

ABSTRACT
Approaches for recovering and analyzing genomes belonging to novel, hitherto-unexplored bacterial lineages have provided invaluable insights into the metabolic capabilities and ecological roles of yet-uncultured taxa. The phylum Acidobacteria is one of the most prevalent and ecologically successful lineages on Earth, yet currently, multiple lineages within this phylum remain unexplored. Here, we utilize genomes recovered from Zodletone Spring, an anaerobic sulfide and sulfur-rich spring in southwestern Oklahoma, as well as from multiple disparate soil and nonsoil habitats, to examine the metabolic capabilities and ecological role of members of family UBA6911 (group 18) Acidobacteria. The analyzed genomes clustered into five distinct genera, with genera Gp18_AA60 and QHZH01 recovered from soils, genus Ga0209509 from anaerobic digestors, and genera Ga0212092 and UBA6911 from freshwater habitats. All genomes analyzed suggested that members of Acidobacteria group 18 are metabolically versatile heterotrophs capable of utilizing a wide range of proteins, amino acids, and sugars as carbon sources, possess respiratory and fermentative capacities, and display few auxotrophies. Soil-dwelling genera were characterized by larger genome sizes, higher numbers of CRISPR loci, an expanded carbohydrate active enzyme (CAZyme) machinery enabling debranching of specific sugars from polymers, possession of a C1 (methanol and methylamine) degradation machinery, and a sole dependence on aerobic respiration. In contrast, nonsoil genomes encoded a more versatile respiratory capacity for oxygen, nitrite, sulfate, and trimethylamine N-oxide (TMAO) respiration, as well as the potential for utilizing the Wood-Ljungdahl (WL) pathway as an electron sink during heterotrophic growth. Our results not only expand our knowledge of the metabolism of a yet-uncultured bacterial lineage but also provide interesting clues on how terrestrialization and niche adaptation drive metabolic specialization within the Acidobacteria.
IMPORTANCE Members of the Acidobacteria are important players in global biogeochemical cycles, especially in soils. A wide range of acidobacterial lineages remain currently unexplored. We present a detailed genomic characterization of genomes belonging to family UBA6911 (also known as group 18) within the phylum Acidobacteria. The genomes belong to different genera and were obtained from soil (genera Gp18_AA60 and QHZH01), freshwater habitats (genera Ga0212092 and UBA6911), and an anaerobic digestor (genus Ga0209509). While all members of the family shared common metabolic features, e.g., heterotrophic respiratory abilities, broad substrate utilization capacities, and few auxotrophies, distinct differences between soil and nonsoil genera were observed. Soil genera were characterized by expanded genomes, higher numbers of CRISPR loci, a larger carbohydrate active enzyme (CAZyme) repertoire enabling monomer extractions from polymer side chains, and methylotrophic (methanol and methylamine) degradation capacities. In contrast, nonsoil genera encoded more versatile respiratory capacities for utilizing nitrite, sulfate, TMAO, and the WL pathway, in addition to oxygen as electron acceptors. Our results not only broaden our understanding of the metabolic capacities within the Acidobacteria but also provide interesting clues on how terrestrialization shaped Acidobacteria evolution and niche adaptation.
KEYWORDS: Acidobacteria, environmental genomics, methylotrophy, sulfate reduction
INTRODUCTION
Our appreciation of the scope of phylogenetic and metabolic diversities within the microbial world is rapidly expanding. Approaches enabling direct recovery of genomes from environmental samples without the need for cultivation allow for deciphering the metabolic capacities and putative physiological preferences of yet-uncultured taxa (1–9). Furthermore, the development of a genome-based taxonomic framework that incorporates environmentally sourced genomes (10) has opened the door for phylocentric (lineage-specific) studies. In such investigations, comparative analysis of genomes belonging to a target lineage is conducted to determine its common defining metabolic traits, the adaptive strategies of its members to various environments, and evolutionary trajectories and patterns of gene gain/loss across this lineage.
Members of the phylum Acidobacteria are one of the most dominant, diverse, and ecologically successful lineages within the bacterial domain (11–16). Originally proposed to accommodate an eclectic group of acidophiles (17), aromatic compound degraders and homoacetogens (18), and iron reducers (19), it was subsequently identified as a soil-dwelling bacterial lineage in early 16S rRNA gene-based diversity surveys (20–22). Subsequent 16S rRNA studies have clearly shown its near-universal prevalence in a wide range of soils, where it represents 5 to 50% of the overall community (23, 24).
Various taxonomic outlines have been proposed for the Acidobacteria. Genome-based classification by the Genome Taxonomy Database (GTDB [r95, October 2020]) (10) splits the phylum into 14 classes, 34 orders, 58 families, and 175 genera. This classification broadly, but not always, corresponds to 16S rRNA gene-based taxonomic schemes in SILVA (25), the 26-group (subdivision) classification scheme (26), and the most recently proposed refined class/order classification scheme (27) (see Table S1 in the supplemental material). Regardless, a strong concordance between habitat and phylogeny was observed within most lineages in the Acidobacteria. Some lineages, e.g., groups 1, 3 (both in class Acidobacteriae in the GTDB), and 6 (class Vicinamibacteria in the GTDB), have been predominantly encountered in soils, while others, e.g., groups 4 (class Blastocatellia in the GTDB), 8 (class Holophagae in the GTDB), and 23 (class Thermoanaerobaculia in the GTDB), are more prevalent in nonsoil habitats (11).
Genomic analysis of representatives of cultured (11) and uncultured metagenome-assembled genomes (MAGs) and single-cell genomes (SAGs) (28–30) of the phylum Acidobacteria has provided valuable insights into their metabolic capacities and lifestyle. However, the majority of genomic (and other -omics) approaches have focused mostly on cultured and yet-uncultured genomes of soil Acidobacteria (31–33). Genomic-based investigations of nonsoil Acidobacteria pure cultures (34–37) or MAGs (38, 39) are more limited, and consequently, multiple lineages within the Acidobacteria remain unexplored.
We posit that genomic analysis of hitherto-unexplored lineages of Acidobacteria would not only expand our knowledge of their metabolic capacities but also enable comparative genomic investigation on how terrestrialization and niche adaptation shaped the evolutionary trajectory and metabolic specialization within the phylum. To this end, we focus on a yet-uncultured lineage in the Acidobacteria: family UBA6911 (subdivision 18 in reference 21, class 1-2 in reference 27, and group 18 in SILVA database release 138.1 [25]). We combine the analysis of genomes recovered from Zodletone Spring, an anaerobic sulfide- and sulfur-rich spring in southwestern Oklahoma, with available genomes from multiple disparate soil and nonsoil habitats. Our goal was to understand the metabolic capacities, physiological preferences, and ecological role of this yet-uncharacterized group and to utilize the observed genus-level niche diversification to identify genomic changes associated with the terrestrialization process.
RESULTS
Ecological distribution patterns of family UBA6911.
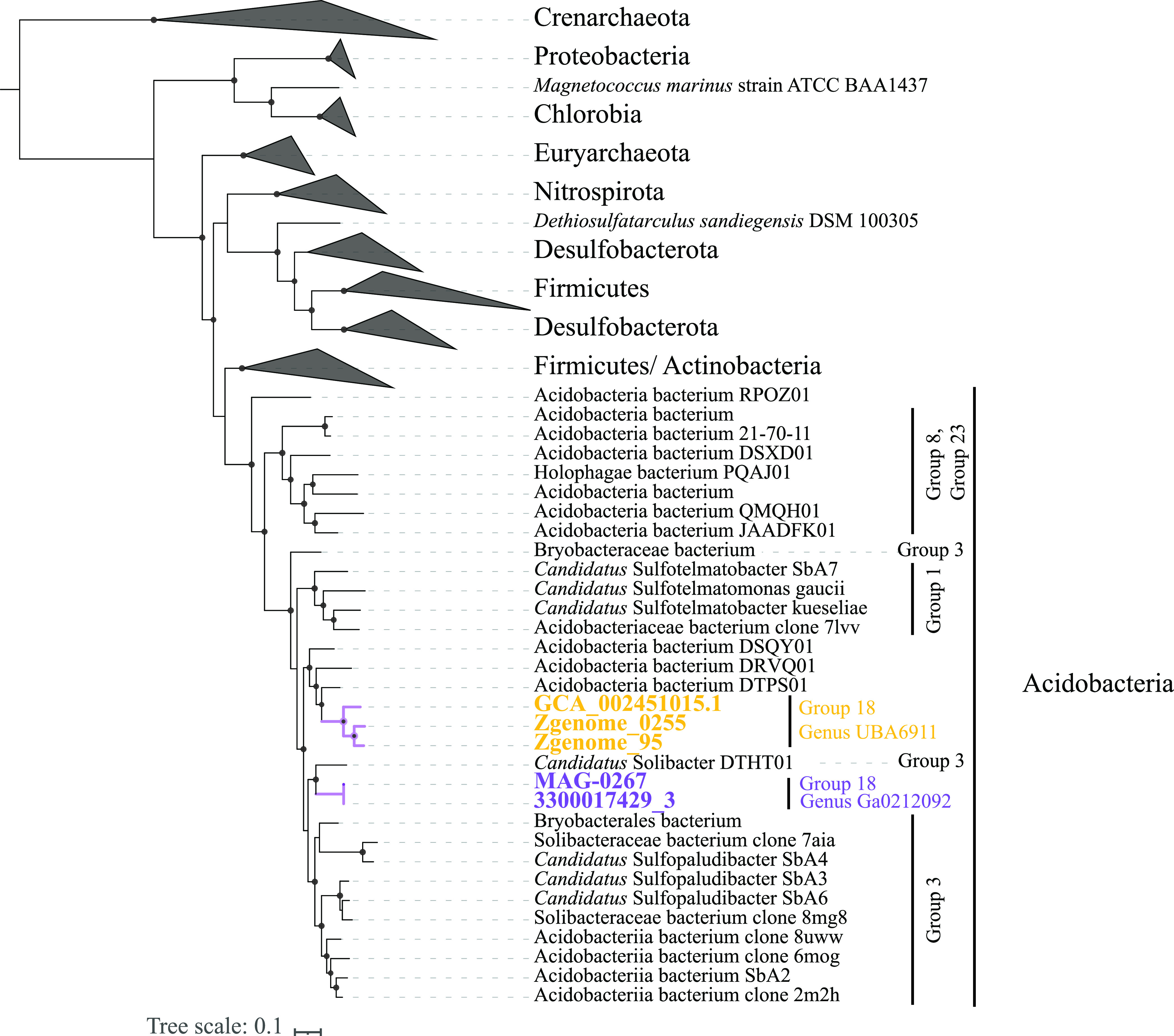
Family UBA6911 genomes clustered into five genera based on amino acid identity (AAI) and shared gene content values (Fig. 1; Table 1). Genomes of the genera Gp18_AA60 (n = 3) and QHZH01 (n = 1) were exclusively binned from a grassland meadow within the Angelo Coastal Range Reserve in Northern California (30). Genus Ga0209509 genomes were mostly (10/11 genomes) binned from an anaerobic gas digestor (Washington, USA) (40). Genus Ga0212092 (n = 2) genomes were binned from a Cone Pool hot spring microbial mat in California, USA. Finally, genus UBA6911 (10 genomes) displayed a broader distribution pattern, as its genomes were recovered from multiple, mostly freshwater habitats, e.g., river and estuary sediments and Zodletone Spring sediment (Table 1; Fig. 1).
FIG 1.
