

Supplemental Digital Content is Available in the Text.
Key Words: bictegravir, HIV, Black Americans, tenofovir alafenamide, INSTI
Background:
With the highest rates of HIV/AIDS in the United States, Black Americans are still underrepresented in HIV medical research.
Setting:
BRAAVE (NCT03631732) is a randomized, phase 3b, multicenter, open-label US study.
Methods:
Adults identifying as Black or African American and virologically suppressed on 2 nucleoside reverse transcriptase inhibitors (NRTIs) plus third agent were randomized (2:1) to switch to open-label bictegravir/emtricitabine/tenofovir alafenamide (B/F/TAF) once daily or stay on baseline regimen (SBR) for 24 weeks, after which SBR had delayed switch to B/F/TAF. Resistance to non-NRTIs, protease inhibitors, and/or NRTIs was permitted; integrase strand transfer inhibitor resistance was exclusionary. Primary endpoint was proportion of participants with HIV-1 RNA ≥50 copies/mL at week 24 (snapshot algorithm; noninferiority margin of 6%).
Results:
Of 558 screened, 495 were randomized/treated (B/F/TAF n = 330; SBR n = 165). Overall, 32% were ciswomen, 2% transwomen, and 10% had an M184V/I mutation. At week 24, 0.6% on B/F/TAF vs 1.8% on SBR had HIV-1 RNA ≥50 copies/mL (difference −1.2%; 95% confidence interval −4.8% to 0.9%), demonstrating noninferiority of B/F/TAF vs SBR. Proportions with HIV-1 RNA <50 copies/mL at week 24 were 96% B/F/TAF and 95% SBR and remained high at week 48. No participant had treatment-emergent resistance to study drug. Treatments were well tolerated. Study drug-related adverse events, mostly grade 1, occurred in 10% of participants on B/F/TAF through week 48 and led to discontinuation in 9 participants through week 48.
Conclusions:
For Black Americans with HIV, switching to B/F/TAF was noninferior to continuing a variety of regimens, including those with pre-existing NRTI mutations.
INTRODUCTION
Among racial and ethnic groups in the United States, Black people have the highest incidence and prevalence of HIV/AIDS.1,2 Black Americans make up about 13% of the US population but account for 42% of HIV infections. In 2018, the rate of diagnosis of new HIV infections among Black Americans was 39.2 per 100,000, as compared with 4.8 among whites and 13.3 in the population as a whole. This disparity was even greater in women: The rate of new HIV diagnoses was 21.3 per 100,000 for Black women, nearly 13 times higher than the rate of 1.7 in white women. Moreover, the risk of death for Black Americans from HIV/AIDS is approximately 9 times that of their white counterparts.1,2 These and other racial disparities show the impacts of structural racism, which contribute to myriad inequities including access to systems of health care, education, and employment. Within the health care system, interpersonal discrimination based on race, gender, and sex also results in disparities in health outcomes.3–5 The social factors and structural inequalities that confound racial health disparities in the United States are challenging to disentangle6 and likewise may complicate clinical decision-making for patients with HIV despite evidence that neither race nor ethnic origin plays a significant role in HIV treatment outcomes when health care is more equitably distributed.7,8 And yet, despite overrepresentation in the US HIV epidemic and clinical need to address health disparities, in clinical trials of HIV treatment regimens, Black people living with HIV (PLWH) are consistently underrepresented.9–11 As a result, the findings of clinical trials may not necessarily be generalizable to Black Americans.
The single-tablet regimen of the integrase strand transfer inhibitor (INSTI) bictegravir plus the nucleoside reverse transcriptase inhibitors (NRTIs) emtricitabine and tenofovir alafenamide (B/F/TAF) is a recommended initial treatment for most people with HIV.12–14 Four randomized international trials have established that switching to B/F/TAF from other combination antiretroviral (ARV) regimens is safe and effective in a general population of virologically suppressed adults with and without resistance to NRTIs.15–18 Although the B/F/TAF clinical trials of treatment-naive PLWH enrolled relatively representative proportion of Black participants in the United States, the inclusion of Black PLWH in treatment-experienced studies was low. There is a clear need for clinical data focusing on the safety and efficacy of B/F/TAF switch in virologically suppressed Black American PLWH.
The primary objective of this phase 3b study was to evaluate the efficacy of switching from a regimen of 2 NRTIs and a third agent to a fixed-dose combination of B/F/TAF vs continuing their baseline regimen in virologically suppressed adults self-identifying as African American or Black.
METHODS
Study Design and Participants
This randomized, open-label, multicenter, active-controlled, phase 3b trial was conducted at 82 outpatient centers in the United States. We enrolled adults (aged 18 years and older) living with HIV who self-described as Black, African American, or mixed race that included Black. Eligible participants had documented plasma HIV-1 RNA levels <50 copies/mL for at least 12 months before screening while on a stable ARV regimen other than B/F/TAF that consisted of any 2 NRTIs along with an allowed third agent for at least 6 months. Participants had an estimated glomerular filtration rate (eGFR) ≥50 mL per minute (calculated by the Cockcroft–Gault equation) and no documented history of resistance to INSTIs or tenofovir (TFV) resistance defined as K65R/E/N mutations, ≥3 thymidine analog mutations, or T69 insertions (See Appendix, Supplemental Digital Content, http://links.lww.com/QAI/B674).
Participants were randomized (2:1 ratio) to either switch to B/F/TAF (50 mg/200 mg/25 mg) administered orally once daily without regard to food or stay on baseline regimen (SBR) (which was not provided by the study) from day 1 until week 24. After week 24, the SBR group switched to B/F/TAF for an additional 24 weeks. Participants who completed the study through week 48 and wished to continue B/F/TAF were given the option to receive B/F/TAF for up to an additional 24 weeks in an extension phase and attend study visits every 12 weeks. Randomization was stratified by the baseline third-agent ARV class at entry.
