

ABSTRACT
Short-read, high-throughput sequencing (HTS) methods have yielded numerous important insights into microbial ecology and function. Yet, in many instances short-read HTS techniques are suboptimal, for example, by providing insufficient phylogenetic resolution or low integrity of assembled genomes. Single-molecule and synthetic long-read (SLR) HTS methods have successfully ameliorated these limitations. In addition, nanopore sequencing has generated a number of unique analysis opportunities, such as rapid molecular diagnostics and direct RNA sequencing, and both Pacific Biosciences (PacBio) and nanopore sequencing support detection of epigenetic modifications. Although initially suffering from relatively low sequence quality, recent advances have greatly improved the accuracy of long-read sequencing technologies. In spite of great technological progress in recent years, the long-read HTS methods (PacBio and nanopore sequencing) are still relatively costly, require large amounts of high-quality starting material, and commonly need specific solutions in various analysis steps. Despite these challenges, long-read sequencing technologies offer high-quality, cutting-edge alternatives for testing hypotheses about microbiome structure and functioning as well as assembly of eukaryote genomes from complex environmental DNA samples.
KEYWORDS: PacBio sequencing, Oxford Nanopore sequencing, synthetic long reads, unique molecular identifiers (UMI), direct RNA sequencing, epigenomics, bioinformatics, nanopore
INTRODUCTION
Modern microbial ecology research takes advantage of techniques that determine the molecular structure of nucleic acids. Because of their relative ease of use and lack of culturing requirements, high-throughput sequencing (HTS) of genomes, marker genes, and metagenomes has largely replaced microarrays and fingerprinting techniques, becoming the method of choice for understanding patterns in microbiome diversity, population structure, and evolution (1–3). HTS-based marker gene sequencing (i.e., metabarcoding) enables in-depth characterization of the community composition of both prokaryotic (bacteria and archaea) and eukaryotic (protists, fungi, and microfauna) microorganisms. Reconstruction of prokaryote genomes from complex environmental samples, such as soil, water, and sediments, using deep sequencing has greatly benefited our understanding about the potential function of so-far uncultivable microbial species and much expanded the tree of life (4, 5). Although the second-generation HTS methods have shed light on fundamental aspects of microbial ecology and function, they suffer from issues associated with short read lengths (up to 550 bp). This has forced a step back in biodiversity research that traditionally uses molecular marker genes exceeding 550 bp in length that provide greater taxonomic resolution (2, 6, 7). Furthermore, short read lengths limit the number of available target-specific primer binding sites that can be utilized when designing a marker gene amplicon. In genome and metagenome analyses, short reads cannot reliably reconstruct long repeats because of uncertainties in mapping reads; although protocols for mate-pair libraries are available, they have rarely been used.
To overcome these shortfalls, synthetic long-read sequencing and third-generation HTS methods have become increasingly used in microbiology (8, 9). The Pacific Biosciences (PacBio) and Oxford Nanopore Technologies (ONT) sequencing platforms both generate 30- to 50-kb reads on average but suffer from low raw read accuracy and higher per-read costs compared to those of short-read platforms (Table 1). Here, we provide an overview of the cutting-edge developments in laboratory and bioinformatics methods for long-read analysis from the microbial ecology perspective. We illustrate situations in which long reads have provided unique answers to research questions related to microbial identification, community and population ecology, and interspecific interactions. Finally, we outline the current limitations of long-read sequencing and evaluate their future prospects in microbiology research.
TABLE 1.
Comparison of recent long-read and short-read sequencing technologiesa
| Sequencing technology (specifications) | Instrument cost | Library prep cost | Cost per lane/cell | Cost per million reads | Cost per billion bases | Runtime (h) | Throughput (k reads at maximum runtime) | Avg read length (maximum) | Read accuracy (%) | Error profile |
|---|---|---|---|---|---|---|---|---|---|---|
| PacBio Sequel I (maximum 8 cells) | 300,000 | 150–400 | 1,000 | 2,000 | 100 | 10–20 | 500 | 20,000 (50,000) | 85–88/99.9 (CCS) | Homopolymers |
| PacBio Sequel II (maximum 16 cells) | 650,000 | 75–400 | 2,000 | 500 | 17 | 10–30 | 4,000 | 30,000 (100,000) | 88–90/99.9 (CCS) | Homopolymers |
| ONT Flongle | 1,300 | 40–90 | 80 | 400 | 13 | 0.1–12 | 200 | 30,000 (60,000) | 96–99 | Homopolymers |
| ONT MinION | 900 | 90–130 | 600 | 600 | 12 | 0.1–48 | 1,000 | 50,000 (2.3M) | 96–99 | Homopolymers |
| ONT GridION (maximum 5 FCs) | 45,000 | 90–130 | 600 | 600 | 12 | 48 | 1,000 | 50,000 (2.3M) | 96–99 | Homopolymers |
| ONT PromethION (maximum 48 FCs) | 176,000 | 90–600 | 1,400 | 250 | 8 | 72 | 6,000 | 30,000 (330,000) | 96–99 | Homopolymers |
| SLR (example of NovaSeq 2 × 150 PE, 30× coverage, for 5 kb) | 900,000 | 30–50 | 5,000 | 1,250 | 140 | 44 | 4,000 | 9,000 (12,000) | 100 | Unequal coverage |
| Illumina NovaSeq S4 (2 × 150 PE; maximum 4 lanes) | 900,000 | 50–100 | 5,000 | 2.5 | 9 | 44 | 2,000,000 | 250 (290) | 99.9 | Low-quality ends |
| Illumina MiSeq (2 × 300 PE; maximum 2 lanes) | 100,000 | 50–100 | 2,000 | 100 | 175 | 56 | 20,000 | 550 (590) | 99.9 | Low-quality ends |
| ION G5-S5 Prime, P550 chip (maximum 2 chips) | 180,000 | 50 | 700 | 5 | 25 | 6.5 | 130,000 | 200 (250) | 99.0–99.5 | Homopolymers |
| ION G5-S5, P530 chip 600 SE | 60,000 | 50 | 500 | 40 | 70 | 7 | 12,000 | 570 (650) | 99.3–99.7 | Homopolymers, low-quality ends |
| MGI Tech DNBSEQ-T7 (2 × 150 PE; maximum 4 cells) | 990,000 | 50–100 | 6,000 | 1.2 | 4.5 | 24 | 5,000,000 | 250 (290) | 99.9 | Low-quality ends |
| MGI Tech DNBSEQ-G400RS (2 × 200 PE; 400 SE maximum 2 cells × 4 lanes) | 480,000 | 50–100 | 1,800 | 4 | 11 | 108 | 450,000 | 350 (400) | 99.9 | Low-quality ends |
Costs are given in EUR. Information is compiled from multiple recent literature sources and price requests from sequencing companies. FC, flow cell; PE, paired-end; SE, single-end.
LONG-READ SEQUENCING METHODS
PacBio sequencing.
PacBio was the first third-generation, long-read HTS technology to enter the market. This method is based on recording fluorescently labeled deoxyribonucleoside triphosphates (dNTPs) during complementary strand synthesis (Fig. 1). In a decade, PacBio raw error rates have declined from 25% to 10–12%, whereas the average sequence length of 2 kb has increased to 30 kb (early 2021). Over the past decade, sequencing throughput has increased from around 50,000 reads (RS I model) to 4 million reads per single-molecule real-time sequencing (SMRT) cell (Sequel II model). PacBio has also developed a much higher accuracy circular consensus sequence (CCS) approach, branded by the company as HiFi sequencing. In CCS sequencing, hairpin adaptors are ligated to the template DNA molecules, permitting individual molecules to be sequenced multiple times in a circular manner (10) (Fig. 1). The multiple “subreads” are then assembled into CCS reads with accuracies of >99.9% at >10-fold consensus.
FIG 1.

Comparison of workflows of the third-generation sequencing platforms for DNA and RNA sequencing. (Left) Pacific BioSciences PacBio SMRT sequencing; (right) Oxford Nanopore Technologies nanopore sequencing.
PacBio CCS sequencing is proven to be efficient for high-resolution marker gene profiling of bacterial and fungal communities. CCS sequencing with the earlier RS II model could produce full-length 16S rRNA gene sequences (1.5 kb) at >99% accuracy that improved species-level identification in bacteria (7, 11–14). In fungi, marker gene sequencing of the 5′ part of the 28S rRNA gene (650 bp) (15) and the full-length rRNA internal transcribed spacer (ITS) region (500 to 700 bp) (16) was the first step toward incorporating long-read HTS into fungal ecology research. Long reads spanning both the variable ITS region and more conserved rRNA genes further added to species-level taxonomic identification in both bacteria (17–20) and fungi (21–24). At present, long PacBio CCS sequences can distinguish bacterial genotypes and rRNA gene copies that differ by a single nucleotide (18, 19, 25, 26).
PacBio has been used for de novo genome assembly in viruses (27), bacteria (28, 29), fungi (30), plants (31), and animals (32). Initially, the best results were obtained when PacBio reads were coassembled with short Illumina reads (33). Since 2019, large-scale genome sequencing initiatives such as the 1000 Fungal Genomes Project use only PacBio sequencing (34). Furthermore, genomes as complex as a human’s no longer require complementary short-read sequencing (9, 35).
PacBio sequencing can be used for the analysis of transcriptomes and metagenomes. PacBio transcriptome sequences are sufficiently accurate and long to distinguish both single-nucleotide mutations and alternatively spliced isoforms (36, 37). Using a metagenomics approach, PacBio sequencing was effectively used for identification of bacterial species in mock communities (38) and complex environments (39) as well as assays of functional genes and the construction of high-quality metagenome-assembled genomes (MAGs) (40). Although early PacBio sequencing enabled direct RNA sequencing by using reverse transcription recording (41), later protocols use cDNA.
Nanopore sequencing.
ONT provides the best known and, so far, the only commercial nanopore-based sequencing method. This technique is based on recording signal disruptions of nucleotide oligomers when DNA molecules are translocated through nanopores (42) (Fig. 1). The mobile phone-sized MinION was the first instrument released in 2014 through an early access program that boosted improvement of laboratory and bioinformatics protocols by the research community. The early R7 series flow cells had an average read length of 4.7 to 6.7 kb (maximum, 40 to 300 kb) (43, 44) and 72 to 74% read accuracy. Currently, modal raw accuracy and sequence length reach 95 to 98% (2021 early access chemistry with 99% accuracy) and 50 kb (maximum, 2.3 Mb), respectively. Accuracy of individual reads can be further improved by various methods during sample preparation, sequencing, and bioinformatics. The outdated methods based on rolling circle amplification—INC-seq (45), NanoAmpli-seq (46), and R2C2 (47)—improve accuracy of ONT reads by up to 97 to 98%. Integration of unique molecular identifiers (UMIs) (see specific subsection below) has enabled researches to increase nanopore sequence accuracy to above 99.99% (26).
The first complete genomes of bacteria (44, 48–50) and viruses (51, 52) constructed from nanopore sequences were published in the mid-2010s. As in PacBio, these resolved many repeat-rich regions, but suffered from indels and hence frame shifts in genes (53). Therefore, ONT has been combined with short-read techniques to generate high-quality complete genomes (54). Other studies reported accurate bacterial species-level identification from mock communities and environmental samples (55, 56). In addition to cDNA-based (meta)transcriptomics (57), ONT enables direct RNA sequencing (58, 59). Furthermore, ONT instruments allow detection of base modifications during sequencing of DNA (60) and RNA (58).
MinION was followed by the benchtop GridION and PromethION models that increased throughput. In 2019, miniature Flongle flow cells were introduced to reduce costs of single-sample analyses. The main benefits of MinION and Flongle include small size, rapid analysis process, and low power requirements that enable analyses in field camps (61) and other nonlaboratory conditions (52, 62, 63).
Synthetic long-read sequencing.
Synthetic long reads (SLRs) represent long DNA molecules that are assembled from multiple short reads based on UMI barcodes located within the reads that link them to the DNA molecule from which they originated (Fig. 2). SLR protocols typically include indexing or capturing individual DNA molecules, a few rounds of amplification with UMI-tagged primers, enzymatic or sonication-based fragmentation, short-read HTS (Illumina, MGI Tech, or Ion Torrent), and bioinformatics-based reconstruction of long DNA molecules. SLR length and quality is limited by polymerase processivity (around 10 to 12 kb) and securing even coverage of UMIs and reads across long molecules. Compared with long-read HTS platforms, SLRs exhibit low capacity to resolve tandem repeats longer than 500 bases.
FIG 2.
