

Abstract
A prominent finding of postmortem and molecular imaging studies on Alzheimer's disease (AD) is the accumulation of neuropathological proteins in brain regions of the default mode network (DMN). Molecular models suggest that the progression of disease proteins depends on the directionality of signaling pathways. At network level, effective connectivity (EC) reflects directionality of signaling pathways. We hypothesized a specific pattern of EC in the DMN of patients with AD, related to cognitive impairment. Metabolic connectivity mapping is a novel measure of EC identifying regions of signaling input based on neuroenergetics. We simultaneously acquired resting‐state functional MRI and FDG‐PET data from patients with early AD (n = 35) and healthy subjects (n = 18) on an integrated PET/MR scanner. We identified two distinct subnetworks of EC in the DMN of healthy subjects: an anterior part with bidirectional EC between hippocampus and medial prefrontal cortex and a posterior part with predominant input into medial parietal cortex. Patients had reduced input into the medial parietal system and absent input from hippocampus into medial prefrontal cortex (p < 0.05, corrected). In a multiple linear regression with unimodal imaging and EC measures (F 4,25 = 5.63, p = 0.002, r 2 = 0.47), we found that EC (β = 0.45, p = 0.012) was stronger associated with cognitive deficits in patients than any of the PET and fMRI measures alone. Our approach indicates specific disruptions of EC in the DMN of patients with AD and might be suitable to test molecular theories about downstream and upstream spreading of neuropathology in AD.
Keywords: default mode network, directional signaling, effective connectivity, energy metabolism, resting state, simultaneous PET/fMRI
1. INTRODUCTION
Alzheimer's disease (AD) is characterized by the regional accumulation of misfolded proteins that spread to widely distributed brain regions along disease progression. For decades, the temporal and spatial distribution of amyloid‐β plaques and tau tangles have been studied with postmortem neuropathology (Braak & Braak, 1991; Price & Morris, 1999). Recently, the topographic progression of disease proteins has been associated with macroscopic brain networks from magnetic resonance imaging (MRI; Buckner et al., 2005; Palop, Chin, & Mucke, 2006; Pievani, de Haan, Wu, Seeley, & Frisoni, 2011; Seeley, Crawford, Zhou, Miller, & Greicius, 2009). In patients with AD, functional (FC) and structural connectivity are primarily disrupted in the default mode network (DMN) spanning medial and lateral parietal regions, medial prefrontal cortex (MPFC) and hippocampus (Greicius, Srivastava, Reiss, & Menon, 2004; Hahn et al., 2013). We and others reported progressive FC disruption in the DMN, starting even in early stages of mild cognitive impairment and preclinical stages (Greicius et al., 2004; Lim et al., 2014; Nuttall, Pasquini, Scherr, & Sorg, 2016; Pasquini et al., 2015; Sorg et al., 2007; Zhou et al., 2010). Together, regions forming the DMN seem to affected early and most prominently in the progression of AD.
Several recent studies tested models of pathological spread in relation to DMN connectivity. Zhou et al. identified lateral parietal regions of the DMN as vulnerability hubs for pathological spread in AD (Zhou, Gennatas, Kramer, Miller, & Seeley, 2012). Similar coincidences were reported for DMN structural connectivity and atrophy (Raj, Kuceyeski, & Weiner, 2012), as well as for DMN FC, hypometabolism, and amyloid‐β burden (Drzezga et al., 2011; Iturria‐Medina et al., 2014; Myers et al., 2014; Pasquini et al., 2017; Scherr et al., 2018). Sepulcre et al. found evidence for network based distribution of both amyloid‐β and tau when relating neuropathological accumulation to gray matter tissue loss (Sepulcre et al., 2016). These findings suggest that distinct features of DMN connectivity are critical for neuropathology spread in AD, but the mechanism of spreading is still unknown.
Cellular and molecular models have proposed that the spatially cascading accumulation of neuropathological changes might be related to the signaling hierarchy in brain networks (Brettschneider, Del Tredici, Lee, & Trojanowski, 2015). According to one idea, misfolded disease proteins accumulate in a prion‐like fashion spreading downstream from an infected to a naïve neuron (Frost & Diamond, 2010). Another theory suggests spreading in an activity‐ or plasticity dependent manner (Mesulam, 2000; Palop & Mucke, 2010; Yamamoto et al., 2015). Together, these models suggest that the signaling directionality in the DMN might be a useful indicator of neuropathological spread in AD. Statistical approaches for fMRI data, such as Granger causality analysis (GCA) and dynamic causal modeling (DCM), exist to identify directional signaling, or effective connectivity (EC), in macroscopic brain networks. GCA revealed altered EC in DMN regions, showing both reductions in connection strength to the posterior cingulate cortex and stronger coupling of the MPFC with bilateral inferior parietal regions, as compared with healthy controls (Zhong et al., 2014). Using DCM, (Agosta et al., 2010) reported that AD patients had significantly reduced EC between the left and right primary somatomotor cortices as compared to healthy control participants.
We recently introduced metabolic connectivity mapping (MCM) as a novel, in vivo method to identify EC in large‐scale networks (Riedl et al., 2016) (Figure 1). According to cellular data from neuroenergetics, up to 75% of glucose metabolism is related to postsynaptic activity, that is, signaling input (Attwell & Iadecola, 2002; Attwell & Laughlin, 2001). We scaled this model to the macroscopic level and identified signaling input along given FC pathways by integrating simultaneously acquired fMRI and FDG‐PET data. In the current study, we applied MCM to the DMN and asked, whether EC is reduced in patients with early AD. First, we identified two DMN subsystems in healthy subjects, an anterior DMN with bidirectional EC between MPFC and hippocampus and a posterior DMN with directed signaling converging into medial parietal cortex (MPC). Both systems were linked only via unilateral EC from anterior into posterior DMN. In patients with early AD, distinct connections of these DMN‐subsystems were disrupted, offering systems level support for distinct molecular spreading theories of AD.
FIGURE 1.
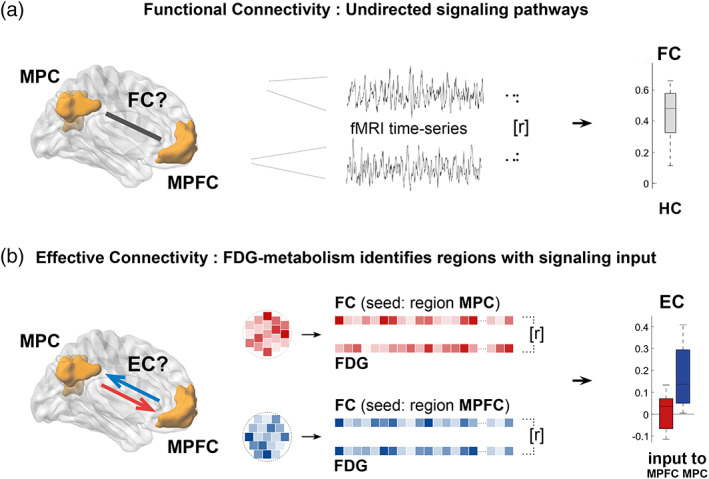
Novel measure of directional signaling in human brain networks derived from simultaneously acquired fMRI and FDG data. (a) Using fMRI only, functional connectivity (FC), the temporal correlation of fMRI‐based voxel time‐series (r), indicates undirected connectivity among macroscopic brain regions of the DMN (here: Medial parietal cortex (MPC) and medial prefrontal cortex (MPFC)). (b) Simultaneously acquired energy consumption from FDG‐PET allows for voxelwise correlation (r) of FC and FDG in each subject. Based on cellular models of neuroenergetics, a positive correlation in a given region indicates signaling input along a FC pathway (here, afferent EC into MPC [blue], but not into MPFC [red]). Applied to all FC pathways among DMN regions, this novel approach reveals directional signaling or effective connectivity (EC) in the DMN
2. METHODS AND MATERIALS
2.1. Subjects
We examined 18 matched healthy controls and 35 patients with early AD, including patients with mild cognitive impairment (MCI; n = 15) or mild dementia (n = 20) both due to AD (AD‐MCI, AD‐dementia; Albert et al., 2011; McKhann et al., 2011). The study was approved by the university ethics committee (Technische Universität München) in line with the institute's Human Research Committee guidelines. All participants provided informed consent. Patients were recruited from the Center for Cognitive Disorders at the Department of Psychiatry and Psychotherapy, healthy controls by word‐of‐mouth advertising. Further details about inclusion criteria can be found in Supporting Information.