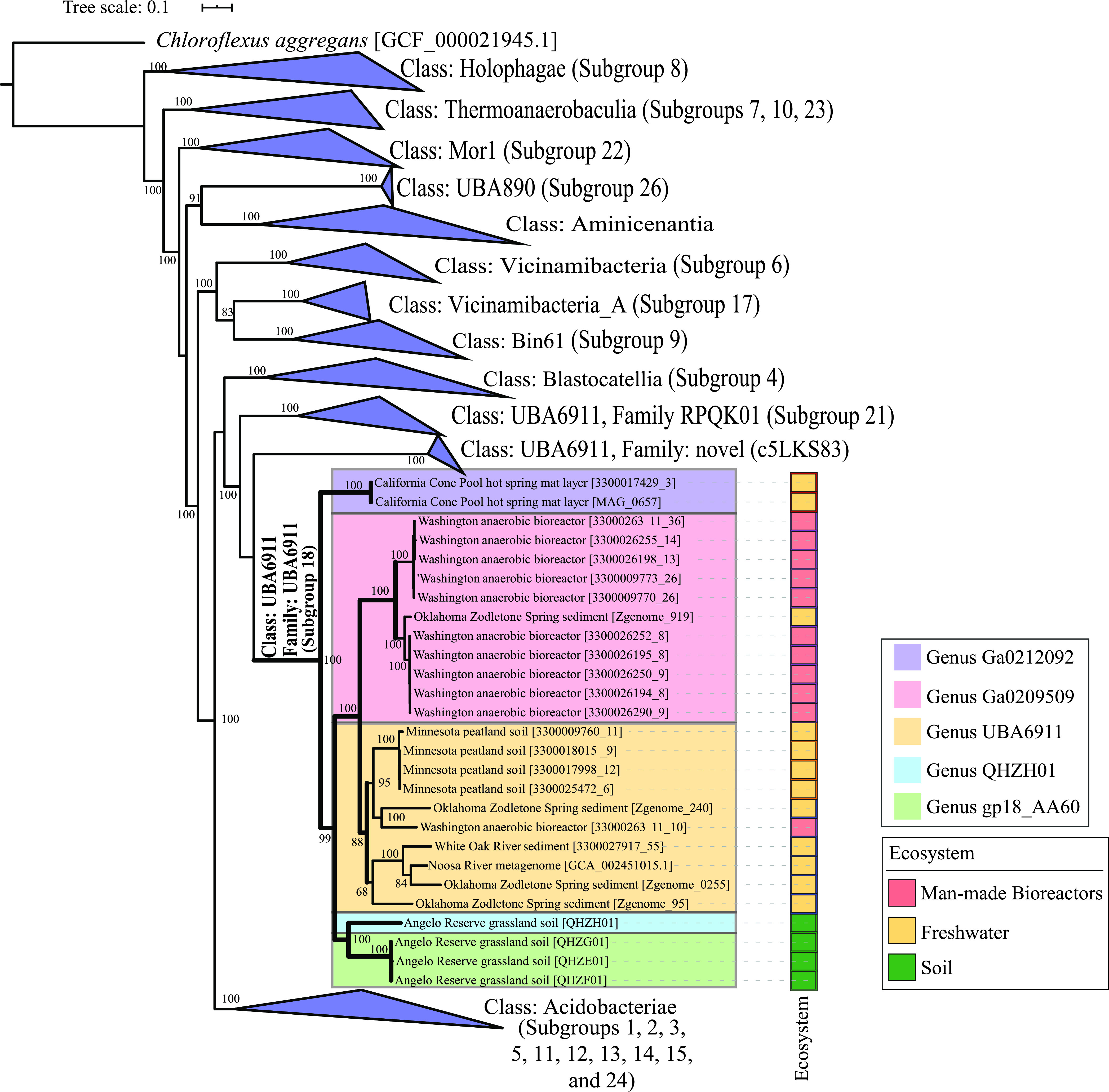
Maximum likelihood phylogenetic tree based on concatenated alignment of 120 single-copy genes from all Acidobacteria classes in GTDB r95. Family UBA6911 (Acidobacteria subgroup 18), is shown unwedged with thick branches. Branches are named by the bin name in brackets (as shown in Table 1) and with the ecosystem from which they were binned. The five genera described here are color coded as shown in the legend. Bootstrap values (from 100 bootstraps) are displayed for branches with ≥70% support. The tree was rooted using Chloroflexus aggregans (GCF_000021945.1) as the outgroup. The tracks to the right of the tree represent the ecosystem from which the genomes were binned (color coded as shown in the legend). All other Acidobacteria classes are shown as wedges with the corresponding subgroup number(s) in parentheses.
TABLE 1.
Binning sources, similarity statistics, and general genomics features for the genomes analyzed in this study
| Genus and bin names | Binning source | Similarity statistics |
Binned genome size (Mbp) | Estimated genome size (Mbp) | % Protein coding bases | GC content (%) | No. of CRISPRs | Avg gene length (bp) | Total no. of genes | Total no. of protein coding genes | Accession no. | Reference(s) | |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| AAI (avg ± SD) | SGC (avg ± SD) | ||||||||||||
| Genus Ga0212092 | |||||||||||||
| 3300022548-MAG_0657 | Cone Pool hot spring mat layer | 54.09 ± 0.98 | 40.34 ± 5.3 | 3.74 | 4.33 | 90.08 | 65.68 | 5 | 1,021 | 3,349 | 3,303 | Ga0212092a | —c |
| 3300017429_3 | 3.82 | 4.56 | 90.05 | 65.60 | 4 | 1,018 | 3,424 | 3,376 | Ga0185343a | — | |||
| Genus QHZH01 | |||||||||||||
| QHZH01 | Angelo Reserve soil | 54.63 ± 5.62 | 35.97 ± 5.12 | 6.00 | 8.01 | 90.41 | 66.73 | 2 | 895 | 6,088 | 6,058 | QHZH01b | 30 |
| Genus Gp18_AA60 | |||||||||||||
| QHZE01 | Angelo Reserve soil | 55.32 ± 5.08 | 37.1 ± 4.46 | 7.54 | 8.12 | 84.85 | 58.70 | 18 | 985 | 6,542 | 6,497 | QHZE01b | 30 |
| QHZF01 | 7.06 | 8.42 | 84.49 | 58.85 | 25 | 916 | 6,546 | 6,508 | QHZF01b | 30 | |||
| QHZG01 | 7.33 | 7.65 | 84.45 | 58.87 | 31 | 980 | 6,360 | 6,314 | QHZG01b | 30 | |||
| Genus UBA6911 | |||||||||||||
| 3300026311_10 | Anaerobic biogas reactor | 53.59 ± 4.66 | 38.83 ± 7.76 | 4.38 | 4.71 | 92.16 | 56.50 | 3 | 1,002 | 4,076 | 4,029 | Ga0209723a | 78 |
| 3300009760_11 | Peatland | 3.10 | 5.17 | 84.98 | 52.10 | 0 | 827 | 3,218 | 3,184 | Ga0116131a | 76, 77 | ||
| 3300017998_12 | 4.18 | 5.15 | 87.18 | 52.70 | 4 | 866 | 4,262 | 4,211 | Ga0187870a | 76, 77 | |||
| 3300025472_6 | 4.15 | 5.61 | 86.20 | 52.60 | 4 | 838 | 4,309 | 4,267 | Ga0208692a | 76, 77 | |||
| 3300018015_9 | 4.69 | 5.91 | 87.26 | 52.80 | 2 | 831 | 4,956 | 4,923 | Ga0187866a | 76, 77 | |||
| 3300027917_55 | White Oak River sediment | 3.87 | 4.43 | 84.76 | 48.90 | 9 | 898 | 3,695 | 3,656 | Ga0209536a | 40 | ||
| GCA_002451015 | Noosa River | 4.94 | 5.72 | 85.35 | 51.60 | 1 | 911 | 4,657 | 4,632 | DKBB01b | 79 | ||
| Zgenome_0255 | Zodletone Spring sediment | 3.75 | 5.12 | 87.68 | 57.60 | 19 | 987 | 3,252 | 3,220 | JAFGAO01b | This study | ||
| Zgenome_240 | 6.51 | 6.81 | 87.10 | 52.30 | 5 | 1,011 | 5,666 | 5,615 | JAFGIY01b | This study | |||
| Zgenome_95 | 5.45 | 5.85 | 86.79 | 51.80 | 7 | 987 | 4,835 | 4,791 | JAFGTD01b | This study | |||
| Genus Ga0209509 | |||||||||||||
| 3300009770_26 | Anaerobic biogas reactor | 54.58 ± 3.99 | 40.93 ± 8.14 | 2.50 | 2.70 | 90.94 | 65.20 | 1 | 1,008 | 2,291 | 2,253 | Ga0123332a | 78 |
| 3300009773_26 | 2.56 | 2.75 | 90.73 | 65.20 | 2 | 1,006 | 2,346 | 2,307 | Ga0123333a | 78 | |||
| 3300026194_8 | 3.76 | 3.88 | 91.48 | 66.60 | 2 | 1,035 | 3,370 | 3,326 | Ga0209509a | 78 | |||
| 3300026195_8 | 3.83 | 3.95 | 91.56 | 66.50 | 5 | 1,041 | 3,418 | 3,371 | Ga0209312a | 78 | |||
| 3300026198_13 | 2.52 | 2.82 | 91.52 | 65.70 | 1 | 997 | 2,344 | 2,310 | Ga0209313a | 78 | |||
| 3300026250_9 | 3.75 | 3.87 | 91.48 | 66.60 | 3 | 1,040 | 3,348 | 3,300 | Ga0209612a | 78 | |||
| 3300026252_8 | 3.75 | 3.86 | 91.47 | 66.60 | 2 | 1,041 | 3,337 | 3,291 | Ga0209722a | 78 | |||
| 3300026255_14 | 2.70 | 2.91 | 91.17 | 65.60 | 2 | 999 | 2,502 | 2,465 | Ga0209613a | 78 | |||
| 3300026290_9 | 3.86 | 3.98 | 91.35 | 66.20 | 3 | 1,045 | 3,425 | 3,376 | Ga0209510a | 78 | |||
| 3300026311_36 | 2.69 | 2.78 | 90.30 | 64.90 | 3 | 1,004 | 2,466 | 2,423 | Ga0209723a | 78 | |||
| Zgenome_919 | Zodletone Spring sediment | 4.75 | 4.98 | 91.97 | 66.50 | 0 | 996 | 3,038 | 3,001 | JAFGSS01b | This study | ||
Accession number corresponds to Gold analysis project number (for MAGs from the recently released 52,515 genomes in the Earth microbiome catalogue collection [76] that were deposited in the IMG/M database).
NCBI (WGS Project) assembly accession number.
—, unpublished, used with the principal investigator’s permission.
To further examine the ecological distribution patterns of family UBA6911, we analyzed 177 near-full-length 16S rRNA gene amplicons generated in multiple culture-independent amplicon-based diversity surveys and available through the SILVA database (r138.1) (Fig. 2). Genus distribution patterns gleaned from the origin of MAGs were generally confirmed: 16S rRNA sequences affiliated with genus Gp18_AA60 were mostly recovered from soil (14/15, 93.3%), those affiliated with genus Ga0209509 were mostly encountered in anaerobic digestors (11/14, 85.7%), and the majority of 16S rRNA sequences affiliated with the genera Ga0212092 (90.9%) and UBA6911 (58.8%) were encountered in a wide range of freshwater environments (e.g., freshwater lake sediments, estuary sediments, and thermal springs). Collectively, the combined MAGs and 16S rRNA amplicon data suggest a preference for soil for genera Gp18_AA60 and QHZH01, a preference for anaerobic digestors for Ga0209509, and a wide occurrence of genera Ga0212092 and UBA6911 in freshwater habitats.
FIG 2.
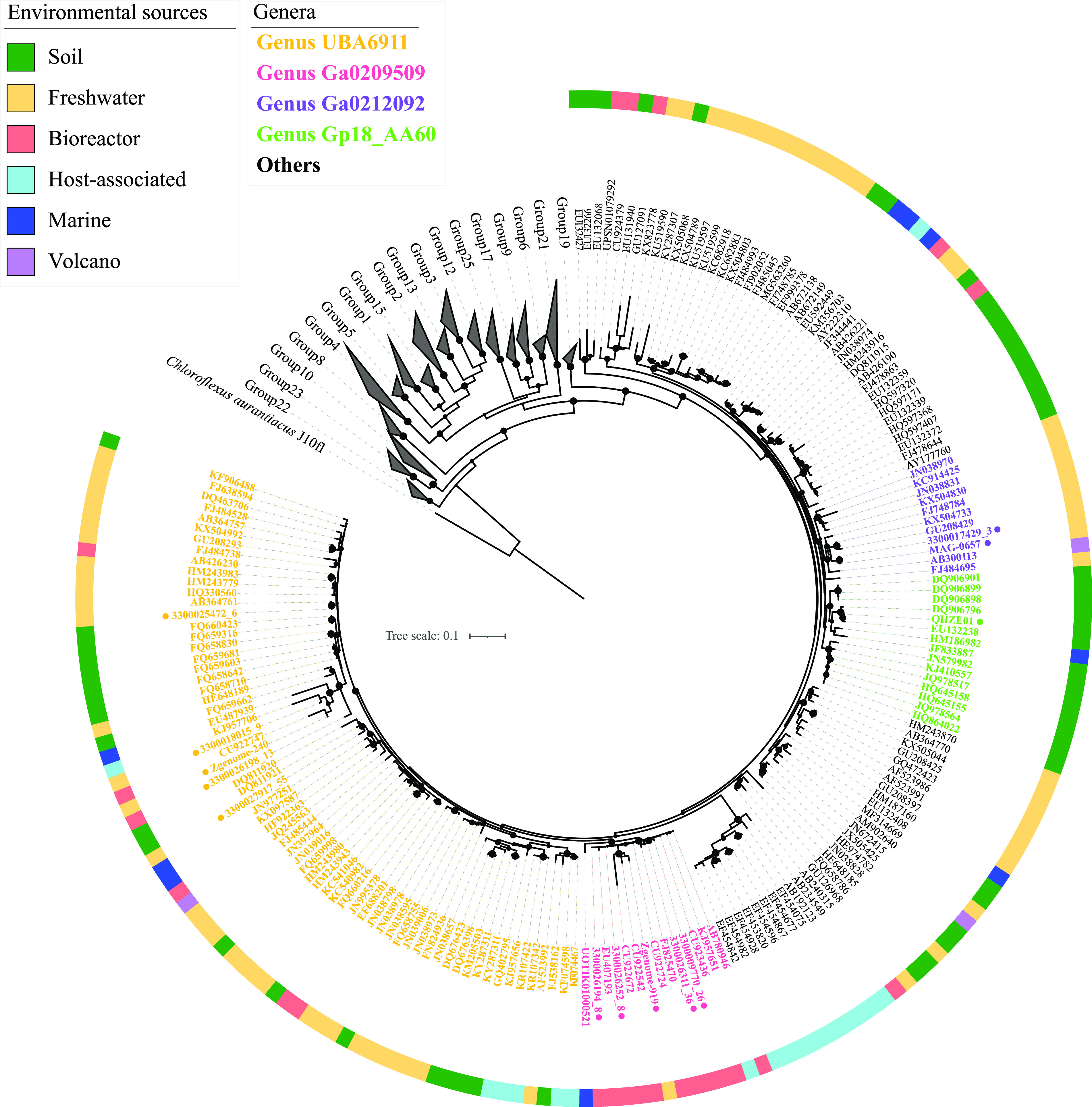
Phylogenetic tree based on 16S rRNA gene for Acidobacteria classes. Groups other than family UBA6911 (subgroup 18) are shown as gray wedges. A total of 177 near-full-length (>1,200-bp) 16S rRNA sequences belonging to Acidobacteria group 18 in the SILVA database are shown with their GenBank accession numbers. Representative 16S rRNA sequences from the analyzed genomes are shown with their corresponding bin name (as in Table 1) and with a colored dot next to the name for ease of recognition. Branch labels are color coded by genus, as shown in the legend (using the same color scheme as that in Fig. 1). The track around the tree shows the environmental classification of the ecosystem from which the sequence was obtained, and the corresponding color codes are shown in the legend. Sequences were aligned using the SINA aligner (97), and the alignment was used to construct a maximum likelihood phylogenetic tree with FastTree (92). Bootstrap values (from 100 bootstraps) are displayed as bubbles for branches with ≥70% support. The tree was rooted using Chloroflexus aggregans (GenBank accession number M34116.1) as the outgroup.
