

ABSTRACT
Acid mine drainage (AMD) is a global problem in which iron sulfide minerals oxidize and generate acidic, metal-rich water. Bioremediation relies on understanding how microbial communities inhabiting an AMD site contribute to biogeochemical cycling. A number of studies have reported community composition in AMD sites from 16S rRNA gene amplicons, but it remains difficult to link taxa to function, especially in the absence of closely related cultured species or those with published genomes. Unfortunately, there is a paucity of genomes and cultured taxa from AMD environments. Here, we report 29 novel metagenome-assembled genomes from Cabin Branch, an AMD site in the Daniel Boone National Forest, Kentucky, USA. The genomes span 11 bacterial phyla and one archaeal phylum and include taxa that contribute to carbon, nitrogen, sulfur, and iron cycling. These data reveal overlooked taxa that contribute to carbon fixation in AMD sites as well as uncharacterized Fe(II)-oxidizing bacteria. These data provide additional context for 16S rRNA gene studies, add to our understanding of the taxa involved in biogeochemical cycling in AMD environments, and can inform bioremediation strategies.
IMPORTANCE Bioremediating acid mine drainage requires understanding how microbial communities influence geochemical cycling of iron and sulfur and biologically important elements such as carbon and nitrogen. Research in this area has provided an abundance of 16S rRNA gene amplicon data. However, linking these data to metabolisms is difficult because many AMD taxa are uncultured or lack published genomes. Here, we present metagenome-assembled genomes from 29 novel AMD taxa and detail their metabolic potential. These data provide information on AMD taxa that could be important for bioremediation strategies, including taxa that are involved in cycling iron, sulfur, carbon, and nitrogen.
KEYWORDS: AMD, metagenome-assembled genome, biogeochemical cycling, bioremediation, MAG, acid mine drainage, iron, metagenome
INTRODUCTION
Acid mine drainage (AMD) is a global environmental problem. Oxidative processes, both biotic and abiotic, release protons and reduced metals from sulfide minerals, resulting in highly acidic and toxic conditions that degrade environmental quality. Due to the toxicity and environmental impact of AMD, bioremediation strategies have become of interest. Research in AMD environments often seeks to understand the biogeochemical cycling occurring in the environment and aims to inform and improve the bioremediation of these sites (1–4). Biotic oxidation of reduced metal sulfides contributes to the formation of AMD, while sulfate and iron reduction can both decrease the concentration of soluble metals and increase pH (5–11). However, the metabolic potential of many taxa in AMD environments remains uncharacterized because these taxa are not closely related to cultured taxa or those with published genomes.
AMD environments are characterized by redox gradients including contrasting concentration of oxygen and reduced metals. They can also vary in heavy metal content and pH. However, 16S rRNA gene surveys reveal that many of the same species inhabit AMD sites across the globe. For example, Ferrovum spp. are found in the Appalachian Coal Belt (12–14), the Iberian Pyrite Belt (15, 16), Wales (17, 18), and southeast and southwest China (19, 20). Archaea within the order Thermoplasmatales are commonly found in AMD sites worldwide, especially those with low pH (9, 21–23). In many instances, 16S rRNA sequences isolated from AMD are only distantly related to cultured Thermoplasmatales. Taxa in the Thermoplasmatales perform diverse metabolic functions, including Fe(II) oxidation (24), obligate heterotrophy (25, 26), and sulfur respiration (27). Therefore, it is difficult to infer their metabolic potential (22). Similarly, taxa within newly discovered phyla such as the Elusimicrobiota (formerly Termite Group 1) and Eremiobacteriota (formerly the WPS-2) inhabit AMD sites (28, 29), but these groups have few if any cultured taxa. Given the widespread distribution of these lineages, these taxa may play an important role in biogeochemical cycling in AMD environments, but without closely related cultured relatives or well-annotated genomes, it is not possible to elucidate their role or potential use in bioremediation strategies. Even in well-studied AMD groups such as the Gammaproteobacteria, multiple closely related taxa may occur in AMD sites but may play different roles from their close relatives. For example, multiple Ferrovum taxa differ in their ability to fix nitrogen (30–33). This intragenus metabolic diversity complicates our ability to understand biogeochemical cycling in AMD environments.
Obtaining a species in pure culture has long been considered the gold standard for determining the biogeochemical role that a taxon may play in the environment. However, characterized isolates from AMD environments are rare. Culturing taxa is inherently time-consuming, especially those that require micro-oxic conditions, and can be difficult because species often require cooccurring taxa. For example, in culture, Ferrovum spp. cooccur with heterotrophic organisms that remove pyruvic acid and other organic material (34). Metagenomic sequencing has proven to be a valuable tool for guiding isolation of common AMD microbes through the recovery of near-complete genomes. Tyson et al. used a metagenome-directed approach to isolate a Leptospirillum ferrooxidans sp. capable of nitrogen fixation (35). Metagenomic approaches also provide valuable information about community structure and diversity. Thus, ’omics-based approaches can complement pure culture studies, provide valuable insight to biogeochemical cycling in AMD environments, and inform bioremediation strategies in the absence of fully characterized isolates.
Here, we present 29 novel, high-quality, metagenome-assembled genomes (MAGs) from Cabin Branch, an acid mine drainage site in the Daniel Boone National Forest, Kentucky, USA, which further elucidate potential biogeochemical cycling in AMD ecosystems. Several of the MAGs encode Cyc2 like cytochrome c involved in Fe(II) oxidation (36–38) and thus represent potential previously uncharacterized Fe(II)-oxidizing bacteria. For example, MAGs within the recently discovered phyla Elusimicrobia and Eremiobacterota encode Cyc2. These taxa have not previously been recognized as Fe(II) oxidizers, but the recovery of Cyc2 in these MAGs further expands our knowledge of the taxonomic diversity of Fe(II) oxidation. Furthermore, MAGs encoding Cyc2 were recovered across sample sites that range in dissolved oxygen and Fe(II) concentration from 77 to 401 μmol/liter and 11 to 882 μmol/liter, respectively, and are present at relative abundances that suggest key roles in community function. Collectively, the data highlight the metabolic potential of a number of microbes commonly recovered in 16S rRNA-based studies of AMD. These genomes will provide additional context for gene amplicon studies in AMD environments, aid in culturing these taxa in the future, and could inform AMD bioremediation strategies.
RESULTS
We sequenced metagenomes from three locations in the outflow channel of Cabin Branch, an acid mine drainage site in the Daniel Boone National Forest in Kentucky. Between 74 and 101 million reads were generated per sample. Of these, between 78% and 81% retained both forward and reverse reads after trimming and quality control. These were assembled into 50,449 to 118,786 contigs with median contig lengths of 1,979 to 3,853 bp. Between 53.9% and 56.0% of reads were successfully mapped back to the assembled contigs. The emergence sample resulted in 38 bins, the outflow, 32, Rose Pool, 66, and the combined assembly, 120. Genome assembly, mapping, and binning statistics are available in File S4 in the supplemental material.
We recovered 256 bins from the metagenomes—38 from the emergence, 32 from the limestone-lined channel, 66 from Rose Pool, and 120 from the coassembly. Of these, 56 were >70% complete with <3% contamination. These bins belonged to 32 unique taxa (Table 1). Here, we present 29 novel, high-quality, metagenome-assembled genomes (MAGs), 4 from the emergence, 7 from the limestone-lined channel, 9 from Rose Pool, and 9 from the coassembled data. The MAGs ranged in relative abundance from ∼4.3% to ∼0.17% (Fig. 1 and 2). The Ferrovum MAGs (MAG 23 and MAG24) were described in reference 33, and MAG 7 is closely related to a previously described genome (39, 40).
TABLE 1.