2.2. Data acquisition
Scanning was performed on an integrated Siemens Biograph mMR scanner (Siemens, Erlangen, Germany) simultaneously acquiring PET‐ and MR‐imaging data using the vendor‐supplied 12‐channel phase‐array head coil. We simultaneously measured FDG activity and blood oxygenation level dependent (BOLD) signals of fMRI during resting condition. fMRI was acquired during the initial 10 min after bolus injection of FDG tracer reflecting highest dynamic of tracer kinetics, followed by structural MRI. An average PET image of saturated FDG distribution was reconstructed from list‐mode data 30–45 min post injection. Subjects were instructed to relax and keep their eyes closed, not to think of anything in particular, and to not fall asleep. Details about scanning parameters can be found in Supporting Information.
2.3. Data processing
2.3.1. Definition of DMN
We derived regions of the DMN from an independent sample of healthy controls (n = 31, mean age 25.4 years ± 3.1). Resting state fMRI data underwent the same preprocessing pipeline as patients and healthy controls (see below). Preprocessed data were analyzed via canonical group‐independent component analysis (see Supporting Information). The component representing the DMN was selected by visual inspection, statistically evaluated (one‐sample t test, p < 0.05 FWE corrected, cluster level > 100), and clusters in MPFC, MPC, left and right parietal cortices (L/RPC), and the hippocampus region (HPC) were defined as separate DMN regions for further analyses. One should note that HPC was only partly included in our DMN component (what is a typical result of ICA‐based network parcellation, see for example (Allen et al., 2011; Nuttall et al., 2016; Sorg et al., 2007). As previous studies identified the whole HPC as part of the DMN, for example, (Andrews‐Hanna, Reidler, Sepulcre, Poulin, & Buckner, 2010), we extended our HPC mask to a publicly available bilateral HPC identified by an automatic surface parcellation of healthy subjects (n = 39) using FreeSurfer (Fischl et al., 2002). Importantly, parahippocampal, and entorhinal cortices are assigned to separate regions in this parcellation atlas and were joined for our HPC mask. All regions‐of‐interest (ROI) were then transformed into individual subject space of patients and controls using the inverse transformation matrix from the normalization procedure. In the following, we used these individualized DMN ROIs to calculate regional metabolism, inter‐regional FC, and most importantly, ROI‐to‐ROI EC based on MCM.
2.3.2. Data preprocessing
We applied standardized preprocessing to fMRI and PET data as previously reported (for details see (Riedl et al., 2016). In brief, the mean FDG‐PET volume was coregistered to the mean EPI volume, resulting in a subject‐specific coregistered multimodal dataset.
For FC analysis, we additionally applied spatial normalization, smoothing, and movement artifact control. In brief, fMRI volumes were normalized to a standard template of the Montréal Neurological Institute (MNI template), spatially smoothed using a Gaussian kernel of full‐width‐half‐maximum of 6 mm, and controlled for excessive head motion (exclusion criteria: cumulative motion translation or rotation >3 mm or 3° and mean point‐to‐point translation or rotation >0.15 mm or 0.1°); in addition movement‐induced artifacts across groups were controlled by comparing movement‐ and signal quality parameters across groups such as frame‐wise displacement (Power, Barnes, Snyder, Schlaggar, & Petersen, 2012), the root‐mean‐square of translational parameters (Van Dijk, Sabuncu, & Buckner, 2012), and signal‐to‐noise ratio of functional MRI data differed across groups (anova, p > 0.05 each). Further details about these preprocessing steps can be found in Supporting Information.
We performed MCM in native space, therefore omitting spatial normalization during preprocessing. Additionally, standard space tissue‐probability maps of gray, white matter and CSF were warped onto single‐subject T1‐weighted images and stored as masks for later use. In order to reduce the partial volume effects during coregistration of fMRI‐ and FDG‐images we first resampled both datasets to common voxel dimensions of 2 × 2 × 2 mm3 and applied spatial smoothing with a kernel of 4 mm. MCM was limited to gray matter voxels using a range of probability thresholds (0.4–0.7) which were obtained in the segmentation process of the T1 weighted images (see Supporting Information Tables S4 and S5 for control analyses).
2.3.3. Region‐to‐region FC
We performed a region‐to‐region FC‐analysis (FCmean) using the REST toolbox (Song et al., 2011). In a pairwise manner, we temporally correlated the mean time courses of all DMN‐regions. Pearson's correlation coefficients were r‐to‐z‐Fisher‐transformed. For each group and region‐region pair, FC was evaluated by one‐sample t tests, and group comparisons (i.e., healthy controls vs. patients [collapsed], and vs. AD‐MCI and AD‐dementia separately) were performed using two‐sample t tests and anova. We indicated all alpha levels surpassing a Bonferroni correction for multiple testing at p < 0.05.
2.3.4. Regional metabolism
After coregistration of the mean FDG‐PET volume to the mean fMRI volume, we scaled PET volume data to normalized FDG activity by whole brain FDG uptake values, and then resampled both datasets to common voxel dimensions of 2 × 2 × 2 mm3 (for more details, see (Riedl et al., 2016). Then we calculated regional metabolism as the averaged FDG‐activity across voxels for each DMN‐region (FDGamp). Data from the control and patient groups (collapsed and AD‐MCI and AD‐dementia groups separately) were compared using two‐sample t tests and anovas (p < 0.05, Bonferroni corrected).
2.3.5. MCM
We recently introduced MCM as a measure of intrinsic EC integrating voxel‐wise values of FC (FCvox) and FDG (FDGvox) activity in single subject space (Riedl et al., 2016). Based on a cellular model of neuroenergetics, we interpret a positive spatial correlation between FCvox and FDGvox in a given region as signaling into this area (target region). Both voxel profiles are extracted in native space and no spatial smoothing is applied. FCvox is calculated for each voxel in the target area as the temporal correlation between each voxel's time series and the averaged time series of a distant (source) region. FDGvox in the target area is derived from the PET‐image. We calculated the EC among all regions of the DMN mask back‐projected into each individual's native space. The statistical significance of a directional signaling pathway among DMN‐regions was calculated using one sample t tests for each group separately (p < 0.05, Bonferroni corrected). Group differences (i.e., healthy controls vs. patients [collapsed], and vs. AD‐MCI and AD‐dementia separately) in the strength of EC were evaluated using two‐sample t tests and ANOVA (p < 0.05, Bonferroni corrected). The variance of both FC (FCvar) and FDG (FDGvar) in a region was calculated as the average squared deviation of all voxel values from the mean value in that region.
2.3.6. Integrated analysis
In a final step, we tested whether EC impairments were associated with cognitive deficits in the patient group. Therefore, we performed a multiple linear regression analysis, with MMSE as dependent variable and the following independent variables: EC, FDGamp, and FCmean. Independent variables were regionally specified due to group difference results. Additional control variables of no‐interest were age, gender, and global gray matter volume. MMSE was chosen as simple reliable index for cognitive impairment for the range from MCI to dementia.