General genomic and structural features of family UBA6911 genomes.
Soil-affiliated genera (Gp18_AA60 and QHZH01) displayed larger estimated genome sizes (average ± SD of 8.05 ± 0.32 Mbp). In comparison, in the freshwater genus UBA6911, these values were 5.45 ± 0.68 Mbp, with only a single genome (Zgenome_240) exceeding 6 Mbp. Even smaller genomes were observed for genus Ga0212092 (4.45 ± 0.17 Mbp) and genus Ga0209509 (3.5 ± 0.75 Mbp) genomes (Table 1). GC content was generally high in all genomes (Table 1), with three genera (QHZH01, Ga0209509, and Ga0212092) exhibiting >65% GC content. Structurally, all genera in family UBA6911 are predicted to be Gram-negative rods with similar predicted membrane phospholipid compositions (Table 2; Table S3). Genes mediating type IV pilus formation, generally involved in a wide range of functions, e.g., adhesion and aggregation, twitching motility, DNA uptake, and protein secretion (41), were observed in most genera. Genes encoding secretion systems I and II were identified in all genomes. Interestingly, genes encoding the type VI secretion system, typically associated with pathogenic Gram-negative bacteria (mostly Proteobacteria) and known to mediate protein transport to adjacent cells as a mean of bacterial antagonism (42), was identified in all but a single (anaerobic digestor Ga0209509) genus. A search of the AnnoTree database (43) identified type VI secretion system genes in only 14 Acidobacteria genomes, all belonging to class Holophagae, suggesting the rare distribution of such trait in the phylum. Finally, genomes of the soil genus Gp18_AA60 possess an exceptionally large number of CRISPR genes (24.67 ± 6.5) (Table 1). In comparison, genomes from all other genera possessed 0 to 9 CRISPR genes per genome, with the notable exception of the Zgenome_0255 genome (genus UBA6911), which possessed 19 CRISPR genes.
TABLE 2.
Salient defining features of family UBA6911 genera
| Feature | Results for indicated genusa |
||||
|---|---|---|---|---|---|
| Gp18_AA60 | QHZH01 | UBA6911 | Ga0209509 | Ga0212092 | |
| Predicted structural features | |||||
| Flagellar motility | n | n | Y | n | Y |
| Type IV pilus assembly | Y | n | Y | Y | Y |
| Chemotaxis | n | n | Y | n | n |
| Type VI secretion system | Y | Y | Y | n | Y |
| CRISPR count | 24.67 ± 6.5 | 2 | 5.4 ± 5.4 | 2.18 ± 1.33 | 4.5 ± 0.71 |
| Biosynthesis: biosynthetic gene clusters | |||||
| Terpenes | Y | Y | Y | Y | Y |
| Phenazine | Y | Y | n | n | n |
| NRPS/PKS | Y | Y | Y | Y | Y |
| Homoserine lactones | n | n | n | Y | n |
| Bacteriocin | n | Y | Y | n | n |
| Heterotrophic substrates predicted to support growth | |||||
| Sugarsb | |||||
| CAZymes (GHs + PLs + CEs) | 98.3 ± 11.7 | 166 | 48.2 ± 22.3 | 45.6 ± 13.4 | 42.5 ± 0.71 |
| Hexoses | Glu, Man, Gal, Fru | Glu, Man | Glu, Man, Gal, Fru | Glu, Man, Gal, Fru | Glu, Man, Gal |
| Hexosamines | Y | Y | Y | Y | Y |
| Hexuronic acids | n | n | Y | Y | n |
| Sialic acid | n | Y | n | n | n |
| Sugar alcohols | Ino, Sorb, Xylitol | Ino, Sorb, Xylitol | Sorb, Xylitol | Sorb, Xylitol | Xylitol |
| Pentoses | Lyx, Xyl | Lyx, Xyl | n | Xyl | n |
| Amino acids | |||||
| Acidic and amides | D, N, E, Q | D, N, E, Q | D, N, E, Q | D, E, Q | D, N, E, Q |
| Aliphatic | A, G | A, G | A, G, V, L, I | A, G | A, G |
| Aromatic | n | n | Y | n | n |
| Basic | H | n | R, K | R | R, H |
| Hydroxy and S containing | S, T | S, T | M | M | S, T, M |
| Cyclic | P | P | P | P | P |
| C1 compounds | |||||
| Methanol | n | Y | n | n | n |
| Methylamines | n | Y | n | n | n |
| Formaldehyde | n | Y | Y | n | n |
| Predicted respiratory capacities | |||||
| Aerobic (low affinity) | Y | Y | Y | Y | Y |
| Aerobic (high affinity) | Y | n | Y | Y | Y |
| Dissimilatory nitrate reduction to ammonium | n | n | Y | Y | |
| Dissimilatory nitrite reduction to ammonium | n | n | Y | n | |
| Dissimilatory sulfate reduction | n | n | Y | n | Y |
| TMAO respiration | n | n | n | n | Y |
| Predicted fermentation products | |||||
| Acetate | Y | Y | Y | Y | n |
| Ethanol | n | Y | Y | Y | Y |
| Formate | n | n | Y | Y | n |
| Acetone | n | n | Y | n | n |
| Acetoin and butanediol | Y | n | Y | Y | Y |
Y, full pathway identified; n, full pathway missing.
Sugars are abbreviated as follows; Glu, glucose; Man, mannose; Gal, galactose; Fru, fructose; Ino, myo-inositol; Sorb, sorbitol; Lyx, lyxose; Xyl, xylose.
Anabolic capabilities in family UBA6911 genomes.
All UBA6911 genomes encoded a fairly extensive anabolic repertoire, with capacity for biosynthesis of the majority of amino acids from precursors (from 15 in genus Ga0212092 to 20 in genus UBA6911) (Table S3). Gluconeogenic capacity was encoded in all genera (Table S3). Cofactor biosynthesis capability was also widespread in all but the freshwater Cone Pool Ga0212092 genomes (Table S3).
A complete assimilatory sulfate reduction pathway for sulfate uptake and assimilation was observed in all genomes. In addition, the presence of genes encoding taurine dioxygenase (in soil genus QHZH01 genome) and alkanesulfonate monooxygenase (in soil genus gp18_AA60 genomes) argue for the capacity for organosulfur compound assimilation. For nitrogen assimilation, in addition to NH4+ and amino acids, multiple pathways for N assimilation from organic substrates were identified. These include the presence of urease genes (ureABC) for urea assimilation in soil genus gp18_AA60 genomes, along with genes encoding urease Ni-delivering chaperones (ureDEFG). As well, genes for N extraction from arylformamides (arylformamidase, EC 3.5.1.9), N,N-dimethylformamide (N,N-dimethylformamidase, EC 3.5.1.56), and N-formylglutamate (formylglutamate deformylase, EC 3.5.1.68) were identified to various degrees (Table S3) in all genera, with the exception of freshwater Cone Pool genus Ga0212092 genomes. No evidence for assimilatory nitrate reduction or nitrogen fixation was identified in any of the genomes.
In addition to the above-described biosynthetic capacities, all examined genomes, regardless of environmental source or genus-level affiliation, included biosynthetic gene clusters (BGCs) ranging in number from 3 to 12. Such clusters encode polyketide synthases (PKS) and nonribosomal peptide synthases (NPRS), homoserine lactones, terpenes, phenazines, and bacteriocin. While the numbers of BGCs per genome did not differ much between genera (Table S4), differences in the gene clusters, and hence putative products, were identified (Table 2; Fig. S1). For example, phenazine biosynthetic clusters were identified only in the soil genera genomes, while BCGs for homoserine lactone biosynthesis were exclusive to the anaerobic digestor Ga0209509 genomes. While not exclusive, terpene biosynthetic clusters appear to be more enriched in anaerobic digestor Ga0209509 genomes, while NRPS and PKS clusters were enriched in the soil genus genomes and UBA6911 genomes. Finally, a bacteriocin biosynthesis cluster was identified only in genus QHZH01 genome and a single genome affiliated with genus UBA6911 (Fig. S1). Finally, we queried NRPS and PKS clusters identified in all 28 genomes against the NCBI nucleotide database. Surprisingly, only 14 genes (3.8%) had significant hits to previously deposited sequences (Table S4).
Substrate utilization patterns in family UBA6911 genomes.
Genomic analysis of all family UBA6911 genomes suggests a heterotrophic lifestyle with robust aminolytic and saccharolytic machineries. Genomes from all genera encoded a broad capacity to metabolize proteins and amino acids. An arsenal of endopeptidase, oligopeptidase, and dipeptidase genes were identified in all genomes (Table S5). Oligopeptide transporters (in UBA6911 genomes), peptide/Ni transporters (all genomes), and dedicated amino acid transporters (e.g., branched-chain amino acid transporters in QHZH01, UBA6911, and Ga0212092 genomes, and trp/tyr transporters in gp18_AA60 genomes) suggest the capacity for amino acid and oligo/dipeptide uptake. Furthermore, all genomes to various degrees showed degradation pathways for a wide range of amino acids, ranging from 9 (in genus Ga0212092) to 15 (in genus UBA6911) (Table 2; Table S3).
Similarly, with the notable exception of members of freshwater Cone Pool genus Ga0212092, a robust machinery for sugar metabolism was identified (Table 2; Table S3). Glucose, mannose, galactose, fructose, hexosamines e.g., N-acetyl hexosamines, uronic acids, sorbitol, and xylitol degradation capacities were identified in all other four genera (Table 2; Table S3). Further, soil genus genomes encoded the full degradation machinery for myo-inositol (iolBCDEG), an abundant soil component (44). Interestingly, the QHZH01 genome also encoded the full machinery for sialic acid (an integral component of soil fungal cell walls [45]) degradation to fructose-6-P.
Analysis of the carbohydrate active enzyme (CAZyme) repertoire was conducted to assess family UBA6911 polysaccharide degradation capacities. A notable expansion of the CAZyme (carbohydrate esterase [CE] plus polysaccharide lyase [PL] plus glycoside hydrolase [GH]) repertoire in soil genomes was observed (166 in QHZH01 genome, 98.3 ±11.7 in gp18_AA60 genomes), in comparison to 46.48 ± 17.01 in all other genera (Table S6). Higher numbers in soil genus genomes were brought about by the expansion of families GH33 (sialidase), GH165 (β-galactosidase), and GH56 (hyaluronidase) in genus QHZH01, of family GH13 (amylase) in gp18_AA, and of family GH109 (N-acetylhexosaminidase) in both genera (Table S6). As described above, genes encoding degradation machineries of the released monomer products for these enzymes (sialic acid, galactose, uronic acid, glucose, and N-acetylhexosamine) are encoded in the soil genomes (Table 2; Table S3).
Overall, all family UBA6911 genomes analyzed in this study displayed a notable absence or extreme paucity of CAZy families encoding/initiating breakdown of large polymers, e.g., endo- and exocellulases (only 6 copies of GH5 genes and 5 copies of GH9 genes in all 28 genomes examined), hemicellulases (only 19 copies of GH10 genes in all 28 genomes examined), and pectin degradation (only 22 copies of GH28 genes in all genomes examined), suggesting the limited ability of all members of the family for degrading these high-molecular-weight polysaccharides (Table S6). Collectively, the CAZyome and sugar degradation patterns of family UBA6911 argue for a lineage specializing in sugar, but not polymer, degradation, with added specialized capacities for debranching specific sugars from polymers in the soil-dwelling genera.
Further, methylotrophic C1 degradation capacity for methanol and methylamine utilization was encoded in the soil QHZH01 genome. The genome encoded a methanol dehydrogenase (EC 1.1.2.10) for methanol oxidation to formaldehyde (Fig. 3 and Table 2; Table S3). QHZH01 methanol dehydrogenase belonged to the lanthanide-dependent pyrroloquinoline quinone (PQQ) methanol dehydrogenase XoxF type. More specifically, QHZH01 XoxF methanol dehydrogenase belonged to clade xoxF-2, previously described for “Candidatus Methylomirabilis” (NC10) (Fig. 4). The accessory xoxG (c‐type cytochrome) and xoxJ (periplasmic binding) genes were fused and encoded in QHZH01 genome downstream of the xoxF gene. A gene encoding quinohemoprotein amine dehydrogenase (EC 1.4.9.1) for methylamine oxidation to formaldehyde (Fig. 3 and 4 and Table 2; Table S3) was also identified in the genome. Furthermore, the genome also encoded subsequent formaldehyde oxidation to formate via the glutathione-independent formaldehyde dehydrogenase (EC 1.2.1.46), as well as formate oxidation to CO2 via formate dehydrogenase (EC 1.17.1.9). Finally, for assimilating formaldehyde into biomass, genes encoding the majority of enzymes of the serine cycle were identified in the genome, as well as the majority of genes encoding the ethylmalonyl coenzyme A (ethylmalonyl-CoA) pathway for glyoxylate regeneration (Fig. 3 and Table 2; Table S3).
