

Isolating protoplasts allows stochastic gene expression to enable regeneration of differentiated cells at a low frequency.
Abstract
Cell pluripotency is fundamental to biology. It has long been known that differentiated somatic plant cells may reacquire pluripotency, but the underlying mechanism remains elusive. In many plant species, a single isolated mesophyll protoplast may regenerate into an entire plant, which is widely used in gene transformation. Here, we identified two transcription factors whose ectopic activation promotes protoplast regeneration. Furthermore, we found that their expression was induced by protoplast isolation but at a very low frequency. Using live imaging and single-cell transcriptomics, we show that isolating protoplasts induces enhanced expression variation at the genome level. Isolating protoplasts also leads to genome-wide increases in chromatin accessibility, which promotes stochastic activation of gene expression and enhances protoplast regeneration. We propose that transcriptome chaos with increased expression variability among cells creates a cellular-level evolutionary driver selecting for regenerating cells.
INTRODUCTION
A fundamental question in biology is how differentiated cells can reprogram themselves to attain pluripotency or totipotency. Plants have a broad capacity to regenerate tissues (1, 2). Damage to root and shoot meristems rapidly triggers adjacent potent cell types to divide and acquire specific cell identities (3, 4); for example, the removal of the entire root stem cell niche allows the respecification of multiple neighboring cell types through the integration of auxin and cytokinin signaling, which results in an embryonic-like development program (5). Outside meristems, certain cell types are totipotent or pluripotent and retain the capacity for regeneration. Root xylem pole pericycle cells, which are parenchymatous cells associated with the vasculature, can form a callus and subsequently produce shoot or root meristems when supplemented with auxin and cytokinin (6, 7). In aerial organs, pericycle-like cells, which express the xylem pole pericycle cell marker J0121 and surround the midvein (7), are also callus percussors. Furthermore, pericycle cells initiate lateral roots and adventitious roots during normal development (8).
The above-mentioned regeneration derives from existing stem cells, i.e., progenitor cells that remain within the meristematic zone or are specialized to retain pluripotency. By contrast, little is known about how differentiated cells can reprogram and regenerate. Plants have the notable ability to regenerate a whole fertile plant from a fully differentiated somatic cell (1, 9). This is well exemplified by mesophyll cell regeneration, in which fully differentiated leaf mesophyll cells, with cell walls removed through protoplast isolation, reenter the cell cycle, proliferate, and form a callus (10). Meristems or somatic embryos are subsequently established from the callus and can regenerate into whole plants. Although protoplast regeneration has been widely used in gene transformation, it remains unclear how totipotency is acquired by differentiated cells (1). Notably, this impressive regeneration ability is stringently suppressed during normal growth. It has been reported that protoplast isolation induces microscopic-level changes in the appearance of the nucleolar domain, including the decondensation of heterochromatin (11–13). However, it remains unclear whether these changes are connected to regeneration.
Here, we identified genes that are not only required for but also greatly promote protoplast regeneration, in particular, callus formation. Their expression can be activated by protoplast isolation at a low frequency, suggesting enhanced gene expression variation. By combining a marker analysis with single-cell gene expression profiling, we show that protoplast isolation induces transcriptome chaos, with widespread stochastic gene expression. This is accompanied by genome-wide increase in chromatin accessibility, which promotes stochastic expression and protoplast regeneration. These results provide insights into stem cell induction and reveal previously unrecognized roles of stochastic gene expression in cell fate reprogramming.
RESULTS
Variable cell division patterns correlate with low regeneration rates
Protoplasts from multiple origins—such as embryos, cotyledons, and suspension-cultured cells—all have regeneration capacity (1). We chose to study leaf mesophyll protoplasts because they are fully differentiated and more homogeneous. We used a protocol for plant regeneration from Arabidopsis thaliana mesophyll cells (14). In this somatic regeneration process, protoplasts derived from fully differentiated mesophyll cells reenter the cell cycle and form microcalli (at least four cells and 70 to 500 μm in diameter), which subsequently form calli (>500 μm in diameter) and can ultimately regenerate into plants (Fig. 1, A and B). To simplify subsequent analyses, we optimized the protoplast isolation procedure to enrich mesophyll cells. Briefly, the mesophyll cells (~96%; see below) of mature Arabidopsis leaves were released in Gly Glc medium (MGG) by an overnight enzymatic digestion of the cell walls. The resulting protoplasts were cultured in an initial protoplast induction medium (PIM), which contains the synthetic auxin 2,4-dichlorophenoxyacetic acid and the synthetic cytokinin thidiazuron, for 11 days. The protoplasts were regenerated into calli after 2 months of culture in callus induction medium 1 and 2 (CIM1 and CIM2). Subsequently, the calli were transformed onto a semisolid shoot induction medium (SIM) and cultured for 30 days, when buds formed. Plantlets were regenerated from the buds after transformation onto a plant development medium (PDM).
Fig. 1. Regeneration of plants from Arabidopsis mesophyll protoplasts.
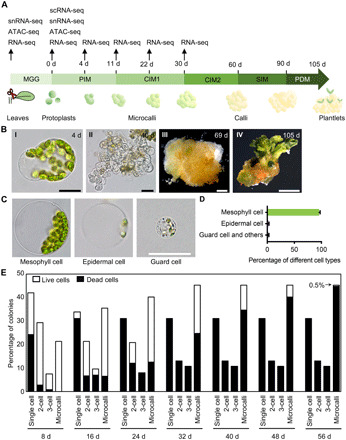
(A) Schematic outline of the protoplast regeneration protocol. Time points of sample collection for genome-wide assays are specified. (B) Key stages of mesophyll protoplast regeneration, showing a cell at 4 days after protoplast isolation, a microcallus at 40 days, a callus at 69 days, and a plantlet at 105 days. Scale bars, 25 μm (I) and (II), 1 mm (III), or 1 cm (IV). (C) Protoplasts derived from three cell types after an overnight enzymatic digestion. Scale bar, 25 μm. (D) Percentage of protoplasts derived from mesophyll cells, epidermal cells, guard cells, and other cell types after an overnight enzymatic digestion (182 ≤ n ≤ 204). Data are presented as means ± SD for more than three independent experiments. (E) Distribution of mesophyll protoplast cells during regeneration, measured at various time points after protoplast isolation (n = 376).
To live image early protoplast regeneration, we embedded protoplasts in alginate following established protocols (15). Mesophyll protoplasts are distinguished from protoplasts derived from other cell types based on their chlorophyll content and cell size (Fig. 1C). In addition, mesophyll cells are less resistant to wall digestion than most other cell types. All epidermal cells and vascular cells, except guard cells, have substantially lower chlorophyll contents than mesophyll cells; moreover, guard cell protoplasts can be distinguished from mesophyll cell protoplasts by its size, with the diameter of guard cell protoplasts being less than one-third of that of the mesophyll cell protoplasts. Under our isolation conditions, 95.8% of the protoplasts were chlorophyllous cells derived from mesophyll cells (Fig. 1D). Furthermore, we observed that, at 30 days, all of the microcalli were derived from chlorophyllous mesophyll protoplasts. Together, these results suggested that mesophyll protoplasts accounted for most regeneration events in our experimental conditions, which was also observed in previous studies (10, 15).
To understand this regeneration process at the cellular resolution, we followed the cell divisions of the mesophyll protoplasts starting from 0 days after plating on PIM (Fig. 1E). After 8 days in PIM, 58% of the cells had divided at least once, including 28.7% having divided twice or more. By 8 days, more than a half of the nondividing cells had been dead, while all dividing cells were viable. By 56 days, most cells were dead; only 0.5% were viable and had formed microcalli greater than 200 μm in diameter. Overall, our results suggested that morphologically identical protoplasts behaved highly variably in culture, and only ~0.5% could regenerate.
Genes enriched during regeneration
To identify genes promoting totipotency acquisition, we followed the transcriptome changes during single protoplast regeneration. In this study, we focused on the initial microcalli formation process. Because nonregenerating cells had limited cell division ability and many were even dead, regenerating cells were enriched after prolonged culture. We reasoned that genes expressed by then would be enriched in totipotency-promoting functions. We obtained transcriptome profiles from the protoplasts or microcalli immediately after cell wall digestion, and after 4, 11, 22, and 30 days of culture, and compared them with those of intact leaf cells (table S1). A total of 7558 genes were differentially expressed after protoplast isolation. In particular, 508 genes were enriched in the samples cultured for 30 days after protoplast isolation. Gene expression profiles significantly changed during the regeneration time course; Gene Ontology (GO) analyses indicated that the genes commonly induced at all time points after protoplast isolation were enriched in stress response and metabolic process functions (fig. S1A). By contrast, the genes commonly induced at a later time point when compared with each earlier time point were enriched in biosynthetic process and development functions (table S2 and fig. S1B). We also compared each sample with freshly obtained protoplasts (table S3 and fig. S1C) and found enrichment of functions including cell growth and metabolic process in the 3978 genes constitutively activated during regeneration (fig. S1D).
