

Abstract
Phylogenetic trees based on genome-wide sequence data may not always represent the true evolutionary history for a variety of reasons. One process that can lead to incorrect reconstruction of species phylogenies is gene flow, especially if interspecific gene flow has affected large parts of the genome. We investigated phylogenetic relationships within a clade comprising eight species of passerine birds (Phylloscopidae, Phylloscopus, leaf warblers) using one de novo genome assembly and 78 resequenced genomes. On the basis of hypothesis-exclusion trials based on D-statistics, phylogenetic network analysis, and demographic inference analysis, we identified ancient gene flow affecting large parts of the genome between one species and the ancestral lineage of a sister species pair. This ancient gene flow consistently caused erroneous reconstruction of the phylogeny when using large amounts of genome-wide sequence data. In contrast, the true relationships were captured when smaller parts of the genome were analyzed, showing that the “winner-takes-all democratic majority tree” is not necessarily the true species tree. Under this condition, smaller amounts of data may sometimes avoid the effects of gene flow due to stochastic sampling, as hidden reticulation histories are more likely to emerge from the use of larger data sets, especially whole-genome data sets. In addition, we also found that genomic regions affected by ancient gene flow generally exhibited higher genomic differentiation but a lower recombination rate and nucleotide diversity. Our study highlights the importance of considering reticulation in phylogenetic reconstructions in the genomic era.[Bifurcation; introgression; recombination; reticulation; Phylloscopus.]
Early phylogenetic reconstructions were typically based on single or a small number of genes, and the resulting phylogenetic trees were widely interpreted as true species trees (Felsenstein 2004). However, it is now widely appreciated that individual gene trees may differ from the species phylogeny due to, for example, gene duplication, idiosyncratic lineage sorting and/or introgressive hybridization (Pamilo and Nei 1988; Maddison 1997; Nichols 2001; Avise and Robinson 2008; Degnan and Rosenberg 2009). Incongruence between the species phylogeny and the gene trees contained within it, as well as among gene trees, is pervasive (even 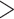 50%) when internodal times are short in relation to the effective population sizes of internodal populations (Degnan and Rosenberg 2006). Under such circumstances, concatenation methods for phylogenetic inference have been shown to be statistically inconsistent, meaning that a wrong tree is more likely to be reconstructed as more data are added (Kubatko and Degnan 2007). “Species tree methods” based on the multispecies coalescent model (Rannala and Yang 2003) are more likely to reconstruct the true species phylogeny, especially in the “anomaly zone” where gene trees incongruent with the phylogeny are more likely than gene trees that agree with the species phylogeny (Edwards 2009; Liu et al. 2019).
50%) when internodal times are short in relation to the effective population sizes of internodal populations (Degnan and Rosenberg 2006). Under such circumstances, concatenation methods for phylogenetic inference have been shown to be statistically inconsistent, meaning that a wrong tree is more likely to be reconstructed as more data are added (Kubatko and Degnan 2007). “Species tree methods” based on the multispecies coalescent model (Rannala and Yang 2003) are more likely to reconstruct the true species phylogeny, especially in the “anomaly zone” where gene trees incongruent with the phylogeny are more likely than gene trees that agree with the species phylogeny (Edwards 2009; Liu et al. 2019).
While species tree methods can overcome the problem of gene tree heterogeneity, most of them cannot effectively handle introgression, as the multispecies coalescent model assumes lack of gene flow among species. A recently published multispecies-coalescent-with-introgression model accommodates introgression, although genomic data may be needed to properly account for this process (Flouri et al. 2020). If gene flow affects large parts of the genome, even the use of genome-scale information will produce trees that largely reflect the reticulation history (Mallet et al. 2016). One such case is the Anopheles mosquito species complex, in which a whole-genome consensus phylogeny reflected rampant introgression on autosomes, while only a small part of the genome, mostly on the sex chromosomes, was shielded from the effects of gene flow and could be used to infer the correct species branching order (Fontaine et al. 2015). Similar cases have been documented in vertebrates. For example, in a study of an Australasian radiation of Lonchura munias, Faust Stryjewski and Sorenson (2017) detected substantial autosomal introgression and multiple cases in which tree inferences based on loci assumed to code for color differences among species differed significantly from inferences based on genome-wide single-nucleotide variants (SNVs). Accordingly, a simple increase in the amount of sequence data may not be effective in dealing with the problem of gene flow when it affects large parts of the genome. Given the pervasive nature of interspecific gene flow in most animal classes (Mallet 2005; Rheindt and Edwards 2011; Payseur and Rieseberg 2016; Ottenburghs et al. 2017), bifurcating trees may provide a poor or even incorrect representation of the real tree of life when possible reticulation is not taken into account (Edelman et al. 2019). To overcome the complications introduced by gene flow, it has been suggested that genomic regions of low recombination can be used to reconstruct true phylogenetic relationships (Edelman et al. 2019; Li et al. 2019; Martin et al. 2019) because these regions are more likely to be shielded from gene flow due to potentially strong linkage to deleterious alleles (Feder et al. 2012; Nachman and Payseur 2012). However, genomic regions of low recombination can also produce misleading tree topologies as a result of selection (Nater et al. 2015).
Leaf warblers (family Phylloscopidae) are an assemblage of small insectivorous songbirds consisting of 80 species distributed from Africa to Australasia, with the greatest diversity in the Sino-Himalayan mountains (Gill et al. 2020). Two genera were previously recognized within this family: Phylloscopus and Seicercus. However, a multilocus phylogenetic analysis comprising all species in the family showed that Seicercus species were placed in two nonsister clades both nested within Phylloscopus (Alström et al. 2018), leading to a synonymization of Seicercus. One of these two clades comprises the eight species Phylloscopus whistleri, Phylloscopus valentini, Phylloscopus omeiensis, Phylloscopus soror, Phylloscopus burkii, Phylloscopus tephrocephalus, Phylloscopus intermedius, and Phylloscopus poliogenys, which have a particularly chequered taxonomic history and uncertain phylogenetic relationships (Alström et al. 2018 and references therein), and which are the focal species group of the present study.