FIG 3.
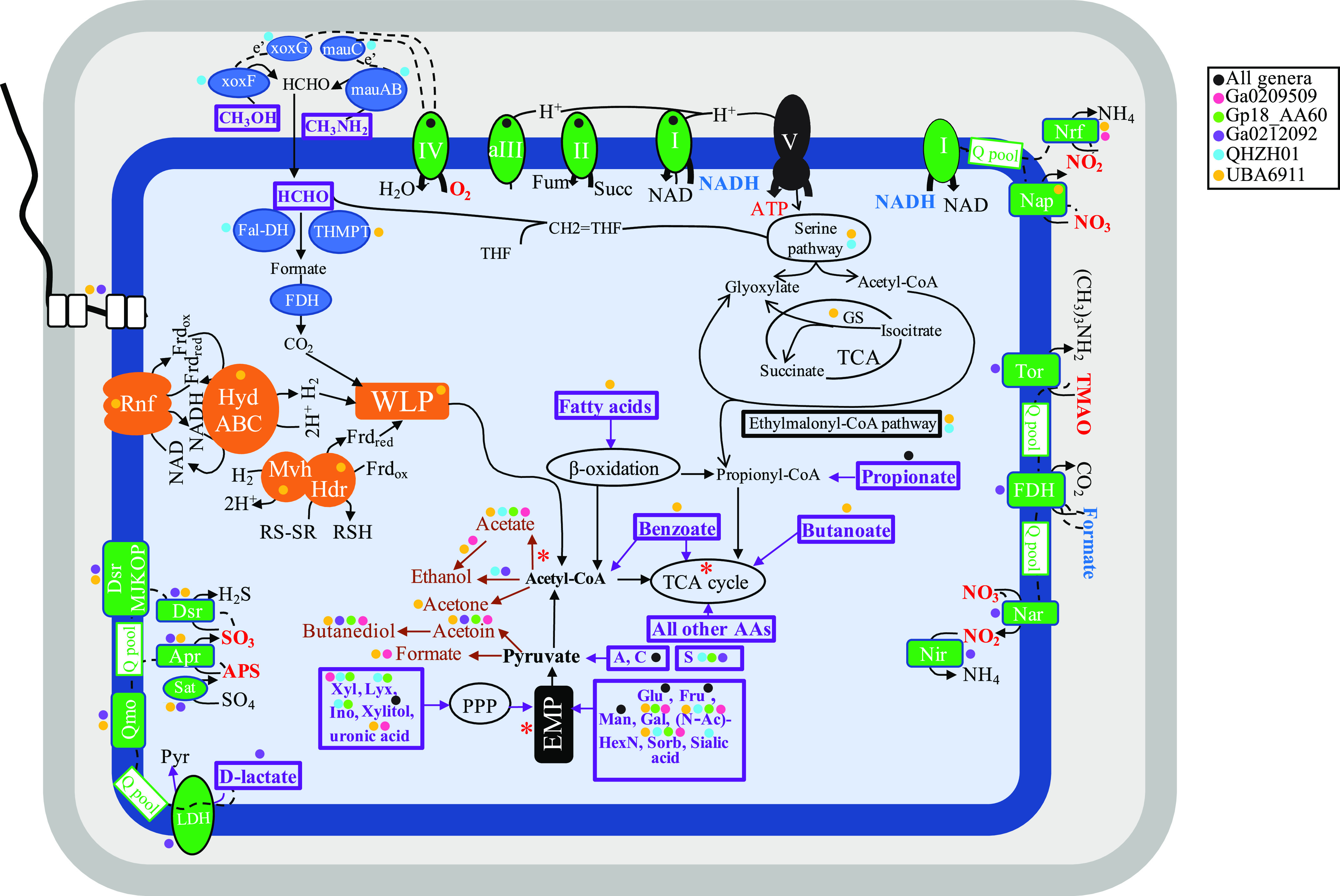
Cartoon depicting different metabolic capabilities encoded in family UBA6911 genomes with capabilities predicted for different genera shown as colored circles (all orders, black; genus Ga0209509, pink; genus Gp18_AA60, green; genus Ga0212092, purple; genus QHZH01, cyan; genus UBA6911, yellow). Enzymes for C1 metabolism are shown in blue. Electron transport components are shown in green, and electron transfer is shown as dotted black lines from electron donors (shown in boldface blue text) to terminal electron acceptors (shown in boldface red text). The sites of proton extrusion to the periplasm are shown as black arrows, as is the F-type ATP synthase (V). Proton motive force generation, as well as electron carrier recycling pathways associated with the operation of the WLP, is shown in orange. All substrates predicted to support growth are shown in boldface purple text within thick purple boxes. Fermentation end products are shown in brown. Sites of substrate-level phosphorylation are shown as red asterisks. A flagellum is depicted, the biosynthetic genes of which were identified in genomes belonging to the genera UBA6911 and Ga0212092. The cell is depicted as rod shaped based on the identification of the rod shape-determining proteins rodA, mreB, and mreC in all genomes. Abbreviations: Apr, the enzyme complex adenylylsulfate reductase (EC 1.8.99.2); APS, adenylyl sulfate; Dsr, dissimilatory sulfite reductase (EC 1.8.99.5); EMP, Embden-Meyerhoff-Parnas pathway; Fal-DH, formaldehyde dehydrogenase; FDH, formate dehydrogenase; Frdox/red, ferredoxin (oxidized/reduced); Fru, fructose; fum, fumarate; Gal, galactose; Glu, glucose; GS, glyoxylate shunt; Hdr, heterodisulfide reductase complex; HydABC, cytoplasmic [Fe Fe] hydrogenase; I, II, aIII, and IV, aerobic respiratory chain comprising complex I, complex II, alternate complex III, and complex IV; Ino, myo-inositol; LDH, l-lactate dehydrogenase; Lyx, lyxose; Man, mannose; mauABC; methylamine dehydrogenase; Mvh, Cytoplasmic [Ni Fe] hydrogenase; Nap, nitrate reductase (cytochrome); Nar, nitrate reductase; Nir, nitrite reductase (NADH); Nrf, nitrite reductase (cytochrome c-552); N-AcHexN, N-acetylhexosamines; PPP, pentose phosphate pathway; Pyr, pyruvate; Q pool, quinone pool; Qmo, quinone-interacting membrane-bound oxidoreductase complex; RNF, membrane-bound RNF complex; RSH/RS-SR, reduced/oxidized disulfide; Sorb, sorbitol; succ, succinate; TCA, tricarboxylic acid cycle; THMPT, tetrahydromethanopterin-linked formaldehyde dehydrogenase; TMAO, trimethylamine N-oxide; Tor, trimethylamine-N-oxide reductase (cytochrome c); V, ATP synthase complex; WLP, Wood-Ljungdahl pathway; xoxFG, methanol dehydrogenase; Xyl, xylose.
FIG 4.
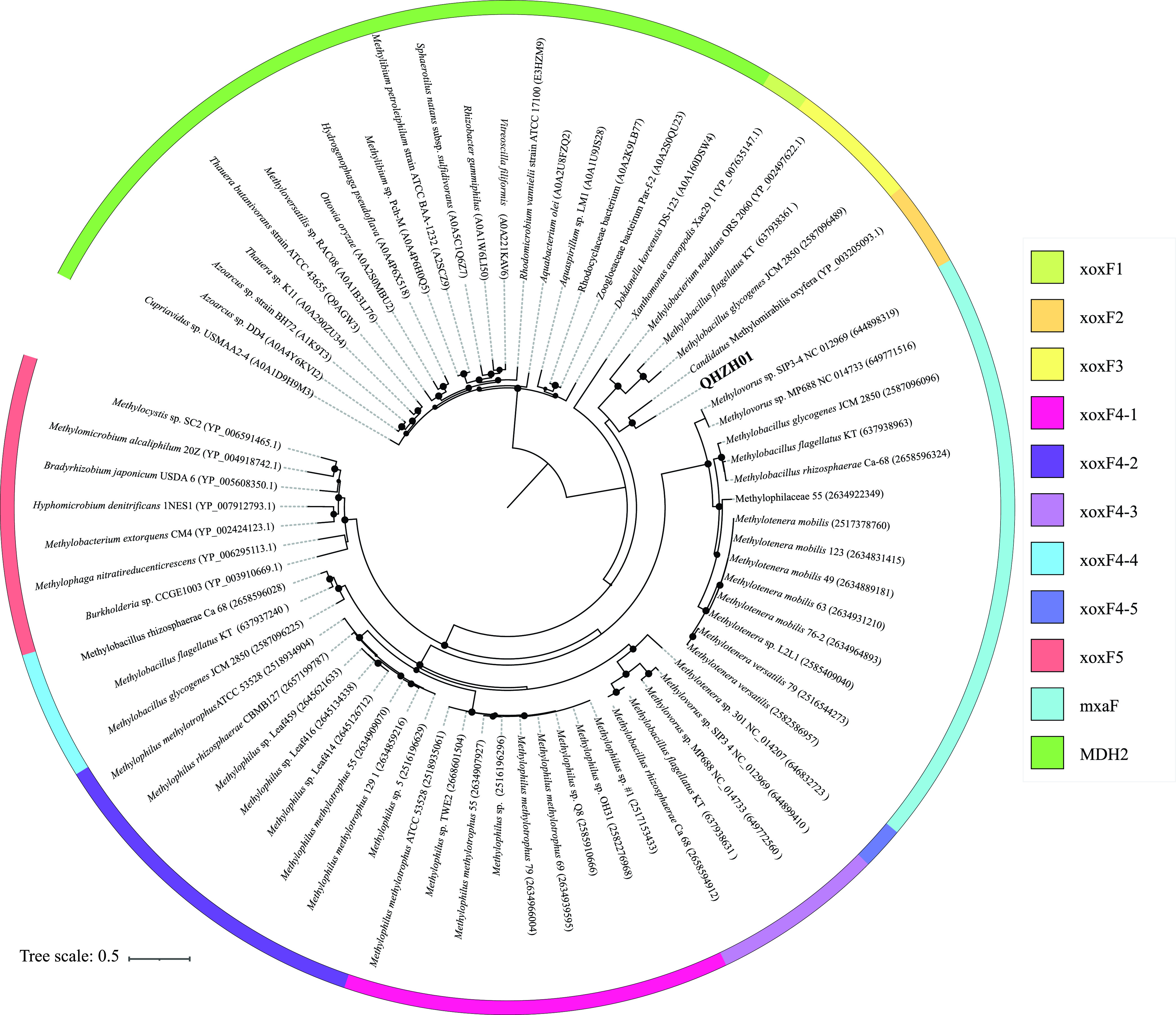
Phylogenetic affiliation for family UBA6911 methanol dehydrogenase (XoxF) in relation to reference sequences. The genus QHZH01 sequence is shown in boldface. The track around the tree depicts the XoxF clade as shown in the key. The tree was rooted using methanol dehydrogenase sequences belonging to the MDH2 group previously described in members of Burkholderiales (100). UniProt, IMG, and/or GenBank accession numbers are shown for reference sequences. Bootstrap support values are based on 100 replicates and are shown for nodes with >70% support.
In addition, several genomes in genus UBA6911 encoded formaldehyde oxidation (via the tetrahydromethopterin-linked pathway, glutathione-independent formaldehyde dehydrogenase [EC 1.2.1.46], and glutathione-dependent formaldehyde dehydrogenase [EC 1.1.1.284, EC 3.1.2.12]) to formate, formate oxidation to CO2 via formate dehydrogenase (EC:1.17.1.9), formaldehyde assimilation into biomass (genes encoding the majority of enzymes of the serine cycle were identified in some genomes), and glyoxylate regeneration (the majority of genes encoding the ethylmalonyl-CoA pathway were identified in some genomes, with only one genome containing glyoxylate shunt genes) (Fig. 3 and Table 2; Table S3). Surprisingly, upstream genes for formaldehyde production from C1 compounds (e.g., methane, methanol, methylamine, C1 sulfur compounds) were missing from all genomes. Phylogenetic analysis of key C1 metabolism genes identified their close affiliation with group 1, 5, and 6 Acidobacteria genomes, previously demonstrated to mediate C1 metabolism and formaldehyde assimilation (30). The closest relatives outside the phylum varied but were members of Chloroflexi, Desulfobacterota, and Actinobacteriota (Table S7).
Finally, in addition to proteins, carbohydrates, and C1 compounds, other potential carbon sources for family UBA6911 were identified. These include long-chain fatty acids (a complete beta-oxidation pathway) in genus UBA6911 genomes, short-chain aliphatic fatty acids (propionate and butyrate degradation to acetyl-CoA) in both UBA6911 and Ga0212092 genomes, and benzoate in genus UBA6911 genomes (Table S3).
Respiratory capacities.