To determine whether any known developmental program is used in protoplast regeneration, as previously reported in other regeneration processes (5–7, 16), we compared the genes enriched during protoplast regeneration with genes enriched in the embryo (17), shoot and root meristem domains (18–20), and guard cell progenitor meristemoid cells (21). Although enrichment was identified for several cell domains (Fig. 2, A to D), we did not find any developmental program commonly enriched during protoplast regeneration. Genes specific to certain cell domains—including leaf axil, lateral root cap, root quiescent center, and premature stomata cells—were enriched in microcalli formation. This suggests that multiple meristem pathways might be coupled and used in protoplast regeneration.
Fig. 2. Gene expression profiles after a prolonged culture.
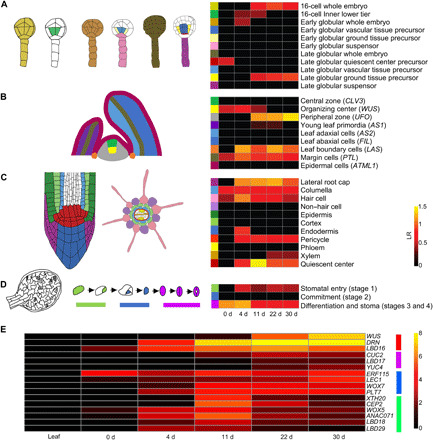
(A to D) Enrichment of meristematic and embryonic cell type–specific genes during protoplast regeneration. Left: Color codes showing cell types. Right: Heatmaps of the log2-scaled odds ratio (LR) for various tissues showing the levels of gene enrichment during regeneration. Only significant categories determined using a hypergeometric test with a false discovery rate correction (adjusted P ≤ 0.05) are shown. (A) Early embryonic cell types. (B) Shoot apex cell types. (C) Root tip cell types. (D) Stomatal meristemoid cell types. (E) Progressive activation of putative regeneration regulators after a prolonged culture. Four groups were clustered according to their expression pattern, as labeled on the right. The heatmap shows log2 (1 + RPKM) values.
We next focused on potential regeneration regulators (1) and established that many of them were enriched during regeneration (fig. S1E). Among them, a subset of 16 genes was commonly activated and gradually enriched after the prolonged culture, suggesting a potential association with totipotency. On the basis of their expression profiles, these genes were clustered into four groups (Fig. 2E). Because WUSCHEL (WUS) and DORNRÖSCHEN (DRN; also known as ENHANCER OF SHOOT REGENERATION1 or ESR1; we used drn mutant alleles and therefore adopted DRN for simplicity) were most prominently enriched after a 30-day culture (Fig. 2E), we selected these two genes and analyzed their expression with live imaging. In samples taken 50 days after protoplast isolation, WUS and DRN were expressed in most microcalli cells (fig. S2). By contrast, we could not detect the expression of BABY BOOM (BBM), CLAVATA3 (CLV3), DRN-LIKE (DRNL; also known as ESR2), WUSCHEL-RELATED HOMEOBOX2 (WOX2), and WOX5 in any microcallus after the 30-day culture (fig. S2). Note that DRNL is highly homologous to DRN and can promote shoot regeneration from root explants (22). We also analyzed the expression of J0121, a pericycle-like cell marker, and could not detect expression after the extended culture (fig. S2).
WUS is required for somatic cell regeneration
Because WUS is broadly expressed in most regenerating microcalli (fig. S2), we next asked whether WUS is required for protoplast regeneration. Protoplasts derived from wus-101, a null allele (23), were less competent in cell division; after 11 days in PIM, 50.8% of the wus-101 cells remained nondividing, while only 21.8% of the Col-0 wild-type cells were nondividing at this stage (Fig. 3A). The number of viable wus-101 cells dropped markedly after 22 days in CIM1. After 30 days, only 2.8% of the wus-101 microcalli contained viable cells, displaying a substantially lower viability than the wild type (76.6%) (Fig. 3A). After 38 days, almost all the wus-101 cells were dead (Fig. 3B). We could not obtain any regenerated callus or plant from wus-101.
Fig. 3. Mesophyll protoplast regeneration requires WUS.
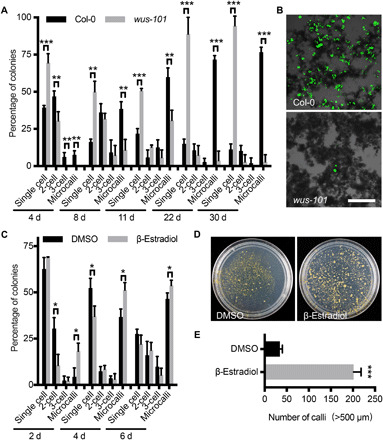
(A) Distribution of Col-0 and wus-101 cells during regeneration. Note the reduction of cell division in wus-101 (10 ≤ n ≤ 140). (B) Viable Col-0 and wus-101 cells after 38 days in culture, as shown by a fluorescein diacetate stain. Scale bar, 200 μm. (C) Distribution of pga6-1 cells during 6 days of culture in a mock treatment [dimethyl sulfoxide (DMSO)] or β-estradiol induction (29 ≤ n ≤ 145). (D) Representative plates showing the callus density of pga6-1 mesophyll protoplasts after 73 days in a DMSO treatment or β-estradiol induction. (E) Statistical analysis of the number of calli derived from the pga6-1 mesophyll protoplasts after 73 days in a DMSO treatment or β-estradiol induction. In (A), (C), and (E), the data are presented as means ± SD for more than three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 (Student’s t test).
We further explored whether constitutive WUS expression promoted protoplast regeneration using two inducible WUS-overexpressing lines. Protoplasts derived from pga6-1 (24), in which β-estradiol induces WUS overexpression, divided more frequently after the β-estradiol induction (Fig. 3C). After 4 days in PIM, we obtained 39.8% more microcalli with continuous induction than in the mock treatment. Samples were continuously induced until transfer onto semisolid SIM without β-estradiol. By then, we obtained five to six times more calli than samples with only mock treatment (Fig. 3, D and E). Calli obtained from protoplasts overexpressing WUS had comparable shoot regeneration potentials to those from the mock-treated protoplasts. Similar results were obtained using an independent inducible line, pUBQ10::WUS-GR (23), which is induced by dexamethasone (Dex) (fig. S3A). Together, these observations indicate that WUS is necessary for protoplast regeneration, and its overexpression enhances regeneration efficiency.
Several WUS paralogs promote meristem functions. In particular, WOX2 is required for the formation of the shoot apical meristem during embryogenesis (25), while WOX5 is required for root meristem activity and pericycle-based tissue regeneration (26). We found that the regeneration of wox2-4 mutant protoplasts is comparable to that of the wild-type protoplasts, while the wox5-1 mutant protoplasts displayed a slightly reduced regeneration capacity (fig. S3, B and C).
DRN is required for somatic cell regeneration
DRN is also broadly expressed in regenerating microcalli (fig. S2). We found that the APETALA2 (AP2) transcription factor DRN also promoted protoplast cell regeneration in a way similar to WUS. Similar to, but more prominent than wus-101, cell division was greatly reduced in protoplasts derived from drn-1. After 8 days in PIM, 79.1% of the drn-1 cells remained nondividing (Fig. 4A), while after 11 days, we could no longer detect any viable drn-1 cells (Fig. 4B), resulting in no callus formation or regeneration at all.
Fig. 4. Mesophyll protoplast regeneration requires DRN.
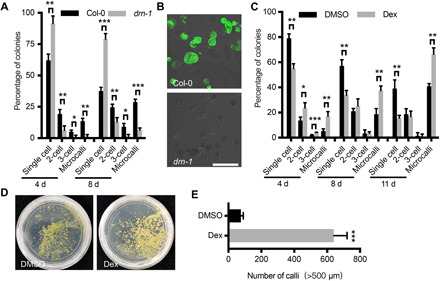
(A) Distribution of Col-0 and drn-1 cells during regeneration over 8 days of culture (26 ≤ n ≤ 75). (B) Viable Col-0 and drn-1 cells after 38 days in culture, as shown by a fluorescein diacetate stain. Scale bar, 100 μm. (C) Distribution of p35S::DRN-GR cells during 11 days of culture in a mock treatment (DMSO) or Dex induction (34 ≤ n ≤ 118). (D) Representative plates showing the callus density of p35S::DRN-GR mesophyll protoplasts 85 days in a DMSO treatment or Dex induction. (E) Statistical analysis of the number of calli following the DMSO treatment or Dex induction. In (A), (C), and (E), the data are presented as means ± SD for more than three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 (Student’s t test).