3. RESULTS
3.1. Spatially distinct patterns of reduced metabolism and reduced FC in the DMN of patients with early AD
We first identified major regions of the DMN in an independent sample of healthy controls (Figure 2). These regions were then used as templates to calculate FDG uptake and FC in our study cohort of patients with early AD and matched controls (Table 1). We identified changes of regional energy metabolism in the DMN of patients, both for the collapsed group including AD‐MCI and AD‐dementia, and for each patient group separately. For the collapsed group, two‐sample t tests revealed a significant decrease of FDGamp in all parietal regions and in the hippocampus of patients (MPC: t 51 = −6.61, p = 8 × 10−8; LPC: t 51 = −7.27, p = 2 × 10−9; RPC: t 51 = −7.36, p = 1 × 10−9; HPC: t 51 = −4.89, p = 1 × 10−5; all p < 0.05 corrected), but not in the prefrontal cortex (MPFC: t 51 = −1.82, p = 0.09). The drop in metabolic activity ranged from 17–30% (see Table S1A for full statistics, and Figure 3(a)). We found similar results in separate groups of AD‐MCI and AD‐dementia with an additional trend toward reduced metabolism in the MPFC of the AD‐dementia group (Supporting Information Table S1B).
FIGURE 2.
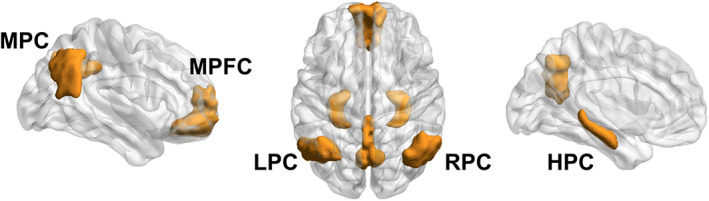
Regions of the DMN as defined in the present study. Key regions of the DMN were derived from an independent component analysis of an independent dataset of healthy subjects and used as regions‐of‐interest for all subsequent analyses of connectivity in this manuscript. Additionally, we added a HPC mask from a publicly available surface parcellation atlas (see Section 2). HPC = Bilateral hippocampus; LPC = left parietal cortex; MPC = medial parietal cortex; MPFC = medial prefrontal cortex; RPC = right parietal cortex
TABLE 1.
Demographics of patients with early AD and healthy controls
| Healthy controls | AD‐MCI | AD‐dementia | p value | |
|---|---|---|---|---|
| n | 18 | 15 | 20 | ‐ |
| Age (y, mean) | 63.2 (8.5) | 66.9 (10.1) | 65.7 (6.1) | >0.05 |
| Sex (female) | 8 | 8 | 10 | >0.05 |
| Education (y, mean) | 10.3 (1.4) | 10.1 (2.0) | 9.7 (1.6) | >0.05 |
| MMSE score (mean) | 29.4 (1.1) | 27.2 (2.3) | 21.9 (5.4) | 0.002 |
| CERAD total (mean) | 87.2 (8.9) | 69.2 (11.2) | 58.0 (13.5) | 0.037 |
p‐values significant for p<0.05.
CERAD = consortium to establish a registry for Alzheimer's disease; MCI = mid cognitive impairment; MMSE = mini‐mental state examination; standard deviations in brackets; statistics derived from ANOVA, except for chi squared test applied to sex.
FIGURE 3.
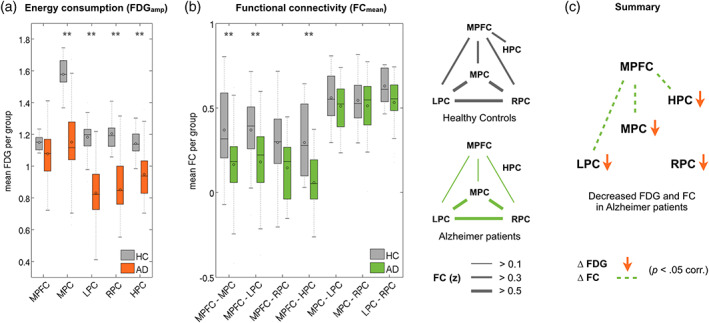
Energy metabolism and functional connectivity in the DMN. (a) Bar plots indicate decreased energy metabolism (FDGamp) in medial (MPC), left (LPC), and right (RPC) parietal cortex, and in hippocampus (HPC) of patients with AD (orange) compared to healthy controls (gray). (b) Left: Bar plots indicate decreased pairwise functional connectivity (FCmean) of fronto‐parietal and fronto‐hippocampal connections in the patient group (green) compared to healthy controls (gray). Right: Graphical illustration of significant FC among all DMN regions in healthy controls and patients. (c) Summary illustrating significantly decreased energy metabolism (orange arrows) and FC (dotted lines) in the DMN of patients with AD. **p < 0.05 Bonferroni corrected. See Supporting Information Tables S1 and S2 for full statistics
We then studied pairwise FC pathways among all DMN‐regions. One‐sample t tests (p < 0.05 corrected) indicated significant FCmean among all pairs of isocortical DMN regions, both in healthy controls and in patients; significant hippocampal FCmean was found only with MPFC in the healthy control group (Table S2A, Figure 3(b)). In group comparisons using two‐sample t tests, all functional connections between the medial prefrontal and all parietal regions were reduced in patients with early AD (MPC: t 51 = −2.94, p = 0.005; LPC: t 51 = −2.84, p = 0.007; HPC: t 51 = −4.75, p = 0.002; all p < 0.05 corrected; RPC: t 51 = −2.75, p = 0.008). The FCmean of parieto‐parietal connections did not differ between patients and controls (Table S2A, Figure 3(b)). In patients with AD‐dementia, additional reductions of FCmean were found between parietal regions (Table S2B). In summary, we found spatially distinct patterns of reduced energy metabolism and reduced FC in patients with AD. Lower energy metabolism occurred locally in hippocampal and parietal regions, while FC was diminished for long‐range connections between parietal and MPFCs (Figure 3(c)).
3.2. Effective connectivity among DMN regions is severely disrupted in patients
In a next step, we studied EC among DMN regions. In healthy controls, one‐sample t tests revealed two subnetworks of reciprocal EC within the DMN (Supporting Information Table S3A, Figure 4(a)). In the anterior part of the DMN, significant unidirectional EC occurred from the hippocampus into MPFC (with a trend to bidirectional EC, see Supporting Information Table S3A); in posterior DMN, unidirectional EC predominated from lateral into MPC. The only link between anterior and posterior DMN was unidirectional EC from MPFC into MPC. In patients with early AD, we observed a reduced pattern of EC among DMN regions. We found measurable EC only into MPC and from MPFC into hippocampus (Table S3A, Figure 4(a)). Group differences based on two‐sample t tests (p < 0.05 corrected) revealed significantly decreased EC in the patient group for signaling from MPFC (t 51 = −3.51, p = 9 × 10−4) and left parietal cortices (t 51 = −3.13, p = 0.003) into MPC, from left to right parietal cortex (t 51 = −3.84, p = 3 × 10−4), and from hippocampus into MPFC (t 51 = −2.81, p = 0.007; Supporting Information Table S3A, Figure 4a). Analyzing EC for AD‐MCI and AD‐dementia groups separately revealed that group differences in early AD were mainly driven by differences in the AD‐dementia group (Supporting Information Table S3B). In summary, we found a specific pattern of EC in the DMN of healthy subjects with predominant EC into MPC and bidirectional EC between MPFC and hippocampus. Anterior and posterior DMN were linked via unidirectional EC from MPFC into MPC. In patients, the EC into MPC and from hippocampus to MPFC was most severely disrupted (Figure 4b).
FIGURE 4.