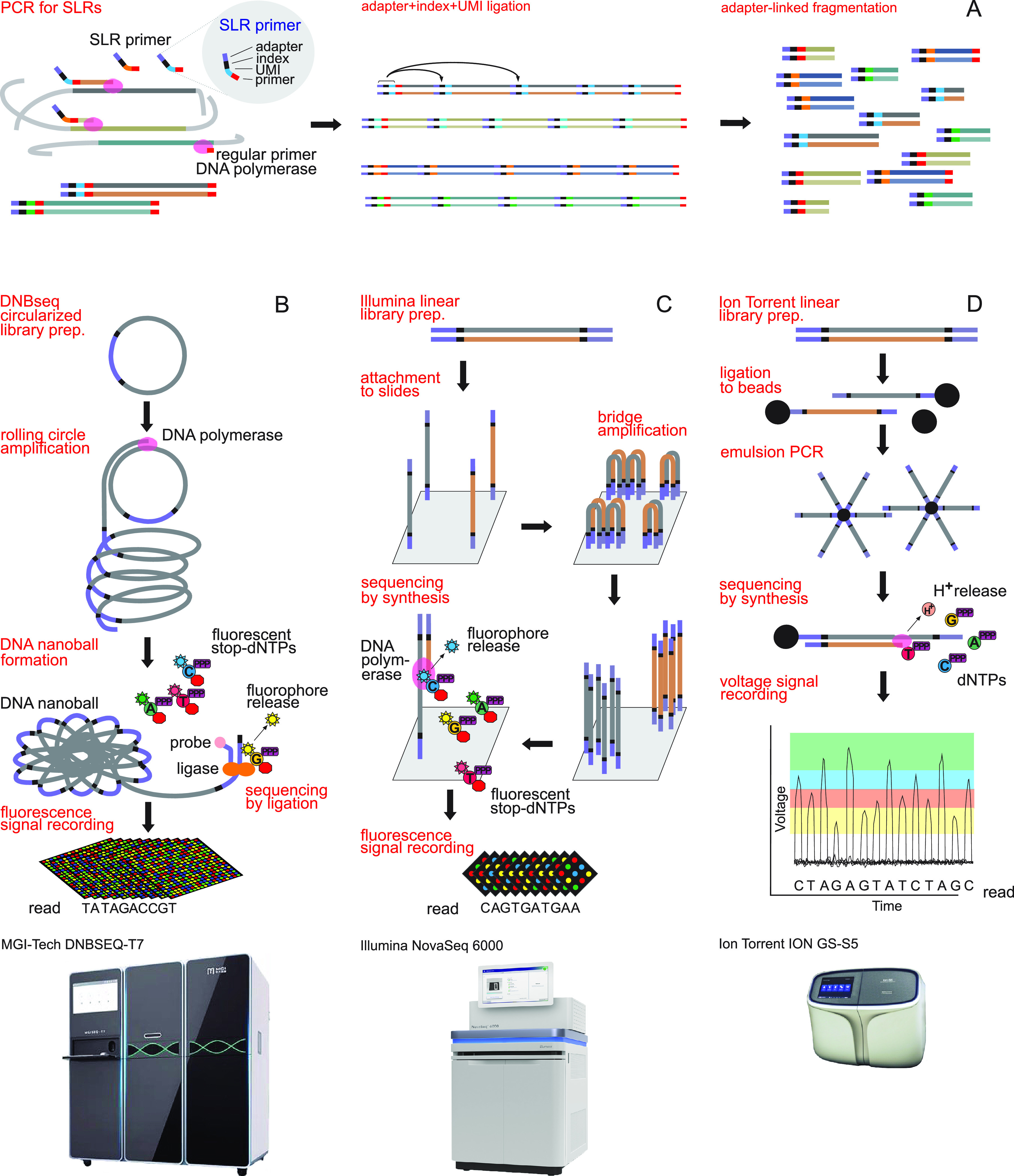
Workflow of producing synthetic long reads (SLRs) using unique molecular identifiers (UMIs) (A) and the following three alternative short-read sequencing platforms: MGI Tech (B), Illumina (C), and Ion Torrent (D). The SLR preparation follows the LOOPseq protocol (74).
SLRs (1 to 3 kb) were initially developed to facilitate genome assembly of humans (64) and viruses (65). SLRs enabled reconstruction of full-length transcripts (66) and 16S rRNA genes from metagenomes (67) and amplicons (68) that exceeded PacBio and ONT sequencing in terms of accuracy, assembly length, and cost (69, 70). Moleculo, Inc. and 10× Genomics, Inc. commercialized methods to separate several up to 12-kb DNA molecules into 384-well plates and microfluidics (100,000 wells) chips, respectively, for compartmentalized sample preparation and Illumina sequencing (71, 72). These methods became limited by the number of wells and the difficulty of distinguishing homologous DNA molecules included in the same compartment. Recently, single-molecule SLR methods have been developed (73, 74). Loop Genomics, Inc. released the LoopSeq method to tag millions of individual molecules with UMI-indexed primers that are unique to each DNA molecule (74). LoopSeq amplicons of the full-length 16S rRNA gene (1.5 kb) had 10-fold higher accuracy compared to PacBio CCS reads and Illumina short reads in bacterial mock communities (75, 76) and maintained >99.99% accuracy (75). The UMI tagging used in the construction of SLRs also facilitates identification and removal of chimeric molecules produced during the PCR (68).
APPLICATIONS OF LONG-READ SEQUENCING METHODS
Identification of specimens and cultures.
Molecular identification of species relies on a suitable genetic marker (i.e., DNA barcode). These marker genes are chosen based on ease of amplification, taxonomic resolution, and the presence of a reference database. Longer marker genes contain more taxonomic and phylogenetic information but can be more difficult to amplify and sequence than shorter fragments (77). In spite of relatively low species-level resolution, the 16S rRNA gene is by far the most broadly used taxonomic marker (Fig. 3A). In fungi, the rRNA ITS region is the most commonly used amplicon for taxonomic classification (Fig. 3B), but 18S and 28S rRNA are alternatively used for several fungal phyla (2). In plants and photosynthetic protists, the ITS region, plastid genes, and their introns are all popular species-level identification markers, whereas various heterotrophic protist and animal groups rely on 18S or 28S rRNA gene, the ITS region, mitochondrial cytochrome c oxidase 1 (COI), or other mitochondrial genes (6). Because rRNA genes usually occur in operons, it is feasible to analyze genetic information from long rRNA gene fragments or the entire mitochondria or plastids for genotype-level identification and building reference information for marker gene sequencing (78, 79).
FIG 3.
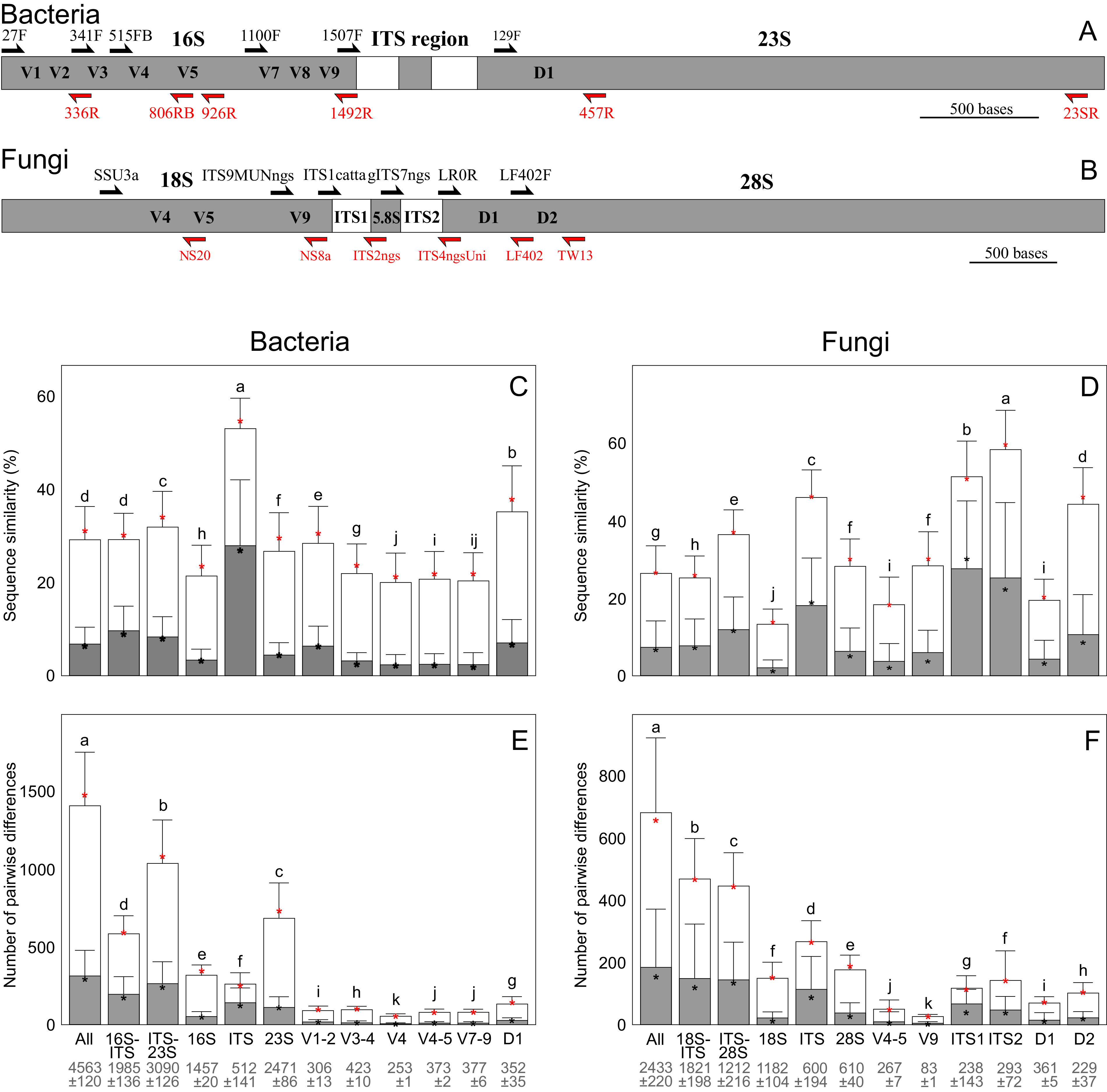
Taxonomic resolution of rRNA marker gene fragments in short-read and long-read analyses. Distribution of taxonomic information in the rRNA operon of bacteria (A) and fungi (B). Relative sequence similarity (C, D) and number of mismatching nucleotides (E, F) in long-read and short-read rRNA gene sequence data of bacteria (left) and fungi (right) at the level of kingdom (open columns) and genus (shaded columns) based on pairwise alignments of fragments. The values are given as mean (box) ± standard deviation (whiskers) and median (asterisk). Different letters above bars depict statistically significant differences among groups (Padj < 0.001). Mean fragment length ± standard deviation is indicated below the fragments. The methods of calculating distances are outlined in Item S1 in the supplemental material.
Subregions of bacterial and fungal rRNA operons differ greatly in the number and proportion of pairwise differences that are informative for discrimination among species. In bacteria, all widely used rRNA short-read markers have much lower taxonomic resolution than the full-length 16S rRNA gene, ITS region, or 23S rRNA gene (Fig. 3C and E). Combining the ITS region with any (or both) of the flanking genes provides the best resolution at the level of strains and species. However, the ITS marker is of highly variable length, which may introduce PCR and sequencing biases. Long fragments covering the ITS region may miss multiple taxa, in which the rRNA genes are not arranged in a conventional manner. In fungi, the 18S rRNA gene subregions and the entire gene has very limited species-level resolution (Fig. 3D and F). On average, ITS1 and ITS2 subregions are nearly equally variable and of comparable length albeit with great among-group variation. In many fungal taxa (e.g., Sordariales, Glomerales, Hypocreales, Helotiales), variability and capacity to identify species are limited to one of the ITS subregions; therefore, full-length ITS sequences provide greater overall taxonomic resolution. Supplementing 18S and/or 28S sequence data with ITS sequences may add some species-level resolution (21), but it mainly improves above order-level identification and allows construction of phylogenies, addressing questions about phylogenetic diversity and community phylogeny.
Sanger sequencing of marker genes is still broadly used for species identification. However, long-read HTS approaches provide more accurate information at the level of alleles and genotypes (80) and enable retrieval of marker gene sequences from samples that are contaminated with cooccurring organisms (81). Using a single PacBio Sequel SMRT cell for sequencing, Hebert et al. (82) recovered high-quality, 658-base COI barcodes from 86% of 9,800 arthropod individuals. Similar results were obtained for analyzing 3,500 fly individuals in a single ONT MinION run (83).
For fungi, protocols covering the entire rRNA operon in one or two amplicons were successfully implemented for 77 samples using both PacBio and ONT sequencing (78). A part of this approach was later used to generate ca. 4,700-base 18S-ITS-28S barcodes for 430 cultures of early diverging fungal lineages, which improved the understanding of molecular diversity, inter- and intraspecific differences, and phylogenetic relations among these taxa (80). ONT sequencing of rRNA intergenic spacer (IGS) sequences (2.5 kb) was suggested as a preferential method for reliably distinguishing among closely related human pathogenic and nonpathogenic species of Cryptococcus (84).
Multiplexing hundreds to thousands of specimen samples in a single HTS run allows cost-savings of up to 100-fold compared with Sanger sequencing. For microorganisms, the high barcoding capacity of third-generation HTS has remained underutilized despite its potential value for routine screening of culture collections and fungaria. Furthermore, long-read marker gene sequencing may simultaneously recover the identity of the target organism along with the associated microbiota and reveal novel biotic interactions (85).
Biodiversity assessment.
Marker gene sequencing (metabarcoding) has become a standard procedure for determining richness and composition of microbial communities (86). At the depth of tens to hundreds of thousands of reads per sample, short reads offer a comprehensive semiquantitative view about the taxa present but usually provide low resolution at species level (Fig. 3). Long reads improve taxonomic resolution to species and genotype levels, especially when covering both variable and conserved loci in fungi (22–24) and bacteria (17, 18, 20). For example, long PacBio reads of the Cyanobacteria rbcL gene improved taxonomic and phylogenetic resolution and facilitated the discovery of two novel cyanobacterial clades associated with hornworts; cyanobacterial communities were driven by dispersal limitation, plant species, and temporal variation (87). The gyrB gene sequencing of rooibos (Aspalathus linearis) root nodulating bacteria showed that the distribution of symbionts is dispersal limited, whereas cooccurring nonnodulating bacteria are influenced by drift (88).