Lineage divergences within this eight-species clade have been estimated at between 6.3 and 2.3 million years ago (Ma) based on a mitochondrial cytochrome 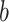 molecular clock calibration (Alström et al. 2018). The monophyly of this clade has been corroborated by multiple studies based on different data sets comprising both mitochondrial and a few nuclear markers (Olsson et al. 2004; Olsson et al. 2005; Johansson et al. 2007; Päckert et al. 2012; Alström et al. 2018) and is also supported by plumage characters (Olsson et al. 2004; Alström et al. 2018). However, its internal phylogenetic relationships remain uncertain, as conflicting tree topologies have been observed in different studies, and even in different analyses of the most comprehensive of these data sets (Alström et al. 2018). Such incongruence may be attributable to lineage sorting effects, such as hemiplasy (Avise and Robinson 2008), or to gene flow. Although there are strong premating reproductive isolating barriers among these eight species (Alström and Olsson 1999; Price 2010), prezygotic isolation may still be ineffective in preventing gene flow according to a simulation study (Irwin 2020). Therefore, both recent and ancient gene flow might explain the high degree of topological incongruence within this clade. Evidence of ancient gene flow, dating back to early stages of lineage divergence, have been found in a growing number of studies based on genomic data (Li et al. 2016; Wen et al. 2016; Zarza et al. 2016; Meier et al. 2017; Burbrink and Gehara 2018; Harris et al. 2018; Thom et al. 2018; Edelman et al. 2019; MacGuigan and Near 2019; Pulido-Santacruz et al. 2020, Rancilhac et al. 2021). Zhang et al. (2019) suggested that the strong mito-nuclear discordance and topological conflicts within another clade of Phylloscopus species were due to “ghost introgression” from an extinct species.
molecular clock calibration (Alström et al. 2018). The monophyly of this clade has been corroborated by multiple studies based on different data sets comprising both mitochondrial and a few nuclear markers (Olsson et al. 2004; Olsson et al. 2005; Johansson et al. 2007; Päckert et al. 2012; Alström et al. 2018) and is also supported by plumage characters (Olsson et al. 2004; Alström et al. 2018). However, its internal phylogenetic relationships remain uncertain, as conflicting tree topologies have been observed in different studies, and even in different analyses of the most comprehensive of these data sets (Alström et al. 2018). Such incongruence may be attributable to lineage sorting effects, such as hemiplasy (Avise and Robinson 2008), or to gene flow. Although there are strong premating reproductive isolating barriers among these eight species (Alström and Olsson 1999; Price 2010), prezygotic isolation may still be ineffective in preventing gene flow according to a simulation study (Irwin 2020). Therefore, both recent and ancient gene flow might explain the high degree of topological incongruence within this clade. Evidence of ancient gene flow, dating back to early stages of lineage divergence, have been found in a growing number of studies based on genomic data (Li et al. 2016; Wen et al. 2016; Zarza et al. 2016; Meier et al. 2017; Burbrink and Gehara 2018; Harris et al. 2018; Thom et al. 2018; Edelman et al. 2019; MacGuigan and Near 2019; Pulido-Santacruz et al. 2020, Rancilhac et al. 2021). Zhang et al. (2019) suggested that the strong mito-nuclear discordance and topological conflicts within another clade of Phylloscopus species were due to “ghost introgression” from an extinct species.
Here, based on one de novo assembled draft genome and 78 resequenced genomes, we explore alternative explanations for the uncertainties surrounding the phylogeny of the clade of these eight Phylloscopus species. We conclude that ancient gene flow affecting large parts of the genome is responsible for past phylogenetic ambiguities based on few markers. We show that such gene flow events may lead to erroneous phylogenetic reconstructions even when incorporating a large amount of genome-wide data, and smaller amounts of data may sometimes avoid the effects of gene flow due to stochastic sampling. Our study also suggests that genomic regions with low recombination are not effective in reconstructing the true phylogeny due to ancient gene flow, and highlights the importance of taking reticulation into account in phylogenetic analysis in the genomic era (Mallet et al. 2016; Edelman et al. 2019; MacGuigan and Near 2019; Pulido-Santacruz et al. 2020).
Materials and Methods
Study Group
We focused on a clade of eight Phylloscopus species that were previously placed in the genus Seicercus: P. whistleri, P. valentini, P. omeiensis, P. soror, P. burkii, P. tephrocephalus, P. intermedius, and P. poliogenys (hereafter the names without the genus name will be used). This eight-species group is monophyletic and has a particularly interesting evolutionary history (Alström et al. 2018 and references therein). We sequenced a total of 79 samples (one de novo assembly and 78 resequenced) covering each of the species as well as multiple populations within some species (Fig. 1; Supplementary Fig. S1 and Table S1 available on Dryad at https://doi.org/10.5061/dryad.fbg79cnsz).
Figure 1.

DNA sampling localities (coloured circles and triangles) and the approximate breeding distributions of the eight Phylloscopus species in the present study. The circles and triangles represent sampling sites. The dashed box in the bottom left of the map represents the study area. The breeding maps are based on BirdLife (https://www.birdlife.org/) and Birds of the World (https://birdsoftheworld.org). Up to four species are sympatric at several localities in China, exceptionally up to five species.
De novo Assembly of the Genome of P. whistleri
We de novo sequenced and assembled the draft genome of a male whistleri (sample id: IOZ_24198; Supplementary Table S1 available on Dryad) using the PacBio Sequel II platform. Raw long reads were processed to remove adapter sequences, low-quality reads and short length reads using in-house scripts. A total of 119.7 Gb quality controlled long reads were obtained. The number of reads was 10,323,141 with an average length of 11.6 Kb. The genome was assembled using canu 2.0 (Koren et al. 2017) with the setting genomeSize 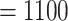 Mb, and leaving other parameters at default. We corrected assembly errors using Gcpp (https://github.com/PacificBiosciences/pbbioconda) by mapping all long reads to the assembled sequences using pbmm2 (https://github.com/PacificBiosciences/pbbioconda).
Mb, and leaving other parameters at default. We corrected assembly errors using Gcpp (https://github.com/PacificBiosciences/pbbioconda) by mapping all long reads to the assembled sequences using pbmm2 (https://github.com/PacificBiosciences/pbbioconda).