| Phylum | MAG | Accession no. | Taxonomy | Completeness (%) | Contamination (%) | Strain heterogeneity | Size (Mbp) | No. of contigs | GC content (%) | No. of protein coding sequences | Carbon fixation | Sulfur cycling | Nitrogen fixation | Fe(II) oxidation |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Thermoplasmatota | MAG 1 | SAMN14771053 | Archaea; Thermoplasmatota; Thermoplasmata; UBA184; UBA184 | 84.36 | 1.6 | 0 | 1.418903 | 217 | 0.67 | 1,457 | No | No | No | No |
| Actinobacteria | MAG 2 | SAMN14771054 | Bacteria; Actinobacteriota; Acidimicrobiia; Acidimicrobiales | 84.62 | 2.14 | 0 | 2.526595 | 340 | 0.45 | 2,362 | No | No | No | No |
| MAG 3 | SAMN14771055 | Bacteria; Actinobacteriota; Acidimicrobiia; Acidimicrobiales; Acidimicrobiaceae | 90.88 | 0.85 | 0 | 2.378105 | 192 | 0.52928 | 2,137 | Yes | No | No | No | |
| MAG 4 | SAMN14771056 | Bacteria; Actinobacteriota; Acidimicrobiia; Acidimicrobiales; Acidimicrobiaceae | 99.15 | 1.28 | 0 | 2.817353 | 95 | 0.55046 | 2,584 | Yes | No | No | No | |
| MAG 5 | SAMN14771057 | Bacteria; Actinobacteriota; Acidimicrobiia; Acidimicrobiales; Acidimicrobiaceae | 85.13 | 1.36 | 33.33 | 2.138544 | 268 | 0.53278 | 1,873 | Yes | No | No | No | |
| MAG 6 | SAMN14771058 | Bacteria; Actinobacteriota; Acidimicrobiia; Acidimicrobiales; Acidimicrobiaceae | 94.02 | 2.14 | 0 | 2.323875 | 280 | 0.50685 | 2,129 | Yes | No | No | No | |
| MAG 7 | Bacteria; Actinobacteriota; Acidimicrobiia; Acidimicrobiales; Acidimicrobiaceae; Acidithrix; Acidithrix ferrooxidans | 70.36 | 2.14 | 33.33 | 2.417212 | 336 | 0.47572 | 2,062 | NA | NA | NA | NA | ||
| MAG 8 | SAMN14771059 | Bacteria; Actinobacteriota; Acidimicrobiia; Acidimicrobiales; RAAP-2; RAAP-2 | 96.3 | 2.99 | 0 | 2.796923 | 246 | 0.59901 | 2,749 | No | No | No | No | |
| MAG 9 | SAMN14771060 | Bacteria; Actinobacteriota; Acidimicrobiia; Acidimicrobiales; RAAP-2; RAAP-2 | 93.08 | 2.23 | 0 | 2.2056 | 175 | 0.6155 | 2,197 | Yes | No | No | No | |
| MAG 10 | SAMN14771061 | Bacteria; Actinobacteriota; Acidimicrobiia; Acidimicrobiales; RAAP-2; RAAP-2; | 94.87 | 2.14 | 0 | 1.873312 | 163 | 0.63812 | 1,856 | No | No | No | No | |
| Bacteroidota | MAG 11 | SAMN14771062 | Bacteria; Bacteroidota; Bacteroidia; AKYH767; Palsa-948 | 84.22 | 2 | 40 | 2.420414 | 335 | 0.46586 | 2,125 | No | No | No | No |
| Dormibacterota | MAG 12 | SAMN14771063 | Bacteria; Dormibacterota; Dormibacteria; UBA8260; Bog-877 | 95.37 | 0.93 | 0 | 2.296128 | 226 | 0.68404 | 2,207 | No | No | No | No |
| Elusimicrobiota | MAG 13 | SAMN14771064 | Bacteria; Elusimicrobiota; Elusimicrobia; UBA1565; UBA9628; GWA2-66-18 | 86.06 | 1.5 | 0 | 3.360875 | 299 | 0.6663 | 3,200 | No | No | Yes | Yes |
| Eremiobacterota | MAG 14 | SAMN14771065 | Bacteria; Eremiobacterota; Eremiobacteria; UBP12; UBA5184 | 71.43 | 2.78 | 100 | 1.980657 | 269 | 0.62541 | 1,976 | No | No | No | No |
| MAG 15 | SAMN14771066 | Bacteria; Eremiobacterota; Eremiobacteria; UBP12; UBA5184 | 79.18 | 1.85 | 50 | 2.083017 | 215 | 0.62452 | 2,102 | No | No | No | Yes | |
| Firmicutes | MAG 16 | SAMN14771067 | Bacteria; Firmicutes_K; Alicyclobacillia; Alicyclobacillales; Acidibacillaceae; | 92.65 | 1.57 | 50 | 2.540112 | 156 | 0.45602 | 2,410 | No | No | No | No |
| MAG 17 | SAMN14771068 | Bacteria; Patescibacteria; Paceibacteria; UBA9983_A; UBA2163; C7867-001 | 71.84 | 0 | 0 | 2.138544 | 268 | 0.53278 | 1,873 | No | No | No | No | |
| Planctomycetota | MAG 18 | SAMN14771069 | Bacteria; Planctomycetota; Phycisphaerae | 73.45 | 0 | 0 | 2.799192 | 419 | 0.56703 | 2,424 | No | No | No | No |
| MAG 19 | SAMN14771070 | Bacteria; Planctomycetota; Phycisphaerae; UBA1161 | 96.32 | 0 | 0 | 3.578576 | 255 | 0.56229 | 2,916 | No | No | No | No | |
| Proteobacteria | MAG 20 | SAMN14771071 | Bacteria; Proteobacteria; Alphaproteobacteria; Acetobacterales; Acetobacteraceae; Acidocella | 92.45 | 2.78 | 57.14 | 2.678786 | 222 | 0.63102 | 2,577 | Yes | No | Yes | Yes |
| MAG 21 | SAMN14771072 | Bacteria; Proteobacteria; Gammaproteobacteria; Burkholderiales | 91.83 | 2.01 | 20 | 2.555995 | 242 | 0.57887 | 2,573 | Yes | Yes | Yes | Yes | |
| MAG 22 | SAMN14771073 | Bacteria; Proteobacteria; Gammaproteobacteria; Burkholderiales | 99.14 | 0 | 0 | 3.695939 | 108 | 0.64413 | 3,442 | Yes | Yes | No | Yes | |
| MAG 23 | Bacteria; Proteobacteria; Gammaproteobacteria; Burkholderiales; Ferrovaceae | 95.76 | 0.5 | 0 | 2.374093 | 102 | 0.55802 | 2,182 | NA | NA | NA | NA | ||
| MAG 24 | Bacteria; Proteobacteria; Gammaproteobacteria; Burkholderiales; Ferrovaceae; Ferrovum | 86.7 | 0.63 | 100 | 1.845635 | 96 | 0.5385 | 1,727 | NA | NA | NA | NA | ||
| MAG 25 | SAMN14771074 | Bacteria; Proteobacteria; Gammaproteobacteria; Burkholderiales; Gallionellaceae; Gallionella | 96.67 | 1.43 | 0 | 2.478987 | 157 | 0.5653 | 2,425 | Yes | No | No | Yes | |
| MAG 26 | SAMN14771075 | Bacteria; Proteobacteria; Gammaproteobacteria; Burkholderiales; Gallionellaceae; Gallionella | 94.44 | 2.41 | 83.33 | 2.296128 | 226 | 0.68404 | 2,207 | Yes | No | No | Yes | |
| MAG 27 | SAMN14771076 | Bacteria; Proteobacteria; Gammaproteobacteria; Pseudomonadales | 91.16 | 1.24 | 14.29 | 2.572997 | 319 | 0.51007 | 2,364 | No | No | Yes | Yes | |
| MAG 28 | SAMN14771077 | Bacteria; Proteobacteria; Gammaproteobacteria; Steroidobacterales; Steroidobacteraceae; | 85.78 | 2.5 | 18.18 | 2.564032 | 320 | 0.65702 | 2,382 | Yes | No | No | Yes | |
| MAG 29 | SAMN14771078 | Bacteria; Proteobacteria; Gammaproteobacteria; UBA1113; UBA1113 | 93.6 | 1.36 | 33.33 | 2.699483 | 337 | 0.39968 | 2,364 | No | No | No | No | |
| Spirochaetota | MAG 30 | SAMN14771079 | Bacteria; Spirochaetota; Spirochaetia; Spirochaetales | 95.4 | 0.4 | 0 | 2.61822 | 142 | 0.50392 | 2,361 | No | No | No | No |
| MAG 31 | SAMN14771080 | Bacteria; Spirochaetota; Spirochaetia; Spirochaetales | 77.93 | 1.6 | 50 | 1.946496 | 327 | 0.53893 | 1,830 | No | No | No | No | |
| Verrucomicrobiota | MAG 32 | SAMN14771081 | Bacteria; Verrucomicrobiota_A; Chlamydiia; Parachlamydiales; Ga0074140 | 87.1 | 1.35 | 50 | 1.881114 | 247 | 0.39274 | 1,615 | No | No | No | No |
Taxa in italic text were not analyzed in this work because they were either closely related to cultured taxa (e.g., MAG 7) or presented previously (MAGs 24 and 24 [33]).
NA, not analyzed.
FIG 1.
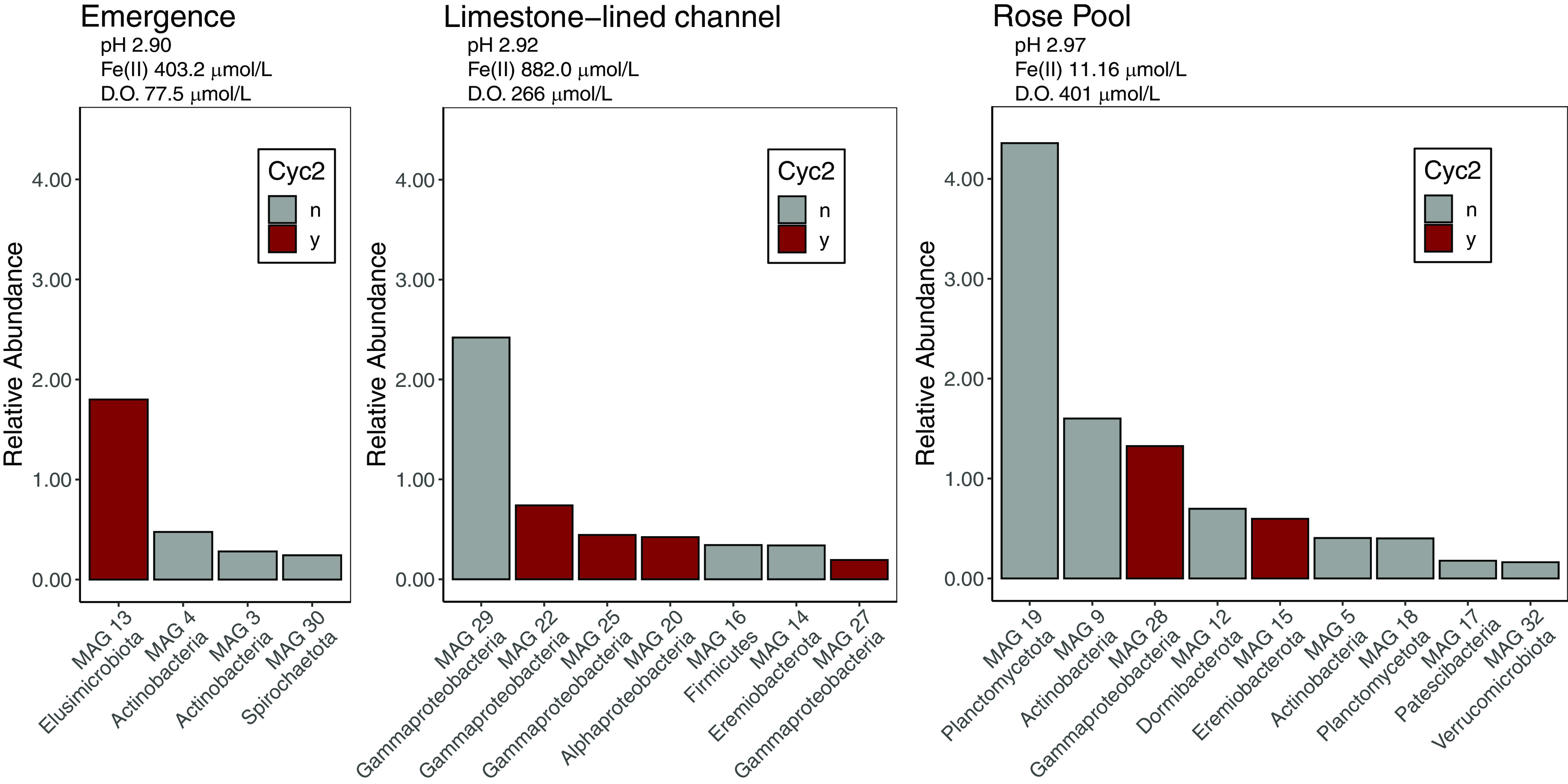
Rank abundance curve of the relative abundance of MAGs recovered from the emergence, the limestone-lined channel, and Rose Pool. pH, Fe(II), and D.O. were measured at the time of sample collection and are reported in reference 33. Red bars indicate MAGs that encode Cyc2. D.O., dissolved oxygen.