On the other hand, overexpressing DRN promoted regeneration. Using a Dex-inducible DRN-overexpressing line, p35S::DRN-GR (20), we observed immediate cell division after Dex induction, which is similar to induced WUS overexpression. After 4 days in PIM with Dex, we observed three times more cell divisions than in the mock treatment control plants (Fig. 4C), while after 11 days of continuous induction, we obtained 62.5% more microcalli than in the controls. After the continuously induced microcalli were transformed onto semisolid SIM without induction, we obtained eight to nine times more calli than cells with only mock treatment (Fig. 4, D and E). The calli obtained after the induction displayed comparable plantlet regeneration potential to those produced from the control protoplasts.
Activation of gene expression by protoplast isolation
As WUS and DRN expression can significantly promote protoplast regeneration, it is essential to determine how these genes are activated. To this end, we used reporters to monitor the gene expression of DRN and WUS. By analyzing more than 10,000 mesophyll cells within mature leaves, we confirmed that neither of these two genes was expressed in mature leaves (fig. S4A). We then followed their expression in mesophyll protoplasts right after their isolation and found that these two genes were expressed after protoplast isolation only at a very low frequency. We observed that 0.09% of the cells expressed WUS and 0.03% expressed DRN. Furthermore, the expression levels were highly variable (Fig. 5). Note that cells expressing WUS or DRN had similar morphology as other cells. The number of WUS- or DRN-expressing cells increased after a few days. This could be attributed to two reasons: (i) Positive cells divided much more frequently and cell masses might dissociate, and (ii) there were de novo activation events.
Fig. 5. Isolating protoplasts induces ectopic gene expression.

After 12 hours of enzymatic digestion, stochastic activation of pWOX5::GFP, pPLT7::PLT7-YFP, pBBM::BBM-YFP, pWUS::3×Venus-N7, and pDRN::mCherry-N7 in mesophyll protoplasts with signal intensity quantifications. The frequencies of expression in a given cell are listed in parentheses. Color bar indicates relative fluorescence intensity (black to red).
We also followed the reporter lines of three other genes, BBM, PLETHORA7 (PLT7), and WOX5. None of these genes was expressed in mature leaves (fig. S4A). Upon protoplast isolation, low frequencies of activation were found for these genes (Fig. 5). Nevertheless, these genes were no longer expressed in regenerating microcalli (fig. S2). Together, we found that genes can be activated, often at a very low frequency, by protoplast isolation.
We further resolved whether gene activation could be attributed to wall digestion or to other mechanical damage to leaves. To this end, we immersed chopped leaves in enzyme-free MGG, soaked overnight, and followed more than 15,000 mesophyll cells in chopped leaves. We could not detect any cell expressing WUS, DRN, PLT7, or WOX5 (fig. S4B). On the other hand, we found two cells expressing BBM (~0.01%), which was lower than after wall digestion (0.09%). Thus, protoplast isolation, i.e., wall digestion, seems to be the major cause of ectopic activation of gene expression.
Last, we followed the expression of WUS and DRN during regeneration using time-lapse imaging. We tracked 46 DRN-expressing cells and observed that three of them formed microcalli after multiple rounds of division. Notably, DRN expression was maintained in the daughter cells (Fig. 6A). Similar observations were made for WUS, with 4 of 46 WUS-expressing cells forming microcalli (Fig. 6A). Notably, we analyzed 42 regenerating calli at 60 days after isolating protoplasts and found that all calli expressed both DRN and WUS. However, they had three expression patterns in calli (Fig. 6B): (i) Both WUS and DRN were widely distributed in calli; (ii) WUS was widely distributed in calli, and DRN was expressed in a few cells; and (iii) WUS and DRN were both expressed in a few cells.
Fig. 6. WUS and DRN are maintained in regeneration microcalli and calli.
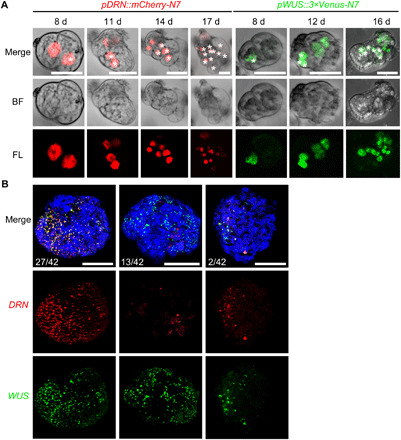
(A) Time-lapse images showing the maintained expression of pDRN::mCherry-N7 (red) and pWUS::3×Venus-N7 (green) in the regenerating mesophyll protoplasts. Asterisks label progenitors of the same cells in corresponding time points. Scale bars, 25 μm. (B) Maximum intensity views of confocal images showing the coexpression of pDRN::mCherry-N7 (red) and pWUS::3×Venus-N7 (green) in representative regenerating calli at 60 days. Three expression patterns are shown. Scale bars, 250 μm.
Genome-wide stochastic expression after protoplast isolation
To understand protoplast isolation-induced ectopic activation of gene expression at the genome level, we used single-cell transcriptome analyses and directly compared the variance in the expression levels between leaf cells before and after isolating protoplasts. Because plant single-cell RNA sequencing (scRNA-seq) analyses require protoplast isolation, we instead used single-nucleus RNA-seq (snRNA-seq). We isolated leaf cell nuclei before and after an overnight enzymatic wall digestion and applied parallel snRNA-seq with droplet technology (27). By this means, we totally sequenced 572 leaf nuclei and 892 protoplast nuclei (table S4 and fig. S5A). To assess the data quality, we combined reads from the single cells and compared them with bulk RNA-seq results. As shown in fig. S5 (B and C), the snRNA-seq data had a high correlation coefficient with the bulk RNA-seq, which was comparable to analyses in animals (28). To exclude nonmesophyll cells, we selected leaf nuclei expressing the mesophyll cell–specific photosynthesis-related genes RUBISCO SMALL SUBUNIT 1A (RBCS1A), RBCS2B, and RBCS3B (29). In addition, we excluded protoplast nuclei expressing guard cell marker genes (SCRM, MYB60, or CYP86A2), vascular tissue marker genes (APL or DOF5.6), or epidermal cell marker genes (ATML1 or TMM). We compared the coefficients of variation (CVs) of expression among the mesophyll cells before and after wall digestion. As shown in Fig. 7A, nuclei from the protoplasts (after digestion) showed a significantly higher variation in their expression levels (P < 0.0001, Wilcoxon rank sum test), and this increase in CV was independent of expression levels (Fig. 7B).
Fig. 7. Genome-wide analyses of gene expression variability using snRNA-seq and scRNA-seq.
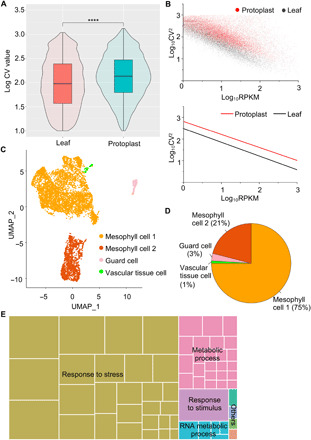
(A) Comparison of the gene expression CVs obtained using snRNA-seq on the leaf and protoplast nuclei. The significance of the difference was determined using a Wilcoxon rank sum test (****P < 0.0001). (B) Comparison of gene expression CV with expression levels obtained by snRNA-seq. Top: Scatterplot of log10-transformed expression CV2s and expression levels in reads per kilobase of transcript per million mapped reads (RPKM) for leaf cells and protoplasts. Bottom: Simple linear fits to the above scatterplot. (C) Visualization of four leaf cell clusters (mesophyll cell 1, mesophyll cell 2, guard cell, and vascular tissue cell) using the uniform manifold approximation and projection (UMAP) algorithm. Dots represent individual cells and colors indicate corresponding cell clusters. (D) Numbers and percentages of four groups of cells in (C). (E) GO analysis of genes with high expression variability. The function-related enrichments were summarized using REVIGO. The aggregate size indicates the significance levels of the GO term, as determined using the Yekutieli test with false discovery rate correction.
