

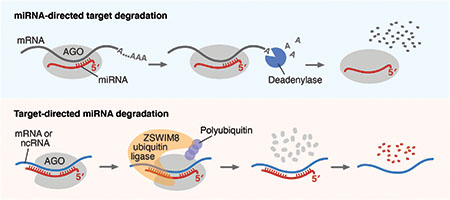
Summary
INTRODUCTION:
MicroRNAs (miRNAs) are short RNAs that direct widespread gene repression in humans and other animals. Each miRNA associates with an Argonaute (AGO) protein to form a silencing complex in which the miRNA pairs to sites within target mRNAs, and AGO recruits machinery that causes destabilization or reduced translation of the targeted transcript.
Most miRNAs are quite long-lived because association with AGO protects them from cellular nucleases. However, some miRNAs are relatively unstable. A potential explanation for the instability of these miRNAs comes from the observation that interactions with certain target sites can promote miRNA destruction, thereby inverting the typical regulatory logic. This phenomenon of target-directed miRNA degradation (TDMD) is exploited by some viruses that produce transcripts that trigger the decay of specific host miRNAs that would otherwise impede viral replication. Moreover, four endogenous transcripts have recently been found to trigger TDMD. These include the CYRANO noncoding RNA, which directs efficient degradation of miR-7. However, the extent to which this phenomenon might accelerate degradation of other miRNAs has been unclear—in part because proteins required for TDMD had not been identified.
RATIONALE:
The unusual target sites that trigger TDMD differ from the typical sites that mediate gene repression in that they not only pair to the 5′ region of the miRNA but also extensively pair to the miRNA 3′ region. This pairing to the 3′ region can cause conformational changes that expose the miRNA 3′ terminus to enzymes that append or remove nucleotides—processes called tailing and trimming, respectively. The tailing and the trimming observed in the presence of sites that trigger TDMD have been proposed to be obligate steps of the TDMD pathway. However, loss of an enzyme responsible for target-directed tailing has little influence on TDMD, suggesting that TDMD might occur through another mechanism. To learn more about this mechanism, we carried out a CRISPRi screen designed to identify proteins required for CYRANO-directed degradation of miR-7.
RESULTS:
Our screen revealed that ZSWIM8 is required for CYRANO-directed miR-7 degradation, as well as for other known examples of TDMD. Moreover, identification of miRNAs that increased after knocking out the ZSWIM8 ortholog in various mammalian cells, as well as in Drosophila S2 cells and nematodes, implicated dozens of additional miRNAs as substrates of endogenous TDMD. Indeed, for cells in which miRNA half-lives were known, TDMD explained the destabilization of most short-lived miRNAs.
ZSWIM8 is the substrate receptor of a Cullin-RING E3 ubiquitin ligase, suggesting an alternative model for TDMD that does not depend on miRNA tailing and trimming. In this model, the ZSWIM8 ubiquitin ligase recognizes the conformational changes that occur upon extensive pairing to the miRNA 3′ region, which leads to polyubiquitination of AGO. Polyubiquitinated AGO is then degraded by the 26S proteasome, thereby exposing the miRNA to cytoplasmic nucleases. Supporting this model, inhibiting of either ubiquitination or proteasome-mediated proteolysis impeded TDMD. Moreover, the miRNAs that were TDMD substrates preferentially associated with polyubiquitinated proteins in the presence of ZSWIM8, and substitutions of lysines on the surface of AGO abrogated TDMD, implicating AGO as the direct target of the ubiquitin ligase.
CONCLUSION:
TDMD requires the ZSWIM8 ubiquitin ligase, supporting a model of TDMD in which target-directed proteolysis of AGO exposes the miRNA for degradation. Furthermore, the implied scope of TDMD extends far beyond the four endogenous examples previously identified in mammals and other vertebrates. Endogenous TDMD appears to influence dozens of miRNAs in mammalian cells, and it extends to Drosophila and nematodes, implying that it has been shaping miRNA levels and dynamics since the last common ancestor of bilaterian animals.
Summary Figure
Molecular models for effects of miRNAs on targets, and vice versa. During miRNA-mediated regulation (top), the miRNA directs AGO to an mRNA, which recruits proteins that shorten the mRNA poly(A) tail, causing more rapid mRNA degradation. During TDMD (bottom), a target site with extensive pairing to the miRNA 3′ region induces conformational changes that are recognized by the ZSWIM8 ubiquitin ligase. This ligase polyubiquitinates AGO, causing its proteolysis, which exposes the miRNA for degradation. ncRNA, noncoding RNA.
Abstract
MicroRNAs (miRNAs) associate with Argonaute (AGO) proteins to direct widespread post-transcriptional gene repression. Although association with AGO typically protects miRNAs from nucleases, extensive pairing to some unusual target RNAs can trigger miRNA degradation. Here we found that this target-directed miRNA degradation (TDMD) required the ZSWIM8 Cullin-RING E3 ubiquitin ligase. This and other findings suggested and supported a mechanistic model of TDMD in which target-directed proteolysis of AGO by the ubiquitin–proteasome pathway exposes the miRNA for degradation. Moreover, loss-of-function studies indicated that the ZSWIM8 Cullin-RING ligase accelerates degradation of numerous miRNAs in cells of mammals, flies, and nematodes, thereby specifying the half-lives of most short-lived miRNAs. These results elucidate the mechanism of TDMD and expand its inferred role in shaping miRNA levels in bilaterian animals.
One-sentence Summary:
Some microRNA targets trigger proteolysis of Argonaute proteins, providing a widely employed mechanism for sculpting microRNA levels.
MicroRNAs (miRNAs) are ~22-nucleotide (nt) RNAs that pair to sites in mRNAs, which causes more rapid degradation and/or translational repression of these mRNA targets (1, 2). In aggregate, miRNAs regulate most human mRNAs (3), and most of the 90 broadly conserved miRNA families found in mammals are required for viability or proper development (1). Compared to mRNAs, which typically decay after only a few hours, most miRNAs persist much longer—often longer than a day and sometimes longer than a week (4–6). This stability is explained by residence of each miRNA within an effector protein Argonaute (AGO), which interacts with the miRNA throughout its length, thereby protecting it from nucleases (7, 8).
Despite the stability of most miRNAs, some can be relatively short-lived, with half-lives ranging from <2 h to 10 h (4, 5, 9). A potential explanation for these shorter half-lives comes from the observation that pairing between a miRNA and an unusually highly complementary target can trigger decay of the miRNA, inverting the typical regulatory logic. This phenomenon, termed target-directed miRNA degradation (TDMD), was initially observed after introducing either antisense miRNA inhibitors or artificial targets with extensive pairing into cells (10–14), or after infection by some herpesviruses, which express transcripts with extensively paired sites that direct the degradation of host antiviral miRNAs (15–18). More recently, four cellular transcripts have also been found to trigger TDMD (19–21). For example, the Cyrano long noncoding RNA (lncRNA) directs efficient degradation of the miR-7 miRNA (21). After either deleting Cyrano or mutating the site within Cyrano that pairs extensively to miR-7 (Fig. 1A), steady-state miR-7 levels increase by as much as 50-fold in some cells and tissues (4, 21). Moreover, after loss of Cyrano, the half-life of miR-7 increases to resemble that of most other miRNAs (4).
Fig. 1. A CRISPRi screen identifies ZSWIM8 as a regulator of miR-7.
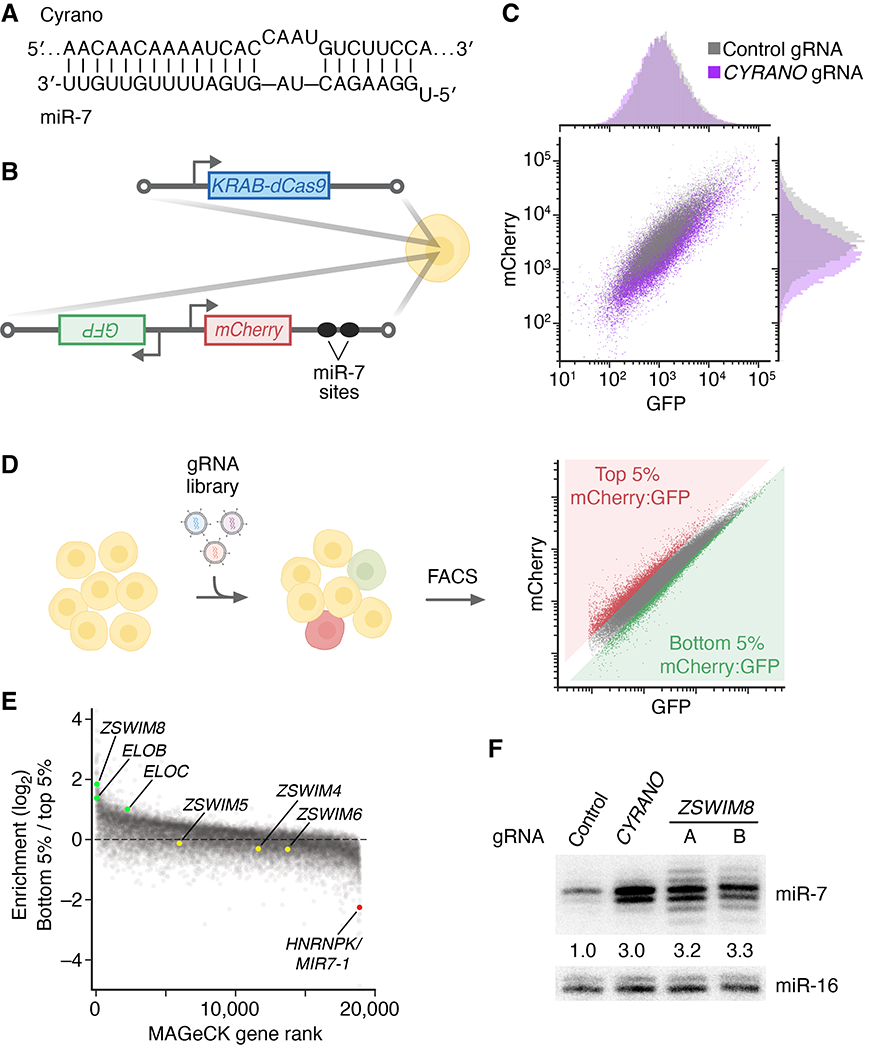
(A) The extensive complementary between Cyrano and miR-7. Vertical lines indicate Watson–Crick complementarity, excluding the first nucleotide of the miRNA, which is not available for pairing (1). (B) The engineered K562 cell line used to perform the CRISPRi screen. To generate this line, two constructs were stably integrated into the genome. One expressed KRAB-dCas9, and the other expressed GFP and mCherry reporter mRNAs from a bidirectional promoter. The mCherry mRNA had two sites in its 3′ UTR that were designed to be susceptible to miR-7–directed slicing. (C) Flow cytometry of cells in (B) transduced with lentiviral constructs expressing either a non-targeting control gRNA or an gRNA targeting CYRANO for knockdown. (D) Schematic of the genome-wide CRISPRi screen. Cells in (B) were transduced with a lentiviral CRISPRi gRNA library, cultured, and sorted with respect to their mCherry:GFP ratios, collecting cells with ratios in the top 5% and bottom 5% of all cells. The abundances of gRNAs in these two populations were then determined by high-throughput sequencing. (E) Results of the screen. Aggregate gRNA enrichment in cells with mCherry:GFP ratios in the bottom 5% compared to that in cells with ratios in the top 5% is plotted as function of MAGeCK rank. (F) The influence of ZSWIM8 on miR-7 accumulation. Shown is a representative RNA blot measuring miR-7 and miR-16 levels in an independently derived K562i line expressing either a non-targeting control gRNA, a gRNA targeting CYRANO, or one of two gRNAs (A and B) targeting ZSWIM8 for CRISPRi-mediated knockdown. miR-7 levels were normalized to those of miR-16, and mean levels relative to that observed for the non-targeting control are shown. n = 3 biological replicates.