This trial was undertaken in accordance with the Declaration of Helsinki and approved by central or site-specific review boards or ethics committees. All participants provided written informed consent.
A computer-generated allocation sequence (block size of 6) was created by Bracket (San Francisco, CA). Study investigators determined eligibility, obtained a participant number, and received automated treatment assignment based on a randomization sequence.
Procedures
Participants had visits on day 1, weeks 4, 12, 24, 36, and 48, with an additional week 28 visit for the SBR group. Laboratory tests included hematological analyses, serum chemistries, cluster of differentiation-4 (CD4) T-cell counts, renal parameters (eGFR) (Covance Laboratories, Indianapolis, IN), and measurement of HIV-1 RNA plasma concentration (Roche TaqMan 2.0; Roche Diagnostics, Indianapolis, IN). Resistance testing by genotyping and phenotyping integrase and reverse transcriptase (Monogram Biosciences, Inc., South San Francisco, CA) was included for any participant with plasma HIV-1 RNA ≥200 copies/mL at confirmed virologic failure and who did not resuppress on study drug.
Safety was assessed by physical examinations, laboratory tests, recording of concomitant drugs, and recording of adverse events, which were coded using the Medical Dictionary for Regulatory Activities (MedDRA, version 22.1). Relatedness of adverse events to study treatment was determined by the investigator.
Outcomes
The primary efficacy endpoint was the proportion of participants with HIV-1 RNA ≥50 copies/mL at week 24 as defined by the US FDA-defined snapshot algorithm. The secondary efficacy endpoints were the proportion of participants with HIV-1 RNA ≥50 copies/mL at week 48, the proportion of participants with HIV-1 RNA <50 copies/mL at weeks 24 and 48, and the change in CD4 cell count from baseline to weeks 24 and 48.
Statistical Analysis
The study was powered using expected efficacy based on a pooled analysis of Black participants in studies of INSTI-based regimens.19 Assuming equal efficacy between the 2 treatment groups, a sample size of 480 participants randomized in a 2:1 ratio (320 in the B/F/TAF group and 160 in the SBR group) would achieve approximately 89% power to detect a prespecified noninferiority margin difference of 6% in the group proportions with HIV-1 RNA ≥50 copies/mL at week 24 (B/F/TAF minus SBR) by FDA snapshot algorithm, using a 1-sided alpha level of 0.025%. All randomized participants who received at least one dose of study treatment (either B/F/TAF or baseline regimen) on or after day 1 and did not have pre-existing INSTI resistance-associated mutations based on historical data were included in the primary efficacy analysis. The proportion with HIV-1 RNA ≥50 copies/mL was also assessed at week 48 as a secondary outcome. The proportions of participants with plasma HIV-1 RNA <50 copies/mL at weeks 24 and 48 by FDA snapshot algorithm were prespecified secondary efficacy outcomes, analyzed similarly to the primary efficacy endpoint but using a noninferiority margin of 10%.
Pre-existing drug resistance was assessed by cumulative historical and/or proviral DNA genotypes performed retrospectively from baseline samples. We performed subgroup analyses of the participants with plasma HIV-1 RNA <50 copies/mL at week 24 based on age, sex, baseline NRTI resistance, and specifically baseline M184V/I resistance. A per-protocol analysis was performed as a secondary outcome to assess proportions of participants with plasma HIV-1 RNA <50 copies/mL at week 24 by FDA snapshot algorithm. Weeks 24 and 48 secondary efficacy endpoints were also analyzed with a plasma HIV-1 RNA threshold of <20 copies/mL by FDA snapshot algorithm. We imputed missing as failure (M = F) and missing as excluded (M = E) to estimate the proportion of participants with plasma HIV-1 RNA <50 copies/mL at weeks 24 and 48.
Differences in percentages of participants with HIV-1 RNA <20 copies/mL, < 50 copies/mL or ≥50 copies/mL between treatment groups and 95% CI were calculated based on exact methods.
An analysis of variance model with treatment group as a fixed effect was used to analyze CD4 count and percentage. Safety was assessed in all randomized participants who received at least one dose of study treatment. Differences between groups in fasting lipids and body weight were tested using the two-sided Wilcoxon rank-sum test. Safety data were collected from the date the study drug was first given for up to 30 days after the last dose of study drug. Treatment satisfaction questionnaire scores were compared between groups using analysis of covariance adjusting for baseline score.
We used SAS Software Version 9.4 (SAS Institute Inc., Cary, NC) for all analyses. This study was conducted according to protocol and is registered with ClinicalTrials.gov, number NCT03631732.
RESULTS
Between August 28, 2018, and March 6, 2019, 558 individuals were screened for enrollment and 496 were randomized: 331 to B/F/TAF and 165 to SBR. One participant randomized to B/F/TAF never received treatment and was excluded from analysis. Of 330 participants randomized and treated with B/F/TAF, 320 (97%) continued treatment after the week 24 primary endpoint, and 163 of 165 (99%) participants randomized to SBR switched to B/F/TAF at week 24 (Fig. 1). Demographics and baseline characteristics were generally balanced between groups (Table 1). The median age overall was 49 years, 32% were ciswomen, 2% transwomen, and 10% had documented baseline M184V/I mutation. Median time on HIV treatment was 10 years, and median time on baseline ARV regimen was 2 years. Most participants (61%) were receiving an INSTI as a third agent at baseline.
FIGURE 1.

CONSORT diagram. B/F/TAF, bictegravir, emtricitabine, and tenofovir alafenamide; SBR, stay on baseline regimen.
TABLE 1.