Assembly errors were further corrected using Pilon 1.23 (Wang et al. 2014) by mapping short paired-end (PE) reads to the assembled sequences. Raw short reads were generated using the Illumina Novaseq platform. Initial reads were processed to remove adapter sequences, low-quality reads (those with over 50% of bases having Phred quality scores 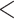 3) and poly-N reads (those with
3) and poly-N reads (those with  3% unidentified nucleotides) using fastp 0.20.0 (Chen et al. 2018), resulting in a total of 62.4 Gb of quality controlled 150 base pair (bp) short PE reads. All short PE reads were mapped to the assembled sequences using Burrows-Wheeler Aligner (BWA) 0.7.12 (Li and Durbin 2009). Merging of the heterozygous contigs was performed using Redundans 0.14c (Pryszcz and Gabaldon 2016) with default parameters. The assembled sequences were blasted to the NCBI nt database using BLASTN 2.2.26 to remove potential contaminating contigs which failed to map to birds or reptiles. The completeness of the genome was estimated by BUSCO 2.0 (Simao et al. 2015). Library preparation, sequencing, and assembly were conducted by Berry Genomics (Beijing, China).
3% unidentified nucleotides) using fastp 0.20.0 (Chen et al. 2018), resulting in a total of 62.4 Gb of quality controlled 150 base pair (bp) short PE reads. All short PE reads were mapped to the assembled sequences using Burrows-Wheeler Aligner (BWA) 0.7.12 (Li and Durbin 2009). Merging of the heterozygous contigs was performed using Redundans 0.14c (Pryszcz and Gabaldon 2016) with default parameters. The assembled sequences were blasted to the NCBI nt database using BLASTN 2.2.26 to remove potential contaminating contigs which failed to map to birds or reptiles. The completeness of the genome was estimated by BUSCO 2.0 (Simao et al. 2015). Library preparation, sequencing, and assembly were conducted by Berry Genomics (Beijing, China).
Sampling and Whole-Genome Resequencing
Total genomic DNA was extracted from muscle tissue or blood using the Tissue/Cell Genomic DNA Extraction Kit (Aidlab Biotechnologies Co. Ltd., Beijing, China) according to the manufacturer’s protocol. DNA libraries with  350 bp insertions were constructed. All libraries were sequenced using the Illumina Novaseq platform with a PE read length of 150 bp on four lanes by Berry Genomics (Beijing, China). Raw reads were quality controlled using fastp 0.20.0 (Chen et al. 2018) according to the same criteria as described above.
350 bp insertions were constructed. All libraries were sequenced using the Illumina Novaseq platform with a PE read length of 150 bp on four lanes by Berry Genomics (Beijing, China). Raw reads were quality controlled using fastp 0.20.0 (Chen et al. 2018) according to the same criteria as described above.
Variant Calling
Quality controlled reads of all 78 samples were mapped to the reference genome of whistleri using BWA 0.7.12 (Li and Durbin 2009). One sample of a congeneric species, Phylloscopus griseolus (Zhang et al. 2019), was used as an outgroup. Polymerase Chain Reaction (PCR) duplicates were removed in SAMtools 0.1.19 (Li et al. 2019). Variants were called in SAMtools 0.1.19 (Li et al. 2019) using the “mpileup” module for all 79 samples. SNVs were filtered using VCFtools 0.1.12b (Danecek et al. 2011) according to the following criteria: i) quality value  30; ii) genotype depth
30; ii) genotype depth  5; iii) only biallelic SNVs were retained; iv) SNVs with
5; iii) only biallelic SNVs were retained; iv) SNVs with 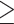 9 missing genotypes across all individuals were removed; v) SNVs differing in the outgroup species but homogeneous within the eight focal species were removed. A total of 35,663,554 SNVs were retained. We further retained only autosomal SNVs because some individuals were females (Supplementary Table S1 available on Dryad). To do so, the reference genome sequences of whistleri were mapped to autosomal chromosomes of Parus major (Laine et al. 2016) using BLAST
9 missing genotypes across all individuals were removed; v) SNVs differing in the outgroup species but homogeneous within the eight focal species were removed. A total of 35,663,554 SNVs were retained. We further retained only autosomal SNVs because some individuals were females (Supplementary Table S1 available on Dryad). To do so, the reference genome sequences of whistleri were mapped to autosomal chromosomes of Parus major (Laine et al. 2016) using BLAST 2.2.26, and only contigs that had a single hit with an e-value
2.2.26, and only contigs that had a single hit with an e-value 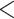 10
10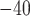 were retained. A total of 1275 autosomal contigs were successfully identified and a total of 30,094,655 autosomal SNVs were retained.
were retained. A total of 1275 autosomal contigs were successfully identified and a total of 30,094,655 autosomal SNVs were retained.
Phylogenetic Analyses
The concatenated autosomal SNVs (30,094,655 SNVs) were used to infer a neighbor-joining (NJ) tree in TreeBeST 1.9.2 (Vilella et al. 2009) with 100 bootstraps. The concatenated autosomal SNVs without missing genotypes (1,875,938 SNVs, IQTREE data set) were used to infer a maximum likelihood (ML) tree in IQTREE 1.6.9 (Nguyen et al. 2015) with 1000 bootstraps and automatic estimation of the substitution model. The concatenated autosomal SNVs without missing genotypes and with a genotype depth 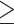 10 (40,980 SNVs, RAxML data set) were used to infer a ML tree in RAxML 8.1 (Stamatakis 2014) using the GTRGAMMA model with 100 bootstraps. Phylloscopus griseolus was used as an outgroup taxon. The consensus tree was generated in TreeAnnotator 1.8 with a burn-in of 10% and visualized in FigTree 1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/).
10 (40,980 SNVs, RAxML data set) were used to infer a ML tree in RAxML 8.1 (Stamatakis 2014) using the GTRGAMMA model with 100 bootstraps. Phylloscopus griseolus was used as an outgroup taxon. The consensus tree was generated in TreeAnnotator 1.8 with a burn-in of 10% and visualized in FigTree 1.3.1 (http://tree.bio.ed.ac.uk/software/figtree/).