The number of genes recovered using snRNA-seq was rather low, precluding more detailed analyses. Compared with snRNA-seq, droplet technology–based scRNA-seq had a substantially higher sequencing depth. We next applied scRNA-seq to more reliably analyze the variation in gene expression between protoplasts, with the results showing high correlation with the bulk RNA-seq results (fig. S5D). By applying the uniform manifold approximation and projection (UMAP) algorithm for data dimension reduction to 6683 captured protoplast cells (table S5 and fig. S5A), we found that the protoplasts were grouped into four major clusters (Fig. 7C), less than the multiple heterogeneous clusters identified in root tip cells (30). On the basis of the limited expression variations, we identified that among the four groups of protoplasts, two small groups represented the guard cells and vascular tissues, respectively, based on their expression of the marker genes, while the other two (comprising 96% of the cells) both expressed mesophyll genes (Fig. 7D). Nevertheless, one group, mesophyll cell 2, contained a small portion of cells expressing other cell identity markers, such as epidermal marker genes (fig. S6). Together, mesophyll protoplasts are highly abundant in isolated protoplasts.
Although individual cells have generally similar expression profiles, there were clear variations. For the genes analyzed using imaging, comparable frequencies and variations of expression were also observed using scRNA-seq (fig. S5, E and F). Among the 92 genes reported to be involved in any type of regeneration in Arabidopsis (1), 86 were expressed in at least one protoplast (fig. S7). Genes that are responsive to wounding and other stresses, such as WOUND INDUCED DEDIFFERENTIATION1 (WIND1) and WIND2 (31), were broadly expressed, whereas most genes were expressed in a limited number of cells. Using the GO analysis, we noted that genes with high expression variability are enriched in stress response, metabolic process, and stimulus response functions (Fig. 7E).
Together, our single-cell transcriptome analyses support that protoplast isolation increases gene expression variation among cells, which is consistent with imaging results. This variation was found for many genes, suggesting enhanced genome-wide stochastic expression. Our previous simulation showed that stochastic allelic expression could recapitulate the observed monoallelic expression patterns in rice mesophyll protoplasts (32), suggesting that stochastic changes in gene expression are common in protoplasts. Nevertheless, stress-responsive genes are more commonly activated.
Increased chromatin accessibility after protoplast isolation
Chromatin is usually open around the start codon of expressed genes (33). To understand potential reasons for the enhanced stochastic gene expression, we used assay for transposase-accessible chromatin sequencing (ATAC-seq) to analyze chromatin accessibility before and after an overnight cell wall digestion (table S6). Notably, chromatin accessibility was generally broadly increased in the protoplasts (Fig. 8A), with approximately fivefold more genes associated with open chromatin in the protoplasts than in the undigested leaf cells (Fig. 8B). Genes with open chromatin after protoplast isolation had enriched GO terms cellular metabolism, stress, and stimulus response (fig. S8A), similar to GO enrichment in the genes with high expression variability. Furthermore, the open chromatin region was much wider in the protoplasts, with an average of 453 nt in the undigested leaves and 556 nt in the protoplasts. In particular, some potential regeneration regulators had enhanced chromatin accessibility following protoplast isolation (fig. S8B). However, chromatin accessibility of the WUS and DRN loci was not significantly changed as assayed by ATAC-seq. WUS locus accessibility remained unchanged, and DRN locus accessibility was even slightly decreased in protoplasts (fig. S8B). We speculate that chromatin accessibility might have cellular-level variations, the elucidation of which requires single-nucleus ATAC-seq. Alternatively, instead of these loci, their upstream regulators are affected in protoplast at the chromatin level. Together, these results show that protoplast isolation increases chromatin accessibility.
Fig. 8. Chromatin accessibility changes after isolating protoplasts, promoting regeneration.
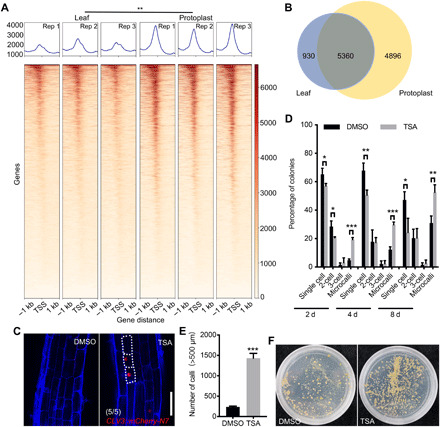
(A) Comparison of chromatin accessibility in undigested leaf cells and protoplasts. Heatmaps showing the chromatin accessibility for each gene, with one line representing one gene. The horizontal axis represents the distance relative to the transcription start site (TSS). The mean values of the accessibility degree at the corresponding sites are represented as a kernel density plot above the heatmap. The color scale bar indicates the relative degree of accessibility. Three replicates (Rep) for each sample are shown. (B) Numbers of highly accessible genes identified in the undigested leaf cells and protoplasts, corresponding to the ATAC-seq peaks. (C) After a 12-hour trichostatin A (TSA) treatment, the pCLV3::mCherry-N7 (red) expression was randomly activated in the root elongation zone cells. The cell walls were stained using FB28 (blue). Scale bar, 50 μm. (D) The TSA treatment increases the cell division and regeneration frequency of Col-0 protoplast cells (30 ≤ n ≤ 102). (E) Statistical analysis of the number of calli formed after 90 days with a mock or TSA treatment. (F) Representative plates showing calli density after 90 days with a mock or TSA treatment. In (A), (D), and (E), the data are presented as means ± SD for more than three independent experiments. *P < 0.05, **P < 0.01, and ***P < 0.001 (Student’s t test).
Increased chromatin accessibility promotes ectopic gene activation and regeneration
To test whether the increased chromatin accessibility resulted in an enhancement of stochastic gene expression, we treated plants with trichostatin A (TSA), a chemical inhibitor of histone deacetylases. TSA treatment broadly increases chromatin accessibility (34). Because root tissues are more transparent, we used roots and followed the selected marker genes that are not normally expressed in root cells. After a 12-hour TSA treatment, we detected ectopic activation of CLV3, but not WUS, SHOOT MERISTEMLESS (STM), CUP-SHAPED COTYLEDON1 (CUC1), and CUC2, in multiple root cell types. Whereas CLV3 was broadly induced by TSA in the root meristematic zone cells, its activation in the elongation zone cell types was more variable between individual plants and even within the same root (Fig. 8C). Notably, the expression level varied among cells of the same type, suggesting that there exists a stochastic expression. Because of the small sample size (five roots), low-frequency activation may not be detected. To test whether TSA enhances stochastic expression in protoplasts, we treated protoplasts with TSA. After a 24-hour TSA treatment, we observed three times more protoplasts expressing WUS than the mock treatment (fig. S9), but no activation for DRN was observed.
The TSA treatment also enhanced protoplast regeneration; the application of a 48-hour TSA treatment immediately after protoplast isolation resulted in four times more microcalli than the mock groups, with six times more calli developing after the transfer onto semisolid SIM (Fig. 8, D to F). Together, these results indicate that increasing chromatin accessibility leads to increased stochastic gene expression and enhanced protoplast regeneration.
DISCUSSION
Key regulators of totipotency
In plants and animals, regeneration is often traced to certain undifferentiated or partially differentiated stem cells, which are primed to acquire pluripotency or totipotency in response to internal or external cues. During plant tissue regeneration, the xylem pole pericycle cells and pericycle-like cells form a callus and, subsequently, adventitious shoots or roots (6, 7). During root and shoot meristem repair, recently differentiated cells can switch the identity to regenerate and replenish the damaged stem cells (3–5, 35–37). Despite these insights, the mechanisms by which fully differentiated somatic cells massively switch cell identity to acquire pluripotency or totipotency remain largely unexplored (38), although protoplast regeneration is a common route for gene transformation. In this study, we used mesophyll protoplast regeneration to study this notable cell identity transition process.
Because regenerating cells are very sparse, it is extremely difficult to investigate this process. By applying time course transcriptome analysis, we identified meristem function–promoting genes that promote the induction of stem cells from differentiated mesophyll protoplasts. Using mutants and overexpression lines, we showed that WUS and DRN were not only required but also greatly promoted the regeneration of differentiated cells. Most WUS clade WOX genes are at least partially functionally exchangeable, as shown by promoter swaps (39); however, we found that only WUS is involved in mesophyll regeneration. During normal embryogenesis, WOX2 and additional WOX genes, but not WUS, are required for shoot apical meristem formation (25), while WUS is subsequently expressed in the meristem organization center. Here, we established that the regeneration of wox2 mutant protoplasts was comparable to that of the wild-type cells, although WOX2 is frequently expressed in protoplasts (fig. S7), suggesting that protoplast-mediated regeneration is not an embryogenesis-like process. WUS also has roles in other regeneration processes; overexpressing WUS causes high-frequency somatic embryo formation from multiple tissues (24), and activating WUS expression is a key step in the induction of shoot growth from a callus (40). As WUS promotes cell division (39), it is conceivable that manipulating cell division could affect protoplast regeneration.