Baseline Demographics and Disease Characteristics
| B/F/TAF (n = 330) |
SBR (Delayed Switch to B/F/TAF) (n = 165) |
|
| Median age, yrs (range) | 49 (18–79) | 49 (19–70) |
| Female sex at birth, n (%) | 101 (31) | 55 (33) |
| Gender identity, n (%) | ||
| Cisgender | 317 (96) | 159 (96) |
| Transgender | 6 (2) | 6 (4) |
| Other gender or prefer not to disclose | 7 (2) | 0 |
| Sexual orientation, n (%) | ||
| Heterosexual (female sex at birth) | 97 (29) | 52 (32) |
| Heterosexual (male sex at birth) | 62 (19) | 41 (25) |
| Gay or bisexual (male sex at birth) | 162 (49) | 67 (41) |
| Ethnicity, n (%) | ||
| Hispanic/latinx | 15 (5) | 5 (3) |
| Median CD4 count, cells/µL (Q1, Q3) | 747 (570, 922) | 758 (494, 969) |
| Median eGFRCG, mL/min (Q1, Q3) | 110 (88, 132) | 107 (86, 132) |
| Median weight, kg (Q1, Q3) | 88 (79, 103) | 89 (76, 104) |
| Median BMI, kg/m2 (Q1, Q3) | 29.2 (25.9, 34.0) | 29.3 (25.7, 34.3) |
| HBV coinfection, n (%) | 15 (5) | 3 (2) |
| Baseline NRTI backbone,* n (%) | ||
| F/TAF | 224 (68) | 107 (65) |
| F/TDF | 56 (17) | 34 (21) |
| Abacavir/lamivudine | 44 (13) | 24 (15) |
| Other | 4 (1) | 0 |
| Baseline 3rd agent,† n (%) | ||
| INSTI | 202 (61) | 99 (60) |
| NNRTI | 100 (30) | 51 (31) |
| PI | 30 (9) | 14 (9) |
| CCR5 antagonist | 0 | 1 (1) |
| Baseline ARV resistance,‡ n (%) | ||
| NRTI resistance | 44 (13) | 26 (16) |
| M184V/I | 31 (9) | 20 (12) |
| NNRTI resistance | 70 (21) | 32 (19) |
| PI resistance | 36 (11) | 26 (16) |
| INSTI resistance | 8 (2) | 3 (2) |
Two participants in the B/F/TAF group did not report a baseline NRTI backbone medication; both were protocol violations.
One participant in the B/F/TAF group reported both PI and INSTI; 1 subject in the B/F/TAF group reported both INSTI and NNRTI; both were protocol violations.
No baseline ARV resistance data B/F/TAF n = 17, SBR n = 7. Resistance assessed by cumulative historical or retrospective baseline proviral DNA genotypes.
BMI, body mass index; CCR5, C-C chemokine receptor type 5; eGFRCG, estimated glomerular filtration rate (calculated by Cockcroft–Gault equation); HBV, hepatitis B virus; NNRTI, non-nucleoside reverse transcriptase inhibitor; PI, protease inhibitor; Q, quartile.
Efficacy
Switching to B/F/TAF was noninferior to SBR for the primary outcome of proportion of participants with plasma HIV-1 RNA ≥50 copies/mL at week 24 as defined by the US FDA snapshot algorithm {0.6% (2 of 328 participants) vs 1.8% (3 of 165), difference −1.2% (95% confidence interval [CI]: −4.8% to 0.9%)} (Table 2, Fig. 2, panel A). Two participants were found to have baseline INSTI resistance based on historical genotype data and were excluded from the primary analysis because of this protocol violation; 1 participant each with Y143C and Q148K mutations, both were randomized to B/F/TAF and had HIV-1 RNA <50 copies/mL at week 24. The secondary efficacy outcome of proportion of participants with plasma HIV-1 RNA <50 copies/mL at week 24 was consistent with the primary endpoint and showed high rates of virologic suppression [B/F/TAF 96.3% (316 of 328 participants) vs SBR 94.5% (156 of 165)] (Table 2).
TABLE 2.
Virologic Outcomes at Week 24, Full Analysis Set
| Week 24 | B/F/TAF (n = 328) |
SBR (No Switch) (n = 165) |
B/F/TAF vs SBR |
| Difference in Percentages (95% CI)* | |||
| HIV-1 RNA <50 copies/mL | 316 (96.3%) | 156 (94.5%) | 1.8% (−2.0% to 6.8%) |
| HIV-1 RNA ≥50 copies/mL | 2 (0.6%) | 3 (1.8%) | −1.2% (−4.8% to 0.9%) |
| HIV-1 RNA ≥50 copies/mL | 2 (0.6%) | 3 (1.8%) | |
| Discontinued because of lack of efficacy | 0 | 0 | |
| Discontinued study drug because of AE/death and last available HIV-1 RNA ≥50 copies/mL | 0 | 0 | |
| Discontinued because of other reasons† and last available HIV-1 RNA ≥50 copies/mL | 0 | 0 | |
| No virologic data in the week 24 window | 10 (3.0%) | 6 (3.6%) | |
| Discontinued because of AE/death and last available HIV-1 RNA <50 copies/mL | 6 (1.8%) | 0 | |
| Discontinued because of other reasons† and last available HIV-1 RNA <50 copies/mL | 4 (2.1%) | 1 (0.6%) | |
| Missing data but on study drug | 0 | 5 (3.0%) | |
| HIV-1 RNA <50 copies/mL by per-protocol snapshot analysis‡ | 304/306 (99.3%) | 145/148 (98.0%) | 1.4% (−1.0% to 5.3%) |
| HIV-1 RNA <50 copies/mL by missing = failure§ | 321/328 (97.9%) | 156/165 (94.5%) | 3.3% (−0.2% to 8.2%) |
| HIV-1 RNA <50 copies/mL by missing = excluded§ | 321/323 (99.4%) | 156/159 (98.1%) | 1.3% (−0.8% to 4.9%) |
| HIV-1 RNA <20 copies/mL | 305/328 (93.0%) | 149/165 (90.3%) | 2.7% (−2.4% to 8.7%) |
| Hepatitis B virus DNA <29 IU/mL by missing = excluded‖ | 6/7 (86%) | 1/1 (100%) |
Data are n (%).