In addition to the phylogenetic analyses on the basis of the concatenated SNVs, we also conducted phylogenetic analyses using concatenated consensus sequences. We used SAMtools 0.1.19 (Li et al. 2019) and bcftools 1.3.1 (Li et al. 2019) on the BAM files to generate 10 Kb consensus sequences for each 50 Kb window, and these consensus sequences were aligned using mafft 7.464 (Nakamura et al. 2018). We randomly selected 100 independent sets, each containing 100 of these 10-Kb sequences concatenated to one another. Every one of these 100 sets was used to infer a ML tree using IQTREE 1.6.9 with 1000 bootstraps and automatic estimation of the substitution model. We also randomly selected 100 sets each containing 10 sequences concatenated to one another; these 100 sets were then used to infer ML trees in IQTREE 1.6.9 with the same settings.
In consideration of the shortcomings of concatenation approaches for phylogenetic analyses (Nater et al. 2015; Edwards et al. 2016; Thawornwattana et al. 2018), we complementarily used an approach based on the multispecies-coalescent model, SNAPP (Bryant et al. 2012) as implemented in BEAST2 (Bouckaert et al. 2014), to infer the species tree. Only five samples were selected for each species. SNVs from the IQTREE data set with a distance of 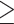 1000 bp and a depth
1000 bp and a depth 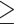 6 (288,355 SNVs) as filtered by VCFtools 0.1.12b (Danecek et al. 2011) were used in SNAPP, and we also performed SNAPP analysis using SNVs from the RAxML data set with a distance of
6 (288,355 SNVs) as filtered by VCFtools 0.1.12b (Danecek et al. 2011) were used in SNAPP, and we also performed SNAPP analysis using SNVs from the RAxML data set with a distance of 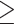 1000 bp (1936 SNVs). We carried out 100,000 runs, sampling every 50 generations. SNAPP trees were displayed in DensiTree 2.2.6 (Bouckaert 2010).
1000 bp (1936 SNVs). We carried out 100,000 runs, sampling every 50 generations. SNAPP trees were displayed in DensiTree 2.2.6 (Bouckaert 2010).
Hypotheses of Phylogenetic Uncertainty Regarding omeiensis
One species, omeiensis, is particularly unstable in its phylogenetic placement across or even within previous studies (Olsson et al. 2004; Päckert et al. 2004; Päckert et al. 2012; Alström et al. 2018), as well as in the present study (see Results), and the resolution of its position is central to disentangling the destabilizing agents in phylogenetic reconstruction. We here propose three alternative hypotheses (H1, H2, and H3) to account for the phylogenetic uncertainty surrounding omeiensis (Fig. 2): H1, in which omeiensis is sister to whistleri/valentini, but gene flow between omeiensis and soror has led to omeiensis and soror forming sister groupings in some phylogenetic reconstructions; H2, in which omeiensis is sister to soror, but ancient gene flow between omeiensis and the most recent common ancestor of whistleri/valentini has led to omeiensis and whistleri/valentini forming sister groups in some phylogenetic reconstructions (using a large amount of data); and H3, in which omeiensis is a hybrid species originating from ancient hybridization between soror and the most recent common ancestor of whistleri/valentini.
Figure 2.

Three possible phylogenetic hypotheses (H1, H2, and H3). Bidirectional arrows indicate bidirectional gene flow; H1 represents omeiensis being sister to whistleri/valentini, with gene flow between omeiensis and soror resulting in tree2 (bifurcating tree topology of H2) based on the genomic regions most affected by this gene flow; H2 represents omeiensis as sister to soror, with gene flow between omeiensis and the ancestral population of whistleri/valentini resulting in tree1 (bifurcating tree topology of H1) based on the genomic regions most affected by this gene flow; H3 represents omeiensis having a hybrid origin between soror and the ancestral population of whistleri/valentini.
H1 predicts that the genomic regions least affected by gene flow between omeiensis and soror would produce tree topology 1 (tree1, bifurcating tree topology of H1) ((omeiensis, whistleri/valentini), soror), while the genomic regions most affected by gene flow between omeiensis and soror would produce tree topology 2 (tree2, bifurcating tree topology of H2) ((omeiensis, soror), whistleri/valentini). H2 predicts that the genomic regions least affected by ancient gene flow between omeiensis and the most recent common ancestor of whistleri/valentini would produce tree2 ((omeiensis, soror), (whistleri, valentini)), while the genomic regions most affected by gene flow would produce tree1 ((omeiensis, (whistleri, valentini)), soror). To test H1 and H2, we estimated the D-statistic (Green et al. 2010; Durand et al. 2011) in 10 Kb nonoverlapping sliding-windows using the Python script egglib_sliding_windows.py (https://github.com/johnomics) (Martin et al. 2015). We defined two taxon sets for testing H1: whistleri or valentini as P1, omeiensis as P2, soror as P3, P. griseolus as outgroup. Along the same lines, we defined two taxon sets for testing H2: soror as P1, omeiensis as P2, whistleri or valentini as P3, P. griseolus as outgroup. For the tests of both H1 and H2, windows with D-statistic values close to zero (absolute D-statistic value  0.01) were regarded as genomic regions least affected by gene flow, while the highest D-statistic values (top 0.5% windows) were regarded as associated with genomic regions most affected by gene flow. The concatenated SNVs in these focal windows were used to reconstruct phylogenetic trees using RAxML 8.1 (Stamatakis 2014) with 100 bootstraps, and the GTRGAMMA substitution model was uniformly applied with P. griseolus as an outgroup. We also used SNAPP (Bryant et al. 2012) for species tree inference on the basis of SNVs with a distance of
0.01) were regarded as genomic regions least affected by gene flow, while the highest D-statistic values (top 0.5% windows) were regarded as associated with genomic regions most affected by gene flow. The concatenated SNVs in these focal windows were used to reconstruct phylogenetic trees using RAxML 8.1 (Stamatakis 2014) with 100 bootstraps, and the GTRGAMMA substitution model was uniformly applied with P. griseolus as an outgroup. We also used SNAPP (Bryant et al. 2012) for species tree inference on the basis of SNVs with a distance of  500 bp for each data set with 100,000 runs and sampling every 50 generations.
500 bp for each data set with 100,000 runs and sampling every 50 generations.
