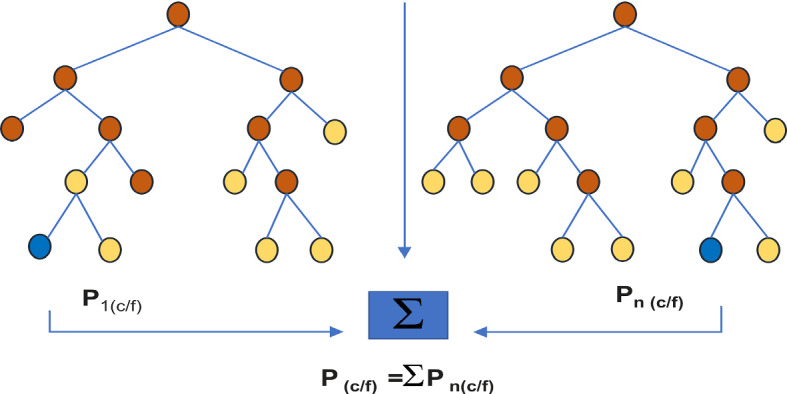
The random forest visually generated a data point decision tree to extract estimations for each sample to determine the best outcomes through voting
The selection of molecular descriptors is seen as an important step in virtual screening to identify bioactive molecules during the drug development process. Because this choice of descriptors shows predictions with lower accuracy. Hence, the random forest technique was used to improve prediction and then select naturally trained molecular descriptors for kinase ligands, hormone receptors, enzymes, etc. (Cano et al. 2017).
In the pharmaceutical industry, when developing drugs, the question that arises naturally is whether a prediction model trained with heterogeneous data is implemented as a similar prediction model. Then the heterogeneity data were compiled for forecasting and model training. In this study, heterogeneity was treated as a problem with the latent distribution, and the covariate-free allocation technique was distributed to be distributed by means of an ensemble leaf node model. In general, an ensemble-based random forest model has incorporated Heterogeneity Aware Random Forest (HARF) and assign specific weights to tree-based categories. Of course, the technique proposed by HARF gives better results than classical random forest, whereas drug feedback with the cancer disease types is something peculiar (Rahman et al. 2017).
Immune network technology is to determine new compounds from drug molecules. Using examples of sulfonamide properties, sulfonamides are divided into various prognostic effects over a period of time. Using a random forest approach, we selected molecular descriptors to achieve better accuracy than the simulation results for compounds designed for the drug (Samigulina and Zarina 2017).
Multilayer perception
The Multilayer perception model is also known as a feed-forward neural network. MLP provides an outcome based on a set of input sources. For training any sort of information, the backpropagation approach is utilized. This model is similar to a directed graph because of the essence of multiple layers as input nodes and output nodes are associated with some weights (Pal et al. 1992). After processing the data, the perceptron can fluctuate each connected weight in the network. In this way, the presence of error in actual output can be compared with the expected output. Consider node in output as degree of error in last data point i.e., Where , the variable developed from the perception. Based on some corrections, weights in each node can be adjusted through decreasing error in the output i.e.,
Also, every weight can be varied through the gradient descent approach i.e.,
where, learning rate and weights can be converted into a response without any oscillations. previous neuron result.
Depending on field, derivative can be calculated. Then, simplified derivative in output node can be
Here cannot be varied itself. Because changing each and every weight in hidden layer becomes difficult; Also, it provides
where is represented as the last node in the output layer. In case, changing any weights in a hidden layer, the activation function can be varied the weights in the output layer. Figure 5 performs specific computations to distinguish few features in input data. It learns optimal weights consequently and afterward input features will be increased with available weights to decide specific neuron was terminated or not. In this way, Multilayer perceptron uses backpropagation strategy with the activation function (Rosenblatt 1961). In this review, a multi-layer perceptron was utilized for predicting action between the drugs. This model has one advantage i.e., it does not require any structural information on compounds because of the fact that it uses experimental data for predicting the accuracy (Stokes et al. 2020). Additionally, MLP was utilized to generate a de-novo drug design. This model having the capability to generate different compounds automatically with some advanced properties (Gómez-Bombarelli et al. 2018).
Fig. 5.
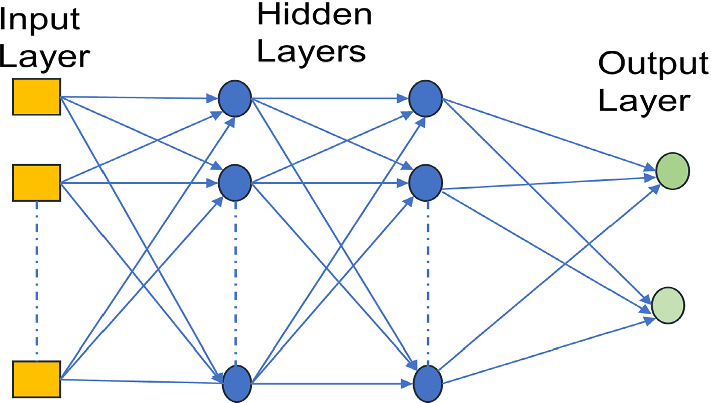
Multilayer Perception Architecture
In general, MLP can be used very easily and very quickly, but fulfilling its duties in training is very difficult, and MLP also does not offer any guarantee of global minimum performance (Gertrudes et al. 2012).
The secondary structure of proteins offers a greater advantage in determining protein function, drug design. In that study, the MLP approach showed greater interest in classification success. However, in the experimental area, determining the secondary structure is more difficult and expensive. Finally, the results from the trained data were reported as a positive success compared to the classification (Yavuz et al. 2018).
Deep learning
Deep learning is a part of machine learning, having the capability to extract a greater level of features through utilization of multiple layers from input data (Deng and Dong 2014). Deep learning is an immense field that is creating massive premiums nowadays. Recently, deep learning techniques have been used in many research fields and have achieved higher profitability in business strikes. But what exactly is deep learning? In general, deep learning is the same neural network architecture that consists of several layers, and data can be transformed between these layers. It’s still a significant popular expression, but the innovation behind it is genuine and very refined. So, models in deep learning can be developed through a strategy called greedy layer-by-layer (Bengio et al. 2007). Figure 6 contrasts the powerful deep learning approaches with pooling layers and figure outs the critical issues and devise the most appropriate solution even problem was in a complex situation. In this review, deep learning algorithms have presented numerous models like DNN, CNN, RNN, Autoencoder in drug discovery areas. The pooling layer is another structure that hinders the neural networks. The capacity of the pooling layer is to reduce the spatial size of the representation to reduce boundary measurement and system computations and work independently on each feature map (channel). The motivation behind why max-pooling layers work so well in various networks is that it enables the system to recognize the features very effectively after down-testing an input structure and it reduces the over-fitting.
Fig. 6.

Deep Learning Architectures
DNN architecture was evolved from an extension of Artificial Neural Network (ANN), contains multiple layers between input and output nodes (Bengio 2009). The DNN architecture traces the outcomes in a mathematical model either it can be a non-linear or linear relationship. Here, each mathematical model expected as a layer, also multiple layers were available in complex DNN, so that network is named as ‘deep’. Deep learning models are introduced in QSAR modeling to retrieve feature extractions and capabilities in chemical characters automatically. Dahl et.al had inspired by Kaggle’s results and improved investigations on multi-task DNN. The results of multi-task DNN have demonstrated incredible execution in learning general features of sharing parameters (Dahl et al. 2014).
Development of candidate drugs plays major desirable property in oral delivery. Molecules in intestinal permeability can be assessed by computational technology through affording rapid and reasonable ways. Multiple studies focused on intestinal intake of chemical composites for predicting the peptide sequence data. ML techniques like artificial neural networks have been adopted for predicting the intestinal permeabilities of peptides. The intestinal permeable of peptides consists of positive controlled data obtained through the peroral phage technique and random sequence data can be prepared through negative controlled data. Multiple statistical indicators like specificity, sensitivity, ROC score, enrichment curves, etc., are validated to produce appropriate predictions. And the statistical results declared that models have good quality and can segregate in between random sequences and permeable with great levels of confidence. Finally, the ANN models demonstrated greater prediction than unpredictable one. So, this model can applicable for intestinal permeable peptide selection to generate peptidomimetics (Jung et al. 2007).
Multi-task neural networks integrated into a platform called ‘DeepChem’, it helps the multi-task neural network to perform in drug development process (Ramsundar et al. 2017). Along with this, networks have assessed performance in the multi-task deep networks was robust. Finally, the performance of deep learning algorithms in QSAR models upgraded the prediction performance. Also, DNN played out a significant role in further research of hit-to-hit lead optimization.
CNN is a subclass of DNN, ordinarily utilized for analyzing the visual images (Valueva et al. 2020). CNN also called shift-invariant ANN because frequently rely on weights. CNN is a regularized version of a multi-layer perceptron. The concept of multi-layer perceptron characterizes fully connected networks, where each neuron in the first layer is associated with the following layer. By using of a fully connected algorithm, a network can conquer the overfitting problem. The CNN algorithm examines the clinical field so that, every neuron in a human cell appears like the visual cortex (Venkatesan and Li 2017). In ligand-protein interaction, many researchers utilized CNN model for predicting affinity in protein-ligand (LeCun et al. 2015; Leelananda and Lindert 2016). The affinity prediction indicated the best correlation in the dataset (Jiménez et al. 2018). In protein-ligand interaction, the CNN algorithm predicted binding affinities which can further increase scoring function but predictive capabilities must upgrade simultaneously.
RNN algorithm is an area of the artificial neural network, connections can occur between the input node and the output node. In this way, a directed graph can be created in the network along with a temporal sequence. Likewise, the RNN network utilizes the internal memory to perform grouping in input variables (Dupond 2019). It also exhibits dynamic performance Miljanovic (2012) because the RNN algorithm struggled for two networks at a time with the general structure. Each network may contain various impulses i.e., finite and infinite impulses.
Determining the functionality of protein structure will play a vital role in secondary and tertiary structures. Previously, numerous algorithms relates to folding prediction have improved to encode in the protein sequence experiment to develop protein structures. So, Visibelli has found signals on a large dataset. To locate specific occurrences in amino acids to characterize the specifications in secondary structure for deciding the helical moieties boundaries. The occurrences are predicted through various ML models for validating the hypothesis equipped with an attention mechanism. This mechanism can interpret the weights of each input, model’s decision for prediction. At last, the similar subsequences show the experimental outcomes, where input code-driven in secondary structure information (Visibelli et al. 2020).
Day by day, it has been turning out to be a challenge in improving affordable and effective treatments to humans without any prescience in drug target information. The deeDTnet is one of the deep learning techniques that were embedded with 15 variations of phenotypic, chemicals, cellular profiles, genomics utilized to accelerate drug repurposing and target identification. Due to the presence of high accuracy, deepDTnet has been approved by U.S. Food and Drug Administration with the identification of novel targets to familiar drugs. Through experimental results, topotecan was an approved inhibitor that can directly be utilized for human retinoic-acid receptors to diminish transitional void in drug development (Zeng et al. 2020).
In virtual screening, RNN utilized to cause new molecular libraries, so it got supportive in finding anticancer agents through molecular fingerprints (Kadurin et al. 2017). In producing the de novo drug design, the prediction must be conducted on biological performance. In this way, the RNN algorithm was utilized for generating molecules (Olivecrona et al. 2017). In the ChEMBL dataset, molecules could be gathered. For sampling, generated molecules must be trained by the RNN algorithm through conditional probability. Various classifiers performed data sampling however RNN with reinforcement learning has given 95% accuracy in scoring function (Mnih et al. 2015).
’Deep Interact’ was an integrative domain-based approach is utilized to predict PPI’s through Deep Neural Network. Assortment of multiple PPIs is extended out from (KUPS) Kansas University Proteomics Service and (DIP) Database of Interacting Proteins. It’s highly fundamental to discover and analyze the cellular components in the specificity of interactions and explicit molecular protein complexes. The significant goal is to develop enormous scope high-throughput experiments through silico approach to improve the uncovering levels in PPI. From a dataset known as Saccharomyces cervisiae, 34,100 PPIs have been validated to return promising results with a sensitivity of 86.85%, an accuracy of 98.31%, a specificity of 98.51%, and an accuracy of 92.67%. At last, the Deep Interact approach concluded to be better performed over existing ML approaches in PPI prediction (Patel et al. 2017).
Autoencoder is a class of artificial neural network, it retrieves information through unsupervised learning (Kramer 1991). Autoencoder objective is to represent the encoding data format in dimensionality reduction for maintaining a strategic distance from the ‘noise’ signal in the network. Along with this, the autoencoder must explore input data and then copied to the output layer. Autoencoder has two areas i.e., Encoder and Decoder; and one hidden layer. Here, the hidden layer is considered as code. Encoder transfers input data to the hidden layer. The decoder can retrieve information for reproducing the signal output. Autoencoders was most appropriate in dimensionality reduction and learning the data from generative models (Kingma and Welling 2013; Larsen et al. 2015).
Considering encoder as and decoder as , such that ,
In first hidden layer, encoder considered input as and maps to . Here, considered as code, as weight matrix, as bias vector, acts as activation function. Basically, biases and weights are randomly utilized and updated through backpropagation technique. Then, decoder maps to with same structure of ,
Decoder consists coefficients may vary in encoder i.e., coefficients. Mainly autoencoders were trained to decrease reconstruction errors (loss).
Here, feature space consists of less dimensionality than input space . Also is a compressed format for input ‘y’. At whatever point, hidden layers are more prominent than or equivalent to the input layer, it offers the adequate capability to learn identity function, finally, it was useless. In Autoencoders, test results despite everything to learn numerous valuable features from training set (Kingma and Welling 2019). In drug discovery, autoencoders utilized as unique architecture to deliver molecules through conducting experiments right into vermin (Zhavoronkov et al. 2019). In designing of de-novo drug design, deep learning model i.e., autoencoder have utilized for generating the molecules. So, the autoencoder approach was employed with various classifiers like multilayer perceptron for generating new compounds automatically with appropriate properties (Gómez-Bombarelli et al. 2018). In many situations, the drug produces invalid SMILES syntax, so as to defeat this issue, grammar variational autoencoder was utilized for developing SMILES syntax with more effectiveness (Pu et al. 2017) (Fig. 7).
Fig. 7.
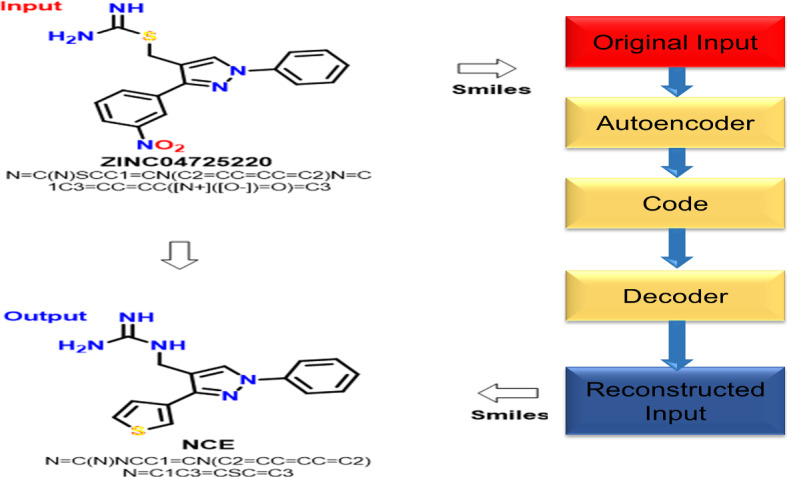
Basic flowchart of an AutoEncoder with an example NCE
Drug design applications
The review of drug discovery is further categorized on the basis of task performing of ML and their applications like target identification, hit discovery, hit to lead, lead optimization techniques are discussed out. The drug design techniques rely on the databases which are inturn developed based on the different ML algorithms. The precise training, validation, and application of ML algorithms in the drug discovery era provide an enthusiastic outcome by easing the complicated error-prone protocols. The ML techniques are introduced in most of the drug design processes to reduce the time as well as manual interference. The best example is QSAR, in which the huge data collection and training of datasets are considered as rate-limiting steps in defining the ligand-based virtual screening protocols and are now replaced by Denovo design techniques. The relationship between drug discovery steps and algorithms is presented in Fig 8.
Fig. 8.

Primary, secondary, tertiary and quaternary structures of the protein highlighted with active site residues. The AmpC beta-lactamase (PDB:6DPZ) as case example is taken and depicted in the above figures
Homology modeling/prediction of protein folding
The folding of secondary structure like and , which is formed by the interaction of side-chain amino acid residues are very critical to regulating the smooth functioning of three-dimensional proteins. An accurate protein folding along with its prerogative active ligand site can be experimentally obtained by X-ray crystallography, NMR-spectroscopy, and Cryogenic electron microscopic techniques (Cryo-EM).
Information about the primary amino acid sequences of proteins/enzymes/receptors, both dissolved / insoluble, is stored on the UNIPROT server along with their targets and cellular functions. Based on medicinal chemistry or pharmacological or biochemical studies, the main role of proteins is identified, and this information is also the basic unit for developing the protein folding prediction studies by software or experimental studies. Whereas, the protein folding predictions in the provided aminoacid (UNIPROT) sequence were compared with its experimentally derived PDB homologues which became a hopeful technique to refine the new protein models computationally and is also termed as ”homology modeling”. The homology modeling or comparative modeling is analyzed by the several algorithms which need to be implemented in either software modules (PRIME) or web servers (EXPASY, SWISS-MODEL) will definitely make a decision to predict the secondary structure folding with high accuracy within provided templates. However, the fine-tuning for the obtained homology models or template-based models are again scrutinized by Ramachandran analysis which can be sorted out by commercial modules (PRIME) or web servers (QMEAN, PROCHEK). For further understanding, the homology of CHIKV nsP2 protease is described here (Fig. 9) which is obtained based on experimentally predicted VEEV nsP2 protease template by using insilico techniques. The inisilico tool utilizes the computational databases to dig the information about the homology templates and provides the best closest match as considering for more practical bioinformatics and medicinal chemistry applications. Figure 8 depicted the alignment of secondary structures such as -helices, -pleated sheets, and loop representations present in tertiary complexes. The surface view also useful for recognizing the hotspots present on the protein to bind with incoming ligands/substrates. The sequence alignment mode also shows the mutations or differences in their primary sequences, it can be employed in different chemo-informatics approaches to identify the mutations similar kinds of viruses or any other pathogenic disorders. The significance of chemo-informatics is playing a crucial role and prevailing as an emerging tool in the current SARS-COV2 pandemic towards the identification of new drug-like molecules (Fig. 10).