In eukaryotes, including fungi, accurate PacBio CCS reads provide a relatively more reliable phylogenetic placement of taxa that remain unidentified to order or phylum level based on sequence similarity comparisons (89, 90). Phylogenies of reads belonging to unculturable taxa revealed multiple novel genera of anaerobic Neocallimastigomycota from feces of herbivores, with a substantial host species effect on richness and composition (91). The ITS-28S 1,500-base amplicons enabled the resolution of much of the phytopathogenic genus Fusarium to species level and revealed that certain cover crops may act as reservoirs of harmful crop pathogens (22). Furthermore, use of such amplicons improved identification of arbuscular mycorrhizal fungi and revealed that genotypes present in commercially inoculated soils may replace native strains (21).
Molecular diagnostics.
For most sequencing platforms, sample preparation and sequencing take days or weeks if an external service is used. Here, ONT offers the following several unique properties suited for rapid diagnostics: low cost of apparatus, portability and low power requirements, technically nondemanding library preparation steps, affordability, and real-time access to sequencing data from DNA or RNA molecules. High-speed, accurate identification greatly benefits detection of pathogenic or hazardous organisms for rapid countermeasures. For a striking example, the Ebola virus was diagnosed from symptomatic patients using ONT MinION instruments in field camps and hospitals in West Africa within a day since sample collection based on cDNA (51, 52). A similar workflow was previously used to trace a salmonellosis outbreak at the pathotype level (43). Furthermore, ONT sequence data were used for accurate real-time identification of antibiotic resistance indicators in Neisseria gonorrhoeae isolates to predict susceptibility to therapeutic antimicrobials (92). Using 3.5- to 6-kb amplicons covering the rRNA operon, putative fungal pathogens were identified from external otitis in dogs (93).
In plant pathology, ONT has been used to detect cDNA of “Candidatus Liberibacter asiaticus” and plum pox virus from infected plants and insect vectors (94). Various fungal pathogens were detected from vegetables and pine needles using marker gene sequencing and metagenomic approaches with variable success (95). Llontop et al. (96) developed a protocol for species-level and occasionally genotype-level identification of bacterial tomato pathogens based on metagenomics data. With respect to other environmental applications, toxic dinoflagellates were identified to species based on a 3,000-bp 18S-ITS-28S rRNA gene amplicon (97). Similarly, invasive bivalves were diagnosed from water samples based on a 600-base mt-16S rRNA gene amplicon (98).
For rapid diagnosis potential, ONT MinION is unrivalled among sequencing platforms. Shortened DNA extraction and library preparation protocols revealed that metagenome-based pathogen diagnosis from plant samples can be achieved within 150 min of sample collection (95). Hundreds of clinical samples can be tested for SARS-CoV-2 using a single instrument in 1 day (99). Unfortunately, ONT sequencing has become routinely established in only a few human and plant pathology laboratories, probably due to lack of standard workflows and unwillingness to implement novel techniques. Also, the benefit of low cost compared with that of other HTS methods has remained poorly utilized, as very few publications using ONT emerge from developing countries.
Population biology.
Long reads are advantageous over short reads in intraspecific population genetics analyses because of their capacity to distinguish among genotypes, phase haplotypes, and resolve repeat-rich regions. PacBio sequencing has been used for developing primers and sequencing long microsatellite-rich markers (100) as well as detecting multiple antiretroviral drug-resistant genotypes in an HIV+ patient (101). SLRs revealed 10 genotypes of the hepatitis B virus in a single patient (65) and population structure of gut bacteria of fruit flies (102). Combined SLR and PacBio shotgun sequencing of Acropora coral holobionts revealed no biogeographic structuring of the coral or its Symbiodiniaceae algal mutualists, but symbiont composition explained much of the vulnerability to bleaching (103). PacBio 16S-ITS sequencing of lake bacteria revealed that different genotypes of the same species dominate in Japan and Europe, but there is very low regional-scale difference in Japan and Central Europe (19). In humans, PacBio-based detection of SNPs in alleles revealed that introgressed genomic fragments of the Denisovans and Neanderthals were positively selected in the evolution of Melanesian population (104). These methods offer great potential for tracking hybridization in microorganisms, such as pathogenic fungi.
In pathogens, all long-read sequencing methods have been successfully used to detect and monitor genotypes and virulence genes. SLRs revealed that long-term use of antibiotics may lead to evolution of Escherichia coli strains with up to 50 antibiotic resistance genes (105). PacBio genomic sequencing of Treponema pallidum isolates revealed >100 different, nonoverlapping copies of the tprk gene that encodes a membrane-bound protein, which prevents efficient resistance and vaccine development (106). PacBio single-cell sequencing revealed >100 karyotypes harboring mosaic aneuploidy (partial polyploidy) among a clonal population of Leishmania, suggesting that local variability in chromosome number contributes to the high adaptability of the pathogen (107). ONT-based phylogenomics of Mycoplasma bovis revealed no genetic differentiation among host ungulates but common cross-continental transportation of strains belonging to geographical subgroups with calves (108).
Long-read HTS has revealed the organization and copy number of rRNA genes and evolutionary mechanisms of their persistence. In strains of the arbuscular mycorrhizal fungal genus Rhizophagus, multiple isoforms with distinct secondary structure of the 28S rRNA genes are present, and their transcription depends on host associations rather than other biotic and abiotic factors, suggesting that these variants may be related to host selectivity in arbuscular mycorrhizal symbiosis (109). Overall, the presence of different ITS copies points to introgression and ineffective concerted evolution; divergent gene copies can be fixed by inbreeding (110). In prokaryotes, long reads have been used to show that unlinked rRNA genes are widespread (111).
Long reads are increasingly used in genome analysis and population genomics (112), with the greatest benefits in diploid and polyploid macroorganisms (9, 113) because of their capacity to solve repeat-rich regions and high variability suitable for distinguishing genotypes. Thus, long-read population genomics tools are useful for assessing functional differences among genotypes in addition to in-depth quantitative recording of genetic differences.
Metagenomics.
Metagenomics is broadly used for analyses of taxonomic and functional potential of microbial communities and recovery of individual viral and prokaryote genomes (i.e., metagenome-assembled genomes [MAGs]). MAG assemblies benefit from long reads by greater continuity in repeat-rich regions and reduced incorporation of contaminant sequences from other organisms (40, 105, 114). Using ONT long-read assemblies polished with short-read data, Singleton et al. (115) generated 1,083 high-quality prokaryote genomes from 23 sewage sludge samples. Many of these MAGs represented the first full genomes of various bacterial orders and candidate phyla, adding unique information about the potential functioning of these enigmatic taxonomic groups. Furthermore, long reads improve assignment of functional genes to specific taxa (116) and reduce the overall proportion of taxonomically and functionally unassigned contigs (117). Finally, long reads with sufficiently low error rates, such as PacBio CCS, allow direct determination of full-length functional genes without the need for assembly, thus offering better sensitivity to rare genes (13, 40).
Quantification accuracy of target groups using long-read approaches may outperform Illumina-based quantification for bacteria depending on GC content and library preparation methods (38, 118). ONT sequencing has been used to uncover the predominately vegan diet of rats (122) and quantify plant species’ contribution to pollen samples (120). However, quantification accuracy is constrained by among-taxon and among-tissue differences in DNA extractability, amount of DNA per biomass, as well as mitochondrial, plastid, and rRNA gene copy numbers (121, 122).
Gene expression.
Sequence-based (meta)transcriptomics methods can be used as proxies to estimate community activity (123). Most RNA molecules, especially in eukaryotes, are alternatively spliced or have highly similar copies; therefore, long reads are more efficient than short reads in recovering these RNA isoforms (36, 57, 124). For example, the PacBio Iso-Seq technique revealed that mutations in viruses cause a variable immune response among cells (125), and alternative splicing of a maize gene secures resistance to the root parasitic worm Diabrotica virgifera (126).
RNA sequencing is traditionally performed by reverse transcription and sequencing of cDNA molecules. Currently, only ONT supports direct RNA sequencing (58). This method revealed that the dominant zooplankton species, their parasites, and transcript profiles may all differ seasonally (127). In double stranded DNA (dsDNA) herpes simplex virus 1 (HSV-1), direct RNA sequencing enabled novel fusion transcripts that encode chimeric proteins (128). Direct RNA sequencing has several potentially important advantages over cDNA approaches because it allows for epigenetic investigation of native RNA and avoids biases related to reverse transcriptase activity potentially leading to codon switch artifacts and exclusion of certain RNA molecules.
Epigenetics.
DNA and RNA methylation influence nucleic acid-protein interactions and affect processes such as gene expression, chromosome stability, and self-recognition. The signal of PacBio and ONT sequencing is inherently affected by modified bases, which can be used for explicit recording of these epigenetic modifications (Fig. 1) (60, 129–131). Through regulating transcription, DNA methylation is related to the development, pathogenicity, and secondary metabolism of antagonists and viruses (131–133). Thus, epigenetics provides a yet little-studied dimension to functionality of populations and species within microbial communities.
Third-generation HTS techniques have become methods of choice in epigenetics, with important discoveries in phage-bacteria and host-pathogen interactions. PacBio analyses revealed that DNA methylation in certain genes is related to expression of phage-encoded Shiga toxin genes in an E. coli pathogenic isolate (134). The m4C methylations in Helicobacter pylori are essential for the bacterial association to host cells (135). In Campylobacter jejuni, methylations affect cell adherence, invasion, and biofilm formation, and their variation among cells promotes overall adaptability of the population (136). Matching methylation signatures may also improve binning of metagenomic contigs among closely related strains and linking plasmids or phages to their host (137, 138).
COMBINING OTHER MOLECULAR TOOLS WITH LONG-READ SEQUENCING
Target enrichment and single-cell methods.
In environmental samples, metagenomics and metatranscriptomics approaches may require enrichment of low-abundance target organisms or genes. Several methods exist to selectively enrich certain community members. DNA or proteins in microbial cells can be selectively labeled with fluorescent or heavy isotopes for stable isotope probing (SIP) (139), fluorescence in situ hybridization (FISH) (140), Raman spectroscopy (141), etc. Sorted cells or small groups of cells can be subjected to separate DNA extraction and molecular analysis in nano-wells or droplets (142, 143). Combined ONT and Illumina single-cell sequencing improved detection of full-length transcript isoforms in various mouse cell lines (144).
Target capture techniques are used to enrich chromosomes, genes, or RNA molecules by linking these targets to biotin-attached probes, followed by separation onto streptavidin-coated beads or wells. For example, the major histocompatibility complex of human DNA (20-kb fragments) was enriched 83-fold, yielding 10% on-target Illumina and PacBio sequences (145). Using large enriched fragment targeted sequencing (LEFTseq), reads from DNA fragments of the symbiotic bacterial genus Wolbachia were enriched 11-fold to 200-fold in invertebrate samples (146). Experiments using biotin-16-aminoallyl-2′-dUTP-labeled probes were used to enrich RNA isoforms (1 to 2.5 kb) around 7,600-fold in hundreds of human genes (147). Combined with SLR HTS, single-cell and target enrichment methods can be used for selective genome or transcriptome sequencing of certain microbial taxa, which may be particularly important for understanding the structure and function of the rare biosphere.
The properties of CAS9, an RNA-guided DNA endonuclease, enable the enrichment of target molecules to yield nearly 200-kb DNA fragments for PacBio and ONT sequencing (148, 149). Given the site-specificity of restrictases, this method is applicable for population-level studies but is more difficult to use for gene or community profiling of environmental samples because of high chances of nontarget restriction and systematic bias in loss of material.
ONT nanopore sequencing offers a unique opportunity for target enrichment by aborting the recording of reads during the sequencing process. The nontarget DNA molecules are recognized by predefined motifs and ejected from the active nanopores. The Read Until (150) and UNCALLED (151) software enable severalfold enrichment of target sequences.
Unique molecular identifiers.
UMIs are short unique index sequences that are integrated into DNA molecules, allowing reads to be reassociated with the same DNA molecule using bioinformatics approaches (68, 152). UMIs are essential to SLR technologies by linking together the short reads derived from a common DNA molecule and thereby allowing the long sequences of that DNA molecule (SLR) to be reconstructed (Fig. 2). Recently UMIs have also been implemented in third-generation HTS to enable >99.99% accuracy and a <0.02% chimera rate (26, 75). Furthermore, as UMIs label individual molecules, they are also useful for improving relative quantification (152).
BIOINFORMATICS TOOLS AND DATABASES
The emergence of long-read sequencing technologies required optimization of bioinformatics tools initially developed for short reads and generation of new software to realize the full potential of long-read sequencing in microbial ecology (153). An overview of bioinformatics tools used for long-read analyses is regularly updated in the Long Read Tools database (https://long-read-tools.org/).