Under normal development, DRN functions redundantly with DRNL to promote embryo patterning, lateral organ formation, and the initiation of axillary meristems (20, 41). In addition, DRN overexpression promotes shoot regeneration from root explants (42). Our findings demonstrate that DRN, but not DRNL, strongly promotes mesophyll protoplast regeneration.
Transcriptome analysis identified more genes that were enriched during callus formation. We expect that other genes may also be involved in callus formation, which can be tested in the future.
Protoplast regeneration as an evolutionary and ecological process
It is widely accepted that mutation and natural selection is the driving force for evolution. As random mutations occur, natural selection decides which mutations will live on. We envision that random activation of expression and selection during culture for protoplast regeneration makes it a comparable process. In this model, protoplast isolation–induced stochastic expression patterns endow cells with heterogeneous fates, and a small portion of the cells could be pluripotent or totipotent. The prolonged cultivation of protoplasts also provides a cellular-level selection for totipotent cells that eventually regenerate into calli and plantlets (Fig. 9). The protoplast regeneration process echoes other scenarios involving cellular-level evolution; for example, it has been proposed that cancers can result from the cellular-level selection of cells that reproduce and spread most quickly (43), whereas heterogeneity is usually caused by different mutations in cancer cells. In yeast cells, stochastic expression can be an efficient strategy for survival in fluctuating environments (44).
Fig. 9. Conceptual model of mesophyll protoplast regeneration.
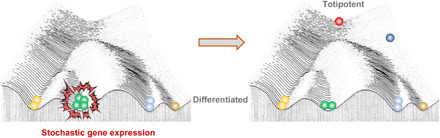
Scheme of mesophyll protoplast fate changes during regeneration on Waddington’s epigenetic landscape. During normal development, cells take on a terminal differentiated fate (down the hill) by progressing from the totipotent and pluripotent state (at the top). Protoplast isolation induces stochastic gene expression, causing a portion of differentiated cells to acquire heterogeneous fates, including totipotency and pluripotency.
Chromatin accessibility affects stochastic expression
What causes the stochastic gene expression? In this study, we revealed a notable increase in chromatin accessibility after protoplast isolation, which provides a mechanistic explanation for the observed stochastic expression. Our observation is consistent with previously reported heterochromatin disruption and decondensation after protoplast isolation (11–13).
This evolutionary perspective points toward new strategies to increase regeneration efficiency, as the further “scrambling” of gene expression may promote regeneration. Alterations to epigenetic modifications are expected to increase stochastic expression (45); we observed that increasing histone acetylation boosted ectopic gene expression and the regeneration rate (Fig. 8, D and E). Consistently, mutants of histone acetylase have reduced callus formation after wounding (46). It is conceivable that protoplasts from these mutants could have reduced regeneration ability. The same principle may also function in the induction of pluripotent mammalian stem cells (47).
MATERIALS AND METHODS
Plant materials
A. thaliana ecotype Col-0 was used as the wild type unless otherwise specified. The seeds were sown on half-strength Murashige and Skoog medium containing 1% sucrose, stratified at 4°C for 2 days and then grown under long-day conditions (16-hour light and 8-hour dark at 22°C) for 11 days before cells were collected from the leaves for use in generating protoplasts. The transgenic lines pPLT7::PLT7-YFP, pWOX5::GFP, pBBM::BBM-YFP, pWUS::3 ×Venus-N7, pDRN::mCherry-N7, pDRNL::3×Venus-N7, pCLV3::mCherry-N7, pWOX2::NLS-DsRed2, pUBQ10::WUS-GR, and p35S::DRN-GR are in the Col-0 background (48–51), while J0121 is in the C24 background (52). The wus-101, drn-1, wox2-4, and wox5-1 mutants are in the Col-0 background (23, 25, 53), and pga6-1 is in the Ws background (24).
Protoplast isolation and culture
We followed a previously described protocol for protoplast isolation and regeneration (14), in which media constituents and preparation are described for MGG, PIM, CIM1, CIM2, SIM, and PDM. Approximately 1 g of leaves from 11-day-old sterile plants were soaked in 10 ml of MGG and then rapidly chopped. The enzymatic digestion was performed overnight in the dark at 24°C. After the cell wall digestion, the protoplast suspension was filtered through an autoclaved 70-μm mesh filter, washed with 15 ml of W5 solution [2 mM MES, 154 mM NaCl, 125 mM CaCl2, and 5 mM KCl (pH 5.6)], and centrifuged for 5 min at 100g before the solution was removed. The washing and centrifugation were performed four more times. After the final centrifugation, the protoplast suspension was diluted in PIM to a concentration of 1 × 105 protoplasts/ml and cultured in the dark. After 11 days, the medium was diluted twofold using CIM1. One month later, the medium was diluted fourfold with CIM2. Two months later, the callus was transplanted onto SIM. After the callus turned green, it was transplanted onto PDM (Fig. 1A).
For control experiments without enzymatic digestion, identical procedures were used except that no enzyme was added. Briefly, we soaked the twice-cut leaves in enzyme-free MGG, which lacked Onozuka R-10, macerozyme, and Driselase, overnight (~12 hours). The samples were then washed with W5 buffer twice before imaging. To embed the protoplasts in alginate, a newly digested protoplast suspension was filtered through an autoclaved 70-μm mesh filter and washed once by 10 ml of MGG, as previously described (14). After the first wash, the pellet was resuspended in 10 ml of MES sucrose calcium medium, and 2 ml of MES mannitol magnesium (MMM) medium was layered on top. After centrifugation for 5 min at 100g, the protoplasts were collected from the interface. The pellet was washed with MMM medium for two additional times. The protoplasts were resuspended in MMM at a density of 105/ml, and the same volume of alginate solution [MgCl2·6H2O (1.02 g/liter), MgSO4·7H2O (1.25 g/liter), mannitol (85 g/liter), MES (1.925 g/liter), and alginate (12 g/liter) (pH 5.6)] was added to the protoplast suspension. The protoplast-alginate mixture with a piece of polypropylene grid was laid on the surface of agar-solidified medium [CaCl2·2H2O (2.94 g/liter), mannitol (85 g/liter), MES (1.952 g/liter), and agar (10 g/liter) (pH 5.6)]. After 20 min, the thin alginate layer was transferred into PIM.
For the chemical treatments, the pCLV3::mCherry-N7 roots were treated with 5 μM TSA (Sigma-Aldrich) for 12 hours. After protoplast isolation, the Col-0 protoplasts were treated with 1 μM TSA in PIM for 48 hours. To induce WUS expression, the pga6-1 protoplasts were treated with 10 μM β-estradiol (Sigma-Aldrich) in PIM, CIM1, and CIM2, while the pUBQ10::WUS-GR protoplasts were treated with 10 μM Dex (Sigma-Aldrich) in PIM for 24 hours. To induce DRN expression, the p35S::DRN-GR protoplasts were treated with 10 μM Dex in PIM, CIM1, and CIM2.
Confocal microscopy and optical microscopy
The excitation and detection window setups for green fluorescent protein (GFP), yellow fluorescent protein (YFP), DsRed, mCherry, and Venus were as described (54). To detect Fluorescent Brighteners 28 (FB28) staining, a 405-nm laser was used for excitation, while the emission was collected by a 425- to 475-nm band-pass filter. The fluorescein diacetate staining was performed as described in (14), with a 488-nm laser used for the excitation and the emission collected by a 500- to 530-nm band-pass filter. Autofluorescence was excited at 488 nm and detected at 660 to 700 nm. All optical photographs were taken with a Nikon SMZ1000 stereoscopic microscope or an Olympus BX60 microscope. The relative fluorescence intensity analysis (as shown in Fig. 6) was semiquantified using ImageJ (55).
To image the internal mesophyll cells, ClearSee was used. The ClearSee solutions were prepared as described in (56). Leaves were fixed with 4% (w/v) paraformaldehyde for 60 min in phosphate-buffered saline (PBS) under vacuum (∼690 mmHg) at 4°C. The fixed leaves were then washed twice for 1 min each in PBS, and the cleared leaves were stained with FB28 (final 100 μg/ml) in ClearSee solution for 1 hour. After staining, the tissues were washed in ClearSee solution for 1 hour before imaging.
