

Abstract
Over the past 25 years, antibody therapeutics have emerged as clinically and commercially successful pharmaceuticals, rapidly approaching 100 Food and Drug Administration approvals with combined annual global sales exceeding $100 billion. Nearly half of the marketed antibody therapeutics are used in oncology. These antibody-based cancer therapies can be broken down into three categories based on their different mechanisms of action, i.e. (i) natural properties, (ii) engagement of cytotoxic T cells, and (iii) delivery of cytotoxic payloads. Both natural and engineered properties of the antibody molecule are founded on its highly stable and modular architecture. In this review we provide an overview and outlook of the rapidly evolving landscape of antibody-based cancer therapy.
Introduction
Since the Food and Drug Administration (FDA) approval of the first mAbs for cancer therapy, CD20-targeting rituximab (Rituxan®) in 1997 and HER2-targeting trastuzumab (Herceptin®) in 1998, mAbs have become highly successful pharmaceuticals. Twenty years later, the top 15 drugs based on global sales across all indications include five cancer mAbs with combined revenues of approximately $40 billion [1]. This list is topped by immune checkpoint inhibitors (ICIs) pembrolizumab (Keytruda®) and nivolumab (Opdivo®) which both target PD1. The current number of FDA-approved and marketed antibody-based cancer therapies is 43 and includes a variety of formats, targets, and indications (Table 1). Initially, the success of mAbs as pharmaceuticals was driven by natural properties of the antibody molecule, such as high affinity and specificity to virtually any antigen, the ability to block receptor-ligand interactions, long circulatory half-life, and engagement of proteins and cells of the innate immune system and in doing so mediate complement-dependent cytotoxicity (CDC), antibody-dependent cellular cytotoxicity (ADCC), and antibody-dependent cellular phagocytosis (ADCP) (marked in green in Table 1). Increasingly, mAbs with engineered properties not found in nature are utilized for cancer therapy. These can be grouped into antibody therapeutics that either engage cytotoxic T cells or deliver cytotoxic payloads (marked in blue and orange, respectively, in Table 1). FDA-approved antibody therapeutics that deploy T cells as their mechanism of action (MOA) include seven ICIs, one T cell-engaging bispecific antibody (T-biAb), and five chimeric antigen receptor T cells (CAR-Ts). FDA-approved payload-delivering antibody therapeutics include one radioimmunoconjugate, nine antibody-drug conjugates (ADCs), and one immunotoxin. A rich pipeline of antibody therapeutics from all three MOA categories is at various stages of preclinical and clinical investigations, with another record number of FDA approvals anticipated for 2021 [2].
Table 1. FDA-approved and marketed antibody-based cancer therapies.
Currently, cancer patients in the United States have access to 43 different antibody therapeutics, with more than half added in the past five years. The differently colored rows indicate MOAs based on natural or enhanced natural properties of mAbs (green), on engaging cytotoxic T cells (blue), and on delivering cytotoxic payloads (orange).
| Name | Format | Payload | Target | Cancer | Approval |
|---|---|---|---|---|---|
| rituximab (Rituxan®) | chimeric mouse/human IgG1κ | none | CD20 | B-NHL, CLL | 1997 |
| trastuzumab (Herceptin®) | humanized IgG1κ | none | HER2 | breast, stomach | 1998 |
| ibritumomab tiuxetan (Zevalin®) | mouse IgG1κ | 90Y | CD20 | B-NHL | 2002 |
| cetuximab (Erbitux®) | chimeric mouse/human IgG1κ | none | EGFR | colorectal, h & n | 2004 |
| bevacizumab (Avastin®) | humanized IgG1κ | none | VEGF | colorectal, lung, brain, kidney, cervical, ovarian, fallopian, peritoneal, liver | 2004 |
| panitumumab (Vectibix®) | human IgG2κ | none | EGFR | colorectal | 2006 |
| ofatumumab (Arzerra®) | human IgG1κ | none | CD20 | CLL | 2009 |
| ipilimumab (Yervoy®) | human IgG1κ | none | CTLA4 | melanoma, kidney, MSI-H/dMMR colorectal, liver, lung, mesothelioma | 2011 |
| brentuximab vedotin (Adcetris®) | chimeric mouse/human IgG1κ | auristatin | CD30 | HL, T-NHL | 2011 |
| pertuzumab (Perjeta®) | humanized IgG1κ | none | HER2 | breast | 2012 |
| ado-trastuzumab emtansine (Kadcyla®) | humanized IgG1κ | maytansine | HER2 | breast | 2013 |
| obinutuzumab (Gazyva®) | humanized IgG1κ (glycoengineered Fc) | none | CD20 | CLL, B-NHL | 2013 |
| ramucirumab (Cyramza®) | human IgG1κ | none | VEGFR2 | stomach, colorectal, liver, lung | 2014 |
| pembrolizumab (Keytruda®) | humanized IgG4κ | none | PD1 | melanoma, lung, h & n, HL, bladder, MSI-H/dMMR, stomach, cervical, B-NHL, liver, kidney, esophageal, endometrial, TMB-H, skin, breast (TNBC) | 2014 |
| blinatumomab (Blincyto®) | mouse (scFv)2 (BiTE) | none | CD19×CD3 | B-ALL | 2014 |
| nivolumab (Opdivo®) | human IgG4κ | none | PD1 | melanoma, lung, kidney, HL, h & n, bladder, MSI-H/dMMR colorectal, liver, esophageal, mesothelioma | 2014 |
| dinutuximab (Unituxin®) | chimeric mouse/human IgG1κ | none | GD2 | neuroblastoma | 2015 |
| daratumumab (Darzalex®) | human IgG1κ | none | CD38 | multiple myeloma | 2015 |
| necitumumab (Portrazza®) | human IgG1κ | none | EGFR | lung | 2015 |
| elotuzumab (Empliciti®) | humanized IgG1κ | none | SLAMF7 | multiple myeloma | 2015 |
| atezolizumab (Tecentriq®) | humanized IgG1κ (aglycosylated Fc) | none | PDL1 | bladder, lung, breast (TNBC), liver, melanoma | 2016 |
| olaratumab (Lartruvo®) | human IgG1κ | none | PDGFRA | sarcoma | 2016 |
| avelumab (Bavencio®) | human IgG1λ | none | PDL1 | Merkel cell carcinoma, bladder, kidney | 2017 |
| durvalumab (Imfinzi®) | human IgG1κ (engineered Fc) | none | PDL1 | lung | 2017 |
| inotuzumab ozogamicin (Besponsa®) | humanized IgG4κ | calicheamicin | CD22 | B-ALL | 2017 |
| tisagenlecleucel (Kymriah®) | mouse scFv-based CAR-T | T cell | CD19 | B-ALL, B-NHL (DLBCL, FL) | 2017 |
| gemtuzumab ozogamicin (Mylotarg®) | humanized IgG4κ | calicheamicin | CD33 | AML | 2017 |
| axicabtagene ciloleucel (Yescarta®) | mouse scFv-based CAR-T | T cell | CD19 | B-NHL (DLBCL, FL) | 2017 |
| mogamulizumab-kpkc (Poteligeo®) | humanized IgG1κ (afucosylated Fc) | none | CCR4 | T-NHL | 2018 |
| moxetumomab pasudotox-tdfk (Lumoxiti®) | mouse dsFv | bacterial toxin | CD22 | B-NHL (hairy cell leukemia) | 2018 |
| cemiplimab-rwlc (Libtayo®) | human IgG4κ (S228P hinge) | none | PD1 | cutaneous squamous cell carcinoma, basal cell carcinoma, lung | 2018 |