Fig. 9.

a Overlap of 3TRK with 2HWK; b surface view of 2HWK; c, d off-surface/ribbon diagram of finest 3TRK model; and e homology validation parameter obtained from SWISS-MODEL
Fig. 10.

The overlap of active site residue of the CHIKV (homology model) (red sticks) and VEEV nsP2 protease (green sticks)
In addition, selecting the best homologous model obtained from the above process is another major task that can be performed with SVQMA (Support-vector-machine Protein single-model Quality Assessment ) servers or ProQ3 or ERRAT, which are operated by the Deep-learning methods. After going through the above steps, the best 3D protein template can be used for any basic drug chemistry study to identify hits that are part of a structure-based virtual screening protocol.
To provide insight for homology modeling, the Q5XXP4 fasta sequence belongs to CHIKV nsP2 protease domain has been employed as a template by overlapping its closest VEEV nsP2 protease solved protein (PDB:2HWK) as reference model using the SWISS-MODEL web server and the results are presented in Fig. 11. for understanding the above-specified concepts. Further, the active site residue position analysis of the finest developed model has been done and is found to have similar to VEEV nsP2 protease residues as shown in Fig. 10. The SWISS-MODEL also provides the information about percentage similarity along with structure alignment, the Fig. 10 shown the overlap of similar active site residues consists of catalytic site (catalytic diad Cys and His). It also represents the conformational changes present in the new template which also considered as an essential parameter for drug interaction studies
Fig. 11.
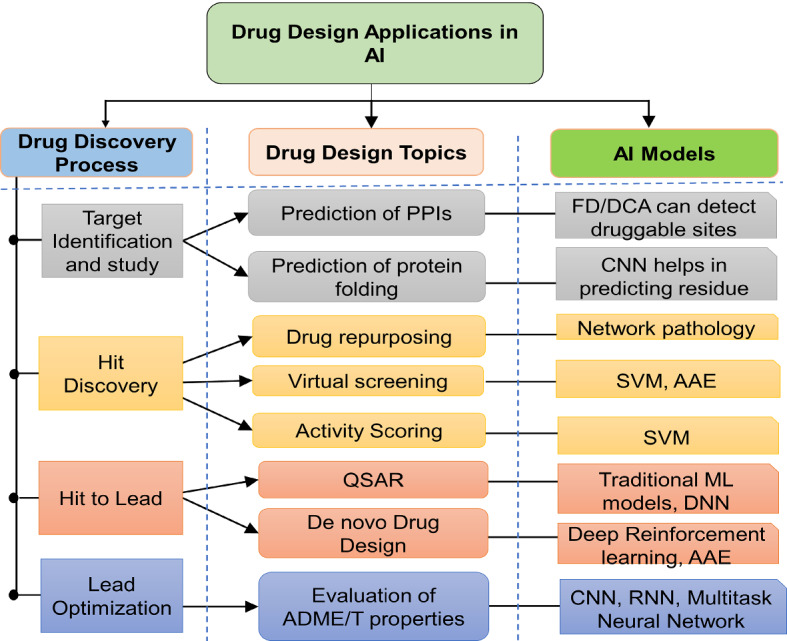
Illustrating drug discovery design techniques and topics with AI models
Target identification
The target identification for NCE’s is an extreme task due to lack of knowledge on their off-targets such as enzymes, ion channels, proteins, or receptors. The binding site recognition for the NCE’s is another key task for computational/bioinformatics experiments where more than one active site has existed in the protein. In the above cases, the predefined most popular web servers (FTMap), as well as specific modules such as ”Sitemap” developed with the help of algorithms, can define the preferential binding site to speed up the drug discovery process. A few other online programs like GHECOM, POCASA, Pocketome, SURFNET, ConCavity, LIGSITE, Q-SiteFinder, Fpocket, and PASS predicts the feasible binding sites located within the provided protein templates. Whereas, the metaPocket 2.0 program utilizes the above platforms to afford the most reliable ligand binding sites present on templates. Further, AI models like FD/DCA can also predict the druggable sites in the provided biological macromolecules. Recently, the DeepDTnet as a new target identifier in drug repurposing has been tested. The DeepDTnet strategy is developed by amalgamating the multi-disease cellular targets, pathogenic genes (genomics), and drugs (chemical spaces) being utilized for their treatment.
Prediction of protein folding
Patients who experienced illnesses can be recognized through protein dysfunctions. Here, active molecules can recognize through a structure-based drug design approach. Time and cost consumption should be required for 3D structural processing, and it is also important to be aware of what algorithms are used to predict the 3D structure of proteins. Because of the essence of the large amount of protein sequence data, it creates a problematic issue in making 3D structure accuracy for de-novo prediction. For retrieving feature extraction capabilities, deep learning approaches must apply prediction in backbone torsion angle (Li et al. 2017), secondary structure (Spencer et al. 2014), and protein residue contacts (Wang et al. 2017). At long last, the goal was to predict the 3D protein structure. Also, deep learning techniques have elaborated this field for improving 3D protein structure.
Prediction of protein–protein structure
PPI’s are essential for biological processes and infections (Falchi et al. 2014; Scott et al. 2016). PPI can be characterized as ‘it performs similar to networks for mathematical representation of physical contacts between cell proteins. Composed contacts between binding regions in proteins have specific biological importance. Also, it obtains the experimental and bioinformatics strategies from PPI’s database (Li and Lai 2007; Szklarczyk et al. 2015). PPI interface is also referred to as a collection of multiple residues (Cukuroglu et al. 2014). In this way, it turns into a new class for drug targets that are different from mainstream pharmaceutical targets like ion channels, coupled receptors, G-protein, etc (Higueruelo et al. 2013; Santos et al. 2017). At that point, a new class will extend the target space for improving small molecule drugs (Shin et al. 2017). When contrasted with traditional drug targets, target PPI’s reduces harmful impacts because of increment in biological selectivity of regulatory impacts (Valkov et al. 2011). It is mandatory to learn fundamental ideas of the PPI interface on the protein-protein structure. Because of the less accessibility of PPI’s data, it contributes many computational techniques for predicting PPI’s interface (Xue et al. 2015). Those techniques are dependent on a template which makes it simple for PPI interface protection (Zhang et al. 2010). For example, a website name “eFindSite” (Maheshwari and Brylinski 2016) utilized for predicting PPI interfaces which consist of templates, residues, and sequence-related features for improving SVM, NBC techniques. If the chance of two interactive protein structures is vacant then it makes it easy for predicting the PPI interface (Vakser 2014) where it mainly relies on complementarity rules of protein-protein docking (Chen et al. 2003) and SymmDock strategies (Schneidman-Duhovny et al. 2005). When two unbound proteins are integrated and converged as one protein, then a difficulty emerges for predicting the conformational change. When an equivalent accent sequence needs to be derived, deep learning models are used to predict PPI and better improvement is achieved compared to machine learning models such as SVM (Du et al. 2016). Searching for druggable sites for interface in the buried zone (in the range of 1500-3000 A2) (Scott et al. 2016) was mandatory. Considering druggable sites as hotspots because of providing an enormous amount of binding free energy to convince the medical chemists (Cukuroglu et al. 2014).
Bai et al. (2016), utilized two techniques i.e., fragment docking and direct coupling analysis for detecting druggable PPI sites. Fragment docking named “iFitDock”, utilized for checking druggable hot spots(problem areas) in the PPI interface. Further improvement for candidate binding locales needs to integrate similar small hot spots. At last, based on the evolutionary conservative level, the scoring function must be located to provide the finest protein-protein binding spots. The PPI interface objective was to improve computational methodologies for locating the best hot spots and significant structure of small modulator targets in the PPI interface.
Prophecy of protein–protein interactions
The Protein-Protein Interactions (PPI) is one of the major biological phenomena through which the basic units of the body (cell) transports the signals, ions, substrates, and energy production components that need to improve the pharmacological responses needed by the body. In another circumstance, the PPI plays a critical role in the pathogenesis of the disease such as various types of cancer, especially colorectal carcinoma. The development of colorectal carcinoma in humans is purely dependent on lifestyle as well as hereditary means. However, the pathogenesis of the colorectal carcinoma is linked with the formation of malignant Adenomatous Polyposis Coli (APC) and its migration in the entire colorectal portion in the body is majorly occurs due to the interaction of APC protein with Asef (guanine nucleotide exchange factor) and with TCF4 component peptide are located in the pathogenic carcinoma cells. The example APC-Asef, -catenin-TCF4 PPI has been illustrated in Fig. 12.
Fig. 12.

a, b Protein-protein interactions of APC-Asef (yellow surface/cartoon-APC & cyan surface/cartoon-Asef); and c, d PPI of -catenin/TCF4 in surface & cartoon forms (yellow surface cartoon- -catenin & cyan surface/cartoon-TCF4)
In recent years, the PPI-based drug discovery programs are experimentally produced a hopeful pharmacological substance, in terms of cancer pathogenesis, APC-Asef PPI inhibitors are the best example which are delivered the basic peptides as an initial point to switch on the medicinal chemistry oriented drug design projects. The importance of PPIs in understanding host pathogenic protein interactions is another extreme task that excites most vaccination programs. Battling against SARS-CoV2 infection is a key paradigm in the current scenario where the scientific community targets a protein spike from SARS-CoV2 that preferentially binds to the human angiotensin converting enzyme-2 (hACE2) to enter into the alveoli mainstream of lungs and cause severe obstruction in respiratory syndrome. However, the time and cost for experimental prediction of PPI are considered as rate limiting barriers. In this regard, the different databases hosted the web servers (few are publicly available) framed by targeting PPI which are prevailing as preliminary PPI identification tools to accelerate the medicinal chemistry research.
Hit discovery
The Hit discovery process is advanced in success which has been taken in drug discovery. In this procedure, small molecules are considered as hits for target binding to identify the best-altered functions. The detection of hit by diverse algorithms is currently prevailing as a robust technique in the current drug discovery paradigm. An application of multivariate parameters (K-nearest neighbors (K-NN) and support vector machine(SVM)) on high-content screening (HCS) analysis in one such method produced a variety of hits against neurological complications.
Drug repurposing
DeepDTnet’s training parameters outperform other existing target identification techniques and rely on a minimum quantity of FDA-approved drugs (732 drugs) to produce beneficial therapeutic effects (human retinoic acid receptor orphan receptor gamma t-ROR-t) of the existing topoisomerase inhibitor Topotecan (TPT). The deepDTnet strategy also transfers several FDA drugs with different chemical scaffolds against GPCR with new targeted pharmacological actions. (See in Figs. 13, 14, 15). The deepDTnet algorithm is considered to be much more advantageous than NetLapRLS and KBMF2K methods as well as Naive Bayes, SVM, KNN, and Random Forest algorithms.
Fig. 13.

The FDA approved drugs under drug-target repurposing applications derived by deepDTnet
Fig. 14.

The FDA approved drugs under drug-target repurposing applications derived by deepDTnet (contd.)
Fig. 15.

Basic overview of molecular docking sampling and scoring flowchart
“Repurpose” refers “reprocess/reused/recycle”. Drug Repurposing is characterized as ‘locating new indications for drugs (Ashburn and Thor 2004; Lotfi Shahreza et al. 2018) which are as of now in the existence stage’. Because it reduces time and hazardous circumstances in drug discovery (Ashburn and Thor 2004). A significant reason for utilizing the drug repurposing concept in drug discovery, because it exceptionally supportive to have multiple targets (Susan et al. 2017) in each drug which corresponds to various impacts. In this way, it provides high diversity in drug-disease relationships. Example: Few drugs extend its life expectancy such as “Metformin” which is an approved medicine to deal with diseases like “type 2 Diabetes”. In repurpose, essential elements are “drugs and diseases” (Cabreiro et al. 2013, De Haes et al. 2014, Martin-Montalvo et al. 2013) utilized. Drug targets and disease genes are other elements utilized in drug repurposing.
In order to show the interactions that have occurred in element (Lotfi Shahreza et al. 2018), this can be performed through the network investigations based on diversity interactions. Nine sorts of networks arranged in drug design concept i.e., Gene regulatory networks, target-disease networks, drug-adverse networks, metabolic networks, protein-protein networks, drug-drug networks, drug-disease networks, disease-disease networks, drug-target networks (Lotfi Shahreza et al. 2018). In general, the network’s model principle was, indistinguishable drugs have similar targets/effects (Yamanishi et al. 2008). If data is less or fragmented, in that situation drug repurposing is necessary. For repurposing, integrating the entire multiple networks to create extraordinary (heterogeneous) networks. At last, consolidate the drug repurposing with drug target prediction to generate drug target (Wang et al. 2014). So, drug target assists with treating the sicknesses. To generate new targets and indications, then utilize the network diffusion algorithm and dimensionality reduction approach (Luo et al. 2017).
Virtual screening
It is an AI strategy utilized in the drug discovery process for locating small molecules to distinguish bind structures for a drug target. In drug development, virtual screening also utilized software as well as algorithms to recognize hits from private chemical collections for retrieving unique hits inefficient way. After identification of new hits, a further step needs to purify compounds with unfavorable scaffolds (framework) (Lavecchia and Di Giovanni 2013). And furthermore incorporates hardly includes few strategies like docking-based, similarity searching (Willett 2006), pharmacore-based (Willett 2006), and machine learning methods (Leelananda and Lindert 2016). Based on the above techniques, classification has taken two strategies i.e., structure-based and ligand-based virtual screening.
When 3D-protein structure was accessible then molecular docking process can be widely utilized (Chen 2015). Many applications related to docking-based virtual screening have built (Talele et al. 2010) effectively without any impacts. May some obstacles are present in this strategy such as the scoring function. A scoring function cannot estimate binding affinities (bond/relationship) with accuracy because insufficient arrangements and entropy impacts (Huang and Zou 2010) have taken protein flexibility which makes it more complicated (Chen 2015). Finally, many docking models considered binding affinities and refuses remained like docking score, distance-time (Copeland 2010; Xing et al. 2017). When compared to docking-based virtual screening, the ligand-based virtual screening cannot confide to the 3D-protein structure. Its goal is to design bioactivity domains from molecular features (Lavecchia and Di Giovanni 2013).
In this concept, the aim is to persistently improve yields and to decrease false hit rates (Leelananda and Lindert 2016; Liew et al. 2009; Melville et al. 2009). To accomplish this objective, the SVM technique was frequently utilized in virtual screening (Ma et al. 2009). DL strategies have been applied to retrieve great classification capacity, low generalization error (LeCun et al. 2015; Thomas et al. 2014) and powerful feature extraction ability. Example: In virtual screening, sparse distribution method wastes a lot of time in searching process (Ma et al. 2009; Segler et al. 2018). So as to conquer this issue, molecule libraries must be provided along with unique training molecules (Thomas et al. 2014) among the Simplified Molecular Input Line Entry Specifications (SMILES) and natural language relies on long short-term memory network architecture. ML techniques like DNN and gradient boosting trees provided the molecular libraries by RNN. Adversarial autoencoder models the molecular fingerprints to locate potential anti-cancer agents (Kadurin et al. 2017).
High throughput virtual screening and scoring in molecular docking techniques
Routine techniques used after target identification are high through virtual screening (HTVS) and molecular docking techniques embedded in free energy perturbations, sampling, and scoring algorithms. The knowledge of active site for the protein/receptor where ligand would bind to mimic/antagonize the physiological role which is an essential task to initiate the HTVS protocol. Similarly, the ligand-based virtual screening (LBVS) considered as another basic method relies on the Physico-chemical properties of chemical databases (Fig. 15).
Activity scoring
In virtual scoring, the scoring function is a fundamental component in molecular docking for assessing binding affinities towards target (Huang and Zou 2010). In machine learning, mapping ability features can yield great accomplishment to extract physical, geometric, and chemical features (Khamis et al. (2015)) to retrieve scores. Based on scores, data-driven black box models which are considered to predict interactions in binding affinities and furthermore avoiding few concepts in docking like physical function are very hard to study (Ain et al. 2015). Random Forest and SVM concepts identified with AI utilization for better performance in the scoring function. For instance, an SVM model can be utilized instead of a linear additive method related to the energy terms concept. Since an SVM can characterize the relationship between experimental binding affinities and own energy terms i.e., can be extracted from docking program eHiTS. Thus, data gives better execution in scoring power and screening power (Kinnings et al. 2011; Zsoldos et al. 2007).
Numerous researchers initiated in utilizing the CNN model in image processing (LeCun et al. 2015) field because CNN demonstrated better performance and protein-ligand interactions providing numerous features to CNN for predicting protein-ligand affinities. In the estimation of protein-ligand affinities, Jimenez et al. worked on the 3D visual representation of CNN model and binding affinities (Jiménez et al. 2018) which have indicated better correlation behavior in data sets. And essentially, deep learning represents its genuine intensity to increase abstract features from primitive features, since it’s necessary to represent fundamental features for a compound-protein structure like molecule types, particle separation (LeCun et al. 2015) etc. A structure Deep VS, reliant on CNN model, got familiar with abstract features from fundamental features to provide docking programs like GLIDE SP (Friesner et al. 2004) and ICM (Abagyan et al. 1994). Thus, the point in activity scoring was, choosing few features among protein-ligand interaction for predicting binding affinities with help of the CNN model, so it increases information scoring function but it upgrades the predictive capabilities.
Hit to lead
It is also referred to as lead generation in the beginning phases of drug discovery. It locates small molecules referred to as hits from the High Throughput Screen (HTS) through deficient optimization to locate promising lead compounds. The practical interface of hit-to-lead optimization approach integrated with chemical synthesis as well as mapping algorithm ”design layer”/Random Forest regression applied to create new biologically active chemical spaces through the utilization of existed kinase inhibitor library (Desai et al. 2013) (Fig. 16).