Single-nucleus RNA sequencing
The nucleus isolation process was performed on ice. Arabidopsis leaves were cut into pieces in nucleus isolation buffer (NIB; Sigma-Aldrich), while the protoplast pellets from Arabidopsis were directly resuspended in NIB. After filtering with a three-layer nylon mesh (Calbiochem), Triton X-100 (Sigma-Aldrich) was added to each sample to a final concentration of 0.3% (v/v). After centrifugation at 100g and 4°C for 10 min, the pellet was gently suspended in 600 μl of NIB, to which 800 μl of 1.25 M sucrose was carefully added. The samples were then centrifuged at 1000g and 4°C for 10 min, and the supernatant was washed with NIB. Last, after centrifugation at 200g and 4°C for 10 min, the nucleus pellet was resuspended in NIB.
The snRNA-seq libraries were constructed using the Dolomite Bio scRNA-seq system, according to the manufacturer’s protocol (Dolomite Bio), and were sequenced with an Illumina HiSeq in the 150-nt paired-end mode. The snRNA-seq reads were processed using UMI-tools (57). After the empty droplets were removed, the barcodes were extracted and the low-quality reads were filtered, the remaining sequences were aligned to the TAIR10 reference genome using STAR (58). featureCounts (59) was used to assign reads to genes. To reduce the effect of sequencing depth of each cell and gene length, the count matrices were normalized to the RPKM (reads per kilobase of transcript per million mapped reads per cell) using our custom scripts (the scripts are available at https://github.com/duqingwei1989/ScRNA-seq). The CVs of the RPKM values of the expressed genes were calculated for each gene as RPKM SDs divided by mean values using custom scripts.
Single-cell RNA sequencing
A. thaliana protoplasts were isolated from the leaves of 11-day-old seedlings, according to a previously described protocol (14). Protoplast pellets were gently resuspended in 0.4 M mannitol and filtered through a 40-μm-diameter cell strainer (Falcon), after which they were washed twice with 0.4 M mannitol. The purity and viability of the protoplasts were determined using trypan blue and acridine orange/propidium iodide staining.
scRNA-seq libraries were constructed using a 10X platform (10X Genomics Single Cell 3′ Reagent Kits v. 2 protocol) and were sequenced with an Illumina HiSeq using the 150-nt paired-end mode. scRNA-seq sequencing reads were processed using CellRanger v. 2.1.0 (https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger). TAIR10 reference genome and annotation files were downloaded from The Arabidopsis Information Resource (www.arabidopsis.org/). The expression matrices of 6683 protoplast cells generated from Cell Ranger were imported into Seurat v. 3.0 (60). The cells were filtered to remove those with fewer than 500 unique features or more than 8000. Cells in which more than 5% of the sequence was mitochondrial DNA or chloroplastal DNA were also removed. The global-scaling normalization method “LogNormalize” was used. Highly variable genes were calculated using the FindVariableFeature function with a mean.cutoff of (0.0125, 3) and a dispersion.cutoff of (1.5, Inf). The three expression matrices were merged using the FindIntegrationAnchors and IntegrateData functions. After scaling the data, a linear dimensional reduction was performed using RunPCA, with the settings npcs = 40, and FindClusters function with “resolution = 1”. Cell clusters were visualized using UMAP with “dims = 40′,” n.neighbors = 30′, and “min.dist = 0.3”.
For the digested protoplast scRNA-seq, 17 cell clusters were reclassified using mesophyll cell marker genes (RBCS1A, RBCS2B, and RBCS3B), epidermal cell marker genes (ATML1 and TMM), vascular tissue marker genes (APL and DOF5.6), and guard cell marker genes (SCRM, MYB60, and CYP86A2). Only the mesophyll cells were retained for further CV analysis. The count matrices of 5000 mesophyll cells were normalized to the RPKM. The CV of the RPKM values for 86 regeneration regulators were calculated for the mesophyll cells and visualized using the pheatmap R package (https://CRAN.R-project.org/package=pheatmap).
Assay for transposase-accessible chromatin sequencing
The same procedures were used to isolate nuclei for ATAC-seq as for snRNA-seq. In addition, fluorescence-activated cell sorting was performed to purify the nuclei from chloroplast and mitochondria contamination. For each sample, 106 purified nuclei were treated with a 50-μl transposase reaction solution for 30 min at 37°C using Nextera reagents (Illumina). The DNA fragments were purified using a MinElute PCR purification kit (Qiagen) and eluted in 11 μl of elution buffer. The samples were amplified using the High-Fidelity PCR Mix (New England Biolabs) for 11 polymerase chain reaction (PCR) cycles. Three independent biological replicates were performed in the same batch. The amplified libraries were purified using AMPure XP beads (Beckman Coulter) and sequenced with an Illumina HiSeq in the 150-nt paired-end mode.
The ATAC-seq reads were aligned to the TAIR10 reference genome using bowtie2, allowing two mismatches (61). After sorting and indexing, the duplicated reads were removed using Picard (http://broadinstitute.github.io/picard/) with default parameters. For the normalization and visualization, deepTools v. 2.0 (62) was used, with a bin size of 1 bp and RPKM normalization. Heatmaps and average plots were generated using the deepTools “computeMatrix” and “plotHeatmap” parameters. The significance of the difference of the relative accessibility degree was tested using Student’s t test. The peaks were called using MACS2 (63), and the peaks of the three replicates were merged using bedtools (64). The annotatePeaks.pl script of HOMER (65) was used to annotate the peaks with gene names. featureCounts was used to assign reads to genes, and the count matrices were normalized to the RPKM values using edgeR (66). The RPKM values of 40 regeneration regulators were visualized using the pheatmap package.
Bulk RNA-seq
The total RNA of the A. thaliana leaves; the protoplasts from the leaf cell digestion (0 day); and the protoplasts cultured for 4, 11, 22, and 30 days were extracted using the Qiagen RNeasy Kit. Three independent biological replicates were performed for each time point. Polyadenylated RNAs were isolated and subjected to a library preparation (67). The libraries were sequenced using an Illumina HiSeq in the 150-nt paired-end mode.
The reads were aligned to the TAIR10 reference genome using STAR. With the low-quality reads removed and the uniquely mapped reads extracted, the count matrices were generated with featureCounts. The differential expression was then determined using the edgeR package with the cutoff value “>2 fold change” and “Benjamini-Hochberg false discovery rate < 0.01.” A heatmap of the regeneration regulators was generated using the pheatmap package. The correlation between the RPKM from the bulk RNA-seq and the CV of the scRNA-seq was visualized using ggplot2, and the Spearman correlation was calculated. Along the CV axis of every 1000 genes, the genes with the top 5% RPKM values were selected as high-CV genes. A GO enrichment analysis was operated using agriGO in Singular Enrichment Analysis (SEA) mode. REVGO was used to summarize and visualize the enrichment results. The domain-specific enrichment was quantified as a log2 odds ratio, while the statistical significance (P value) of the enrichment was assessed using a hypergeometric test.
Acknowledgments
We thank C. Li, L. Xu, Z. Zhao, and J. Zuo for providing seeds. Funding: Funding was from National Key R&D Program of China grant 2019YFA0903901 (to Y.J.), National Key R&D Program of China grant 2019YFA0903902 (to Y.W.), National Natural Science Foundation of China grant 31825002 (to Y.J.), National Natural Science Foundation of China grant 31871245 (to Y.W.), Key Research Project of the Frontier Science of CAS grant ZDBS-LY-SM012 (to Y.J.), Newton Advanced Fellow of the Royal Society award NAF\R1\180125 (to Y.J.), National Natural Science Foundation of China grant 31970805 (to C.T.), and Youth Innovation Promotion Association of CAS award 2017139 (to C.T.). Author contributions: Conceptualization: Y.J. and Y.W. Methodology: M.X., Q.D., C.T., Y.W., and Y.J. Investigation: M.X., Q.D., and C.T. Supervision: Y.J. and Y.W. Writing—original draft: Y.J. and M.X. Writing—review and editing: Y.J., Y.W., M.X., and Q.D. Competing interests: The authors declare that they have no competing interests. Data and materials availability: All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The raw sequencing data have been deposited in the NCBI SRA database under the BioProject number PRJNA648028 (SRR12588186-SRR12588214) and the National Genomics Data Center (https://bigd.big.ac.cn) database under the accession number PRJCA004852.