In analysis of metagenomes, the recently developed metaFlye outperforms software generated for short-read genome assembly (154). For gene-level analyses of metagenomics data, the use of highly-accurate long reads allows assembly to be avoided entirely because most sequenced genes are covered by a single read, simplifying bioinformatics workflows and reducing assembly and binning biases (155, 156). New taxonomic classification methods, such as MetaMaps, outperform simple applications of short-read tools (157, 158). Purpose-built classification methods may be most valuable for ONT metagenomic reads due to the potential application of that technology to real-time surveillance for pathogens or antimicrobial resistance (96, 159).
In long-read marker gene sequencing, bioinformatics tools developed for short reads have been updated to handle long sequences (7). High-resolution “denoising” tools such as DADA2 and unoise3 have been extended for highly accurate long reads (25, 160, 161). New taxonomic assignment approaches have shown marginally higher classification performance from the common full-length 16S or ITS gene targets (162, 163) than standard short-read methods, but a void still exists in methods that can exploit the below-species level resolution that might be achievable from such data.
Most popular marker gene databases contain sequences that are of variable length but often shorter than long-read amplicons, which limits the taxonomic resolution that can be obtained. SILVA contains near full-length sequences of small and large subunits of rRNA operon of prokaryotes and eukaryotes, but these are insufficient for amplicons spanning the ITS region. Based on genomic data in RefSeq (https://ftp.ncbi.nlm.nih.gov/genomes/refseq/), Graf et al. (18) constructed the Athena database, which comprises around 56,000 sequences spanning >2,000 bases, i.e., much of the 16S rRNA gene, ITS, and around 600 bases of 23S rRNA gene. Since high-quality long reads often distinguish between different strains within a species and between different alleles of multicopy marker genes within a genome, reference databases need to account for such infraspecific variation. Such genotype-level information about bacterial 16S rRNA genes has been gathered in more local and habitat-specific databases (161, 164). For fungi and other eukaryotes, the UNITE database includes user-annotated information about multiple marker genes (2) with a specific reference subset developed for long 18S-ITS-28S sequences in version 9. The EukRef holds nuclear 18S rRNA and plastid 16S rRNA sequences for most eukaryotes, with a specific focus on protists (165).
LIMITATIONS OF LONG-READ SEQUENCING
One of the principal challenges to incorporating long-read HTS technologies is the relatively high requirements on the quality and quantity of the input DNA or RNA, which can be difficult to obtain from some samples. The higher rate of chimera formation in long amplicons and high raw error-rate of nanopore and PacBio sequencing represent another challenge, but this can be circumvented through the use of UMIs and proper bioinformatics tools (26).
Long-read sequencing methods are still relatively new compared to Illumina short-read sequencing. As standard practices continue to evolve, we strongly recommend that researchers routinely include quality control measures, such as mock communities comprising organisms with high-quality sequenced genomes (e.g., ZymoBIOMICS Microbial Community DNA Standard), to evaluate the technical biases and quantitative performance of their long-read sequencing protocols. In this rapidly progressing field, continued evaluation and improvement of long-read workflows for microbial ecology are of vital importance.
CHOICE OF SEQUENCING PLATFORMS
Both short-read and long-read technologies advance rapidly, as several sequencing instruments and important supporting methodological advances become available every year. Upon choosing the optimal method, microbial ecologists need to consider the required accuracy, amount of data needed, and expenses afforded (Table 2). Although the long-read sequencing is currently cheaper than the obsolete 454 pyrosequencing and early versions of Illumina sequencing, short-read sequencing costs have dropped >10-fold in the last decade. Because generation of sequencing libraries and data are costlier for long reads, short-read technologies may be preferable, for example, in marker gene sequencing, when short reads are expected to possess sufficient resolution, or when high species-level taxonomic resolution is of limited concern.
TABLE 2.
Feasibility of DNA and RNA sequencing using various sequencing platforms, their combination with UMIs (in parentheses), and hybrid approaches using short reads and long reads based on cost-efficiency, read length, and accuracya
| Sequencing capability | Illumina/MGI Tech | SLRs (30×) | PacBio CCS (+UMIs) | ONT (+UMIs) | Hybrid approach |
|---|---|---|---|---|---|
| Rapid diagnostics | + | − | − (−) | +++ (++) | − |
| (Meta)genomes/ transcriptomes <0.5 kb | +++ | − | + (−) | − (+) | − |
| DNA (meta)barcoding <0.5 kb | +++ | − | + (−) | − (+) | − |
| (Meta)genomes/ transcriptomes 0.5–3.5 kb | + | ++ | +++ (+) | ++ (++) | + |
| DNA (meta)barcoding 0.5–3.5 kb | + | ++ | +++ (+) | − (+++) | − |
| (Meta)genomes 3.5–10 kb | − | +++ | ++ (+) | ++ (++) | ++ |
| DNA (meta)barcoding 3.5–10 kb | − | +++ | ++ (++) | − (+++) | + |
| (Meta)genomes >10 kb | − | ++ | ++ (++) | +++ (+) | +++ |
| Allele/isoform detection, phasing | + | ++ | ++ (+) | ++ (++) | +++ |
| Base methylation | − | − | +++ (−) | +++ (−) | − |
| Direct RNA sequencing | − | − | − (−) | +++ (−) | − |
+++, preferred; ++, suboptimal; +, useable but not recommended; −, not applicable or not competitive.
Besides the unique direct RNA sequencing and epigenome sequencing options, long-read protocols offer several advantages over short reads in many routine applications. With careful DNA extraction and library preparation, ONT and PacBio platforms can produce ultralong reads (>100 kb) that exceed those produced by short-read instruments by 3 to 4 orders of magnitude. These ultralong reads may be critical for circularizing bacterial chromosomes and solving long repeats such as entire rRNA loci in eukaryotes (40, 166). Currently, the ONT PromethION in-house application produces the largest amount of data per euro, which enables reconstruction of hundreds of high-quality bacterial genomes from metagenomes (115). However, the MGI Tech DNBSEQ-T7 short-read platform is the cheapest commercial solution in terms of throughput per base and per read (Table 1). Given the amounts of data and sequence accuracy, MGI Tech sequencing of SLRs is currently the most cost-efficient solution for high-quality sequencing of fragments >3.5 kb (73). UMI-based methods outperform other methods when there is scant starting material or a need for ultrahigh accuracy (>99.99%) of a single long read, e.g., for detecting rare genotypes and frameshifts (74, 75). Because of its relatively lower raw read length compared with ONT, PacBio Sequel II may be best suited for sequencing DNA fragments of 0.5 kb to 3.5 kb to take full advantage of the CCS option for the quality of individual reads (Table 2). We anticipate that any major shift in quality, throughput, or read length of HTS platforms may strongly alter the method preferred for testing specific scientific hypotheses.
CONCLUSIONS
Long-read HTS offers multiple cutting-edge options for understanding microbiome structure and functioning. Direct RNA sequencing has proved its efficiency in detecting transcriptomes of various DNA viruses (167) and discovery of single-stranded RNA (ssRNA) viruses (168), including coronaviruses (169), without the reverse transcriptase bias. Direct RNA sequencing has a potential to recover the diversity and function of double-stranded RNA (dsRNA) viruses and establish the roles of small RNA molecules in the functioning of organisms and interacting with viruses or microbial pathogens and symbionts. cDNA sequencing and potentially direct RNA sequencing offer a possibility to detect the rRNA gene products of highly divergent organisms that have issues with primer matching and introns (70). DNA and RNA methylome sequencing has been successfully used to link plasmids and associated phages to bacterial organisms (137, 167), with a further potential for detecting various epigenomic modifications in eukaryotes that are little understood thus far (170). We speculate that using these epigenetic patterns along with GC content, codon use, intron types, and relative read coverage may improve assembly of chromosome-sized scaffolds and linking chromosomes to reconstruct nearly full eukaryote genomes from complex environmental samples in the near future. Beside natural base modifications, nucleotide analogues such as bromodeoxyuridine and biotin-tagged DNA molecules can be directly recorded using the ONT technology to track DNA replication (171), suggesting that these methods may offer great promise for real-time detection of functionally enriched taxa based on DNA and RNA. Although long-read HTS methods have a potential to outperform short-read approaches in nearly all subfields of microbiology in terms of analytical performance, the requirement for relatively large amounts of input material and higher costs (compared to short-read sequencing) may remain major bottlenecks for certain research applications.
ACKNOWLEDGMENTS
This work was supported by the Norway-Baltic financial mechanism (EMP442), Estonian Research Council (PRG632, MOBTP198), Danish-Estonian collaboration program (Silva Nova), and National Institutes of Health (R35GM133745).
We declare no conflict of interest.
Footnotes
Supplemental material is available online only.
Contributor Information
Leho Tedersoo, Email: leho.tedersoo@ut.ee.
Irina S. Druzhinina, Nanjing Agricultural University
REFERENCES
- 1.Knight R, Vrbanac A, Taylor BC, Aksenov A, Callewaert C, Debelius J, Gonzalez A, Kosciolek T, McCall L-I, McDonald D, Melnik AV, Morton JT, Navas J, Quinn RA, Sanders JG, Swafford AD, Thompson LR, Tripathi A, Xu ZZ, Zaneveld JR, Zhu Q, Caporaso JG, Dorrestein PC. 2018. Best practices for analysing microbiomes. Nat Rev Microbiol 16:410–422. 10.1038/s41579-018-0029-9. [DOI] [PubMed] [Google Scholar]
- 2.Nilsson R, Anslan S, Bahram M, Wurzbacher C, Baldrian P, Tedersoo L. 2019. Mycobiome diversity: high-throughput sequencing and identification of fungi. Nat Rev Microbiol 17:95–109. 10.1038/s41579-018-0116-y. [DOI] [PubMed] [Google Scholar]
- 3.Zhu Q, Mai U, Pfeiffer W, Janssen S, Asnicar F, Sanders JG, Belda-Ferre P, Al-Ghalith GA, Kopylova E, McDonald D, Kosciolek T, Yin JB, Huang S, Salam N, Jiao J-Y, Wu Z, Xu ZZ, Cantrell K, Yang Y, Sayyari E, Rabiee M, Morton JT, Podell S, Knights D, Li W-J, Huttenhower C, Segata N, Smarr L, Mirarab S, Knight R. 2019. Phylogenomics of 10,575 genomes reveals evolutionary proximity between domains Bacteria and Archaea. Nat Commun 10:5477. 10.1038/s41467-019-13443-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Hug LA, Baker BJ, Anantharaman K, Brown CT, Probst AJ, Castelle CJ, Butterfield CN, Hernsdorf AW, Amano Y, Ise K, Suzuki Y, Dudek N, Relman DA, Finstad KM, Amundson R, Thomas BC, Banfield JF. 2016. A new view of the tree of life. Nat Microbiol 1:16048. 10.1038/nmicrobiol.2016.48. [DOI] [PubMed] [Google Scholar]