In contrast to the extensively paired sites that trigger TDMD (e.g., Fig. 1A), most miRNA regulatory sites pair only to the miRNA seed region (2–8 nucleotides from the miRNA 5′ terminus) (22). Some sites also possess supplementary pairing, but this pairing is less common (conserved in only 5% of the preferentially conserved sites) and rarely approaches the miRNA 3′ terminus (22), which explains why most regulatory sites do not trigger TDMD.
Some sites that trigger TDMD also cause addition of untemplated nucleotides to the miRNA 3′ terminus and/or removal of 3′-terminal nucleotides, processes known as tailing and trimming, respectively (8, 12–14, 17, 19–21, 23–26). Mechanistically, the extensive 3′ pairing characteristic of these sites is proposed to promote conformational changes that separate the 3′ end of the miRNA from its binding pocket in the PAZ domain of AGO, thereby exposing it to cytosolic terminal nucleotidyltransferases and exonucleases (12). Supporting this proposal, tailing and trimming each occur while the affected miRNA is still bound by AGO (8, 23, 25). Moreover, crystal structures reveal conformational changes in both the protein and the miRNA that accompany extensive pairing to the 3′ portion of the miRNA and indicate that these changes leave the miRNA 3′ terminus potentially vulnerable to enzymatic modification (8).
Taken together with the role of tailing in marking other RNA substrates for degradation (27–35), these findings have led to a model of TDMD whereby tailing marks the miRNA for trimming, and ultimately degradation by cellular nucleases (8, 12). However, any model that mechanistically couples target-directed tailing and trimming (TDTT) to TDMD is challenged by the observations that some miRNA sites trigger TDTT without detectable miRNA degradation (8, 12), whereas others trigger TDMD without detectable tailing and trimming (12). Moreover, deletion of the gene encoding the PAPD4 (TENT2/TUT2/GLD2) terminal nucleotidyltransferase eliminates miR-7 tailing attributable to Cyrano yet has no detectable effect on Cyrano-directed miR-7 degradation (21).
A better understanding of the TDMD mechanism would benefit from the identification of proteins required for this phenomenon. Although TDMD was first described more than a decade ago, no such proteins have been reported. Because many candidates, including known terminal nucleotidyltransferases and nucleases, had already been tested for roles in either TDMD or miRNA turnover more generally (9, 21, 25, 26, 36–38), we set out to use an unbiased approach to identify proteins that function in the TDMD pathway.
ZSWIM8 is required for CYRANO-directed miR-7 degradation
To find factors required for TDMD, we developed a CRISPR screen designed to identify protein-coding genes needed to reduce the level of miR-7. The screen used CRISPR interference (CRISPRi), a gene-knockdown method in which KRAB-dCas9 [catalytically defective Cas9 (dCas9) fused at its N terminus to the Krüppel associated box (KRAB) transcriptional repression domain] is ectopically expressed in cells and directed to the vicinity of transcription start sites by co-expressed guide RNAs (gRNAs) (39). After engineering K562 cells (a human erythroleukemia cell line) to express KRAB-dCas9, we further modified these cells to express two reporter mRNAs from a bidirectional promoter (Fig. 1B). One reporter was designed to report on cellular miR-7 activity; it contained two sites predicted to undergo miR-7–directed slicing and coded for the mCherry fluorescent protein. The other reporter was designed to normalize for expression independent of miR-7; it contained no miR-7 sites and coded for green fluorescent protein (GFP).
To confirm the utility of these cells, we transduced them with either a lentivirus expressing a control gRNA or one that expressed a gRNA that recruits KRAB-dCas9 to the CYRANO promoter and silences its transcription (21). As expected if cellular fluorescence reported on the efficacy of CYRANO-directed miR-7 degradation, transduction with the CYRANO gRNA caused mCherry:GFP ratios to drop (Fig. 1C), which presumably reflected reduced CYRANO-directed miR-7 degradation, leading to increased miR-7 accumulation and thus reduced mCherry expression. Having confirmed the desired response, albeit with relatively low magnitude, we went forward with the screen. The reporter cells were transduced with the CRISPRi v2 lentiviral library designed to express 10 gRNAs per protein-coding gene (Fig. 1D) (40). After 10 days of culture, cells were sorted, collecting populations at the bottom 5% and the top 5% with respect to mCherry:GFP ratio (Fig. 1D), which were expected to be enriched and depleted, respectively, in cells with increased miR-7 accumulation. The gRNA cassettes from these two populations were sequenced, and genes that were preferentially targeted in the bottom 5% relative to the top 5% were identified by MAGeCK, which statistically scores gRNA enrichment for each gene (41) (Fig. 1E, fig S1B, and Data S1).
Among the 70 genes with false discovery rate (FDR) ≤ 0.3 (Data S1), 56 candidates were selected for a secondary screen that more directly queried the influence on miR-7 levels. For each candidate, gRNAs were expressed in K562i cells (an independent K562 cell line competent for CRISPRi) (42), and an RNA blot was used to detect an increase in miR-7 without a change in either the pre-miRNA hairpin or a control miRNA (fig. S1A). This secondary screen validated a single candidate: Knockdown of ZSWIM8 with either gRNA caused an increase in miR-7, with magnitude resembling that of the CYRANO knockdown (Fig. 1F and fig. S1A). Likewise, Cas9-mediated knockout of ZSWIM8 caused increased miR-7, and as expected, expression of wild-type ZSWIM8 in the knockout background restored the normal miR-7 level (fig. S1C; DNA lesions of this and other clonal knockout lines are listed in table S1A).
ZSWIM8 acts in miR-7 degradation, downstream of CYRANO
High-throughput sequencing of small RNAs isolated from K562i cells further established the similarities between the functions of ZSWIM8 and CYRANO, showing that as observed upon CYRANO knockdown, miR-7 levels specifically increased upon either ZSWIM8 knockdown or ZSWIM8 knockout, as also observed upon CYRANO knockdown (Fig. 2A, fig. S2A, table S1A, and Data S2). These small-RNA sequencing (sRNA-seq) results also provided the opportunity to monitor the effect of ZSWIM8 on the level of the miR-7 passenger strand. MicroRNA biogenesis culminates in the production of a miRNA duplex, consisting of a miRNA and its passenger strand. This duplex associates with AGO in a manner that retains one strand—typically the one annotated as the miRNA—as the guide strand and expels the passenger strand, which is rapidly degraded. The sRNA-seq showed that, despite the increase in miR-7 observed upon ZSWIM8 knockdown, the level of its passenger strand did not significantly change (Fig. 2A). This increase in miRNA, with no change in its passenger strand, was also observed after loss of CYRANO (Fig. 2A) (21) and would be expected after knockdown of a TDMD factor because TDMD acts after the miRNA is loaded into AGO and thus affects only the guide strand. In contrast, most other processes that influence miRNA levels (such as differential transcription or processing) affect accumulation of both guide and passenger strands. Thus, this behavior of miR-7 and its passenger strand supported the idea that our CRISPRi screen had identified a protein required for CYRANO-directed miR-7 degradation in K562 cells.
Fig. 2. ZSWIM8 is required for target-directed miR-7 degradation, acting downstream of CYRANO.