Fig. 16.
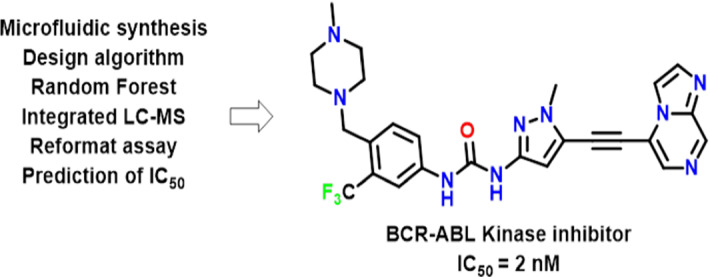
Abl kinase inhibitor obtained from Hit-to-lead optimization protocol linked with ML algorithms
QSAR
QSAR analysis was used in the hit-to-lead optimization process to find potential lead compounds from the hit analogs with the prediction of bioactivity analogs (Esposito et al. 2004). And primarily utilized in mathematical concepts to study quantitative mapping with physicochemical or structural objects and biological activities. QSAR analysis taken apart in foundation of mathematical models, selection and making the progression of molecular descriptions, evaluation and interpretation methods, utilization techniques (Myint and Xie 2010). Here, mathematical models and chemical structure representations are considered issues in QSAR demonstration. When descriptors are chosen, then locating mathematical models is necessary to fit relationships in the structure-activity technique. In the year 1964, Hansch equation was suggested by Hansch et al. For clarifying the 2D structure-activity relationship, utilize the parameters like physicochemical descriptors and linear regression models for presenting QSAR study as another section (Hansch and Fujita 1964).
In the same year, Free-Wilson model suggested by Free et al. He formulated the bioactivity description and chemical structure relationships have hypothesis concept to contribute substituent in compound activities (Free and Wilson 1964). Contrasted with the Hansch method, the Free-Wilson method can encode the chemical structures since it predicts legitimately from the chemical structure without any physiochemical parameters. Random Forest and SVM are machine learning procedures, used in mathematical models (A Dobchev et al. 2014; Dudek et al. 2006; Ning and Karypis 2011).
Likewise, QSAR modeling utilized deep learning techniques to retrieve capabilities in chemical strings and automatically extracts the features. Merck Molecular Activity challenge was held in 2012 and a team called George Dahl’s won the challenge in ensemble methods like gaussian progress regression, multi-task DNN, and gradient boosting machine (Ma et al. 2015). Kaggle inspired the results in multi-task DNN. Along with this, Dahl et al. proceeded to work on the multi-task DNN concept and shown excellent performance in single-task neural systems.
Due to multi-task strategy, neural networks learn features from different parameters however tasks can be similar (Dahl et al. 2014). Ramsundar et al. (2017) utilized multi-task neural structures in drug development to assess the performance and finally, excellent results appeared in the random forests algorithm. Since multi-task neural structures consolidated towards platform called Deeepchem. Subramanian utilized canvas descriptors for employing DNN. Prediction in binding affinities needs to reinforce the regression and classification model to gain results in human -secretase-1 inhibitors (Subramanian et al. 2016). Usage of DNN model gives great results in validation set i.e., classification capability gives 0.82 accuracy, it exhibits regression ability with 0.74, MAE (Mean Absolute Error) is 0.52. DNN model utilizes the 2D descriptors and indicated better results when compared with force-field-based strategies because of the utilization of partial capability models in deep learning. At last, QSAR models rely upon deep learning techniques which allots the better results in the future prediction role of hit-to-lead optimization research.
De novo drug architecture
De novo Drug Architecture progressed unique chemical structures by adjusting or balancing the target interest (Hartenfeller and Schneider 2010). To introduce a new molecule from scratch using a popular De novo model called the fragment-based approach. If at this point there are impracticalities and complexities in the molecular structure (Schneider et al. 2017), the risk arises in the development of the structure and becomes difficult in the assessment of bioactivity. Deep learning models utilized powerful knowledge and generative capabilities to introduce a new structure with appropriate properties (Mullard 2017).
In the De novo drug design process, the deep learning models acts as autoencoder to generate an appropriate format for new chemical entities (NCE’s). Therefore, an embedment of autoencoder with multilayer perceptron classifier is also a value-added technique in the generation of NCE’s with predefined physicochemical properties. The syntax of the drug/chemical structure is produced in SMILES format which might be difficult to understand in many circumstances and grammar variational autoencoder (VAE) overcomes this problem to accelerate the process (Fig. 17).
Fig. 17.
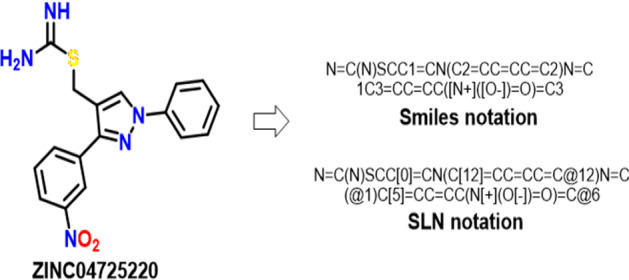
Smiles/SLN notation of antiviral compound
Deep reinforcement learning technique extended by Olivecrona et al. for predicting biological activities to develop new molecules by adjusting RNN model (Olivecrona et al. 2017). To obtain SMILES syntax, RNN model to be trained; where molecules can collect from chemBL. In reinforcement learning, agents act through actions in activities under certain conditions. At this point, if the agent gets a positive reward, the actions made by the agent’s trend can be renewed (Mnih et al. 2015). To acquire a high reward for activity scoring, then utilize the SVM technique to enhance few approaches relying upon ligands concept in the training set. Generate few molecules against dopamine receptor 2-type for employing deep reinforcement learning model with RNN model. Along with this, it observed predictions have taken over 95% for structures in the bioactive region through the scoring capacity of SVM. By utilizing deep learning techniques, unique molecules can be created through the auto-encoders technique. To generate new molecules automatically with appropriate properties then, Gomez-Bombarelli et al. (2018) integrated multilayer perceptron (MLP) and variational autoencoder (VAE).
In PPI prediction, numerous tackles have taken placed due to (i) spending low expenditure in protein information, (ii) lack of known PPI to learn about the explicit virus, (iii) inefficient strategies due to sequence dissimilarity in viral families. The de-novo methodology motivation is to predict innovative PPI virus with its host. De-novo was a sequence-based negative examining framework that learns the diverse viruses in PPI to predict the innovative one, where the shared host proteins can exploit. For assessing generalization, de-novo has endeavored to test the PPI’s with various domains. At last, the De novo approach retrieved 81% accuracy in reducing the noisy negative associations and 86% accuracy in the viral protein prediction that utilized in the training period respectively. De-novo strategy accomplished more comparable in intra-species and single virus-host prediction cases. In this way, it turns to be difficult to predict the PPI for a contaminated person and optimal accuracy is obtained when carrying out tests for the human-bacteria interactions (Eid et al. 2016).
To develop biological and chemical prospects, multi-objective optimization technique and AI has given promising outcomes through entrusting an automated De-novo compound structure like a human-creative mechanism. In this study, innovative perception pair, which relies on multi-objective technology, is to apply the RNN algorithm to automate unique molecules with a de-novo structure build on common properties found among constant physicochem properties for leading trade-offs. In this view, multiple chemical libraries related to de-novo structure targeting acetylcholinesterase and neuraminidase. For assessing chemical feasibility, validity, drug-likeness, and diversity content were employed through numerous quality metrics. In the de-novo generative molecules, molecular docking has taken place for the evaluation of posing and scoring through X-ray cognate ligands with similar molecular counterparts. At last, multi-objective optimization and AI are provided to use easily for customizable design techniques which especially effective for lead advancement and generation (Domenico et al. 2020).
For the most part, the network consists of 3 segments i.e., encoder, decoder, and predictor. Encoder plays a significant role in changing strings called discrete SMILES into latent (inactive) space, where vectors are considered as constants. The decoder role was considering vectors back to the past string stage i.e., discrete SMILES. In the predictor stage, Multi-Layer Perceptron (MLP) approach is used for predicting the molecules. For retrieving a high prediction ratio in constant vectors, then utilize the gradient-based technique. To locate new molecules rapidly with appropriate properties, then utilize 2 techniques i.e., Bayesian inference and gradient-based approach. By using both approaches, a significant advantage was delivering a high predictive ratio consequently, where humans can comprehend the chemical structure. It does not correlate to chemical structure when SMILES syntax is invalid. To maintain a strategic distance from such difficulties, make the result source more constrained; Pu et al. used variational autoencoder (VAE) for characterizing SMILES syntax (Pu et al. 2017).
For creating molecular fingerprints, Kadurin et al. have utilized the AAE model, were later referred as druGAN. While using the AAE technique, it demonstrated excellent performance in the VAE model in areas of generation ability, error in reconstruction area, further extraction ability (Kadurin et al. 2017). Coley et al. (2018) suggested locating whether the generated molecule was synthetically accessed or not. Depending upon the reaction database, the neural network was trained because of the availability of excellent approximation capabilities for retrieving synthetic complexity metrics. The fundamental explanation behind synthetic reaction is to increase the reactant complexities i.e., the score in product complexity must be greater than reactant (Andras 2017). Coley strived numerous attempts to build scoring function through encoding chemicals response into product pair and reactant pair for clarifying correlation inequalities between product and reactant complexities. To become familiar with any scoring capacity at that point, neural networks need to be trained where Coley utilized reactant and product pairs in a scope of 22 million. Along with this, the outcome determined with huge complexities in the synthesis process. At long last, generative models not just clarify drug activities in inverse synthetic planning yet additionally discloses synthetic complexities due to disposing of the non-realistic molecules.
Lead optimization
The lead optimization is an essential step of the drug discovery process in which the best medicinally active fragment hits are considered leads to extend the medicinal chemistry projects. The main aim of the lead optimization is to eliminate the side effects/notorious effects of the existing active analogues by a minimal structural modification to yield a better and safer scaffold. One such example is the optimization of Autotaxin inhibitors such as GLPG1690 clinical agent which is advanced in human clinical trials to combat pulmonary fibrosis. Another example is to increase the potency by tailor-made approaches to provide better active analogue. Here, the various properties of ADME/T like Chemical and physical properties, Absorption, distribution, metabolism and excretion, Toxicity, and the ADME/T multi-task neural networks are discussed in the following sections.
Chemical and physical properties
In the drug discovery pipeline, physical and chemical properties have been utilized to reduce significant failures. At that point, deep learning models are utilized lead optimization techniques to improve unique methodologies (Lusci et al. 2013). Duvenaud et al. (2015) extracted data from molecular graph directly by adopting the CNN-ANN concept to perform prediction i.e., (MAE is 0.53+0.07) due to relied upon interpretability concept. Coley et al. inspired Duvenaud’s work and begun working for better results in molecular aqueous concepts. And furthermore used the tensor-based convolutional technique and gave better outcomes as MAE (0.424+0.005).
It’s necessary to clarify molecular graph attribution since tensor-based techniques need to integrate features like a bond, atom levels. For predicting molecular aqueous solution, Coley’s employed an enormous number of atom level information compared to Duvenaud’s model (Coley et al. 2017). Establishing a great correlation between Caco-2 permeability coefficients and oral drug absorption (P app) for predicting the candidate drug (P app) (Artursson and Karlsson 1991; Hubatsch et al. 2007) in the estimation of pharmacokinetic properties. To fabricate prediction templates with 30 descriptors (Wang et al. 2016) at that point, Wang et al. composed 1,272 components for permeability information of Caco-2 including models like SVM regression, boosting. In the testing set, the boosting model demonstrated the best outcomes with great expectation capability. It follows QSAR principles from OECD (Organization for Economic Co-operation and Development). So as to persuade reliability and rationality, then follow the sequence of OECD standards.
Absorption, distribution, metabolism and excretion
Entering medicines or drugs into veins of the human body under some activity site known as drug absorption. For examining the degree of absorptions utilize the bioavailability parameter. Numerous clinical departments clarified optimization of absorption properties with a prediction of bioavailability molecules (Tian et al. 2011). In the usage of the MLR model, Tian et al. employed 1,014 molecules for bioavailability prediction through molecular assets and structural fingerprints. By utilizing the genetic function technique, excellent results appeared in predictive performance as RMSE = 0.2355 and correlation coefficient is 0.71 respectively. Conveying drugs or medicine into the human body i.e., intracellular and interstitial fluids along with few drug absorption (Sim 2015) properties called as drug distribution. Drug distribution at steady state (VDss) is a proportion of dosage from vivo stage into plasma reaction. The steady phase in drug distribution is the significant index for evaluating the drug distribution process. Thus, VDss must be predicted; Lombardo and Jing have created PLS and Random Forest techniques along with 1,096 molecules (Lombardo and Jing 2016). Here, board members are not satisfied with prediction results because 50% of molecules are accessible in twofold error. VDss may influence by the presence of obscure factors. To defeat this issue, intently taken as a challenge for VDss value in molecular structural data. If a drug or drug enters the human body under the conditions applied, the drug itself tries to produce the current toxic metabolite in order to successfully structure the metabolism. To ensure the strength of the metabolic structure, use structural optimization techniques to encourage the metabolism to make predictions with high accuracy. Many AI strategies adopted a huge amount of drug metabolism information to predict unique metabolic enzymes like UDP-glucuronosyltransferases (UGT’s), cytochrome P450s, etc. Furthermore, neural networks trained in UGT metabolism at Xenosite (Matlock et al. 2015; Zaretzki et al. 2013) platform for predicting the UGT metabolism (Dang et al. 2016). Eliminating dosage from drugs and also metabolites from the human body referred to as drug excretion. Drug metabolites are wiped out from the human body either with the usage of water (i.e., some drugs can be soluble in water) or it directly eliminated through the absence of metabolism. For retrieving excellent results in unique mechanisms, Lombardo et al. utilized the PCA technique with an expectation pace of 84% (Lombardo et al. 2014) accuracy.
Toxicity and the ADME/T multi-task neural networks
In clinical and preclinical damage accomplishment was reduced the adequacy of about 33% of significant molecules in drug localization, optimizing the significant molecules reducing risk hazards by predicting toxicity (Guengerich 2010). Prediction can perform through techniques called structural alerts and rule-based expert knowledge for toxicity profiles like kidney and liver. Here, deep learning models are required to produce better results in toxicity prediction. Along these, Xu et al. created a prediction model named acute-oral toxicity, for predicting results on molecular graph encoding CNN (MGE-CNN). Predicted outcomes indicated as better when compared with SVM model (Youjun et al. 2017). Therefore, the MGE-CNN model succussed because of feature extraction, model development, molecular encoding is similar in training for neural networks. The advantage was, the issue can alter through molecular fingerprints because of accessibility of flexibility in the MGE-CNN model. For acquiring great fragments relates to structural alerts, Xu et al. utilized toxic features for fingerprints which characterizes TOX Alerts (Sushko et al. 2012). If parameters were comparative, then it’s necessary to correlate with trained multi-task neural networks and performance demonstrated better results contrasted with single task neural networks (Mayr et al. 2016) because of sharing parameters and more supportive towards multiple tasks for retrieving similar features. At last, some information is provided to the human body when drug absorption, distribution, metabolism, and excretion has handled and prediction improved through performing multi-tasking neural networks. Here, single-task and multi was tasks contrasted by Kearnes et al. with ADME/T experimental data, and outcome demonstrated better performance in multi-task model (Kearnes et al. 2016).
ML in e-Resources for drug discovery
The AI and ML algorithms prevailed as the main computational scoring functions for evaluation when a predicted value was added as a parameter, which is involved in the basic drug discovery paradigm (Stork et al. 2020), it illustrated in 18. The detailed applications of the ML algorithms specified in the e-resource are described in the following sections (Fig. 18).
Fig. 18.
ML in e-Resources of drug discovery platform
ML in Pan-assay interference screening (PAINS)
The precise information about hits can be obtained from primary or secondary biological screening assays of purchasable/commercially available databases which were the most important parameters before starting medicinal chemistry projects. Thus, elimination of the compounds has been exhibited its presence in different cellular biological assays considered as pan-assay derived hits could reduce the cost and time of the medicinal chemists. The pan assay information can be accessed from the PAINS database on request. Therefore, the Hit Dexter 2.0 web server has been launched compiled from Pubchem library and screening assays. The Hit Dexter 2.0 could be initially utilized to know the biological properties of the newly designed compound and thus anyone can easily eliminate the pan-assay interfering compound at the initial stage itself (Stork et al. 2019).
ML in drug metabolite and metabolic site prediction
The identification of metabolic site for any kind of drug or new chemical entity is very essential before its administration into the human body. The prediction of drug metabolism can be done by animal models (preclinical studies) which was a rate-limiting step as well as costly and it is mandatory to retrieve therapeutic approval of new chemical entities. The site of metabolism can be predicted by several modules among ”ADMET Predictor” of SimulationsPlus tools have gained attention and is pure works on the models compiled by the artificial intelligence algorithms. The FAME3 is one of the online servers which predicts the region for the given drug/compound which undergoes metabolism validated databases gathering phase-1/phase-2 metabolic parameters associated with several databases validated by comparing with Matthews correlation coefficient (MCC) (Stork et al. 2020). It is also important to have an overview of the chemical modification of drugs/NCE’s which are undergone the metabolism and thus can be used in calculating dosage regimen, dosage frequency, toxicity, and other beneficial side effects. The online services such as GLORY/GLORYx provides the precise information about the possibilities of new metabolite and their relevant formation data with respect to mitochondrial cytochromeP450 enzyme and conjugations (de Bruyn Kops et al. 2019).
ML in skin sensitivity parameter prediction
The prediction of skin sensitivity is one of the essential criteria for assessing safety parameters of the new drugs/compounds and it is patient to patient specifications. In this regard, the AI models such as Random Forest based MACCS (RF_MACCS) and support vector machine (SVM) based PaDEL (SVM_PaDEL) algorithms trained with approximately 1400 ligands linked with local lympho node assay (LLNA) information (Stork et al. 2020; Vranic et al. 2019).