| polatuzumab vedotin-piiq (Polivy®) | humanized IgG1κ | auristatin | CD79B | B-NHL (DLBCL) | 2019 |
| enfortumab vedotin-ejfv (Padcev®) | human IgG1κ | auristatin | NECTIN4 | bladder | 2019 |
| fam-trastuzumab deruxtecan-nxki (Enhertu®) | humanized IgG1κ | camptothecin | HER2 | breast, stomach | 2019 |
| isatuximab-irfc (Sarclisa®) | chimeric IgG1κ | none | CD38 | multiple myeloma | 2020 |
| sacituzumab govitecan-hziy (Trodelvy®) | humanized IgG1κ | camptothecin | TROP2 | breast (TNBC), bladder | 2020 |
| brexucabtagene autoleucel (Tecartus®) | mouse scFv-based CAR-T | T cell | CD19 | B-NHL (mantle cell lymphoma) | 2020 |
| tafasitamab-cxix (Monjuvi®) | humanized IgG1κ (engineered Fc) | none | CD19 | B-NHL (DLBCL) | 2020 |
| belantamab mafodotin-blmf (Blenrep®) | humanized IgG1κ (afucosylated Fc) | auristatin | BCMA | multiple myeloma | 2020 |
| naxitamab-gqgk (Danyelza®) | humanized IgG1κ | none | GD2 | neuroblastoma | 2020 |
| margetuximab-cmkb (Margenza®) | chimeric IgG1κ (engineered Fc) | none | HER2 | breast | 2020 |
| lisocabtagene maraleucel (Breyanzi®) | mouse scFv-based CAR-T | T cell | CD19 | B-NHL (DLBCL, FL) | 2021 |
| idecabtagene vicleucel (Abecma®) | mouse scFv-based CAR-T | T cell | BCMA | multiple myeloma | 2021 |
Antibodies as pharmaceuticals
At the core of its success as a pharmaceutical is the highly stable and modular architecture of the antibody molecule, reflecting its evolution to a key guardian of the vertebrate immune system adapted to physically, chemically, and biologically harsh extracellular environments. Its building block, the immunoglobulin (Ig) fold, is a β-sandwich composed of two disulfide-linked antiparallel β-sheets with protruding β-turns. Both variable and constant domains of the antibody molecule are Ig folds. In variable domains, three of the β-turns serve as complementarity determining regions (CDRs) with hypervariable amino acid sequences. The most common format of both natural and synthetic human antibodies is the IgG1 molecule (Figure 1). Its concentration in the blood is 5–10 g/L, comprising more than half of all immunoglobulins. Of the 43 FDA-approved antibody-based cancer therapies 30 have an IgG1 format (Table 1). The ~150-kDa IgG1 molecule is composed of two identical ~25-kDa light chains and two identical ~50-kDa heavy chains that are covalently connected by four interchain disulfide bridges. Both light and heavy chain have an N-terminal variable domain (VL and VH, respectively), each contributing three CDRs which collectively comprise the paratope of the ~25-kDa Fv fragment that binds antigens with high affinity and specificity, followed by one and three constant domains, respectively. The ~50-kDa Fab fragment encompasses the Fv fragment and the adjacent constant domain of the light chain (CL) and first constant domain of the heavy chain (CH1). The two C-terminal constant domains of the heavy chain (CH2 and CH3) form the Fc fragment of the IgG1 molecule which mediates its prolonged circulatory half-life through interaction with the neonatal Fc receptor (FcRn) and its effector functions through interaction with complement component C1q (triggering CDC) and Fcγ receptors (FcγRs) I, IIA, and IIIA (triggering ADCC and ADCP) (Figure 1). The only conserved N-glycosylation site is an asparagine in CH2. The two branched glycans of the IgG1 molecule and their composition impact the effector functions, specifically the interaction of the Fc fragment with FcγRs and C1q, and as such have been subjected to glycoengineering [3]. For example, the absence of fucose on the glycans increases binding to FcγRIIIA, enhancing ADCC. Both stability and modularity of the antibody molecule are founded on the Ig fold itself and its intramolecular and intermolecular assembly into multidomain and multichain proteins. First, the robust tertiary structure of the Ig fold facilitates rational design and directed evolution. An example of rational design is the grafting of the six CDRs from a nonhuman to a human scaffold, a process known as humanization. The randomization of amino acid sequences in the CDRs, scaffold, or both, followed by their selection by phage display, yeast display, or other techniques, is an example of directed evolution toward affinity maturation. Second, the quaternary structure of the IgG1 molecule is based on strong variable (VL-VH) and constant domain (CL-CH1 and CH3-CH3) interactions that are further stabilized by interchain disulfide bridges (Figure 1). This highly defined assembly has enabled chimerization, the combination of nonhuman variable domains with human constant domains as well as numerous other synthetic assemblies to tailor valency, avidity, specificity, circulatory half-life, and effector functions.
Figure 1. Structural and functional modularity of the antibody molecule.
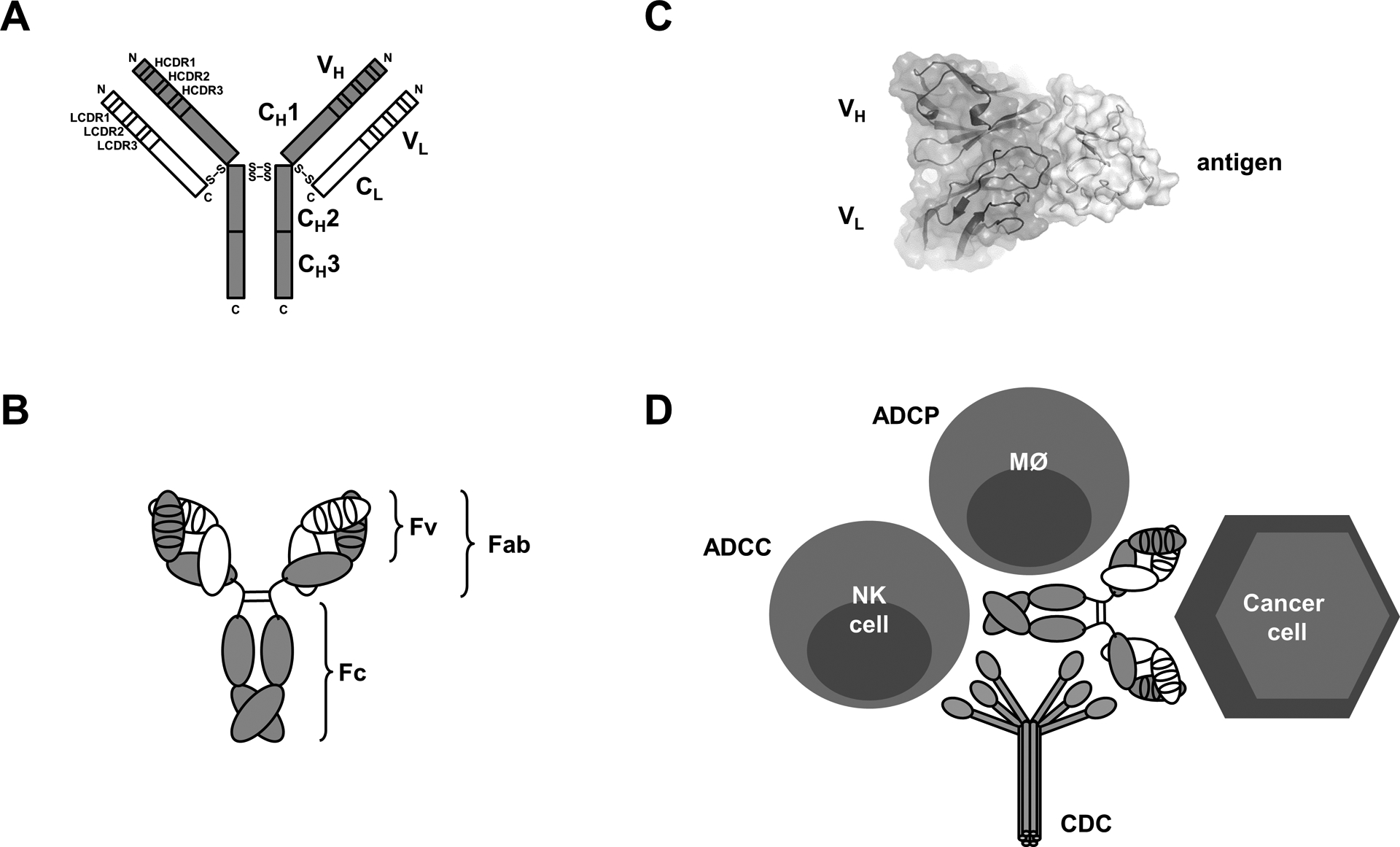
Two-dimensional (A) and three-dimensional (B) depictions of the light and heavy chain assembly of the IgG1 molecule. The Y-shaped IgG1 format is composed of two Fab arms linked through the Fc stem and consists of a total of twelve Ig domains organized into two light chains (white) and two heavy chains (gray) that are connected by a total of four interchain disulfide bridges (S-S). The four N-termini and four C-termini are indicated as N and C, respectively. The N-terminal four Ig domains are known as the variable domains; VL for the light chain and VH for the heavy chain, each consisting of three CDRs depicted as smaller rectangles or ovals. Together, VL and VH and their six CDRs form the Fv that harbors the unique paratope that binds a distinct antigen with high affinity and specificity. (C) The antibody-antigen interaction is defined by complementary shapes and charges of paratope (on the antibody) and epitope (on the antigen), shown here in a co-crystal structure determined by X-ray crystallography at 1.4-Å resolution (PDB: 6OSV) [54]. The interaction of paratope (left) and epitope (right) typically involves a large buried surface area, which was determined to be 720-Å2 in this example. (D) The Fc stem is formed by the two C-terminal constant domains of the heavy chain (CH2 and CH3) and mediates the prolonged circulatory half-life of the IgG1 molecule through interaction with FcRn and its various effector functions (CDC, ADCC, and ADCP) through interactions with complement component C1q as well as NK cells, macrophages, and other FcγR-expressing myeloid and lymphoid cells. In cancer therapy, the natural effector functions are triggered upon arrayed engagement of the mAb with cancer cell surface antigens.