FIG 2.

Rank abundance curve of the relative abundance of MAGs recovered from the coassembled data in each site—the emergence, the limestone-lined channel, and Rose Pool. Asterisks denote MAGs that encode Cyc2.
Below, we examine functions that are most relevant to AMD ecosystems, including aerobic respiration, carbon fixation, nitrogen cycling, and biogeochemical cycling of sulfur and iron in each MAG by phylogenetic group. For sulfur cycling, we focus on dissimilatory sulfate reduction and sulfur oxidation by examining the presence or absence of dsr and sox genes. The metabolic potential for ferrous iron oxidation was based on the presence of Cyc2-like genes that may be involved in this process (36, 37). None of the MAGs contain complete genomes, and a gene that is absent in the MAG may be present in the taxon. Therefore, these data indicate the genes present in, not absent from, a taxon. A summary of these taxa is available in File S5. More complete genome descriptions are available in the supplemental material, and the METABOLIC and DRAM results are provided in File S6 and Data Set S1.
Bacteria.
(i) Actinobacteriota (Actinobacteria). We retrieved nine actinobacterial MAGs, all of which belong to the order Acidimicrobiales (Fig. 3). MAGs 3 to 7 belonged to the Acidimicrobiaceae, but MAGs 3 to 6 were unidentified below this level. MAGs 8 to 10 were affiliated with the family and genus RAAP-2 but were unidentified at the species level. Five taxa (MAGs 3 to 6 and 9) encode genes for carbon fixation. Actinobacteria MAGs were recovered from the emergence and Rose Pool (Fig. 1) as well as the coassembly (Fig. 2).
FIG 3.

Concatenated single-copy marker gene tree constructed using genes from reference 46. The tree contains 83 taxa. MAGs from Cabin Branch are indicated in bold. Shapes indicate the metabolic potential of the MAGs. Carbon fixation is represented by blue circles. Dissimilatory sulfur oxidation is represented by yellow triangles with points up and reduction, by triangles with points down, nitrogen fixation is represented by green squares, and Fe(II) oxidation is represented by red pentagons. The concatenated single-copy marker gene tree is provided in Newick format in File S1.
(a) MAG 2. MAG 2 belonged to the order Acidimicrobiales (taxonomy was not resolved below this level) (Fig. 1). The only bin from this taxon was retrieved from the coassembly. It contained 2.53 Mbp in 340 contigs with 2,362 protein-coding genes. The MAG was 84.6% complete with 2.14% contamination, and the GC content was 45% (Table 1). It encodes many of the genes necessary for oxidative phosphorylation with oxygen as a terminal electron acceptor using a caa3-type cytochrome c oxidase. It also contains the genes necessary for conversion of nitrate to nitrite and the reverse reaction (File S6 and Data Set S1).
(b) MAG 3. The only bin from MAG 3 was retrieved from the coassembly. It contained 2.38 Mbp in 192 contigs with 2,137 protein-coding genes. It was 90.9% complete with 0.85% contamination. The GC content was 53% (Table 1). The genome encodes homologs of the genes necessary for carbon fixation, including those for form I RuBisCO and for the Wood Ljungdahl pathway. It also encodes many of the genes necessary for oxidative phosphorylation with oxygen as a terminal electron acceptor using caa3- and cbb3-type cytochrome c oxidases (Data Set S1 and File S6).
(c) MAG 4. MAG 4 was retrieved from metagenomes from the emergence, Rose Pool, and the coassembly. The most complete bin from this taxon was retrieved from the emergence. It contained 2.82 Mbp in 95 contigs with 2,584 protein-coding genes. The MAG was 99.2% complete with 1.28% contamination, and the GC content was 55% (Table 1). MAG 4 encodes the genes necessary for oxidative phosphorylation with a caa3- or cbb3-type cytochrome c oxidase and carbon fixation with form I RuBisCO. It also contains homologs of the genes necessary for mercury reduction (File S6 and Data Set S1).
(d) MAG 5. The only bin from MAG 5 was retrieved from Rose Pool. It contained 2.14 Mbp in 268 contigs with 1,873 protein-coding genes. The MAG was 85.1% complete with 1.36% contamination, and the GC content was 53% (Table 1). MAG 5 encodes homologs of the genes necessary for oxidative phosphorylation using either a caa3- or a bd-type cytochrome c oxidation as a terminal electron acceptor. It also encodes many of those necessary for carbon fixation using form I RuBisCO and via the 3 hydroxypropionate cycle (File S6 and Data Set S1).
(e) MAG 6. Bins of MAG 6 were retrieved from Rose Pool and the coassembly. The most complete bin from this taxon was retrieved from the coassembly. It contained 2.32 Mbp in 280 contigs with 2,129 protein-coding genes. The MAG was 94% complete with 2.14% contamination, and the GC content was 51% (Table 1). MAG 6 encodes homologs of many of the genes necessary for oxidative phosphorylation using a caa3- or cbb3-type cytochrome c oxidase as a terminal electron acceptor (File S6 and Data Set S1).
(f) MAG 7. MAG 7 was closely related to Acidithrix ferrooxidans (96.96% average nucleotide identity [ANI]). This taxon was described elsewhere (1, 2), and therefore we do not discuss it here.
(g) MAG 8. Bins of MAG 8 were retrieved from Rose Pool and the coassembly. The most complete bin from MAG 8 was retrieved from the coassembly. It contained 2.80 Mbp in 246 contigs with 2,749 protein-coding genes. The MAG was 96.3% complete with 2.99% contamination, and the GC content was 60% (Table 1). MAG 8 encodes many of the genes necessary for oxidative phosphorylation using caa3-, cbb3-, and bd-type cytochrome c oxidases (File S6 and Data Set S1).
(h) MAG 9. The only bin of MAG 9 was retrieved from Rose Pool. It contained 2.20 Mbp in 175 contigs with 2,197 protein-coding genes. The MAG was 93.1% complete with 2.23% contamination, and the GC content was 62% (Table 1). It contains homologs of some of the genes necessary for oxidative phosphorylation using cbb3- and bd type cytochrome c oxidases (File S6 and Data Set S1).
(i) MAG 10. The only bin of MAG 10 was retrieved from the coassembly. It contained 1.87 Mbp in 163 contigs with 1,856 protein-coding genes. The MAG was 94.9% complete with 2.14% contamination, and the GC content was 64% (Table 1). MAG 10 encodes homologs for some of the genes necessary for oxidative phosphorylation using caa3- and cbb3-type cytochrome c oxidases (Data Set S1 and File S6).
(ii) Bacteroidota.
We retrieved a single taxon from the phylum Bacterioidota (Bacteroidetes; MAG 11). Bins from MAG 11 were retrieved from the emergence and coassembly. It was affiliated with the class Bacteroidia, order AKH767, and family Palsa-948 and was unclassified below this level. The most closely related taxon was retrieved from thawing permafrost (41). The best bin from this taxon was retrieved from the coassembly. The most complete bin contained 2.42 Mbp in 335 contigs with 2,125 protein-coding genes. The MAG was 84.2% complete with 2.0% contamination, and the GC content was 47% (Table 1). This taxon encodes some of the genes necessary for oxidative phosphorylation but does not encode any genes for terminal oxidases. It also encodes the genes necessary for arsenate reduction (Data Set S1 and File S6).
(iii) Dormibacteraeota.
We retrieved one taxon from the phylum Dormibacteraeota (MAG 12). Bins from MAG 12 were retrieved from Rose Pool and coassembly. This taxon was affiliated with the class Dormibacteria, the order UBA8260, and the family Bog-877 and is most closely related to a taxon from thawing permafrost (41). The most complete bin from this taxon was retrieved from Rose Pool. It contained 2.30 Mbp in 226 contigs with 2,207 protein-coding genes. The MAG was 95.4% complete with 0.93% contamination, and the GC content was 68% (Table 1). This taxon encodes some of the genes necessary for oxidative phosphorylation using a caa3-type cytochrome c oxidase as a terminal electron acceptor. It did not encode genes for carbon fixation, N2 fixation, denitrification, dissimilatory sulfur cycling, or Fe(II) oxidation (Data Set S1 and File S6).
(iv) Elusimicrobiota.
We retrieved a single taxon, MAG 13, from the phylum Elusimicrobiota (formerly Termite Group 1). Bins of MAG 13 were retrieved from the emergence and coassembly. In the emergence, it was present with a relative abundance of ∼1.8%. It was affiliated with the class Elusimicrobia, order UBA1565, family UBA9628, and genus GWA2-66-18. This taxon is most closely related to one from an aquifer system (42). The most complete bin from this taxon was retrieved from the emergence. It contained 3.36 Mbp in 299 contigs with 3,200 protein-coding genes. It was 86.1% complete with 1.5% contamination. The GC content was 67% (Table 1). MAG 13 encodes many of the genes necessary for oxidative phosphorylation with caa3-, cbb3-, and bd-type cytochrome c oxidases. It encodes the genes necessary for N2 fixation and nitric oxide reduction (Table 1). It also encodes the genes necessary for arsenate reduction (Data Set S1 and File S6) and encodes a Cyc2 like cytochrome that it may use for Fe(II) oxidation (File S2).
(v) Eremiobacteriota.
We retrieved two taxa, MAGs 14 and 15, within the phylum Eremiobacteriota (formerly WPS-2). Both taxa were affiliated with the class Eremiobacteria, order UPB12, and family UBA5184. Neither bin contained genes for carbon fixation, N2 fixation, denitrification, dissimilatory sulfur cycling, or Fe(II) oxidation. Both taxa appear to be capable of heterotrophic metabolism, including aromatics degradation and acetogenesis. MAG 14 was recovered from the limestone-lined channel while MAG 15 was present in Rose Pool (Fig. 1).
(a) MAG 14. The only bin from MAG 14 was retrieved from the limestone-lined channel. It contained 1.98 Mbp in 269 contigs with 1,976 protein-coding genes. The MAG was 71.4% complete with 2.78% contamination, and the GC content was 63% (Table 1). MAG 14 encodes many of the genes necessary for oxidative phosphorylation using caa3- and bd-type cytochrome c oxidases (Data Set S1 and File S6).
(b) MAG 15. The only bin from MAG 15 was retrieved from Rose Pool. It contained 2.08 Mbp in 215 contigs with 2,102 protein-coding genes. The MAG was 79.2% complete with 1.85% contamination, and the GC content was 62% (Table 1). MAG 15 encodes many of the genes necessary for oxidative phosphorylation using a bd-type cytochrome c oxidase (Data Set S1 and File S6). It also appears to encode a Cyc2-like cytochrome that it may use for Fe(II) oxidation (File S2).