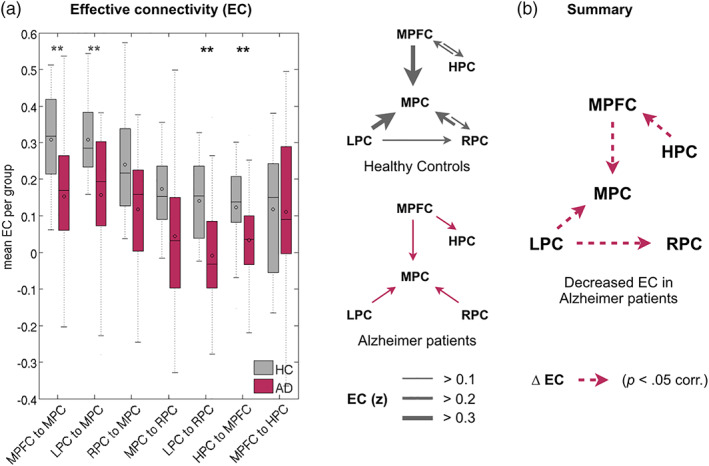
Effective connectivity in the DMN. (a) Left: Bar plots indicate pathways with decreased directional signaling, particularly connectivity into medial (MPC) and right (RPC) parietal cortex and from hippocampus into prefrontal cortex (MPFC). Right: Graphical illustration of significant EC among DMN regions in healthy controls and patients. (b) Summary illustrating significantly decreased EC (dotted lines) in the DMN of patients with AD. **p < .05 Bonferroni corrected. See Supporting InformationTable S3 for full statistics
In control analyses we tested the stability of our finding with respect to region size and signal variance. Previously, we had demonstrated stable results of EC, even when reducing the number of voxels by up to 80% (Riedl et al., 2016). In the current study, we replicated the EC‐analysis on DMN‐regions with a balanced number of voxels (range within 10%) across all subjects (Supporting Information Table S4, right columns) yielding identical results with deviations of less than 4% for the strength of EC within each group (Supporting Information Table S5). Furthermore, EC depends on the variance of FDG (FDGvar) and FC (FCvar) values within each DMN‐region. We therefore tested whether FDGvar and FCvar differed across groups (Supporting Information Figure S1). Two‐sample t tests revealed a significant difference for FDGvar in lateral parietal and hippocampal regions but not in medial parietal or prefrontal regions. Our major finding of reduced EC into medial parietal and prefrontal cortices was therefore not related to differences in variance of FDG in these regions.
3.3. Cognitive deficits were associated with multimodal EC but not with unimodal imaging measures
In a final step, we tested whether any of the uni‐ or multimodal imaging parameters were associated with cognitive deficits as measured with MMSE. As only MPC showed changes in any of the three parameters, we initially focused on this region. We performed a multiple linear regression analysis in patients with early AD, with MMSE being the dependent variable and the following independent variables: “EC into MPC,” “FDGamp in MPC,” and “FCmean of MPC.” Control variables were age, gender, and global gray matter volume. Furthermore, we defined an interaction variable EC × FDGamp, to test for an influence of EC and FDGamp on cognitive performance. This model significantly explained variance in MMSE scores (F 4,25 = 5.63, p = 0.002) with an r 2 = 0.47. Among the individual explanatory variables, only “EC into MPC” (standardized β = 0.54, p = 0.012) was significantly associated with MMSE scores (Supporting Information Figure S2). FDGamp (std. β = 0.39, p = 0.06) showed a trend toward significance, while FCmean (std. β = 0.05, p = 0.76) was not significant when assuming a corrected alpha level of p = 0.017. The interaction between EC and FDGamp was not significant (std. β = −0.12, p = 0.63), indicating independent influence of EC on MMSE. To further control for interrelatedness among independent variables of the model, we also tested for relationships among the explanatory imaging variables by calculating collinearity via variance inflation factor (VIF). Importantly, no collinearity occurred, particularly among the significant variables FDGamp and “EC into MPC” (VIFEC: 1.01, VIFFDG: 1.0, VIFFC: 1.2).
Although MPC was the only region with changes in any of the imaging parameters, the diminished FC of the MPFC might be a more representative explanatory variable of cognitive deficits. We therefore repeated the multiple regression analysis but replaced “FCmean of MPC” with “FCmean of MPFC.” The overall model equally well explained MMSE variance (F 4,25 = 5.59, p = 0.002, r 2 = 0.47), but the contribution of FCmean was again not significant (std. β = 0.004, p = 0.98).
4. DISCUSSION
Using a novel, multimodal imaging measure of directional signaling in the brain, we identified a unique pattern of EC in the DMN of healthy subjects and specific disruptions of EC in patients with AD. Particularly, frontal and parietal input to MPC and hippocampal input to MPFC was reduced.
4.1. Localized and network effects in the DMN of patients with AD
In our study, unimodal analyses of energy metabolism and FC revealed both regional and network‐wide changes in patients with AD. FDG‐PET showed hypometabolism in parietal cortices and hippocampus, while network analysis of fMRI data revealed disruptions of long‐range FC between medial prefrontal and all parietal areas of the DMN. Both findings are in line with prior studies on AD using different imaging modalities. Amyloid‐β accumulation in medial and lateral parietal cortices coincides with hypometabolism in the same regions even in prodromal stages of AD (Grothe et al., 2016). Tau pathology accumulates first in the entorhinal cortex and hippocampus (Braak & Braak, 1991; Schöll et al., 2016; Sepulcre et al., 2016) while amyloid‐β originates in the neocortex (Grothe et al., 2016; Klunk et al., 2004; Sepulcre et al., 2016). Complementing these localized effects, network analyses of fMRI, EEG, and MEG data point to widespread connectivity changes in AD among DMN regions particularly affecting long‐range connections (Greicius et al., 2004; Stam et al., 2005, 2006; Zhang et al., 2010). Together, molecular imaging identifies brain regions most affected by neuropathology, and network imaging detects disruptions of connectivity pathways between these regions. Our multimodal EC approach might offer a framework to integrate localized and network changes in AD.
4.2. Directional signaling in the DMN of healthy subjects
Based on a cellular model of neuroenergetics, we recently introduced a simultaneous functional imaging approach to detect directional signaling in large‐scale brain networks (Riedl et al., 2014, 2016). Here, we first identified EC in the DMN of healthy subjects. MCM revealed two distinct patterns of directional signaling in the DMN extending the more unspecific pattern of FC characterized by long‐range connections between medial prefrontal and all parietal regions. We found unidirectional EC converging onto MPC in the posterior DMN, and bidirectional EC between MPFC and hippocampus in the anterior DMN. These two subnetworks were linked via unidirectional EC from medial prefrontal into MPC. This means that we found no evidence for EC directly connecting lateral parietal and MPFCs. This finding extends prior network analyses based on undirected functional and structural connectivity measures. Network analyses with low‐dimensional independent component analysis identified a singular DMN, but this network split into an anterior and a posterior DMN with higher component decompositions (Allen et al., 2011; Fransson, 2006). Similarly, seed‐based FC analyses have identified a prefronto‐medial temporal and a parietal subsystem of the DMN (Andrews‐Hanna et al., 2010; Uddin, Clare Kelly, Biswal, Xavier Castellanos, & Milham, 2009). In whole‐brain graph analyses the medial fronto‐parietal axis has been identified as a major network hub in functional and structural connectivity analyses (Hagmann et al., 2008; van den Heuvel & Sporns, 2011). Moreover, these hub regions consume relatively large amounts of energy (Tomasi, Wang, & Volkow, 2013), which is consistent with high demands on information integration in these regions from an energetics point of view.