Virology outcomes are based on snapshot algorithm unless otherwise specified.
The week 24 window is between days 127 and 210 (inclusive).
Per-protocol analysis excluded patients in full analysis set who were off study drug at week 24 or had low adherence, that is, adherence ≤2.5th percentile among those in study or did not meet the inclusion criteria for baseline ARV medication or met inclusion/exclusion criteria for mutations detected before baseline or after baseline.
The difference in percentages of participants between treatment groups and their 95% CIs were calculated based on exact methods.
Other reasons include participants who discontinued study drug because of investigator's discretion, subject decision, lost to follow-up, noncompliance with study drug, protocol violation, pregnancy, and study terminated by sponsor.
The per-protocol analysis was performed in a similar manner to the primary endpoint but excluded participants with low adherence (below the 2.5th percentile), violation of key entry criteria, or who did not have a plasma HIV-1 RNA value in the week 24 analysis window because of reasons other than lack of efficacy.
Difference in percentages and 95% CI were based on a dichotomized response: HIV-1 RNA <50 copies/mL vs HIV-1 RNA ≥50 copies/mL or missing for missing = failure approach and HIV-1 RNA <50 copies/mL vs HIV-1 RNA ≥50 copies/mL for missing = excluded approach.
At baseline, 18 participants had HBV coinfection: 15 (5%) in the B/F/TAF group and 3 (2%) in the SBR group. At baseline, 13 of 15 in the B/F/TAF group and 2 of 3 in the SBR group had HBV DNA <29 IU per mL. The denominator for percentage is the number of participants in the full analysis set with HIV/HBV coinfection and with nonmissing HBV DNA at week 24.
FIGURE 2.
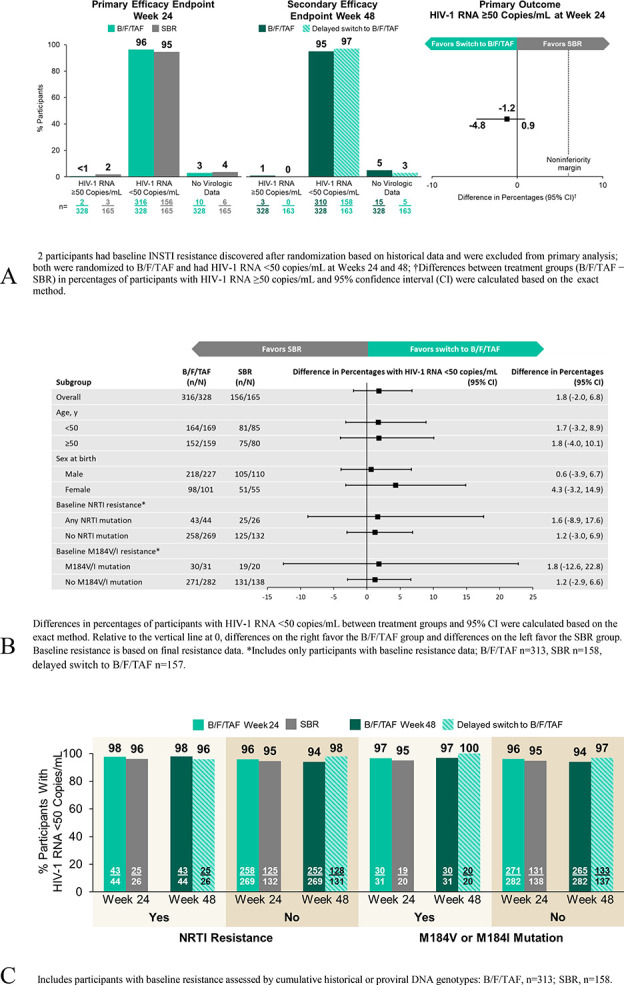
Virologic outcomes. A, FDA Snapshot algorithm. B, Treatment differences based on prespecified subgroups at week 24: HIV-1 RNA <50 copies/mL. C, Virologic outcomes at weeks 24 and 48 by baseline resistance profile.
After 48 weeks of receiving B/F/TAF, 95% (310 of 328) in the group randomized to B/F/TAF had HIV-1 RNA <50 copies/mL (Fig. 2, panel A; Appendix Table 4, Supplemental Digital Content, http://links.lww.com/QAI/B674). Two had HIV-1 RNA ≥50 copies/mL. One individual had an HIV-1 RNA level of 55 copies/mL at week 48 and switched back to the prestudy regimen. The other had an HIV-1 RNA level of 455 copies/mL at week 48, which resuppressed on continued treatment with B/F/TAF; no treatment-emergent resistance was detected. Of 163 participants randomized to SBR who switched to B/F/TAF at week 24, no participants had HIV-1 RNA ≥50 copies/mL; 158 (97%) had HIV-1 RNA <50 copies/mL at week 48.
Results from the per-protocol, M = F, and M = E analyses conducted at weeks 24 and 48 were consistent with the primary outcome (Table 2). Analysis of the proportion of participants with plasma HIV-1 RNA <20 copies/mL showed high and similar rates of virologic suppression at this lower threshold. Efficacy between B/F/TAF and SBR was similar across prespecified subgroups (Fig. 2, panel B), and testing for homogeneity found no significant interactions between treatment and subgroup. We observed no treatment differences in changes from baseline in CD4 cell count and CD4 percentage at week 24 (see Appendix Table 1, Supplemental Digital Content, http://links.lww.com/QAI/B674).