All family UBA6911 genomes encoded respiratory capacities. Genomic analysis suggested that the soil genera Gp18_AA60 and QHZH01 could potentially utilize O2 as their sole electron acceptor, based on their possession of a respiratory chain comprising complex I, complex II, alternate complex III, and complex IV (cytochrome oxidase aa3) (Table 2; Table S3). On the other hand, all anaerobic digestor genus Ga0209509 genomes could mediate dissimilatory nitrite reduction to ammonium, based on the presence of nitrite reductase (cytochrome c-552) (EC 1.7.2.2), plus respiratory complexes I and II. Only 6 out of the 11 genomes in this genus possessed a complete aerobic respiratory chain. Freshwater genus Ga0212092 genomes from Cone Pool microbial sediments were notable in encoding a complete aerobic respiratory chain as well as the complete machinery for dissimilatory sulfate reduction. An additional plausible anaerobic electron acceptor for genus Ga0212092 is trimethylamine N-oxide (TMAO). Here, electron transfer is thought to occur from formate via the membrane-bound formate dehydrogenase (EC 1.17.1.9) through the quinone pool onto TMAO reductase (EC 1.7.2.3), eventually reducing trimethylamine N-oxide to trimethylamine (Fig. 3 and Table 2; Table S3), as previously shown in Escherichia coli (46) and Rhodopseudomonas capsulata (47). Finally, members of the freshwater genus UBA6911 demonstrated the most versatile respiratory capacities, with evidence for aerobic respiration (in all genomes except a single Oak River estuary sediment MAG), dissimilatory nitrite reduction to ammonium in genomes from Washington anaerobic gas digestor and Zodletone Spring sediment, and dissimilatory sulfate reduction to sulfide in Noosa River sediment and Zodletone Spring sediment genomes (Fig. 3 and Table 2; Table S3).
Sulfate reduction machinery identified in the genomes of freshwater genera UBA6911 and Ga0212092 included sulfate adenylyltransferase (Sat; EC 2.7.7.4) for sulfate activation to adenylyl sulfate (APS), the enzyme complex adenylylsulfate reductase (AprAB; EC 1.8.99.2) for APS reduction to sulfite, the quinone-interacting membrane-bound oxidoreductase complex (QmoABC) for electron transfer, the enzyme dissimilatory sulfite reductase (DsrAB; EC 1.8.99.5) and its cosubstrate DsrC for dissimilatory sulfite reduction to sulfide, and the sulfite reduction-associated membrane complex DsrMKJOP for linking cytoplasmic sulfite reduction to energy conservation (Fig. 3). Phylogenetic affiliation using a concatenated alignment of DsrA and DsrB proteins placed genus UBA6911 sequences close to Thermoanaerobaculia acidobacterial sequences from hydrothermal vents (Fig. 5), while genus Ga0212092 sequences were close to group 3 Acidobacteria sequences from hydrothermal vents and peatland soil (Fig. 5).
FIG 5.
Maximum likelihood phylogenetic tree based on the concatenated alignment of the alpha and beta subunits of dissimilatory sulfite reductase (DsrAB) from family UBA6911 in relation to reference sequences. Genus UBA6911 sequences are shown in yellow, while genus Ga0212092 sequences are shown in purple. Reference sequences from phyla other than Acidobacteria are shown as gray wedges. Acidobacteria references are labeled by the subgroup number. Bootstrap support values are based on 100 replicates and are shown for nodes with >70% support.
Finally, in addition to inorganic electron acceptors, the majority of genus UBA6911 genomes encoded a complete Wood-Ljungdahl pathway (WLP) (Fig. 3). WLP is a versatile widespread pathway that is incorporated in the metabolic schemes of a wide range of phylogenetically disparate anaerobic prokaryotes, e.g., homoacetogenic bacteria, hydrogenotrophic methanogens, autotrophic sulfate‐reducing prokaryotes, heterotrophic sulfate‐reducing bacteria, and syntrophic acetate‐oxidizing (SAO) bacteria, as well as acetoclastic methanogens. When operating in the reductive direction, the pathway can be used for carbon dioxide fixation and energy conservation during autotrophic growth or as an electron sink during heterotrophic fermentative metabolism. When operating in the oxidative direction, the pathway is used for acetate catabolism (48–50). Syntrophic acetate oxidizers employing the oxidative WLP usually possess high-affinity acetate transporters to allow for the uptake of small concentrations of acetate, a competitive advantage in the presence of acetoclastic methanogens (51). The absence of genes encoding high-affinity acetate transporters in any of the genomes argues against the involvement of WLP in syntrophic acetate catabolism. The possibility of its operation for autotrophic CO2 fixation is also unlikely, due to the absence of evidence for utilization of an inorganic electron donor (e.g., molecular H2). Its most plausible function, therefore, is acting as an electron sink to reoxidize reduced ferredoxin, as previously noted in “Candidatus Bipolaricaulota” genomes (51). The Rhodobacter nitrogen fixation (RNF) complex encoded in the majority of genus UBA6911 genomes would allow the reoxidation of reduced ferredoxin at the expense of NAD, with the concomitant export of protons to the periplasm, thus achieving redox balance between heterotrophic substrate oxidation and the WLP function as the electron sink. Additional ATP production via oxidative phosphorylation following the generation of the proton motive force is expected to occur via the F-type ATP synthase encoded in all genomes. Recycling of electron carriers would further be achieved by the cytoplasmic electron-bifurcating mechanisms HydABC and MvhAGD‐HdrABC, both of which are encoded in the genomes (Fig. 3).
Fermentative capacities.
In addition to respiration, elements of fermentation were also encoded in all genomes. All MAGs encoded pyruvate dehydrogenase, as well as 2-oxoacid ferredoxin oxidoreductase for pyruvate oxidation to acetyl-CoA. Genes encoding fermentative enzymes included the acetate production genes (acetyl-CoA synthase [EC 6.2.1.1] in the soil genus and UBA6911 genomes, acetate:CoA ligase [ADP forming] [EC 6.2.1.13] in UBA6911 and Ga0209509 genus genomes, and phosphate acetyltransferase [EC 2.3.1.8] and acetate kinase [EC 2.7.2.1] [the latter two-gene pathway is associated with substrate-level phosphorylation and was encoded only in UBA6911 genomes]), genes for ethanol production from acetate (aldehyde dehydrogenase [EC 1.2.1.3] and alcohol dehydrogenases) in UBA6911 and Ga0209509 genus genomes, genes for ethanol production from acetyl-CoA (aldehyde dehydrogenase [EC 1.2.1.10] and alcohol dehydrogenases) in QHZH01 and Ga0212092 genomes, genes for formate production from pyruvate (formate C-acetyltransferase [EC 2.3.1.54] and its activating enzyme) in UBA6911 and Ga0209509 genus genomes, genes for acetoin and butanediol production from pyruvate in all but QHZH01 genomes, and genes for acetone production from acetyl-CoA (acetyl-CoA C-acetyltransferase [EC 2.3.1.9], acetate CoA/acetoacetate CoA-transferase alpha subunit [EC 2.8.3.8], and acetoacetate decarboxylase [EC 4.1.1.4]) in UBA6911 genomes (Fig. 3 and Table 2; Table S3).
DISCUSSION
Our analysis reveals multiple defining features in all examined family UBA9611 genomes regardless of their origin or genus-level affiliation. All members of the family are predicted to be heterotrophs, with an expected robust anabolic capacity as well as a high level of catabolic versatility. Catabolism of a wide range of sugars, proteins, and amino acids by members of this family is predicted. In addition, all members of this family appear to possess respiratory capacities. Such traits are similar to known metabolic capacities of the majority of examined genomes within the Acidobacteria (15, 33, 52). However, our analysis uncovered multiple interesting differences between the examined genomes, all of which appear to be genus specific and given the strong preference of various genera for specific habitats. Such differences appear to differentiate the two soil genera from the three nonsoil (freshwater and anaerobic digestor) genera (Table 2 and Fig. 3; Table S3) and hence provide clues to the genus-level adaptation to the terrestrialization process within family UBA6911. Three main differences are notable: genomic architecture, substrate-level preferences, and respiratory/electron-accepting processes.
Soil genera gp18_AA60 and QHZH01 possess significantly larger genome sizes (8.05 ± 0.32 Mbp), than freshwater genomes (5.28 ± 0.73 Mbp) and anaerobic digestor genomes (3.5 ± 0.75 Mbp) (Table 1). Soil Acidobacteria usually (33, 52, 53), but not always (33, 54, 55), exhibit large genome sizes, compared to nonsoil Acidobacteria (for examples, see references 35 and 36; for a review, see reference 56), strongly arguing that genome expansion is associated with terrestrialization in the Acidobacteria. The association between genome size expansion and adaptation to soils has been identified as a general trait across the microbial world (57), including Acidobacteria (52, 56). Such expansion has been attributed to a larger number of paralogous genes as a mechanism for generation of new functions or optimal resource utilization under the different environmental conditions occurring in the highly complex and spatiotemporally dynamic soil ecosystem (58). Our analysis suggests that such expansion in gp18_AA60 and QHZH01 genomes is due to a broad increase in the number of genes in soil genera across a wide range of cellular/metabolic functions, rather than gene duplication or differences in coding density. In addition to genome size expansion, soil genus gp18_AA60 exhibited a high number of CRISPR loci (24.67 ± 6.5) (Table 1). In bacteria, CRISPR repeats arise from invading genetic elements that are incorporated into the host's CRISPR locus. These short sequence tags (spacer sequences) are subsequently transcribed into small RNAs to guide the destruction of foreign genetic material (59). Within the Acidobacteria, an examination of 177 genomes (including MAGs and SAGs) affiliated with the phylum Acidobacteria in the IMG database revealed that 67 possessed at least 1 CRISPR count, and only 4 of these possessed >5 CRISPRs. The expansion in the number of CRISPR loci in gp18_AA60 genomes is possibly a protective mechanism against higher potential for viral infection by Acidobacteria-specific viruses in soil, which is expected, given the higher relative abundance of members of this phylum in soil (23) in comparison to their low abundance in other habitats.
Multiple differences in expected catabolic capacities between soil and nonsoil family UBA6911 genera were identified in this study. First, a larger CAZyome was identified in the soil genera gp18_AA60 and QHZH01. This was mainly driven by the expansion of specific GH families, e.g., GH33 (sialidase), GH165 (β-galactosidase), and GH56 (hyaluronidase) in genus QHZH01, GH13 (amylase) in gp18_AA60, and GH109 (N-acetylhexosaminidase) in both genera. It is notable that the capacity for degradation of all released monomers (sialic acid, galactose, uronic acids, glucose, and N-acetylhexosamines) is indeed encoded in these soil genomes. As such, acquisition of these specific CAZymes appears to be important for niche adaptation in family UBA6911 soil genomes. Second, the capacity for methanol and methylamine metabolism was predicted in members of the soil genus QHZH01. As previously noted (60), methylotrophy requires the possession of three metabolic modules for C1 oxidation to formaldehyde, formaldehyde oxidation to CO2, and formaldehyde assimilation. QHZH01 contains genes for all three modules. Formaldehyde assimilation via the serine cycle requires regeneration of glyoxylate from acetyl-CoA to restore glycine and close the cycle. In addition to all three modules described above, the QHZH01 genome also encodes the ethylmalonyl-CoA pathway for glyoxylate regeneration. Soil represents a major source of global methanol emissions (61), where demethylation reactions associated with pectin and other plant polysaccharide degradation contribute to the soil methanol pool. QHZH01 methanol dehydrogenase belongs to the XoxF family (Fig. 4), previously detected in 187 genomes, including Acidobacteria SD1, SD5, and SD6 genomes (30), binned from Angelo Reserve soil, and identified as one of the most abundant proteins in a proteomics study from the same site (62). In contrast, some genus UBA6911 genomes possessed capacities for formaldehyde degradation and assimilation but lacked any genes or modules for conversion of other C1 substrates to formaldehyde. The functionality and value of such truncated C1 machinery remain unclear. Formaldehyde in freshwater environments could be present as a contaminant in aquaculture facility effluent (63) or as a product of C1 metabolism by other members of the community.
Finally, while all family UBA6911 genomes encoded respiratory chains components, distinct differences in predicted respiratory capacities were observed. Soil genera gp18_AA60 and QHZH01 encoded only an aerobic respiratory electron transport chain. In contrast, all anaerobic digestor genus Ga0209509 genomes contained genes for dissimilatory nitrite reduction to ammonium, suggesting a capacity and/or preference to grow in strict anaerobic settings. Freshwater genera Ga0212092 and UBA6911 were the most diverse, encoding aerobic capacity (in all but one genome), dissimilatory nitrite reduction to ammonium (in four UBA6911 genomes), trimethylamine N-oxide respiration (in both Cone Pool Ga0212092 genomes), potential use of WLP for electron acceptance (in genus UBA6911 genomes), and dissimilarity sulfate reduction capacities (in both Cone Pool Ga0212092 and some UBA6911 genomes). This versatility might be a reflection of the relatively broader habitats where these genomes were binned, many of which exhibit seasonal and diel fluctuation in O2 and other electron acceptors levels. Of note is the presence of the complete machinery for dissimilatory sulfate reduction, a process long thought of as a specialty for Deltaproteobacteria (now Desulfobacterota [64]), some Firmicutes, and a few thermophilic bacteria and archaea (65). However, with the recent accumulation of metagenomics data, genes for dissimilatory sulfate reduction were identified in a wide range of phyla (2, 64). In the Acidobacteria, the presence and activity of genes for sulfate reduction have been reported from peatland samples (subdivisions 1 and 3) (28). Subsequently, dissimilarity sulfate reduction capacities were identified in more Acidobacteria MAGs (subdivision 23) from mine drainage (2) and, recently, from marine fjord sediments of Svalbard (38). Our study adds Acidobacteria family UBA6911 (subgroup 18) to the list of the dissimilatory sulfite reductase (DSR)-harboring Acidobacteria and suggests a role in freshwater habitats. The concurrent occurrence of dissimilatory sulfate reduction and aerobic respiration in the same MAG is notable but has been previously reported in the subdivision 23 Acidobacteria MAGs from marine fjord sediments of Svalbard (38). All Acidobacteria DsrAB sequences (subdivisions 1, 3, 18, and 23) cluster within the reductive bacterial type branches in DsrAB concatenated phylogenetic trees, away from the oxidative Chlorobia and Proteobacteria (Fig. 5). Also, the absence of DsrEFH homologs, known to be involved in the reverse DSR pathway, argue for the involvement of Acidobacteria DsrAB in the reductive dissimilatory sulfate reduction.