Additionally, we found strong FC and EC among MPFC and hippocampus which is supported by imaging and tracer approaches (Kahn, Andrews‐Hanna, Vincent, Snyder, & Buckner, 2008; Strange, Witter, Lein, & Moser, 2014). In humans, FC between MPFC and hippocampus is present during rest (Kahn et al., 2008; Uddin et al., 2009) and increases during memory consolidation (van Kesteren, Fernández, Norris, & Hermans, 2010). Moreover, axonal tracing studies in rats identified the same pathway using structural connectivity methods (Strange et al., 2014). However, fMRI studies have also reported equal coupling of the hippocampus with posterior parts of the DMN (Dunn et al., 2014; Greicius, Supekar, Menon, & Dougherty, 2009; Wang et al., 2010) that we could not identify in our approach. While anterior hippocampus is particularly connected with prefrontal cortex, posterior parts are more strongly connected with the posterior DMN and particularly via parahippocampal structures (Ward et al., 2014). However, spatial overlap exists along this anterior–posterior axis (Ranganath & Ritchey, 2012; Strange et al., 2014; Zarei et al., 2013). In the current study, we aimed for balanced ROI‐size across regions and subjects as well as for a sufficient number of voxels per ROI to achieve accurate MCM‐results (see stability of MCM‐pathways with avg. ROI size of around 500 voxels in (Riedl et al., 2016)). We therefore included voxels associated with hippocampus, entorhinal, and parahippocampal cortex in the hippocampus mask to achieve sufficient extent. This, on the other hand, might explain why we were not able to identify significant signal and parietal pathways from the hippocampus in the average ROI signal.
4.3. Directional signaling in the DMN of patients with AD
In patients with early AD, we found EC patterns that were generally consistent with healthy subjects. The EC into MPC from lateral parietal and MPFCs was preserved, as well as the medial prefrontal input to hippocampus. However, the strength of all connections was diminished, particularly in the subgroup with AD‐dementia, most notably the EC into medial and right parietal cortices and the hippocampal input to MPFC. This means that our novel approach identified similar patterns of EC in the DMN across both populations but strongly reduced EC between specific brain regions in patients with AD.
Compared to fMRI‐only approaches, such as GCA and DCM, our multimodal attempt might compensate for several pitfalls inherent to these statistical approaches. GCA might be prone to spurious “causality” that is in fact the result of naturally occurring time‐lags among different brain regions. For example, GCM applied to simulated fMRI time‐series data was shown to perform relatively poor, which “suggests that the directionality results may not be trustworthy” (Smith et al., 2011). DCM, on the other hand, integrates a hemodynamic forward model that describes the transformation from neural activity to the measured BOLD signal. A growing body of evidence indicates that Aβ not only affects neurons but also cerebral blood vessels (Zhang, Gordon, & Goldberg, 2017). Due to the damaged vasculature in AD, important assumptions of DCM, that is, the mapping between neuronal activity (hidden states) and measured BOLD response, might therefore be violated in these patients. Here, we substantially extend prior results from fMRI data because our approach on EC is not confounded by violated preconditions of Granger causality and DCM. In contrast to GCM, our fMRI analysis relies on more conventional 0‐lag FC instead of lagged FC. In contrast to DCM, our approach is model‐free and relies on the integration of two independently measured brain signals. However, future studies are necessary to demonstrate which specific aspect of EC are captured by MCM.
Finally, we tested whether any of the imaging parameters was associated with cognitive deficits in patients and whether our integrated measure of EC might explain more variance than unimodal FDG or FC alone. The overall model including all three parameters explained 47% variance of MMSE in patients with early AD. However, only EC significantly contributed to the regression. While FDG alone showed a trend to significance, its contribution in the interaction term with EC was negligible. Prior studies using FDG have identified decline in local energy metabolism of parietal regions as a key marker for AD (Buckner et al., 2005; Grothe et al., 2016, 2017; Scherr et al., 2018). Still, our regression model suggests that including further information about distant connectivity and interpreting energy metabolism in terms of directional signaling might be better suited to explain cognitive deficits in patients.
4.4. EC and two molecular theories about neuropathological spreading in AD
AD begins with dysfunction in discrete regions but involves much larger brain areas with disease progression. Two pathophysiological spreading mechanisms have been suggested based on molecular findings. A “nodal stress” model suggests disease progression in an upstream manner starting in highly connected hub regions with strong synaptic plasticity (Palop & Mucke, 2010; Zhou et al., 2012). This model is based on recent data that identified amyloid‐β as an activity‐dependent regulator of postsynaptic activity in the healthy brain (Walsh et al., 2002). Pathological accumulation of amyloid‐β in highly active brain regions would therefore trigger feedback suppression of upstream regions. This is in line with hypometabolism and amyloid‐β accumulation in parietal cortices of the DMN (Buckner et al., 2005). Following a prion‐like mechanism, another model suggests downstream spreading to connected cells (Frost & Diamond, 2010; Jucker & Walker, 2011). This model is based on findings of transneuronal spread where the transfer of intracellular tau aggregates has been observed between cocultured cells in vitro (Frost, Jacks, & Diamond, 2009).
On the systems level, we here found impaired signaling pathways in the DMN of patients that might relate to both neuropathological models. Given the preference for accumulation of amyloid‐β in neocortical regions and of tau in the hippocampus, we propose the following working hypothesis for both upstream and downstream pathological influences. The presence of EC converging into MPC might best support the theory about activity‐dependent accumulation of amyloid‐β. The unilateral decrease of EC from hippocampus into MPFC, on the other hand, might best relate to a prion‐like spread of tau starting from medial temporal lobes. Another startling yet consistent imaging finding might also relate to distinct spreading models. Hypometabolism and atrophy are most consistently observed in hippocampal and parietal regions, together with accumulation of tau and amyloid‐β respectively. However, MPFC is strongly affected by impaired FC yet largely spared by hypometabolism and atrophy (La Joie et al., 2012). This discrepancy in the overlap of distinct imaging parameters might be related to distinct influences of neuropathologies in subcircuits of the DMN and to a secondary impairment of prefrontal regions by both downstream and upstream pathological processes. While it is likely that diverse factors and interactions between tau and amyloid‐β pathology contribute to the pathogenesis of AD, our hypothesis about protein‐dependent spreading mechanisms could be directly tested in vivo using pathology‐specific PET tracers (Sepulcre et al., 2016).
4.5. Limitations
Our MCM based measure of EC might be confounded by altered vascular‐hemodynamic properties in patients. Recently, we demonstrated that impaired perfusion contributes to impaired FC in AD (Göttler et al., 2018), and it is well known that FDG‐PET‐based measures reflect not only metabolism but also hemodynamics (Raichle & Mintun, 2006). As both FC and FDG‐PET‐based metabolism contribute to EC, altered hemodynamics might confound EC changes in patients. Future studies, therefore should control for these hemodynamic‐vascular aspects by additional perfusion‐based imaging and subsequent control. Furthermore, FC measures might be confounded by group differences in vigilance fluctuations (Chang et al., 2016). Although we immediately asked participants after rs‐fMRI about falling asleep, more subtle vigilance changes and reliability of self‐reports should be added in future studies.
Supporting information
Figure S1 A) Variance of FDG (FDGvar) and B) FC (FC var). A) Two‐sample t tests (** p < .05, Bonferroni corrected) indicate different variance of voxel FDG values in lateral parietal DMN‐regions and hippocampus. FDG‐variance in medial prefrontal and parietal regions did not differ. B) Two‐sample t tests (** p < .05, Bonferroni corrected) indicate different variance of FCvox values in medial parietal (only for MPFC seed) and in right parietal (for LPC seed). Variance of FCvox for all other connections with group differences in EC did not differ.
Supplementary Figure 2. Linear regression of the relationship between MMSE and ‘EC into MPC’ in patients. This plot illustrates the relationship of MMSE with the only significant explanatory variable from the multiple linear regression analysis (see main text), i.e., average EC into MPC, across all patients. While the multiple regression model is only significant for the subgroup of AD patients (F4,25 = 3.27, p = .05 with an r 2 = 0.74; ‘EC into MPC’: standardized β = 0.65, p = .042), the plot indicates a continuous transition from MCI to AD. Green: AD, blue: MCI.