At baseline, 13% (44 of 328) in the B/F/TAF group and 16% (26 of 165) in the SBR group had NRTI resistance, including 9% (31 of 328) and 12% (20 of 165) with M184V/I mutations in the B/F/TAF and SBR groups, respectively. Efficacy by snapshot analysis was similar in participants with or without NRTI resistance (Fig. 2, panel C). Among participants with NRTI resistance at baseline 98% (43 of 44) on B/F/TAF and 96% (25 of 26) on SBR had HIV-1 RNA <50 copies/mL at week 24. Among participants with M184V or I mutations at baseline 97% (30 of 31) on B/F/TAF and 95% (19 of 20) on SBR had HIV-1 RNA <50 copies/mL at week 24. One participant with M184V on B/F/TAF discontinued after the baseline visit and did not have virologic data at week 24. In addition, 2% (8 of 328) in the B/F/TAF group and 2% (3 of 165) in the SBR group had primary INSTI resistance detected at baseline, all of whom had HIV-1 RNA <50 copies/mL at week 24.
Altogether, 68 participants with baseline NRTI resistance (including 50 with M184V/I) received B/F/TAF during the study and had postswitch virologic data. At study week 48, all 68 (100%) had HIV-1 RNA <50 copies/mL; 43 randomized to B/F/TAF had 48 weeks of treatment, and 25 with a delayed switch from SBR had 24 weeks of treatment with B/F/TAF. In addition, a total of 11 participants with baseline INSTI resistance received B/F/TAF, and all 11 (100%) had HIV-1 RNA <50 copies/mL at week 48.
One participant in each treatment group met criteria for resistance testing through week 24. No treatment-emergent resistance to any components of the regimens was detected in either group during the study.
Safety
Through week 24, 64% of participants who switched to B/F/TAF (210 of 330) experienced at least one adverse event compared with 52% (85 of 165) of those on SBR (Table 3). The most common adverse events in the B/F/TAF and SBR groups were upper respiratory infection, diarrhea, cough, arthralgia, and bronchitis. The proportion of participants who experienced at least one grade 3 or 4 adverse event was the same in both groups (5%) as was the proportion of participants experiencing a serious adverse event (4%). Investigators attributed events to treatment in 11% (36 participants) in the B/F/TAF group and none in the SBR group. For most participants who had adverse events attributed to treatment, events were either mild (25 of 36) or moderate (8 of 36) and resolved on treatment. Three of these 36 participants experienced grade 3 events: one (somnolence) that resolved on study drug and 2 that led to study drug discontinuation (headache n = 1, agitation, psychomotor hyperactivity, and anxiety n = 1). In total, 7 (2%) of participants in the B/F/TAF group and none in the SBR group had adverse events leading to discontinuation up to week 24. No deaths or pregnancies were reported.
TABLE 3.
Safety Through Week 24
| B/F/TAF (n = 330) |
SBR (No Switch) (n = 165) |
|
| Any adverse event | 210 (64) | 85 (52) |
| Adverse events in >3% of participants | ||
| Upper respiratory tract infection | 20 (6) | 6 (4) |
| Diarrhea | 11 (3) | 5 (3) |
| Cough | 9 (3) | 6 (4) |
| Arthralgia | 11 (3) | 2 (1) |
| Bronchitis | 5 (2) | 7 (4) |
| Grade 3 or 4 adverse event | 17 (5) | 8 (5) |
| Serious adverse event | 13 (4) | 7 (4) |
| Adverse event leading to study drug discontinuation* | 7 (2) | 0 |
| Treatment-related adverse event | 36 (11) | 0 |
| Death | 0 | 0 |
| Laboratory abnormalities | ||
| Of any grade | 215 (65) | 111 (67) |
| Grade 3 | 24 (7) | 10 (6) |
| Grade 4 | 3 (1) | 0 |
Data are n (%).
Adverse events leading to study drug discontinuation in the B/F/TAF group included nightmare† (n = 1); headache† (n = 1); acute kidney injury (n = 1); diarrhea,† dry mouth,† psychomotor hyperactivity,† agitation,† anxiety,† and insomnia† (n = 1); abdominal distension† and flatulence† (n = 1); diarrhea† (n = 1); and migraine† (n = 1).
Reported as treatment related.
Incidence of grade 3 or 4 laboratory abnormalities through week 24 was comparable between groups: 8% (27 of 329) B/F/TAF and 6% (10 of 165) SBR (see Appendix Table 2, Supplemental Digital Content, http://links.lww.com/QAI/B674). Median [interquartile range (IQR)] change from baseline in eGFR at week 24 was −4.6 (−12.1, 2.6) mL/min for B/F/TAF (n = 318) vs 1.1 (−7.1, 7.8) mL/min for SBR (n = 160); P < 0.001; consistent with the previously described inhibition of tubular secretion of creatinine without affecting renal glomerular function by bictegravir. In the B/F/TAF group, one participant with prostate cancer discontinued study drug because of acute kidney injury secondary to urinary obstruction with hydronephrosis; this adverse event was judged by the investigator as not related to study drug. There were no discontinuations because of renal adverse effects in the SBR group and no reported cases of proximal renal tubulopathy in either group.
A cumulative analysis of safety was performed for all 493 participants treated with B/F/TAF when the last participant reached the week 48 endpoint. At the time of the analysis, the median (IQR) exposure to B/F/TAF was of 61 (56–68) weeks among those randomized to B/F/TAF and 38 (32–44) weeks for those with delayed switch to B/F/TAF; B/F/TAF was well tolerated during the study. Overall, 367 (74%) had at least one adverse event (see Appendix Table 5, Supplemental Digital Content, http://links.lww.com/QAI/B674). Three individuals who received B/F/TAF discontinued treatment after week 24 because of adverse events. Grade 3 or 4 laboratory abnormalities were reported for 60 participants (12%). One death was reported because of a gunshot wound; no pregnancies were reported.