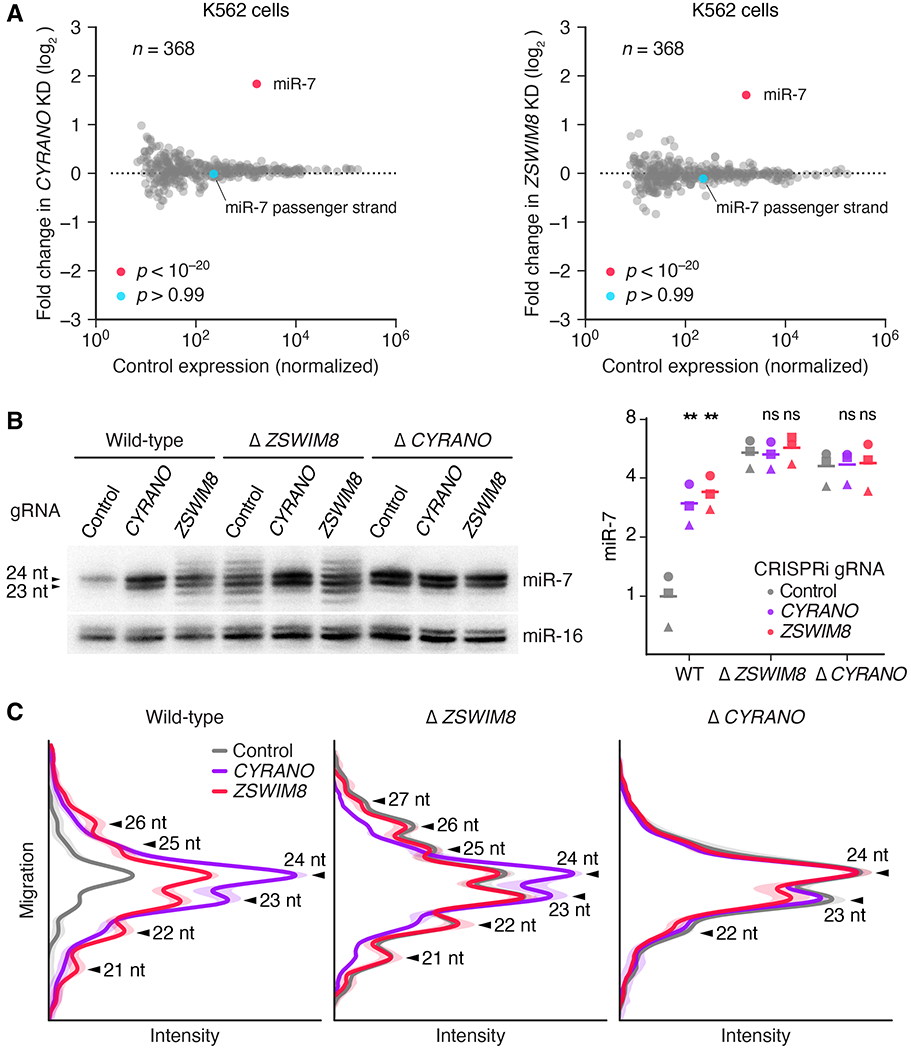
(A) The influence of CYRANO and ZSWIM8 on miRNA levels in K562i cells. Shown are fold changes in mean miRNA levels observed upon CRISPRi-mediated knockdown of either CYRANO (left) or ZSWIM8 (right). Relative miRNA levels measured by sRNA-seq after knockdown were compared with those observed after expressing a control gRNA (n = 3 biological replicates). The point for the miRNA that significantly changed (adjusted p value < 10−7, as determined by DESeq2) is indicated (red), as is the point for its passenger strand (blue). (B) Genetic interaction between CYRANO and ZSWIM8. On the left is an RNA blot measuring miR-7 and miR-16 levels in clonal K562i lines in which the indicated gene had been knocked out (wild-type, Δ CYRANO, and Δ ZSWIM8; table S1A) and either a non-targeting control gRNA, a gRNA targeting CYRANO, or a gRNA targeting ZSWIM8 (gRNA B) was expressed for CRISPRi-mediated knockdown (fig. S2C). On the right are the results of quantification of this blot (squares), measuring miR-7 normalized to miR-16, plotted together with results from two additional biological replicates, each performed with independent control or knockout clonal lines (circles and triangles; means, horizontal lines). All values were normalized to the loading control (miR-16), then to hybridization standards, and finally to the mean of WT expressing the control gRNA. Statistical significance of fold changes between cells expressing targeting and control gRNAs within the same genetic background are indicated (**, p < 0.005; ns, not significant, two-tailed paired ratio t-test). (C) The influence of CYRANO and ZSWIM8 on miR-7 length variation. Plotted are intensity values (arbitrary units) as a function of gel migration (arbitrary units) measured by line densitometry of RNA blots in (B) and its biological replicate. Peaks correspond to lengths of miR-7 isoforms, as indicated (arrowheads). Shaded areas denote 95% confidence intervals across biological replicates (n = 2).
One way that ZSWIM8 might influence CYRANO-directed miR-7 degradation is by affecting CYRANO expression. For example, if ZSWIM8 were required for transcription of CYRANO, then knockdown of ZSWIM8 would phenocopy knockdown of CYRANO. However, RNA-sequencing (RNA-seq) analyses showed that ZSWIM8 knockdown had no detectable effect on CYRANO levels, and vice versa, thereby excluding the possibility that ZSWIM8 affects CYRANO expression (fig. S2B). To investigate the hypothesis that ZSWIM8 and CYRANO nonetheless act in the same genetic pathway, we knocked down ZSWIM8 in CYRANO-knockout cells and reciprocally knocked down CYRANO in ZSWIM8-knockout cells (fig. S2C). Whereas individual loss of either ZSWIM8 or CYRANO function caused miR-7 levels to increase, neither combination of perturbations caused any additional increase (Fig. 2B). Thus, ZSWIM8 and CYRANO each required the presence of the other to influence miR-7 levels and are part of the same pathway.
Although ZSWIM8 and CYRANO each had a similar effect on the total level of miR-7, the ZSWIM8 knockdown did not precisely phenocopy the CYRANO knockdown. The distribution of miR-7 isoforms observed upon ZSWIM8 knockdown was much broader than that observed upon CYRANO knockdown (Fig. 1F and Fig. 2, B and C). Analysis of sRNA-seq results confirmed that this length variability was specific to the miR-7 guide strand and occurred at its 3′ end—a hallmark of tailing and trimming (fig. S3). These distinct molecular phenotypes provided the opportunity to perform epistasis analyses to order CYRANO and ZSWIM8 in the pathway. The reduced extent of tailing and trimming observed upon combining a CYRANO knockout with a ZSWIM8 knockdown (or a CYRANO knockdown with a ZSWIM8 knockout) resembled that observed for the knockout (or knockdown) of only CYRANO (Fig. 2, B and C). Thus, CYRANO lies upstream of ZSWIM8 in the pathway, and CYRANO but not ZSWIM8 is required for miR-7 tailing and trimming.
We also explored the effects of other genes on the extensive tailing and trimming observed upon ZSWIM8 knockout. Knockdown of PARN and DIS3L2 in ZSWIM8 knockout cells did not reveal a substantial role for these nucleases in miR-7 trimming (fig. S2D). However, knockdown of PAPD4 in ZSWIM8 knockout cells caused tailed species to decrease and trimmed species to increase, with no change in overall levels of miR-7. This finding confirmed the role of the PAPD4 terminal nucleotidyltransferase in miR-7 tailing (21), and indicated that trimmed species were not degradation intermediates downstream of tailing (fig. S2D). Moreover, building on the observation that a miRNA loaded into any of the four AGO paralogs (AGO1–4) can undergo TDTT (8), we found that miR-7 loaded into any of the four AGO paralogs could undergo ZSWIM8-dependent TDMD (fig. S2E).
ZSWIM8 and TDMD have widespread influence
Having found that ZSWIM8 is required for CYRANO-directed miR-7 degradation, we examined whether it might also be required for other instances of TDMD. ZSWIM8 mRNA is broadly expressed in human tissues, with a median of 45.2 TPM (transcripts per million) in a human transcript-expression atlas (Fig. S4A) (43), implying that it could be acting in many cell types to cause target-directed degradation of other miRNAs.
The founding biological example of TDMD is directed by Herpesvirus saimiri HSUR1, a 143-nt viral noncoding RNA that triggers the decay of host miR-27a (8, 15). To examine the potential requirement of ZSWIM8 for HSUR1-directed miR-27a degradation, we knocked out ZSWIM8 in BJAB cells (a human Burkitt lymphoma B cell line) engineered to express HSUR1 (8, 15). Polyclonal knockout of ZSWIM8 restored miR-27a levels in HSUR1-expressing cells but had no detectable effect on miR-27a levels in control lines that expressed either no additional RNA (empty vector) or a mutant HSUR1 unable to direct miR-27a degradation (Fig. 3A; efficiencies of these and other polyclonal knockouts are listed in table S1B). Probing for other miRNAs showed that, as expected, ZSWIM8 influenced miR-7 levels regardless of whether HSUR1 was present and had no influence on miR-20, which was not a suspected TDMD substrate in BJAB cells (Fig. 3A). Similar experiments indicated that NREP-directed degradation of miR-29b, the founding example of TDMD triggered by an endogenous mRNA (19), also depended on ZSWIM8 (fig. S4B).
Fig. 3. ZSWIM8 is generally required for TDMD and limits accumulation of many miRNAs in diverse species.