Collectively, the structural and functional modularity of the antibody molecule has served as a preferred canvas for protein engineers. However, when compared to small molecules, antibodies were initially not considered suitable pharmaceuticals due to their size and variability, the latter caused by numerous posttranslational modifications (PTMs). Over the past two decades, antibodies have been routinely manufactured at >1,000-kg scale along with high precision PTM mapping. Thus, both antibody engineering and manufacturing have driven the success of the antibody molecule as a pharmaceutical with broad therapeutic utility in oncology and non-oncology indications.
MOA: Natural properties
High affinity and specificity of the paratope of the Fv fragment to a virtually unlimited number and variety of antigens is achieved by a highly sophisticated natural sequence diversification encompassing VL-JL and VH-D-JH recombination of light and heavy chain germlines, respectively, in primary lymphoid tissues (such as human bone marrow) and their stringent selection paired with further sequence diversification by somatic hypermutation in germinal centers within secondary lymphoid tissues (such as human lymph nodes and spleen) [4]. This primary, immunogen-independent, and secondary, immunogen-dependent diversification gives rise to the naïve and immune antibody repertoires, respectively. A typical in vivo evolved antibody features a paratope with a large footprint (Figure 1) that engages non-self-antigens with 100 pM to 100 nM monovalent affinity (and 10–100-fold higher bivalent avidity) but does not cross-react with self-antigens. A recent extensive deep sequencing study of circulating B cells from ten individuals revealed that the human antibody repertoire is extremely diverse and individualized with an estimated 1016–1018 unique Fv sequences [5]. This massive paratope space serves as a highly effective, adaptive, and memorizing defense system against infectious diseases caused by pathogenic viruses, bacteria, fungi, and parasites. Although a role of the naïve and immune antibody repertoire in cancer prevention is less established, it clearly can target non-self-antigens on the cancer cell surface.
The majority of FDA-approved antibody therapeutics contains an in vivo evolved paratope that originated from mouse or human immune antibody repertoires following immunization with the antigen of interest. Initially hybridoma technology and increasingly B-cell cloning have provided access to mouse and human monoclonal antibodies (mAbs) that contain the original Fv, i.e. original VH and VL domain pairing [6]. Chimerization (e.g., rituximab) and humanization (e.g., trastuzumab) of mouse mAbs enabled their tolerization by the human immune system, a prerequisite for therapeutic utility. Access to human mAbs from transgenic mice with human Ig genes or directly from humans has delivered mAbs that are indistinguishable from human antibodies. Not included in the majority of FDA-approved antibody therapeutics that contain an in vivo evolved paratope are four human mAbs that were generated by phage display technology from naïve or synthetic human antibody libraries and randomly combine VH and VL domains [6]. These are ramucirumab (Cyramza®), necitumumab (Portrazza®), atezolizumab (Tecentriq®), and avelumab (Bavencio®) (Table 1). In addition to de novo mAb generation, directed evolution technologies such as phage display and yeast display have been used to improve natural and synthetic paratopes by affinity maturation, as was done for moxetumomab pasudotox-tdfk (Lumoxiti®) (Table 1). Numerous additional human mAbs with in vitro evolved paratopes are in clinical trials [7].
Although not yet among FDA-approved antibody-based cancer therapies, bispecific antibodies (biAbs) that target two different cancer cell surface antigens, such as EGFR×MET biAb amivantamab, which is in pivotal clinical trials for the treatment of non-small cell lung cancer (NSCLC) [2], have confronted the “one-antibody, one-antigen” paradigm and revealed improved targeting properties when compared to the monospecific antibodies on their own or in combination [8]. Nonetheless, the MOAs of such cis-acting biAbs directed against two different antigens in the same cell membrane are still based on natural properties.
The size, stability, diversity, and adaptability of their paratope bestow antibody molecules not only with exceptional affinity and specificity but also with the ability to interfere with protein-protein interactions that involve large buried surface areas and as such are difficult to block with small molecules [9]. To clear pathogens, antibodies not only need to bind tightly and selectively but also recruit proteins and cells of the innate immune system using CDC, ADCC, and ADCP as their effector functions. Combining Fab-mediated recognition with Fc-mediated eradication constitutes a highly precise and effective extracellular interception system. Optimized for fighting infectious diseases, the natural properties of the antibody molecule have been exploited by antibody-based cancer therapies. While these can be collectively defined as pharmaceuticals that target antigens through antibody-based recognition, their MOAs differ widely. Natural effector functions can be tailored by using different IgG isotypes [10], for example IgG4 for mitigating CDC, ADCC, and ADCP, or by subjecting the Fc fragment to protein or carbohydrate engineering [11]. All of these modulations have resulted in FDA-approved mAbs. Of the 19 mAbs with natural MOAs (marked in green in Table 1), four have an engineered Fc fragment, including three of the last five FDA approvals. It can be anticipated that this trend will continue for both new mAbs to new targets and new mAbs, also known as biobetters, to old targets. Beyond maintaining, reducing, or increasing natural effector functions as their MOAs, antibody-based cancer therapies can deploy synthetic MOAs not found in nature. These are discussed in the following two sections.
MOA: Engagement of cytotoxic T cells
While the natural properties of antibodies are confined to deploying the innate immune system, the discovery and development of antibodies that engage components of the adaptive immune system, in particular cytotoxic T cells, constitutes a major advancement for cancer therapy (highlighted in blue in Table 1). This is founded on the realization that cancer and the immune system are in a constant battle involving immunosurveillance, immunosuppression, and immunoediting [12]. As depicted in Figure 2, FDA-approved antibody therapeutics that deploy T cells as their MOA include seven ICIs, one T-biAb, and five CAR-Ts.
Figure 2. Structural and functional diversity of FDA-approved T-cell engaging antibodies.