Participants randomized to the B/F/TAF group had higher baseline levels of fasting total and low-density lipoprotein cholesterol and triglycerides compared with those randomized to SBR (P = 0.006 P = 0.02, and P = 0.03, respectively) (see Appendix Table 3, Supplemental Digital Content, http://links.lww.com/QAI/B674). At week 24, treatment differences in changes from baseline were small but statistically significant for fasting total cholesterol (−5-mg/dL B/F/TAF vs −2-mg/dL SBR; P = 0.006), high-density lipoprotein cholesterol (−2-mg/dL B/F/TAF vs −1-mg/dL SBR; P = 0.05), and triglycerides (−5 vs 3 mg/dL; P = 0.01). No significant treatment differences were noted for changes from baseline in low-density lipoprotein cholesterol or total cholesterol:high-density lipoprotein ratio at week 24. A similar proportion of participants in each group were taking lipid-lowering medication at baseline and initiated lipid-lowering agents through week 24. In sensitivity analyses excluding participants on lipid-lowering medications, changes from baseline in fasting lipids were consistent with the full analyses.
Median (IQR) weight at baseline was 88 (79–103) kg in the B/F/TAF group and 89 (76–104) kg in the SBR group (P = 0.66), corresponding to a median (IQR) body mass index of 29 (26–34) in each group. Median (IQR) weight change from baseline at week 24 was +0.9 (−1.3, +3.0) kg in the B/F/TAF group and +0.2 (−1.7, +2.0) kg in the SBR group (P = 0.09). At week 48, median (IQR) change in body weight remained at 0.9 kg (−1.5, 4.1) in the B/F/TAF arm. Post hoc exploratory analyses of the change in weight were performed in subgroups by sex at birth for all participants and by baseline NRTIs in those switched to B/F/TAF (see Appendix Fig. 1, Supplemental Digital Content, http://links.lww.com/QAI/B674). In the male sex at birth subanalysis, median change in weight from baseline was similar between groups. Among female sex at birth participants, a treatment difference was noted in median (IQR) change in weight from baseline at week 24 [+1.0 (−1.4, 2.7) kg B/F/TAF, −0.4 (−1.8, 1.2) kg SBR; P = 0.03]. Participants who switched to B/F/TAF from a TFV disoproxil fumarate (TDF)-containing regimen had median (IQR) weight gain of +1.8 (−0.8, 4.3) kg at week 24 compared with +0.8 (−1.3, 2.5) kg and +0.7 (−2.0, 3.0) kg for those taking TAF- and abacavir (ABC)-containing regimens respectively (P = 0.0235 TDF vs TAF; P = 0.89 ABC vs TAF).
Among the 18 HIV/HBV-coinfected participants, there were no hepatic adverse events, no grade 3 or 4 adverse events, and no participants interrupted or discontinued their study regimen because of an adverse event.
Based on responses using the HIV Treatment Satisfaction Questionnaire, median (IQR) scores were higher with B/F/TAF than SBR at week 24 [29 (24–30) vs 28 (7–30), P < 0.001], indicating a small but significant difference favoring switch to B/F/TAF.
DISCUSSION
In this randomized, open-label, multicenter trial, we found that virologically suppressed Black adults with HIV in the United States can switch from a regimen of 2 NRTIs plus a third agent to B/F/TAF with no loss of virologic efficacy. Few of those who switched to B/F/TAF had HIV RNA ≥50 copies/mL through week 48, and the rate of virologic suppression was high in those with pre-existing resistance to NRTIs. None of the participants experienced treatment-emergent resistance. Treatment satisfaction was higher for participants after switching to B/F/TAF.
Community advisors who participated in the study design and oversight set goals of inclusion based on sex at birth, gender, and sexual orientation to ensure representative inclusion of Black Americans with HIV. Study sites were specifically selected to mirror the HIV epidemic among Black PLWH in the United States. In doing so, more than 62% of participants were enrolled in Southern states, as defined by the CDC, with others mainly from urban areas with a high prevalence of HIV among Black people. Despite meeting the enrollment goals, the size of some individual subgroups of participants, particularly transgender or other nonbinary gender groups in the study, remains small and insufficient for meaningful subanalyses. The impacts of structural racism and inequities in access to health care and medical research for Black Americans20 are magnified among Black transgender PLWH, and better data on which to inform their clinical care should remain a priority.
Our study enrolled participants on a variety of treatment regimens and included those with a history of treatment failure or with documented resistance to NRTIs, both patient groups are commonly seen in clinical practice but often excluded from clinical trials. Pre-existing NRTI resistance, including the M184V/I mutation, did not impact the efficacy of switching to B/F/TAF.
Participants in both groups gained weight while on study, and there was no difference in weight gain from baseline between those randomized to B/F/TAF vs SBR. For those starting B/F/TAF, weight gain was 0.9 kg in the first 24 weeks and plateaued thereafter. In subgroup analyses, cisgender women and those previously on TDF-containing regimens switching to B/F/TAF had greater weight gain compared with those on SBR; treatment differences in weight gain were not significant for men. TDF has been associated with weight suppression in studies of both treatment and HIV pre-exposure prophylaxis or PrEP.18,22,23–26 The large variety of baseline regimens precluded meaningful description of the influence each may have on weight. Altogether, these findings do not suggest a substantial weight gain for Black men and women changing to B/F/TAF.
Limitations to this trial include its open-label design. Although this design is unlikely to have affected the primary endpoint, which is a laboratory value not subject to bias, it may have biased the reporting of adverse events, both by participants and investigators.