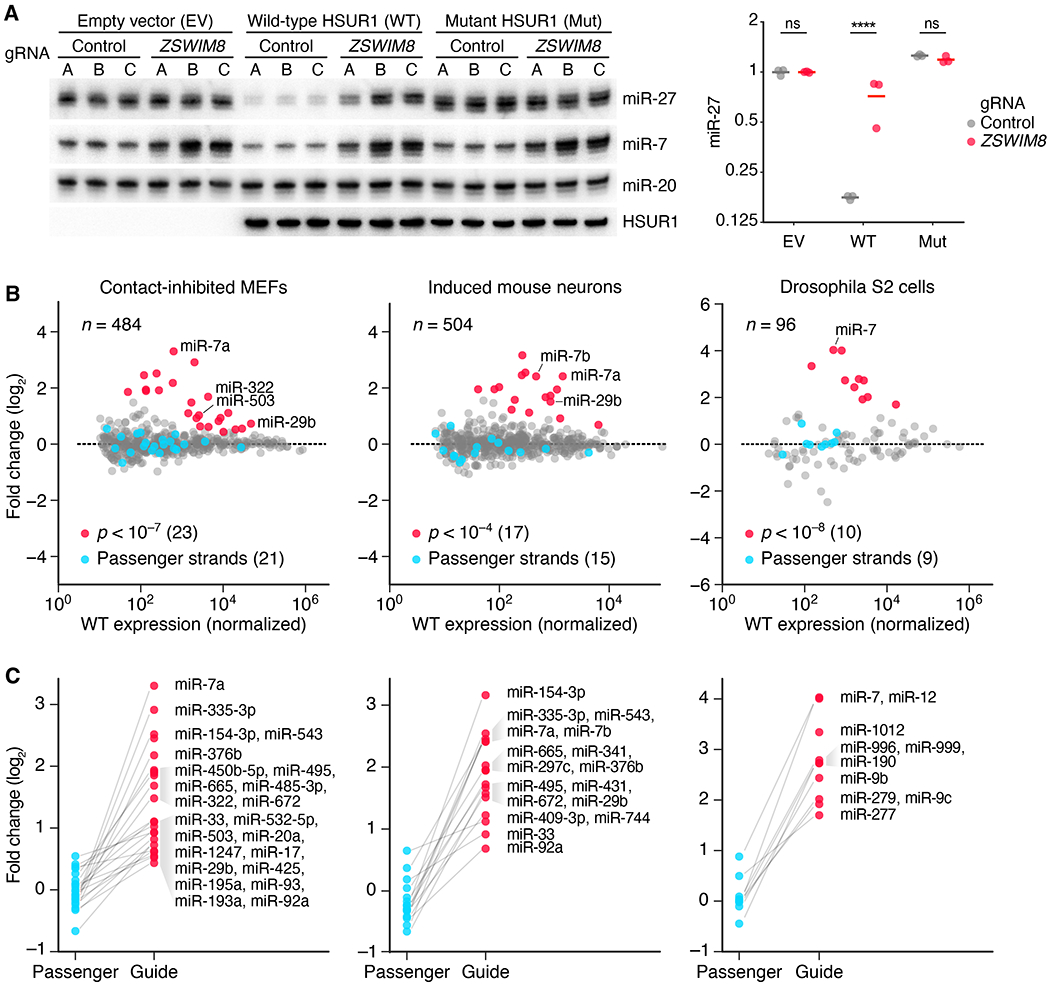
(A) The requirement of ZSWIM8 for HSUR1-directed miR-27a degradation. The RNA blot measures levels of the indicated RNAs in BJAB cells stably expressing either HSUR1, mutant HSUR1, or empty vector—each also expressing Cas9 and either one of three control gRNAs (A–C) or one of three gRNAs targeting ZSWIM8 (A–C) for polyclonal knockout (table S1B). The plot shows relative levels of miR-27 after normalizing to that of miR-20 and then to the mean level of cells expressing empty vector (EV) and control gRNAs (means, horizontal lines; ****, p < 0.0001, two-way ANOVA followed by Bonferroni’s multiple-comparisons test). (B) The influence of ZSWIM8 on miRNA levels in cells from diverse species, as measured by sRNA-seq. Plotted are fold changes in miRNA levels observed upon polyclonal knockout of Zswim8 in MEFs and induced mouse neurons, and clonal knockout of the Zswim8 ortholog in Drosophila S2 cells (table S1). miRNA fold changes with significance exceeding the indicated p-value threshold are red, and fold changes for their corresponding passenger strands are blue. miR-29b in MEFs and miR-7a in induced neurons each had two quantifiable passenger strands, whereas some other miRNAs did not have a passenger strand that exceeded our threshold for accurate quantification (Data S2). Adjusted p values were determined by DESeq2. n = 3, 2, and 3 biological replicates for MEFs, induced mouse neurons, and S2 cells, respectively. Each replicate was performed with independent knockout and control lines (table S1A). (C) The asymmetric influence of ZSWIM8 on miRNA guide and passenger strands. Shown are the miRNA fold changes highlighted in (B), each paired with the fold change of its passenger strand(s). Unpaired points correspond to miRNAs for which passenger strands did not exceed the threshold for reliable quantification.
The finding that ZSWIM8 was required for known instances of TDMD raised the prospect that additional miRNAs might be implicated as TDMD substrates on the basis of their increased accumulation following ZSWIM8 loss. Reasoning that the most affected miRNAs would be those with the shortest half-lives, we first tested miR-503, which has a relatively short half-life in mouse fibroblasts (4, 9). miR-503 levels increased following polyclonal knockout of Zswim8 in contact-inhibited NIH 3T3 cells, as would be expected if it was a TDMD substrate (fig. S4C). We then looked more broadly for evidence of additional miRNAs subject to TDMD, using sRNA-seq to quantify changes in miRNA accumulation after polyclonal knockout of Zswim8 in mouse embryonic fibroblasts (MEFs). To identify ZSWIM8-sensitive miRNAs, we considered different significance thresholds for differential expression, choosing p < 10−7 as a value sufficiently permissive to capture increases in abundance upon Zswim8 knockout, yet sufficiently stringent to avoid capturing any decreases. This approach identified 23 ZSWIM8-sensitive miRNAs. For these 23 miRNAs, 21 passenger strands could be reliably quantified, and in each case, the guide strand increased without a corresponding increase in its passenger strand(s)—as would be expected if each of these guides was a TDMD substrate (Fig. 3, B and C, Data S2). These 23 ZSWIM8-sensitive miRNAs included miR-7a (the murine miR-7 paralog expressed in MEFs) and miR29b, as well as another 21 miRNAs not known to be TDMD substrates (including miR-503 and miR-322, which is related to miR-503 and reported to have similar expression dynamics (9)). Moreover, for most (20/21) of these additional ZSWIM8-sensitive miRNAs, the fraction of tailed and trimmed isoforms increased upon ZSWIM8 knockout (fig. S4, D and E). Such increases would be expected if these miRNAs, like miR-7 and other known TDMD substrates (Fig. 2B and fig. S3A) (8), also pair to triggers in a manner that exposes their 3′ termini to tailing and trimming activities.
The identification of 23 ZSWIM8-sensitive miRNAs in MEFs contrasted with the single ZSWIM8-sensitive miRNA observed in K562 cells, suggesting that the scope of TDMD is much broader in some cells than it is in others. To examine a third mammalian cell type, we investigated the effects of polyclonal Zswim8 knockout in neurons induced from mouse embryonic stem cells (mESCs). This analysis revealed 17 ZSWIM8-sensitive miRNAs, including miR-7a, miR-7b, miR-29b, and another nine miRNAs found to be ZSWIM8-sensitive in MEFs, as well as five additional newly identified ZSWIM8-sensitive miRNAs (Fig. 3, B and C, fig. S5A, Data S2). Similar analyses identified ten ZSWIM8-sensitive miRNAs in human cancer lines, of which six were identified in the HeLa line, six in the MCF7 line, and four in the A549 line (originating from epithelia of cervix, breast, and lung, respectively) (fig. S5B, Data S2). These ten included miR-7, miR-29b and orthologs of five other ZSWIM8-sensitive miRNAs found in mouse cells (fig. S5A). In sum, 32 ZSWIM8-sensitive miRNAs were identified in these mammalian cells (fig. S5A), which when added to three TDMD substrates identified in other mammalian contexts (miR-30b, miR-30c, and miR-671) (20, 21), brought the number of inferred TDMD substrates to 35.
The observation that some miRNAs were identified as TDMD substrates in some cells but not in others presumably reflected not only differential production of miRNAs in different cells but also differential expression of their TDMD triggers. Supporting this scenario, differential expression of NREP (the mRNA that triggers miR-29b degradation) explained why miR-29b was identified as ZSWIM8-sensitive in some cells but not in others (Fig. S5C).
We then extended our analyses beyond mammals, identifying 10 miRNAs sensitive to knockout of the ZSWIM8 ortholog (CG34401) in Drosophila S2 cells, each of which increased without a corresponding increase in the passenger strand (Fig. 3B and C, and Data S2). These 10 included the Drosophila miR-7 ortholog, suggesting that targeted degradation of miR-7 might have been conserved since the bilaterian ancestor of flies and mammals. For each of these inferred TDMD substrates, the fraction of tailed and trimmed isoforms increased upon CG34401 knockout (fig. S4E), which suggested that, as in mammals, endogenous TDMD in flies is triggered by targets that pair in a manner that exposes the miRNA 3′ termini to cytoplasmic tailing and trimming activities.
In Caenorhabditis elegans adult animals (which contain embryos), a null mutation in ebax-1, the ZSWIM8 ortholog in nematodes (44), caused an increase in levels of 10 miRNAs, with no corresponding change in levels of their passenger strands (fig. S5D, Data S2). These ebax-1–sensitive miRNAs included members of the miR-35 family. When considering that expression of this family rapidly diminishes after embryos progress beyond the 4-cell stage (45), our results implied that TDMD destabilizes members of the miR-35 family to help clear these miRNAs in older embryos. Other ebax-1–sensitive miRNAs are reported to increase during heat shock of stage-4 larva (fig. S5D) (46), which suggested that heat shock might inhibit the target-directed degradation normally occurring for these miRNAs.
In MEFs and S2 cells, two cell types in which miRNA half-lives have been globally measured (4, 5), we found that the ZSWIM8-sensitive miRNAs tended to have shorter half-lives (Fig. 4A). Indeed, for these ZSWIM8-sensitive miRNAs, half-life values inversely correlated with ZSWIM8 sensitivity (r2 = 0.79 in MEFs; r2 = 0.45 in S2 cells), which supported the idea that ZSWIM8-mediated TDMD largely determines the half-lives of these short-lived miRNAs. To explore a causal relationship between ZSWIM8 and half-life, we examined the influence of ZSWIM8 on the decay of miR-503 and miR-322. When NIH 3T3 cells re-enter the cell cycle after cell-cycle arrest, the production of these miRNAs drops, and because they have relatively short half-lives, their levels decline rapidly (9). In Zswim8 polyclonal knockout cells, levels of both miRNAs declined much less rapidly upon cell-cycle re-entry, which indicated that ZSWIM8 largely determines the half-lives of these two short-lived miRNAs (Fig. 4B).
Fig. 4. TDMD explains the instability of most short-lived miRNAs.