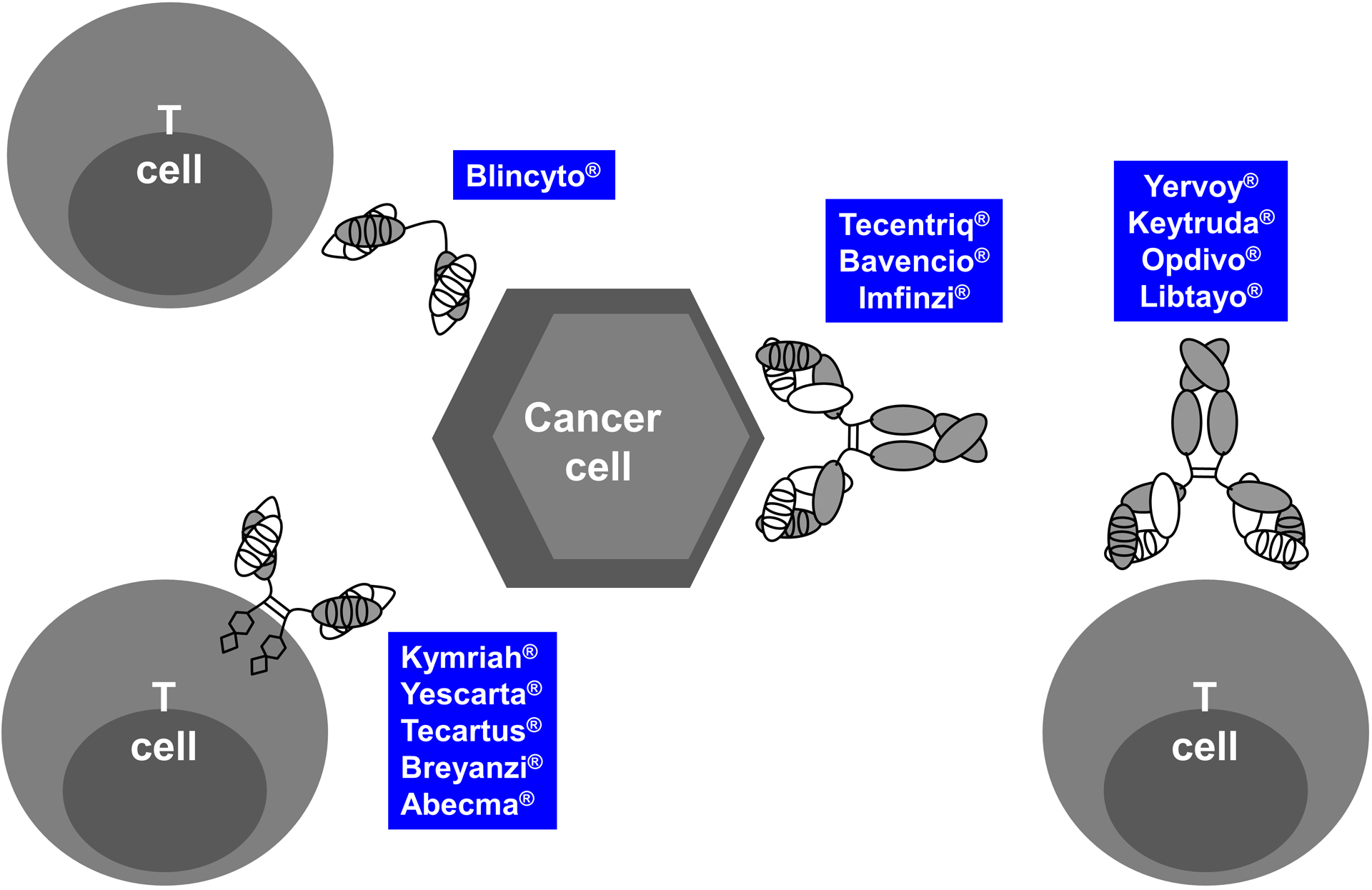
T-cell engaging antibodies, which fall into the category of IO drugs, can have numerous different structures and functions. Shown here are the 13 FDA-approved and marketed antibody-based cancer therapies that directly interface with T cells in one of four principal ways, including as T-biAbs (upper left), CAR-Ts (lower left), or ICIs that target inhibitory receptors on T cells (right) or their ligands on cancer cells (middle) and APCs in the TME (not shown).
The FDA-approved ICIs targeting (i) T cells via CTLA4 (ipilimumab (Yervoy®)) or PD1 (pembrolizumab (Keytruda®), nivolumab (Opdivo®), and cemiplimab-rwlc (Libtayo®)) and (ii) cancer cells and antigen-presenting cells (APCs) in the tumor microenvironment (TME), such as macrophages and dendritic cells, via PDL1 ((atezolizumab (Tecentriq®), avelumab (Bavencio®), and durvalumab (Imfinzi®)) have broad utility in solid malignancies (Table 1) and act by suppressing the cancer cell and APC-mediated inhibition of tumor-infiltrating cytotoxic T cells. As such, they provide a powerful boost to the cancer patients’ immune system, albeit at the risk of evoking autoimmune disease [13]. Immune checkpoint targeting antibody therapeutics in pivotal clinical trials [2] include 13 mAbs to first-generation targets CTLA4, PD1, and PDL1, along with four mAbs to next-generation targets LAG3, NKG2A, TIGIT, and TIM3, whose MOAs may involve both T cells and NK cells. Notably, this list also includes biAbs that simultaneously engage two different immune checkpoints, namely PD1×CTLA4, PD1×LAG3, and PDL1×CTLA4 [2]. These dual targeting approaches were prompted by findings that revealed overall survival benefits of advanced melanoma patients treated with a combination of two ICIs [14, 15]. Combining two ICIs in one biAb as an alternative to a mixture of two mAbs has the potential of saving costs but the disadvantage of losing freedom for discordant dosing of the two mAbs, which may be important for optimizing the therapeutic index [16]. Mixtures of anti-CTLA4 mAb ipilimumab (Yervoy®) and anti-PD1 mAb nivolumab (Opdivo®) have now received FDA approval for the treatment of several different cancers. ICIs, i.e. mAbs antagonizing inhibitory receptors [17], along with mAbs agonizing activating receptors, such as ICOS, CD28, and CD137 [18], and their various combinations will likely continue to dominate the preclinical and clinical pipelines of immuno-oncology (IO) drugs for the treatment of a broad range of solid malignancies. Notably, this includes cancers that are defined by specific molecular anomalies rather than by their tissue of origin. In 2017, pembrolizumab (Keytruda®) became the first tissue-agnostic cancer therapy by receiving FDA approval for microsatellite instability-high (MSI-H) or mismatch repair deficient (dMMR) solid malignancies (Table 1) [19]. Despite their promise, only a small subset of cancer patients responds to the FDA-approved ICIs. T-cell-inflamed (“hot”) tumors with high mutational burden that triggers cytotoxic T-cell infiltration, such as melanoma and NSCLC, respond better than non-T-cell-inflamed (“cold”) tumors. Triggering immunogenic cell death (ICD) in cold tumors can promote cytotoxic T-cell infiltration and as such is being investigated in combination with IO drugs [20]. Potential ICD-triggering regimens include chemotherapy, radiotherapy, and oncolytic viruses [21]. Selective chemotherapy mediated by ADCs holds substantial promise for combination with ICIs [22] and is investigated in several clinical trials [23].
Beyond T cells and NK cells, ICIs have also been used to engage macrophages by interfering with the CD47-SIRPα axis [24]. When expressed on the cancer cell surface, CD47, commonly known as a “don’t eat me” signal, inhibits phagocytosis by immunosurveilling macrophages. On their own and in combination with other antibody therapeutics, anti-CD47 mAbs and SIRPα-Fc fusion proteins that block this immunoescape mechanism are in early to advanced clinical trials for the treatment of both hematologic and solid malignancies [24]. In general, it can be anticipated that our increasing understanding of the TME and its role in supporting and suppressing tumor growth will be reflected by an increasing number of TME-engaging antibody therapeutics in preclinical and clinical pipelines.