SUPPLEMENTARY MATERIALS
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/33/eabg8466/DC1
REFERENCES AND NOTES
- 1.Ikeuchi M., Favero D. S., Sakamoto Y., Iwase A., Coleman D., Rymen B., Sugimoto K., Molecular mechanisms of plant regeneration. Annu. Rev. Plant Biol. 70, 377–406 (2019). [DOI] [PubMed] [Google Scholar]
- 2.Sang Y. L., Cheng Z. J., Zhang X. S., iPSCs: A comparison between animals and plants. Trends Plant Sci. 23, 660–666 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Marhava P., Hoermayer L., Yoshida S., Marhavý P., Benková E., Friml J., Re-activation of stem cell pathways for pattern restoration in plant wound healing. Cell 177, 957–969.e13 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reinhardt D., Frenz M., Mandel T., Kuhlemeier C., Microsurgical and laser ablation analysis of interactions between the zones and layers of the tomato shoot apical meristem. Development 130, 4073–4083 (2003). [DOI] [PubMed] [Google Scholar]
- 5.Efroni I., Mello A., Nawy T., Ip P. L., Rahni R., DelRose N., Powers A., Satija R., Birnbaum K. D., Root regeneration triggers an embryo-like sequence guided by hormonal interactions. Cell 165, 1721–1733 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Atta R., Laurens L., Boucheron-Dubuisson E., Guivarc’h A., Carnero E., Giraudat-Pautot V., Rech P., Chriqui D., Pluripotency of Arabidopsis xylem pericycle underlies shoot regeneration from root and hypocotyl explants grown in vitro. Plant J. 57, 626–644 (2009). [DOI] [PubMed] [Google Scholar]
- 7.Sugimoto K., Jiao Y., Meyerowitz E. M., Arabidopsis regeneration from multiple tissues occurs via a root development pathway. Dev. Cell 18, 463–471 (2010). [DOI] [PubMed] [Google Scholar]
- 8.Sheng L., Hu X., Du Y., Zhang G., Huang H., Scheres B., Xu L., Non-canonical WOX11-mediated root branching contributes to plasticity in Arabidopsis root system architecture. Development 144, 3126–3133 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vasil V., Hildebrandt A. C., Differentiation of tobacco plants from single, isolated cells in microcultures. Science 150, 889–892 (1965). [DOI] [PubMed] [Google Scholar]
- 10.Takebe I., Labib G., Melchers G., Regeneration of whole plants from isolated mesophyll protoplasts of tobacco. Naturwissenschaften 58, 318–320 (1971). [Google Scholar]
- 11.Williams L., Zhao J., Morozova N., Li Y., Avivi Y., Grafi G., Chromatin reorganization accompanying cellular dedifferentiation is associated with modifications of histone H3, redistribution of HP1, and activation of E2F-target genes. Dev. Dyn. 228, 113–120 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Zhao J., Morozova N., Williams L., Libs L., Avivi Y., Grafi G., Two phases of chromatin decondensation during dedifferentiation of plant cells: Distinction between competence for cell fate switch and a commitment for S phase. J. Biol. Chem. 276, 22772–22778 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Tessadori F., Chupeau M. C., Chupeau Y., Knip M., Germann S., van Driel R., Fransz P., Gaudin V., Large-scale dissociation and sequential reassembly of pericentric heterochromatin in dedifferentiated Arabidopsis cells. J. Cell Sci. 120, 1200–1208 (2007). [DOI] [PubMed] [Google Scholar]
- 14.Chupeau M.-C., Granier F., Pichon O., Renou J. P., Gaudin V., Chupeau Y., Characterization of the early events leading to totipotency in an Arabidopsis protoplast liquid culture by temporal transcript profiling. Plant Cell 25, 2444–2463 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Damm B., Willmitzer L., Regeneration of fertile plants from protoplasts of different Arabidopsis thaliana genotypes. Mol. Gen. Genet. 213, 15–20 (1988). [Google Scholar]
- 16.Ishihara H., Sugimoto K., Tarr P. T., Temman H., Kadokura S., Inui Y., Sakamoto T., Sasaki T., Aida M., Suzuki T., Inagaki S., Morohashi K., Seki M., Kakutani T., Meyerowitz E. M., Matsunaga S., Primed histone demethylation regulates shoot regenerative competency. Nat. Commun. 10, 1786 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Palovaara J., Saiga S., Wendrich J. R., van ‘t Wout Hofland N., van Schayck J. P., Hater F., Mutte S., Sjollema J., Boekschoten M., Hooiveld G. J., Weijers D., Transcriptome dynamics revealed by a gene expression atlas of the early Arabidopsis embryo. Nat. Plants 3, 894–904 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Brady S. M., Orlando D. A., Lee J. Y., Wang J. Y., Koch J., Dinneny J. R., Mace D., Ohler U., Benfey P. N., A high-resolution root spatiotemporal map reveals dominant expression patterns. Science 318, 801–806 (2007). [DOI] [PubMed] [Google Scholar]
- 19.Tian C., Wang Y., Yu H., He J., Wang J., Shi B., Du Q., Provart N. J., Meyerowitz E. M., Jiao Y., A gene expression map of shoot domains reveals regulatory mechanisms. Nat. Commun. 10, 141 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Tian C., Zhang X., He J., Yu H., Wang Y., Shi B., Han Y., Wang G., Feng X., Zhang C., Wang J., Qi J., Yu R., Jiao Y., An organ boundary-enriched gene regulatory network uncovers regulatory hierarchies underlying axillary meristem initiation. Mol. Syst. Biol. 10, 755 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Adrian J., Chang J., Ballenger C. E., Bargmann B. O., Alassimone J., Davies K. A., Lau O. S., Matos J. L., Hachez C., Lanctot A., Vaten A., Birnbaum K. D., Bergmann D. C., Transcriptome dynamics of the stomatal lineage: Birth, amplification, and termination of a self-renewing population. Dev. Cell 33, 107–118 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ikeda Y., Banno H., Niu Q. W., Howell S. H., Chua N. H., The ENHANCER OF SHOOT REGENERATION 2 gene in Arabidopsis regulates CUP-SHAPED COTYLEDON 1 at the transcriptional level and controls cotyledon development. Plant Cell Physiol. 47, 1443–1456 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Daum G., Medzihradszky A., Suzaki T., Lohmann J. U., A mechanistic framework for noncell autonomous stem cell induction in Arabidopsis. Proc. Natl. Acad. Sci. U.S.A. 111, 14619–14624 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Zuo J., Niu Q. W., Frugis G., Chua N. H., The WUSCHEL gene promotes vegetative-to-embryonic transition in Arabidopsis. Plant J. 30, 349–359 (2002). [DOI] [PubMed] [Google Scholar]
- 25.Zhang Z., Tucker E., Hermann M., Laux T., A molecular framework for the embryonic initiation of shoot meristem stem cells. Dev. Cell 40, 264–277.e4 (2017). [DOI] [PubMed] [Google Scholar]
- 26.Kim J.-Y., Yang W., Forner J., Lohmann J. U., Noh B., Noh Y.-S., Epigenetic reprogramming by histone acetyltransferase HAG1/AtGCN5 is required for pluripotency acquisition in Arabidopsis. EMBO J. 37, e98726 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Habib N., Avraham-Davidi I., Basu A., Burks T., Shekhar K., Hofree M., Choudhury S. R., Aguet F., Gelfand E., Ardlie K., Weitz D. A., Rozenblatt-Rosen O., Zhang F., Regev A., Massively parallel single-nucleus RNA-seq with DroNc-seq. Nat. Methods 14, 955–958 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kolodziejczyk A. A., Kim J. K., Tsang J. C., Ilicic T., Henriksson J., Natarajan K. N., Tuck A. C., Gao X., Buhler M., Liu P., Marioni J. C., Teichmann S. A., Single cell RNA-sequencing of pluripotent states unlocks modular transcriptional variation. Cell Stem Cell 17, 471–485 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim J. Y., Yuan Z., Jackson D., Developmental regulation and significance of KNOX protein trafficking in Arabidopsis. Development 130, 4351–4362 (2003). [DOI] [PubMed] [Google Scholar]
- 30.Ryu K. H., Huang L., Kang H. M., Schiefelbein J., Single-cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiol. 179, 1444–1456 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Iwase A., Mitsuda N., Koyama T., Hiratsu K., Kojima M., Arai T., Inoue Y., Seki M., Sakakibara H., Sugimoto K., Ohme-Takagi M., The AP2/ERF transcription factor WIND1 controls cell dedifferentiation in Arabidopsis. Curr. Biol. 21, 508–514 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Han Y., Chu X., Yu H., Ma Y.-K., Wang X.-J., Qian W., Jiao Y., Single-cell transcriptome analysis reveals widespread monoallelic gene expression in individual rice mesophyll cells. Sci. Bull. 62, 1304–1314 (2017). [DOI] [PubMed] [Google Scholar]