ML in natural product identification
The ML trained with 265000 natural product isolates and synthetic libraries validated by MCC is being used as a basic predictive model NP Scout online server will reveal the probable identity of the newly discovered analogs. The application of NP Scout in the prediction of sources for the query molecule might provide information about their natural product sources and could become a part of natural product-based drug discovery (An et al. 2019).
Drug discovery problems
In drug development and discovery, numerous clinicians and specialists confronted challenges towards target validation, computational pathology data, identification of prognostic biomarkers in clinical preliminaries.
Target validation
By regulating the molecular target activity, drugs can be developed through the utilization of ultimate methodologies in drug discovery for altering the infection state. By inaugurating a program in drug development, target identification requires a therapeutic hypothesis for modulating target regulation in the outcome of the infection state. When available evidence is identified for that target, it can be considered as target identification. Based on fundamental decisions, in vivo and ex vivo models are utilized to validate the target disease. In target validation, outcomes can be retrieved through clinical preliminaries, yet it’s necessary to concentrate on target validation efforts for successful projects. The diseases incorporate metabolomic, transcriptomic, proteomic profiles that are available in-patient clinical material. With the clinical database, the capability of re-utilizing data through public databases provides the primitive target identification and target validation. For predicting target identification, it requires appropriate strategies for yielding legitimate statistical models.
ML approaches are used in target identification because of the increment of data-driven target identification experiments. In target identification, recognizing causal confederation among disease and target is the initial step. Target disease modulates either naturally or artificially (experimental). By using ML approaches, prediction can be taken placed on known properties of targets, causalities, driven targets. ML techniques can apply from various perspectives in the target identification field. For predicting genes with dysphoria, a decision-tree classifier need to be trained on a protein-protein localization network (Costa et al. 2010). So, distinguished few key parameters in decision-tree inspection i.e., extracellular path, transcription factors, metabolic paths. John et al. improved a classifier model called SVM with genomic details for classifying proteins towards non-drug and drug spots in ovarian and breast cancer (Jeon et al. 2014). mRNA expression, network topology, protein-protein interaction, DNA copy numbers are the key segments in classification and recognized 122 cancer targets globally. Targets identified as 462, 266, and 355 related to pancreatic, breast, and ovarian tumors. Peptide inhibitors were validated through the prediction of two targets. Outcomes in the cell culture approach were identified as more prominent anti-proliferative effects. Although, in pancreatic tumors, usage of inhibitors shown twice greater inhibition on cells.
To distinguish transcriptional changes in Huntington’s disease, Ament et al. developed a model called mouse transcription factor site with transcriptome information (Ament et al. 2018). By utilizing LASSO and regression models in mouse striatum, a genome-scale has been created for 718 transcription factors. Transcriptional factor modules are recognized to provide treatment in the early phases of Huntington’s disease. In tissue-related anti-aging treatments, Mamoshina et al. (2018) identified molecular targets for comparing gene-expression signature with old and new muscles. When contrasted with supervised machine learning models, SVM exposed feature selection and linear kernels are generally appropriate for identifying biomarkers. Predicted targets can be developed through ML i.e., blind drugs can furtherly be utilized for therapeutic assumptions. For identifying affiliations like gene-disease, drug-disease, target-drugs, then apply NLP kernel strategies in Medline concept (Bravo et al. 2015). Many supervised learning techniques rely upon EU-ADR [European Union Adverse Drug Reaction] database for disease genes identification in the Medline concept. NLP technique is used in the extraction of biological entity events (Kim et al. 2017).
For identifying therapeutic treatment through novel targets, ML is the best extension for understanding biological aspects. The splicing signal model is an example had in curing Alzheimer’s disease. DL splicing signal model is utilized to predict alternate signal (Leung et al. 2014). Binding the integrative splicing signals (Jha et al. 2017) like RNA sequencing data and CLIP-seq splicing data indicated knock-down results. To identify variations in Alzheimer disease (Vaquero-Garcia et al. 2016), then code models like complex variants and de-novo designs must integrate for prediction. ML can predict cancer-related drug impacts (Iorio et al. 2016). So that, ML investigated how DNA-methylation, somatic mutation data, genome-wide data impacts the drug feedback. To identify molecular features, then utilize logical models, ANOVA, and machine learning models like random forests for predicting the drug response.
Gene expression, DNA methylation are recognized as the best predictive data types in cancer regions. Data utilized from RNAi screens to locate molecular features from 501 cancer lines, so it predicts 769 genes from cancer cells (Tsherniak et al. 2017). 171 chemicals are necessary to locate in genetic affiliations because targetable vulnerabilities revealed as oncotypes don’t influence cancer therapy (McMillan et al. 2018). The models used in predictive data types how therapy in cancer-intrinsic medicine. Many queries emerge for developers i.e., how specific drugs are developed for the given target. For identifying targets in small molecular design, proteins suggested integrating with small molecules for delivering drugs. In this way, a random forest algorithm must train on genomic attributes like physicochemical and cavities of 1,187 compounds in non-drug adhesive sites against 99 protein collection (Nayal and Honig 2006). Additionally, length and configuration are considered significant features in surface cavities. For predicting drug targets, distinctive physicochemical properties from protein sequences applied SVM’s (Li and Lai 2007; Bakheet and Doig 2009) DL model (Bakheet and Doig 2009). Proteins occupy explicit locations in PPI network to associate exceptionally (Jeon et al. 2014; Costa et al. 2010; Kandoi et al. 2015). ML algorithms utilized newly developed targets to predict blind drugs for reducing search space, but drug target requires more endorsements. Predicting the clinical trial success in drug targets is a complicated goal for target validation and identification. Along ML approaches, omics information utilized 332 drug targets, so it can come up short or accomplishment in the third phase of clinical trials through multivariate compound selection (Rouillard et al. 2018).
Gene-expression data is identified as successful prediction across tissue layers with high variance and less RNA mean expression in clinical trials. In this way, the drug target was confirmed that specific disease expression can influence tissue region (Kumar et al. 2016). For predicting de-novo therapeutic drug targets, (Koscielny et al. 2017) ML classifiers should train from open platform (Ferrero et al. 2017). Significant indications are key data types such as genetic data, gene expression for predicting therapeutic drug targets. In such cases, ML approaches constrained because of data absence and sparse data are fundamental purposes behind failure in drug development programs. Practically, to initiate any drug in the market, it considers the length of time period due to more advancement in technology, new models like biologics (antibodies were included) can accessible and small molecular drug design may not same as today. Additional constraints are developed to predict medicine because it can fail or succeed with accessible metadata in public space.
Prognostic biomarkers
Using the ML approach, biomarker discovery is used to improve clinical trial performance by differentiating drugs and understanding drug mechanisms for reasonable patients (Li et al. 2015; van Gool et al. 2017; Kraus 2018). It consumes a lot of time and cost in the final stages of clinical trials. To defeat this issue, necessary to apply, build and validate predicted models in the early stages of clinical trials. Usage of ML algorithms allows predicting translational biomarkers in preclinical data assortment. After data validation, corresponding biomarkers and models must investigate the patient indications and lastly propose the medication. In literature, several papers provided information relates to predictive models and biomarkers, and last, few were utilized in clinical trials. Various factors like model rebuilding, designing, data accessing, data quality and software, model selection are necessary for a clinical setting. The principal issue was, ML approaches assess community endeavors for developing regression and classification models. Many years ago, in US FDA (Food and Drug Administration) led (MAQC II) MicroArray Quality Control evaluated ML algorithms for predicting gene expression data (Shi et al. 2010) in the final stage of clinical trials. In this project, 6 microarray data collections were analyzed by 36 independent groups to develop predictive models for classifying in the end stage of clinical sites. For modelling appropriate approaches in a clinical trial, information incorporates data quality, skilled scientists, control processes. Multiple myeloma is a poor prediction in patients and cut-off within 24 months due to partially applied. Here, the regression-based approach is appropriate for prediction because multiple myeloma and gene expression are continuous variables. By utilizing Cox regression models, it confirmed to predict (Zhan et al. 2006) patient risk factors through gene expression signature. In this review, the advantage was, utilizing regression models (Shaughnessy et al. 2007; Zhan et al. 2008; Decaux et al. 2008; Mulligan et al. 2007) can be highlighted due to the absence of predefined classes that can perform prediction in clinical trials. To evaluate regression models, NCI (National Cancer Institute) challenge is to build drug predictive models (Costello et al. 2014). Each group must utilize the best model with key parameters in training data collection (i.e., treating 35 breast tumor cells with 31 drugs) and models ought to be verified through similar blind testing data collection (i.e., treating 18 breast tumor cells with similar 31 drugs). For generating more predictive techniques, six sorts of data profiles are considered i.e., RNA sequencing, RNA microarray, reverse protein phase array, SNP (Single Nucleotide Polymorphism) array, DNA methylation status, exome sequencing for 44 groups are utilized for applying multiple regression models like sparse linear regression, kernel methods, regression trees, principal component methods. In MAQC II results, individual groups performed well and other groups utilized similar models. In differentiating, few groups maintained technical details like feature selection, quality control, data reduction, tuning ML parameters, splitting strategy, and biological data like gene expression data to improve the predictive model. Numerous drugs are convenient in the development of the predictive model when compared to other strategies.
Challenge of NCI-DREAM needs to maintain a data collection and outcomes for evaluating, improving group factor analyses in validation (Bunte et al. 2016), Random forest framework (Rahman et al. 2017) and other approaches (Huang et al. (2017); Hejase and Chan (2015)). Predictive ML models were published in several papers where biomarkers play a significant role in drug development and discovery. A conference was conducted in utilizing the tumor cell screen data to create drug sensitivity models (i.e., sorafenib and erlotinib) (Li et al. 2015). In BATTLE clinical trials (Kim et al. 2011), improved models ought to apply to patients for finalizing whether these approaches are drug-specific and predictive. In this case, study, utilizing ML models helps in recognizing key parameters in drug sensitivity sites across tumors in tissue cells. PD1 (Programmed cell Death 1) inhibitor endorsed by FDA in 2017, at that situation, genetic biomarkers utilized s pembrolizumab as inhibitors for tumors. It was the first endorsement made by FDA that relates to genetic biomarkers other than tumor type (Boyiadzis et al. 2018), which can highlight the biomarker disclosure. Recently, predictive biomarkers indicated improvement in ML other than different oncology data types. For improving drug responses in patients, ML algorithms ought to apply multi-omics data (Tasaki et al. 2018). And gradient regression tree is utilized for improving polygenic risk scores in predicting clinical trials (Paré et al. 2017). Tested outcomes in UK Biobank explained the presentation of SNP model is indicated polygenic variance as 46.9% for height, 32.7% for BMI. For distinguishing high complexes in individuals such as cardiac arrests, breast cancers, inflammatory bowel cancers, at that point, genome-wide scored data must develop (Khera et al. 2018).
RNA sequencing for single-cell innovation is widely utilized in advanced biomarker discoveries and gene clustering. This technique is utilized to locate lineages of trace development, determining cell states, novel cell varieties. Here, reducing estimations in gene expression from thousand cells into the low-dimensional regions was the unresolved issue. For reducing high-dimensional into low dimensional form, Ding et al introduced probabilistic generative structure in gene expression of single-cell data accompanied by unpredictable estimations (Ding et al. 2018). Here, a probabilistic model is widely utilized to examine RNA sequencing for four single cells data. Along with, it develops 2D structure in the multi-dimensional regions for distinguishing cell patterns in RNA sequencing single-cell data. Transformation of RNA sequencing single-cell data into the encoded feature of latent space, VAE’s (Variational autoencoders) utilized for determining subpopulations in hidden tumour (Sabrina et al. 2019). Encoded features assessed few relationships in gene cell subpopulations. This strategy contains a data pre-processing technique since it relies upon unsupervised learning. RNA sequencing of single-cell data utilized the VASC model for data visualization (Wang and Jin 2018).
When testing was conducted on 20 informational sets, results indicated more superior to VASC model other than SIMLR (Wang et al. 2017) and ZIFA (Pierson and Yau 2015) reduction models. By utilizing ML approaches, feature selection received huge advancements in biomarker discovery. For extracting appropriate structures in clusters (Tan et al. 2016), many specialists have claimed unsupervised deep learning methods. To locate explicit structures in VAE encoded features, then the VAE technique must compete with TCGA (The Cancer Genome Atlas) data in RNA sequencing (Way and Greene 2017). To upgrade identifications in carcinoma disease, Beck et al. (2011) explained data integration techniques, image analysis with gene expression data to identify the squamous cells in lungs. And CNN model showed better execution in predicting the cardiac failures i.e., (AUC=0.97) from endomyocardial biopsy data other than (AUC=0.73 and 0.75) trained samples (Nirschl et al. 2018). From the above examples, the usage of ML approaches has shown success in biomarker discovery and still, numerous issues need to be rectified. A few issues considered as; one classifier must understandable by end-users for clinical adoptions. Another key issue was, every approach needs to validate the multi-institutional, multi-site data sets for determining the generalizability approach. Many community parties tended to key issues and providing a quick advancement like model extraction and interpretations in biological sites (Finnegan and Song 2017), key optimization and training algorithms (Angermueller et al. 2016), model reproducib0ility (Hutson 2018).
Digital pathology
The word pathology refers to a realistic field, each pathologist clarified what can see from a glass slide through visual assessment. A lot of information is produced through glass slides for example, which cell type is arranged in tissue layer and spatial context. In this way, it is generally imperative to examine relationships between immune cells and immune-oncology cancers. In clinical trials, before choosing a patient to test with thousands of compounds, pharmaceutical industries must realize how the particular drug can treat patient cells and tissues in the body. Because of rapid advancements in clinical trials, locating biomarkers became more significant for victims i.e., who can ready to react to the therapy. Fast improvement in digital pathology can discover new biomarkers with more reasonable, precise, and high-throughput behavior for reducing time in drug development, and also victims can access therapy very fast. Prior to applying deep learning models, many algorithms related to image analysis propelled me to collaborate with pathologists. For classifying tissue layers, numerous computer scientists are required to handcraft graphical features in computers.
The objective of digital pathology study is to recognize etymological descriptors largely utilized in hematoxylin and easin (H&E) structures. Here, Nuclear morphometry is an implementation in the digital study for explaining relationships between prognosis (Veltri et al. 2000) and features created by PCs. From the spatial context, Beck et al. (2011) identified tissues in stroma cancer and stroma survival features in breast cancer. Recently, the Nuclear orientation structure was explained by LU et al. (2017) for clarifying survival features in oral cancers and breast cancers (Cheng et al. 2018). In many conditions, antibodies utilized immunohistochemical stains for targeting image proteins. With the absence of deep learning tools, morphology can detect tissues in sophisticated data. Investigation of immuno-oncology permits ML approaches for generating high throughput features to explain thousands of cells associated with a spatial context, and impossible tasks given for pathologists. Usage of DL methods shows improvement more precisely for tissue and cell detection in cancer environments. Many different features are explained spatial context associations for cells and tissues through scale estimations. Understanding heterogeneity concept in breast-cancer population to utilize lymphocytes in biomarkers (Mani et al. 2016). The cell-cell relationship was examined and delivered outcomes through cell locations like CD8+, PD1+ and cell densities for distinguishing carcinoma Merkel cell to respond in pembrolizumab (Giraldo et al. 2017). For leading a trial, utilized the number of tissues for each stain. If thousands of features are examined, then cell-cell interaction increases in each stain. In this circumstance, ML models and feature selections must be incorporated to predict the therapeutic response.
The CNN model is well applicable for digital pathology works since a single biopsy was utilized to train feasible pixels. So, DL models automatically learn structured features from various classification tasks (Janowczyk and Madabhushi 2016). Here model was, M-CNN (Multi-scale CNN) considered as a supervised learning technique for phenotyping images with high-content cells (Godinez et al. 2017), where it restricts a few models with their customized steps. Converting image pixel values to phenotype images, then the M-CNN approach demonstrated more accuracy at classification levels. For creating objectives in image analysis, numerous DL methods utilized in tubules (Romo-Bucheli et al. 2016), lymphocytes (Saltz et al. 2018; Corredor et al. 2019), mitotic activity (Romo-Bucheli et al. 2016), cancer tumours (Sharma et al. 2017; Korbar et al. 2017; Bychkov et al. 2018; Cruz-Roa et al. 2017) situated in lung and breast cancers. In digital pathology, DL models provide information related to other methodologies. Utilization of deep learning models can stimulate data acquisition (Cohen et al. 2018) of MRI (Magnetic Resonance Imaging) or it diminishes dosage for radiation in CT (Computed Tomography) image process (Chen et al. 2017). The quality of images improved a lot in noise signal ratio, spatial resolution; so, applications like victim stratification, disease prediction, image qualification have correspondingly improved. The deep learning framework is another study (Coudray et al. 2018) which determines to predict the usage of mutated genes called lung cancers from hematoxylin & eosin (H & E)-stained images.
In image analysis, numerous deep learning procedures are required to perform explicit tasks; So, integration of image analysis and deep learning algorithms can be accommodated for problem-solving. In numerous issues, usage of DL techniques can outperform the results, however, it was not an image analysis tool because of lack of flexibility. Likewise, many scientific experts are accessible for any classification tasks. However, it consumes a lot of money to generate. To defeat this challenge (Turkki et al. 2016) immunohistochemistry staining would utilize to mitigate this problem. Due to community tasks, it provides more data for pathologists to build annotations for many use-cases. The transparency issue is another challenge to digital pathology. Black-box is a known methodology in deep learning strategies. In classification tasks, decision-making is unclear. For understanding numerous mechanisms in drug development, interpretable outcomes can be accommodating in locating potential biomarkers and drug targets for predictive response in therapy. Additionally, trust should be improved in generating assembled features with interpretability. In clinical trials, the large sample size required to apply DL techniques legitimately for predictive response in therapy is a further challenge. The DL requires countless sample examples in clinical trials. Sometimes, integrating data in clinical trials can be possible however the existence of bias can make the outcomes difficult for interpretation. Corredor et al. (2019) and Saltz et al. (2018) explained numerous models related to image analysis and DL models for predictive response in therapy, at that point CNN model used to identify features in sub-sequent graph and lymphocytes situated in H&E-stained cells. In the future, DL consists of more capabilities to replace nuclear detection and traditional segmentation algorithms for providing spatial context features (Table 2).