While the FDA-approved ICIs suppress inhibitory receptors on T cells or their ligands, T-biAbs bind activating receptors on T cells with one arm and cancer cell surface antigens with the other arm (Figure 2), effectively bridging cytotoxic T cells and cancer cells to enable major histocompatibility complex (MHC)-independent recognition and destruction [25]. As such, T-biAbs are trans-acting biAbs that bridge two different cell membranes. The currently only FDA-approved T-biAb, blinatumomab, binds to CD19 on malignant B cells of acute lymphocytic leukemia (ALL) and CD3ε, a T-cell receptor (TCR) component. Numerous additional T-biAbs are in preclinical and clinical pipelines [16, 26], including flotetuzumab (CD123×CD3) and mosunetuzumab (CD20×CD3) in pivotal clinical trials for, respectively, acute myeloid leukemia (AML) and follicular lymphoma, a B-cell non-Hodgkin lymphoma (B-NHL) subtype [2]. The above-mentioned ICD-triggering regimens for overcoming immunosuppression in the TME may facilitate an expansion of the therapeutic utility of T-biAbs from hematologic to solid malignancies. In terms of formats, T-biAbs are exceptionally diverse [27, 28], taking full advantage of the modularity of the antibody molecule to tailor the valency and affinity of their T cell-engaging and cancer cell-engaging arms along with their distance and flexibility. All of these parameters influence cytolytic synapse formation between T cell and cancer cell [29]. Notably, to address manufacturing challenges and short circulatory half-lives of certain T-biAb formats, preclinical investigations have revealed that they can also be delivered as mRNA or DNA for in situ generation in the cancer patient [26].
Conceptually related to T-biAbs are CAR-Ts of which five have been approved by the FDA. Four of these, i.e. tisagenlecleucel (Kymriah®), axicabtagene ciloleucel (Yescarta®), brexucabtagene autoleucel (Tecartus®), and lisocabtagene maraleucel (Breyanzi®), are targeting CD19 in relapsed or refractory (r/r) B-ALL or B-NHL (Figure 2). In addition, the anti-BCMA CAR-T idecabtagene vicleucel (Abecma®) was recently approved by the FDA for the therapy of r/r multiple myeloma. CAR-Ts are formally categorized as cell therapy (-cel suffix in their generic name) rather than antibody therapy (-mab suffix). However, they can be considered antibody-based cancer therapies based on our above definition as pharmaceuticals that target antigens through antibody-based recognition. They are built by retrovirally transducing autologous T cells from cancer patients with chimeric antigen receptors (CARs) that fuse an extracellular antibody fragment, typically a scFv, to a transmembrane segment, followed by the cytoplasmic signaling domain of a T cell costimulatory receptor (typically CD28 or 4-1BB) and the cytoplasmic signaling domain of CD3ζ of the TCR complex. As such, a CAR-T links antibody-mediated MHC-independent recognition of cancer cell surface antigens to cytotoxic T-cell engagement [30]. CAR-Ts have cured previously incurable r/r B-cell malignancies and are being pursued as paradigm-changing therapy in other cancers [31, 32]. The ability to engineer autologous or allogeneic T cells ex vivo, provides an opportunity for equipping CAR-Ts with a plethora of features that promote their efficacy and safety in vivo. This includes logic gate CAR-Ts whose activity is controlled by two different cancer cell surface antigens and switchable CAR-Ts that use a universal CAR that can be turned on and off with an adaptor molecule [33]. CRISPR-Cas9-edited CAR-Ts to remove the TCR α and β chains as well as PD1 have also entered clinical trials [34]. Combinations of CAR-Ts with ICD-triggering regimens and ICIs have the potential of expanding the therapeutic utility of CAR-Ts from hematologic to solid malignancies [35]. Nonetheless, as living drugs, CAR-Ts face unique impediments in terms of manufacturing, logistics, and pharmacology, prompting increasing interest in side-by-side comparisons of CAR-Ts and T-biAbs in clinical trials [26].
Life-threatening adverse events, particularly cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS), have remained key challenges for both CAR-Ts and T-biAbs and necessitate hospitalization for close monitoring and immediate intervention. Notably, simultaneously with the FDA approval of the first CAR-T, tisagenlecleucel (Kymriah®), in 2017, the anti-IL6 receptor mAb tocilizumab (Actemra®) received FDA approval for CAR-T-induced CRS [36]. Switching from T cells to NK cells, i.e. pursuing NK-biAbs and CAR-NKs [37, 38], has potential to mitigate toxicity while imitating activity of T-biAb and CAR-T treatments. This is a rapidly advancing area of high promise for cancer immunotherapy.
Engaging immune effector cells can also be achieved with so called immunocytokines. Here, a cancer cell surface antigen-targeting antibody is fused to a cytokine to enable its targeted delivery to the TME [39]. This approach effectively increases the therapeutic index of cytokines, such as IL-2, which on its own was the first FDA-approved cancer immunotherapy [40] but can only be administered at low doses due to systemic toxicity. Although immunocytokines have not received FDA approval for cancer therapy yet, a diverse panel of antibody-cytokine fusion proteins is being investigated preclinically and clinically. This includes Daromun, a combination of two immunocytokines that fuse a scFv directed to the extra domain B of fibronectin to IL-2 and TNF-α, respectively. In a pivotal clinical trial for the therapy of melanoma, Daromun is tested as neoadjuvant intratumoral injection followed by surgery.
MOA: Delivery of cytotoxic payloads
Highlighted in orange in Table 1, eleven antibody-based cancer therapies feature the delivery of a cytotoxic payload as their MOA. Serving as a carrier of cytotoxic payloads effectively bestows antibodies with the ability to deliver chemotherapy or radiotherapy selectively to cancer cells while sparing healthy cells and tissues. Among the FDA-approved cytotoxic payloads are a radioisotope, various small molecules, and a bacterial toxin. Attached to an antibody carrier, the ensuing hybrid molecules are referred to as radioimmunoconjugates, ADCs, and immunotoxins, respectively.
With only one radioimmunoconjugate and one immunotoxin currently available to cancer patients, the nine FDA-approved ADCs (Figure 3) clearly dominate this category and will continue to do so based on a rich clinical and preclinical pipeline [23]. This includes seven ADCs in pivotal clinical trials, six of which are investigated for the therapy of solid malignancies. Among these, tusamitamab ravtansine (NSCLC), mirvetuximab soravtansine (ovarian cancer), and tisotumab vedotin (cervical cancer) target new indications not covered by the nine FDA-approved ADCs [2]. All ADCs are composed of an antibody, a linker, and a small molecule payload, i.e. the drug. Although the antibody of first-generation ADCs is exclusively based on an IgG1 or IgG4 format, they differ widely with respect to the composition of linker and drug (Figure 3). Linkers can be categorized as cleavable or non-cleavable. Cleavable linkers permit release of the drug in endosomes and lysosomes following receptor-mediated endocytosis of the ADC. Release is triggered by acidic or reducing conditions or enzymatically. Non-cleavable linkers stay attached to the drug and to at least one amino acid of the antibody following its proteolysis in lysosomes. For example, the active drug of ado-trastuzumab emtansine (Kadcyla®) is the lysine-linker-drug catabolite (Figure 3). As for the drugs and specifically their MOAs, they can be broadly classified into natural product derivatives that interfere with tubulin polymerization (maytansine and auristatin used in 5 FDA-approved ADCs) or damage DNA (calicheamicin and camptothecin used in 4 FDA-approved ADCs). First-generation ADCs are stochastic assemblies of antibody and drug, using surface lysine and hinge cysteine residues for conjugation. The ensuing mixtures are composed of a variety of ADC species with different drug-to-antibody ratios (DAR) and different pharmacokinetic and pharmacodynamic properties. This heterogeneity has been addressed in two recently FDA-approved ADCs, fam-trastuzumab deruxtecan-nxki (Enhertu®) and sacituzumab govitecan-hziy (Trodelvy®), in which all hinge cysteine residues involved in interchain disulfide bridges in the IgG1 molecule are conjugated to the linker-drug for a uniform DAR of 8. Next-generation ADCs, which typically have a DAR of 2 and are highly homogeneous, use a variety of methods for site-specific antibody-drug conjugation which can be grouped into chemical and enzymatic assemblies. In addition to engineered natural and unnatural amino acids, the glycans of the CH2 domain have been used for site-specific drug conjugation [41]. In addition to the ability to build molecularly defined ADCs with high precision, novel drugs with different MOAs have the potential to further broaden the therapeutic utility of ADCs by overcoming cancer resistance. Advances in antibody engineering have enabled the generation of bispecific ADCs as well as ADCs carrying two different payloads to further enhance their specificity and potency, respectively. In addition, as mentioned above, combinations of ADCs with ICIs are widely investigated preclinically and clinically. Collectively, next-generation ADCs are among the most promising pharmaceuticals for broadening and deepening cancer therapy. As for other antibody-based cancer therapies, their therapeutic utility relies on the identification of suitable targets. Specifically, ADCs require cancer cell surface antigens that are efficiently internalized and trafficked to lysosomes. Despite their promise, a number of ADCs in preclinical and clinical pipelines have been discontinued due to encountering on-target-off-tumor and off-target-off-tumor toxicities [23]. Extremely potent investigational payloads, such as the DNA damaging pyrrolobenzodiazepine dimers, require careful tailoring to indication, target, linker, and antibody in order to achieve an acceptable safety profile.
