

Abstract
The design of new functional polymers depends on the successful navigation of their structure-function landscapes. Advances in combinatorial polymer chemistry and machine learning provide exciting opportunities for the engineering of fit-for-purpose polymeric materials.
Polymeric materials with high-performance characteristics can be achieved by replicating monomeric units of often simple chemistry into functional macromolecules with desirable properties. Indeed, biology demonstrates the immense potential of this approach by using amino acids and sugars as the building blocks of diverse and hierarchical polymeric materials in the form of proteins and polysaccharides. Like proteins, synthetic polymers possess innumerable monomer combinations that may translate to favourable structure-function relationships. From an engineering perspective, the combinatorial complexity of polymeric materials manifests itself in the curse of dimensionality, making the rational design of high-performance features (for example, ionic conductivity, photoconversion efficiency, shape-memory response and self-healing) difficult. Alternatively, combinatorial polymer chemistry provides efficient and informed surveys of high-dimensional polymer design spaces.
The emergence of the fourth paradigm of science, that is, data-intensive scientific discovery, may open the door to new forays of combinatorial polymer chemistry in materials science. Indeed, artificial intelligence (AI) and machine learning (ML) are increasingly used in the physical sciences and engineering, as highlighted by the recent performance of DeepMind’s AlphaFold2 in the 2020 Critical Assessment of Protein Structure Prediction (CASP) competition. Using ML methods trained with data of over 170,000 known protein structures, AlphaFold2 demonstrated unprecedented accuracy in predicting single-chain protein folding, a grand challenge in structural biology. This achievement underlines the promise of ML in scientific applications and, in particular, for polymeric materials. After all, proteins are polymers made of amino acids, whose primary sequence ultimately determines their structure and therefore function.
Converging ideas
The accessibility of benchtop and automated combinatorial polymer chemistry, advances in molecular modelling and the increasing availability of flexible machine learning software present new possibilities for data-driven exploration of structure-function relationships in polymers.
Combinatorial and automated polymer chemistry.
Starting in the 1990s, laboratory synthesizers have enabled the use of combinatorial and automated polymer chemistry in polymer research and material design1,2. However, the intolerance of polymerization reactions to ambient air (that is, oxygen and water) have long limited efficient and automated high-throughput experimentation owing to the requirement of sealed reaction vessels purged with inert atmosphere or freeze–pump–thaw cycling. Air-tolerant chemistries can address this limitation by allowing controlled living radical polymerizations to proceed in open air, including in well plates3. Therefore, combinatorial libraries can easily be prepared on the benchtop by simple addition of starting reagents in routine labware. Moreover, open platform liquid handling robotics can be applied for fully or semi-automated polymer synthesis, opening a new era of high-throughput combinatorial polymer chemistry4.
The evolution of molecular modelling.
Molecular modelling has long been a valuable tool in materials design, complementing experimental work by providing detailed theoretical characterizations to reveal mechanistic features and design principles. Recently, spurred by computing advances, algorithmic developments and the impetus of the Materials Genome Initiative, high-throughput calculations and virtual screening have emerged as cost-effective in silico design paradigms5. However, these approaches have mainly been applied for small-molecule drug compounds and inorganic materials thus far and less so for polymers. This is partly because density functional theory (DFT), the work-horse of high-throughput molecular theory, is impractical or ill-suited for characterizing polymers because they are often typified by large, disordered systems with properties that depend on weak interactions and conformational heterogeneity. Molecular dynamics (MD) modelling is typically more suitable but computationally challenging for macromolecular systems at atomistic resolution. However, coarse-grained (CG) modelling, which sacrifices chemical resolution for computational tractability, may provide a practical solution to the quandary of approaching theoretical polymer characterization at scales needed for ML6,7.
Data nexus of experimentation and modelling.
Combinatorial polymer chemistry is poised to harness ML by combining high-throughput experimentation and modelling. An example of how theory can lead experimentation was the identification of organic light-emitting diodes (LEDs) by hundreds of thousands of time-dependent DFT calculations and ML; however, experimental calibrations and human assessment were needed for the final selection of candidates8. ML-based polymer property prediction has been demonstrated using ML models trained on theoretical calculations, which show good correspondence to ML models benchmarked against available experimental data9. If data-generation capabilities are mismatched between theory and experiment, transfer learning may provide a pathway toward high-fidelity ML models that combine datasets from disparate sources.
Small steps to going big
Complex laboratory automation and exhaustive calculations often require enormous capital and human resources. However, the application of new data-centric tools can be achieved by implementing small but significant steps, enabling the widespread use of these tools in all polymer science laboratories.
The robo-chemist.