Table 2.
Different ML methods related to various tasks in Drug discovery
| Method | Element/features | Task | Refs. |
|---|---|---|---|
| MACCS | Molecular fingerprints | Locating anti-cancer molecules | Kadurin et al. (2017) |
| CNN | Molecular graph | Identifying graph convolutional fingerprints | Duvenaud et al. (2015), Coley et al. (2017) |
| CNN | Subsequent graph- based features HE-stained tissues | To predict the disease response in lymphocytes | Saltz et al. (2018), Corredor et al. (2019) |
| CNN | 2D chemical structure image | Biological Activity/toxicity | Pu et al. (2017) |
| RNN | Molecular Graph | Generating molecules with predicted biological activity | Olivecrona et al. (2017) |
| RNN | SMILES | To predict molecular properties | Pu et al. (2017) |
| RNN | Molecular fingerprints and protein sequence | Compound protein interaction | Wang et al. (2016) |
| RNN | SMILES | Generating novel molecules | Olivecrona et al. (2017) |
| RNAi | 501 cancer cell lines | To identify molecular markers for predicting cancer dependencies | Tsherniak et al. (2017) |
| DNN | Atom pair descriptor and donor-acceptor pair descriptor | To predict molecular bioactivity | Ma et al. (2015) |
| DNN | Molecular descriptors and protein features | Drug target interactions | Wang et al. (2014) |
| M-DNN | Molecular descriptor | To predict the chemical descriptors | Mayr et al. (2016) |
| Univariate Cox regression | Gene expression signatures | It identifies the predicted high-risk subgroup of victims | Zhan et al. (2006) |
| Gradient boosted regression trees | Genome-wide polygenic scores | To identify high-risks of breast cancer, coronary artery type 2 diabetes diseases in patients. | Khera et al. (2018) |
| VAEs | RNA sequencing dataset | Differentiating hidden cancer subpopulations with effectively | Sabrina et al. (2019) |
| Auto Encoder | Fingerprints | Virtual screening | Kadurin et al. (2017) |
Challenges
Many challenges are there in Drug discovery, most of the challenges can be solved by using Machine Learning Techniques. Here, some of the challenges are being given with possible suggestions.
-
Numerous ML strategies produced precise results, despite the fact that a couple of parameters and structures lead to trouble during the training period. Especially when data is insufficient during the training period, the particular algorithm cannot fulfill the accuracy and local optimum.
To defeat this issue, a deep belief architecture, which is an unsupervised pre-trained model needs to be implemented for improving parameters, so the results can be created with more effectiveness Ghasemi et al. (2018)).
-
The transparency issue is another challenge in drug discovery. Because decision-making is unclear in different classification models. In drug development, numerous mechanisms need to comprehend for interpreting the outcomes. So, it makes more supportive in locating new drug targets and multiple assembled features need to improve trust in interpretability Vamathevan et al. (2019)).
In drug development, numerous mechanisms like SVM, MLR, RF, and Deep learning techniques can be implemented to comprehend for interpreting the outcomes. So, it makes more supportive in locating new drug targets and multiple assembled features for developing trust in interpretability.
-
Integrated data can accessible from many references, especially from the ‘omics’ region. It’s turning out to be more challenging in day-by-day, because not only expanding the data as well as this data type contains profoundly heterogeneity in pharmaceutical companies (Searls 2005).
Public databases are available like ZINC, BindingDB, PUBCHEM, Drugbank, and REAL chemical databases, developers need to create a pipeline architecture to integrate these heterogeneous data sources. However, the Data warehousing tools which work based on ETL (Extract Transform and Load) are Integrated Genomic Database, Adaptable Clinical Trail Database, DataFoundry, SWISS-PROT, SCoP, and dbEST. Genome Information Management System, BIOMOLQUEST, PDB, SWISS-PORT, ENZIME and CATH data (Cornell et al. 2001; Bukhman and Skolnick 2001).
-
Additionally, Homogeneous data can generate integration challenges, commencing with testing and logical issues, cross-platform normalization, and statistical issues can expand enormous heterogeneity information (Searls 2005).
So, ML with Big data analytic can be utilized for integrating homogenous data sources. Some Ontology-based integration tools are available like Ontology Web Language, Extensive Markup Language (XML), RDF Schema or Resource Description Language (RDF), Unified Medical Language System, etc (A Seoane et al. 2013). Some weblink based integration tools available like Sequence Retrieval System (Etzold et al. 1996, ChEMBL (Gaulton et al. 2012, NCBI Entrez, PubChem, Integr8, DisaseCard and EMBL-EBI search and Sequence analysis (A Seoane et al. 2013; Madeira et al. 2019). Some visualization tools are also available like Microsoft Power BI, IBM Cognos, Tableau, Zoho Analytics, Sisense, SAS Business Intelligence, etc. Because integration and visualization tools help in identifying bottlenecks and potential problems before which affects important processes (Soukup and Davidson 2002).
-
In pharmaceutical companies, research was stretched out from huge molecules to individuals, and generally relied upon integration of heterogenous data which sustain its own challenges in varying contexts and scales (Searls 2005).
A high level of artificial intelligence needs to be obtained for managing various sources and must be improved with a better understanding of the gathered data. So that, modern data connectors are suggested to centralize the dissimilar data and at last, these data connectors help in allotting original data.
Conclusion and future directions
The AI technology is utilized in pharmaceutical industries including ML algorithms and deep learning techniques in daily life. ML techniques in drug development regions and health service centers have encountered numerous conflicts, especially in image analysis and omics data. In medical science, ML models predict the trained data in a known framework i.e., the compound structure can perform alternative tools like PPT inhibitors, macrocycles with traditional algorithms. Additionally, deep learning models can be considered the chemical structures and QSAR models from pharmaceutical data which was pertinent for molecules with appropriate properties, because to the forward success rate in clinical trials. AI technology has taken a forward step in entering into computer-aided drug development to retrieve the powerful capabilities in data mining. Some issues still existed i.e.,
The performance of deep learning methods can directly influence the innovation of data mining because multiple deep neural networks are effectively trained on a large volume of data. The main aim is to tackle the transfer learning automatic problem.
“Black-Box” model became confused in deep learning concepts. The Local Interpretable Model-Explanations (LIME) is an example of a counterfactual probe. LIME was utilized to unlock the black-box model (Voosen 2017). Here, restricted data was mandatory to explain through deep learning models (Tishby and Zaslavsky 2015). However, revealing data by deep learning techniques perform only in the initial stages.
Many parameters are adjusted during the training period of neural networks but some theoretical and practical frameworks are out of reach to optimize these models.
Future directions
Web innovation was integrated with medical science to improve predictive power in decision-making and deep learning algorithms about biomarkers, side effects in therapies, therapeutic benefits. In clinical trials, success is achieved through the utilization of particular applications. So, motivation is performed for future investment in pharmaceutical companies. In the future, drug discovery and development, looking forward to covering all aspects by AI technology. Automated AI needs to coordinate theoretical results such as chemistry information, omics data, and medical data for emerging. Also, we are anticipating that more confirmations should be rebuilt for the medication revelation campaign.
Biographies
Suresh Dara,
currently working as Professor in B V Raju Institute of Technology, Narsapur, Medak -502313, India. He has completed his Ph.D. from Indian Institute of Technology (ISM), Dhanbad, India in 2015, completed his Masters in CST from Andhra University, India in 2008. He has published many research articles in reputed journals, International conferences, Books and Book chapters. He served as reviewer for many reputed journals. His current research interests are Soft computing, Machine Learning, Deep Learning, Evolutionary Computation, and Bioinformatics.
Swetha Dhamercherla,
currently Post Graduate student and working towards her academic Project. She did her Under Graduate in Computer Science and Engineering from JNTU Hyderabad, India in 2019. Her research interests include Machine Learning, Deep Learning, Evolutionary Computation, and Bioinformatics.
Surender Singh Jadav,
currently working as Scientist in Centre for Molecular Cancer Research (CMCR), Hyderabad, India, and Visiting Professor at Vishnu Institute of Pharmaceutical Education and Research (VIPER), Narsapur, Medak-502313, Telangana. He has done his Ph.D. from Birla Institute of Technology, Mesra, India in 2015. He has published many research articles in reputed journals, International conferences, Books and Book chapters. His current research interests are Drug Discovery, Machine Learning, Deep Learning and Bioinformatics.
CH Madhu Babu,
currently working as Professor and HOD/CSE in B V Raju Institute of Technology, Narsapur, Medak-502313, India. He has completed his Ph.D. from Acharya Nagarjuna University, India in 2017. He has published many research articles in reputed journals, International conferences, Books and Book chapters. His current research interests are Software Engineering, Machine Learning, Deep Learning and Bioinformatics.
Mohamed Jawed Ahsan
has earned his B. Pharmacy and M. Pharmacy (Pharmaceutical Chemistry) from Jamia Hamdard University, New Delhi; and Ph.D. (Pharmaceutical Sciences) from NIMS University, Jaipur, India. Presently he is working as Professor and Head, Department of Pharmaceutical Chemistry, Maharishi Arvind College of Pharmacy, Jaipur. He has a remarkable research profile in the area of Pharmaceutical and Medicinal Chemistry. His area of interest is in design and discovery of novel molecules against cancer and tuberculosis.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Contributor Information
Suresh Dara, Email: darasuresh@live.in.
Swetha Dhamercherla, Email: swetha.07031998@gmail.com.
Surender Singh Jadav, Email: jawedpharma@gmail.com.
CH Madhu Babu, Email: madhubabu.ch@bvrit.ac.in.
Mohamed Jawed Ahsan, Email: jadavmedchem@gmail.com.
References
- Abagyan R, Totrov M, Kuznetsov D. Icm–a new method for protein modeling and design: applications to docking and structure prediction from the distorted native conformation. J Comput Chem. 1994;15(5):488–506. doi: 10.1002/jcc.540150503. [DOI] [Google Scholar]
- Ain QU, Aleksandrova A, Roessler FD, Ballester PJ. Machine-learning scoring functions to improve structure-based binding affinity prediction and virtual screening. Wiley Interdisciplinary Reviews. Comput Molec Sci. 2015;5(6):405–424. doi: 10.1002/wcms.1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alpaydin E. Introduction to machine learning. Cambridge: MIT press; 2020. [Google Scholar]
- Ament SA, Pearl JR, Cantle JP, Bragg RM, Skene PJ, Coffey SR, Bergey DE, Wheeler VC, MacDonald ME, Baliga NS, et al. Transcriptional regulatory networks underlying gene expression changes in huntington’s disease. Mol Syst Biol. 2018;14(3):e7435. doi: 10.15252/msb.20167435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- An H, Li M, Gao J, Zhang Z, Ma S, Chen Y. Incorporation of biomolecules in metal-organic frameworks for advanced applications. Coord Chem Rev. 2019;384:90–106. doi: 10.1016/j.ccr.2019.01.001. [DOI] [Google Scholar]
- Andras P. High-dimensional function approximation with neural networks for large volumes of data. IEEE Trans Neural Netw Learn Syst. 2017;29(2):500–508. doi: 10.1109/TNNLS.2017.2651985. [DOI] [PubMed] [Google Scholar]
- Angermueller C, Pärnamaa T, Parts L, Stegle O. Deep learning for computational biology. Mol Syst Biol. 2016;12(7):878. doi: 10.15252/msb.20156651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artursson P, Karlsson J. Correlation between oral drug absorption in humans and apparent drug permeability coefficients in human intestinal epithelial (caco-2) cells. Biochem Biophys Res Commun. 1991;175(3):880–885. doi: 10.1016/0006-291X(91)91647-U. [DOI] [PubMed] [Google Scholar]
- Ashburn TT, Thor KB. Drug repositioning: identifying and developing new uses for existing drugs. Nat Rev Drug Discovery. 2004;3(8):673–683. doi: 10.1038/nrd1468. [DOI] [PubMed] [Google Scholar]
- Asher M. The drug-maker’s guide to the galaxy. Nature News. 2017;549(7673):445. doi: 10.1038/549445a. [DOI] [PubMed] [Google Scholar]
- Bai F, Morcos F, Cheng RR, Jiang H, Onuchic JN. Elucidating the druggable interface of protein- protein interactions using fragment docking and coevolutionary analysis. Proc Natl Acad Sci. 2016;113(50):E8051–E8058. doi: 10.1073/pnas.1615932113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bakheet TM, Doig AJ. Properties and identification of human protein drug targets. Bioinformatics. 2009;25(4):451–457. doi: 10.1093/bioinformatics/btp002. [DOI] [PubMed] [Google Scholar]
- Beck AH, Sangoi AR, Leung S, Marinelli RJ, Nielsen TO, Van De Vijver MJ, West RB, Van De Rijn M, Koller D. Systematic analysis of breast cancer morphology uncovers stromal features associated with survival. Sci Trans Med. 2011;3(108):108ra113–108ra113. doi: 10.1126/scitranslmed.3002564. [DOI] [PubMed] [Google Scholar]
- Bengio Y. Learning deep architectures for AI. Norwell: Now Publishers Inc; 2009. [Google Scholar]
- Bengio Y, Lamblin P, Popovici D, Larochelle H (2007) Greedy layer-wise training of deep networks advances in neural information processing systems
- Boyiadzis MM, Kirkwood JM, Marshall JL, Pritchard CC, Azad NS, Gulley JL. Significance and implications of fda approval of pembrolizumab for biomarker-defined disease. J Immunother Cancer. 2018;6(1):1–7. doi: 10.1186/s40425-018-0342-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bravo À, Piñero J, Queralt-Rosinach N, Rautschka M, Furlong LI. Extraction of relations between genes and diseases from text and large-scale data analysis: implications for translational research. BMC Bioinform. 2015;16(1):55. doi: 10.1186/s12859-015-0472-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bukhman YV, Skolnick J. Biomolquest: integrated database-based retrieval of protein structural and functional information. Bioinformatics. 2001;17(5):468–478. doi: 10.1093/bioinformatics/17.5.468. [DOI] [PubMed] [Google Scholar]
- Bundela S, Sharma A, Bisen PS. Potential compounds for oral cancer treatment: resveratrol, nimbolide, lovastatin, bortezomib, vorinostat, berberine, pterostilbene, deguelin, andrographolide, and colchicine. PLoS ONE. 2015;10(11):e0141719. doi: 10.1371/journal.pone.0141719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunte K, Leppäaho E, Saarinen I, Kaski S. Sparse group factor analysis for biclustering of multiple data sources. Bioinformatics. 2016;32(16):2457–2463. doi: 10.1093/bioinformatics/btw207. [DOI] [PubMed] [Google Scholar]
- Bychkov D, Linder N, Turkki R, Nordling S, Kovanen PE, Verrill C, Walliander M, Lundin M, Haglund C, Lundin J. Deep learning based tissue analysis predicts outcome in colorectal cancer. Sci Rep. 2018;8(1):1–11. doi: 10.1038/s41598-018-21758-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cabreiro F, Au C, Leung K-Y, Vergara-Irigaray N, Cochemé HM, Noori T, Weinkove D, Schuster E, Greene NDE, Gems D. Metformin retards aging in c. elegans by altering microbial folate and methionine metabolism. Cell. 2013;153(1):228–239. doi: 10.1016/j.cell.2013.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cano G, Garcia-Rodriguez J, Garcia-Garcia A, Perez-Sanchez H, Benediktsson JA, Thapa A, Barr A. Automatic selection of molecular descriptors using random forest: Application to drug discovery. Expert Syst Appl. 2017;72:151–159. doi: 10.1016/j.eswa.2016.12.008. [DOI] [Google Scholar]
- Chen Y-C. Beware of docking! Trends Pharmacol Sci. 2015;36(2):78–95. doi: 10.1016/j.tips.2014.12.001. [DOI] [PubMed] [Google Scholar]
- Chen H, Engkvist O, Wang Y, Olivecrona M, Blaschke T. The rise of deep learning in drug discovery. Drug Discovery Today. 2018;23(6):1241–1250. doi: 10.1016/j.drudis.2018.01.039. [DOI] [PubMed] [Google Scholar]
- Chen R, Li L, Weng Z. Zdock: an initial-stage protein-docking algorithm. Proteins Struct Funct Bioinf. 2003;52(1):80–87. doi: 10.1002/prot.10389. [DOI] [PubMed] [Google Scholar]