Figure 3. Composition of FDA-approved ADCs.
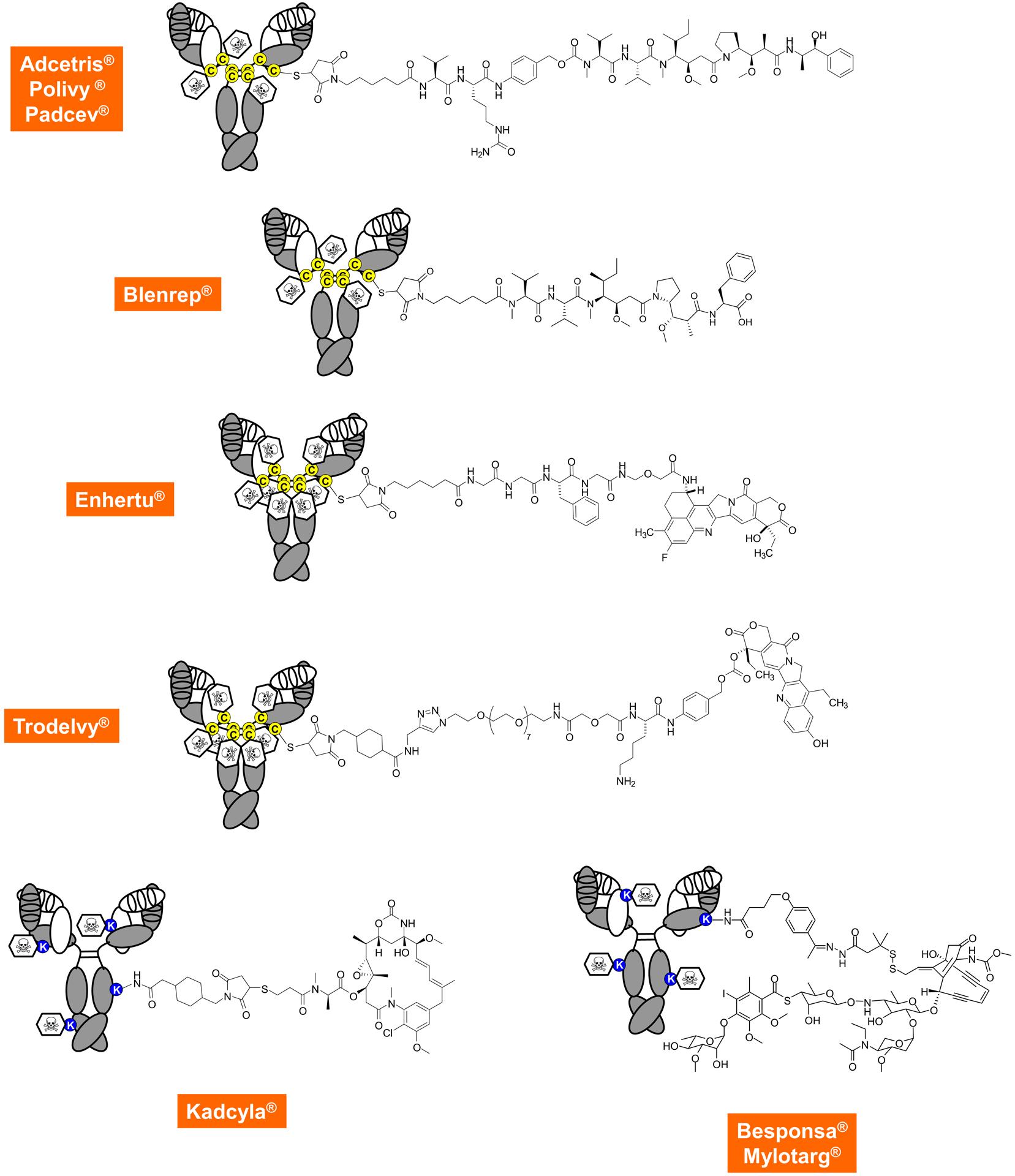
The top row shows first-generation heterogeneous ADCs targeting CD30, CD79B, and NECTIN4, respectively, in which the drug is stochastically linked to reduced cysteine residues in the hinge region using a thiosuccinimide, a caproyl spacer, an enzymatically cleavable unit (a valine-citrulline dipeptide fused to a para-aminobenzylcarbamate), followed by the cytotoxic payload which is monomethylauristatin E (MMAE), a subnanomolar inhibitor of tubulin polymerization. The mean DAR is approximately 4, ranging from 0 to 8. The second row shows another auristatin-based and stochastically assembled ADC that targets BCMA and links monomethylauristatin F (MMAF) through a non-cleavable linker to hinge cysteine residues. Recently, the first homogeneous ADCs shown in the third and fourth row received FDA approval. They target HER2 and TROP2, respectively, and use the same thiosuccinimide connector to hinge cysteine residues but rather than doing this stochastically, all eight cysteines are involved. This requires the use of more hydrophilic linkers but, due to their high DAR, also affords slightly less cytotoxic payloads, which are two different camptothecin derivatives that inhibit topoisomerase I. The bottom row shows the three first-generation ADCs that target HER2, CD22, and CD33, respectively, and use surface lysine residues for drug attachment. With 80–90 of these in the average antibody molecule, the location and distribution is even more stochastic. Cytotoxic payloads include tubulin polymerization inhibitor maytansine linked through a non-cleavable linker (left) and the DNA damaging calicheamicin linked through a hydrazone-disulfide linker that gets cleaved in acidic or reducing conditions (right).
The overall success of ADCs has fueled other antibody-based cancer therapies that rely on the delivery of cytotoxic payloads as their MOA. Moxetumomab pasudotox-tdfk (Lumoxiti®) is an FDA-approved immunotoxin for the therapy of hairy cell leukemia, a B-NHL subtype. It consists of a CD22-targeting Fv recombinantly fused to PE38, a fragment of a highly cytotoxic bacterial protein, Pseudomonas exotoxin A, which inhibits translation in mammalian cells [42]. Closing in on FDA approval for the therapy of bladder cancer is oportuzumab monatox, which combines an EPCAM-targeting scFv with PE38. Due to the use of a bacterial protein, immunotoxins have to overcome immunogenicity that prevents repeated administration. Numerous approaches for removing B-cell and T-cell epitopes of the bacterial protein as well as using concomitant immunosuppressive treatment have been developed to improve the therapeutic utility of next-generation immunotoxins [43].