The safety profile of B/F/TAF was consistent with that observed in other phase 3 studies. In the randomized phase, incidence of any adverse event and treatment-related adverse events was higher with B/F/TAF than SBR; however, this finding is not unexpected in our open-label trial and common in open-label studies in which a switch to a new drug is compared with continuing a previously well-tolerated baseline regimen because of reporting bias on the part of participants as well as ascertainment or attribution bias on the part of investigators.16,21 Most events were mild, and the proportions of individuals who experienced grade 3 or 4 adverse events and serious adverse events were the same in both groups. Less than 2% of all individuals (9 of 493) who switched to B/F/TAF had adverse events leading to discontinuation of treatment.
Overall, our results demonstrate that, among Black Americans living with HIV, switching to B/F/TAF from a variety of baseline suppressive regimens was efficacious and safe. Results from this study complement those from other large clinical trials that demonstrated the efficacy, tolerability, and lack of resistance of B/F/TAF. Our results draw us closer to fulfilling a critical need to evaluate infectious disease treatments in a disproportionately affected population.27
CONCLUSION
B/F/TAF is safe and effective for Black Americans with HIV switching from a variety of regimens, including those with TFV sensitivity but pre-existing NRTI resistance.
NOTES
Community advisors representing HIV advocacy and care organizations across North America reviewed the protocol, provided input into study design, contributed to training for study personnel, and set goals for inclusion across participant groups according to sex at birth, gender identity, and sexual orientation. A study advisory committee composed of 3 community advisors, and 3 clinical site personnel provided ongoing monitoring and input throughout the study.
Supplementary Material
ACKNOWLEDGMENTS
The authors thank the individuals who participated in this trial and their partners and families, the BRAAVE principal investigators (see Appendix page 1, Supplemental Digital Content, http://links.lww.com/QAI/B674) and their staff, the Study Advisory Committee members Martez Smith-University of Rochester, Danielle Campbell—ATAC/LA Women's HIV/AIDS Task Force, PJ Moton-Poole—AIDS United, Linda Kirkman, and Dr. A.K.W., and the Gilead study staff. Editorial assistance was provided under the direction of the authors by David McNeel, MS, and Anna Kido, BS, of Gilead Sciences and was funded by Gilead. This work was funded by Gilead Sciences.
Footnotes
Presented in part at CROI 2020 (virtual); March 8–11 2020, Boston, MA, Oral 2979; and IDWeek 2020 (virtual); October 21–25, 2020, Virtual Event, Poster 1046.
P.K. reports grants from Gilead Sciences, Inc. (Gilead), GlaxoSmithKline (GSK), Merck, and Theratechnologies, has received personal fees for participating in advisory boards for GSK, Merck, and Theratechnologies, and is a shareholder of stock from Gilead, GSK, Johnson & Johnson, Merck, and Pfizer. M.S. has received research grants and support awarded to his institution from Gilead, Merck, and ViiV Healthcare. A.K.W. has received research grants from Gilead, Janssen, Pfizer, and GSK and has served on advisory boards for Gilead and Janssen. I.B. has received speakers' bureau honoraria from Gilead, ViiV, and Janssen. C.O.H. has received grant support from the National Institutes of Health (K23HL116209) and has served as consultant to Gilead Sciences. M.N.R. has served on the speakers' bureau for Gilead, Janssen, AbbVie, and Allergan and has consulted for Gilead, ViiV, and Merck. C.M. reports receiving research grants from Gilead, Janssen, Merck, and ViiV and speaker's bureau income from Gilead and Merck. C.B., K.A., S.E.C., D.M.B., and H.M. are employees of Gilead and shareholders of Gilead stock. The remaining authors have no conflicts of interest to disclose.
Supplemental digital content is available for this article. Direct URL citations appear in the printed text and are provided in the HTML and PDF versions of this article on the journal's Web site (www.jaids.com).
REFERENCES
- 1.Centers for Disease Control and Prevention. HIV Surveillance Report, 2018. Vol 31. 2020. Available at: http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html. Accessed June 26, 2020. [Google Scholar]
- 2.Centers for Disease Control and Prevention. Estimated HIV Incidence and Prevalence in the United States, 2014–2018. HIV Surveillance Supplemental Report 2020. Vol 25. 2020. Available at: http://www.cdc.gov/hiv/library/reports/hiv-surveillance.html. Accessed June 26, 2020. [Google Scholar]
- 3.Oramasionwu CU, Brown CM, Lawson KA, et al. Differences in national antiretroviral prescribing patterns between black and white patients with HIV/AIDS, 1996–2006 [author manuscript]. South Med J. 2011;104:794–800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Institute of Medicine (US) Committee on Understanding and Eliminating Racial and Ethnic Disparities in Health Care, Smedley BD, Stith AY, Nelson AR, eds. Unequal Treatment: Confronting Racial and Ethnic Disparities in Health Care. Washington (DC): National Academies Press (US); 2003. [PubMed] [Google Scholar]
- 5.Paradies Y, Truong M, Priest N. A systematic review of the extent and measurement of healthcare provider racism. J Gen Intern Med. 2014;29:364–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Ribaudo HJ, Smith KY, Robbins GK, et al. Racial differences in response to antiretroviral therapy for HIV infection: an AIDS clinical trials group (ACTG) study analysis. Clin Infect Dis. 2013;57:1607–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Jensen-Fangel S, Pedersen L, Pedersen C, et al. The effect of race/ethnicity on the outcome of highly active antiretroviral therapy for human immunodeficiency virus type 1-infected patients. Clin Infect Dis. 2002;35:1541–1548. [DOI] [PubMed] [Google Scholar]
- 8.Moore RD, Keruly JC, Bartlett JG. Improvement in the health of HIV-infected persons in care: reducing disparities. Clin Infect Dis. 2012;55:1242–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Walmsley SL, Antela A, Clumeck N, et al. Dolutegravir plus abacavir-lamivudine for the treatment of HIV-1 infection. N Engl J Med. 2013;369:1807–1818. [DOI] [PubMed] [Google Scholar]
- 10.Orkin C, Arasteh K, Górgolas Hernández-Mora M, et al. Long-acting cabotegravir and rilpivirine after oral induction for HIV-1 infection. N Engl J Med. 2020;382:1124–1135. [DOI] [PubMed] [Google Scholar]
- 11.Molina JM, Squires K, Sax PE, et al. Doravirine versus ritonavir-boosted darunavir in antiretroviral-naive adults with HIV-1 (DRIVE-FORWARD): 48-week results of a randomised, double-blind, phase 3, non-inferiority trial. Lancet HIV. 2018;5:e211–e220. [DOI] [PubMed] [Google Scholar]
- 12.Panel on Antiretroviral Guidelines for Adults and Adolescents. Guidelines for the Use of Antiretroviral Agents in Adults and Adolescents With HIV, DHHS. Available at: http://www.aidsinfo.nih.gov/ContentFiles/AdultandAdolescentGL.pdf. Accessed June 26, 2020. [Google Scholar]
- 13.European AIDS Clinical Society Guidelines Version 10, November 2019. Available at: https://www.eacsociety.org/guidelines/eacs-guidelines/eacs-guidelines.html. Accessed June 26, 2020.