Suppl Table 1A: Regional metabolism in DMN‐regions – HC and early AD
Suppl Table 1B: Regional metabolism in DMN‐regions – anova for HC, AD‐MCI, and AD‐Dementia
Suppl Table 2A. FC among regions of the DMN ‐ HC and early AD.
Suppl Table 2B. FC among regions of the DMN – anova for HC, AD‐MCI, and AD‐Dementia.
Suppl Table 3A. EC among regions of the DMN in healthy controls and patients with early Alzheimer's disease.
Suppl Table 3B. EC among regions of the DMN – anova in HC, AD‐MCI, and AD‐Dementia
Supplementary Table 4. ROI‐sizes of EC analysis
Supplementary Table 5. EC among DMN‐regions with balanced ROI‐size
ACKNOWLEDGMENTS
The authors wish to thank all patients and their relatives for their participation in the study. Furthermore, the authors thank the staff of the Departments of Psychiatry and Psychotherapy and also Nuclear Medicine for their help in recruitment and data collection. The authors declare no conflict of interest with respect to the current study. This study was supported by the German Federal Ministry of Education and Science (BMBF 01EV0710 to A.M.W.). The manuscript was prepared with partial support from the Technische Universität Muenchen––Institute for Advanced Study, funded by the German Excellence Initiative and the European Union Seventh Framework Programme under grant agreement n 291763 (J.P.R.). V.R. was funded by the German Research Foundation (DFG) grant 273427765.
Scherr M, Utz L, Tahmasian M, et al. Effective connectivity in the default mode network is distinctively disrupted in Alzheimer's disease—A simultaneous resting‐state FDG‐PET/fMRI study. Hum Brain Mapp. 2021;42:4134–4143. 10.1002/hbm.24517
Funding information Bundesministerium für Bildung und Forschung, Grant/Award Number: 01EV0710; Deutsche Forschungsgemeinschaft, Grant/Award Number: 273427765; German Research Foundation, Grant/Award Number: 273427765; European Union Seventh Framework Programme, Grant/Award Number: n 291763; Technische Universität Muenchen—Institute for Advanced Study; Federal Ministry of Education and Science, Grant/Award Number: BMBF 01EV0710
REFERENCES
- Agosta, F., Rocca, M. A., Pagani, E., Absinta, M., Magnani, G., Marcone, A., … Filippi, M. (2010). Sensorimotor network rewiring in mild cognitive impairment and Alzheimer's disease. Human Brain Mapping, 31, 515–525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albert, M. S., DeKosky, S. T., Dickson, D., Dubois, B., Feldman, H. H., Fox, N. C., … Phelps, C. H. (2011). The diagnosis of mild cognitive impairment due to Alzheimer's disease: Recommendations from the National Institute on Aging‐Alzheimer's Association workgroups on diagnostic guidelines for Alzheimer's disease. Alzheimer's & Dementia, 7, 270–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allen, E. A., Erhardt, E. B., Damaraju, E., Gruner, W., Segall, J. M., Silva, R. F., … Calhoun, V. D. (2011). A baseline for the multivariate comparison of resting‐state networks. Frontiers in Systems Neuroscience, 5, 2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews‐Hanna, J. R., Reidler, J. S., Sepulcre, J., Poulin, R., & Buckner, R. L. (2010). Functional‐anatomic fractionation of the brain's default network. Neuron, 65, 550–562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Attwell, D., & Iadecola, C. (2002). The neural basis of functional brain imaging signals. Trends in Neurosciences, 25, 621–625. [DOI] [PubMed] [Google Scholar]
- Attwell, D., & Laughlin, S. B. (2001). An energy budget for signaling in the grey matter of the brain. Journal of Cerebral Blood Flow and Metabolism, 21, 1133–1145. [DOI] [PubMed] [Google Scholar]
- Braak, H., & Braak, E. (1991). Neuropathological stageing of Alzheimer‐related changes. Acta Neuropathologica, 82, 239–259. [DOI] [PubMed] [Google Scholar]
- Brettschneider, J., Del Tredici, K., Lee, V. M. Y., & Trojanowski, J. Q. (2015). Spreading of pathology in neurodegenerative diseases: A focus on human studies. Nature Reviews. Neuroscience, 16, 109–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buckner, R. L., Snyder, A. Z., Shannon, B. J., LaRossa, G., Sachs, R., Fotenos, A. F., … Mintun, M. A. (2005). Molecular, structural, and functional characterization of Alzheimer's disease: Evidence for a relationship between default activity, amyloid, and memory. The Journal of Neuroscience, 25, 7709–7717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang, C., Leopold, D. A., Schölvinck, M. L., Mandelkow, H., Picchioni, D., Liu, X., … Duyn, J. H. (2016). Tracking brain arousal fluctuations with fMRI. Proceedings of the National Academy of Sciences of the United States of America, 113, 4518–4523. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drzezga, A., Becker, J. A., Van Dijk, K. R. A., Sreenivasan, A., Talukdar, T., Sullivan, C., … Sperling, R. A. (2011). Neuronal dysfunction and disconnection of cortical hubs in non‐demented subjects with elevated amyloid burden. Brain, 134, 1635–1646. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunn, C. J., Duffy, S. L., Hickie, I. B., Lagopoulos, J., Lewis, S. J. G., Naismith, S. L., & Shine, J. M. (2014). Deficits in episodic memory retrieval reveal impaired default mode network connectivity in amnestic mild cognitive impairment. NeuroImage: Clinical, 4, 473–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fischl, B., Salat, D. H., Busa, E., Albert, M., Dieterich, M., Haselgrove, C., … Dale, A. M. (2002). Whole brain segmentation. Neuron, 33, 341–355. [DOI] [PubMed] [Google Scholar]
- Fransson, P. (2006). How default is the default mode of brain function? Further evidence from intrinsic BOLD signal fluctuations. Neuropsychologia, 44, 2836–2845. [DOI] [PubMed] [Google Scholar]
- Frost, B., & Diamond, M. I. (2010). Prion‐like mechanisms in neurodegenerative diseases. Nature Reviews. Neuroscience, 11, 155–159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frost, B., Jacks, R. L., & Diamond, M. I. (2009). Propagation of tau misfolding from the outside to the inside of a cell. The Journal of Biological Chemistry, 284, 12845–12852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göttler, J., Stephan K., Michael, K., & Isabel, W., Hans‐Henning, E., Claus Z., Christian, S., Christine, P., and Fahmeed, H., (2008). “Flow‐Metabolism Uncoupling in Patients with Asymptomatic Unilateral Carotid Artery Stenosis Assessed by Multi‐Modal Magnetic Resonance Imaging”. Journal of Cerebral Blood Flow & Metabolism. 10.1177/0271678X18783369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Göttler, J., Preibisch, C., Riederer, I., Pasquini, L., Alexopoulos, P., Bohn, K. P., … Sorg, C. (2018). Reduced blood oxygenation level dependent connectivity is related to hypoperfusion in Alzheimer's disease. Journal of Cerebral Blood Flow and Metabolism:271678X18759182, 0271678X1875918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius, M., Supekar, K., Menon, V., & Dougherty, R. (2009). Resting‐state functional connectivity reflects structural connectivity in the default mode network. Cerebral Cortex, 19, 72–78. 10.1093/cercor/bhn059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greicius, M. D., Srivastava, G., Reiss, A. L., & Menon, V. (2004). Default‐mode network activity distinguishes Alzheimer's disease from healthy aging: Evidence from functional MRI. Proceedings of the National Academy of Sciences of the United States of America, 101, 4637–4642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe, M. J., Barthel, H., Sepulcre, J., Dyrba, M., Sabri, O., & Teipel, S. J. (2017). In vivo staging of regional amyloid deposition. Neurology, 89, 2031–2038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grothe, M. J., Teipel, S. J., & Alzheimer´s Disease Neuroimaging Initiative . (2016). Spatial patterns of atrophy, hypometabolism, and amyloid deposition in Alzheimer's disease correspond to dissociable functional brain networks. Human Brain Mapping, 37, 35–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hagmann, P., Cammoun, L., Gigandet, X., Meuli, R., Honey, C. J., Wedeen, V. J., & Sporns, O. (2008). Mapping the structural core of human cerebral cortex. PLoS Biology, 6, e159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahn, K., Myers, N., Prigarin, S., Rodenacker, K., Kurz, A., Förstl, H., … Sorg, C. (2013). Selectively and progressively disrupted structural connectivity of functional brain networks in Alzheimer's disease ‐ revealed by a novel framework to analyze edge distributions of networks detecting disruptions with strong statistical evidence. NeuroImage, 81, 96–109. [DOI] [PubMed] [Google Scholar]