- 5.Stewart R, Auffret M, Warr A, Walker A, Roehe R, Watson M. 2019. Compendium of 4,941 rumen metagenome-assembled genomes for rumen microbiome biology and enzyme discovery. Nat Biotechnol 37:953–961. 10.1038/s41587-019-0202-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Pawlowski J, Audic S, Adl S, Bass D, Belbahri L, Berney C, Bowser SS, Cepicka I, Decelle J, Dunthorn M, Fiore-Donno AM, Gile GH, Holzmann M, Jahn R, Jirků M, Keeling PJ, Kostka M, Kudryavtsev A, Lara E, Lukeš J, Mann DG, Mitchell EAD, Nitsche F, Romeralo M, Saunders GW, Simpson AGB, Smirnov AV, Spouge JL, Stern RF, Stoeck T, Zimmermann J, Schindel D, de Vargas C. 2012. CBOL Protist Working Group: barcoding eukaryotic richness beyond the animal, plant, and fungal kingdoms. PLoS Biol 10:e1001419. 10.1371/journal.pbio.1001419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Schloss PD, Jenior ML, Koumpouras CC, Westcott SL, Highlander SK. 2016. Sequencing 16S rRNA gene fragments using the PacBio SMRT DNA sequencing system. PeerJ 4:e1869. 10.7717/peerj.1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Kraft F, Kurth I. 2020. Long-read sequencing to understand genome biology and cell function. Int J Biochem Cell Biol 126:105799. 10.1016/j.biocel.2020.105799. [DOI] [PubMed] [Google Scholar]
- 9.Logsdon G, Vollger M, Eichler E. 2020. Long-read human genome sequencing and its applications. Nat Rev Genet 21:597–614. 10.1038/s41576-020-0236-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, et al. 2009. Real-time DNA sequencing from single polymerase molecules. Science 323:133–138. 10.1126/science.1162986. [DOI] [PubMed] [Google Scholar]
- 11.Mosher JJ, Bowman B, Bernberg EL, Shevchenko O, Kan J, Korlach J, Kaplan LA. 2014. Improved performance of the PacBio SMRT technology for 16S rDNA sequencing. J Microbiol Methods 104:59–60. 10.1016/j.mimet.2014.06.012. [DOI] [PubMed] [Google Scholar]
- 12.Franzen O, Hu J, Bao X, Itzkowitz S, Peter I, Bashir A. 2015. Improved OTU-picking using long-read 16S rRNA gene amplicon sequencing and generic hierarchical clustering. Microbiome 3:43. 10.1186/s40168-015-0105-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Singer E, Andreopoulos B, Bowers RM, Lee J, Deshpande S, Chiniquy J, Ciobanu D, Klenk H-P, Zane M, Daum C, Clum A, Cheng J-F, Copeland A, Woyke T. 2016. Next generation sequencing data of a defined microbial mock community. Sci Data 3:160081. 10.1038/sdata.2016.81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Wagner J, Coupland P, Browne H, Lawley T, Francis S, Parkhill J. 2016. Evaluation of PacBio sequencing for full-length bacterial 16S rRNA gene classification. BMC Microbiol 16:274. 10.1186/s12866-016-0891-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cline L, Zak D. 2015. Initial colonization, community assembly and ecosystem function: fungal colonist traits and litter biochemistry mediate decay rate. Mol Ecol 24:5045–5058. 10.1111/mec.13361. [DOI] [PubMed] [Google Scholar]
- 16.James T, Marino J, Perfecto I, Vandermeer J. 2016. Identification of putative coffee rust mycoparasites via single-molecule DNA sequencing of infected pustules. Appl Environ Microbiol 82:631–639. 10.1128/AEM.02639-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Martijn J, Lind A, Schön M, Spiertz I, Juzokaite L, Bunikis I, Pettersson O, Ettema T. 2019. Confident phylogenetic identification of uncultured prokaryotes through long read amplicon sequencing of the 16S‐ITS‐23S rRNA operon. Environ Microbiol 21:2485–2498. 10.1111/1462-2920.14636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Graf J, Ledala N, Caimano M, Jackson E, Gratalo D, Fasulo D, Driscoll M, Coleman S, Matson A. 2020. Tracking closely related enteric bacteria at high resolution in fecal samples of premature infants using a novel rRNA amplicon. medRxiv 10.1101/2020.09.26.20201608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Okazaki Y, Fujinaga S, Salcher M, Callieri C, Tanaka A, Kohzu A, Oyagi H, Tamaki H, Nakano S. 2020. Microdiversity and phylogeographic diversification of bacterioplankton in pelagic freshwater systems revealed through long-read amplicon sequencing. bioRxiv 10.1101/2020.06.03.133140. [DOI] [PMC free article] [PubMed]
- 20.Wang J, Bleich R, Zarmer S, Arthur J. 2020. Long-read sequencing to interrogate strain-level variation among adherent-invasive Escherichia coli isolated from human intestinal tissue. bioRxiv 10.1101/2020.03.10.985440. [DOI] [PMC free article] [PubMed]
- 21.Schlaeppi K, Bender S, Mascher F, Russo G, Patrignani A, Camenzind T, Hempel S, Rillig M, Heijden M. 2016. High‐resolution community profiling of arbuscular mycorrhizal fungi. New Phytol 212:780–791. 10.1111/nph.14070. [DOI] [PubMed] [Google Scholar]
- 22.Walder F, Schlaeppi K, Wittwer R, Held A, Vogelgsang S, van der Heijden MG. 2017. Community profiling of Fusarium in combination with other plant-associated fungi in different crop species using SMRT sequencing. Front Plant Sci 8:2019. 10.3389/fpls.2017.02019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Heeger F, Bourne E, Baschien C, Yurkov A, Bunk B, Spröer C, Overmann J, Mazzoni C, Monaghan M. 2018. Long-read DNA metabarcoding of ribosomal rRNA in the analysis of fungi from aquatic environments. Mol Ecol Resour 18:1500–1514. 10.1111/1755-0998.12937. [DOI] [PubMed] [Google Scholar]
- 24.Tedersoo L, Tooming-Klunderud A, Anslan S. 2018. PacBio metabarcoding of fungi and other eukaryotes: errors, biases and perspectives. New Phytol 217:1370–1385. 10.1111/nph.14776. [DOI] [PubMed] [Google Scholar]
- 25.Callahan B, Wong J, Heiner C, Oh S, Theriot C, Gulati A, McGill S, Dougherty M. 2019. High-throughput amplicon sequencing of the full-length 16S rRNA gene with single-nucleotide resolution. Nucleic Acids Res 47:e103. 10.1093/nar/gkz569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Karst S, Ziels R, Kirkegaard R, Sørensen E, McDonald D, Zhu Q, Knight R, Albertsen M. 2021. High-accuracy long-read amplicon sequences using unique molecular identifiers with nanopore or PacBio sequencing. Nat Methods 18:165–169. 10.1038/s41592-020-01041-y. [DOI] [PubMed] [Google Scholar]
- 27.Tombacz D, Sharon D, Oláh P, Csabai Z, Snyder M, Boldogkői Z. 2014. Strain Kaplan of pseudorabies virus genome sequenced by PacBio single-molecule real-time sequencing technology. Genome Announc 2:e00628-14. 10.1128/genomeA.00628-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ee R, Lim Y, Yin W, Chan K. 2014. De novo assembly of the quorum-sensing Pandoraea sp. strain RB-44 complete genome sequence using PacBio single-molecule real-time sequencing technology. Genome Announc 2:e00245-14. 10.1128/genomeA.00245-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Satou K, Shiroma A, Teruya K, Shimoji M, Nakano K, Juan A, Tamotsu H, Terabayashi Y, Aoyama M, Teruya M, Suzuki R. 2014. Complete genome sequences of eight Helicobacter pylori strains with different virulence factor genotypes and methylation profiles, isolated from patients with diverse gastrointestinal diseases on Okinawa Island, Japan, determined using PacBio single-molecule real-time technology. Genome Announc 2:e00286-14. 10.1128/genomeA.00286-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bao J, Chen M, Zhong Z, Tang W, Lin L, Zhang X, Jiang H, Zhang D, Miao C, Tang H, Zhang J, Lu G, Ming R, Norvienyeku J, Wang B, Wang Z. 2017. PacBio sequencing reveals transposable elements as a key contributor to genomic plasticity and virulence variation in Magnaporthe oryzae. Mol Plant 10:1465–1468. 10.1016/j.molp.2017.08.008. [DOI] [PubMed] [Google Scholar]
- 31.Rothfels C, Pryer K, Li F. 2017. Next‐generation polyploid phylogenetics: rapid resolution of hybrid polyploid complexes using PacBio single‐molecule sequencing. New Phytol 213:413–429. 10.1111/nph.14111. [DOI] [PubMed] [Google Scholar]
- 32.Wen M, Ng J, Zhu F, Chionh Y, Chia W, Mendenhall I, Lee B, Irving A, Wang L. 2018. Exploring the genome and transcriptome of the cave nectar bat Eonycteris spelaea with PacBio long-read sequencing. GigaScience 7:giy116. 10.1093/gigascience/giy116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim KE, Peluso P, Babayan P, Yeadon PJ, Yu C, Fisher WW, Chin C-S, Rapicavoli NA, Rank DR, Li J, Catcheside DEA, Celniker SE, Phillippy AM, Bergman CM, Landolin JM. 2014. Long-read, whole-genome shotgun sequence data for five model organisms. Sci Data 1:140045. 10.1038/sdata.2014.45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Miyauchi S, Kiss E, Kuo A, Drula E, Kohler A, Sánchez-García M, Morin E, Andreopoulos B, Barry KW, Bonito G, Buée M, Carver A, Chen C, Cichocki N, Clum A, Culley D, Crous PW, Fauchery L, Girlanda M, Hayes RD, Kéri Z, LaButti K, Lipzen A, Lombard V, Magnuson J, Maillard F, Murat C, Nolan M, Ohm RA, Pangilinan J, Pereira M.dF, Perotto S, Peter M, Pfister S, Riley R, Sitrit Y, Stielow JB, Szöllősi G, Žifčáková L, Štursová M, Spatafora JW, Tedersoo L, Vaario L-M, Yamada A, Yan M, Wang P, Xu J, Bruns T, Baldrian P, Vilgalys R, et al. 2020. Large-scale genome sequencing of mycorrhizal fungi provides insights into the early evolution of symbiotic traits. Nat Commun 11:5125. 10.1038/s41467-020-18795-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vollger MR, Logsdon GA, Audano PA, Sulovari A, Porubsky D, Peluso P, Wenger AM, Concepcion GT, Kronenberg ZN, Munson KM, Baker C, Sanders AD, Spierings DCJ, Lansdorp PM, Surti U, Hunkapiller MW, Eichler EE. 2020. Improved assembly and variant detection of a haploid human genome using single-molecule, high-fidelity long reads. Ann Hum Genet 84:125–140. 10.1111/ahg.12364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Sharon D, Tilgner H, Grubert F, Snyder M. 2013. A single-molecule long-read survey of the human transcriptome. Nat Biotechnol 31:1009–1014. 10.1038/nbt.2705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gordon SP, Tseng E, Salamov A, Zhang J, Meng X, Zhao Z, Kang D, Underwood J, Grigoriev IV, Figueroa M, Schilling JS, Chen F, Wang Z. 2015. Widespread polycistronic transcripts in fungi revealed by single-molecule mRNA sequencing. PLoS One 10:e0132628. 10.1371/journal.pone.0132628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Singer E, Bushnell B, Coleman-Derr D, Bowman B, Bowers RM, Levy A, Gies EA, Cheng J-F, Copeland A, Klenk H-P, Hallam SJ, Hugenholtz P, Tringe SG, Woyke T. 2016. High-resolution phylogenetic microbial community profiling. ISME J 10:2020–2032. 10.1038/ismej.2015.249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Charalampous T, Richardson H, Kay G, Baldan R, Jeanes C, Rae D, Grundy S, Turner D, Wain J, Leggett R, Livermore D. 2018. Rapid diagnosis of lower respiratory infection using nanopore-based clinical metagenomics. bioRxiv 10.1101/387548. [DOI] [PubMed]