MATERIALS AND METHODS
Sample collection, DNA extraction, and metagenomic sequencing.
Samples were collected from Zodletone Spring, a sulfide- and sulfur-rich spring in western Oklahoma’s Anadarko Basin (N34.99562° W98.68895°). The ecology, geochemistry, and phylogenetic diversity of the various locations within the spring have been previously described (66–69). Samples were collected 5 cm deep into the black sediments by completely filling sterile 50-ml polypropylene plastic tubes. Ten different samples were collected. The tubes were capped, sealed, and kept on ice until brought back to the lab (∼2-h drive), where they were immediately processed. DNA extraction was conducted on 0.5 g sediment from each of the 10 replicate samples using the DNeasy PowerSoil kit (Qiagen, Valencia, CA, USA) according to the manufacturer’s protocols. On average, 0.15 to 0.3 µg DNA was obtained per extraction. All extractions were pooled and used for the preparation of sequencing libraries by use of the Nextera XT DNA library prep kit (Illumina, San Diego, CA, USA) per the manufacturer’s instructions. Sequencing was conducted on the Illumina HiSeq 2500 platform and 150-bp pair-end technology using the services of a commercial provider (Novogene, Beijing, China). Sequencing produced 281.0 Gbp of raw data. Metagenomic reads were assessed for quality using FastQC, followed by quality filtering and trimming using Trimmomatic v0.38 (70). High-quality reads were assembled into contigs using MegaHit (v.1.1.3) (71) with a minimum kmer of 27, maximum kmer of 127, kmer step of 10, and minimum contig length of 1,000 bp. Bowtie2 was used to calculate sequencing coverage of each contig by mapping the raw reads back to the contigs. Contigs were binned into draft genomes using both MetaBAT (72) and MaxBin2 (73), followed by selection of the highest-quality bins using DasTool (74). GTDB-Tk (75) (v1.3.0) was used for the taxonomic classification of the bins using the classification workflow option -classify_wf, and 4 bins belonging to Acidobacteria family UBA6911 were selected for further analysis. In addition, 18 genomes belonging to family UBA6911 were selected from the recently released 52,515 genomes in the Earth microbiome catalogue collection (76) available through the IMG/M database. These genomes were binned from peatland soils in Minnesota, USA (4 genomes) (77, 78), an anaerobic biogas reactor in Washington, USA (11 genomes) (40), a Cone Pool hot spring microbial mat in California, USA (2 genomes), and White Oak River estuary sediment in North Carolina, USA (1 genome) (79). Five other genomes belonging to family UBA6911 were available from GenBank and were obtained in previous studies. These included 4 genomes binned from the Angelo Coastal Range Reserve in Northern California (30) and 1 genome binned from Noosa River sediments (Queensland, Australia) (80).
Genome quality assessment and general genomic features.
Genome completeness and contamination were assessed using CheckM (v1.0.13) (81) by employing the lineage-specific workflow (lineage_wf flag). All genomes included in this study were of medium or high quality with >70% completion and <10% contamination (Table S2). Designation as medium- or high-quality drafts was based on the criteria set forth by MIMAGs (82). The 5S, 16S, and 23S rRNA sequences were identified using Barrnap 0.9 (https://github.com/tseemann/barrnap). tRNA sequences were identified and enumerated with tRNAscan-SE (v2.0.6, May 2020) (83). Genomes were mined for CRISPR and Cas proteins using the CRISPR/CasFinder (84).
Phylogenomic analysis.
Taxonomic classification using the GTDB taxonomic framework was conducted using the classification workflow option -classify_wf within the GTDB-Tk (75). Phylogenomic analysis was conducted using the concatenated alignment of 120 single-copy marker genes (10) that is generated by GTDB-Tk (75). Maximum likelihood phylogenetic trees were constructed in RAxML (v8.2.8) (85) with the PROTGAMMABLOSUM62 model and default settings. Representatives of all other Acidobacteria classes were included in the analysis (Fig. 1), and Chloroflexus aggregans (GCF_000021945.1) was used as the outgroup. As expected, all 28 genomes were classified to the family UBA6911 within the UBA6911 class of the Acidobacteria, but only 14 genomes were classified by the GTDB to the genus level into two genera, gp18_AA60 and UBA6911, while the remaining genomes were unclassified at the genus level. To further assign these genomes to putative genera, average amino acid identity (AAI) and shared gene content (SGC) were calculated using the AAI calculator (http://enve-omics.ce.gatech.edu/). Based on these values, we propose assigning these 14 genomes to three putative novel genera. We propose the names QHZH01 (1 genome from grassland soil), Ga0212092 (2 genomes from a Cone Pool microbial mat), and Ga0209509 (10 genomes from an anaerobic gas digestor [Washington, USA] and 1 Zodletone sediment genome) based on the assembly accession number of the most complete genome within each genus (Table 1; Table S2).
Functional annotation.
Protein coding genes were annotated using Prodigal (v2.50) (86). BlastKOALA (87) was used to assign KEGG orthologies (KO) to protein coding genes, followed by metabolic pathway visualization in KEGG mapper (88). In addition, all genomes were queried with custom-built HMM profiles for sulfur metabolism, electron transport chain components (for alternate complex III), C1 metabolism, and hydrogenases. To construct hmm profiles, UniProt reference sequences for genes with an assigned KO number were downloaded and aligned with Mafft (89), and the alignment was used to construct hmm profiles using the hmmbuild function of HMMer (v3.1b2) (90). For genes without a designated KO number, a representative protein was queried against the KEGG gene database using BLASTP, and hits with E values of <1e−80 were downloaded, aligned, and used to construct an hmm profile as described above. Hydrogenase hmm profiles were built using alignments downloaded from the Hydrogenase Database (HydDB) (91). The hmmscan function of HMMer (90) was used with the constructed profiles and a thresholding option of -T 100 to scan the protein coding genes for possible hits. Further confirmation was achieved through phylogenetic assessment and tree building procedures. For that, putatively identified Acidobacteria sequences were aligned with Mafft (89) against the reference sequences used to build the HMM database, and the alignment was then used to construct a maximum likelihood phylogenetic tree using FastTree (v2.1.10) (92). Sequences that clustered with reference sequences were deemed to be true hits and were assigned a corresponding KO number or function. Carbohydrate active enzymes (CAZymes) (including glycoside hydrolases [GHs], polysaccharide lyases [PLs], and carbohydrate esterases [CEs]) were identified by searching all open reading frames (ORFs) from all genomes against the dbCAN hidden Markov models V9 (93, 94) (downloaded from the dbCAN web server in September 2020) using hmmscan. AntiSMASH 3.0 (95) was used with default parameters to predict biosynthetic gene clusters in the genomes. Canonical correspondence analysis (CCA) was used to identify the correlation between the genus, environmental source, and types of BGCs predicted in the genomes, using the function cca in the R package Vegan (https://cran.r-project.org/web/packages/vegan/index.html).
To evaluate the novelty of the NRPS and PKS clusters identified in family UBA6911 genomes, we queried the synthetic genes from these clusters against the NCBI nucleotide database (downloaded in April 2020). A threshold of 75% identity over 50% of the query length was used to determine the novelty of these genes.
Phylogenetic analysis of XoxF methanol dehydrogenase and dissimilatory sulfite reductase DsrAB.
Family UBA6911 predicted XoxF methanol dehydrogenase and dissimilatory sulfite reductase subunits A and B were compared to reference sequences for phylogenetic placement. Family UBA6911 predicted XoxF protein sequences were aligned to corresponding reference sequences from other methylotrophic taxa, while the dissimilatory sulfite reductase subunits A and B were aligned to corresponding subunits from sulfate-reducing taxa using Mafft (89). DsrA and DsrB alignments were concatenated in Mega X (96). The XoxF alignment and the DsrAB concatenated alignment were used to construct maximum-likelihood phylogenetic trees using FastTree (v2.1.10) (92).
Ecological distribution.
To further examine the ecological distribution of family UBA6911 genera, we analyzed 177 near-full-length 16S rRNA gene sequences (>1,200 bp) associated with this lineage in the SILVA database (r138.1) (25) (SILVA classification, Bacteria;Acidobacteriota;Subgroup 18). A nearly complete 16S rRNA gene from each genus (with the exception of genus QHZH01 represented by a single genomic assembly that unfortunately lacked a 16S rRNA gene) was selected as a representative and was included in the analysis. Sequences were aligned using the SINA aligner (97), and the alignment was used to construct maximum-likelihood phylogenetic trees with FastTree (92). The environmental source of hits clustering with the appropriate reference sequences was then classified with a scheme based on the GOLD ecosystem classification scheme (98). All phylogenetic trees were visualized and annotated in iTol (99).
Data availability.
Metagenomic raw reads for Zodletone Spring sediment are available under SRA accession no. SRX9813571. The Zodletone Spring whole-genome shotgun project was submitted to GenBank under Bioproject ID PRJNA690107 and Biosample ID SAMN17269717. The individual assembled Acidobacteria MAGs analyzed in this study have been deposited in DDBJ/ENA/GenBank under accession no. JAFGAO000000000, JAFGSS000000000, JAFGIY000000000, and JAFGTD000000000.
ACKNOWLEDGMENT
This work was supported by National Science Foundation grant no. 2016423 to N.H.Y. and M.S.E.
Footnotes
Supplemental material is available online only.
Contributor Information
Noha H. Youssef, Email: Noha@okstate.edu.