- 14.Saag MS, Benson CA, Gandhi RT, et al. Antiretroviral drugs for treatment and prevention of HIV infection in adults: 2018 recommendations of the International Antiviral Society-USA panel. JAMA. 2018;320:379–396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Molina JM, Ward D, Brar I, et al. Switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from dolutegravir plus abacavir and lamivudine in virologically suppressed adults with HIV-1: 48 week results of a randomised, double-blind, multicentre, active-controlled, phase 3, non-inferiority trial. Lancet HIV. 2018;5:e357–e365. [DOI] [PubMed] [Google Scholar]
- 16.Daar ES, DeJesus E, Ruane P, et al. Efficacy and safety of switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide from boosted protease inhibitor-based regimens in virologically suppressed adults with HIV-1: 48 week results of a randomised, open-label, multicentre, phase 3, non-inferiority trial. Lancet HIV. 2018;5:e347–e356. [DOI] [PubMed] [Google Scholar]
- 17.Kityo C, Hagins D, Koenig E, et al. Switching to fixed-dose bictegravir, emtricitabine, and tenofovir alafenamide (B/F/TAF) in virologically suppressed HIV-1 infected women: a randomized, open-label, multicenter, active-controlled, phase 3, noninferiority trial. J Acquir Immune Defic Syndr. 2019;82:321–328. [DOI] [PubMed] [Google Scholar]
- 18.Sax PE, Rockstroh JK, Luetkemeyer AF, et al. Switching to bictegravir, emtricitabine, and tenofovir alafenamide in virologically suppressed adults with HIV. Clin Infect Dis. 2020:ciaa988. doi: 10.1093/cid/ciaa988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.DeJesus E, Villanueva JFA, Lopez JR, et al. Tenofovir Alafenamide (TAF) Versus Tenofovir Disoproxil Fumarate (TDF) in Hispanic/Latinx and Black Participants: Efficacy, Bone and Renal Safety Results from a Pooled Analysis of 7 Clinical Trials. Poster presented at Poster 318: IDWeek 2019; October 2–6, 2019; Washington, DC.
- 20.Chen A, Wright H, Itana H, et al. Representation of women and minorities in clinical trials for new molecular entities and original therapeutic biologics approved by FDA CDER from 2013 to 2015. J Womens Health (Larchmt). 2018;27:418–429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Trottier B, Lake JE, Logue K, et al. Dolutegravir/abacavir/lamivudine versus current ART in virally suppressed patients (STRIIVING): a 48-week, randomized, non-inferiority, open-label, phase IIIb study. Antivir Ther. 2017;22:295–305. [DOI] [PubMed] [Google Scholar]
- 22.Sax PE, Erlandson KM, Lake JE, et al. Weight gain following initiation of antiretroviral therapy: risk factors in randomized comparative clinical trials. Clin Infect Dis. 2020;71:1379–1389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cahn P, Madero JS, Arribas JR, et al. Dolutegravir plus lamivudine versus dolutegravir plus tenofovir disoproxil fumarate and emtricitabine in antiretroviral-naive adults with HIV-1 infection (GEMINI-1 and GEMINI-2): week 48 results from two multicentre, double-blind, randomised, non-inferiority, phase 3 trials. Lancet. 2019;393:143–155. [DOI] [PubMed] [Google Scholar]
- 24.Glidden DV, Mulligan K, McMahan V, et al. Metabolic effects of preexposure prophylaxis with coformulated tenofovir disoproxil fumarate and emtricitabine. Clin Infect Dis. 2018;67:411–419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Grant RM, Lama JR, Anderson PL, et al. Preexposure chemoprophylaxis for HIV prevention in men who have sex with men. N Engl J Med. 2010;363:2587–2599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Mayer KH, Molina JM, Thompson MA, et al. Emtricitabine and tenofovir alafenamide vs emtricitabine and tenofovir disoproxil fumarate for HIV pre-exposure prophylaxis (DISCOVER): primary results from a randomised, double-blind, multicentre, active-controlled, phase 3, non-inferiority trial. Lancet. 2020;396:239–254. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Evens MK. Covid's color line—infectious disease, inequity, and racial justice. N Engl J Med. 2020;383:408–410. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.