- 40.Somerville V, Lutz S, Schmid M, Frei D, Moser A, Irmler S, Frey J, Ahrens C. 2019. Long-read based de novo assembly of low-complexity metagenome samples results in finished genomes and reveals insights into strain diversity and an active phage system. BMC Microbiol 19:143. 10.1186/s12866-019-1500-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Saletore Y, Meyer K, Korlach J, Vilfan I, Jaffrey S, Mason C. 2012. The birth of the Epitranscriptome: deciphering the function of RNA modifications. Genome Biol 13:175. 10.1186/gb-2012-13-10-175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Jain M, Olsen H, Paten B, Akeson M. 2016. The Oxford Nanopore MinION: delivery of nanopore sequencing to the genomics community. Genome Biol 17:239. 10.1186/s13059-016-1103-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Quick J, Ashton P, Calus S, Chatt C, Gossain S, Hawker J, Nair S, Neal K, Nye K, Peters T, De Pinna E, Robinson E, Struthers K, Webber M, Catto A, Dallman TJ, Hawkey P, Loman NJ. 2015. Rapid draft sequencing and real-time nanopore sequencing in a hospital outbreak of Salmonella. Genome Biol 16:114. 10.1186/s13059-015-0677-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Ip CLC, Loose M, Tyson JR, de Cesare M, Brown BL, Jain M, Leggett RM, Eccles DA, Zalunin V, Urban JM, Piazza P, Bowden RJ, Paten B, Mwaigwisya S, Batty EM, Simpson JT, Snutch TP, Birney E, Buck D, Goodwin S, Jansen HJ, O'Grady J, Olsen HE, MinION Analysis and Reference Consortium. 2015. MinION analysis and reference consortium: phase 1 data release and analysis. F1000Res 4:1075. 10.12688/f1000research.7201.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Li C, Chng K, Boey E, Ng A, Wilm A, Nagarajan N. 2016. INC-Seq: accurate single molecule reads using nanopore sequencing. GigaScience 5:34. 10.1186/s13742-016-0140-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Calus S, Ijaz U, Pinto A. 2018. NanoAmpli-Seq: a workflow for amplicon sequencing for mixed microbial communities on the nanopore sequencing platform. GigaScience 7:giy140. 10.1093/gigascience/giy140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Volden R, Palmer T, Byrne A, Cole C, Schmitz R, Green R, Vollmers C. 2018. Improving nanopore read accuracy with the R2C2 method enables the sequencing of highly multiplexed full-length single-cell cDNA. Proc Natl Acad Sci U S A 115:9726–9731. 10.1073/pnas.1806447115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Quick J, Quinlan A, Loman N. 2014. A reference bacterial genome dataset generated on the MinION portable single-molecule nanopore sequencer. GigaSci 3:22. 10.1186/2047-217X-3-22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Loman N, Quick J, Simpson J. 2015. A complete bacterial genome assembled de novo using only nanopore sequencing data. Nat Methods 12:733–735. 10.1038/nmeth.3444. [DOI] [PubMed] [Google Scholar]
- 50.Ashton P, Nair S, Dallman T, Rubino S, Rabsch W, Mwaigwisya S, Wain J, O'Grady J. 2015. MinION nanopore sequencing identifies the position and structure of a bacterial antibiotic resistance island. Nat Biotechnol 33:296–300. 10.1038/nbt.3103. [DOI] [PubMed] [Google Scholar]
- 51.Hoenen T, Groseth A, Rosenke K, Fischer RJ, Hoenen A, Judson SD, Martellaro C, Falzarano D, Marzi A, Squires RB, Wollenberg KR, de Wit E, Prescott J, Safronetz D, van Doremalen N, Bushmaker T, Feldmann F, McNally K, Bolay FK, Fields B, Sealy T, Rayfield M, Nichol ST, Zoon KC, Massaquoi M, Munster VJ, Feldmann H. 2016. Nanopore sequencing as a rapidly deployable Ebola outbreak tool. Emerg Infect Dis 22:331–334. 10.3201/eid2202.151796. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Quick J, Loman NJ, Duraffour S, Simpson JT, Severi E, Cowley L, Bore JA, Koundouno R, Dudas G, Mikhail A, Ouédraogo N, Afrough B, Bah A, Baum JH, Becker-Ziaja B, Boettcher J-P, Cabeza-Cabrerizo M, Camino-Sanchez A, Carter LL, Doerrbecker J, Enkirch T, Dorival IGG, Hetzelt N, Hinzmann J, Holm T, Kafetzopoulou LE, Koropogui M, Kosgey A, Kuisma E, Logue CH, Mazzarelli A, Meisel S, Mertens M, Michel J, Ngabo D, Nitzsche K, Pallash E, Patrono LV, Portmann J, Repits JG, Rickett NY, Sachse A, Singethan K, Vitoriano I, Yemanaberhan RL, Zekeng EG, Trina R, Bello A, Sall AA, Faye O, et al. 2016. Real-time, portable genome sequencing for Ebola surveillance. Nature 530:228–232. 10.1038/nature16996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Watson M, Warr A. 2019. Errors in long-read assemblies can critically affect protein prediction. Nat Biotechnol 37:124–126. 10.1038/s41587-018-0004-z. [DOI] [PubMed] [Google Scholar]
- 54.Daims H, Lebedeva EV, Pjevac P, Han P, Herbold C, Albertsen M, Jehmlich N, Palatinszky M, Vierheilig J, Bulaev A, Kirkegaard RH, von Bergen M, Rattei T, Bendinger B, Nielsen PH, Wagner M. 2015. Complete nitrification by Nitrospira bacteria. Nature 528:504–509. 10.1038/nature16461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Benitez-Paez A, Portune K, Sanz Y. 2016. Species-level resolution of 16S rRNA gene amplicons sequenced through the MinION portable nanopore sequencer. GigaScience 5:4. 10.1186/s13742-016-0111-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shin J, Lee S, Go M, Lee S, Kim S, Lee C, Cho B. 2016. Analysis of the mouse gut microbiome using full-length 16S rRNA amplicon sequencing. Sci Rep 6:29681. 10.1038/srep29681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bolisetty M, Rajadinakaran G, Graveley B. 2015. Determining exon connectivity in complex mRNAs by nanopore sequencing. Genome Biol 16:204. 10.1186/s13059-015-0777-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Garalde DR, Snell EA, Jachimowicz D, Sipos B, Lloyd JH, Bruce M, Pantic N, Admassu T, James P, Warland A, Jordan M, Ciccone J, Serra S, Keenan J, Martin S, McNeill L, Wallace EJ, Jayasinghe L, Wright C, Blasco J, Young S, Brocklebank D, Juul S, Clarke J, Heron AJ, Turner DJ. 2018. Highly parallel direct RNA sequencing on an array of nanopores. Nat Methods 15:201–206. 10.1038/nmeth.4577. [DOI] [PubMed] [Google Scholar]
- 59.Smith M, Ersavas T, Ferguson J, Liu H, Lucas M, Begik O, Bojarski L, Barton K, Novoa E. 2020. Molecular barcoding of native RNAs using nanopore sequencing and deep learning. Genome Res 30:1345–1353. 10.1101/gr.260836.120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Simpson J, Workman R, Zuzarte P, David M, Dursi L, Timp W. 2017. Detecting DNA cytosine methylation using nanopore sequencing. Nat Methods 14:407–410. 10.1038/nmeth.4184. [DOI] [PubMed] [Google Scholar]
- 61.Edwards A, Debbonaire A, Sattler B, Mur L, Hodson A. 2016. Extreme metagenomics using nanopore DNA sequencing: a field report from Svalbard, 78°N. bioRxiv 10.1101/073965. [DOI]
- 62.Stahl-Rommel S, Jain M, Nguyen HN, Arnold RR, Aunon-Chancellor SM, Sharp GM, Castro CL, John KK, Juul S, Turner DJ, Stoddart D, Paten B, Akeson M, Burton AS, Castro-Wallace SL. 2021. Real-time culture-independent microbial profiling onboard the international space station using nanopore sequencing. Genes 12:106. 10.3390/genes12010106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Gowers G-OF, Vince O, Charles J-H, Klarenberg I, Ellis T, Edwards A. 2019. Entirely off-grid and solar-powered DNA sequencing of microbial communities during an ice cap traverse expedition. Genes 10:902. 10.3390/genes10110902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Peters BA, Kermani BG, Sparks AB, Alferov O, Hong P, Alexeev A, Jiang Y, Dahl F, Tang YT, Haas J, Robasky K, Zaranek AW, Lee J-H, Ball MP, Peterson JE, Perazich H, Yeung G, Liu J, Chen L, Kennemer MI, Pothuraju K, Konvicka K, Tsoupko-Sitnikov M, Pant KP, Ebert JC, Nilsen GB, Baccash J, Halpern AL, Church GM, Drmanac R. 2012. Accurate whole-genome sequencing and haplotyping from 10 to 20 human cells. Nature 487:190–195. 10.1038/nature11236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Hong LZ, Hong S, Wong HT, Aw PPK, Cheng Y, Wilm A, de Sessions PF, Lim SG, Nagarajan N, Hibberd ML, Quake SR, Burkholder WF. 2014. BAsE-Seq: a method for obtaining long viral haplotypes from short sequence reads. Genome Biol 15:517. 10.1186/PREACCEPT-6768001251451949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Stapleton JA, Kim J, Hamilton JP, Wu M, Irber LC, Maddamsetti R, Briney B, Newton L, Burton DR, Brown CT, Chan C, Buell CR, Whitehead TA. 2016. Haplotype-phased synthetic long reads from short-read sequencing. PLoS One 11:e0147229. 10.1371/journal.pone.0147229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Sharon I, Kertesz M, Hug LA, Pushkarev D, Blauwkamp TA, Castelle CJ, Amirebrahimi M, Thomas BC, Burstein D, Tringe SG, Williams KH, Banfield JF. 2015. Accurate, multi-kb reads resolve complex populations and detect rare microorganisms. Genome Res 25:534–543. 10.1101/gr.183012.114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Burke C, Darling A. 2016. A method for high precision sequencing of near full-length 16S rRNA genes on an Illumina MiSeq. PeerJ 4:e2492. 10.7717/peerj.2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.White R, Bottos E, Chowdhury T, Zucker J, Brislawn C, Nicora C, Fansler S, Glaesemann K, Glass K, Jansson J. 2016. Moleculo long-read sequencing facilitates assembly and genomic binning from complex soil metagenomes. mSystems 1:e00045-16. 10.1128/mSystems.00045-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Karst S, Dueholm M, McIlroy S, Kirkegaard R, Nielsen P, Albertsen M. 2018. Retrieval of a million high‐quality, full-length microbial 16S and 18S rRNA gene sequences without primer bias. Nat Biotechnol 36:190–195. 10.1038/nbt.4045. [DOI] [PubMed] [Google Scholar]
- 71.Bishara A, Liu Y, Weng Z, Kashef-Haghighi D, Newburger D, West R, Sidow A, Batzoglou S. 2015. Read clouds uncover variation in complex regions of the human genome. Genome Res 25:1570–1580. 10.1101/gr.191189.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zheng GXY, Lau BT, Schnall-Levin M, Jarosz M, Bell JM, Hindson CM, Kyriazopoulou-Panagiotopoulou S, Masquelier DA, Merrill L, Terry JM, Mudivarti PA, Wyatt PW, Bharadwaj R, Makarewicz AJ, Li Y, Belgrader P, Price AD, Lowe AJ, Marks P, Vurens GM, Hardenbol P, Montesclaros L, Luo M, Greenfield L, Wong A, Birch DE, Short SW, Bjornson KP, Patel P, Hopmans ES, Wood C, Kaur S, Lockwood GK, Stafford D, Delaney JP, Wu I, Ordonez HS, Grimes SM, Greer S, Lee JY, Belhocine K, Giorda KM, Heaton WH, McDermott GP, Bent ZW, Meschi F, Kondov NO, Wilson R, Bernate JA, Gauby S, et al. 2016. Haplotyping germline and cancer genomes with high-throughput linked-read sequencing. Nat Biotechnol 34:303–311. 10.1038/nbt.3432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Wang O, Chin R, Cheng X, Wu MKY, Mao Q, Tang J, Sun Y, Anderson E, Lam HK, Chen D, Zhou Y, Wang L, Fan F, Zou Y, Xie Y, Zhang RY, Drmanac S, Nguyen D, Xu C, Villarosa C, Gablenz S, Barua N, Nguyen S, Tian W, Liu JS, Wang J, Liu X, Qi X, Chen A, Wang H, Dong Y, Zhang W, Alexeev A, Yang H, Wang J, Kristiansen K, Xu X, Drmanac R, Peters BA. 2019. Efficient and unique cobarcoding of second-generation sequencing reads from long DNA molecules enabling cost-effective and accurate sequencing, haplotyping, and de novo assembly. Genome Res 29:798–808. 10.1101/gr.245126.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wu I, Kim HS, Ben-Yehezkel T. 2019. A single-molecule long-read survey of human transcriptomes using LoopSeq synthetic long read sequencing. bioRxiv 10.1101/532135. [DOI]
- 75.Callahan B, Grinevich D, Thakur S, Balamotis M, Yehezkel T. 2020. Ultra-accurate microbial amplicon sequencing with synthetic long reads. bioRxiv 10.1101/2020.07.07.192286. [DOI] [PMC free article] [PubMed]
- 76.Chung N, Van GMW, Preston M, Lhota F, Cerna L, Garcia-Pichel F, Fernandes V, Giraldo-Silva A, Kim H, Hurowitz E, Balamotis M. 2020. Accurate microbiome sequencing with synthetic long read sequencing. bioRxiv 10.1101/2020.10.02.324038. [DOI]
- 77.Coissac E, Hollingsworth P, Lavergne S, Taberlet P. 2016. From barcodes to genomes: extending the concept of DNA barcoding. Mol Ecol 25:1423–1428. 10.1111/mec.13549. [DOI] [PubMed] [Google Scholar]
- 78.Wurzbacher C, Larsson E, Bengtsson-Palme J, Van den Wyngaert S, Svantesson S, Kristiansson E, Kagami M, Nilsson R. 2019. Introducing ribosomal tandem repeat barcoding for fungi. Mol Ecol Resour 19:118–127. 10.1111/1755-0998.12944. [DOI] [PubMed] [Google Scholar]