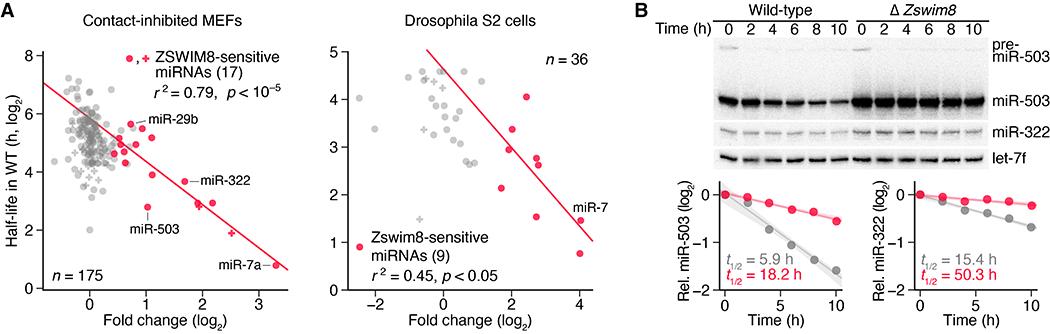
(A) The relationship between miRNA half-life and ZSWIM8 sensitivity. For each miRNA guide strand reliably quantified in contact-inhibited MEFs (left) or Drosophila S2 cells (right), its half-life in wild-type cells (4, 5) is plotted as function of the fold change in its mean level observed upon loss of ZSWIM8, as quantified in Fig. 3B. If both strands of a miRNA duplex accumulated to similar levels (within 5-fold of each other in Zswim8 knockout cells), implying that both strands commonly served as guide strands, then both strands were included in the analysis, with points indicated (+ instead of •). Points for ZSWIM8-sensitive miRNAs are colored red, as in Fig. 3B, and the line is the least-squares fit to these points (r2, coefficient of determination; p value indicated). (B) The influence of ZSWIM8 on miRNA decay. At the top is an RNA blot measuring miR-503, miR-322, and let-7f levels in NIH 3T3 polyclonal lines expressing Cas9 and either a non-targeting control gRNA (wild-type) or a Zswim8-targeting gRNA (table S1B). Serum was re-introduced to serum-starved cultures, and samples were taken at the indicated times. Plotted below are miR-503 and miR-322 levels in wild-type and Δ Zswim8 cells (gray and red, respectively), after normalization to the loading control (let-7f) and the 0 h timepoint of each cell line. Lines show the least-squares fit to the data (shading, 95% confidence interval), with apparent half-lives (t1/2 values) derived from the fit also indicated. n = 1 biological replicate.
Together, our global analyses of the effects of ZSWIM8 disruption extended the inferred scope of endogenous TDMD far beyond the four miRNA families known as TDMD substrates in mammals and fish (19–21) to include dozens of miRNA families in mammals, flies, and worms. Indeed, in cells with measured miRNA half-lives, endogenous TDMD explained the rapid decay of most short-lived miRNAs.
The E3 model of TDMD
Although ZSWIM8 mediates TDMD, it does not appear to be a nuclease. Instead, ZSWIM8 is the substrate receptor of a Cullin-RING E3 ubiquitin ligase (CRL) (44). CRLs make up the largest class of E3 ubiquitin ligases, which often function to target specific proteins for ubiquitin-dependent degradation by the 26S proteasome (47). Each CRL contains a Cullin protein, which acts as a scaffold to link the substrate receptor with a RING-finger protein that binds E2 ubiquitin-conjugating enzymes, thereby promoting ubiquitination of the substrate (48). The ZSWIM8 CRL falls into an abundant subclass of CRLs that use Elongin B (ELOB/TCEB2) and Elongin C (ELOC/TCEB1) as adaptor proteins to help link the substrate receptor to a Cullin protein (44, 49). Accordingly, ZSWIM8 has a Cullin box and a BC box, required for binding Cullin and the two Elongin proteins, respectively (Fig. 5A) (44). In addition, ZSWIM8 has a zinc-finger SWIM domain (a 35–amino acid domain initially recognized in the SWI/SNF2 chromatin remodeler and MuDR transposase), for which the protein is named, although no molecular function has been reported for this domain in ZSWIM8 (50). The Cullin box, BC box, and SWIM domain all fall within the first 208 amino acids of ZSWIM8, which spans 1837 amino acids in human (Fig. 5A). Although the remainder of the protein has no recognizable folds or motifs, ZSWIM8 is conserved broadly in animals throughout most of its length, with multiple expansive regions of high predicted order (Fig. 5A). This conserved and putatively structured remainder of the protein might help recognize substrates and orient them for ubiquitination.
Fig. 5. The E3 model of TDMD and its interplay with TDTT.
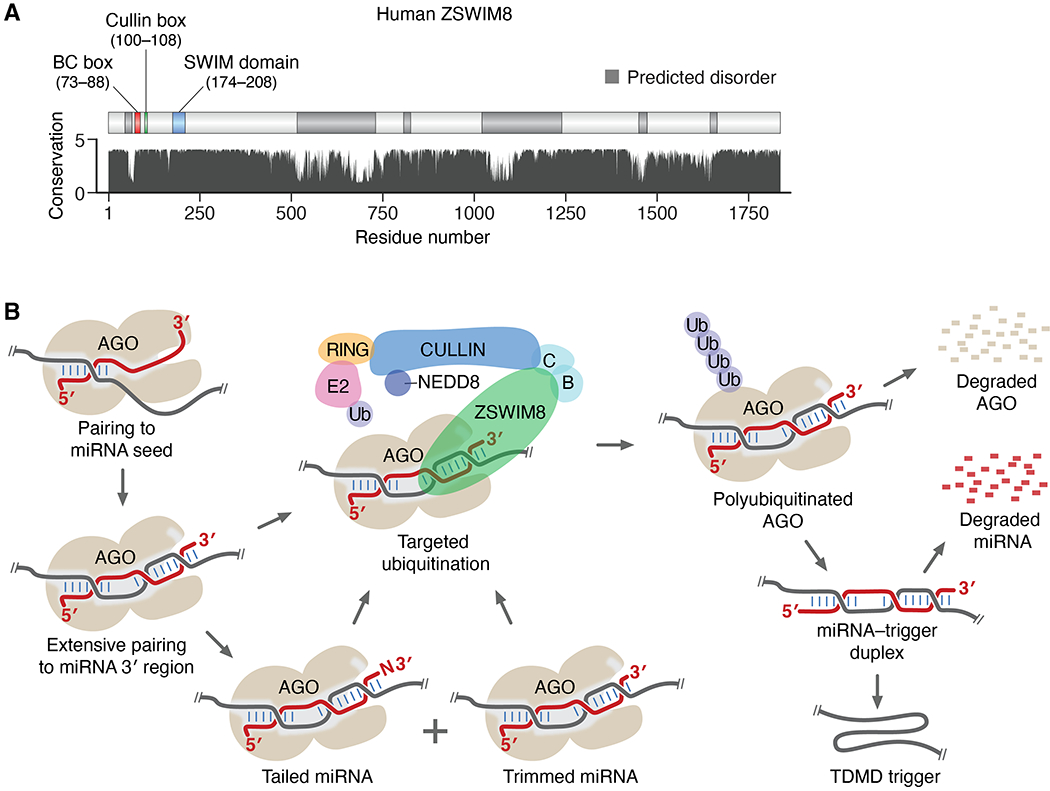
(A) The domain structure and conservation of human ZSWIM8. Annotated are the SWIM domain and known interaction motifs (44, 49, 50), predicted structural order and disorder (58), and relative amino acid conservation (arbitrary units) (59). (B) Schematic of the E3 model of TDMD and its interplay with TDTT (B, ELOB; C, ELOC; E2, Ubiquitin-conjugating enzyme; RING, RING-finger protein; Ub, ubiquitin). See main text for description.
The biological functions of ZSWIM8 have been examined most extensively in C. elegans, where the ZSWIM8 ortholog is named EBAX-1 (Elongin BC-binding axon regulator). EBAX-1 is required for proper axon guidance, a function attributed to EBAX-1–directed degradation of misfolded SAX-3/Robo receptor in developing neurons (44). Our finding that EBAX-1 is also required for TDMD (fig. S5D) suggests that miRNA misregulation might also contribute to this phenotype. In flies, the ZSWIM8 ortholog is required for viability, as indicated by the isolation of two lethal point substitutions (a missense allele and a nonsense allele) in a large-scale screen, which have not been further characterized (51). In human cells, ZSWIM8-dependent degradation of Robo, analogous to the EBAX-1–dependent degradation of C. elegans SAX-3, has been reported (44), but other functional analyses have been limited to genome-wide screens in cell lines. For example, results of CRISPR screens conducted in many cancer lines show that ZSWIM8 is not required for cell viability but typically does impart a fitness advantage (52).
The discovery that ZSWIM8 is required for TDMD, together with the established function of ZSWIM8 as a CRL substrate receptor, suggested a revised model for the molecular mechanism of TDMD (Fig. 5B). In this model, ZSWIM8 specifically recognizes the conformational changes that occur as the TDMD trigger extensively pairs to the miRNA 3′ region, thereby directing the polyubiquitination of AGO, causing it to be degraded by the proteasome. As AGO is proteolyzed, the miRNA is exposed to unknown cellular exonucleases and rapidly degraded, freeing the intact trigger, which resists degradation because it has either a cap and poly(A) tail (e.g., Cyrano) or other stabilizing features (e.g., HSUR1).
Although it differs substantially from the previous model of TDMD, in which tailing marks the miRNA for trimming and ultimately degradation by cellular nucleases without degradation of AGO (8, 12), the E3 model of TDMD is consistent with previous observations concerning both TDMD and TDTT. Like the previous model, the E3 model depends on conformational changes observed upon extensive pairing to the miRNA 3′ region, in which the miRNA 3′ terminus dissociates from its binding pocket in the PAZ domain of AGO and this domain rotates as the channel within the protein widens so that it can accommodate the additional pairing (8). However, in the E3 model, the key consequence of these changes is to create a platform for ZSWIM8 association, thereby presenting a degron. Although these changes also expose the miRNA 3′ terminus for TDTT, tailing is not required for degradation in the E3 model, consistent with Cyrano-directed miR-7 degradation in the absence of PAPD4-catalyzed tailing (21) (fig. S2D). The E3 model also explains why a specific cellular nuclease required for TDMD has yet to be identified; as AGO is degraded, the exposed miRNA would be the substrate of multiple exonucleases acting redundantly. Finally, release of the intact trigger explains why Cyrano can act with multiple turnover (21).
Additional support for the E3 model of TDMD
To investigate a role for other components of the ZSWIM8 CRL in TDMD, we knocked down ELOB and ELOC. ELOB and ELOC had respectively scored in the top 0.2% and 12% in our CRISPRi screen (Fig. 1E), but neither was selected as a candidate for secondary screening. Knockdown of each of these adaptor proteins caused miR-7 accumulation to increase, with tailing and trimming resembling that observed after knocking down ZSWIM8 (Fig. 6A and fig. S6A). Likewise, knockdown of the gene encoding Cullin 3 caused increased miR-7 accumulation, suggesting that this protein is also a component of the ZSWIM8 CRL (fig. S6A). As also predicted by the model, general inhibition of ubiquitination activity by TAK-243 (53) resulted in the increased accumulation of miR-7 relative to that of a control miRNA (miR-16) (Fig. 6B). Likewise, inhibition of neddylation, which is required for Cullin-mediated ubiquitination, by MLN4924 (54) specifically increased miR-7 accumulation, as did inhibition of proteasome-mediated proteolysis by MG-132 (Fig. 6B). Furthermore, the effects of these inhibitors on miR-7 entirely depended on ZSWIM8 function. Thus, ZSWIM8 acts as a component of a CRL to promote TDMD via the ubiquitin–proteasome system.