The robo-chemist could become a new colleague in many polymer science laboratories. Fully automated robotics driven by AI will undoubtedly have a major impact on material discovery and design. With the emergence of air-tolerant polymer chemistry3, a high number of new polymer designs within a combinatorial library (tens of polymers per library) can be produced by simple manual pipetting in well plates. Low-cost instruments with user-friendly interfaces can automate simple tasks (for example, reagent additions, plate-to-plate transfers and serial dilutions), enabling the production of even more polymer designs within a library without requiring much training or programming skills (hundreds of polymers per library). Such systems allow low-to-medium-throughput polymer chemistry, which is also amenable to data-driven techniques. Robotic systems can dramatically improve high-throughput workflows both in terms of scale (thousands of polymers per library) and complexity. Ultimately, experimentalists should rationally approach these options according to realistic needs, leaving room for future upgrades. For example, some low-cost instruments can easily be incorporated into future fully automated workflows through open-source application programming interfaces (APIs).
Learning with less.
Polymer datasets derived by experiment or simulation remain relatively small by ML standards; however, dataset size may not be a limiting factor for sequence-based design of polymers. For example, accurate ML models describing polymer conformation have been trained from only a few hundred randomly chosen but distinct polymers6. In addition, active learning approaches are a promising route toward judicious dataset construction. Here, ML models are iteratively trained with data points that are optimally selected according to an acquisition function. This strategy led to successful identification of oligopeptides that self-assemble into nanoaggregates from only 186 CG simu lations7. Although these examples are in the con text of modelling, the numbers are well within the capabilities of experimental combinatorial polymer libraries.
Organizing disorder.
Critical to the success of AlphaFold2 was the existence of the protein data bank (PDB). Despite the construction of several polymer databases (for example, Polymer Genome, CHEMnetBASE-Polymers, Polymer Property Predictor and Database), polymer characterization data are generally not accessible in standardized and downloadable formats for data mining and ML. Moreover, available data are potentially obfuscated by a variety of variables (for example, molecular weight, processing history and characterization protocol) and mostly correspond to simple homopolymers. The question remains whether it is feasible, or necessary, to create orderly databases of combinatorial polymer chemistry for diverse applications. Many ML applications for polymers will likely use in-house generated data, which may be informally shared amongst research teams and deposited in repositories, such as the Materials Data Facility. Nevertheless, discussions on data organization and representing polymers, such as through BigSMILES language10, must continue. Relatedly, open-access datasets for monomeric units, akin to the QM9 dataset for small-molecule research, would greatly benefit polymer ML development.
Opportunities in polymer data science
Translating the chemical landscape of monomer combinations as polymers into distinct structure-function relationships remains challenging. The emergence of AI and ML, in tandem with advances in combinatorial chemistry, may provide a route toward the data-enabled design of polymeric materials. In the future, we imagine the use of AI-driven robotics to plan and optimize entire experiments (Fig. 1). Advances in systematic polymer modelling will encourage tightly integrated workflows that make use of in silico and experimental characterizations, guided and selected by efficient active learning paradigms. These tools, driven by human innovation, will enable the autonomous design and engineering of new polymer materials with optimized application-specific properties in fashion.