(vi) Firmicutes.
We retrieved a single taxon from the phylum Firmicutes (MAG 16) from the limestone-lined channel and the coassembly (Fig. 1). This taxon was affiliated with the class Alicyclobacillia, the order Alicyclobacillales, and the family Acidibacillaceae. This taxon was most closely related to Acidobacillus ferrooxidans, a Fe(II) and sulfide mineral oxidizing species isolated from an AMD environment (Fig. 1) (43). The most complete bin from this taxon was retrieved from the limestone-lined channel. It contained 2.54 Mbp in 156 contigs with 2,410 protein-coding genes. The MAG was 92.7% complete with 1.57% contamination, and the GC content was 46% (Table 1). The genome encodes some of the genes necessary for oxidative phosphorylation using bo-, bd- and caa3-type cytochrome c oxidases. It also encodes the genes necessary for denitrification (Data Set S1 and File S6). Unlike its closest relative, it does not encode the genes necessary for Fe(II) or sulfide mineral oxidation.
(vii) Patescibacteria.
We retrieved a single taxon, MAG 17, from the phylum Patescibacteria (formerly Candidate Phylum Radiation) from Rose Pool (Fig. 1). This taxon was affiliated with the class Paceibacteria, the order UBA9983_A, the family UBA2163, and the genus C7867-001. It contained 2.13 Mbp in 268 contigs with 1,873 protein-coding genes. It was 71.84% complete with 0% contamination. The GC content was 53% (Table 1).
(viii) Planctomycetota (Planctomycetes). We recovered two taxa from the phylum Planctomycetota (Planctomycetes), MAGs 18 and 19. Both taxa were affiliated with the class Phycisphaerae. One, MAG 18, was unclassified below this level. The other, MAG 19, was affiliated with the order UBA1161. Both MAG 18 and 19 were recovered from Rose Pool, where MAG 19 was more abundant (4.3% and 0.4%, respectively, Fig. 1).
(a) MAG 18. The only bin from this taxon was retrieved from Rose Pool. It contained 2.80 Mbp in 419 contigs with 2,424 protein-coding genes. It was 73.5% complete with 0% contamination. The GC content was 57% (Table 1). MAG 18 encodes some of the genes necessary for oxidative phosphorylation using a caa3- or bd-type cytochrome c oxidase (Data Set S1 and File S6).
(b) MAG 19. Bins from this taxon were recovered from Rose Pool and the coassembly. The most complete bin from this taxon was retrieved from Rose Pool. It contained 3.58 Mbp in 255 contigs with 2,916 protein-coding genes. The MAG was 96.3% complete with 0% contamination, and the GC content was 56% (Table 1). The MAG encodes some of the genes necessary for oxidative phosphorylation using a caa3- or bd-type cytochrome c oxidase (Data Set S1 and File S6).
(ix) Proteobacteria.
We recovered 10 taxa from the Proteobacteria. One, MAG 20, is a member of the Alphaproteobacteria. Nine are from the gammaproteobacterial order Burkholderiales. Two of these were described previously and were not analyzed here (33). Three encode genes for N2 fixation (MAGs 20, 21, and 27), six encode genes for carbon fixation, (MAGs 20 to 22, 25, 26, and 28), seven encode genes for Fe(II) oxidation (MAGs 20 to 22 and 25 to 28), and three encode genes for partial sulfate reduction (MAGs 22, 25, and 26). Gammaproteobacterial MAGs were recovered from the limestone-lined channel, Rose Pool, and the coassembly (Fig. 1 and 2). MAG 21 from the coassembly was particularly abundant at the emergence (3.3%), while MAG 29 was abundant in the limestone-lined channel (2.4%).
(a) MAG 20. MAG 20 is from the alphaproteobacterial order Acetobacterales and is affiliated with the family Acetobacteraceae and the genus Acidocella. The only bin from this taxon was retrieved from the limestone-lined channel. It contained 2.67 Mbp in 222 contigs with 2,577 protein-coding genes. The MAG was 92.5% complete with 2.78% contamination, and the GC content was 63% (Table 1). MAG 20 encodes the genes necessary for oxidative phosphorylation using caa3- and bd-type cytochrome c oxidases. It encodes homologs of genes necessary for N2 fixation. It also encodes the genes necessary for mercury reduction (Data Set S1 and File S6). It may also encode a Cyc2-like cytochrome that it may use for Fe(II) oxidation (File S2).
(b) MAG 21. MAG 21 is from the gammaproteobacterial order Burkholderiales but is unclassified below this level. Bins from this taxon were retrieved from the emergence and the coassembly. The best bin from this taxon was retrieved from the coassembly. It contained 2.55 Mbp in 242 contigs with 2,573 protein-coding genes. The MAG was 91.8% complete with 2.01% contamination, and the GC content was 57% (Table 1). MAG 21 encodes the genes necessary for oxidative phosphorylation using a bo- or bd-type cytochrome c oxidase. This taxon encodes homologs of genes for a form II RuBisCO for carbon fixation and encodes the genes necessary for N2 fixation. The genome encoded homologs of dsrA, dsrB, and aprA, indicating that it may be capable of dissimilatory sulfate reduction, at least from APS (adenosine 5′-phosphosulfate) to sulfide. It also encodes the genes necessary for the breakdown of halogenated compounds and arsenate reduction (Data Set S1 and File S6). It may also encode a Cyc2-like cytochrome that it may use for Fe(II) oxidation (File S2).
(c) MAG 22. Bins from this taxon were retrieved from the emergence, limestone-lined channel, and coassembly. The best bin from this taxon was retrieved from the limestone-lined channel. It contained 3.70 Mbp in 108 contigs with 3,442 protein-coding genes. The MAG was 99.1% complete with 0% contamination, and the GC content was 64% (Table 1). MAG 22 encodes homologs of the genes necessary for oxidative phosphorylation using caa3- and bd-type cytochrome c oxidases. The MAG contains genes necessary for carbon fixation with a form I RuBisCO. It also encodes the genes necessary for the oxidation of thiosulfate to sulfate (soxB, soxY, and soxC) and for mercury reduction (File S5). MAG 22 also appears to encode a Cyc2-like cytochrome that it may use for Fe(II) oxidation (Data Set S1 and File S6).
(d) MAGs 23 and 24. These taxa were members of the genus Ferrovum and were described elsewhere (33). Therefore, we do not discuss them here.
(e) MAG 25. Bins from this taxon were retrieved from the emergence, limestone-lined channel, and coassembly. The best bin from this taxon was retrieved from the limestone-lined channel. It contained 2.48 Mbp in 157 contigs with 2,425 protein-coding genes. The MAG was 96.7% complete with 1.43% contamination, and the GC content was 57% (Table 1). MAG 25 encodes the genes necessary for oxidative phosphorylation but does not encode any terminal oxidases. It also contains the genes necessary for carbon fixation, including type I and II RuBisCOs (Data Set S1 and File S6). It appears to encode a Cyc2 like cytochrome that it may use for Fe(II) oxidation (File S2).
(f) MAG 26. Bins from this taxon were retrieved from the emergence and coassembly. The most complete bin from this taxon was retrieved from the coassembly. It contained 2.30 Mbp in 226 contigs with 2,207 protein-coding genes. The MAG was 94.4% complete with 2.41% contamination, and the GC content was 68% (Table 1). It encodes the genes necessary for oxidative phosphorylation using cbb3- and bd-type cytochrome c oxidases. It also contains the genes for type I and II RuBisCO for carbon fixation (Data Set S1 and File S6). It appears to encode a Cyc2-like cytochrome that it may use for Fe(II) oxidation (File S2).
(g) MAG 27. The only bin from this taxon was retrieved from the limestone-lined channel. It contained 2.57 Mbp in 319 contigs with 2,364 protein-coding genes. The MAG was 91.1% complete with 1.24% contamination, and the GC content was 51% (Table 1). MAG 27 encodes the genes for oxidative phosphorylation using a cbb3-type cytochrome c oxidase. It encodes the genes necessary for N2 fixation (Data Set S1 and File S6). It may also encode a Cyc2 like cytochrome that it may use for Fe(II) oxidation (File S2).
(h) MAG 28. Bins from this taxon were retrieved from Rose Pool and the coassembly. The best bin from this taxon was retrieved from Rose Pool. It contained 2.56 Mbp in 320 contigs with 2,382 protein-coding genes. The MAG was 85.8% complete with 2.50% contamination, and the GC content was 66% (Table 1). MAG 28 encodes the genes necessary for oxidative phosphorylation using caa3- and bo-type cytochrome c oxidases (Data Set S1 and File S6). It may also encode a Cyc2-like cytochrome that it may use for Fe(II) oxidation (File S2).
(i) MAG 29. Bins from this taxon were retrieved from the limestone-lined channel and coassembly. The most complete bin from this taxon was retrieved from the limestone-lined channel. It contained 2.70 Mbp in 337 contigs with 2,364 protein-coding genes. The MAG was 93.6% complete with 1.36% contamination, and the GC content was 40% (Table 1). MAG 29 encodes most of the genes necessary for oxidative phosphorylation using bo- and bd-type cytochrome c oxidases (Data Set S1 and File S6). It does not encode the genes necessary for carbon fixation or any other genes of interest.
(x) Spirochaetota (Spirochaetes).
We recovered two taxa from the phylum Spirochaetota (Spirochaetes; MAGs 30 and 31) from Rose Pool (Fig. 1). MAG 30 was recovered from the emergence, and MAG 31 was present in the coassembly with low relative abundance across all sites (Fig. 1 and 2). Both bins were members of the class Spirochaetia and the order Spirochaetales. One encodes the genes necessary for nitrate reduction to ammonia.
(a) MAG 30. Bins from this taxon were retrieved from the emergence, limestone-lined channel, and coassembly. The best bin from this taxon was retrieved from the emergence. It contained 2.62 Mbp in 142 contigs with 2,361 protein-coding genes. The MAG was 95.4% complete with 0.4% contamination, and the GC content was 50% (Table 1). MAG 30 encodes the genes for bo- and bd type cytochrome c oxidases but does not contain other genes associated with oxidative phosphorylation. It encodes the genes necessary for nitrification and denitrification (Data Set S1 and File S6).