- 33.Maher K. A., Bajic M., Kajala K., Reynoso M., Pauluzzi G., West D. A., Zumstein K., Woodhouse M., Bubb K., Dorrity M. W., Queitsch C., Bailey-Serres J., Sinha N., Brady S. M., Deal R. B., Profiling of accessible chromatin regions across multiple plant species and cell types reveals common gene regulatory principles and new control modules. Plant Cell 30, 15–36 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Görisch S. M., Wachsmuth M., Tóth K. F., Lichter P., Rippe K., Histone acetylation increases chromatin accessibility. J. Cell Sci. 118, 5825–5834 (2005). [DOI] [PubMed] [Google Scholar]
- 35.Xu J., Hofhuis H., Heidstra R., Sauer M., Friml J., Scheres B., A molecular framework for plant regeneration. Science 311, 385–388 (2006). [DOI] [PubMed] [Google Scholar]
- 36.Sena G., Wang X., Liu H. Y., Hofhuis H., Birnbaum K. D., Organ regeneration does not require a functional stem cell niche in plants. Nature 457, 1150–1153 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zhou W., Lozano-Torres J. L., Blilou I., Zhang X., Zhai Q., Smant G., Li C., Scheres B., A jasmonate signaling network activates root stem cells and promotes regeneration. Cell 177, 942–956.e14 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Steward F. C., Mapes M. O., Mears K., Growth and organized development of cultured cells. II. Organization in cultures grown from freely suspended cell. Am. J. Bot. 45, 705–708 (1958). [Google Scholar]
- 39.Dolzblasz A., Nardmann J., Clerici E., Causier B., van der Graaff E., Chen J., Davies B., Werr W., Laux T., Stem cell regulation by Arabidopsis WOX genes. Mol. Plant 9, 1028–1039 (2016). [DOI] [PubMed] [Google Scholar]
- 40.Sugimoto K., Temman H., Kadokura S., Matsunaga S., To regenerate or not to regenerate: Factors that drive plant regeneration. Curr. Opin. Plant Biol. 47, 138–150 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Chandler J. W., Cole M., Flier A., Grewe B., Werr W., The AP2 transcription factors DORNRÖSCHEN and DORNRÖSCHEN-LIKE redundantly control Arabidopsis embryo patterning via interaction with PHAVOLUTA. Development 134, 1653–1662 (2007). [DOI] [PubMed] [Google Scholar]
- 42.Banno H., Ikeda Y., Niu Q. W., Chua N. H., Overexpression of Arabidopsis ESR1 induces initiation of shoot regeneration. Plant Cell 13, 2609–2618 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Sprouffske K., Merlo L. M., Gerrish P. J., Maley C. C., Sniegowski P. D., Cancer in light of experimental evolution. Curr. Biol. 22, R762–R771 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Acar M., Mettetal J. T., van Oudenaarden A., Stochastic switching as a survival strategy in fluctuating environments. Nat. Genet. 40, 471–475 (2008). [DOI] [PubMed] [Google Scholar]
- 45.Toyoda S., Kawaguchi M., Kobayashi T., Tarusawa E., Toyama T., Okano M., Oda M., Nakauchi H., Yoshimura Y., Sanbo M., Hirabayashi M., Hirayama T., Hirabayashi T., Yagi T., Developmental epigenetic modification regulates stochastic expression of clustered protocadherin genes, generating single neuron diversity. Neuron 82, 94–108 (2014). [DOI] [PubMed] [Google Scholar]
- 46.Rymen B., Kawamura A., Lambolez A., Inagaki S., Takebayashi A., Iwase A., Sakamoto Y., Sako K., Favero D. S., Ikeuchi M., Suzuki T., Seki M., Kakutani T., Roudier F., Sugimoto K., Histone acetylation orchestrates wound-induced transcriptional activation and cellular reprogramming in Arabidopsis. Commun. Biol. 2, 404 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Graf T., Stadtfeld M., Heterogeneity of embryonic and adult stem cells. Cell Stem Cell 3, 480–483 (2008). [DOI] [PubMed] [Google Scholar]
- 48.Blilou I., Xu J., Wildwater M., Willemsen V., Paponov I., Friml J., Heidstra R., Aida M., Palme K., Scheres B., The PIN auxin efflux facilitator network controls growth and patterning in Arabidopsis roots. Nature 433, 39–44 (2005). [DOI] [PubMed] [Google Scholar]
- 49.Hofhuis H., Laskowski M., Du Y., Prasad K., Grigg S., Pinon V., Scheres B., Phyllotaxis and rhizotaxis in Arabidopsis are modified by three PLETHORA transcription factors. Curr. Biol. 23, 956–962 (2013). [DOI] [PubMed] [Google Scholar]
- 50.Luo L., Zeng J., Wu H., Tian Z., Zhao Z., A molecular framework for auxin-controlled homeostasis of shoot stem cells in Arabidopsis. Mol. Plant 11, 899–913 (2018). [DOI] [PubMed] [Google Scholar]
- 51.Zhang T.-Q., Lian H., Zhou C.-M., Xu L., Jiao Y., Wang J.-W., A two-step model for de novo activation of WUSCHEL during plant shoot regeneration. Plant Cell 29, 1073–1087 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Laplaze L., Parizot B., Baker A., Ricaud L., Martiniere A., Auguy F., Franche C., Nussaume L., Bogusz D., Haseloff J., GAL4-GFP enhancer trap lines for genetic manipulation of lateral root development in Arabidopsis thaliana. J. Exp. Bot. 56, 2433–2442 (2005). [DOI] [PubMed] [Google Scholar]
- 53.Kirch T., Simon R., Grunewald M., Werr W., The DORNRÖSCHEN/ENHANCER OF SHOOT REGENERATION1 gene of Arabidopsis acts in the control of meristem cell fate and lateral organ development. Plant Cell 15, 694–705 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Shi B., Zhang C., Tian C., Wang J., Wang Q., Xu T., Xu Y., Ohno C., Sablowski R., Heisler M. G., Theres K., Wang Y., Jiao Y., Two-step regulation of a meristematic cell population acting in shoot branching in Arabidopsis. PLOS Genet. 12, e1006168 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Schneider C. A., Rasband W. S., Eliceiri K. W., NIH Image to ImageJ: 25 years of image analysis. Nat. Methods 9, 671–675 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kurihara D., Mizuta Y., Sato Y., Higashiyama T., ClearSee: A rapid optical clearing reagent for whole-plant fluorescence imaging. Development 142, 4168–4179 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Smith T., Heger A., Sudbery I., UMI-tools: Modeling sequencing errors in Unique Molecular Identifiers to improve quantification accuracy. Genome Res. 27, 491–499 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Dobin A., Davis C. A., Schlesinger F., Drenkow J., Zaleski C., Jha S., Batut P., Chaisson M., Gingeras T. R., STAR: Ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Liao Y., Smyth G. K., Shi W., featureCounts: An efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930 (2014). [DOI] [PubMed] [Google Scholar]
- 60.Butler A., Hoffman P., Smibert P., Papalexi E., Satija R., Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat. Biotechnol. 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Langmead B., Salzberg S. L., Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ramírez F., Ryan D. P., Grüning B., Bhardwaj V., Kilpert F., Richter A. S., Heyne S., Dündar F., Manke T., deepTools2: A next generation web server for deep-sequencing data analysis. Nucleic Acids Res. 44, W160–W165 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Feng J., Liu T., Qin B., Zhang Y., Liu X. S., Identifying ChIP-seq enrichment using MACS. Nat. Protoc. 7, 1728–1740 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Quinlan A. R., Hall I. M., BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 26, 841–842 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Heinz S., Benner C., Spann N., Bertolino E., Lin Y. C., Laslo P., Cheng J. X., Murre C., Singh H., Glass C. K., Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Robinson M. D., McCarthy D. J., Smyth G. K., edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 26, 139–140 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.He J., Jiao Y., Next-generation sequencing applied to flower development: RNA-seq. Methods Mol. Biol. 1110, 401–411 (2014). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary material for this article is available at http://advances.sciencemag.org/cgi/content/full/7/33/eabg8466/DC1