M. Julia Pettinari, University of Buenos Aires.
REFERENCES
- 1.Doud DFR, Bowers RM, Schulz F, De Raad M, Deng K, Tarver A, Glasgow E, Vander Meulen K, Fox B, Deutsch S, Yoshikuni Y, Northen T, Hedlund BP, Singer SW, Ivanova N, Woyke T. 2020. Function-driven single-cell genomics uncovers cellulose-degrading bacteria from the rare biosphere. ISME J 14:659–675. 10.1038/s41396-019-0557-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Anantharaman K, Hausmann B, Jungbluth SP, Kantor RS, Lavy A, Warren LA, Rappé MS, Pester M, Loy A, Thomas BC, Banfield JF. 2018. Expanded diversity of microbial groups that shape the dissimilatory sulfur cycle. ISME J 12:1715–1728. 10.1038/s41396-018-0078-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Becraft ED, Woyke T, Jarett J, Ivanova N, Godoy-Vitorino F, Poulton N, Brown JM, Brown J, Lau MCY, Onstott T, Eisen JA, Moser D, Stepanauskas R. 2017. Rokubacteria: genomic giants among the uncultured bacterial phyla. Front Microbiol 8:2264. 10.3389/fmicb.2017.02264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rinke R, Rubino F, Messer LF, Youssef N, Parks DH, Chuvochina M, Brown M, Jeffries T, Tyson GW, Seymour JR, Hugenholtz P. 2019. A phylogenomic and ecological analysis of the globally abundant Marine Group II archaea (Ca. Poseidoniales ord. nov.). ISME J 13:663–675. 10.1038/s41396-018-0282-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Farag IF, Davis JP, Youssef NH, Elshahed MS. 2014. Global patterns of abundance, diversity and community structure of the Aminicenantes (candidate phylum OP8). PLoS One 9:e92139. 10.1371/journal.pone.0092139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hu P, Dubinsky EA, Probst AJ, Wang J, Sieber CMK, Tom LM, Gardinali PR, Banfield JF, Atlas RM, Andersen GL. 2017. Simulation of Deepwater Horizon oil plume reveals substrate specialization within a complex community of hydrocarbon degraders. Proc Natl Acad Sci U S A 114:7432–7437. 10.1073/pnas.1703424114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhou Z, Tran PQ, Kieft K, Anantharaman K. 2020. Genome diversification in globally distributed novel marine Proteobacteria is linked to environmental adaptation. ISME J 14:2060–2077. 10.1038/s41396-020-0669-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Rinke C, Schwientek P, Sczyrba A, Ivanova NN, Anderson IJ, Cheng JF, Darling A, Malfatti S, Swan BK, Gies EA, Dodsworth JA, Hedlund BP, Tsiamis G, Sievert SM, Liu WT, Eisen JA, Hallam SJ, Kyrpides NC, Stepanauskas R, Rubin EM, Hugenholtz P, Woyke T. 2013. Insights into the phylogeny and coding potential of microbial dark matter. Nature 499:431–437. 10.1038/nature12352. [DOI] [PubMed] [Google Scholar]
- 9.Beam JP, Becraft ED, Brown KM, Schulz F, Jarett JK. 2020. Ancestral absence of electron transport chains in Patescibacteria and DPANN. Front Microbiol 11:1848. 10.3389/fmicb.2020.01848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil P-A, Hugenholtz P. 2018. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 36:996–1004. 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- 11.Kielak AM, Barreto CC, Kowalchuk GA, van Veen JA, Kuramae EE. 2016. The ecology of Acidobacteria: moving beyond genes and genomes. Front Microbiol 7:744. 10.3389/fmicb.2016.00744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Quaiser A, Ochsenreiter T, Lanz C, Schuster SC, Treusch AH, Eck J, Schleper C. 2003. Acidobacteria form a coherent but highly diverse group within the bacterial domain: evidence from environmental genomics. Mol Microbiol 50:563–575. 10.1046/j.1365-2958.2003.03707.x. [DOI] [PubMed] [Google Scholar]
- 13.Fierer N, Jackson JA, Vilgalys R, Jackson RB. 2005. Assessment of soil microbial community structure by use of taxon-specific quantitative PCR assays. Appl Environ Microbiol 71:4117–4120. 10.1128/AEM.71.7.4117-4120.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kim J-S, Sparovek G, Longo RM, De Melo WJ, Crowley D. 2007. Bacterial diversity of terra preta and pristine forest soil from the Western Amazon. Soil Biol Biochem 39:684–690. 10.1016/j.soilbio.2006.08.010. [DOI] [Google Scholar]
- 15.Kielak A, Pijl AS, van Veen JA, Kowalchuk GA. 2009. Phylogenetic diversity of Acidobacteria in a former agricultural soil. ISME J 3:378–382. 10.1038/ismej.2008.113. [DOI] [PubMed] [Google Scholar]
- 16.Zhang Y, Cong J, Lu H, Li G, Qu Y, Su X, Zhou J, Li D. 2014. Community structure and elevational diversity patterns of soil Acidobacteria. J Environ Sci (China) 26:1717–1724. 10.1016/j.jes.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 17.Kishimoto N, Kosako Y, Tano T. 1991. Acidobacterium capsulatum gen. nov., sp. nov.: an acidophilic chemoorganotrophic bacterium containing menaquinone from acidic mineral environment. Curr Microbiol 22:1–7. 10.1007/BF02106205. [DOI] [PubMed] [Google Scholar]
- 18.Liesack W, Bak F, Kreft JU, Stackebrandt E. 1994. Holophaga foetida gen. nov., sp. nov., a new, homoacetogenic bacterium degrading methoxylated aromatic compounds. Arch Microbiol 162:85–90. 10.1007/BF00264378. [DOI] [PubMed] [Google Scholar]
- 19.Coates JD, Ellis DJ, Gaw CV, Lovley DR. 1999. Geothrix fermentans gen. nov., sp. nov., a novel Fe(III)-reducing bacterium from a hydrocarbon-contaminated aquifer. Int J Syst Bacteriol 49:1615–1622. 10.1099/00207713-49-4-1615. [DOI] [PubMed] [Google Scholar]
- 20.Borneman J, Triplett EW. 1997. Molecular microbial diversity in soils from eastern Amazonia: evidence for unusual microorganisms and microbial population shifts associated with deforestation. Appl Environ Microbiol 63:2647–2653. 10.1128/aem.63.7.2647-2653.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Barns SM, Takala SL, Kuske CR. 1999. Wide distribution and diversity of members of the bacterial kingdom Acidobacterium in the environment. Appl Environ Microbiol 65:1731–1737. 10.1128/AEM.65.4.1731-1737.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Dunbar J, Takala S, Barns SM, Davis JA, Kuske CR. 1999. Levels of bacterial community diversity in four arid soils compared by cultivation and 16S rRNA gene cloning. Appl Environ Microbiol 65:1662–1669. 10.1128/AEM.65.4.1662-1669.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Janssen PH. 2006. Identifying the dominant soil bacterial taxa in libraries of 16S rRNA and 16S rRNA genes. Appl Environ Microbiol 72:1719–1728. 10.1128/AEM.72.3.1719-1728.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Liles MR, Manske BF, Bintrim SB, Handelsman J, Goodman RM. 2003. A census of rRNA genes and linked genomic sequences within a soil metagenomic library. Appl Environ Microbiol 69:2684–2691. 10.1128/AEM.69.5.2684-2691.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Quast C, Pruesse E, Yilmaz P, Gerken J, Schweer T, Yarza P, Peplies J, Glöckner FO. 2013. The SILVA ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Res 41:D590–D596. 10.1093/nar/gks1219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barns SM, Cain EC, Sommerville L, Kuske CR. 2007. Acidobacteria phylum sequences in uranium-contaminated subsurface sediments greatly expand the known diversity within the phylum. Appl Environ Microbiol 73:3113–3116. 10.1128/AEM.02012-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dedysh SN, Yilmaz P. 2018. Refining the taxonomic structure of the phylum Acidobacteria. Int J Syst Evol Microbiol 68:3796–3806. 10.1099/ijsem.0.003062. [DOI] [PubMed] [Google Scholar]
- 28.Hausmann B, Pelikan C, Herbold CW, Köstlbacher S, Albertsen M, Eichorst SA, Glavina del Rio T, Huemer M, Nielsen PH, Rattei T, Stingl U, Tringe SG, Trojan D, Wentrup C, Woebken D, Pester M, Loy A. 2018. Peatland Acidobacteria with a dissimilatory sulfur metabolism. ISME J 12:1729–1742. 10.1038/s41396-018-0077-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Crits-Christoph A, Diamond S, Butterfield CN, Thomas BC, Banfield JF. 2018. Novel soil bacteria possess diverse genes for secondary metabolite biosynthesis. Nature 558:440–444. 10.1038/s41586-018-0207-y. [DOI] [PubMed] [Google Scholar]
- 30.Diamond S, Andeer PF, Li Z, Crits-Christoph A, Burstein D, Anantharaman K, Lane KR, Thomas BC, Pan C, Northen TR, Banfield JF. 2019. Mediterranean grassland soil C–N compound turnover is dependent on rainfall and depth, and is mediated by genomically divergent microorganisms. Nat Microbiol 4:1356–1367. 10.1038/s41564-019-0449-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Podar M, Turner J, Burdick LH, Pelletier DA. 2019. Complete genome sequence of Terriglobus albidus strain ORNL, an acidobacterium isolated from the Populus deltoides rhizosphere. Microbiol Resour Announc 8:e01065-19. 10.1128/MRA.01065-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Rawat SR, Männistö MK, Bromberg Y, Häggblom MM. 2012. Comparative genomic and physiological analysis provides insights into the role of Acidobacteria in organic carbon utilization in Arctic tundra soils. FEMS Microbiol Ecol 82:341–355. 10.1111/j.1574-6941.2012.01381.x. [DOI] [PubMed] [Google Scholar]
- 33.Ward NL, Challacombe JF, Janssen PH, Henrissat B, Coutinho PM, Wu M, Xie G, Haft DH, Sait M, Badger J, Barabote RD, Bradley B, Brettin TS, Brinkac LM, Bruce D, Creasy T, Daugherty SC, Davidsen TM, DeBoy RT, Detter JC, Dodson RJ, Durkin AS, Ganapathy A, Gwinn-Giglio M, Han CS, Khouri H, Kiss H, Kothari SP, Madupu R, Nelson KE, Nelson WC, Paulsen I, Penn K, Ren Q, Rosovitz MJ, Selengut JD, Shrivastava S, Sullivan SA, Tapia R, Thompson LS, Watkins KL, Yang Q, Yu C, Zafar N, Zhou L, Kuske CR. 2009. Three genomes from the phylum Acidobacteria provide insight into the lifestyles of these microorganisms in soils. Appl Environ Microbiol 75:2046–2056. 10.1128/AEM.02294-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Anderson I, Held B, Lapidus A, Nolan M, Lucas S, Tice H, Del Rio TG, Cheng JF, Han C, Tapia R, Goodwin LA, Pitluck S, Liolios K, Mavromatis K, Pagani I, Ivanova N, Mikhailova N, Pati A, Chen A, Palaniappan K, Land M, Brambilla EM, Rohde M, Spring S, Göker M, Detter JC, Woyke T, Bristow J, Eisen JA, Markowitz V, Hugenholtz P, Klenk HP, Kyrpides NC. 2012. Genome sequence of the homoacetogenic bacterium Holophaga foetida type strain (TMBS4(T)). Stand Genomic Sci 6:174–184. 10.4056/sigs.2746047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Garcia Costas AM, Liu Z, Tomsho LP, Schuster SC, Ward DM, Bryant DA. 2012. Complete genome of Candidatus Chloracidobacterium thermophilum, a chlorophyll-based photoheterotroph belonging to the phylum Acidobacteria. Environ Microbiol 14:177–190. 10.1111/j.1462-2920.2011.02592.x. [DOI] [PubMed] [Google Scholar]
- 36.Stamps BW, Losey NA, Lawson PA, Stevenson BS. 2014. Genome sequence of Thermoanaerobaculum aquaticum MP-01T, the first cultivated member of Acidobacteria subdivision 23, isolated from a hot spring. Genome Announc 2:e00570-14. 10.1128/genomeA.00570-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ward LM, McGlynn SE, Fischer WW. 2017. Draft genome sequence of Chloracidobacterium sp. CP2_5A, a phototrophic member of the phylum Acidobacteria recovered from a Japanese hot spring. Genome Announc 5:e00821-17. 10.1128/genomeA.00821-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Flieder M, Buongiorno J, Herbold CW, Hausmann B, Rattei T, Lloyd KG, Loy A, Wasmund K. 2020. Novel taxa of Acidobacteriota involved in seafloor sulfur cycling. bioRxiv 10.1101/2020.10.01.322446:2020.10.01.322446. [DOI] [PMC free article] [PubMed]
- 39.Wegner CE, Liesack W. 2017. Unexpected dominance of elusive Acidobacteria in early industrial soft coal slags. Front Microbiol 8:1023. 10.3389/fmicb.2017.01023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ziels RM, Sousa DZ, Stensel HD, Beck DAC. 2018. DNA-SIP based genome-centric metagenomics identifies key long-chain fatty acid-degrading populations in anaerobic digesters with different feeding frequencies. ISME J 12:112–123. 10.1038/ismej.2017.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ligthart K, Belzer C, de Vos WM, Tytgat HLP. 2020. Bridging bacteria and the gut: functional aspects of type IV pili. Trends Microbiol 28:340–348. 10.1016/j.tim.2020.02.003. [DOI] [PubMed] [Google Scholar]
- 42.Coulthurst S. 2019. The type VI secretion system: a versatile bacterial weapon. Microbiology (Reading) 165:503–515. 10.1099/mic.0.000789. [DOI] [PubMed] [Google Scholar]
- 43.Mendler K, Chen H, Parks DH, Lobb B, Hug LA, Doxey AC. 2019. AnnoTree: visualization and exploration of a functionally annotated microbial tree of life. Nucleic Acids Res 47:4442–4448. 10.1093/nar/gkz246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Courtney G, Barbara C-M, Jane H. 2011. The inositol phosphates in soils and manures: abundance, cycling, and measurement. Canadian J Soil Science 91:397–416. [Google Scholar]
- 45.Gruteser N, Marin K, Krämer R, Thomas GH. 2012. Sialic acid utilization by the soil bacterium Corynebacterium glutamicum. FEMS Microbiol Lett 336:131–138. 10.1111/j.1574-6968.2012.02663.x. [DOI] [PubMed] [Google Scholar]
- 46.Abaibou H, Giordano G, Mandrand-Berthelot MA. 1997. Suppression of Escherichia coli formate hydrogenlyase activity by trimethylamine N-oxide is due to drainage of the inducer formate. Microbiology (Reading) 143:2657–2664. 10.1099/00221287-143-8-2657. [DOI] [PubMed] [Google Scholar]
- 47.Cox JC, Madigan MT, Favinger JL, Gest H. 1980. Redox mechanisms in “oxidant-dependent” hexose fermentation by Rhodopseudomonas capsulata. Arch Biochem Biophys 204:10–17. 10.1016/0003-9861(80)90002-8. [DOI] [PubMed] [Google Scholar]
- 48.Müller B, Sun L, Schnürer A. 2013. First insights into the syntrophic acetate-oxidizing bacteria—a genetic study. Microbiologyopen 2:35–53. 10.1002/mbo3.50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ragsdale SW, Pierce E. 2008. Acetogenesis and the Wood-Ljungdahl pathway of CO(2) fixation. Biochim Biophys Acta 1784:1873–1898. 10.1016/j.bbapap.2008.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Schuchmann K, Müller V. 2016. Energetics and application of heterotrophy in acetogenic bacteria. Appl Environ Microbiol 82:4056–4069. 10.1128/AEM.00882-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Youssef NH, Farag IF, Rudy S, Mulliner A, Walker K, Caldwell F, Miller M, Hoff W, Elshahed M. 2019. The Wood-Ljungdahl pathway as a key component of metabolic versatility in candidate phylum Bipolaricaulota (Acetothermia, OP1). Environ Microbiol Rep 11:538–547. 10.1111/1758-2229.12753. [DOI] [PubMed] [Google Scholar]