Fig. 6. Additional support for the E3 model of TDMD.
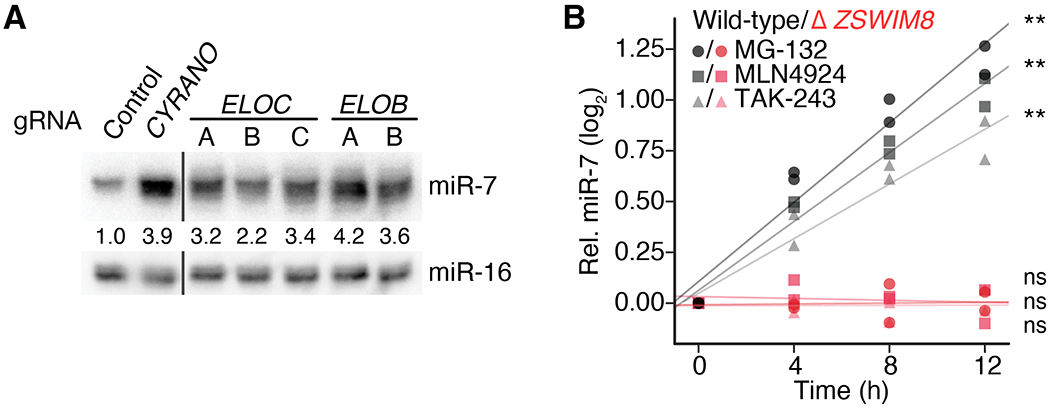
(A) The influence of ELOB and ELOC on miR-7 levels in K562i cells. Shown is an RNA blot measuring miR-7 and miR-16 levels in cells expressing either a non-targeting control gRNA or a gRNA targeting either CYRANO, ELOB, or ELOC for CRISPRi-mediated knockdown (fig. S5A). Omitted irrelevant lanes are marked by black lines. Otherwise, as in Fig. 1F. (B) Importance of the ubiquitin–proteasome system and the Cullin–RING ligase activity to TDMD. Shown are analyses of RNA blots that measured miR-7 and miR-16 levels after inhibitor treatment of either wild-type (gray) or knockout (Δ ZSWIM8, red) clonal K562i lines (table S1A). For each time point of each biological replicate, the signal for miR-7 was normalized to that of miR-16 and then to the miR-7 signal at 0 h. Lines show the least-squares fit to the results of each treatment (**, p < 0.001; ns, not significant). MG-132 inhibits the 26S proteasome; MLN4924 inhibits Nedd8-activating enzyme (NAE); TAK-243 inhibits ubiquitin-activating enzyme (UAE/E1). n = 2 biological replicates.
Another feature of the E3 model is the notion that TDMD does not depend on TDTT. To investigate this idea, we examined the effect of modifying the terminal 2′-OH of miR-7 with a methyl group, which inhibits miRNA tailing and trimming (12). Although terminal 2′-O-methylation reduced tailing and trimming of miR-7, it did not reduce the rate of ZSWIM8-dependent miR-7 degradation, which indicated that, as proposed by the model, TDMD does not depend on TDTT (fig. S6B).
To investigate the proposal that the ZSWIM8 CRL polyubiquitinates AGO, we co-immunoprecipitated stabilized polyubiquitinated proteins from both wild-type and zswim8-knockout S2 cells using tagged TUBE (tandem ubiquitin-binding entity) protein (55) that we had expressed ectopically in the cells. Ago1 protein (the Drosophila AGO ortholog that primarily associates with miRNAs) was not detected by immunoblot analysis of the pulldown samples (fig. S6C), which was attributable, at least in part, to the small fraction of Ago1 loaded with TDMD substrates. Nonetheless, high-throughput sequencing of small RNAs from the polyubiquitinated and input fractions revealed significant enrichment of Zswim8-sensitive miRNAs in the polyubiquitinated sample from wild-type cells compared to that from Zswim8-knockout cells (Fig. 7A). Moreover, a strong correlation (r2 = 0.82) was observed between Zswim8 sensitivity and differential TUBE enrichment (Fig. 7A). Thus, ZSWIM8 causes preferential association of TDMD substrates with polyubiquitinated proteins (fig. S6D)—a key feature of the E3 model of TDMD.
Fig. 7. Evidence for AGO polyubiquitination.
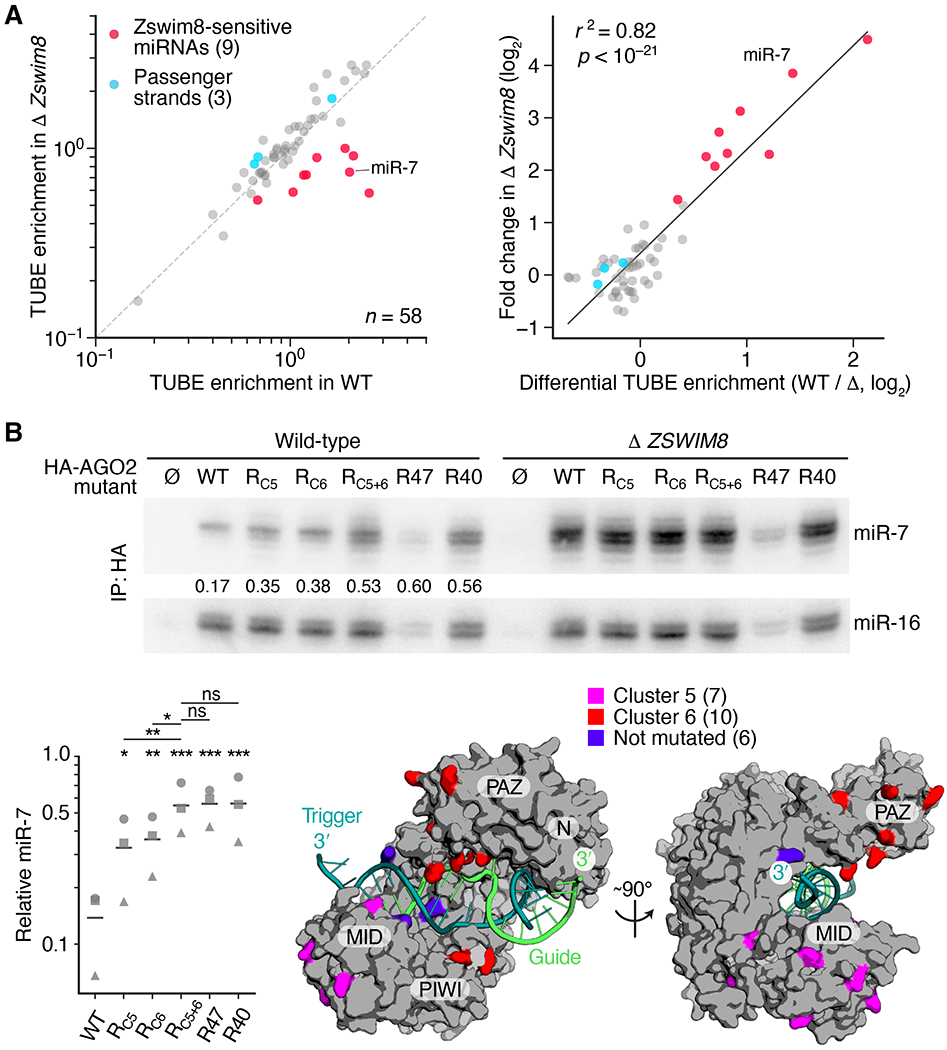
(A) A physical association between TDMD substrates and polyubiquitinated proteins in S2 cells. On the left are enrichments of miRNAs co-purifying with polyubiquitinated proteins, as measured by sRNA-seq of TUBE pulldown and input samples. Results are shown for all miRNAs passing the threshold required for reliable quantification, which included nine ZSWIM8-sensitive miRNAs (red) and three of their passenger strands (blue), plotting mean enrichment in a clonal CG34401-knockout S2 line (Δ Zswim8) as a function of mean enrichment in an analogously derived clonal wild-type line (WT) (table S1A). n = 3 biological replicates. On the right is the relationship between Zswim8 sensitivity and the mean enrichment observed in TUBE pulldowns from wild-type cells (WT) divided by that observed from Δ Zswim8 cells (Δ). The line shows the least-squares fit to all data (r2, coefficient of determination; p value indicated). (B) The importance of AGO2 surface lysines for TDMD. On top is an RNA blot measuring miR-7 and miR-16 levels in immunoprecipitations from either wild-type or Δ ZSWIM8 K562i clonal cell lines (table S1A) expressing the indicated 3XHA-tagged protein (∅, HA-tagged GFP; WT, HA-tagged AGO2; RX, AGO2 variant with lysine-to-arginine substitutions described in the text). For each AGO2 variant, the ratio of miR-7:miR-16 observed in a clonal wild-type cell line was normalized to the mean of that observed in three clonal Δ ZSWIM8 cell lines, and these normalized ratios are shown within each wild-type lane and plotted at the bottom left (squares). Circles and triangles show results of replicates performed with two other clonal lines (means, horizontal lines; *, p < 0.05; **, p < 0.01; ***, p < 0.001; ns, not significant, 2-way ANOVA followed by Tukey’s multiple-comparisons test). At the bottom right is human AGO2 in the TDMD conformation (PDB ID: 6MDZ, (8)), highlighting cluster 5, cluster 6, and unsubstituted lysines. The miRNA (green), TDMD trigger (teal), and AGO domains (N, PAZ, MID, and PIWI) are indicated.