To complement these hypothesis-exclusion trials, we conducted coalescent-based demographic inference using fastsimcoal2 2.6.0.3 (Excoffier et al. 2013). Variants with no missing sites were converted into a joint site frequency spectrum (SFS) using the Python script easySFS.py (https://github.com/isaacovercast/easySFS). H1 was subdivided into 9 demographic models (Supplementary Fig. S2a–i available on Dryad): bidirectional or unidirectional gene flow between omeiensis and soror occurring before T20 (the divergence time between omeiensis and whistleri/valentini) (Supplementary Fig. S2a–c available on Dryad); bidirectional or unidirectional gene flow between omeiensis and soror occurring before T10 (the divergence time between whistleri and valentini) (Supplementary Fig. S2d–f available on Dryad); bidirectional or unidirectional gene flow between omeiensis and soror occurring between T10 and T20 (Supplementary Fig. S2g–i available on Dryad). H2 was subdivided into three demographic models (Supplementary Fig. S2j–l available on Dryad): bidirectional or unidirectional gene flow between omeiensis and the ancestral lineage of whistleri/valentini between T10 (the divergence time between whistleri and valentini) and T20 (the divergence time between omeiensis and soror). H3 was also tested by setting omeiensis as a hybrid species between soror and the ancestral lineage of whistleri/valentini at time T20 (Supplementary Fig. S2m available on Dryad). Two demographic models identical to H1 and H2 but without gene flow were also tested (Supplementary Fig. S2n,o available on Dryad). In all models, we set T10  T20 and T20
T20 and T20  T30 (the root divergence time for these four species). We set a search range of T10 from 100,000 to 2,000,000 generations, T20 from 200,000 to 4,000,000 generations and T30 from 300,000 to 9,000,000 generations, respectively (based on previous estimations of divergence time). The effective population sizes for all modern and ancestral populations were set between 1000 and 1,000,000. A generation time of 1.7 years (Bensch et al. 1999) and a mutation rate of 0.33% per million years (Zhang et al. 2014) were applied. We performed 500,000 coalescent simulations (
T30 (the root divergence time for these four species). We set a search range of T10 from 100,000 to 2,000,000 generations, T20 from 200,000 to 4,000,000 generations and T30 from 300,000 to 9,000,000 generations, respectively (based on previous estimations of divergence time). The effective population sizes for all modern and ancestral populations were set between 1000 and 1,000,000. A generation time of 1.7 years (Bensch et al. 1999) and a mutation rate of 0.33% per million years (Zhang et al. 2014) were applied. We performed 500,000 coalescent simulations ( n 500,000) to approximate the expected SFS in each cycle and ran 50 optimization cycles (
n 500,000) to approximate the expected SFS in each cycle and ran 50 optimization cycles ( L 50) to estimate parameters. For each model, 50 independent runs were conducted to obtain the best run with the highest likelihood values. As different models may have different numbers of estimated parameters, we used the Akaike information criterion (AIC) to determine the best-fit model.
L 50) to estimate parameters. For each model, 50 independent runs were conducted to obtain the best run with the highest likelihood values. As different models may have different numbers of estimated parameters, we used the Akaike information criterion (AIC) to determine the best-fit model.
D-Statistic Analysis in Dsuite
On the basis of what we hypothesized to be the true species tree topology (see Results), we estimated the overall D-statistic for the eight species in Dsuite (Malinsky et al. 2020) across all possible combinations using P. griseolus as the outgroup. Dsuite is able to estimate the D-statistic across all possible combinations at one single time. The data set of the 30,094,655 autosomal SNVs was used for this analysis. The heatmap of the D-statistic values was plotted using the Ruby script plot_d.rb (https://github.com/mmatschiner/tutorials/tree/master/analysis_of_introgression_with_snp_data). Only combinations with the unadjusted 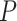 values = 0 were retained. We introduced a parsimony principle to determine possible gene flow events: if all terminal taxa in clade A showed evidence of gene flow with all the terminal taxa in clade B, we parsimoniously assumed a single gene flow event between the ancestral lineages of clades A and B (Pulido-Santacruz et al. 2020).
values = 0 were retained. We introduced a parsimony principle to determine possible gene flow events: if all terminal taxa in clade A showed evidence of gene flow with all the terminal taxa in clade B, we parsimoniously assumed a single gene flow event between the ancestral lineages of clades A and B (Pulido-Santacruz et al. 2020).
Demographic Inference for Species Pairs
We used  a
a i 1.6.3 (Gutenkunst et al. 2009) to infer the best divergence model for the four sister species pairs (tephrocephalus–burkii, poliogenys–intermedius, whistleri–valentini, and omeiensis–soror). Demographic inferences were also conducted for another two species pairs (valentini–soror and valentini–omeiensis) in
i 1.6.3 (Gutenkunst et al. 2009) to infer the best divergence model for the four sister species pairs (tephrocephalus–burkii, poliogenys–intermedius, whistleri–valentini, and omeiensis–soror). Demographic inferences were also conducted for another two species pairs (valentini–soror and valentini–omeiensis) in  a
a i as gene flow had been detected between the species in these two pairs using the D-statistic. Four divergence models were tested: i) strict isolation without gene flow; ii) isolation with bidirectional gene flow; iii) secondary contact with bidirectional gene flow (gene flow in recent times only); and iv) ancient bidirectional gene flow (gene flow in ancient times but ceased in recent times) (Supplementary Fig. S3 available on Dryad).
i as gene flow had been detected between the species in these two pairs using the D-statistic. Four divergence models were tested: i) strict isolation without gene flow; ii) isolation with bidirectional gene flow; iii) secondary contact with bidirectional gene flow (gene flow in recent times only); and iv) ancient bidirectional gene flow (gene flow in ancient times but ceased in recent times) (Supplementary Fig. S3 available on Dryad).
Phylogenetic Network Analysis
Phylogenetic network analysis on the basis of gene trees was carried out in PhyloNet 3.8.2 (Wen et al. 2018) using a maximum pseudolikelihood (MPL) algorithm with the command “InferNetwork_MPL,” allowing for 1–3 reticulations and performing 10 independent searches for each reticulation setting to avoid local optima. The MPL algorithm in PhyloNet is able to estimate phylogenies with a reticulation history by accounting for both incomplete lineage sorting and hybridization. We generated 10 Kb consensus sequences for each 50 Kb window following the procedures described above. A total of 4000 randomly selected aligned sequences were used to infer ML gene trees in IQTREE 1.6.9 (Nguyen et al. 2015) with 1000 bootstraps and P. griseolus as an outgroup. These gene trees were then analyzed in PhyloNet, and a bootstrap support threshold of 90 was applied using  b flag, with other parameters at default settings. The optimal networks were displayed in Dendroscope 2.7.4 (Huson et al. 2007).
b flag, with other parameters at default settings. The optimal networks were displayed in Dendroscope 2.7.4 (Huson et al. 2007).