The first two FDA-approved antibody-based cancer therapies that delivered a cytotoxic payload were both radioimmunoconjugates. CD20-targeting 131iodine-tositumomab (Bexxar®), which was withdrawn from the market in 2014, and 90yttrium-ibritumomab tiuxetan (Zevalin®), proved to be safe and efficacious for B-NHL therapy but failed wider adoption in clinical practice due to logistic and other obstacles [44]. A renaissance of radioimmunoconjugates is seen in ongoing pivotal clinical trials of B7H3-targeting 131iodine-omburtamab for the therapy of neuroblastoma and CD45-targeting 131iodine-apamistamab for conditioning of AML patients prior to allogeneic hematopoietic stem cell transplantation [2]. Separating the administration of antibody and radioisotope in so called pretargeted radioimmunotherapy can lower systemic exposure to irradiation from circulating conventional radioimmunoconjugates [44]. One approach is the utilization of bispecific antibodies that combine a cancer cell surface antigen-binding antibody module with a hapten-binding antibody module. Following accumulation of the slower clearing bispecific antibody at the tumor site, a faster clearing hapten-derivatized chelate loaded with a radioisotope is administered [45].
Another approach is near-infrared photoimmunotherapy [46, 47]. Here, the antibody is conjugated to a small molecule dye. Unlike in ADCs and radioimmunoconjugates, the payload is not toxic on its own. Rather, following binding of the photoimmunoconjugate to a cancer cell surface antigen, the dye is activated with a near infrared light source and induces cell membrane perforations followed by ICD. An EGFR-targeting photoimmunoconjugate, cetuximab sarotalocan, has reached pivotal clinical trials for the therapy of head and neck cancer [2]. Rapid induction of ICD makes photoimmunotherapy attractive for combination with ICIs, T-biAbs, and CAR-Ts.
Outlook
Fueled by its remarkable track record of clinical and commercial viability, the landscape of antibody-based cancer therapy is rapidly evolving. What are the limiting factors of this success and progress?
First of all, antibodies are only as good as their antigens, i.e. they rely on the existence of tumor-specific antigens (TSAs) and tumor-associated antigens (TAAs) with different levels of spatially and temporarily restricted expression patterns. Finding additional cancer cell surface antigens that permit to apply antibody-based cancer therapy with high precision and to tailor a suitable MOA are achievable objectives. Other campaigns build on targeting antigen combinations to improve selective intervention. An exciting alternative approach is conditionally active antibody-based cancer therapy that takes advantage of unique properties of the TME to confine the activity of antibody therapeutics to the tumor site. Examples include pro-antibody therapeutics that only engage their antigens after activation by proteases [48], acidic pH [49], or ATP [50] in the TME. Notably, this approach is applicable to essentially all antibody therapeutics regardless of their MOA. Furthermore, the ability to generate antibodies to MHC-peptide complexes has the potential to expand antibody-based cancer therapy from cell surface antigens to intracellular antigens. Also known as TCR-like antibodies [51], their recognition and destruction of cancer cells is MHC-dependent. A recent study expanded the target range of TCR-like antibodies to neoantigens of cancer cells, such as a common mutation of tumor suppressor p53, and demonstrated that the corresponding T-biAbs can potently and selectively kill cancer cells that have fewer than 10 copies of the MHC-peptide complex presenting the neoantigen [52].
Second, like most cancer therapies, contemporary antibody therapeutics are countered by cancer refraction and resistance and often only have modest benefits in terms of overall survival. There is unexploited potential for biobetters with tailored valency, avidity, specificity, circulatory half-life, and effector functions to improve upon first-generation antibody therapeutics with respect to both pharmacokinetics and pharmacodynamics. Preclinical investigations of next-generation antibody therapeutics now include rigorous in vitro and in vivo comparisons of antibodies and antibody fragments with a variety of different compositions and epitopes. In addition, moving from monoclonal to oligoclonal antibody-based cancer therapies and at the same time combining different MOAs has the potential to prolong complete responses in currently incurable cancers.
Another point to consider is our categorization of all FDA-approved and clinically investigated antibody-based cancer therapies into three MOAs. It is likely that other MOAs will enter clinical trials in the near future. One new category is emerging from cis-acting biAbs that exert activity by crosslinking two different antigens in the same cell membrane. For example, cis-acting biAbs can mediate the ubiquitination and subsequent degradation of a cancer cell surface antigen through its crosslinking to a transmembrane E3 ligase, as was recently shown for a PDL1×RNF43 biAb [53].
Overall, antibody-based cancer therapies have seen a steady increase from preclinical and clinical pipelines to market share. Next-generation antibody engineering, conjugation, production, and administration technologies along with innovative combination treatments tailored to cancer patients will continue to fuel the success of these pharmaceuticals.
Acknowledgements
We acknowledge support by predoctoral stipends from the Klorfine Foundation and the Frenchman’s Creek Women for Cancer Research (to R.S.G.) and National Institutes of Health Grants R01 CA174844, R01 CA181258, R01 CA204484, and R21 CA229961 (to C.R.). We thank Dr. HaJeung Park from the X-Ray Crystallography Core of The Scripps Research Institute (Jupiter, FL) for contributing Fig. 1C.
Footnotes
Conflict of interest
There is no conflict of interest to disclose.
References
- 1. https://www.pharmacompass.com/radio-compass-blog/top-drugs-and-pharmaceutical-companies-of-2019-by-revenues.
- 2.Kaplon H, Reichert JM. Antibodies to watch in 2021. MAbs 2021; 13: 1860476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Wang LX, Tong X, Li C, Giddens JP, Li T. Glycoengineering of antibodies for modulating functions. Annu Rev Biochem 2019; 88: 433–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rajewsky K. Clonal selection and learning in the antibody system. Nature 1996; 381: 751–758. [DOI] [PubMed] [Google Scholar]
- 5.Briney B, Inderbitzin A, Joyce C, Burton DR. Commonality despite exceptional diversity in the baseline human antibody repertoire. Nature 2019; 566: 393–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Beerli RR, Rader C. Mining human antibody repertoires. MAbs 2010; 2: 365–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alfaleh MA, Alsaab HO, Mahmoud AB, Alkayyal AA, Jones ML, Mahler SM et al. Phage display derived monoclonal antibodies: from bench to bedside. Front Immunol 2020; 11: 1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng S, Moores S, Jarantow S, Pardinas J, Chiu M, Zhou H et al. Cross-arm binding efficiency of an EGFR x c-Met bispecific antibody. MAbs 2016; 8: 551–561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott DE, Bayly AR, Abell C, Skidmore J. Small molecules, big targets: drug discovery faces the protein-protein interaction challenge. Nat Rev Drug Discov 2016; 15: 533–550. [DOI] [PubMed] [Google Scholar]
- 10.Brezski RJ, Georgiou G. Immunoglobulin isotype knowledge and application to Fc engineering. Curr Opin Immunol 2016; 40: 62–69. [DOI] [PubMed] [Google Scholar]
- 11.Liu R, Oldham RJ, Teal E, Beers SA, Cragg MS. Fc-engineering for modulated effector functions-improving antibodies for cancer treatment. Antibodies 2020; 9: 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011; 331: 1565–1570. [DOI] [PubMed] [Google Scholar]
- 13.Dougan M, Pietropaolo M. Time to dissect the autoimmune etiology of cancer antibody immunotherapy. J Clin Invest 2020; 130: 51–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017; 377: 1345–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Larkin J, Chiarion-Sileni V, Gonzalez R, Grob JJ, Rutkowski P, Lao CD et al. Five-year survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2019; 381: 1535–1546. [DOI] [PubMed] [Google Scholar]