- Chen H, Zhang Y, Kalra MK, Lin F, Chen Y, Liao P, Zhou J, Wang G. Low-dose ct with a residual encoder-decoder convolutional neural network. IEEE Trans Med Imaging. 2017;36(12):2524–2535. doi: 10.1109/TMI.2017.2715284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Lewis JS, Dupont WD, Plummer WD, Janowczyk A, Madabhushi A. An oral cavity squamous cell carcinoma quantitative histomorphometric-based image classifier of nuclear morphology can risk stratify patients for disease-specific survival. Mod Pathol. 2017;30(12):1655–1665. doi: 10.1038/modpathol.2017.98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L, Romo-Bucheli D, Wang X, Janowczyk A, Ganesan S, Gilmore H, Rimm D, Madabhushi A. Nuclear shape and orientation features from h&e images predict survival in early-stage estrogen receptor-positive breast cancers. Lab Invest. 2018;98(11):1438–1448. doi: 10.1038/s41374-018-0095-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen O, Zhu B, Rosen MS. Mr fingerprinting deep reconstruction network (drone) Magn Reson Med. 2018;80(3):885–894. doi: 10.1002/mrm.27198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coley CW, Barzilay R, Green WH, Jaakkola TS, Jensen KF. Convolutional embedding of attributed molecular graphs for physical property prediction. J Chem Inf Model. 2017;57(8):1757–1772. doi: 10.1021/acs.jcim.6b00601. [DOI] [PubMed] [Google Scholar]
- Coley CW, Rogers L, Green WH, Jensen KF. Scscore: synthetic complexity learned from a reaction corpus. J Chem Inf Model. 2018;58(2):252–261. doi: 10.1021/acs.jcim.7b00622. [DOI] [PubMed] [Google Scholar]
- Copeland RA. The dynamics of drug-target interactions: drug-target residence time and its impact on efficacy and safety. Expert Opin Drug Discov. 2010;5(4):305–310. doi: 10.1517/17460441003677725. [DOI] [PubMed] [Google Scholar]
- Cornell M, Paton NW, Wu S, Goble CA, Miller CJ, Kirby P, Eilbeck K, Brass A, Hayes A, Oliver SG (2001) Gims-a data warehouse for storage and analysis of genome sequence and functional data. In: Proceedings 2nd Annual IEEE International Symposium on Bioinformatics and Bioengineering (BIBE 2001). IEEE, pp 15–22
- Corredor G, Xiangxue Wang Yu, Zhou CL, Pingfu F, Syrigos K, Rimm DL, Yang M, Romero E, Schalper KA, et al. Spatial architecture and arrangement of tumor-infiltrating lymphocytes for predicting likelihood of recurrence in early-stage non-small cell lung cancer. Clin Cancer Res. 2019;25(5):1526–1534. doi: 10.1158/1078-0432.CCR-18-2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Costa PR, Acencio ML, Lemke N (2010) A machine learning approach for genome-wide prediction of morbid and druggable human genes based on systems-level data. In: BMC genomics, vol 11. Springer, Berlin, p S9 [DOI] [PMC free article] [PubMed]
- Costello JC, Heiser LM, Georgii E, Gönen M, Menden MP, Wang NJ, Bansal M, Hintsanen P, Khan SA, Mpindi J-P, et al. A community effort to assess and improve drug sensitivity prediction algorithms. Nat Biotechnol. 2014;32(12):1202–1212. doi: 10.1038/nbt.2877. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coudray N, Ocampo PS, Sakellaropoulos T, Narula N, Snuderl M, Fenyö D, Moreira AL, Razavian N, Tsirigos A. Classification and mutation prediction from non-small cell lung cancer histopathology images using deep learning. Nat Med. 2018;24(10):1559–1567. doi: 10.1038/s41591-018-0177-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cruz-Roa A, Gilmore H, Basavanhally A, Feldman M, Ganesan S, Shih NNC, Tomaszewski J, González FA, Madabhushi A. Accurate and reproducible invasive breast cancer detection in whole-slide images: A deep learning approach for quantifying tumor extent. Sci Rep. 2017;7:46450. doi: 10.1038/srep46450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cukuroglu E, Engin HB, Gursoy A, Keskin O. Hot spots in protein-protein interfaces: towards drug discovery. Prog Biophys Mol Biol. 2014;116(2–3):165–173. doi: 10.1016/j.pbiomolbio.2014.06.003. [DOI] [PubMed] [Google Scholar]
- Dahl GE, Jaitly N, Salakhutdinov R (2014) Multi-task neural networks for qsar predictions. arXiv preprint arXiv:1406.1231
- Dang NL, Hughes TB, Krishnamurthy V, Swamidass SJ. A simple model predicts ugt-mediated metabolism. Bioinformatics. 2016;32(20):3183–3189. doi: 10.1093/bioinformatics/btw350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Bruyn KC, Stork C, Šícho M, Kochev N, Svozil D, Jeliazkova N, Kirchmair J. Glory: generator of the structures of likely cytochrome p450 metabolites based on predicted sites of metabolism. Front Chem. 2019;7:402. doi: 10.3389/fchem.2019.00402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Haes W, Frooninckx L, Van Assche R, Smolders A, Depuydt G, Billen J, Braeckman BP, Schoofs L, Temmerman L. Metformin promotes lifespan through mitohormesis via the peroxiredoxin prdx-2. Proc Natl Acad Sci. 2014;111(24):E2501–E2509. doi: 10.1073/pnas.1321776111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Decaux O, Lodé L, Magrangeas F, Charbonnel C, Gouraud W, Jézéquel P, Attal M, Harousseau J-L, Moreau P, Bataille R, et al. Prediction of survival in multiple myeloma based on gene expression profiles reveals cell cycle and chromosomal instability signatures in high-risk patients and hyperdiploid signatures in low-risk patients: a study of the intergroupe francophone du myelome. J Clin Oncol. 2008;26(29):4798–4805. doi: 10.1200/JCO.2007.13.8545. [DOI] [PubMed] [Google Scholar]
- Deng L, Dong Yu. Deep learning: methods and applications. Found Trends Sign Process. 2014;7(3–4):197–387. doi: 10.1561/2000000039. [DOI] [Google Scholar]
- Desai B, Dixon K, Farrant E, Feng Q, Gibson KR, van Hoorn WP, Mills J, Morgan T, Parry DM, Ramjee MK, et al. Rapid discovery of a novel series of abl kinase inhibitors by application of an integrated microfluidic synthesis and screening platform. J Med Chem. 2013;56(7):3033–3047. doi: 10.1021/jm400099d. [DOI] [PubMed] [Google Scholar]
- DiMasi JA, Grabowski HG, Hansen RW. Innovation in the pharmaceutical industry: new estimates of r&d costs. J Health Econ. 2016;47:20–33. doi: 10.1016/j.jhealeco.2016.01.012. [DOI] [PubMed] [Google Scholar]
- Ding J, Condon A, Shah SP. Interpretable dimensionality reduction of single cell transcriptome data with deep generative models. Nat Commun. 2018;9(1):1–13. doi: 10.1038/s41467-018-04368-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobchev DA, G Pillai G, Karelson M. In silico machine learning methods in drug development. Curr Top Med Chem. 2014;14(16):1913–1922. doi: 10.2174/1568026614666140929124203. [DOI] [PubMed] [Google Scholar]
- Domenico A, Nicola G, Daniela T, Fulvio C, Nicola A, Orazio N (2020) De novo drug design of targeted chemical libraries based on artificial intelligence and pair based multi-objective optimization. J Chem Inform Model [DOI] [PubMed]
- Du T, Liao L, Wu CH, Sun B. Prediction of residue-residue contact matrix for protein–protein interaction with fisher score features and deep learning. Methods. 2016;110:97–105. doi: 10.1016/j.ymeth.2016.06.001. [DOI] [PubMed] [Google Scholar]
- Duch W, Swaminathan K, Meller J. Artificial intelligence approaches for rational drug design and discovery. Curr Pharm Des. 2007;13(14):1497–1508. doi: 10.2174/138161207780765954. [DOI] [PubMed] [Google Scholar]
- Duda RO, Hart PE, Stork DG. Pattern classification. New Jersy: John Wiley & Sons; 2012. [Google Scholar]
- Dudek AZ, Arodz T, Gálvez J. Computational methods in developing quantitative structure-activity relationships (qsar): a review. Comb Chem High Throughput Screen. 2006;9(3):213–228. doi: 10.2174/138620706776055539. [DOI] [PubMed] [Google Scholar]
- Dupond S. A thorough review on the current advance of neural network structures. Annu Rev Control. 2019;14:200–230. [Google Scholar]
- Duvenaud DK, Maclaurin D, Iparraguirre J, Bombarell R, Hirzel T, Aspuru-Guzik A, Adams RP (2015) Convolutional networks on graphs for learning molecular fingerprints. In: Advances in neural information processing systems, pp 2224–2232
- Eid F-E, ElHefnawi M, Heath LS. Denovo: virus-host sequence-based protein–protein interaction prediction. Bioinformatics. 2016;32(8):1144–1150. doi: 10.1093/bioinformatics/btv737. [DOI] [PubMed] [Google Scholar]
- Engelbrecht AP. Computational intelligence: an introduction. New Jersy: John Wiley & Sons; 2007. [Google Scholar]
- Esposito EX, Hopfinger AJ, Madura JD (2004) Methods for applying the quantitative structure-activity relationship paradigm. In: Chemoinformatics. Springer, pp 131–213 [DOI] [PubMed]
- Etzold T, Ulyanov A, Argos P. [8] srs: information retrieval system for molecular biology data banks. Methods Enzymol. 1996;266:114–128. doi: 10.1016/S0076-6879(96)66010-8. [DOI] [PubMed] [Google Scholar]
- Falchi F, Caporuscio F, Recanatini M. Structure-based design of small-molecule protein–protein interaction modulators: the story so far. Future Med Chem. 2014;6(3):343–357. doi: 10.4155/fmc.13.204. [DOI] [PubMed] [Google Scholar]
- Ferrero E, Dunham I, Sanseau P. In silico prediction of novel therapeutic targets using gene-disease association data. J Transl Med. 2017;15(1):182. doi: 10.1186/s12967-017-1285-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finnegan A, Song JS. Maximum entropy methods for extracting the learned features of deep neural networks. PLoS Comput Biol. 2017;13(10):e1005836. doi: 10.1371/journal.pcbi.1005836. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Free SM, Wilson JW. A mathematical contribution to structure-activity studies. J Med Chem. 1964;7(4):395–399. doi: 10.1021/jm00334a001. [DOI] [PubMed] [Google Scholar]
- Friedman J, Hastie T, Tibshirani R. The elements of statistical learning, volume 1. 1. New York: Springer series in statistics; 2001. [Google Scholar]
- Friesner RA, Banks JL, Murphy RB, Halgren TA, Klicic JJ, Mainz DT, Repasky MP, Knoll EH, Shelley M, Perry JK, et al. Glide: a new approach for rapid, accurate docking and scoring. 1. method and assessment of docking accuracy. J Med Chem. 2004;47(7):1739–1749. doi: 10.1021/jm0306430. [DOI] [PubMed] [Google Scholar]
- Gaulton A, Bellis LJ, Bento AP, Chambers J, Davies M, Hersey A, Light Y, McGlinchey S, Michalovich D, Al-Lazikani B, et al. Chembl: a large-scale bioactivity database for drug discovery. Nucleic Acids Res. 2012;40(D1):D1100–D1107. doi: 10.1093/nar/gkr777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gertrudes JC, Maltarollo VG, Silva RA, Oliveira PR, Honorio KM, Da Silva ABF. Machine learning techniques and drug design. Curr Med Chem. 2012;19(25):4289–4297. doi: 10.2174/092986712802884259. [DOI] [PubMed] [Google Scholar]
- Ghasemi F, Mehridehnavi A, Fassihi A, Pérez-Sánchez H. Deep neural network in qsar studies using deep belief network. Appl Soft Comput. 2018;62:251–258. doi: 10.1016/j.asoc.2017.09.040. [DOI] [Google Scholar]
- Giraldo NA, Kaunitz GJ, Cottrell TR, Berry S, Sunshine JC, Nguyen P, Xu H, Orgutsova A, Church CD, Miller NJ et al. (2017) The differential association of pd-1, pd-l1, and cd8+ cells with response to pembrolizumab and presence of merkel cell polyomavirus (mcpyv) in patients with merkel cell carcinoma (mcc)
- Godinez WJ, Hossain I, Lazic SE, Davies JW, Zhang X. A multi-scale convolutional neural network for phenotyping high-content cellular images. Bioinformatics. 2017;33(13):2010–2019. doi: 10.1093/bioinformatics/btx069. [DOI] [PubMed] [Google Scholar]
- Goodfellow I, Bengio Y, Courville A, Bengio Y. Deep learning. Cambridge: MIT press; 2016. [Google Scholar]
- Gopal M. Applied machine learning. Chennai: McGraw-Hill Education; 2018. [Google Scholar]
- Guengerich FP (2010) Mechanisms of drug toxicity and relevance to pharmaceutical development. Drug metabolism and pharmacokinetics, p 1010210090 [DOI] [PMC free article] [PubMed]
- Guney E, Menche J, Vidal M, Barábasi A-L. Network-based in silico drug efficacy screening. Nat Commun. 2016;7(1):1–13. doi: 10.1038/ncomms10331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gunther EC, Stone DJ, Gerwien RW, Bento P, Heyes MP. Prediction of clinical drug efficacy by classification of drug-induced genomic expression profiles in vitro. Proc Natl Acad Sci. 2003;100(16):9608–9613. doi: 10.1073/pnas.1632587100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo Y, Lezheng Yu, Wen Z, Li M. Using support vector machine combined with auto covariance to predict protein-protein interactions from protein sequences. Nucleic Acids Res. 2008;36(9):3025–3030. doi: 10.1093/nar/gkn159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S, Chaudhary K, Kumar R, Gautam A, Nanda JS, Dhanda SK, Brahmachari SK, Raghava GPS. Prioritization of anticancer drugs against a cancer using genomic features of cancer cells: A step towards personalized medicine. Sci Rep. 2016;6(1):1–11. doi: 10.1038/srep23857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gómez-Bombarelli R, Wei JN, Duvenaud D, Hernández-Lobato JM, Sánchez-Lengeling B, Sheberla D, Aguilera-Iparraguirre J, Hirzel TD, Adams RP, Aspuru-Guzik A. Automatic chemical design using a data-driven continuous representation of molecules. ACS Cent Sci. 2018;4(2):268–276. doi: 10.1021/acscentsci.7b00572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hansch C, Fujita T. Additions and corrections-analysis. a method for the correlation of biological activity and chemical structure. J Am Chem Soc. 1964;86(24):5710. doi: 10.1021/ja01078a623. [DOI] [Google Scholar]
- Hartenfeller M, Schneider G (2010) De novo drug design. In: Chemoinformatics and computational chemical biology. Springer, Berlin, pp 299–323
- Hassan Baig M, Ahmad K, Roy S, Mohammad Ashraf J, Adil M, Haris Siddiqui M, Khan S, Amjad Kamal M, Provazník I, Choi I. Computer aided drug design: success and limitations. Curr Pharm Des. 2016;22(5):572–581. doi: 10.2174/1381612822666151125000550. [DOI] [PubMed] [Google Scholar]
- Hejase HA, Chan C. Improving drug sensitivity prediction using different types of data. CPT: Pharmacometrics Syst Pharmacol. 2015;4(2):98–105. doi: 10.1002/psp4.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Higueruelo AP, Jubb H, Blundell TL. Protein-protein interactions as druggable targets: recent technological advances. Curr Opin Pharmacol. 2013;13(5):791–796. doi: 10.1016/j.coph.2013.05.009. [DOI] [PubMed] [Google Scholar]
- Hinton G. Deep learning–a technology with the potential to transform health care. JAMA. 2018;320(11):1101–1102. doi: 10.1001/jama.2018.11100. [DOI] [PubMed] [Google Scholar]
- Ho Tin K (1995) Random decision forests. In: Proceedings of 3rd international conference on document analysis and recognition, vol 1. IEEE, pp 278–282
- Huang S, Cai N, Pacheco PP, Narrandes S, Wang Y, Wayne X. Applications of support vector machine (svm) learning in cancer genomics. Cancer Genom Proteom. 2018;15(1):41–51. doi: 10.21873/cgp.20063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Mezencev R, McDonald JF, Vannberg F. Open source machine-learning algorithms for the prediction of optimal cancer drug therapies. PLoS ONE. 2017;12(10):e0186906e0186906. doi: 10.1371/journal.pone.0186906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang S-Y, Zou X. Inclusion of solvation and entropy in the knowledge-based scoring function for protein–ligand interactions. J Chem Inf Model. 2010;50(2):262–273. doi: 10.1021/ci9002987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hubatsch I, Ragnarsson EGE, Artursson P. Determination of drug permeability and prediction of drug absorption in caco-2 monolayers. Nat Protoc. 2007;2(9):2111. doi: 10.1038/nprot.2007.303. [DOI] [PubMed] [Google Scholar]
- Hutson M (2018) Artificial intelligence faces reproducibility crisis [DOI] [PubMed]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, et al. A landscape of pharmacogenomic interactions in cancer. Cell. 2016;166(3):740–754. doi: 10.1016/j.cell.2016.06.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Janowczyk A, Madabhushi A (2016) Deep learning for digital pathology image analysis: a comprehensive tutorial with selected use cases. J Pathol Inform 7 [DOI] [PMC free article] [PubMed]
- Jeon J, Nim S, Teyra J, Datti A, Wrana JL, Sidhu SS, Moffat J, Kim PM. A systematic approach to identify novel cancer drug targets using machine learning, inhibitor design and high-throughput screening. Genome Med. 2014;6(7):1–18. doi: 10.1186/s13073-014-0057-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jha A, Gazzara MR, Barash Y. Integrative deep models for alternative splicing. Bioinformatics. 2017;33(14):i274–i282. doi: 10.1093/bioinformatics/btx268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiménez J, Skalic M, Martinez-Rosell G, De Fabritiis G. K deep: Protein-ligand absolute binding affinity prediction via 3d-convolutional neural networks. J Chem Inf Model. 2018;58(2):287–296. doi: 10.1021/acs.jcim.7b00650. [DOI] [PubMed] [Google Scholar]