Sliding-Window Analyses of Phylogenetic Topology and Population Genomic Parameters
On account of the relatively fragmented reference genome, the 10 longest autosomal contigs, each with a length 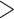 10 Mb (in total 144.3 Mb, accounting for 14.4% of autosomes), were selected to explore the distribution pattern of phylogenetic signal across the genome. SAMtools 0.1.19 (Li et al. 2019) and bcftools 1.3.1 (Li et al. 2019) were used to generate 50 Kb consensus sequences for every 50 Kb nonoverlapping sliding-window using each individual’s BAM files. After alignment with mafft 7.464 (Nakamura et al. 2018), we used IQTREE 1.6.9 (Nguyen et al. 2015) to conduct phylogenetic analyses for each of these windows. Only four species, soror, omeiensis, whistleri, and valentini, which were found to form a clade in all other analyses (see Results), were included in this analysis, and P. griseolus was set as an outgroup.
10 Mb (in total 144.3 Mb, accounting for 14.4% of autosomes), were selected to explore the distribution pattern of phylogenetic signal across the genome. SAMtools 0.1.19 (Li et al. 2019) and bcftools 1.3.1 (Li et al. 2019) were used to generate 50 Kb consensus sequences for every 50 Kb nonoverlapping sliding-window using each individual’s BAM files. After alignment with mafft 7.464 (Nakamura et al. 2018), we used IQTREE 1.6.9 (Nguyen et al. 2015) to conduct phylogenetic analyses for each of these windows. Only four species, soror, omeiensis, whistleri, and valentini, which were found to form a clade in all other analyses (see Results), were included in this analysis, and P. griseolus was set as an outgroup.
Our analyses indicated that tree2 reflects the true phylogenetic relationships (see Results). Therefore, genomic regions producing tree1 would be more influenced by gene flow between omeiensis and the most recent common ancestor of whistleri/valentini than genomic regions producing tree2. We used the D-statistic and D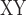 decrease (see below) to identify ancestral gene flow, with higher values of the D-statistic and D
decrease (see below) to identify ancestral gene flow, with higher values of the D-statistic and D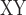 decrease indicating higher levels of gene flow. To test for gene flow between omeiensis and the most recent common ancestor of whistleri/valentini using the D-statistic, we defined soror as P1, omeiensis as P2, whistleri (used to represent whistleri/valentini) as P3, and P. griseolus as the outgroup. D
decrease indicating higher levels of gene flow. To test for gene flow between omeiensis and the most recent common ancestor of whistleri/valentini using the D-statistic, we defined soror as P1, omeiensis as P2, whistleri (used to represent whistleri/valentini) as P3, and P. griseolus as the outgroup. D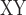 decrease was defined as the D
decrease was defined as the D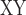 between soror and whistleri (used to represent whistleri/valentini) minus D
between soror and whistleri (used to represent whistleri/valentini) minus D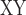 between omeiensis and whistleri (used to represent whistleri/valentini). We estimated the D-statistic and D
between omeiensis and whistleri (used to represent whistleri/valentini). We estimated the D-statistic and D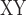 decrease in 50 Kb nonoverlapping sliding-windows using the Python script egglib_sliding_windows.py (https://github.com/johnomics) (Martin et al. 2015).
decrease in 50 Kb nonoverlapping sliding-windows using the Python script egglib_sliding_windows.py (https://github.com/johnomics) (Martin et al. 2015).
We used the whistleri genome to approximate the recombination rate across all eight species. We estimated the population recombination rate (Rho) for whistleri in FastEPRR 2.0 (Gao et al. 2016) using 50 Kb nonoverlapping sliding-windows. FastEPRR is an R package for the rapid estimation of population recombination rates based on intraspecific DNA polymorphisms. As genomic diversity and genomic differentiation patterns across the genome are highly comparable across a speciation continuum of closely related birds (Burri et al. 2015; Van Doren et al. 2017; Vijay et al. 2017), we estimated nucleotide diversity (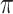 ) for whistleri and omeiensis and genomic differentiation (mean F
) for whistleri and omeiensis and genomic differentiation (mean F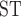 ) between them to approximate
) between them to approximate 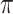 and F
and F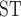 for this clade in VCFtools 0.1.12b (Danecek et al. 2011) using 50 Kb nonoverlapping sliding-windows.
for this clade in VCFtools 0.1.12b (Danecek et al. 2011) using 50 Kb nonoverlapping sliding-windows.
Time-Calibrated Species Tree Estimation
On the basis of hypothesis H2, 259 windows including 105,186 SNVs were identified as genomic regions least affected by gene flow between omeiensis and the ancestral lineage of whistleri and valentini. We filtered 4444 SNVs from the 259 windows with a distance of 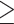 500 bp using VCFtools 0.1.12b (Danecek et al. 2011). These SNVs were used to estimate time calibrated species trees in SNAPP (Bryant et al. 2012) as implemented in BEAST2 (Bouckaert et al. 2014) following Stange et al. (2018). We placed normally distributed priors at the root of the eight species, with a mean divergence time of 6.2 Ma and standard deviation of 1.2 Ma (Alström et al. 2018). We carried out 100,000 runs, sampling every 50 generations. The consensus tree was generated in TreeAnnotator 1.8 with a burn-in of 10% and visualized in FigTree 1.3.1. Only five samples for each species were used in this analysis.
500 bp using VCFtools 0.1.12b (Danecek et al. 2011). These SNVs were used to estimate time calibrated species trees in SNAPP (Bryant et al. 2012) as implemented in BEAST2 (Bouckaert et al. 2014) following Stange et al. (2018). We placed normally distributed priors at the root of the eight species, with a mean divergence time of 6.2 Ma and standard deviation of 1.2 Ma (Alström et al. 2018). We carried out 100,000 runs, sampling every 50 generations. The consensus tree was generated in TreeAnnotator 1.8 with a burn-in of 10% and visualized in FigTree 1.3.1. Only five samples for each species were used in this analysis.