Fig. 1 |. Robotic systems for autonomous structure-function testing in combinatorial polymer libraries.
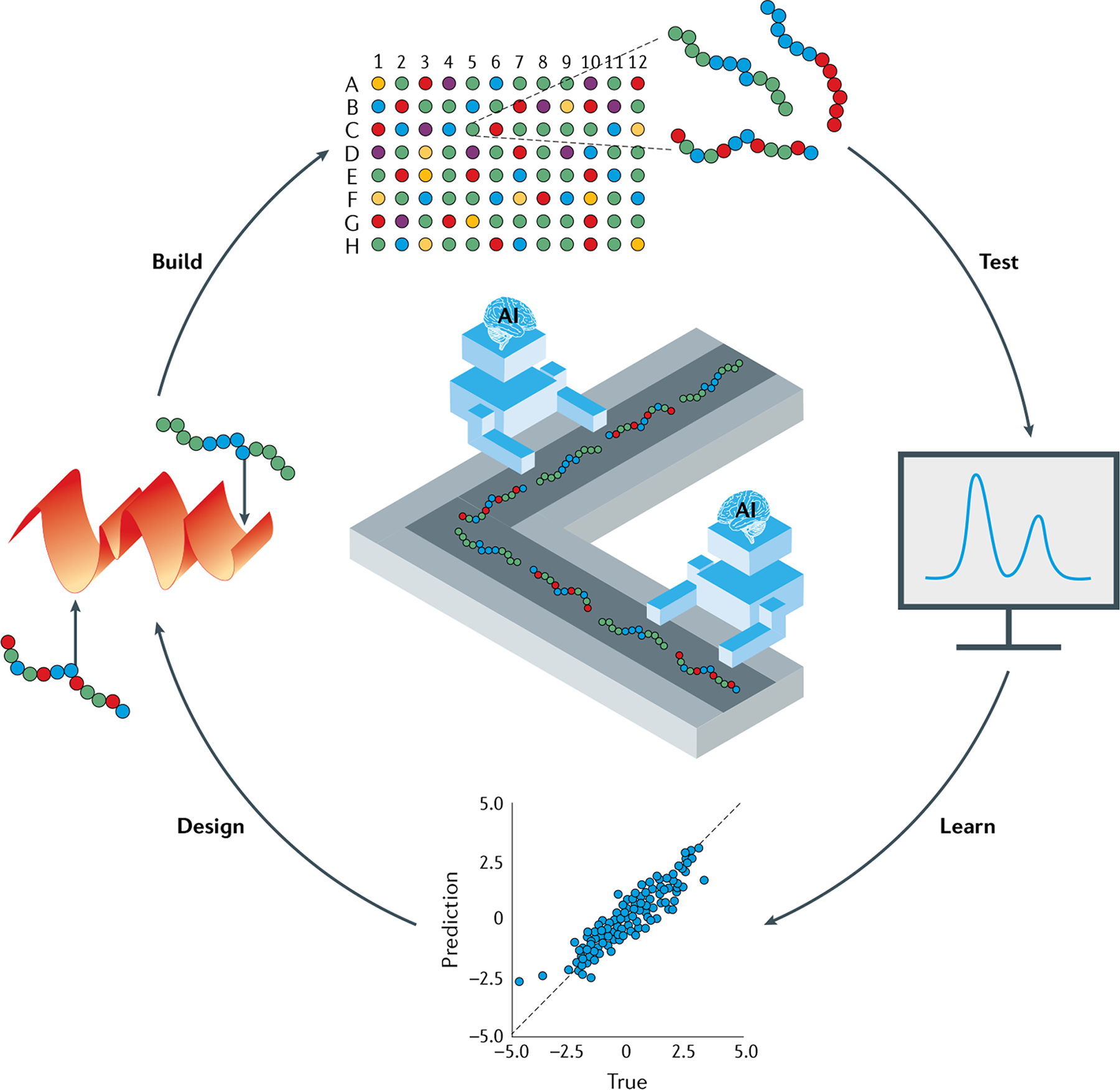
Automated robotic systems driven by artificial intelligence (AI) and modelling enable design–build–test–learn workflows for a series of chemically distinct systems. New and historic data, generated experimentally and/or in silico, are used to train machine learning (ML) models that allow the prediction of application-specific properties across the combinatorial chemical space. The developed ML models facilitate the identification of new, optimal polymer chemistries based on active learning paradigms, which are subsequently synthesized and tested, entering another round of the autonomous cycle.
Acknowledgements
A.J.G. was supported by NSF CBET award number 2009942 and NIH NIGMS award number 1R35GM138296-01. M.A.W. acknowledges support from Princeton University.
Footnotes
Competing interests
The authors declare no competing interests.
References
- 1.Hoogenboom R, Meier MAR & Schubert US Combinatorial methods, automated synthesis and high-throughput screening in polymer research: past and present. Macromol. Rapid Comm 24, 15–32 (2003). [Google Scholar]
- 2.Anderson DG, Lynn DM & Langer R Semi-automated synthesis and screening of a large library of degradable cationic polymers for gene delivery. Angew. Chem. Int. Ed 42, 3153–3158 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Yeow J, Chapman R, Gormley AJ & Boyer C Up in the air: oxygen tolerance in controlled/living radical polymerization. Chem. Soc. Rev 47, 4357–4387 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tamasi M, Kosuri S, DiStefano J, Chapman R & Gormley AJ Automation of controlled/living radical polymerization. Adv. Intell. Syst 2, 1900126 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Jain A et al. Commentary: The Materials Project: a materials genome approach to accelerating materials innovation. APL Mater. 1, 011002 (2013). [Google Scholar]
- 6.Webb MA, Jackson NE, Gil PS & de Pablo JJ Targeted sequence design within the coarse-grained polymer genome. Sci. Adv 6, eabc6216 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shmilovich K et al. Discovery of self-assembling π-conjugated peptides by active learning-directed coarse-grained molecular simulation. J. Phys. Chem. B 124, 3873–3891 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Gómez-Bombarelli R et al. Design of efficient molecular organic light-emitting diodes by a high-throughput virtual screening and experimental approach. Nat. Mater 15, 1120–1127 (2016). [DOI] [PubMed] [Google Scholar]
- 9.Chen L et al. Frequency-dependent dielectric constant prediction of polymers using machine learning. npj Comput. Mater 6, 61 (2020). [Google Scholar]
- 10.Lin TS et al. BigSMILES: a structurally-based line notation for describing macromolecules. ACS Cent. Sci 5, 1523–1531 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Related linkS
CHemnetBASe: http://poly.chemnetbase.com/
Critical Assessment of Protein Structure Prediction Competition: https://predictioncenter.org
materials data facility: https://materialsdatafacility.org/
materials Genome Initiative: https://www.mgi.gov/
PoLyInfo: https://polymer.nims.go.jp/en/
Polymer Genome: https://www.polymergenome.org/
Polymer Property Predictor and Database: https://pppdb.uchicago.edu/
Protein Data Bank: http://www.wwpdb.org/