- Jung E, Kim J, Kim M, Jung DH, Rhee H, Shin J-M, Choi K, Kang S-K, Kim M-K, Yun C-H, et al. Artificial neural network models for prediction of intestinal permeability of oligopeptides. BMC Bioinform. 2007;8(1):245. doi: 10.1186/1471-2105-8-245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kadurin A, Aliper A, Kazennov A, Mamoshina P, Vanhaelen Q, Khrabrov K, Zhavoronkov A. The cornucopia of meaningful leads: Applying deep adversarial autoencoders for new molecule development in oncology. Oncotarget. 2017;8(7):10883. doi: 10.18632/oncotarget.14073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kandoi G, Acencio ML, Lemke N. Prediction of druggable proteins using machine learning and systems biology: a mini-review. Front Physiol. 2015;6:366. doi: 10.3389/fphys.2015.00366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kapoorb R, Haganb M, Paltab J, Ghosha P (2020) Artificial intelligence methods in computer-aided diagnostic tools and decision support analytics for clinical informatics. Artif Intell Prec Health From Conc Appl, p 31
- Kearnes S, Goldman B, Pande V (2016) Modeling industrial admet data with multitask networks. arXiv preprint arXiv:1606.08793
- Khamis MA, Gomaa W, Ahmed WF. Machine learning in computational docking. Artif Intell Med. 2015;63(3):135–152. doi: 10.1016/j.artmed.2015.02.002. [DOI] [PubMed] [Google Scholar]
- Khera AV, Chaffin M, Aragam KG, Haas ME, Roselli C, Choi SH, Natarajan P, Lander ES, Lubitz SA, Ellinor PT, et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat Genet. 2018;50(9):1219–1224. doi: 10.1038/s41588-018-0183-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim ES, Herbst RS, Wistuba II, Lee JJ, Blumenschein GR, Tsao A, Stewart DJ, Hicks ME, Erasmus J, Gupta S, et al. The battle trial: personalizing therapy for lung cancer. Cancer Discov. 2011;1(1):44–53. doi: 10.1158/2159-8274.CD-10-0010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, Kim J, Lee H. An analysis of disease-gene relationship from medline abstracts by digsee. Sci Rep. 2017;7(1):1–13. doi: 10.1038/srep40154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kingma DP, Welling M (2013) Auto-encoding variational bayes. arXiv preprint arXiv:1312.6114
- Kingma DP, Welling M (2019) An introduction to variational autoencoders. arXiv preprint arXiv:1906.02691
- Kinnings SL, Liu N, Tonge PJ, Jackson RM, Xie L, Bourne PE. A machine learning-based method to improve docking scoring functions and its application to drug repurposing. J Chem Inf Model. 2011;51(2):408–419. doi: 10.1021/ci100369f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konar A. Computational intelligence: principles, techniques and applications. Berlin: Springer Science & Business Media; 2006. [Google Scholar]
- Korbar B, Olofson AM, Miraflor AP, Nicka CM, Suriawinata MA, Torresani L, Suriawinata AA, Hassanpour S (2017) Deep learning for classification of colorectal polyps on whole-slide images. J Pathol Inform 8 [DOI] [PMC free article] [PubMed]
- Koscielny G, An P, Carvalho-Silva D, Cham JA, Fumis L, Gasparyan R, Hasan S, Karamanis N, Maguire M, Papa E, et al. Open targets: a platform for therapeutic target identification and validation. Nucleic Acids Res. 2017;45(D1):D985–D994. doi: 10.1093/nar/gkw1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kramer MA. Nonlinear principal component analysis using autoassociative neural networks. AIChE J. 1991;37(2):233–243. doi: 10.1002/aic.690370209. [DOI] [Google Scholar]
- Kraus VB. Biomarkers as drug development tools: discovery, validation, qualification and use. Nat Rev Rheumatol. 2018;14(6):354–362. doi: 10.1038/s41584-018-0005-9. [DOI] [PubMed] [Google Scholar]
- Kumar V, Sanseau P, Simola DF, Hurle MR, Agarwal P. Systematic analysis of drug targets confirms expression in disease-relevant tissues. Sci Rep. 2016;6:36205. doi: 10.1038/srep36205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larsen ABL, Sønderby SK (2015) Generating faces with torch. URL http://torch.ch/blog/2015/11/13/gan. html
- Lavecchia A, Di Giovanni C. Virtual screening strategies in drug discovery: a critical review. Curr Med Chem. 2013;20(23):2839–2860. doi: 10.2174/09298673113209990001. [DOI] [PubMed] [Google Scholar]
- LeCun Y, Bengio Y, Hinton G. Deep learning. Nature. 2015;521(7553):436–444. doi: 10.1038/nature14539. [DOI] [PubMed] [Google Scholar]
- Leelananda SP, Lindert S. Computational methods in drug discovery. Beilstein J Org Chem. 2016;12(1):2694–2718. doi: 10.3762/bjoc.12.267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leung MKK, Xiong HY, Lee LJ, Frey BJ. Deep learning of the tissue-regulated splicing code. Bioinformatics. 2014;30(12):i121–i129. doi: 10.1093/bioinformatics/btu277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Hou J, Adhikari B, Lyu Q, Cheng J. Deep learning methods for protein torsion angle prediction. BMC Bioinform. 2017;18(1):417. doi: 10.1186/s12859-017-1834-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Q, Lai L. Prediction of potential drug targets based on simple sequence properties. BMC Bioinform. 2007;8(1):353. doi: 10.1186/1471-2105-8-353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Shin H, Gulbekyan G, Pustovalova O, Nikolsky Y, Hope A, Bessarabova M, Schu M, Kolpakova-Hart E, Merberg D, et al. Development of a drug-response modeling framework to identify cell line derived translational biomarkers that can predict treatment outcome to erlotinib or sorafenib. PLoS ONE. 2015;10(6):e0130700e0130700. doi: 10.1371/journal.pone.0130700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li L, Wang B, Meroueh SO. Support vector regression scoring of receptor-ligand complexes for rank-ordering and virtual screening of chemical libraries. J Chem Inf Model. 2011;51(9):2132–2138. doi: 10.1021/ci200078f. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liew CY, Ma XH, Liu X, Yap CW. Svm model for virtual screening of lck inhibitors. J Chem Inf Model. 2009;49(4):877–885. doi: 10.1021/ci800387z. [DOI] [PubMed] [Google Scholar]
- Lombardo F, Jing Y. In silico prediction of vol of distribution in humans. extensive data set and the exploration of linear and nonlinear methods coupled with molecular interaction fields descriptors. J Chem Inf Model. 2016;56(10):2042–2052. doi: 10.1021/acs.jcim.6b00044. [DOI] [PubMed] [Google Scholar]
- Lombardo F, Obach RS, Varma MV, Stringer R, Berellini G. Clearance mechanism assignment and total clearance prediction in human based upon in silico models. J Med Chem. 2014;57(10):4397–4405. doi: 10.1021/jm500436v. [DOI] [PubMed] [Google Scholar]
- Lotfi Shahreza M, Ghadiri N, Mousavi SR, Varshosaz J, Green JR. A review of network-based approaches to drug repositioning. Brief Bioinform. 2018;19(5):878–892. doi: 10.1093/bib/bbx017. [DOI] [PubMed] [Google Scholar]
- Luo Y, Zhao X, Zhou J, Yang J, Zhang Y, Kuang W, Peng J, Chen L, Zeng J. A network integration approach for drug-target interaction prediction and computational drug repositioning from heterogeneous information. Nat Commun. 2017;8(1):1–13. doi: 10.1038/s41467-017-00680-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lusci A, Pollastri G, Baldi P. Deep architectures and deep learning in chemoinformatics: the prediction of aqueous solubility for drug-like molecules. J Chem Inf Model. 2013;53(7):1563–1575. doi: 10.1021/ci400187y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma XH, Jia J, Zhu F, Xue Y, Li ZR, Chen YZ. Comparative analysis of machine learning methods in ligand-based virtual screening of large compound libraries. Comb Chem High Throughput Screen. 2009;12(4):344–357. doi: 10.2174/138620709788167944. [DOI] [PubMed] [Google Scholar]
- Ma J, Sheridan RP, Liaw A, Dahl GE, Svetnik V. Deep neural nets as a method for quantitative structure-activity relationships. J Chem Inf Model. 2015;55(2):263–274. doi: 10.1021/ci500747n. [DOI] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD, et al. The embl-ebi search and sequence analysis tools apis in 2019. Nucleic Acids Res. 2019;47(W1):W636–W641. doi: 10.1093/nar/gkz268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maheshwari S, Brylinski M. Template-based identification of protein–protein interfaces using efindsiteppi. Methods. 2016;93:64–71. doi: 10.1016/j.ymeth.2015.07.017. [DOI] [PubMed] [Google Scholar]
- Maltarollo VG, Kronenberger T, Espinoza GZ, Oliveira PR, Honorio KM. Advances with support vector machines for novel drug discovery. Expert Opin Drug Discov. 2019;14(1):23–33. doi: 10.1080/17460441.2019.1549033. [DOI] [PubMed] [Google Scholar]
- Mamoshina P, Volosnikova M, Ozerov IV, Putin E, Skibina E, Cortese F, Zhavoronkov A. Machine learning on human muscle transcriptomic data for biomarker discovery and tissue-specific drug target identification. Front Genet. 2018;9:242. doi: 10.3389/fgene.2018.00242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mani NL, Schalper KA, Hatzis C, Saglam O, Tavassoli F, Butler M, Chagpar AB, Pusztai L, Rimm DL. Quantitative assessment of the spatial heterogeneity of tumor-infiltrating lymphocytes in breast cancer. Breast Cancer Res. 2016;18(1):78. doi: 10.1186/s13058-016-0737-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin-Montalvo A, Mercken EM, Mitchell SJ, Palacios HH, Mote PL, Scheibye-Knudsen M, Gomes AP, Ward TM, Minor RK, Blouin M-J, et al. Metformin improves healthspan and lifespan in mice. Nat Commun. 2013;4(1):1–9. doi: 10.1038/ncomms3192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matlock MK, Hughes TB, Swamidass SJ. Xenosite server: a web-available site of metabolism prediction tool. Bioinformatics. 2015;31(7):1136–1137. doi: 10.1093/bioinformatics/btu761. [DOI] [PubMed] [Google Scholar]
- Matsumoto A, Aoki S, Ohwada H. Comparison of random forest and svm for raw data in drug discovery: prediction of radiation protection and toxicity case study. Int J Mach Learn Comput. 2016;6(2):145. doi: 10.18178/ijmlc.2016.6.2.589. [DOI] [Google Scholar]
- Mayr A, Klambauer G, Unterthiner T, Hochreiter S. Deeptox: toxicity prediction using deep learning. Front Environ Sci. 2016;3:80. doi: 10.3389/fenvs.2015.00080. [DOI] [Google Scholar]
- McMillan EA, Ryu M-J, Diep CH, Mendiratta S, Clemenceau JR, Vaden RM, Kim J-H, Motoyaji T, Covington KR, Peyton M, et al. Chemistry-first approach for nomination of personalized treatment in lung cancer. Cell. 2018;173(4):864–878. doi: 10.1016/j.cell.2018.03.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melville JL, Burke EK, Hirst JD. Machine learning in virtual screening. Comb Chem High Throughput Screen. 2009;12(4):332–343. doi: 10.2174/138620709788167980. [DOI] [PubMed] [Google Scholar]
- Miljanovic M. Comparative analysis of recurrent and finite impulse response neural networks in time series prediction. Indian J Comput Sci Eng. 2012;3(1):180–191. [Google Scholar]
- Mnih V, Kavukcuoglu K, Silver D, Rusu AA, Veness J, Bellemare MG, Graves A, Riedmiller M, Fidjeland AK, Ostrovski G, et al. Human-level control through deep reinforcement learning. Nature. 2015;518(7540):529–533. doi: 10.1038/nature14236. [DOI] [PubMed] [Google Scholar]
- Morita A, Ariyasu S, Wang B, Asanuma T, Onoda T, Sawa A, Tanaka K, Takahashi I, Togami S, Nenoi M, et al. As-2, a novel inhibitor of p53-dependent apoptosis, prevents apoptotic mitochondrial dysfunction in a transcription-independent manner and protects mice from a lethal dose of ionizing radiation. Biochem Biophys Res Commun. 2014;450(4):1498–1504. doi: 10.1016/j.bbrc.2014.07.037. [DOI] [PubMed] [Google Scholar]
- Mulligan G, Mitsiades C, Bryant B, Zhan F, Chng WJ, Roels S, Koenig E, Fergus A, Huang Y, Richardson P, et al. Gene expression profiling and correlation with outcome in clinical trials of the proteasome inhibitor bortezomib. Blood. 2007;109(8):3177–3188. doi: 10.1182/blood-2006-09-044974. [DOI] [PubMed] [Google Scholar]
- Myint KZ, Xie X-Q. Recent advances in fragment-based qsar and multi-dimensional qsar methods. Int J Mol Sci. 2010;11(10):3846–3866. doi: 10.3390/ijms11103846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nayal M, Honig B. On the nature of cavities on protein surfaces: application to the identification of drug-binding sites. Proteins Struct Funct Bioinf. 2006;63(4):892–906. doi: 10.1002/prot.20897. [DOI] [PubMed] [Google Scholar]
- Ning X, Karypis G. In silico structure-activity-relationship (sar) models from machine learning: a review. Drug Dev Res. 2011;72(2):138–146. doi: 10.1002/ddr.20410. [DOI] [Google Scholar]
- Nirschl JJ, Janowczyk A, Peyster EG, Frank R, Margulies KB, Feldman MD, Madabhushi A. A deep-learning classifier identifies patients with clinical heart failure using whole-slide images of h&e tissue. PLoS ONE. 2018;13(4):e0192726. doi: 10.1371/journal.pone.0192726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noble WS. What is a support vector machine? Nat Biotechnol. 2006;24(12):1565–1567. doi: 10.1038/nbt1206-1565. [DOI] [PubMed] [Google Scholar]
- Olivecrona M, Blaschke T, Engkvist O, Chen H. Molecular de-novo design through deep reinforcement learning. J Cheminform. 2017;9(1):48. doi: 10.1186/s13321-017-0235-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pal SK, Mitra S (1992) Multilayer perceptron, fuzzy sets, classifiaction [DOI] [PubMed]
- Paré G, Mao S, Deng WQ. A machine-learning heuristic to improve gene score prediction of polygenic traits. Sci Rep. 2017;7(1):1–11. doi: 10.1038/s41598-017-13056-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel S, Tripathi R, Kumari V, Varadwaj P. Deepinteract: deep neural network based protein-protein interaction prediction tool. Curr Bioinform. 2017;12(6):551–557. doi: 10.2174/1574893611666160815150746. [DOI] [Google Scholar]
- Patil K, Jordan EJ, Park JH, Suresh K, Smith CM, Lemmon AA, Mossé Yaël P, Lemmon MA, Radhakrishnan R (2021) Computational studies of anaplastic lymphoma kinase mutations reveal common mechanisms of oncogenic activation. Proc Natl Acad Sci 118(10) [DOI] [PMC free article] [PubMed]
- Pierson E, Yau C. Zifa: Dimensionality reduction for zero-inflated single-cell gene expression analysis. Genome Biol. 2015;16(1):1–10. doi: 10.1186/s13059-015-0805-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Polamuri S (2017) How the random forest algorithm works in machine learning. Retrieved December, 21
- Poole D, Mackworth A, Goebel R (1998) Computational intelligence
- Pu Y, Wang W, Henao R, Chen L, Gan Z, Li C, Carin L (2017) Adversarial symmetric variational autoencoder. In: Advances in neural information processing systems, pp 4330–4339
- Rahman R, Matlock K, Ghosh S, Pal R. Heterogeneity aware random forest for drug sensitivity prediction. Sci Rep. 2017;7(1):1–11. doi: 10.1038/s41598-017-11665-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rahman R, Otridge J, Pal R. Integratedmrf: random forest-based framework for integrating prediction from different data types. Bioinformatics. 2017;33(9):1407–1410. doi: 10.1093/bioinformatics/btw765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramsundar B, Liu B, Zhenqin W, Verras A, Tudor M, Sheridan RP, Pande V. Is multitask deep learning practical for pharma? J Chem Inf Model. 2017;57(8):2068–2076. doi: 10.1021/acs.jcim.7b00146. [DOI] [PubMed] [Google Scholar]
- Rolan P, Danhof M, Stanski D, Peck C. Current issues relating to drug safety especially with regard to the use of biomarkers: A meeting report and progress update. Eur J Pharm Sci. 2007;30(2):107–112. doi: 10.1016/j.ejps.2006.11.007. [DOI] [PubMed] [Google Scholar]
- Romo-Bucheli D, Janowczyk A, Gilmore H, Romero E, Madabhushi A. Automated tubule nuclei quantification and correlation with oncotype dx risk categories in er+ breast cancer whole slide images. Sci Rep. 2016;6:32706. doi: 10.1038/srep32706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosenblatt F (1961) Principles of neurodynamics. perceptrons and the theory of brain mechanisms. Technical report, Cornell Aeronautical Lab Inc Buffalo NY
- Rouillard AD, Hurle MR, Agarwal P. Systematic interrogation of diverse omic data reveals interpretable, robust, and generalizable transcriptomic features of clinically successful therapeutic targets. PLoS Comput Biol. 2018;14(5):e1006142. doi: 10.1371/journal.pcbi.1006142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabrina R, Sohrab S, Ziv BJ, Ravi P (2019) Dhaka: variational autoencoder for unmasking tumor heterogeneity from single cell genomic data. Bioinformatics [DOI] [PMC free article] [PubMed]
- Saltz J, Gupta R, Hou L, Kurc T, Singh P, Nguyen V, Samaras D, Shroyer KR, Zhao T, Batiste R, et al. Spatial organization and molecular correlation of tumor-infiltrating lymphocytes using deep learning on pathology images. Cell Rep. 2018;23(1):181–193. doi: 10.1016/j.celrep.2018.03.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Samigulina G, Zarina S (2017) Immune network technology on the basis of random forest algorithm for computer-aided drug design. In: International Conference on Bioinformatics and Biomedical Engineering. Springer, pp 50–61