(b) MAG 31. The only bin from this taxon was retrieved from the coassembly. It contained 1.95 Mbp in 327 contigs with 1,830 protein-coding genes. The MAG was 77.9% complete with 1.6% contamination, and the GC content was 54% (Table 1). MAG 31 encodes the genes necessary for a caa3-type cytochrome c oxidase but does not contain other genes associated with oxidative phosphorylation (Data Set S1 and File S6).
(xi) Verrucomicrobiota (Verrucomicrobia). We recovered a single taxon from the Verrucomicrobiota (Verrucomicrobia; MAG 32).
Bins from this taxon were retrieved from Rose Pool and the coassembly. The most complete bin from this taxon was retrieved from Rose Pool. It contained 1.88 Mbp in 247 contigs with 1,615 protein-coding genes. The MAG was 87.1% complete with 1.35% contamination, and the GC content was 39% (Table 1). MAG 32 encodes some of the genes necessary for oxidative phosphorylation using bo- or bd-type cytochrome c oxidases (Data Set S1 and File S6).
Archaea.
We recovered a single MAG classified as Archaea (MAG 1). The MAG belonged to the Thermoplasmata and was most closely related to Methanomassiliicoccus spp. (Fig. 1). The most complete bin contained 1.42 Mbp in 217 contigs with 1,457 protein-coding genes. It was 84.3% complete with 1.6% contamination, and the GC content was 67% (Table 1). It did not encode genes associated with carbon fixation, N2 fixation, Fe(II) oxidation, or dissimilatory sulfur cycling. MAG 1 was recovered from the coassembly and was most abundant in the emergence (Fig. 2).
DISCUSSION
Carbon fixation.
Lithotrophic carbon fixation can be a significant source of primary productivity in AMD ecosystems (44). At Cabin Branch, Ferrovum spp. are abundant, ranging from 5 to 33%, and likely contribute to primary productivity (33). Here, we recovered 7 MAGs that encode the genes necessary for carbon fixation. These autotrophs include those that are closely related to known lithoautotrophic organisms, including Gallionella (MAGs 25 and 26) and other Burkholderiales (e.g., MAGs 21 and 22), as well as Actinobacteria (e.g., MAGs 3, 4 and 5). This indicates that primary productivity in AMD sites may be driven, in part, by organisms that have not been considered in the past.
Sulfur cycling.
Sulfur cycling is an important process in AMD ecosystems. Bioremediation may rely on dissimilatory sulfate reduction, especially in constructed wetlands (4). Dissimilatory sulfate reduction combats AMD by generating alkalinity, can lead to the formation of ferrous sulfide minerals in sediments, and decreases the concentration of soluble metals (5–11). Conversely, biological sulfur oxidation generates AMD by oxidizing sulfur in iron sulfide minerals. MAG 21 encodes homologs of dsrA, dsrB, and aprA, indicating that it may be capable of dissimilatory sulfate reduction, at least from APS to sulfide. Therefore, this taxon may play an important role in constructed wetland bioremediation.
AMD occurs naturally when weathering processes expose sulfide mineral-bearing rocks to oxygen-rich water. The result is the oxidation of these sulfide minerals, which produces sulfuric acid (H2SO4) and dissolved metals. Iron-sulfide minerals such as pyrite can also be oxidized by biological sulfur oxidation (45, 46). The oxidation of iron sulfide minerals may occur either at a pyrite vein or in freshly deposited sediments that are exposed to oxygen. We recovered a single MAG, MAG 22, that contains the genes necessary for sulfur oxidation from thiosulfate to sulfate. This taxon is unlikely to cause the oxidation of sulfide-bearing minerals but may play a role in aqueous sulfur cycling in the environment.
Nitrogen fixation.
Common Fe(II)-oxidizing organisms in AMD environments, such as Ferrovum myxofaciens, are capable of nitrogen fixation (30–33, 47) and may provide fixed nitrogen to AMD communities. Four of the MAGs recovered here (MAGs 13, 20, 21, and 27) contain the genes necessary for nitrogen fixation. These organisms may serve as a source of bioavailable nitrogen in AMD ecosystems and, in so doing, increase the productivity of their communities.
Fe(II) oxidation.
Fe(II) oxidation is a key process in AMD environments for bioremediation and as a source of energy to drive primary productivity. Indeed, our previous analyses recovered multiple abundant Ferrovum MAGs whose genomes are consistent with carbon fixation coupled to Fe oxidation (33). Here, we identified 9 MAGs that encoded homologs of the Cyc2 protein involved in Fe(II) oxidation (37) (Files S2 and S5). These MAGs were recovered across sample sites that range in dissolved oxygen and Fe(II) concentration and are present at relative abundances that suggest key roles in community function (Fig. 1 and 2). Seven of the MAGs that encode Cyc2 belong to proteobacterial lineages with other known Fe oxidizers. Additionally, MAGs within the recently discovered phyla Elusimicrobia (MAG 13) and Eremiobacterota (MAG 15) appear to encode Cyc2 (File S2). These taxa have not previously been recognized as Fe(II) oxidizers, but the recovery of Cyc2 in these MAGs further expands our knowledge of the taxonomic diversity of Fe(II) oxidation. MAG 13 was abundant at the emergence, where oxygen and Fe(II) concentrations were 77.5 μmol/liter and 403.2 μmol/liter, respectively. MAG 15 was recovered from the retention pond (Rose Pool), where oxygen and Fe(II) concentrations were 401 μmol/liter and 11.16 μmol/liter, respectively. Rapid iron oxidation rates in low-pH AMD environments have been attributed to Ferrovum spp. (which are abundant in Cabin Branch [13, 37]), and thus, these taxa are of particular interest for bioremediation of Fe(II)-impacted water. However, Ferrovum spp. have proven difficult to culture and characterize. The recovery of multiple Cyc2-containing MAGs across oxygen and Fe(II) gradients highlights the potential for developing alternative target taxa for bioremediation of AMD-impacted waters.
Phylogenetic relatedness and metabolism.
In the absence of characterized isolates, we often rely on phylogenetic relationships between taxa found at AMD sites and their closest cultured relatives to infer their role in biogeochemical cycling (13, 22, 48, 49). This approach leverages the use of 16S rRNA gene amplicon data, which is relatively inexpensive in terms of time and cost, at the expense of the metabolic insights inferred from expensive and time-consuming ’omics approaches or validated by culture-based approaches. This approach—inferring physiology from 16S rRNA gene sequences—can be informative for major metabolic pathways when taxa are closely related to their nearest cultured relative. For example, like Gallionella ferruginea, the metabolic potential of the Gallionella MAGs (MAGs 25 and 26) recovered here is consistent with chemolithoautotrophy fueled by Fe(II) oxidation. These relationships are less robust with increasing phylogenetic distance.
Inferring metabolism from16S rRNA gene sequences becomes more difficult as the number of available genomes from similar environments decreases. For example, AMD environments often host organisms within the archaeal order Thermoplasmatales (9, 22, 23). However, there is a paucity of Thermoplasmatales genomes available from AMD environments. This order also contains taxa with diverse metabolisms, including Fe(II) oxidation (24), obligate heterotrophy (25, 26), and sulfur respiration (27). The lack of genomes and culture representatives from AMD environments coupled to the physiological diversity of Thermoplasmatales makes it difficult to interpret the role of these archaeal AMD environments. The Thermoplasmatales MAG presented here appears to be a heterotroph capable of aerobic respiration.
The role of taxa affiliated with uncultivated or recently discovered phyla in biogeochemical cycling in AMD is particularly difficult to predict. Here, we presented MAGs from four such phyla—the Dormibacterota, the Elusimicrobiota, the Eremiobacterota, and the Patescibacteria. Dormibacterota and Patescibacteria are not widely reported in AMD. Eremiobacterota inhabit multiple mining-impacted sites, including stalactites in a mining cave (50), neutral mine drainage in Brazil (51), and AMD in the eastern United States (28). The abundances of the Eremiobacterota correlated with total organic carbon in an AMD site in China (71) and were recovered from Rose Pool, where dissolved organic carbon is present (188.0 μmol/liter) (33). One of the Eremiobacterota MAGs (MAG 15) at Cabin Branch encodes a Cyc-2 like protein that may be involved in Fe(II) oxidation. Therefore, this taxon may play an important and underappreciated role in Fe cycling in AMD environments.
Elusimicrobiota have also been found in AMD environments across the globe, including in Spain (50, 52), France (29), and Svalbard (53), but it is not abundant in these environments. The only cultivated taxa from this phylum are strictly anaerobic (54–56). The Elusimicrobiota MAG from Cabin Branch encodes genes for three terminal oxidases and likely employs aerobic respiration. The Elusimicrobiota MAG (MAG 13) was abundant in the emergence, where dissolved oxygen is present, albeit below saturation (77.5 μmol/liter). The MAG also contains the genes necessary for nitrogen fixation and may encode a Cyc2-like protein that it may use for Fe(II) oxidation. Therefore, it likely plays an important role in nitrogen cycling in AMD environments and may also contribute to iron cycling.
A combination of ’omics-based approaches and cultivation can increase our ability to correlate function with taxonomy from 16S rRNA amplicon studies. Here, we present high-quality MAGs from AMD sites to increase our current understanding of community composition and function. These data reveal previously unrecognized taxa that contribute to carbon, nitrogen, and Fe(II) cycling in AMD. In particular, these data underscore roles for previously uncharacterized Gammaproteobacteria in Fe(II) oxidation in addition to uncultivated or recently discovered phyla, the prevalence of Actinobacteria across AMD sites that range in oxygen and Fe(II) concentration, and taxa with high relative abundance whose function remains unclear. These data provide a framework to assist in culturing taxa of interest as well as additional target organisms for AMD bioremediation strategies.
MATERIALS AND METHODS
Site location.
Cabin Branch is an acid mine drainage site in the Daniel Boone National Forest in Kentucky, near the border with Tennessee. Detailed geochemistry, isotopic analyses, 13C uptake experimental results, and the results of community analysis using16S rRNA gene amplicons were described previously (13, 33). Briefly, groundwater at Cabin Branch emerges at pH 2.90 and flows down a limestone-lined channel with a pH of 2.92 (installed as a passive remediation strategy) and enters a pool (Rose Pool) which has a pH of 2.97. Dissolved oxygen increases down the drainage site (77.5 μmol/liter at the emergence, 401 μmol/liter in Rose Pool), and the Fe(II) concentration is 403 μmol/liter at the emergence, 882.0 μmol/liter in the limestone-lined channel, and 11.16 μmol/liter in Rose Pool (33). The concentration of dissolved inorganic carbon was highest at the emergence (1.67 mmol/liter) and lowest (0.30 mmol/liter) at Rose Pool. The concentration of dissolved organic carbon was low at all sites (36.8 to 44.2 μmol/liter). The concentration of NH4(T) was highest at the outflow site (50.0 μmol/liter) and lowest at Rose Pool (10.7 μmol/liter). The concentration of P was highest at the emergence (9.55 μmol/liter) and decreased to below detection limits at Rose Pool.