- Iturria‐Medina, Y., Sotero, R. C., Toussaint, P. J., Evans, A. C., & Alzheimer´s Disease Neuroimaging Initiative . (2014). Epidemic spreading model to characterize misfolded proteins propagation in aging and associated neurodegenerative disorders. PLoS Computational Biology, 10, e1003956. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jucker, M., & Walker, L. C. (2011). Pathogenic protein seeding in Alzheimer disease and other neurodegenerative disorders. Annals of Neurology, 70, 532–540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahn, I., Andrews‐Hanna, J. R., Vincent, J. L., Snyder, A. Z., & Buckner, R. L. (2008). Distinct cortical anatomy linked to subregions of the medial temporal lobe revealed by intrinsic functional connectivity. Journal of Neurophysiology, 100, 129–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klunk, W. E., Engler, H., Nordberg, A., Wang, Y., Blomqvist, G., Holt, D. P., … Långström, B. (2004). Imaging brain amyloid in Alzheimer's disease with Pittsburgh compound‐B. Annals of Neurology, 55, 306–319. [DOI] [PubMed] [Google Scholar]
- La Joie, R., Perrotin, A., Barré, L., Hommet, C., Mézenge, F., Ibazizene, M., … Chetelat, G. (2012). Region‐specific hierarchy between atrophy, hypometabolism, and β‐amyloid (Aβ) load in Alzheimer's disease dementia. The Journal of Neuroscience, 32, 16265–16273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lim, Y. Y., Maruff, P., Pietrzak, R. H., Ames, D., Ellis, K. A., Harrington, K., … AIBL Research Group . (2014). Effect of amyloid on memory and non‐memory decline from preclinical to clinical Alzheimer's disease. Brain, 137, 221–231. [DOI] [PubMed] [Google Scholar]
- McKhann GM, Knopman DS, Chertkow H, Hyman BT, Jack CR, Kawas CH, Klunk WE, Koroshetz WJ, Manly JJ, Mayeux R, Mohs RC, Morris JC, Rossor MN, Scheltens P, Carrillo MC, Thies B, Weintraub S, Phelps CH (2011): The diagnosis of dementia due to Alzheimer's disease: recommendations from the National Institute on Aging‐Alzheimer‘s Association workgroups on diagnostic guidelines for Alzheimer's disease. In: Vol. 7, pp 263–269. [DOI] [PMC free article] [PubMed]
- Mesulam, M. M. (2000). A plasticity‐based theory of the pathogenesis of Alzheimer's disease. Annals of the New York Academy of Sciences, 924, 42–52. [DOI] [PubMed] [Google Scholar]
- Myers, N., Pasquini, L., Göttler, J., Grimmer, T., Koch, K., Ortner, M., … Sorg, C. (2014). Within‐patient correspondence of amyloid‐β and intrinsic network connectivity in Alzheimer's disease. Brain, 137, 2052–2064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuttall, R., Pasquini, L., Scherr, M., & Sorg, C. (2016). Degradation in intrinsic connectivity networks across the Alzheimer's disease spectrum. Alzheimers Dement (Amst), 5, 35–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palop, J. J., Chin, J., & Mucke, L. (2006). A network dysfunction perspective on neurodegenerative diseases. Nature, 443, 768–773. [DOI] [PubMed] [Google Scholar]
- Palop, J. J., & Mucke, L. (2010). Amyloid‐beta‐induced neuronal dysfunction in Alzheimer's disease: From synapses toward neural networks. Nature Neuroscience, 13, 812–818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pasquini, L., Benson, G., Grothe, M. J., Utz, L., Myers, N. E., Yakushev, I., … Sorg, C. (2017). Individual correspondence of amyloid‐beta and Intrinsic connectivity in the posterior default mode network across stages of Alzheimer's disease. Journal of Alzheimer's Disease, 58, 763–773. [DOI] [PubMed] [Google Scholar]
- Pasquini, L., Scherr, M., Tahmasian, M., Meng, C., Myers, N. E., Ortner, M., … Sorg, C. (2015). Link between hippocampus' raised local and eased global intrinsic connectivity in AD. Alzheimers Dement, 11, 475–484. [DOI] [PubMed] [Google Scholar]
- Pievani, M., de Haan, W., Wu, T., Seeley, W. W., & Frisoni, G. B. (2011). Functional network disruption in the degenerative dementias. The Lancet Neurology, 10, 829–843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power, J. D., Barnes, K. A., Snyder, A. Z., Schlaggar, B. L., & Petersen, S. E. (2012). Spurious but systematic correlations in functional connectivity MRI networks arise from subject motion. NeuroImage, 59, 2142–2154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price, J. L., & Morris, J. C. (1999). Tangles and plaques in nondemented aging and “preclinical” Alzheimer's disease. Annals of Neurology, 45, 358–368. [DOI] [PubMed] [Google Scholar]
- Raichle, M. E., & Mintun, M. A. (2006). Brain work and brain imaging. Annual Review of Neuroscience, 29, 449–476. [DOI] [PubMed] [Google Scholar]
- Raj, A., Kuceyeski, A., & Weiner, M. (2012). A network diffusion model of disease progression in dementia. Neuron, 73, 1204–1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranganath, C., & Ritchey, M. (2012). Two cortical systems for memory‐guided behaviour. Nature Reviews. Neuroscience, 13, 713–726. [DOI] [PubMed] [Google Scholar]
- Riedl, V., Bienkowska, K., Strobel, C., Tahmasian, M., Grimmer, T., Forster, S., … Drzezga, A. (2014). Local activity determines functional connectivity in the resting human brain: A simultaneous FDG‐PET/fMRI study. The Journal of Neuroscience, 34, 6260–6266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedl, V., Utz, L., Castrillón, G., Grimmer, T., Rauschecker, J. P., Ploner, M., … Sorg, C. (2016). Metabolic connectivity mapping reveals effective connectivity in the resting human brain. Proceedings of the National Academy of Sciences of the United States of America, 113, 428–433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scherr, M., Pasquini, L., Benson, G., Nuttall, R., Gruber, M., Neitzel, J., … Alzheimer´s Disease Neuroimaging Initiative . (2018). Decoupling of local metabolic activity and functional connectivity links to amyloid in Alzheimer's disease. Journal of Alzheimer's Disease, 64, 405–415. [DOI] [PubMed] [Google Scholar]
- Schöll, M., Lockhart, S. N., Schonhaut, D. R., O'Neil, J. P., Janabi, M., Ossenkoppele, R., … Jagust, W. J. (2016). PET imaging of tau deposition in the aging human brain. Neuron, 89, 971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seeley, W. W., Crawford, R. K., Zhou, J., Miller, B. L., & Greicius, M. D. (2009). Neurodegenerative diseases target large‐scale human brain networks. Neuron, 62, 42–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sepulcre, J., Schultz, A. P., Sabuncu, M., Gomez‐Isla, T., Chhatwal, J., Becker, A., … Johnson, K. A. (2016). In vivo tau, amyloid, and gray matter profiles in the aging brain. Journal of Neuroscience, 36, 7364–7374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith, S. M., Miller, K. L., Salimi‐Khorshidi, G., Webster, M., Beckmann, C. F., Nichols, T. E., … Woolrich, M. W. (2011). Network modelling methods for FMRI. NeuroImage, 54, 875–891. [DOI] [PubMed] [Google Scholar]