- 79.Misas E, Gómez O, Botero V, Muñoz J, Teixeira M, Gallo J, Clay O, McEwen J. 2020. Updates and comparative analysis of the mitochondrial genomes of Paracoccidioides spp. using Oxford Nanopore MinION sequencing. Front Microbiol 11:1751. 10.3389/fmicb.2020.01751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Simmons D, Bonds A, Castillo B, Clemons R, Glasco A, Myers J, Thapa N, Letcher P, Powell M, Longcore J, James T. 2020. The Collection of Zoosporic Eufungi at the University of Michigan (CZEUM): introducing a new repository of barcoded Chytridiomyceta and Blastocladiomycota cultures. IMA Fung 11:20. 10.1186/s43008-020-00041-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Liu S, Yang C, Zhou C, Zhou X. 2017. Filling reference gaps via assembling DNA barcodes using high-throughput sequencing—moving toward barcoding the world. GigaScience 6:gix104. 10.1093/gigascience/gix104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Hebert PDN, Braukmann TWA, Prosser SWJ, Ratnasingham S, deWaard JR, Ivanova NV, Janzen DH, Hallwachs W, Naik S, Sones JE, Zakharov EV. 2018. A sequel to Sanger: amplicon sequencing that scales. BMC Genomics 19:219. 10.1186/s12864-018-4611-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Srivathsan A, Baloğlu B, Wang W, Tan W, Bertrand D, Ng A, Boey E, Koh J, Nagarajan N, Meier R. 2018. A MinION™‐based pipeline for fast and cost‐effective DNA barcoding. Mol Ecol Resour 18:1035–1049. 10.1111/1755-0998.12890. [DOI] [PubMed] [Google Scholar]
- 84.Morrison G, Fu J, Lee G, Wiederhold N, Cañete-Gibas C, Bunnik E, Wickes B. 2020. Nanopore sequencing of the fungal intergenic spacer sequence as a potential rapid diagnostic assay. J Clin Microbiol 58:e01972-20. 10.1128/JCM.01972-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gueidan C, Elix J, McCarthy P, Roux C, Mallen-Cooper M, Kantvilas G. 2019. PacBio amplicon sequencing for metabarcoding of mixed DNA samples from lichen herbarium specimens. MycoKeys 53:73–91. 10.3897/mycokeys.53.34761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Taberlet P, Bonin A, Coissac E, Zinger L. 2018. Environmental DNA: for biodiversity research and monitoring. Oxford University Press, Oxford, England. [Google Scholar]
- 87.Nelson J, Hauser D, Li F. 2020. Symbiotic cyanobacteria communities in hornworts across time, space, and host species. bioRxiv 10.1101/2020.06.18.160382. [DOI]
- 88.Ramoneda J, Le RJJ, Frossard E, Frey B, Gamper H. 2020. Experimental assembly reveals ecological drift as a major driver of root nodule bacterial diversity in a woody legume crop. FEMS Microbiol Ecol 96:fiaa083. 10.1093/femsec/fiaa083. [DOI] [PubMed] [Google Scholar]
- 89.Jamy M, Foster R, Barbera P, Czech L, Kozlov A, Stamatakis A, Bending G, Hilton S, Bass D, Burki F. 2020. Long‐read metabarcoding of the eukaryotic rDNA operon to phylogenetically and taxonomically resolve environmental diversity. Mol Ecol Resour 20:429–443. 10.1111/1755-0998.13117. [DOI] [PubMed] [Google Scholar]
- 90.Tedersoo L, Anslan S, Bahram M, Kõljalg U, Abarenkov K. 2020. Identifying the ‘unidentified’ fungi: a global-scale long-read third-generation sequencing approach. Fung Div 103:273–293. 10.1007/s13225-020-00456-4. [DOI] [Google Scholar]
- 91.Hanafy R, Johnson B, Youssef N, Elshahed M. 2020. Assessing anaerobic gut fungal (Neocalliamstigomycota) diversity using PacBio D1/D2 LSU rRNA amplicon sequencing and multi-year isolation. bioRxiv 10.1101/2020.03.24.005967. [DOI] [PubMed]
- 92.Golparian D, Donà V, Sánchez-Busó L, Foerster S, Harris S, Endimiani A, Low N, Unemo M. 2018. Antimicrobial resistance prediction and phylogenetic analysis of Neisseria gonorrhoeae isolates using the Oxford Nanopore MinION sequencer. Sci Rep 8:17596. 10.1038/s41598-018-35750-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.D'Andreano S, Cuscó A, Francino O. 2020. Rapid and real-time identification of fungi up to the species level with long amplicon Nanopore sequencing from clinical samples. bioRxiv 10.1101/2020.02.06.936708. [DOI] [PMC free article] [PubMed]
- 94.Badial BA, Sherman D, Stone A, Gopakumar A, Wilson V, Schneider W, King J. 2018. Nanopore sequencing as a surveillance tool for plant pathogens in plant and insect tissues. Plant Dis 20:1648–1652. 10.1094/PDIS-04-17-0488-RE. [DOI] [PubMed] [Google Scholar]
- 95.Loit K, Adamson K, Bahram M, Puusepp R, Anslan S, Kiiker R, Drenkhan R, Tedersoo L. 2019. Relative performance of MinION (Oxford Nanopore Technologies) versus Sequel (Pacific Biosciences) third-generation sequencing instruments in identification of agricultural and forest fungal pathogens. Appl Environ Microbiol 85:e01368-19. 10.1128/AEM.01368-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Llontop MEM, Sharma P, Aguilera Flores M, Yang S, Pollok J, Tian L, Huang C, Rideout S, Heath L, Li S, Vinatzer B. 2020. Strain-level identification of bacterial tomato pathogens directly from metagenomic sequences. Phytopathology 110:768–779. 10.1094/PHYTO-09-19-0351-R. [DOI] [PubMed] [Google Scholar]
- 97.Hatfield R, Batista F, Bean T, Fonseca V, Santos A, Turner A, Lewis A, Dean K, Martinez-Urtaza J. 2020. The application of Nanopore sequencing technology to the study of Dinoflagellates: a proof of concept study for rapid sequence-based discrimination of potentially harmful algae. Front Microbiol 11:844. 10.3389/fmicb.2020.00844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Egeter B, Veríssimo J, Lopes-Lima M, Chaves C, Pinto J, Riccardi N, Beja P, Fonseca N. 2020. Speeding up the detection of invasive aquatic species using environmental DNA and nanopore sequencing. bioRxiv 10.1101/2020.06.09.142521. [DOI] [PubMed]
- 99.Peto L, Rodger G, Carter D, Osman K, Yavuz M, Johnson K, Raza M, Parker M, Wyles M, Andersson M, Justice A. 2020. Diagnosis of SARS-CoV-2 infection with LamPORE, a high-throughput platform combining loop-mediated isothermal amplification and nanopore sequencing. MedRxiv 10.1101/2020.09.18.20195370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Grohme M, Soler R, Wink M, Frohme M. 2013. Microsatellite marker discovery using single molecule real-time circular consensus sequencing on the Pacific Biosciences RS. Biotechniques 55:253–256. 10.2144/000114104. [DOI] [PubMed] [Google Scholar]
- 101.Huang D, Riley C, Jiang M, Zheng X, Liang D, Rehman M, Highbarger H, Jiao X, Sherman B, Ma L, Chen X, Skelly T. 2016. Towards better precision medicine: PacBio single-molecule long reads resolve the interpretation of HIV drug resistant mutation profiles at explicit quasispecies (haplotype) level. J Data Mining Genomics Proteomics 7:182. 10.4172/2153-0602.1000182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Deutscher A, Burke C, Darling A, Riegler M, Reynolds O, Chapman T. 2018. Near full-length 16S rRNA gene next-generation sequencing revealed Asaia as a common midgut bacterium of wild and domesticated Queensland fruit fly larvae. Microbiome 6:85. 10.1186/s40168-018-0463-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Fuller ZL, Mocellin VJL, Morris LA, Cantin N, Shepherd J, Sarre L, Peng J, Liao Y, Pickrell J, Andolfatto P, Matz M, Bay LK, Przeworski M. 2020. Population genetics of the coral Acropora millepora: toward genomic prediction of bleaching. Science 369:eaba4674. 10.1126/science.aba4674. [DOI] [PubMed] [Google Scholar]
- 104.Hsieh P, Vollger MR, Dang V, Porubsky D, Baker C, Cantsilieris S, Hoekzema K, Lewis AP, Munson KM, Sorensen M, Kronenberg ZN, Murali S, Nelson BJ, Chiatante G, Maggiolini FAM, Blanché H, Underwood JG, Antonacci F, Deleuze J-F, Eichler EE. 2019. Adaptive archaic introgression of copy number variants and the discovery of previously unknown human genes. Science 366:eaax2083. 10.1126/science.aax2083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Bishara A, Moss EL, Kolmogorov M, Parada AE, Weng Z, Sidow A, Dekas AE, Batzoglou S, Bhatt AS. 2018. High quality genome sequences of uncultured microbes by assembly of read clouds. Nat Biotechnol 36:1067–1075. 10.1038/nbt.4266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Addetia A, Tantalo L, Lin M, Xie H, Huang M, Marra C, Greninger A. 2020. Comparative genomics and full-length Tprk profiling of Treponema pallidum subsp. pallidum reinfection. PLoS Negl Trop Dis 14:e0007921. 10.1371/journal.pntd.0007921. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Negreira G, Monsieurs P, Imamura H, Maes I, Kuk N, Yagoubat A, Van den Broeck F, Sterkers Y, Dujardin J, Domagalska M. 2020. Exploring the evolution and adaptive role of mosaic aneuploidy in a clonal Leishmania donovani population using high throughput single-cell genome sequencing. bioRxiv 10.1101/2020.03.05.976233. [DOI] [PMC free article] [PubMed]
- 108.Kumar R, Register K, Christopher-Hennings J, Moroni P, Gioia G, Garcia-Fernandez N, Nelson J, Jelinski MD, Lysnyansky I, Bayles D, Alt D, Scaria J. 2020. Population genomic analysis of Mycoplasma bovis elucidates geographical variations and genes associated with host-types. Microorganisms 8:1561. 10.3390/microorganisms8101561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Maeda T, Kobayashi Y, Nakagawa T, Ezawa T, Yamaguchi K, Bino T, Nishimoto Y, Shigenobu S, Kawaguchi M. 2020. Conservation and host-specific expression of non-tandemly repeated heterogenous ribosome RNA gene in arbuscular mycorrhizal fungi. bioRxiv 10.1101/2020.05.14.095489. [DOI]
- 110.Tremble K, Suz L, Dentinger B. 2020. Lost in translation: population genomics and long-read sequencing reveals relaxation of concerted evolution of the ribosomal DNA cistron. Mol Phylogenet Evol 148:106804. 10.1016/j.ympev.2020.106804. [DOI] [PubMed] [Google Scholar]
- 111.Brewer T, Albertsen M, Edwards A, Kirkegaard R, Rocha E, Fierer N. 2020. Unlinked rRNA genes are widespread among bacteria and archaea. ISME J 14:597–608. 10.1038/s41396-019-0552-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Koren S, Phillippy A. 2015. One chromosome, one contig: complete microbial genomes from long-read sequencing and assembly. Curr Opin Microbiol 23:110–120. 10.1016/j.mib.2014.11.014. [DOI] [PubMed] [Google Scholar]
- 113.Dumschott K, Schmidt M, Chawla H, Snowdon R, Usadel B. 2020. Oxford Nanopore Sequencing: new opportunities for plant genomics? J Exp Bot 71:5313–5322. 10.1093/jxb/eraa263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Warwick-Dugdale J, Solonenko N, Moore K, Chittick L, Gregory A, Allen M, Sullivan M, Temperton B. 2019. Long-read viral metagenomics captures abundant and microdiverse viral populations and their niche-defining genomic islands. PeerJ 7:e6800. 10.7717/peerj.6800. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Singleton C, Petriglieri F, Kristensen J, Kirkegaard R, Michaelsen T, Andersen M, Kondrotaite Z, Karst S, Dueholm M, Nielsen P, Albertsen M. 2020. Connecting structure to function with the recovery of over 1000 high-quality activated sludge metagenome-assembled genomes encoding full-length rRNA genes using long-read sequencing. bioRxiv 10.1101/2020.05.12.088096. [DOI] [PMC free article] [PubMed]
- 116.Leggett RM, Alcon-Giner C, Heavens D, Caim S, Brook TC, Kujawska M, Martin S, Peel N, Acford-Palmer H, Hoyles L, Clarke P, Hall LJ, Clark MD. 2020. Rapid MinION profiling of preterm microbiota and antimicrobial-resistant pathogens. Nat Microbiol 5:430–442. 10.1038/s41564-019-0626-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Reddington K, Eccles D, O'Grady J, Drown DM, Hansen LH, Nielsen TK, Ducluzeau A-L, Leggett RM, Heavens D, Peel N, Snutch TP, Bayega A, Oikonomopoulos S, Ragoussis J, Barry T, van der Helm E, Jolic D, Richardson H, Jansen H, Tyson JR, Jain M, Brown BL. 2020. Metagenomic analysis of planktonic riverine microbial consortia using nanopore sequencing reveals insight into river microbe taxonomy and function. GigaScience 9:giaa053. 10.1093/gigascience/giaa053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sato M, Ogura Y, Nakamura K, Nishida R, Gotoh Y, Hayashi M, Hisatsune J, Sugai M, Takehiko I, Hayashi T. 2019. Comparison of the sequencing bias of currently available library preparation kits for Illumina sequencing of bacterial genomes and metagenomes. DNA Res 26:391–398. 10.1093/dnares/dsz017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Reference deleted.