The microbial communities at Cabin Branch have been studied and described previously (13, 33). The communities are dominated by the Fe(II)-oxidizing genus Ferrovum. Lithotrophic metabolisms, likely Fe(II) oxidation, drive primary productivity, and Ferrovum spp. encode the enzymes necessary for nitrogen fixation and thus are a potential source of fixed nitrogen (13, 33). Methods for sample collection, DNA extraction and sequencing, and metagenome assembly and binning were described previously (33), and we include brief descriptions below (13, 37). Methods for sample collection, DNA extraction and sequencing, and metagenome assembly and binning were described previously (33), and we include brief descriptions below.
Molecular analyses.
Sample collection, DNA extraction, and sequencing. Triplicate samples from each site were collected for DNA extraction and were flash-frozen and stored at –80°C until processed. DNA was extracted from each replicate sample using a DNeasy PowerSoil kit (Qiagen, Carlsbad, CA, USA) and quantified using a Qubit 3.0 fluorometer (Invitrogen, Burlington, ON, Canada). Extractions were pooled and submitted to the University of Minnesota Genomics Center for metagenomic sequencing and sequenced using HiSeq 2500 high-output 2 × 125-bp chemistry. Three samples were sequenced per lane.
Metagenomic analysis. Trimmed, quality-controlled sequences were assembled using MegaHit v. 1.0.6 (57) using standard parameters except for the minimum contig length, which was set at 1,000 bp. Reads were mapped to the assembly using bowtie2 v. 1.2.2 (58), and the depth was calculated using the jgi_summarize_bam_contig_depths command in Anvi’o v. 6.1 (59). Binning was performed in MetaBAT v. 2.12.1 using default parameters (60) and CheckM v. 1.0.7 was used to determine bin completeness (61). Bins that were >70% complete with <3% contamination were selected for further analysis. The average nucleotide identity (ANI) across the surviving bins was calculated using anvi-compute-ani in Anvi’o v. 6.1, which uses the PyANI algorithm to compute ANI (59, 62). Bins that shared >99% ANI across the genome were considered to be the same taxon. For each taxon, the bin with the highest completion was selected for further analysis. Bins were uploaded to KBASE and annotated using the “annotate assembly and re-annotate genomes with prokka” app v. 1.12 (63) and classified using the GTDB app (64–67).
Single-copy, ribosomal protein sequences from Campbell et al. (68) were retrieved from the MAGs and reference genomes, concatenated, and aligned in Anvi’o (69). Anvi’o uses MUSCLE to align the concatenated sequences (70). Maximum likelihood trees were constructed using RAxML-HPC2 v. 8.2.12 on XSEDE in the CIPRES Science Gateway using standard parameters as follows: 100 bootstrap iterations, a Protein CAT model, DAYHOFF protein substitution matrix, and no correction for ascertainment bias (71, 72). Trees were visualized and rooted in the Interactive Tree of Life (73). A Newick-formatted tree file is available in the supplemental material (File S1).
The relative abundance of each MAG was determined by mapping reads from each metagenome against each MAG using BBMap (74). The pileup tool within BBMap was used to summarize mapped reads, and the relative abundance was calculated from the total number of mapped reads divided by the total number of reads in the metagenome (75). Unmapped reads and reads mapping to more than one region were removed using SAMtools (76) prior to pileup.
Metabolic pathways for carbon, nitrogen, and sulfur cycling were predicted in each MAG using METABOLIC v. 3.0 (77). Each MAG was also analyzed using DRAM v 1.06, which generates and summarizes gene annotation data from multiple databases (78).
Cyc2 genes were identified using BLAST to identify genes homologous to the Cyc2-like cytochrome c involved in Fe(II) oxidation (36–38). A BLAST database was constructed using the Cyc2 retrieved previously (37), and the search was performed using an E value of 1E-5. To ensure that the sequences retrieved with the BLAST search were homologous to the Cyc2-like protein involved in Fe(II) oxidation, retrieved sequences and those described previously (37) were aligned in MAFFT v. 7.471, portions of the alignment that contained >50% gaps were removed using TrimAI v. 1.2.59, the best model was selected using ModelTest-NG on XSEDE v. 0.1.5, and a maximum likelihood tree was constructed as described above except using the LG + G4 model selected by ModelTest-NG. The Cyc2 tree is displayed in File S2 and available as a Newick-formatted tree in File S3.
Data availability.
Quality-controlled, unassembled, metagenomic data are available in the NCBI Sequence Read Archive under accession numbers SRR9677580 to SRR9677585. The metagenome-assembled genomes used for analysis are also available in NCBI under accession numbers SAMN14771053 to SAMN14771081. Metagenome assembly and mapping statistics are provided in File S4.
ACKNOWLEDGMENTS
We are grateful to the staff of the National Forest Service and Daniel Boone National Forest, especially Margueritte Wilson and Claudia Cotton, for the advice and insight regarding mine locations. We thank A. Gangidine, M. Berberich, R. Jain, and C. Schuler for assistance in field sampling and processing samples in the laboratory. We thank Eric Kees for assistance with MAG analyses and Clara Chan and Sean McAllister for sharing their alignment and trees for Cyc2 sequences.
We acknowledge the Minnesota Supercomputing Institute (MSI) at the University of Minnesota for providing resources that contributed to the research results reported within this paper.
C.L.G and T.L.H designed the study, completed the analyses, and wrote the paper.
We declare no competing financial interests.
Footnotes
Supplemental material is available online only.
Contributor Information
Christen L. Grettenberger, Email: clgrett@ucdavis.edu.
Trinity L. Hamilton, Email: trinityh@umn.edu.
Haruyuki Atomi, Kyoto University.
REFERENCES
- 1.Gupta A, Dutta A, Sarkar J, Panigrahi MK, Sar P. 2018. Low-abundance members of the Firmicutes facilitate bioremediation of soil impacted by highly acidic mine drainage from the Malanjkhand Copper Project, India. Front Microbiol 9:2882. 10.3389/fmicb.2018.02882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Johnson DB, Hallberg KB. 2005. Biogeochemistry of the compost bioreactor components of a composite acid mine drainage passive remediation system. Sci Total Environ 338:81–93. 10.1016/j.scitotenv.2004.09.008. [DOI] [PubMed] [Google Scholar]
- 3.Neculita C-M, Zagury GJ, Bussière B. 2007. Passive treatment of acid mine drainage in bioreactors using sulfate-reducing bacteria. J Environ Qual 36:1–16. 10.2134/jeq2006.0066. [DOI] [PubMed] [Google Scholar]
- 4.Sánchez-Andrea I, Sanz JL, Bijmans MFM, Stams AJM. 2014. Sulfate reduction at low pH to remediate acid mine drainage. J Hazard Mater 269:98–109. 10.1016/j.jhazmat.2013.12.032. [DOI] [PubMed] [Google Scholar]
- 5.Baker BJ, Banfield JF. 2003. Microbial communities in acid mine drainage. FEMS Microbiol Ecol 44:139–152. 10.1016/S0168-6496(03)00028-X. [DOI] [PubMed] [Google Scholar]
- 6.Bijmans MFM, de Vries E, Yang C-H, Buisman CJN, Lens PNL, Dopson M. 2010. Sulfate reduction at pH 4.0 for treatment of process and wastewaters. Biotechnol Progr 26:1029–1037. 10.1002/btpr.400. [DOI] [PubMed] [Google Scholar]
- 7.Bijmans MFM, van Helvoort P-J, Dar SA, Dopson M, Lens PNL, Buisman CJN. 2009. Selective recovery of nickel over iron from a nickel-iron solution using microbial sulfate reduction in a gas-lift bioreactor. Water Res 43:853–861. 10.1016/j.watres.2008.11.023. [DOI] [PubMed] [Google Scholar]
- 8.Church CD, Wilkin RT, Alpers CN, Rye RO, McCleskey RB. 2007. Microbial sulfate reduction and metal attenuation in pH 4 acid mine water. Geochem T 8:10. 10.1186/1467-4866-8-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Druschel GK, Baker BJ, Gihring TM, Banfield JF. 2004. Acid mine drainage biogeochemistry at Iron Mountain. California Geochem Trans 5:13. 10.1186/1467-4866-5-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Giloteaux L, Duran R, Casiot C, Bruneel O, Elbaz-Poulichet F, Goñi-Urriza M. 2013. Three-year survey of sulfate-reducing bacteria community structure in Carnoulès acid mine drainage (France), highly contaminated by arsenic. FEMS Microbiol Ecol 83:724–737. 10.1111/1574-6941.12028. [DOI] [PubMed] [Google Scholar]
- 11.Kaksonen AH, Franzmann PD, Puhakka JA. 2004. Effects of hydraulic retention time and sulfide toxicity on ethanol and acetate oxidation in sulfate-reducing metal-precipitating fluidized-bed reactor. Biotechnol Bioeng 86:332–343. 10.1002/bit.20061. [DOI] [PubMed] [Google Scholar]
- 12.Grettenberger CL, Pearce AR, Bibby KJ, Jones DS, Burgos WD, Macalady JL. 2017. Efficient low-pH iron removal by a microbial iron oxide mound ecosystem at scalp level run. Appl Environ Microbiol 83:e00015-17. 10.1128/AEM.00015-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Havig JR, Grettenberger C, Hamilton TL. 2017. Geochemistry and microbial community composition across a range of acid mine drainage impact and implications for the Neoarchean‐Paleoproterozoic transition. J Geophys Res Biogeosci 122:1404–1422. 10.1002/2016JG003594. [DOI] [Google Scholar]