- Santos R, Ursu O, Gaulton A, Bento AP, Donadi RS, Bologa CG, Karlsson A, Al-Lazikani B, Hersey A, Oprea TI, et al. A comprehensive map of molecular drug targets. Nat Rev Drug Discovery. 2017;16(1):19–34. doi: 10.1038/nrd.2016.230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmidhuber J. Deep learning in neural networks: an overview. Neural Netw. 2015;61:85–117. doi: 10.1016/j.neunet.2014.09.003. [DOI] [PubMed] [Google Scholar]
- Schneider G, Funatsu K, Okuno Y, Winkler D. De novo drug design-ye olde scoring problem revisited. Mol Inf. 2017;36(1–2):1681031. doi: 10.1002/minf.201681031. [DOI] [PubMed] [Google Scholar]
- Schneidman-Duhovny D, Inbar Y, Nussinov R, Wolfson HJ. Patchdock and symmdock: servers for rigid and symmetric docking. Nucleic Acids Res. 2005;33(suppl-2):W363–W367. doi: 10.1093/nar/gki481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discovery. 2016;15(8):533. doi: 10.1038/nrd.2016.29. [DOI] [PubMed] [Google Scholar]
- Searls DB. Data integration: challenges for drug discovery. Nat Rev Drug Discovery. 2005;4(1):45–58. doi: 10.1038/nrd1608. [DOI] [PubMed] [Google Scholar]
- Segler MHS, Kogej T, Tyrchan C, Waller MP. Generating focused molecule libraries for drug discovery with recurrent neural networks. ACS Cent Sci. 2018;4(1):120–131. doi: 10.1021/acscentsci.7b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seoane JA, Aguiar-Pulido V, R Munteanu C, Rivero D, R Rabunal J, Dorado J, Pazos A. Biomedical data integration in computational drug design and bioinformatics. Curr Comput Aided Drug Des. 2013;9(1):108–117. doi: 10.2174/15734099112089990011. [DOI] [PubMed] [Google Scholar]
- Sharma H, Zerbe N, Klempert I, Hellwich O, Hufnagl P. Deep convolutional neural networks for automatic classification of gastric carcinoma using whole slide images in digital histopathology. Comput Med Imag Graph. 2017;61:2–13. doi: 10.1016/j.compmedimag.2017.06.001. [DOI] [PubMed] [Google Scholar]
- Shaughnessy JD, Jr, Zhan F, Burington BE, Huang Y, Colla S, Hanamura I, Stewart JP, Kordsmeier B, Randolph C, Williams DR, et al. A validated gene expression model of high-risk multiple myeloma is defined by deregulated expression of genes mapping to chromosome 1. Blood. 2007;109(6):2276–2284. doi: 10.1182/blood-2006-07-038430. [DOI] [PubMed] [Google Scholar]
- Shi L, Campbell G, Jones W, Campagne F, Wen Z, Walker S, Su Z, Chu T, Goodsaid F, Pusztai L, et al. (2010) The maqc-ii project: a comprehensive study of common practices for the development and validation of microarray-based predictive models [DOI] [PMC free article] [PubMed]
- Shin W-H, Christoffer CW, Kihara D. In silico structure-based approaches to discover protein-protein interaction-targeting drugs. Methods. 2017;131:22–32. doi: 10.1016/j.ymeth.2017.08.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sim, DSM (2015) Drug distribution. In: Pharmacological Basis of Acute Care, Springer, Berlin, pp 27–36
- Sistare FD, Dieterle F, Troth S, Holder DJ, Gerhold D, Andrews-Cleavenger D, Baer W, Betton G, Bounous D, Carl K, et al. Towards consensus practices to qualify safety biomarkers for use in early drug development. Nat Biotechnol. 2010;28(5):446–454. doi: 10.1038/nbt.1634. [DOI] [PubMed] [Google Scholar]
- Smola AJ, Schölkopf B. A tutorial on support vector regression. Stat Comput. 2004;14(3):199–222. doi: 10.1023/B:STCO.0000035301.49549.88. [DOI] [Google Scholar]
- Soukup T, Davidson I. Visual data mining: techniques and tools for data visualization and mining. New Jersey: John Wiley & Sons; 2002. [Google Scholar]
- Spencer M, Eickholt J, Cheng J. A deep learning network approach to ab initio protein secondary structure prediction. IEEE/ACM Trans Comput Biol Bioinf. 2014;12(1):103–112. doi: 10.1109/TCBB.2014.2343960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stokes A, Hum W, Zaslavsky J (2020) A minimal-input multilayer perceptron for predicting drug-drug interactions without knowledge of drug structure. arXiv preprint arXiv:2005.10644
- Stork C, Chen Y, Sicho M, Kirchmair J. Hit dexter 2.0: machine-learning models for the prediction of frequent hitters. J Chem Inf Model. 2019;59(3):1030–1043. doi: 10.1021/acs.jcim.8b00677. [DOI] [PubMed] [Google Scholar]
- Stork C, Embruch G, Šícho M, de Bruyn Kops C, Chen Y, Svozil D, Kirchmair J. Nerdd: A web portal providing access to in silico tools for drug discovery. Bioinformatics. 2020;36(4):1291–1292. doi: 10.1093/bioinformatics/btz695. [DOI] [PubMed] [Google Scholar]
- Subramanian G, Ramsundar B, Pande V, Denny RA (2016) Computational modeling of -secretase 1 (bace-1) inhibitors using ligand based approaches. J Chem Inf Model 56(10):1936–1949 [DOI] [PubMed]
- Susan K, Stephanie H, Mathias W, Harald P, Binje V, Paul-Albert K, Maria R, Benjamin R, Svenja P, Chen M et al (2017) The target landscape of clinical kinase drugs. Science 358(6367) [DOI] [PMC free article] [PubMed]
- Sushko I, Salmina E, Potemkin VA, Poda G, Tetko IV (2012) Toxalerts: a web server of structural alerts for toxic chemicals and compounds with potential adverse reactions [DOI] [PMC free article] [PubMed]
- Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, Simonovic M, Roth A, Santos A, Tsafou KP, et al. String v10: protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015;43(D1):D447–D452. doi: 10.1093/nar/gku1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Talele TT, Khedkar SA, Rigby AC. Successful applications of computer aided drug discovery: moving drugs from concept to the clinic. Curr Top Med Chem. 2010;10(1):127–141. doi: 10.2174/156802610790232251. [DOI] [PubMed] [Google Scholar]
- Tan J, Hammond JH, Hogan DA, Greene Casey S (2016) Adage-based integration of publicly available pseudomonas aeruginosa gene expression data with denoising autoencoders illuminates microbe-host interactions. MSystems 1(1) [DOI] [PMC free article] [PubMed]
- Tasaki S, Suzuki K, Kassai Y, Takeshita M, Murota A, Kondo Y, Ando T, Nakayama Y, Okuzono Y, Takiguchi M, et al. Multi-omics monitoring of drug response in rheumatoid arthritis in pursuit of molecular remission. Nat Commun. 2018;9(1):1–12. doi: 10.1038/s41467-018-05044-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas U, Andreas M, Günter K, Marvin S, Wegner Jörg K, Hugo C, Sepp H. Deep learning as an opportunity in virtual screening. Proc Deep Learn Workshop NIPS. 2014;27:1–9. [Google Scholar]
- Tian S, Li Y, Wang J, Zhang J, Hou T. Adme evaluation in drug discovery. 9. prediction of oral bioavailability in humans based on molecular properties and structural fingerprints. Mol Pharm. 2011;8(3):841–851. doi: 10.1021/mp100444g. [DOI] [PubMed] [Google Scholar]
- Tishby N, Zaslavsky N (2015) Deep learning and the information bottleneck principle. In: 2015 IEEE Information Theory Workshop (ITW). IEEE, pp 1–5
- Tsherniak A, Vazquez F, Montgomery PG, Weir BA, Kryukov G, Cowley GS, Gill S, Harrington WF, Pantel S, Krill-Burger JM, et al. Defining a cancer dependency map. Cell. 2017;170(3):564–576. doi: 10.1016/j.cell.2017.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turkki R, Linder N, Kovanen PE, Pellinen T, Lundin J (2016) Antibody-supervised deep learning for quantification of tumor-infiltrating immune cells in hematoxylin and eosin stained breast cancer samples. J Pathol Inform 7 [DOI] [PMC free article] [PubMed]
- Turner JR. New drug development: an introduction to clinical trials. Berlin: Springer Science & Business Media; 2010. [Google Scholar]
- Vakser IA. Protein-protein docking: From interaction to interactome. Biophys J. 2014;107(8):1785–1793. doi: 10.1016/j.bpj.2014.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valkov E, Sharpe T, Marsh M, Greive S, Hyvönen M (2011) Targeting protein–protein interactions and fragment-based drug discovery. In: Fragment-Based Drug Discovery and X-Ray Crystallography. Springer, pp 145–179 [DOI] [PubMed]
- Valueva MV, Nagornov NN, Lyakhov PA, Valuev GV, Chervyakov NI (2020) Application of the residue number system to reduce hardware costs of the convolutional neural network implementation. Math Comput Simul
- Vamathevan J, Clark D, Czodrowski P, Dunham I, Ferran E, Lee G, Li B, Madabhushi A, Shah P, Spitzer M, et al. Applications of machine learning in drug discovery and development. Nat Rev Drug Discovery. 2019;18(6):463–477. doi: 10.1038/s41573-019-0024-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Gool AJ, Bietrix F, Caldenhoven E, Zatloukal K, Scherer A, Litton J-E, Meijer G, Blomberg N, Smith A, Mons B, et al. Bridging the translational innovation gap through good biomarker practice. Nat Rev Drug Discovery. 2017;16(9):587–588. doi: 10.1038/nrd.2017.72. [DOI] [PubMed] [Google Scholar]
- Vaquero-Garcia J, Barrera A, Gazzara MR, Gonzalez-Vallinas J, Lahens NF, Hogenesch JB, Lynch KW, Barash Y. A new view of transcriptome complexity and regulation through the lens of local splicing variations. Elife. 2016;5:e11752. doi: 10.7554/eLife.11752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veltri RW, Partin AW, Miller MC. Quantitative nuclear grade (qng): A new image analysis-based biomarker of clinically relevant nuclear structure alterations. J Cell Biochem. 2000;79(S35):151–157. doi: 10.1002/1097-4644(2000)79:35+<151::AID-JCB1139>3.0.CO;2-7. [DOI] [PubMed] [Google Scholar]
- Venkatesan R, Li B. Convolutional neural networks in visual computing: a concise guide. London: CRC Press; 2017. [Google Scholar]
- Vinod CSS, Anad Hareendran S. Artificial intelligence: a practitioner’s approach. Delhi: PHI Learning Pvt Ltd; 2021. [Google Scholar]
- Vinod CSS, Anand Hareendran S. Machine learning: a practitioner’s approach. Delhi: PHI Learning Pvt Ltd; 2021. [Google Scholar]
- Visibelli A, Bongini P, Rossi A, Niccolai N, Bianchini M (2020) A deep attention network for predicting amino acid signals in the formation of [formula: see text]-helices. J Bioinform Comput Biol:2050028 [DOI] [PubMed]
- Vohora D, Singh G. Pharmaceutical medicine and translational clinical research. London: Academic Press; 2018. [Google Scholar]
- Volkamer A, Kuhn D, Grombacher T, Rippmann F, Rarey M. Combining global and local measures for structure-based druggability predictions. J Chem Inf Model. 2012;52(2):360–372. doi: 10.1021/ci200454v. [DOI] [PubMed] [Google Scholar]
- Voosen P (2017) The ai detectives [DOI] [PubMed]
- Vranic S, Shimada Y, Ichihara S, Kimata M, Wenting W, Tanaka T, Boland S, Tran L, Ichihara G. Toxicological evaluation of sio2 nanoparticles by zebrafish embryo toxicity test. Int J Mol Sci. 2019;20(4):882. doi: 10.3390/ijms20040882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang N-N, Dong J, Deng Y-H, Zhu M-F, Wen M, Yao Z-J, Ai-Ping L, Wang J-B, Cao D-S. Adme properties evaluation in drug discovery: prediction of caco-2 cell permeability using a combination of nsga-ii and boosting. J Chem Inf Model. 2016;56(4):763–773. doi: 10.1021/acs.jcim.5b00642. [DOI] [PubMed] [Google Scholar]
- Wang Q, Feng YH, Huang JC, Wang TJ, Cheng GQ. A novel framework for the identification of drug target proteins: Combining stacked auto-encoders with a biased support vector machine. PLoS ONE. 2017;12(4):e0176486. doi: 10.1371/journal.pone.0176486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Jin G. Vasc: dimension reduction and visualization of single-cell rna-seq data by deep variational autoencoder. Genom Proteom Bioinform. 2018;16(5):320–331. doi: 10.1016/j.gpb.2018.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang C, Kurgan L. Survey of similarity-based prediction of drug-protein interactions. Curr Med Chem. 2020;27(35):5856–5886. doi: 10.2174/0929867326666190808154841. [DOI] [PubMed] [Google Scholar]
- Wang S, Sun S, Li Z, Zhang R, Jinbo X. Accurate de novo prediction of protein contact map by ultra-deep learning model. PLoS Comput Biol. 2017;13(1):e1005324. doi: 10.1371/journal.pcbi.1005324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang W, Yang S, Zhang X, Li J. Drug repositioning by integrating target information through a heterogeneous network model. Bioinformatics. 2014;30(20):2923–2930. doi: 10.1093/bioinformatics/btu403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, Zhu J, Pierson E, Ramazzotti D, Batzoglou S. Visualization and analysis of single-cell rna-seq data by kernel-based similarity learning. Nat Methods. 2017;14(4):414–416. doi: 10.1038/nmeth.4207. [DOI] [PubMed] [Google Scholar]
- Warmuth MK, Liao J, Rätsch G, Mathieson M, Putta S, Lemmen C. Active learning with support vector machines in the drug discovery process. J Chem Inf Comput Sci. 2003;43(2):667–673. doi: 10.1021/ci025620t. [DOI] [PubMed] [Google Scholar]
- Way GP, Greene CS (2017) Extracting a biologically relevant latent space from cancer transcriptomes with variational autoencoders. BioRxiv, p 174474 [PMC free article] [PubMed]
- Webb AR. Statistical pattern recognition. New Jersy: John Wiley & Sons; 2003. [Google Scholar]
- Willett P. Similarity-based virtual screening using 2d fingerprints. Drug Discovery Today. 2006;11(23–24):1046–1053. doi: 10.1016/j.drudis.2006.10.005. [DOI] [PubMed] [Google Scholar]
- Xia Z, Wu L-Y, Zhou X, Wong STC (2010) Semi-supervised drug-protein interaction prediction from heterogeneous biological spaces. In: BMC systems biology, vol 4. BioMed Central, pp 1–16 [DOI] [PMC free article] [PubMed]
- Xing J, Wenchao L, Liu R, Wang Y, Xie Y, Zhang H, Shi Z, Jiang H, Liu Y-C, Chen K, et al. Machine-learning-assisted approach for discovering novel inhibitors targeting bromodomain-containing protein 4. J Chem Inf Model. 2017;57(7):1677–1690. doi: 10.1021/acs.jcim.7b00098. [DOI] [PubMed] [Google Scholar]
- Xue LC, Dobbs D, Bonvin AMJJ, Honavar V. Computational prediction of protein interfaces: A review of data driven methods. FEBS Lett. 2015;589(23):3516–3526. doi: 10.1016/j.febslet.2015.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamanishi Y, Araki M, Gutteridge A, Honda W, Kanehisa M. Prediction of drug–target interaction networks from the integration of chemical and genomic spaces. Bioinformatics. 2008;24(13):i232–i240. doi: 10.1093/bioinformatics/btn162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yavuz BÇ, Yurtay N, Ozkan O (2018) Prediction of protein secondary structure with clonal selection algorithm and multilayer perceptron. IEEE Access 6:45256–45261
- Youjun X, Pei J, Lai L. Deep learning based regression and multiclass models for acute oral toxicity prediction with automatic chemical feature extraction. J Chem Inf Model. 2017;57(11):2672–2685. doi: 10.1021/acs.jcim.7b00244. [DOI] [PubMed] [Google Scholar]
- Zaretzki J, Matlock M, Swamidass SJ. Xenosite: accurately predicting cyp-mediated sites of metabolism with neural networks. J Chem Inf Model. 2013;53(12):3373–3383. doi: 10.1021/ci400518g. [DOI] [PubMed] [Google Scholar]
- Zeng X, Zhu S, Weiqiang L, Liu Z, Huang J, Zhou Y, Fang J, Huang Y, Guo H, Li L, et al. Target identification among known drugs by deep learning from heterogeneous networks. Chem Sci. 2020;11(7):1775–1797. doi: 10.1039/C9SC04336E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zernov VV, Balakin KV, Ivaschenko AA, Savchuk NP, Pletnev IV. Drug discovery using support vector machines. the case studies of drug-likeness, agrochemical-likeness, and enzyme inhibition predictions. J Chem Inf Comput Sci. 2003;43(6):2048–2056. doi: 10.1021/ci0340916. [DOI] [PubMed] [Google Scholar]
- Zhan F, Barlogie B, Mulligan G, Shaughnessy JD, Jr, Bryant B. High-risk myeloma: a gene expression-based risk-stratification model for newly diagnosed multiple myeloma treated with high-dose therapy is predictive of outcome in relapsed disease treated with single-agent bortezomib or high-dose dexamethasone. Blood J Am Soc Hematol. 2008;111(2):968–969. doi: 10.1182/blood-2007-10-119321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhan F, Huang Y, Colla S, Stewart JP, Hanamura I, Gupta S, Epstein J, Yaccoby S, Sawyer J, Burington B, et al. The molecular classification of multiple myeloma. Blood. 2006;108(6):2020–2028. doi: 10.1182/blood-2005-11-013458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang QC, Petrey D, Norel R, Honig BH. Protein interface conservation across structure space. Proc Natl Acad Sci. 2010;107(24):10896–10901. doi: 10.1073/pnas.1005894107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhavoronkov A, Ivanenkov YA, Aliper A, Veselov MS, Aladinskiy VA, Aladinskaya AV, Terentiev VA, Polykovskiy DA, Kuznetsov MD, Asadulaev A, et al. Deep learning enables rapid identification of potent ddr1 kinase inhibitors. Nat Biotechnol. 2019;37(9):1038–1040. doi: 10.1038/s41587-019-0224-x. [DOI] [PubMed] [Google Scholar]
- Zhou H, Gao M, Skolnick J. Comprehensive prediction of drug-protein interactions and side effects for the human proteome. Sci Rep. 2015;5(1):1–13. doi: 10.1038/srep11090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zsoldos Z, Reid D, Simon A, Sadjad SB, Johnson AP. ehits: a new fast, exhaustive flexible ligand docking system. J Mol Graph Model. 2007;26(1):198–212. doi: 10.1016/j.jmgm.2006.06.002. [DOI] [PubMed] [Google Scholar]