To test further the idea that these polyubiquitinated proteins were AGO proteins, as would be expected if the ZSWIM8 CRL directly targets AGO proteins during TDMD, we examined the effects of mutating AGO lysines that might serve as sites of ubiquitination. Tagged AGO2 variants possessing lysine-to-arginine substitutions were ectopically expressed in K562i cells, and levels of both miR-7 and a control miRNA (miR-16) that co-immunoprecipitated with each variant were measured. We reasoned that if critical sites of ubiquitination were substituted, then TDMD would be slowed and the relative level of co-purifying miR-7 would increase. Of the 53 lysines in human AGO2, 47 were substituted (table S2). The six others, which included three that contact the miRNA 5′ phosphate and three that contact either the miRNA or its target within the seed region (8), were not substituted out of concern that they might be required for binding of the miRNA or its targets.
When no lysines were substituted (WT), the ratio of miR-7:miR-16 co-purifying from wild-type cells was substantially lower than that observed in ZSWIM8-knockout cells (Fig. 7B), as expected from our analyses of cellular miRNA levels (e.g., Fig. 1F). When the 47 lysines were simultaneously substituted (variant R47), the ratio of miR-7:miR-16 co-purifying from wild-type cells increased to more closely resemble that observed in ZSWIM8-knockout cells, which indicated that at least one of the 47 substituted lysines was required for efficient degradation of miR-7 (Fig. 7B). The absolute amount of co-purifying miRNAs substantially decreased in this highly substituted variant, which was attributed to reduced generic miRNA association rather than reduced production or stability of the protein because this and other lysine-substitution variants each accumulated to a level resembling that of wild-type AGO2 (Fig. S7A).
Further analyses examined the effects of substituting clusters of lysines mapping to non-overlapping regions of the protein surface (clusters 1–6, fig. S7B, and table S2). These analyses identified a cluster of seven substitutions (cluster 1) in R47 that when reverted to lysines (variant R40) restored miRNA association without restoring miR-7 degradation (Fig. 7B). Furthermore, the lysines required for efficient miR-7 degradation mapped to clusters 5 and 6 (Fig. 7B, fig. S7C, variants RC5, RC6, and RC5+6) yet were not restricted to 10 lysines previously reported to be ubiquitinated (56), which had no effect on miR-7 when substituted (fig. S7C, variant R10). Thus, multiple lysines on the AGO2 surface—at least one within cluster 5 and at least one within cluster 6—are important for TDMD. Most importantly, the ability of these lysine-to-arginine substitutions to phenocopy the ZSWIM8 knockout with respect to the specific buildup of miR-7, a TDMD substrate, supported our proposal that the ZSWIM8 CRL mediates TDMD through the ubiquitination of AGO. Accordingly, we renamed CG34401 (the Drosophila ZSWIM8 ortholog) Dorado (Dora), after a fish that preys on octopuses from the Argonauta genus, following the precedent of the naming of Iruka (Iru), an E3 ubiquitin ligase that targets empty Ago1 (57).
Implications
One of the more surprising features of the E3 mechanism is that it decouples TDMD from TDTT. Despite this uncoupling, we anticipate some interplay between TDTT and TDMD. To the extent that these two phenomena act on overlapping substrates they might compete. Moreover, because slight sequence differences at the miRNA 3′ end can substantially influence the efficiency of TDMD (8), some TDTT-mediated 3′-end modifications are expected to either increase or decrease TDMD efficacy.
Our analyses showing that loss of ZSWIM8 caused increased accumulation of 32 miRNAs in MEFs, neurons, or cancer cell lines substantially expanded the implied scope of endogenous TDMD in mammalian cells, and analogous experiments in Drosophila cells and nematodes extended this scope to diverse bilaterian animals. Nonetheless, loss of ZSWIM8 did not influence accumulation of most miRNAs in each of these contexts, which suggested that in most contexts, most miRNAs, which have relatively long half-lives, turn over through another mechanism. Supporting this conclusion, the half-life of AGO2 protein is substantially longer than that of bulk miRNA, implying that for most miRNA–AGO complexes, the miRNA decays first and the protein is recycled (4)—a series of events incompatible with that of the E3 TDMD mechanism. Nonetheless, our results indicate that most relatively short-lived miRNAs of each cell type decay through the E3 mechanism.
To the extent that different subsets of miRNAs are short-lived in different cell types, the actual scope of TDMD might be considerably broader than that implied from our study of just a few cell types. In this scenario, different transcripts that trigger TDMD for different miRNAs would be expressed in different cell types, carving away accumulation of specific miRNAs in specific cell types or in response to specific stimuli, thereby creating a more complex and more dynamic landscape of miRNA levels than that which would have been achieved by differential miRNA production alone. Perhaps adding to this complexity are three potential paralogs of ZSWIM8 in mammals (ZSWIM4–6). Although these are more related to each other than to ZSWIM8 and appear to be more lowly expressed than is ZSWIM8 (median TPM 4.3, 3.3, 12.9, and 45.2 for ZSWIM4, −5, −6, and −8 respectively) (43), we are intrigued by the notion that one or more of these potential paralogs might also mediate TDMD, perhaps recognizing unique miRNA–AGO–trigger conformations.
We suspect that each ZSWIM8-sensitive miRNA has at least one target that specifies its degradation. Identifying these TDMD triggers is required for validating the expanded scope of TDMD implied by our results and critical for exploring the biological roles of the degradation that is specified. The discovery of ZSWIM8 as an essential mediator of TDMD provides a powerful biochemical and genetic tool for identifying these transcripts that trigger TDMD in different cellular contexts and thereby sculpt miRNA levels throughout the animal.
Methods summary
Gene knockdowns were achieved through CRISPRi. For all knockdown experiments, excluding the CRISPRi screen, stable polyclonal cell lines were generated by transducing K562i cells with a lentivirus that expressed a gRNA and a puromycin-resistance marker under constitutive promoters. Both selection for puromycin resistance and induction of KRAB-dCas9 began 24 h after transduction and continued for at least 10 d before cells were harvested. Knockdown efficiency was monitored using either RT-qPCR or RNA-seq. Polyclonal knockouts were achieved by transduction with a lentivirus that expressed Cas9, a gRNA, and a puromycin-resistance marker. Selection for puromycin resistance began 24 h after transduction and continued for at least 10 d before cells were harvested. Polyclonal knockout efficiency was determined by sequencing of a PCR amplicon spanning the targeted region. Clonal knockout cell lines were generated by expression of Cas9 and a gRNA from a transiently transfected (or nucleofected) plasmid, followed by single-cell sorting and then culture and screening of clones.
For RNA-blot analyses, total RNA was resolved on a 15% polyacrylamide denaturing gel, blotted to a membrane, and then chemically crosslinked to the membrane. Membranes were probed with either a 32P-radiolabeled locked nucleic acid (LNA) corresponding to miR-7 or a 32P-radiolabeled DNA corresponding to another miRNA. The radioactivity was then visualized and quantified using a phosphorimager. For sRNA-seq, sRNAs were gel-purified from each sample and 3′ and 5′ adaptors were sequentially ligated to these sRNAs, which enabled reverse transcription, amplification, and high-throughput sequencing. To reduce biases and enhance reproducibility, the adapter-ligation was performed using an optimized protocol, in which efficiency was monitored using radiolabeled internal standards. The details of each of these methods are described in the supplementary materials.
Supplementary Material
Acknowledgments:
We thank S. McGeary and X. Wu for helpful discussions; A. Liu, A. Whipple, J. Steitz, T. Pham, J. Weissman, P. Sharp, D. Trono, D. Sabatini, S. Lindquist, M. Taipale, Y. Doyon, and Y. Saeki for cell lines and plasmids; Y. Jin and S. Mitani for C. elegans strains; the Whitehead Functional Genomics Core for support with the CRISPRi screen; and the Whitehead Genome Technology Core for sequencing.
Funding:
This work was supported by NIH grant GM118135 (D.P.B.) and a National Science Foundation pre-doctoral GRFP fellowship (E.R.K.). D.H.L. is an HHMI fellow of the Damon Runyon Cancer Research Foundation (DRG-2345-18). D.P.B. is an investigator of the Howard Hughes Medical Institute.
Footnotes
Data and materials availability:
Sequencing data and associated analyses were deposited in Gene Expression Omnibus (accession number GSE160304), and all plasmids generated in this study were submitted to Addgene.