Results
Genome Assembly of Phylloscopus whistleri and Whole-Genome Resequencing of Eight Species
The assembled genome of whistleri is 1.11 Gb in length, and contains 1749 contigs (1731 contigs 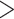 10 Kb, 1167 contigs
10 Kb, 1167 contigs 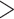 50 Kb and 710 contigs
50 Kb and 710 contigs 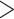 100 Kb) with an N50 and N90 of 3.80 Mb and 380 Kb, respectively. The average contig length is 636 Kb with the largest at 30.63 Mb, and the completeness of the assembly is 94.4%. We sampled 78 individuals from across eight species (Supplementary Table S1 available on Dryad). Mapping to the reference genome of whistleri and removing PCR duplicates resulted in an average sequencing depth and breadth of coverage at 11.1 and 97.0%, respectively (Supplementary Table S1 available on Dryad).
100 Kb) with an N50 and N90 of 3.80 Mb and 380 Kb, respectively. The average contig length is 636 Kb with the largest at 30.63 Mb, and the completeness of the assembly is 94.4%. We sampled 78 individuals from across eight species (Supplementary Table S1 available on Dryad). Mapping to the reference genome of whistleri and removing PCR duplicates resulted in an average sequencing depth and breadth of coverage at 11.1 and 97.0%, respectively (Supplementary Table S1 available on Dryad).
Phylogenies of Genome-Wide Data
The NJ tree based on 30,094,655 concatenated genome-wide autosomal SNVs recovered two main clades (Fig. 3). In clade I, valentini was sister to whistleri, and omeiensis was sister to these two species, while soror was sister to all the three. Within clade II, two bifurcating subclades were recovered: intermedius  poliogenys and tephrocephalus
poliogenys and tephrocephalus  burkii, respectively (Fig. 3a). All nodes had 100% bootstrap support. The ML tree estimated with IQTREE based on 1,875,938 concatenated genome-wide SNVs was identical in topology and bootstrap support to the NJ tree (Fig. 3b). In contrast, the ML tree estimated with RAxML based on 40,980 concatenated genome-wide SNVs recovered soror and omeiensis as sisters with modest bootstrap support (70%) (Fig. 3c). We found that 5 out of 100 ML trees, each constructed using sets of 100 concatenated sequences of 10 Kb genomic windows, recovered a phylogenetic topology identical to Figure 3c for interspecific nodes, with an average bootstrap support of 90.6% for the sister relationship between soror and omeiensis. The same topology was recovered by 30 of the 100 trees constructed from sets of 10 concatenated sequences of 10 Kb genomic windows. In each case, the remaining trees showed a topology more similar to Figure 3a,b for interspecific nodes.
burkii, respectively (Fig. 3a). All nodes had 100% bootstrap support. The ML tree estimated with IQTREE based on 1,875,938 concatenated genome-wide SNVs was identical in topology and bootstrap support to the NJ tree (Fig. 3b). In contrast, the ML tree estimated with RAxML based on 40,980 concatenated genome-wide SNVs recovered soror and omeiensis as sisters with modest bootstrap support (70%) (Fig. 3c). We found that 5 out of 100 ML trees, each constructed using sets of 100 concatenated sequences of 10 Kb genomic windows, recovered a phylogenetic topology identical to Figure 3c for interspecific nodes, with an average bootstrap support of 90.6% for the sister relationship between soror and omeiensis. The same topology was recovered by 30 of the 100 trees constructed from sets of 10 concatenated sequences of 10 Kb genomic windows. In each case, the remaining trees showed a topology more similar to Figure 3a,b for interspecific nodes.
Figure 3.

Phylogenetic trees based on genome-wide SNVs. a) NJ tree based on 30,094,655 concatenated autosomal SNVs; b) ML tree estimated with IQTREE based on 1,875,938 concatenated autosomal SNVs without missing genotypes; c) ML tree estimated with RAxML based on 40,980 concatenated autosomal SNVs without missing genotypes and with a genotype depth  10. Asterisks represent 100% bootstrap support.
10. Asterisks represent 100% bootstrap support.
The species tree estimated with SNAPP using 288,355 SNVs filtered from the IQTREE data set (Fig. 4a) was identical to the NJ and IQTREE trees (Fig. 3a,b) in topology. In contrast, the species tree estimated with SNAPP using 1936 SNVs filtered from the RAxML data set (Fig. 4b) was identical to the RAxML tree (Fig. 3c) in topology, although with an elevated posterior probability of 1.00 for the sister relationship between omeiensis and soror.
Figure 4.

Species trees estimated using SNAPP (including five samples per species). a) Species tree based on a subset of SNVs (288,355 SNVs) from the IQTREE data set with a distance of  1000 bp; b) species tree based on a subset of SNVs (1936 SNVs) from the RAxML data set with a distance of
1000 bp; b) species tree based on a subset of SNVs (1936 SNVs) from the RAxML data set with a distance of  1000 bp. All nodes are supported by a posterior probability of 1.00.
1000 bp. All nodes are supported by a posterior probability of 1.00.
Test for Alternative Tree Topologies
Using D-statistic values interpreted on the basis of H1 (Fig. 2), a total of 190 sequence windows with 37,737 SNVs (2933 SNVs with a distance of 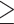 500 bp) were identified as genomic regions that were most affected by gene flow, and a total of 176 sequence windows with 75,887 SNVs (3143 SNVs with a distance of
500 bp) were identified as genomic regions that were most affected by gene flow, and a total of 176 sequence windows with 75,887 SNVs (3143 SNVs with a distance of 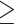 500 bp) were identified as genomic regions least affected by gene flow. The former and latter data sets would produce tree topology H2 and H1, respectively, if hypothesis H1 were true (Fig. 2). However, we observed that both ML and species tree analyses produced tree topology H1 for the former data set, that is, using regions most affected by gene flow (Fig. 5a). In contrast, the latter data set, that is, sequence windows representing regions least affected by gene flow, produced both tree topology H1 (under ML and some species tree subsets) and tree topology H2 (for other species tree subsets) (Fig. 5b). As a consequence, H1 cannot represent the true species tree topology.