- 52.Eichorst SA, Trojan D, Roux S, Herbold C, Rattei T, Woebken D. 2018. Genomic insights into the Acidobacteria reveal strategies for their success in terrestrial environments. Environ Microbiol 20:1041–1063. 10.1111/1462-2920.14043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Huang S, Vieira S, Bunk B, Riedel T, Spröer C, Overmann J. 2016. First complete genome sequence of a subdivision 6 Acidobacterium strain. Genome Announc 4:e00469-16. 10.1128/genomeA.00469-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Domeignoz-Horta LA, DeAngelis KM, Pold G. 2019. Draft genome sequence of Acidobacteria group 1 Acidipila sp. strain EB88, isolated from forest soil. Microbiol Resour Announc 8:e01464-18. 10.1128/MRA.01464-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Eichorst SA, Trojan D, Huntemann M, Clum A, Pillay M, Palaniappan K, Varghese N, Mikhailova N, Stamatis D, Reddy TBK, Daum C, Goodwin LA, Shapiro N, Ivanova N, Kyrpides N, Woyke T, Woebken D. 2020. One complete and seven draft genome sequences of subdivision 1 and 3 Acidobacteria isolated from soil. Microbiol Resour Announc 9:e01087-19. 10.1128/MRA.01087-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kalam S, Basu A, Ahmad I, Sayyed RZ, El-Enshasy HA, Dailin DJ, Suriani NL. 2020. Recent understanding of soil acidobacteria and their ecological significance: a critical review. Front Microbiol 11:580024. 10.3389/fmicb.2020.580024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Maistrenko OM, Mende DR, Luetge M, Hildebrand F, Schmidt TSB, Li SS, Rodrigues JFM, von Mering C, Pedro Coelho L, Huerta-Cepas J, Sunagawa S, Bork P. 2020. Disentangling the impact of environmental and phylogenetic constraints on prokaryotic within-species diversity. ISME J 14:1247–1259. 10.1038/s41396-020-0600-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Challacombe JF, Eichorst SA, Hauser L, Land M, Xie G, Kuske CR. 2011. Biological consequences of ancient gene acquisition and duplication in the large genome of Candidatus Solibacter usitatus Ellin6076. PLoS One 6:e24882. 10.1371/journal.pone.0024882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Karginov FV, Hannon GJ. 2010. The CRISPR system: small RNA-guided defense in bacteria and archaea. Mol Cell 37:7–19. 10.1016/j.molcel.2009.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Chistoserdova L. 2011. Modularity of methylotrophy, revisited. Environ Microbiol 13:2603–2622. 10.1111/j.1462-2920.2011.02464.x. [DOI] [PubMed] [Google Scholar]
- 61.Kolb S. 2009. Aerobic methanol-oxidizing bacteria in soil. FEMS Microbiol Lett 300:1–10. 10.1111/j.1574-6968.2009.01681.x. [DOI] [PubMed] [Google Scholar]
- 62.Butterfield CN, Li Z, Andeer PF, Spaulding S, Thomas BC, Singh A, Hettich RL, Suttle KB, Probst AJ, Tringe SG, Northen T, Pan C, Banfield JF. 2016. Proteogenomic analyses indicate bacterial methylotrophy and archaeal heterotrophy are prevalent below the grass root zone. PeerJ 4:e2687. 10.7717/peerj.2687. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Lalonde BA, Ernst W, Garron C. 2015. Formaldehyde concentration in discharge from land based aquaculture facilities in Atlantic Canada. Bull Environ Contam Toxicol 94:444–447. 10.1007/s00128-015-1493-9. [DOI] [PubMed] [Google Scholar]
- 64.Waite DW, Chuvochina M, Pelikan C, Parks DH, Yilmaz P, Wagner M, Loy A, Naganuma T, Nakai R, Whitman WB, Hahn MW, Kuever J, Hugenholtz P. 2020. Proposal to reclassify the proteobacterial classes Deltaproteobacteria and Oligoflexia, and the phylum Thermodesulfobacteria into four phyla reflecting major functional capabilities. Int J Syst Evol Microbiol 70:5972–6016. 10.1099/ijsem.0.004213. [DOI] [PubMed] [Google Scholar]
- 65.Müller AL, Kjeldsen KU, Rattei T, Pester M, Loy A. 2015. Phylogenetic and environmental diversity of DsrAB-type dissimilatory (bi)sulfite reductases. ISME J 9:1152–1165. 10.1038/ismej.2014.208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Elshahed MS, Senko JM, Najar FZ, Kenton SM, Roe BA, Dewers TA, Spear JR, Krumholz LR. 2003. Bacterial diversity and sulfur cycling in a mesophilic sulfide-rich spring. Appl Environ Microbiol 69:5609–5621. 10.1128/AEM.69.9.5609-5621.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Senko JM, Campbell BS, Henriksen JR, Elshahed MS, Dewers TA, Krumholz LR. 2004. Barite deposition mediated by photootrophic sulfide-oxidizing bacteria. Geochim Cosmochim Acta 68:773–780. 10.1016/j.gca.2003.07.008. [DOI] [Google Scholar]
- 68.Buhring SI, Sievert SM, Jonkers HM, Ertefai T, Elshahed MS, Krumholz LR, Hinrichs K-U. 2011. Insights into chemotaxonomic composition and carbon cycling of phototrophic communities in an artesian sulfur‐rich spring (Zodletone, Oklahoma, USA), a possible analog for ancient microbial mat systems. Geobiology 9:166–179. 10.1111/j.1472-4669.2010.00268.x. [DOI] [PubMed] [Google Scholar]
- 69.Spain AM, Najar FZ, Krumholz LR, Elshahed MS. 2015. Metatranscriptomic analysis of a high-sulfide aquatic spring reveals insights into sulfur cycling and unexpected aerobic metabolism. Peer J 3:e1259. 10.7717/peerj.1259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Bolger AM, Lohse M, Usadel B. 2014. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30:2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 72.Kang DD, Li F, Kirton E, Thomas A, Egan R, An H, Wang Z. 2019. MetaBAT 2: an adaptive binning algorithm for robust and efficient genome reconstruction from metagenome assemblies. PeerJ 7:e7359. 10.7717/peerj.7359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wu YW, Simmons BA, Singer SW. 2016. MaxBin 2.0: an automated binning algorithm to recover genomes from multiple metagenomic datasets. Bioinformatics 32:605–607. 10.1093/bioinformatics/btv638. [DOI] [PubMed] [Google Scholar]
- 74.Sieber CMK, Probst AJ, Sharrar A, Thomas BC, Hess M, Tringe SG, Banfield JF. 2018. Recovery of genomes from metagenomes via a dereplication, aggregation and scoring strategy. Nat Microbiol 3:836–843. 10.1038/s41564-018-0171-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. 2019. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinformatics 36:1925–1927. 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Nayfach S, Roux S, Seshadri R, Udwary D, Varghese N, Schulz F, Wu D, Paez-Espino D, Chen I-M, Huntemann M, Palaniappan K, Ladau J, Mukherjee S, Reddy TBK, Nielsen T, Kirton E, Faria JP, Edirisinghe JN, Henry CS, Jungbluth SP, Chivian D, Dehal P, Wood-Charlson EM, Arkin AP, Tringe SG, Visel A, IMG/M Data Consortium, Woyke T, Mouncey NJ, Ivanova NN, Kyrpides NC, Eloe-Fadrosh EA. 2020. A genomic catalogue of Earth’s microbiomes. Nat Biotechnol 39:499–509. 10.1038/s41587-020-0718-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Kluber LA, Yip DZ, Yang ZK, Schadt CW. 2018. SPRUCE deep peat heating (DPH) to whole ecosystem warming (WEW) metagenomes for peat samples collected June 2016. Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge, TN. [Google Scholar]
- 78.Kluber LA, Yang ZK, Schadt CW. 2016. SPRUCE deep peat heat (DPH) metagenomes for peat samples collected June 2015. Oak Ridge National Laboratory, U.S. Department of Energy, Oak Ridge, TN. [Google Scholar]
- 79.Baker BJ, Lazar CS, Teske AP, Dick GJ. 2015. Genomic resolution of linkages in carbon, nitrogen, and sulfur cycling among widespread estuary sediment bacteria. Microbiome 3:14. 10.1186/s40168-015-0077-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Parks DH, Rinke C, Chuvochina M, Chaumeil P-A, Woodcroft BJ, Evans PN, Hugenholtz P, Tyson GW. 2017. Recovery of nearly 8,000 metagenome-assembled genomes substantially expands the tree of life. Nat Microbiol 2:1533–1542. 10.1038/s41564-017-0012-7. [DOI] [PubMed] [Google Scholar]
- 81.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Bowers RM, Kyrpides NC, Stepanauskas R, Harmon-Smith M, Doud D, Reddy TBK, Schulz F, Jarett J, Rivers AR, Eloe-Fadrosh EA, Tringe SG, Ivanova NN, Copeland A, Clum A, Becraft ED, Malmstrom RR, Birren B, Podar M, Bork P, Weinstock GM, Garrity GM, Dodsworth JA, Yooseph S, Sutton G, Glöckner FO, Gilbert JA, Nelson WC, Hallam SJ, Jungbluth SP, Ettema TJG, Tighe S, Konstantinidis KT, Liu W-T, Baker BJ, Rattei T, Eisen JA, Hedlund B, McMahon KD, Fierer N, Knight R, Finn R, Cochrane G, Karsch-Mizrachi I, Tyson GW, Rinke C, Lapidus A, Meyer F, Yilmaz P, Parks DH, Eren AM, Genome Standards Consortium, et al. 2017. Minimum information about a single amplified genome (MISAG) and a metagenome-assembled genome (MIMAG) of bacteria and archaea. Nat Biotechnol 35:725–731. 10.1038/nbt.3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Chan PP, Lowe TM. 2019. tRNAscan-SE: searching for tRNA. Methods Mol Biol 1962:1–14. 10.1007/978-1-4939-9173-0_1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, Rocha EPC, Vergnaud G, Gautheret D, Pourcel C. 2018. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res 46:W246–W251. 10.1093/nar/gky425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. 10.1093/bioinformatics/btu033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hyatt D, Chen G-L, Locascio PF, Land ML, Larimer FW, Hauser LJ. 2010. Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11:119. 10.1186/1471-2105-11-119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kanehisa M, Sato Y, Morishima K. 2016. BlastKOALA and GhostKOALA: KEGG tools for functional characterization of genome and metagenome sequences. J Mol Biol 428:726–731. 10.1016/j.jmb.2015.11.006. [DOI] [PubMed] [Google Scholar]
- 88.Kanehisa M, Sato Y. 2020. KEGG Mapper for inferring cellular functions from protein sequences. Protein Sci 29:28–35. 10.1002/pro.3711. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Nakamura T, Yamada KD, Tomii K, Katoh K. 2018. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34:2490–2492. 10.1093/bioinformatics/bty121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Mistry J, Finn RD, Eddy SR, Bateman A, Punta M. 2013. Challenges in homology search: HMMER3 and convergent evolution of coiled-coil regions. Nucleic Acids Res 41:e121. 10.1093/nar/gkt263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Søndergaard D, Pedersen CN, Greening C. 2016. HydDB: a web tool for hydrogenase classification and analysis. Sci Rep 6:34212. 10.1038/srep34212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Price MN, Dehal PS, Arkin AP. 2010. FastTree 2—approximately maximum-likelihood trees for large alignments. PLoS One 5:e9490. 10.1371/journal.pone.0009490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhang H, Yohe T, Huang L, Entwistle S, Wu P, Yang Z, Busk PK, Xu Y, Yin Y. 2018. dbCAN2: a meta server for automated carbohydrate-active enzyme annotation. Nucleic Acids Res 46:W95–W101. 10.1093/nar/gky418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Huang L, Zhang H, Wu P, Entwistle S, Li X, Yohe T, Yi H, Yang Z, Yin Y. 2018. dbCAN-seq: a database of carbohydrate-active enzyme (CAZyme) sequence and annotation. Nucleic Acids Res 46:D516–D521. 10.1093/nar/gkx894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Medema MH, Blin K, Cimermancic P, de Jager V, Zakrzewski P, Fischbach MA, Weber T, Takano E, Breitling R. 2011. antiSMASH: rapid identification, annotation and analysis of secondary metabolite biosynthesis gene clusters in bacterial and fungal genome sequences. Nucleic Acids Res 39:W339–W346. 10.1093/nar/gkr466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kumar S, Stecher G, Li M, Knyaz C, Tamura K. 2018. MEGA X: molecular evolutionary genetics analysis across computing platforms. Mol Biol Evol 35:1547–1549. 10.1093/molbev/msy096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Pruesse E, Peplies J, Glöckner FO. 2012. SINA: accurate high-throughput multiple sequence alignment of ribosomal RNA genes. Bioinformatics 28:1823–1829. 10.1093/bioinformatics/bts252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Mukherjee S, Stamatis D, Bertsch J, Ovchinnikova G, Katta HY, Mojica A, Chen I-MA, Kyrpides NC, Reddy TBK. 2019. Genomes OnLine database (GOLD) v.7: updates and new features. Nucleic Acids Res 47:D649–D659. 10.1093/nar/gky977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Letunic I, Bork P. 2016. Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kalyuzhnaya MG, Hristova KR, Lidstrom ME, Chistoserdova L. 2008. Characterization of a novel methanol dehydrogenase in representatives of Burkholderiales: implications for environmental detection of methylotrophy and evidence for convergent evolution. J Bacteriol 190:3817–3823. 10.1128/JB.00180-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Download AEM.00947-21-s0001.pdf, PDF file, 0.07 MB (75.7KB, pdf)
Table S1. Download AEM.00947-21-s0002.xlsx, XLSX file, 0.02 MB (19.9KB, xlsx)
Table S2. Download AEM.00947-21-s0003.xlsx, XLSX file, 0.01 MB (12.7KB, xlsx)
Table S3. Download AEM.00947-21-s0004.xlsx, XLSX file, 0.02 MB (22KB, xlsx)
Table S4. Download AEM.00947-21-s0005.xlsx, XLSX file, 0.01 MB (11.9KB, xlsx)
Table S5. Download AEM.00947-21-s0006.xlsx, XLSX file, 0.02 MB (20.6KB, xlsx)
Table S6. Download AEM.00947-21-s0007.xlsx, XLSX file, 0.02 MB (21.1KB, xlsx)
Table S7. Download AEM.00947-21-s0008.xlsx, XLSX file, 0.01 MB (9.7KB, xlsx)
Data Availability Statement
Metagenomic raw reads for Zodletone Spring sediment are available under SRA accession no. SRX9813571. The Zodletone Spring whole-genome shotgun project was submitted to GenBank under Bioproject ID PRJNA690107 and Biosample ID SAMN17269717. The individual assembled Acidobacteria MAGs analyzed in this study have been deposited in DDBJ/ENA/GenBank under accession no. JAFGAO000000000, JAFGSS000000000, JAFGIY000000000, and JAFGTD000000000.