References and Notes:
- 1.Bartel DP, Metazoan MicroRNAs. Cell 173, 20–51 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Jonas S, Izaurralde E, Towards a molecular understanding of microRNA-mediated gene silencing. Nat Rev Genet 16, 421–433 (2015). [DOI] [PubMed] [Google Scholar]
- 3.Friedman RC, Farh KK-H, Burge CB, Bartel DP, Most mammalian mRNAs are conserved targets of microRNAs. Genome Res 19, 92–105 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kingston ER, Bartel DP, Global analyses of the dynamics of mammalian microRNA metabolism. Genome Res 29, 1777–1790 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Reichholf B et al. , Time-Resolved Small RNA Sequencing Unravels the Molecular Principles of MicroRNA Homeostasis. Mol Cell 75, 756–768.e757 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eisen TJ et al. , The Dynamics of Cytoplasmic mRNA Metabolism. Mol Cell 77, 786–799.e710 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Sheu-Gruttadauria J, MacRae IJ, Structural Foundations of RNA Silencing by Argonaute. J Mol Biol 429, 2619–2639 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Sheu-Gruttadauria J et al. , Structural Basis for Target-Directed MicroRNA Degradation. Mol Cell 75, 1243–1255.e1247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Rissland OS, Hong S-J, Bartel DP, MicroRNA destabilization enables dynamic regulation of the miR-16 family in response to cell-cycle changes. Mol Cell 43, 993–1004 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krutzfeldt J et al. , Specificity, duplex degradation and subcellular localization of antagomirs. Nucleic Acids Res 35, 2885–2892 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Krutzfeldt J et al. , Silencing of microRNAs in vivo with ‘antagomirs’. Nature 438, 685–689 (2005). [DOI] [PubMed] [Google Scholar]
- 12.Ameres SL et al. , Target RNA-directed trimming and tailing of small silencing RNAs. Science 328, 1534–1539 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Baccarini A et al. , Kinetic analysis reveals the fate of a microRNA following target regulation in mammalian cells. Curr Biol 21, 369–376 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie J et al. , Long-term, efficient inhibition of microRNA function in mice using rAAV vectors. Nat Methods 9, 403–409 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cazalla D, Yario T, Steitz JA, Steitz J, Down-regulation of a host microRNA by a Herpesvirus saimiri noncoding RNA. Science 328, 1563–1566 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Libri V et al. , Murine cytomegalovirus encodes a miR-27 inhibitor disguised as a target. Proc Natl Acad Sci U S A 109, 279–284 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Marcinowski L et al. , Degradation of cellular mir-27 by a novel, highly abundant viral transcript is important for efficient virus replication in vivo. PLoS Pathog 8, e1002510 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lee S et al. , Selective degradation of host MicroRNAs by an intergenic HCMV noncoding RNA accelerates virus production. Cell Host Microbe 13, 678–690 (2013). [DOI] [PubMed] [Google Scholar]
- 19.Bitetti A et al. , MicroRNA degradation by a conserved target RNA regulates animal behavior. Nat Struct Mol Biol 25, 244–251 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Ghini F et al. , Endogenous transcripts control miRNA levels and activity in mammalian cells by target-directed miRNA degradation. Nat Commun 9, 3119 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kleaveland B, Shi CY, Stefano J, Bartel DP, A Network of Noncoding Regulatory RNAs Acts in the Mammalian Brain. Cell 174, 350–362.e317 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartel DP, MicroRNAs: target recognition and regulatory functions. Cell 136, 215–233 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.de la Mata M et al. , Potent degradation of neuronal miRNAs induced by highly complementary targets. EMBO Rep 16, 500–511 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Denzler R et al. , Impact of MicroRNA Levels, Target-Site Complementarity, and Cooperativity on Competing Endogenous RNA-Regulated Gene Expression. Mol Cell 64, 565–579 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Haas G et al. , Identification of factors involved in target RNA-directed microRNA degradation. Nucleic Acids Res 44, 2873–2887 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pawlica P, Sheu-Gruttadauria J, MacRae IJ, Steitz JA, How Complementary Targets Expose the microRNA 3’ End for Tailing and Trimming during Target-Directed microRNA Degradation. Cold Spring Harb Symp Quant Biol, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Heo I et al. , Lin28 mediates the terminal uridylation of let-7 precursor MicroRNA. Mol Cell 32, 276–284 (2008). [DOI] [PubMed] [Google Scholar]
- 28.Vanacova S et al. , A new yeast poly(A) polymerase complex involved in RNA quality control. PLoS Biol 3, e189 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.LaCava J et al. , RNA degradation by the exosome is promoted by a nuclear polyadenylation complex. Cell 121, 713–724 (2005). [DOI] [PubMed] [Google Scholar]
- 30.Wyers F et al. , Cryptic pol II transcripts are degraded by a nuclear quality control pathway involving a new poly(A) polymerase. Cell 121, 725–737 (2005). [DOI] [PubMed] [Google Scholar]
- 31.Rissland OS, Norbury CJ, Decapping is preceded by 3’ uridylation in a novel pathway of bulk mRNA turnover. Nat Struct Mol Biol 16, 616–623 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lim J et al. , Uridylation by TUT4 and TUT7 marks mRNA for degradation. Cell 159, 1365–1376 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Mullen TE, Marzluff WF, Degradation of histone mRNA requires oligouridylation followed by decapping and simultaneous degradation of the mRNA both 5’ to 3’ and 3’ to 5’. Genes Dev 22, 50–65 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Song M-G, Kiledjian M, 3’ Terminal oligo U-tract-mediated stimulation of decapping. Rna 13, 2356–2365 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Li J, Yang Z, Yu B, Liu J, Chen X, Methylation protects miRNAs and siRNAs from a 3’-end uridylation activity in Arabidopsis. Curr Biol 15, 1501–1507 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Burroughs AM et al. , A comprehensive survey of 3’ animal miRNA modification events and a possible role for 3′ adenylation in modulating miRNA targeting effectiveness. Genome Res 20, 1398–1410 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Mansur F et al. , Gld2-catalyzed 3’ monoadenylation of miRNAs in the hippocampus has no detectable effect on their stability or on animal behavior. Rna 22, 1492–1499 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Shukla S, Bjerke GA, Muhlrad D, Yi R, Parker R, The RNase PARN Controls the Levels of Specific miRNAs that Contribute to p53 Regulation. Mol Cell 73, 1204–1216.e1204 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gilbert LA et al. , CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Horlbeck MA et al. , Compact and highly active next-generation libraries for CRISPR-mediated gene repression and activation. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Li W et al. , MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Gilbert LA et al. , Genome-Scale CRISPR-Mediated Control of Gene Repression and Activation. Cell 159, 647–661 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Consortium G. T. et al. , The Genotype-Tissue Expression (GTEx) project. Nat Genet 45, 580–585 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wang Z et al. , The EBAX-type Cullin-RING E3 ligase and Hsp90 guard the protein quality of the SAX-3/Robo receptor in developing neurons. Neuron 79, 903–916 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Stoeckius M et al. , Large-scale sorting of C. elegans embryos reveals the dynamics of small RNA expression. Nat Methods 6, 745–751 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Schreiner WP et al. , Remodeling of the Caenorhabditis elegans non-coding RNA transcriptome by heat shock. Nucleic Acids Res 47, 9829–9841 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Enchev RI, Schulman BA, Peter M, Protein neddylation: beyond cullin-RING ligases. Nat Rev Mol Cell Biol 16, 30–44 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hua Z, Vierstra RD, The cullin-RING ubiquitin-protein ligases. Annu Rev Plant Biol 62, 299–334 (2011). [DOI] [PubMed] [Google Scholar]
- 49.Mahrour N et al. , Characterization of Cullin-box sequences that direct recruitment of Cul2-Rbx1 and Cul5-Rbx2 modules to Elongin BC-based ubiquitin ligases. J Biol Chem 283, 8005–8013 (2008). [DOI] [PubMed] [Google Scholar]
- 50.Makarova KS, Aravind L, Koonin EV, SWIM, a novel Zn-chelating domain present in bacteria, archaea and eukaryotes. Trends Biochem Sci 27, 384–386 (2002). [DOI] [PubMed] [Google Scholar]
- 51.Yamamoto S et al. , A drosophila genetic resource of mutants to study mechanisms underlying human genetic diseases. Cell 159, 200–214 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.DepMap, DepMap 20Q2 Public (2020) p. , doi: 10.6084/m9.figshare.12280541.v3. [DOI] [Google Scholar]
- 53.Hyer ML et al. , A small-molecule inhibitor of the ubiquitin activating enzyme for cancer treatment. Nat Med 24, 186–193 (2018). [DOI] [PubMed] [Google Scholar]
- 54.Soucy TA et al. , An inhibitor of NEDD8-activating enzyme as a new approach to treat cancer. Nature 458, 732–736 (2009). [DOI] [PubMed] [Google Scholar]
- 55.Yoshida Y et al. , A comprehensive method for detecting ubiquitinated substrates using TR-TUBE. Proc Natl Acad Sci U S A 112, 4630–4635 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Hornbeck PV et al. , PhosphoSitePlus: a comprehensive resource for investigating the structure and function of experimentally determined post-translational modifications in man and mouse. Nucleic Acids Res 40, D261–270 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Kobayashi H, Shoji K, Kiyokawa K, Negishi L, Tomari Y, Iruka Eliminates Dysfunctional Argonaute by Selective Ubiquitination of Its Empty State. Mol Cell 73, 119–129.e115 (2019). [DOI] [PubMed] [Google Scholar]
- 58.Piovesan D et al. , MobiDB 3.0: more annotations for intrinsic disorder, conformational diversity and interactions in proteins. Nucleic Acids Res 46, D471–D476 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Berezin C et al. , ConSeq: the identification of functionally and structurally important residues in protein sequences. Bioinformatics 20, 1322–1324 (2004). [DOI] [PubMed] [Google Scholar]
- 60.Rideout WM 3rd et al. , Generation of mice from wild-type and targeted ES cells by nuclear cloning. Nat Genet 24, 109–110 (2000). [DOI] [PubMed] [Google Scholar]
- 61.Brenner S, The genetics of Caenorhabditis elegans. Genetics 77, 71–94 (1974). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Brinkman EK, Chen T, Amendola M, van Steensel B, Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bassett AR, Tibbit C, Ponting CP, Liu J-L, Mutagenesis and homologous recombination in Drosophila cell lines using CRISPR/Cas9. Biol Open 3, 42–49 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Adelmann CH, Wang T, Sabatini DM, Lander ES, Genome-Wide CRISPR/Cas9 Screening for Identification of Cancer Genes in Cell Lines. Methods Mol Biol 1907, 125–136 (2019). [DOI] [PubMed] [Google Scholar]
- 65.Ashburner M et al. , Gene ontology: tool for the unification of biology. The Gene Ontology Consortium. Nat Genet 25, 25–29 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Dobin A et al. , STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Anders S, Pyl PT, Huber W, HTSeq--a Python framework to work with high-throughput sequencing data. Bioinformatics 31, 166–169 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Seitz H, Ghildiyal M, Zamore PD, Argonaute loading improves the 5’ precision of both MicroRNAs and their miRNA* strands in flies. Curr Biol 18, 147–151 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Agarwal V, Bell GW, Nam J-W, Bartel DP, Predicting effective microRNA target sites in mammalian mRNAs. Elife 4, (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Jan CH, Friedman RC, Ruby JG, Bartel DP, Formation, regulation and evolution of Caenorhabditis elegans 3’UTRs. Nature 469, 97–101 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Fromm B et al. , MirGeneDB 2.0: the metazoan microRNA complement. Nucleic Acids Res 48, D1172 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.McGeary SE et al. , The biochemical basis of microRNA targeting efficacy. Science 366, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Schirle NT, Sheu-Gruttadauria J, MacRae IJ, Structural basis for microRNA targeting. Science 346, 608–613 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hauptmann J et al. , Biochemical isolation of Argonaute protein complexes by Ago-APP. Proc Natl Acad Sci U S A 112, 11841–11845 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Kim B, Jeong K, Kim VN, Genome-wide Mapping of DROSHA Cleavage Sites on Primary MicroRNAs and Noncanonical Substrates. Mol Cell 66, 258–269.e255 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Sequencing data and associated analyses were deposited in Gene Expression Omnibus (accession number GSE160304), and all plasmids generated in this study were submitted to Addgene.