- Song, X.‐W., Dong, Z.‐Y., Long, X.‐Y., Li, S.‐F., Zuo, X.‐N., Zhu, C.‐Z., … Zang, Y.‐F. (2011). REST: A toolkit for resting‐state functional magnetic resonance imaging data processing. PLoS One, 6, e25031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sorg, C., Riedl, V., Mühlau, M., Calhoun, V. D., Eichele, T., Läer, L., … Wohlschläger, A. M. (2007). Selective changes of resting‐state networks in individuals at risk for Alzheimer's disease. Proceedings of the National Academy of Sciences of the United States of America, 104, 18760–18765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stam, C. J., Jones, B. F., Manshanden, I., van Cappellen van Walsum, A. M., Montez, T., Verbunt, J. P. A., … Scheltens, P. (2006). Magnetoencephalographic evaluation of resting‐state functional connectivity in Alzheimer's disease. NeuroImage, 32, 1335–1344. [DOI] [PubMed] [Google Scholar]
- Stam, C. J., Montez, T., Jones, B. F., Rombouts, S. A. R. B., van der Made, Y., Pijnenburg, Y. A. L., & Scheltens, P. (2005). Disturbed fluctuations of resting state EEG synchronization in Alzheimer's disease. Clinical Neurophysiology, 116, 708–715. [DOI] [PubMed] [Google Scholar]
- Strange, B. A., Witter, M. P., Lein, E. S., & Moser, E. I. (2014). Functional organization of the hippocampal longitudinal axis. Nature Reviews. Neuroscience, 15, 655–669. [DOI] [PubMed] [Google Scholar]
- Tomasi, D., Wang, G.‐J., & Volkow, N. D. (2013). Energetic cost of brain functional connectivity. Proceedings of the National Academy of Sciences of the United States of America, 110, 13642–13647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uddin, L. Q., Clare Kelly, A. M., Biswal, B. B., Xavier Castellanos, F., & Milham, M. P. (2009). Functional connectivity of default mode network components: Correlation, anticorrelation, and causality. Human Brain Mapping, 30, 625–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Heuvel, M. P., & Sporns, O. (2011). Rich‐club organization of the human connectome. The Journal of Neuroscience, 31, 15775–15786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Dijk, K. R. A., Sabuncu, M. R., & Buckner, R. L. (2012). The influence of head motion on intrinsic functional connectivity MRI. NeuroImage, 59, 431–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Kesteren, M. T. R., Fernández, G., Norris, D. G., & Hermans, E. J. (2010). Persistent schema‐dependent hippocampal‐neocortical connectivity during memory encoding and postencoding rest in humans. Proceedings of the National Academy of Sciences of the United States of America, 107, 7550–7555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walsh, D. M., Klyubin, I., Fadeeva, J. V., Cullen, W. K., Anwyl, R., Wolfe, M. S., … Selkoe, D. J. (2002). Naturally secreted oligomers of amyloid |[beta]| protein potently inhibit hippocampal long‐term potentiation in vivo. Nature, 416, 535–539. [DOI] [PubMed] [Google Scholar]
- Wang, L., LaViolette, P., O'Keefe, K., Putcha, D., Bakkour, A., Van Dijk, K. R. A., … Sperling, R. A. (2010). Intrinsic connectivity between the hippocampus and posteromedial cortex predicts memory performance in cognitively intact older individuals. NeuroImage, 51, 910–917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward, A. M., Schultz, A. P., Huijbers, W., Van Dijk, K. R. A., Hedden, T., & Sperling, R. A. (2014). The parahippocampal gyrus links the default‐mode cortical network with the medial temporal lobe memory system. Human Brain Mapping, 35, 1061–1073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamamoto, K., Tanei, Z.‐I., Hashimoto, T., Wakabayashi, T., Okuno, H., Naka, Y., … Iwatsubo, T. (2015). Chronic optogenetic activation augments abeta pathology in a mouse model of Alzheimer disease. Cell Reports, 11, 859–865. [DOI] [PubMed] [Google Scholar]
- Zarei, M., Beckmann, C. F., Binnewijzend, M. A. A., Schoonheim, M. M., Oghabian, M. A., Sanz‐Arigita, E. J., … Barkhof, F. (2013). Functional segmentation of the hippocampus in the healthy human brain and in Alzheimer's disease. NeuroImage, 66, 28–35. [DOI] [PubMed] [Google Scholar]
- Zhang, H.‐Y., Wang, S.‐J., Liu, B., Ma, Z.‐L., Yang, M., Zhang, Z.‐J., & Teng, G.‐J. (2010). Resting brain connectivity: Changes during the progress of Alzheimer disease. Radiology, 256, 598–606. [DOI] [PubMed] [Google Scholar]
- Zhang, N., Gordon, M. L., & Goldberg, T. E. (2017). Cerebral blood flow measured by arterial spin labeling MRI at resting state in normal aging and Alzheimer's disease. Neuroscience and Biobehavioral Reviews, 72, 168–175. [DOI] [PubMed] [Google Scholar]
- Zhong, Y., Huang, L., Cai, S., Zhang, Y., Deneen, v. K. M., Ren, A., & Ren, J. (2014). Altered effective connectivity patterns of the default mode network in Alzheimer's disease: An fMRI study. Neuroscience Letters, 578, 171–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J., Gennatas, E. D., Kramer, J. H., Miller, B. L., & Seeley, W. W. (2012). Predicting regional neurodegeneration from the healthy brain functional connectome. Neuron, 73, 1216–1227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou, J., Greicius, M. D., Gennatas, E. D., Growdon, M. E., Jang, J. Y., Rabinovici, G. D., … Seeley, W. W. (2010). Divergent network connectivity changes in behavioural variant frontotemporal dementia and Alzheimer's disease. Brain, 133, 1352–1367. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1 A) Variance of FDG (FDGvar) and B) FC (FC var). A) Two‐sample t tests (** p < .05, Bonferroni corrected) indicate different variance of voxel FDG values in lateral parietal DMN‐regions and hippocampus. FDG‐variance in medial prefrontal and parietal regions did not differ. B) Two‐sample t tests (** p < .05, Bonferroni corrected) indicate different variance of FCvox values in medial parietal (only for MPFC seed) and in right parietal (for LPC seed). Variance of FCvox for all other connections with group differences in EC did not differ.
Supplementary Figure 2. Linear regression of the relationship between MMSE and ‘EC into MPC’ in patients. This plot illustrates the relationship of MMSE with the only significant explanatory variable from the multiple linear regression analysis (see main text), i.e., average EC into MPC, across all patients. While the multiple regression model is only significant for the subgroup of AD patients (F4,25 = 3.27, p = .05 with an r 2 = 0.74; ‘EC into MPC’: standardized β = 0.65, p = .042), the plot indicates a continuous transition from MCI to AD. Green: AD, blue: MCI.
Suppl Table 1A: Regional metabolism in DMN‐regions – HC and early AD
Suppl Table 1B: Regional metabolism in DMN‐regions – anova for HC, AD‐MCI, and AD‐Dementia
Suppl Table 2A. FC among regions of the DMN ‐ HC and early AD.
Suppl Table 2B. FC among regions of the DMN – anova for HC, AD‐MCI, and AD‐Dementia.
Suppl Table 3A. EC among regions of the DMN in healthy controls and patients with early Alzheimer's disease.
Suppl Table 3B. EC among regions of the DMN – anova in HC, AD‐MCI, and AD‐Dementia
Supplementary Table 4. ROI‐sizes of EC analysis
Supplementary Table 5. EC among DMN‐regions with balanced ROI‐size