- 16.Labrijn AF, Janmaat ML, Reichert JM, Parren P. Bispecific antibodies: a mechanistic review of the pipeline. Nat Rev Drug Discov 2019; 18: 585–608. [DOI] [PubMed] [Google Scholar]
- 17.Pardoll DM. The blockade of immune checkpoints in cancer immunotherapy. Nat Rev Cancer 2012; 12: 252–264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Mayes PA, Hance KW, Hoos A. The promise and challenges of immune agonist antibody development in cancer. Nat Rev Drug Discov 2018; 17: 509–527. [DOI] [PubMed] [Google Scholar]
- 19.Lemery S, Keegan P, Pazdur R. First FDA Approval agnostic of cancer site - when a biomarker defines the indication. N Engl J Med 2017; 377: 1409–1412. [DOI] [PubMed] [Google Scholar]
- 20.Galluzzi L, Buque A, Kepp O, Zitvogel L, Kroemer G. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol 2017; 17: 97–111. [DOI] [PubMed] [Google Scholar]
- 21.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol 2013; 31: 51–72. [DOI] [PubMed] [Google Scholar]
- 22.Müller P, Kreuzaler M, Khan T, Thommen DS, Martin K, Glatz K et al. Trastuzumab emtansine (T-DM1) renders HER2+ breast cancer highly susceptible to CTLA-4/PD-1 blockade. Sci Transl Med 2015; 7: 315ra188. [DOI] [PubMed] [Google Scholar]
- 23.Beck A, Goetsch L, Dumontet C, Corvaia N. Strategies and challenges for the next generation of antibody-drug conjugates. Nat Rev Drug Discov 2017; 16: 315–337. [DOI] [PubMed] [Google Scholar]
- 24.Feng M, Jiang W, Kim BYS, Zhang CC, Fu YX, Weissman IL. Phagocytosis checkpoints as new targets for cancer immunotherapy. Nat Rev Cancer 2019; 19: 568–586. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clynes RA, Desjarlais JR. Redirected T cell cytotoxicity in cancer therapy. Annu Rev Med 2019; 70: 437–450. [DOI] [PubMed] [Google Scholar]
- 26.Rader C. Bispecific antibodies in cancer immunotherapy. Curr Opin Biotechnol 2020; 65: 9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wu X, Demarest SJ. Building blocks for bispecific and trispecific antibodies. Methods 2019; 154: 3–9. [DOI] [PubMed] [Google Scholar]
- 28.Brinkmann U, Kontermann RE. The making of bispecific antibodies. MAbs 2017; 9: 182–212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ellerman D. Bispecific T-cell engagers: towards understanding variables influencing the in vitro potency and tumor selectivity and their modulation to enhance their efficacy and safety. Methods 2019; 154: 102–117. [DOI] [PubMed] [Google Scholar]
- 30.Gross G, Waks T, Eshhar Z. Expression of immunoglobulin-T-cell receptor chimeric molecules as functional receptors with antibody-type specificity. Proc Natl Acad Sci U S A 1989; 86: 10024–10028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.June CH, O’Connor RS, Kawalekar OU, Ghassemi S, Milone MC. CAR T cell immunotherapy for human cancer. Science 2018; 359: 1361–1365. [DOI] [PubMed] [Google Scholar]
- 32.Sadelain M, Riviere I, Riddell S. Therapeutic T cell engineering. Nature 2017; 545: 423–431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Hong M, Clubb JD, Chen YY. Engineering CAR-T cells for next-generation cancer therapy. Cancer Cell 2020; 38: 473–488. [DOI] [PubMed] [Google Scholar]
- 34.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E et al. CRISPR-engineered T cells in patients with refractory cancer. Science 2020; 367: eaba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Srivastava S, Furlan SN, Jaeger-Ruckstuhl CA, Sarvothama M, Berger C, Smythe KS et al. Immunogenic chemotherapy enhances recruitment of CAR-T cells to lung tumors and improves antitumor efficacy when combined with checkpoint blockade. Cancer Cell 2021; 39: 193–208 e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Maude SL, Laetsch TW, Buechner J, Rives S, Boyer M, Bittencourt H et al. Tisagenlecleucel in children and young adults with B-cell lymphoblastic leukemia. N Engl J Med 2018; 378: 439–448. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Daher M, Rezvani K. Outlook for new CAR-based therapies with a focus on CAR NK cells: what lies beyond CAR-engineered T cells in the race against cancer. Cancer Discov 2021; 11: 45–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hodgins JJ, Khan ST, Park MM, Auer RC, Ardolino M. Killers 2.0: NK cell therapies at the forefront of cancer control. J Clin Invest 2019; 129: 3499–3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Neri D, Sondel PM. Immunocytokines for cancer treatment: past, present and future. Curr Opin Immunol 2016; 40: 96–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rosenberg SA. IL-2: the first effective immunotherapy for human cancer. J Immunol 2014; 192: 5451–5458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Walsh SJ, Bargh JD, Dannheim FM, Hanby AR, Seki H, Counsell AJ et al. Site-selective modification strategies in antibody-drug conjugates. Chem Soc Rev 2021; 50: 1305–1353. [DOI] [PubMed] [Google Scholar]
- 42.Kreitman RJ, Pastan I. Antibody fusion proteins: anti-CD22 recombinant immunotoxin moxetumomab pasudotox. Clin Cancer Res 2011; 17: 6398–6405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Mazor R, Pastan I. Immunogenicity of immunotoxins containing pseudomonas exotoxin A: causes, consequences, and mitigation. Front Immunol 2020; 11: 1261. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Green DJ, Press OW. Whither radioimmunotherapy: to be or not to be? Cancer Res 2017; 77: 2191–2196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kraeber-Bodere F, Faivre-Chauvet A, Ferrer L, Vuillez JP, Brard PY, Rousseau C et al. Pharmacokinetics and dosimetry studies for optimization of anti-carcinoembryonic antigen x anti-hapten bispecific antibody-mediated pretargeting of Iodine-131-labeled hapten in a phase I radioimmunotherapy trial. Clin Cancer Res 2003; 9: 3973S–3981S. [PubMed] [Google Scholar]
- 46.Kobayashi H, Choyke PL. Near-infrared photoimmunotherapy of cancer. Acc Chem Res 2019; 52: 2332–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Mitsunaga M, Ogawa M, Kosaka N, Rosenblum LT, Choyke PL, Kobayashi H. Cancer cell-selective in vivo near infrared photoimmunotherapy targeting specific membrane molecules. Nat Med 2011; 17: 1685–1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Desnoyers LR, Vasiljeva O, Richardson JH, Yang A, Menendez EE, Liang TW et al. Tumor-specific activation of an EGFR-targeting probody enhances therapeutic index. Sci Transl Med 2013; 5: 207ra144. [DOI] [PubMed] [Google Scholar]
- 49.Johnston RJ, Su LJ, Pinckney J, Critton D, Boyer E, Krishnakumar A et al. VISTA is an acidic pH-selective ligand for PSGL-1. Nature 2019; 574: 565–570. [DOI] [PubMed] [Google Scholar]
- 50.Kamata-Sakurai M, Narita Y, Hori Y, Nemoto T, Uchikawa R, Honda M et al. Antibody to CD137 activated by extracellular adenosine triphosphate is tumor selective and broadly effective in vivo without systemic immune activation. Cancer Discov 2021; 11: 158–175. [DOI] [PubMed] [Google Scholar]
- 51.Denkberg G, Cohen CJ, Lev A, Chames P, Hoogenboom HR, Reiter Y. Direct visualization of distinct T cell epitopes derived from a melanoma tumor-associated antigen by using human recombinant antibodies with MHC-restricted T cell receptor-like specificity. Proc Natl Acad Sci U S A 2002; 99: 9421–9426. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsiue EH-C, Wright KM, Douglass J, Hwang MS, Mog BJ, Pearlman AH et al. Targeting a neoantigen derived from a common TP53 mutation. Science 2021; 371: eabc8697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Cotton AD, Nguyen DP, Gramespacher JA, Seiple IB, Wells JA. Development of antibody-based PROTACs for the degradation of the cell-surface immune checkpoint protein PD-L1. J Am Chem Soc 2021; 143: 593–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Goydel RS, Weber J, Peng H, Qi J, Soden J, Freeth J et al. Affinity maturation, humanization, and co-crystallization of a rabbit anti-human ROR2 monoclonal antibody for therapeutic applications. J Biol Chem 2020; 295: 5995–6006. [DOI] [PMC free article] [PubMed] [Google Scholar]