- 14.Jones DS, Kohl C, Grettenberger C, Larson LN, Burgos WD, Macalady JL. 2015. Geochemical niches of iron-oxidizing acidophiles in acidic coal mine drainage. Appl Environ Microbiol 81:1242–1250. 10.1128/AEM.02919-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.González-Toril E, Santofimia E, López-Pamo E, Omoregie EO, Amils R, Aguilera Á. 2013. Microbial ecology in extreme acidic pit lakes from the Iberian Pyrite Belt (SW Spain). Adv Mat Res 825:23–27. 10.4028/www.scientific.net/AMR.825.23. [DOI] [Google Scholar]
- 16.Santofimia E, González-Toril E, López-Pamo E, Gomariz M, Amils R, Aguilera A. 2013. Microbial diversity and its relationship to physicochemical characteristics of the water in two extreme acidic pit lakes from the Iberian Pyrite Belt (SW Spain). PLoS One 8:e66746. 10.1371/journal.pone.0066746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Hallberg KB, Coupland K, Kimura S, Johnson DB. 2006. Macroscopic streamer growths in acidic, metal-rich mine waters in North Wales consist of novel and remarkably simple bacterial communities. Appl Environ Microbiol 72:2022–2030. 10.1128/AEM.72.3.2022-2030.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kay C, Rowe O, Rocchetti L, Coupland K, Hallberg K, Johnson D. 2013. Evolution of microbial “streamer” growths in an acidic, metal-contaminated stream draining an abandoned underground copper mine. Life (Basel) 3:189–210. 10.3390/life3010189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kuang J-L, Huang L-N, Chen L-X, Hua Z-S, Li S-J, Hu M, Li J-T, Shu W-S. 2013. Contemporary environmental variation determines microbial diversity patterns in acid mine drainage. ISME J 7:1038–1050. 10.1038/ismej.2012.139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sun W, Xiao T, Sun M, Dong Y, Ning Z, Xiao E, Tang S, Li J. 2015. Diversity of the sediment microbial community in the Aha Watershed (Southwest China) in response to acid mine drainage pollution gradients. Appl Environ Microbiol 81:4874–4884. 10.1128/AEM.00935-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.González-Toril E, Aguilera A, Souza-Egipsy V, Ercilla MD, López-Pamo E, Sánchez-España FJ, Amils R. 2009. Comparison between acid mine effluents, La Zarza-Perrunal and Río Tinto (Iberian Pyritic Belt). AMR 71–73:113–116. 10.4028/www.scientific.net/AMR.71-73.113. [DOI] [Google Scholar]
- 22.Grettenberger CL, Rench RLM, Gruen DS, Mills DB, Carney C, Brainard J, Hamasaki H, Ramirez R, Watanabe Y, Amaral-Zettler LA, Ohmoto H, Macalady JL. 2020. Microbial population structure in a stratified, acidic pit lake in the Iberian Pyrite Belt. Geomicrobiol J 37:623–634. 10.1080/01490451.2020.1751748. [DOI] [Google Scholar]
- 23.Qiu G, Wan M, Qian L, Huang Z, Liu K, Liu X, Shi W, Yang Y. 2008. Archaeal diversity in acid mine drainage from Dabaoshan Mine, China. J Basic Microbiol 48:401–409. 10.1002/jobm.200800002. [DOI] [PubMed] [Google Scholar]
- 24.Golyshina OV, Pivovarova TA, Karavaiko GI, Kondratéva TF, Moore ER, Abraham WR, Lünsdorf H, Timmis KN, Yakimov MM, Golyshin PN. 2000. Ferroplasma acidiphilum gen. nov., sp. nov., an acidophilic, autotrophic, ferrous-iron-oxidizing, cell-wall-lacking, mesophilic member of the Ferroplasmaceae fam. nov., comprising a distinct lineage of the Archaea. Int J Syst Evol Microbiol 50:997–1006. 10.1099/00207713-50-3-997. [DOI] [PubMed] [Google Scholar]
- 25.Golyshina OV, Lünsdorf H, Kublanov IV, Goldenstein NI, Hinrichs K-U, Golyshin PN. 2016. The novel extremely acidophilic, cell-wall-deficient archaeon Cuniculiplasma divulgatum gen. nov., sp. nov. represents a new family, Cuniculiplasmataceae fam. nov., of the order Thermoplasmatales. Int J Syst Evol Microbiol 66:332–340. 10.1099/ijsem.0.000725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Itoh T, Yoshikawa N, Takashina T. 2007. Thermogymnomonas acidicola gen. nov., sp. nov., a novel thermoacidophilic, cell wall-less archaeon in the order Thermoplasmatales, isolated from a solfataric soil in Hakone, Japan. Int J Syst Evol Microbiol 57:2557–2561. 10.1099/ijs.0.65203-0. [DOI] [PubMed] [Google Scholar]
- 27.Segerer A, Langworthy TA, Stetter KO. 1988. Thermoplasma acidophilum and Thermoplasma volcanium sp. nov. from Solfatara Fields. Syst Appl Microbiol 10:161–171. 10.1016/S0723-2020(88)80031-6. [DOI] [Google Scholar]
- 28.Brantner JS, Haake ZJ, Burwick JE, Menge CM, Hotchkiss ST, Senko JM. 2014. Depth-dependent geochemical and microbiological gradients in Fe(III) deposits resulting from coal mine-derived acid mine drainage. Front Microbiol 5:215. 10.3389/fmicb.2014.00215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Volant A, Bruneel O, Desoeuvre A, Héry M, Casiot C, Bru N, Delpoux S, Fahy A, Javerliat F, Bouchez O, Duran R, Bertin PN, Elbaz‐Poulichet F, Lauga B. 2014. Diversity and spatiotemporal dynamics of bacterial communities: physicochemical and other drivers along an acid mine drainage. FEMS Microbiol Ecol 90:247–263. 10.1111/1574-6941.12394. [DOI] [PubMed] [Google Scholar]
- 30.Ullrich SR, Poehlein A, Daniel R, Tischler JS, Vogel S, Schlömann M, Mühling M. 2015. Comparative genomics underlines the functional and taxonomic diversity of novel “Ferrovum” related iron oxidizing bacteria acteria. Adv Mat Res 1130:15–18. 10.4028/www.scientific.net/AMR.1130.15. [DOI] [Google Scholar]
- 31.Ullrich SR, González C, Poehlein A, Tischler JS, Daniel R, Schlömann M, Holmes DS, Mühling M. 2016. Gene loss and horizontal gene transfer contributed to the genome evolution of the extreme acidophile “Ferrovum”. Front Microbiol 7:797. 10.3389/fmicb.2016.00797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ullrich SR, Poehlein A, Tischler JS, González C, Ossandon FJ, Daniel R, Holmes DS, Schlömann M, Mühling M. 2016. Genome analysis of the biotechnologically relevant acidophilic iron oxidising strain JA12 indicates phylogenetic and metabolic diversity within the novel genus “Ferrovum”. PLoS One 11:e0146832. 10.1371/journal.pone.0146832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Grettenberger CL, Havig JR, Hamilton TL. 2020. Metabolic diversity and co-occurrence of multiple Ferrovum species at an acid mine drainage site. BMC Microbiol 20:119. 10.1186/s12866-020-01768-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Johnson DB, Hallberg KB, Hedrich S. 2014. Uncovering a microbial enigma: isolation and characterization of the streamer-generating, iron-oxidizing, acidophilic bacterium “Ferrovum myxofaciens”. Appl Environ Microbiol 80:672–680. 10.1128/AEM.03230-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Tyson GW, Lo I, Baker BJ, Allen EE, Hugenholtz P, Banfield JF. 2005. Genome-directed isolation of the key nitrogen fixer Leptospirillum ferrodiazotrophum sp. nov. from an acidophilic microbial community. Appl Environ Microbiol 71:6319–6324. 10.1128/AEM.71.10.6319-6324.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chan C, McAllister SM, Garber A, Hallahan BJ, Rozovsky S. 2018. Fe oxidation by a fused cytochrome-porin common to diverse Fe-oxidizing bacteria. Biorxiv 10.1101/228056. [DOI] [PMC free article] [PubMed]
- 37.McAllister SM, Polson SW, Butterfield DA, Glazer BT, Sylvan JB, Chan CS. 2020. Validating the Cyc2 neutrophilic iron oxidation pathway using meta-omics of Zetaproteobacteria iron mats at marine hydrothermal vents. MSystems 5:e00553-19. 10.1128/mSystems.00553-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol 215:403–410. 10.1016/S0022-2836(05)80360-2. [DOI] [PubMed] [Google Scholar]
- 39.Jones RM, Johnson DB. 2015. Acidithrix ferrooxidans gen. nov., sp. nov.; a filamentous and obligately heterotrophic, acidophilic member of the Actinobacteria that catalyzes dissimilatory oxido-reduction of iron. Res Microbiol 166:111–120. 10.1016/j.resmic.2015.01.003. [DOI] [PubMed] [Google Scholar]
- 40.Eisen S, Poehlein A, Johnson DB, Daniel R, Schlömann M, Mühling M. 2015. Genome sequence of the acidophilic ferrous iron-oxidizing isolate Acidithrix ferrooxidans strain Py-F3, the proposed type strain of the novel actinobacterial genus Acidithrix. Genome Announc 3:e00382-15. 10.1128/genomeA.00382-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Woodcroft BJ, Singleton CM, Boyd JA, Evans PN, Emerson JB, Zayed AAF, Hoelzle RD, Lamberton TO, McCalley CK, Hodgkins SB, Wilson RM, Purvine SO, Nicora CD, Li C, Frolking S, Chanton JP, Crill PM, Saleska SR, Rich VI, Tyson GW. 2018. Genome-centric view of carbon processing in thawing permafrost. Nature 560:49–54. 10.1038/s41586-018-0338-1. [DOI] [PubMed] [Google Scholar]
- 42.Anantharaman K, Brown CT, Hug LA, Sharon I, Castelle CJ, Probst AJ, Thomas BC, Singh A, Wilkins MJ, Karaoz U, Brodie EL, Williams KH, Hubbard SS, Banfield JF. 2016. Thousands of microbial genomes shed light on interconnected biogeochemical processes in an aquifer system. Nat Commun 7:13219. 10.1038/ncomms13219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Dall’Agnol H, Ñancucheo I, Johnson DB, Oliveira R, Leite L, Pylro VS, Holanda R, Grail B, Carvalho N, Nunes GL, Tzotzos G, Fernandes GR, Dutra J, Orellana SC, Oliveira G. 2016. Draft genome sequence of “Acidibacillus ferrooxidans” ITV01, a novel acidophilic firmicute isolated from a chalcopyrite mine drainage site in Brazil. Genome Announc 4:e01748-15. 10.1128/genomeA.01748-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Havig JR, Hamilton TL. 2019. Productivity and community composition of low biomass/high silica precipitation hot springs: a possible window to Earth’s early biosphere? Life Basel Switz 9:64. 10.3390/life9030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Nordstrom DK, Southam G. 1997. Geomicrobiology of sulfide mineral oxidation, p 361–390. In Banfield J, Nealson K (ed), Geomicrobiology, interactions between microbes and minerals. Mineralogical Society of America, Washington, DC. [Google Scholar]