- 120.Peel N, Dicks L, Clark M, Heavens D, Percival‐Alwyn L, Cooper C, Davies R, Leggett R, Yu D. 2019. Semi‐quantitative characterisation of mixed pollen samples using MinION sequencing and reverse metagenomics (RevMet). Methods Ecol Evol 10:1690–1701. 10.1111/2041-210X.13265. [DOI] [Google Scholar]
- 121.McLaren M, Willis A, Callahan B. 2019. Consistent and correctable bias in metagenomic sequencing experiments. Elife 8:e46923. 10.7554/eLife.46923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Freed N, Pearman W, Smith A, Breckell G, Dale J, Freed N, Silander O. 2020. New tools for diet analyses: nanopore sequencing of metagenomic DNA from rat stomach contents to quantify diet. bioRxiv 10.1101/363622. [DOI]
- 123.Lowe R, Shirley N, Bleackley M, Dolan S, Shafee T. 2017. Transcriptomics technologies. PLoS Comput Biol 13:e1005457. 10.1371/journal.pcbi.1005457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Picelli S, Bjorklund A, Faridani O, Sagasser S, Winberg G, Sandberg R. 2013. Smart-seq2 for sensitive full-length transcriptome profiling in single cells. Nat Methods 10:1096–1098. 10.1038/nmeth.2639. [DOI] [PubMed] [Google Scholar]
- 125.Russell A, Elshina E, Kowalsky J, te Velthuis AJW, Bloom J. 2019. Single-cell virus sequencing of influenza infections that trigger innate immunity. J Virol 93:e00500-19. 10.1128/JVI.00500-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Zhao Z, Elsik C, Hibbard B, Shelby K. 2020. Detection of alternative splicing in Diabrotica virgifera virgifera LeConte, in association with Bt resistance using RNA-seq and PacBio Iso-Seq. bioRxiv 10.1101/2020.08.03.234682. [DOI] [PubMed]
- 127.Semmouri I, De Schamphelaere KA, Mees J, Janssen CR, Asselman J. 2020. Evaluating the potential of direct RNA nanopore sequencing: metatranscriptomics highlights possible seasonal differences in a marine pelagic crustacean zooplankton community. Mar Environ Res 153:104836. 10.1016/j.marenvres.2019.104836. [DOI] [PubMed] [Google Scholar]
- 128.Depledge D, Srinivas K, Sadaoka T, Bready D, Mori Y, Placantonakis D, Mohr I, Wilson A. 2019. Direct RNA sequencing on nanopore arrays redefines the transcriptional complexity of a viral pathogen. Nat Commun 10:754. 10.1038/s41467-019-08734-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Flusberg B, Webster D, Lee J, Travers K, Olivares E, Clark T, Korlach J, Turner S. 2010. Direct detection of DNA methylation during single-molecule, real-time sequencing. Nat Methods 7:461–465. 10.1038/nmeth.1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Rand A, Jain M, Eizenga J, Musselman-Brown A, Olsen H, Akeson M, Paten B. 2017. Mapping DNA methylation with high-throughput nanopore sequencing. Nat Methods 14:411–413. 10.1038/nmeth.4189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Beaulaurier J, Schadt E, Fang G. 2019. Deciphering bacterial epigenomes using modern sequencing technologies. Nat Rev Genet 20:157–172. 10.1038/s41576-018-0081-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mondo SJ, Dannebaum RO, Kuo RC, Louie KB, Bewick AJ, LaButti K, Haridas S, Kuo A, Salamov A, Ahrendt SR, Lau R, Bowen BP, Lipzen A, Sullivan W, Andreopoulos BB, Clum A, Lindquist E, Daum C, Northen TR, Kunde-Ramamoorthy G, Schmitz RJ, Gryganskyi A, Culley D, Magnuson J, James TY, O'Malley MA, Stajich JE, Spatafora JW, Visel A, Grigoriev IV. 2017. Widespread adenine N6-methylation of active genes in fungi. Nat Genet 49:964–968. 10.1038/ng.3859. [DOI] [PubMed] [Google Scholar]
- 133.He C, Zhang Z, Li B, Tian S. 2020. the pattern and function of DNA methylation in fungal plant pathogens. Microorganisms 8:227. 10.3390/microorganisms8020227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fang G, Munera D, Friedman DI, Mandlik A, Chao MC, Banerjee O, Feng Z, Losic B, Mahajan MC, Jabado OJ, Deikus G, Clark TA, Luong K, Murray IA, Davis BM, Keren-Paz A, Chess A, Roberts RJ, Korlach J, Turner SW, Kumar V, Waldor MK, Schadt EE. 2012. Genome-wide mapping of methylated adenine residues in pathogenic Escherichia coli using single-molecule real-time sequencing. Nat Biotechnol 30:1232–1239. 10.1038/nbt.2432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kumar S, Karmakar B, Nagarajan D, Mukhopadhyay A, Morgan R, Rao D. 2018. N4-cytosine DNA methylation regulates transcription and pathogenesis in Helicobacter pylori. Nucleic Acids Res 46:3429–3445. 10.1093/nar/gky126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Anjum A, Brathwaite K, Aidley J, Connerton P, Cummings N, Parkhill J, Connerton I, Bayliss C. 2016. Phase variation of a Type IIG restriction-modification enzyme alters site-specific methylation patterns and gene expression in Campylobacter jejuni strain NCTC11168. Nucleic Acids Res 44:4581–4594. 10.1093/nar/gkw019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Beaulaurier J, Zhu S, Deikus G, Mogno I, Zhang X-S, Davis-Richardson A, Canepa R, Triplett EW, Faith JJ, Sebra R, Schadt EE, Fang G. 2018. Metagenomic binning and association of plasmids with bacterial host genomes using DNA methylation. Nat Biotechnol 36:61–69. 10.1038/nbt.4037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Cao J, Zhang Y, Dai M, Xu J, Chen L, Zhang F, Zhao N, Wang J. 2020. Profiling of human gut virome with Oxford Nanopore Technology. Med Microecol 4:100012. 10.1016/j.medmic.2020.100012. [DOI] [Google Scholar]
- 139.Berry D, Loy A. 2018. Stable-isotope probing of human and animal microbiome function. Trends Microbiol 26:999–1007. 10.1016/j.tim.2018.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Specht E, Braselmann E, Palmer A. 2017. A critical and comparative review of fluorescent tools for live-cell imaging. Annu Rev Physiol 79:93–117. 10.1146/annurev-physiol-022516-034055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Wang D, He P, Wang Z, Li G, Majed N, Gu A. 2020. Advances in single cell Raman spectroscopy technologies for biological and environmental applications. Curr Opin Biotechnol 64:218–229. 10.1016/j.copbio.2020.06.011. [DOI] [PubMed] [Google Scholar]
- 142.Zheng GXY, Terry JM, Belgrader P, Ryvkin P, Bent ZW, Wilson R, Ziraldo SB, Wheeler TD, McDermott GP, Zhu J, Gregory MT, Shuga J, Montesclaros L, Underwood JG, Masquelier DA, Nishimura SY, Schnall-Levin M, Wyatt PW, Hindson CM, Bharadwaj R, Wong A, Ness KD, Beppu LW, Deeg HJ, McFarland C, Loeb KR, Valente WJ, Ericson NG, Stevens EA, Radich JP, Mikkelsen TS, Hindson BJ, Bielas JH. 2017. Massively parallel digital transcriptional profiling of single cells. Nat Commun 8:14049. 10.1038/ncomms14049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Kashima Y, Sakamoto Y, Kaneko K, Seki M, Suzuki Y, Suzuki A. 2020. Single-cell sequencing techniques from individual to multiomics analyses. Exp Mol Med 52:1419–1427. 10.1038/s12276-020-00499-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Byrne A, Beaudin A, Olsen H, Jain M, Cole C, Palmer T, DuBois R, Forsberg E, Akeson M, Vollmers C. 2017. Nanopore long-read RNAseq reveals widespread transcriptional variation among the surface receptors of individual B cells. Nat Commun 8:16027. 10.1038/ncomms16027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Dapprich J, Ferriola D, Mackiewicz K, Clark PM, Rappaport E, D'Arcy M, Sasson A, Gai X, Schug J, Kaestner KH, Monos D. 2016. The next generation of target capture technologies-large DNA fragment enrichment and sequencing determines regional genomic variation of high complexity. BMC Genomics 17:486. 10.1186/s12864-016-2836-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Lefoulon E, Vaisman N, Frydman HM, Sun L, Voland L, Foster JM, Slatko BE. 2019. Large enriched fragment targeted sequencing (LEFT-SEQ) applied to capture of Wolbachia genomes. Sci Rep 9:5939. 10.1038/s41598-019-42454-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Sheynkman GM, Tuttle KS, Laval F, Tseng E, Underwood JG, Yu L, Dong D, Smith ML, Sebra R, Willems L, Hao T, Calderwood MA, Hill DE, Vidal M. 2020. ORF Capture-Seq as a versatile method for targeted identification of full-length isoforms. Nat Commun 11:2326. 10.1038/s41467-020-16174-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tsai Y, Greenberg D, Powell J, Höijer I, Ameur A, Strahl M, Ellis E, Jonasson I, Pinto R, Wheeler V, Smith M. 2017. Amplification-free, CRISPR-Cas9 targeted enrichment and SMRT sequencing of repeat-expansion disease causative genomic regions. bioRxiv 10.1101/203919. [DOI]
- 149.Gilpatrick T, Lee I, Graham J, Raimondeau E, Bowen R, Heron A, Downs B, Sukumar S, Sedlazeck F, Timp W. 2020. Targeted nanopore sequencing with Cas9-guided adapter ligation. Nat Biotechnol 38:433–438. 10.1038/s41587-020-0407-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Loose M, Malla S, Stout M. 2016. Real time selective sequencing using nanopore technology. Nat Methods 13:751–754. 10.1038/nmeth.3930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Kovaka S, Fan Y, Ni B, Timp W, Schatz M. 2020. Targeted nanopore sequencing by real-time mapping of raw electrical signal with UNCALLED. bioRxiv 10.1101/2020.02.03.931923. [DOI] [PMC free article] [PubMed]
- 152.Kivioja T, Vähärautio A, Karlsson K, Bonke M, Enge M, Linnarsson S, Taipale J. 2011. Counting absolute numbers of molecules using unique molecular identifiers. Nat Methods 9:72–74. 10.1038/nmeth.1778. [DOI] [PubMed] [Google Scholar]
- 153.Amarasinghe S, Su S, Dong X, Zappia L, Ritchie M, Gouil Q. 2020. Opportunities and challenges in long-read sequencing data analysis. Genome Biol 21:30. 10.1186/s13059-020-1935-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kolmogorov M, Bickhart D, Behsaz B, Gurevich A, Rayko M, Shin S, Kuhn K, Yuan J, Polevikov E, Smith T, Pevzner P. 2020. metaFlye: scalable long-read metagenome assembly using repeat graphs. Nat Methods 17:1103–1110. 10.1038/s41592-020-00971-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Fritz A, Hofmann P, Majda S, Dahms E, Dröge J, Fiedler J, Lesker TR, Belmann P, DeMaere MZ, Darling AE, Sczyrba A, Bremges A, McHardy AC. 2019. CAMISIM: simulating metagenomes and microbial communities. Microbiome 7:17. 10.1186/s40168-019-0633-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Chen L, Anantharaman K, Shaiber A, Eren A, Banfield J. 2020. Accurate and complete genomes from metagenomes. Genome Res 30:315–333. 10.1101/gr.258640.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dilthey A, Jain C, Koren S, Phillippy A. 2019. Strain-level metagenomic assignment and compositional estimation for long reads with MetaMaps. Nat Commun 10:3066. 10.1038/s41467-019-10934-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Maric J, Krizanovic K, Riondet S, Nagarajan N, Sikic M. 2020. Benchmarking metagenomic classification tools for long-read sequencing data. bioRxiv 10.1101/2020.11.25.397729. [DOI]
- 159.Břinda K, Callendrello A, Ma KC, MacFadden DR, Charalampous T, Lee RS, Cowley L, Wadsworth CB, Grad YH, Kucherov G, O'Grady J, Baym M, Hanage WP. 2020. Rapid inference of antibiotic resistance and susceptibility by genomic neighbour typing. Nat Microbiol 5:455–464. 10.1038/s41564-019-0656-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Edgar R. 2016. UNOISE2: improved error-correction for Illumina 16S and ITS amplicon sequencing. bioRxiv 10.1101/081257. [DOI]
- 161.Dueholm M, Andersen K, McIlroy S, Kristensen J, Yashiro E, Karst S, Albertsen M, Nielsen P. 2020. Generation of comprehensive ecosystem-specific reference databases with species-level resolution by high-throughput full-length 16S rRNA gene sequencing and automated taxonomy assignment (AutoTax). mBio 11:e01557-20. 10.1128/mBio.01557-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Edgar R. 2016. SINTAX: a simple non-Bayesian taxonomy classifier for 16S and ITS sequences. bioRxiv 10.1101/074161. [DOI]
- 163.Murali A, Bhargava A, Wright E. 2018. IDTAXA: a novel approach for accurate taxonomic classification of microbiome sequences. Microbiome 6:140. 10.1186/s40168-018-0521-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Escapa I, Huang Y, Chen T, Lin M, Kokaras A, Dewhirst F, Lemon K. 2020. Construction of habitat-specific training sets to achieve species-level assignment in 16S rRNA gene datasets. Microbiome 8:65. 10.1186/s40168-020-00841-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Del Campo J, Kolisko M, Boscaro V, Santoferrara LF, Nenarokov S, Massana R, Guillou L, Simpson A, Berney C, de Vargas C, Brown MW, Keeling PJ, Wegener Parfrey L. 2018. EukRef: phylogenetic curation of ribosomal RNA to enhance understanding of eukaryotic diversity and distribution. PLoS Biol 16:e2005849. 10.1371/journal.pbio.2005849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Jain M, Koren S, Miga KH, Quick J, Rand AC, Sasani TA, Tyson JR, Beggs AD, Dilthey AT, Fiddes IT, Malla S, Marriott H, Nieto T, O'Grady J, Olsen HE, Pedersen BS, Rhie A, Richardson H, Quinlan AR, Snutch TP, Tee L, Paten B, Phillippy AM, Simpson JT, Loman NJ, Loose M. 2018. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat Biotechnol 36:338–345. 10.1038/nbt.4060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Beaulaurier J, Luo E, Eppley J, Den Uyl P, Dai X, Burger A, Turner D, Pendelton M, Juul S, Harrington E, DeLong E. 2020. Assembly-free single-molecule sequencing recovers complete virus genomes from natural microbial communities. Genome Res 30:437–446. 10.1101/gr.251686.119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Wongsurawat T, Jenjaroenpun P, Taylor MK, Lee J, Tolardo AL, Parvathareddy J, Kandel S, Wadley TD, Kaewnapan B, Athipanyasilp N, Skidmore A, Chung D, Chaimayo C, Whitt M, Kantakamalakul W, Sutthent R, Horthongkham N, Ussery DW, Jonsson CB, Nookaew I. 2019. Rapid sequencing of multiple RNA viruses in their native form. Front Microbiol 10:260. 10.3389/fmicb.2019.00260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Viehweger A, Krautwurst S, Lamkiewicz K, Madhugiri R, Ziebuhr J, Hölzer M, Marz M. 2019. Direct RNA nanopore sequencing of full-length coronavirus genomes provides novel insights into structural variants and enables modification analysis. Genome Res 29:1545–1554. 10.1101/gr.247064.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Sood A, Viner C, Hoffman M. 2019. DNAmod: the DNA modification database. J Cheminform 11:30. 10.1186/s13321-019-0349-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Georgieva D, Liu Q, Wang K, Egli D. 2020. Detection of base analogs incorporated during DNA replication by nanopore sequencing. Nucleic Acids Res 48:e88. 10.1093/nar/gkaa517. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Item S1. Download AEM.00626-21-s0001.pdf, PDF file, 1.1 MB (116.3KB, pdf)