- 46.Evangelou VP, Zhang YL. 1995. A review: pyrite oxidation mechanisms and acid mine drainage prevention. Crit Rev Env Sci Tec 25:141–199. 10.1080/10643389509388477. [DOI] [Google Scholar]
- 47.Moya-Beltrán A, Cárdenas JP, Covarrubias PC, Issotta F, Ossandon FJ, Grail BM, Holmes DS, Quatrini R, Johnson DB. 2014. Draft genome sequence of the nominated type strain of “Ferrovum myxofaciens,” an acidophilic, iron-oxidizing Betaproteobacterium. Genome Announc 2:e00834-14. 10.1128/genomeA.00834-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sun W, Xiao E, Krumins V, Dong Y, Xiao T, Ning Z, Chen H, Xiao Q. 2016. Characterization of the microbial community composition and the distribution of Fe-metabolizing bacteria in a creek contaminated by acid mine drainage. Appl Microbiol Biotechnol 100:8523–8535. 10.1007/s00253-016-7653-y. [DOI] [PubMed] [Google Scholar]
- 49.Senko JM, Wanjugi P, Lucas M, Bruns MA, Burgos WD. 2008. Characterization of Fe(II) oxidizing bacterial activities and communities at two acidic Appalachian coalmine drainage-impacted sites. ISME J 2:1134–1145. 10.1038/ismej.2008.60. [DOI] [PubMed] [Google Scholar]
- 50.Méndez-García C, Mesa V, Sprenger RR, Richter M, Diez MS, Solano J, Bargiela R, Golyshina OV, Manteca Á, Ramos JL, Gallego JR, Llorente I, dos Santos VAPM, Jensen ON, Peláez AI, Sánchez J, Ferrer M. 2014. Microbial stratification in low pH oxic and suboxic macroscopic growths along an acid mine drainage. ISME J 8:1259–1274. 10.1038/ismej.2013.242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Pereira LB, Vicentini R, Ottoboni LMM. 2014. Changes in the bacterial community of soil from a neutral mine drainage channel. PLoS One 9:e96605. 10.1371/journal.pone.0096605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mesa V, Gallego JLR, González-Gil R, Lauga B, Sánchez J, Méndez-García C, Peláez AI. 2017. Bacterial, archaeal, and eukaryotic diversity across distinct microhabitats in an acid mine drainage. Front Microbiol 8:1756. 10.3389/fmicb.2017.01756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.García-Moyano A, Austnes AE, Lanzén A, González-Toril E, Aguilera Á, Øvreås L. 2015. Novel and unexpected microbial diversity in acid mine drainage in Svalbard (78° N), revealed by culture-independent approaches. Microorganisms 3:667–694. 10.3390/microorganisms3040667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Geissinger O, Herlemann DPR, MöRschel E, Maier UG, Brune A. 2009. The ultramicrobacterium “Elusimicrobium minutum” gen. nov., sp. nov., the first cultivated representative of the termite group 1 phylum. Appl Environ Microbiol 75:2831–2840. 10.1128/AEM.02697-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Herlemann DPR, Geissinger O, Ikeda-Ohtsubo W, Kunin V, Sun H, Lapidus A, Hugenholtz P, Brune A. 2009. Genomic analysis of “Elusimicrobium minutum,” the first cultivated representative of the phylum “Elusimicrobia” (formerly termite group 1). Appl Environ Microbiol 75:2841–2849. 10.1128/AEM.02698-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Zheng H, Dietrich C, Radek R, Brune A. 2016. Endomicrobium proavitum, the first isolate of Endomicrobia class. nov. (phylum Elusimicrobia): an ultramicrobacterium with an unusual cell cycle that fixes nitrogen with a Group IV nitrogenase. Environ Microbiol 18:191–204. 10.1111/1462-2920.12960. [DOI] [PubMed] [Google Scholar]
- 57.Li D, Liu C-M, Luo R, Sadakane K, Lam T-W. 2015. MEGAHIT: an ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 31:1674–1676. 10.1093/bioinformatics/btv033. [DOI] [PubMed] [Google Scholar]
- 58.Langmead B, Salzberg SL. 2012. Fast gapped-read alignment with Bowtie 2. Nat Methods 9:357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Eren AM, Esen ÖC, Quince C, Vineis JH, Morrison HG, Sogin ML, Delmont TO. 2015. Anvi’o: an advanced analysis and visualization platform for ’omics data. Peerj 3:e1319. 10.7717/peerj.1319. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Kang DD, Froula J, Egan R, Wang Z. 2015. MetaBAT, an efficient tool for accurately reconstructing single genomes from complex microbial communities. Peerj 3:e1165. 10.7717/peerj.1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Parks DH, Imelfort M, Skennerton CT, Hugenholtz P, Tyson GW. 2015. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25:1043–1055. 10.1101/gr.186072.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Pritchard L, Glover RH, Humphris S, Elphinstone JG, Toth IK. 2016. Genomics and taxonomy in diagnostics for food security: soft-rotting enterobacterial plant pathogens. Anal Methods 8:12–24. 10.1039/C5AY02550H. [DOI] [Google Scholar]
- 63.Seemann T. 2014. Prokka: rapid prokaryotic genome annotation. Bioinformatics 30:2068–2069. 10.1093/bioinformatics/btu153. [DOI] [PubMed] [Google Scholar]
- 64.Arkin AP, Cottingham RW, Henry CS, Harris NL, Stevens RL, Maslov S, Dehal P, Ware D, Perez F, Canon S, Sneddon MW, Henderson ML, Riehl WJ, Murphy-Olson D, Chan SY, Kamimura RT, Kumari S, Drake MM, Brettin TS, Glass EM, Chivian D, Gunter D, Weston DJ, Allen BH, Baumohl J, Best AA, Bowen B, Brenner SE, Bun CC, Chandonia J-M, Chia J-M, Colasanti R, Conrad N, Davis JJ, Davison BH, DeJongh M, Devoid S, Dietrich E, Dubchak I, Edirisinghe JN, Fang G, Faria JP, Frybarger PM, Gerlach W, Gerstein M, Greiner A, Gurtowski J, Haun HL, He F, Jain R, Joachimiak MP, Keegan KP, Kondo S, et al. 2018. KBase: the United States Department of Energy Systems Biology Knowledgebase. Nat Biotechnol 36:566–569. 10.1038/nbt.4163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Allen B, Drake M, Harris N, Sullivan T. 2017. Using KBase to assemble and annotate prokaryotic genomes. Curr Protoc Microbiol 46:1E.13.1–1E.13.18. 10.1002/cpmc.37. [DOI] [PubMed] [Google Scholar]
- 66.Parks DH, Chuvochina M, Waite DW, Rinke C, Skarshewski A, Chaumeil P-A, Hugenholtz P. 2018. A standardized bacterial taxonomy based on genome phylogeny substantially revises the tree of life. Nat Biotechnol 36:996–1004. 10.1038/nbt.4229. [DOI] [PubMed] [Google Scholar]
- 67.Chaumeil P-A, Mussig AJ, Hugenholtz P, Parks DH. 2019. GTDB-Tk: a toolkit to classify genomes with the Genome Taxonomy Database. Bioinform Oxf Engl 36:1925–1927. 10.1093/bioinformatics/btz848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Campbell JH, O’Donoghue P, Campbell AG, Schwientek P, Sczyrba A, Woyke T, Söll D, Podar M. 2013. UGA is an additional glycine codon in uncultured SR1 bacteria from the human microbiota. Proc Natl Acad Sci U S A 110:5540–5545. 10.1073/pnas.1303090110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Campbell BJ, Yu L, Heidelberg JF, Kirchman DL. 2011. Activity of abundant and rare bacteria in a coastal ocean. Proc Natl Acad Sci U S A 108:12776–12781. 10.1073/pnas.1101405108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Edgar RC. 2004. MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32:1792–1797. 10.1093/nar/gkh340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Miller MA, Pfeiffer W, Schwartz T. 2010. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees, p 1–8. In Proceedings of the Gateway Computing Environments Workshop (GCE). IEEE, Piscataway, NJ. [Google Scholar]
- 72.Stamatakis A. 2006. RAxML-VI-HPC: maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics 22:2688–2690. 10.1093/bioinformatics/btl446. [DOI] [PubMed] [Google Scholar]
- 73.Letunic I, Bork P. 2016. Interactive Tree of Life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res 44:W242–W245. 10.1093/nar/gkw290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Bushnell B. 2014. BBMap: A fast, accurate, splice-aware aligner. https://www.osti.gov/servlets/purl/1241166.
- 75.Hua Z-S, Wang Y-L, Evans PN, Qu Y-N, Goh KM, Rao Y-Z, Qi Y-L, Li Y-X, Huang M-J, Jiao J-Y, Chen Y-T, Mao Y-P, Shu W-S, Hozzein W, Hedlund BP, Tyson GW, Zhang T, Li W-J. 2019. Insights into the ecological roles and evolution of methyl-coenzyme M reductase-containing hot spring Archaea. Nat Commun 10:4574. 10.1038/s41467-019-12574-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R, 1000 Genome Project Data Processing Subgroup. 2009. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Zhou Z, Tran P, Liu Y, Kieft K, Anantharaman K. 2019. METABOLIC: a scalable high-throughput metabolic and biogeochemical functional trait profiler based on microbial genomes. Biorxiv 10.1101/761643. [DOI]
- 78.Shaffer M, Borton MA, McGivern BB, Zayed AA, La Rosa SL, Solden LM, Liu P, Narrowe AB, Rodríguez-Ramos J, Bolduc B, Gazitúa MC, Daly RA, Smith GJ, Vik DR, Pope PB, Sullivan MB, Roux S, Wrighton KC. 2020. DRAM for distilling microbial metabolism to automate the curation of microbiome function. Nucleic Acids Res 48:8883–8900. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Files S1 to S6. Download AEM.00772-21-s0001.pdf, PDF file, 0.6 MB (583.8KB, pdf)
Data Set S1. Download AEM.00772-21-s0002.xlsx, XLSX file, 0.4 MB (413.9KB, xlsx)
Data Availability Statement
Quality-controlled, unassembled, metagenomic data are available in the NCBI Sequence Read Archive under accession numbers SRR9677580 to SRR9677585. The metagenome-assembled genomes used for analysis are also available in NCBI under accession numbers SAMN14771053 to SAMN14771081. Metagenome assembly and mapping statistics are provided in File S4.