500 bp) were identified as genomic regions least affected by gene flow. The former and latter data sets would produce tree topology H2 and H1, respectively, if hypothesis H1 were true (Fig. 2). However, we observed that both ML and species tree analyses produced tree topology H1 for the former data set, that is, using regions most affected by gene flow (Fig. 5a). In contrast, the latter data set, that is, sequence windows representing regions least affected by gene flow, produced both tree topology H1 (under ML and some species tree subsets) and tree topology H2 (for other species tree subsets) (Fig. 5b). As a consequence, H1 cannot represent the true species tree topology.
Figure 5.

Phylogenetic trees on the basis of hypotheses H1 and H2 (see Fig. 2) using sequence windows with different D-statistic values. a) ML and species trees generated from 190 sequence windows with top 0.5% D values based on the tree topology of tree1; b) ML and species trees generated from 176 windows with absolute D values 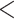 0.01 based on the tree topology of tree1; c) ML and species trees generated from 196 windows with top 0.5% D values based on the tree topology of tree2; (d) ML and species trees generated from 259 sequence windows with absolute D values
0.01 based on the tree topology of tree1; c) ML and species trees generated from 196 windows with top 0.5% D values based on the tree topology of tree2; (d) ML and species trees generated from 259 sequence windows with absolute D values 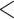 0.01 based on the tree topology of tree2. Each pair of trees in a–d shows ML trees generated by RAxML on the left, with asterisks representing 100% bootstrap support, and species trees generated by SNAPP with blue, red and green indicating majority to minority trees, respectively. All nodes of the species trees in (a), (c), and (d) are supported by posterior probability (PP) 1.00, whereas all nodes of the species trees in (b) are supported by PP 1.00 except for soror
0.01 based on the tree topology of tree2. Each pair of trees in a–d shows ML trees generated by RAxML on the left, with asterisks representing 100% bootstrap support, and species trees generated by SNAPP with blue, red and green indicating majority to minority trees, respectively. All nodes of the species trees in (a), (c), and (d) are supported by posterior probability (PP) 1.00, whereas all nodes of the species trees in (b) are supported by PP 1.00 except for soror 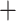 omeiensis (PP
omeiensis (PP 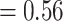 ), omeiensis
), omeiensis 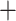 whistleri/valentini (PP
whistleri/valentini (PP 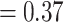 ), and soror
), and soror 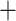 whistleri/valentini (PP
whistleri/valentini (PP 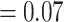 ).
).
Using D-statistic values interpreted on the basis of H2, a total of 196 sequence windows with 39,065 SNVs (3038 SNVs with a distance of 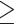 500 bp) were identified as genomic regions most affected by gene flow, and a total of 259 sequence windows with 105,186 SNVs (4444 SNVs with a distance of
500 bp) were identified as genomic regions most affected by gene flow, and a total of 259 sequence windows with 105,186 SNVs (4444 SNVs with a distance of 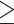 500 bp) were identified as genomic regions least affected by gene flow. The former and latter data sets would produce tree1 and tree2, respectively, if hypothesis H2 were true (Fig. 2). In support of this hypothesis, we observed that the former and latter data sets did produce tree1 and tree2, respectively, for both ML and species trees (Fig. 5c,d). Therefore, we accept tree2 as the true species tree topology.
500 bp) were identified as genomic regions least affected by gene flow. The former and latter data sets would produce tree1 and tree2, respectively, if hypothesis H2 were true (Fig. 2). In support of this hypothesis, we observed that the former and latter data sets did produce tree1 and tree2, respectively, for both ML and species trees (Fig. 5c,d). Therefore, we accept tree2 as the true species tree topology.
Using fastsimcoal2, we tested 15 different models and found that 3 models (model j, k, and l; Supplementary Fig. S2 available on Dryad), each representing H2, generally presented higher likelihood values than other models (Supplementary Table S2 available on Dryad). Among these, models j and k were the best-fit models as determined by the lowest AIC values. According to model k, which assumed unidirectional gene flow from omeiensis to the ancestral lineage of whistleri/valentini, the T10, T20, and T30 divergence events were estimated at 149,033 generations (253,356 years), 262,355 generations (446,004 years), and 375,490 generations (638,333 years) ago, respectively, and the migration rate from omeiensis to the ancestral lineage of whistleri/valentini was estimated at 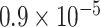 (Supplementary Table S3 available on Dryad). Model j, which assumed bidirectional gene flow between omeiensis and the ancestral lineage of whistleri/valentini, provided similar estimates to model k except that gene flow from omeiensis to the ancestral lineage of whistleri/valentini was estimated to be approximately an order of magnitude higher (
(Supplementary Table S3 available on Dryad). Model j, which assumed bidirectional gene flow between omeiensis and the ancestral lineage of whistleri/valentini, provided similar estimates to model k except that gene flow from omeiensis to the ancestral lineage of whistleri/valentini was estimated to be approximately an order of magnitude higher (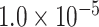 ) than in the opposite direction (
) than in the opposite direction (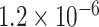 ) (Supplementary Table S3 available on Dryad).
) (Supplementary Table S3 available on Dryad).
Dsuite and  a
a i Analyses
i Analyses
Gene flow between omeiensis and valentini or whistleri emerged as the most significant gene flow event, with extremely high D-statistic values when soror was set as P1, omeiensis was set as P2 and valentini or whistleri were set as P3 (D-statistic values 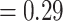 ) (Fig. 6; Supplementary Table S5 available on Dryad). Significant gene flow was also detected between valentini and omeiensis or soror with low D-statistic values when whistleri was set as P1, valentini was set as P2, and omeiensis or soror were set as P3 (D-statistic values ranging from 0.025 to 0.037). We also detected significant gene flow between intermedius or poliogenys and all four species in clade I, with low D-statistic values (ranging from 0.022 to 0.032) (Fig. 6; Supplementary Table S5 available on Dryad). Therefore, according to the parsimony principle described above, three gene flow events were inferred: one between omeiensis and the ancestral lineage of whistleri and valentini; one between the ancestral lineage of all four species in clade I and the ancestral lineage of intermedius and poliogenys; and one between valentini and the ancestral lineage of soror and omeiensis. In consideration of the recent divergence between whistleri and valentini and the much older divergence between soror and omeiensis (see Fig. 9), the third of these gene flow events is not likely, instead suggesting that the D-statistic values reflect a less parsimonious interpretation of two independent gene flow events from valentini into modern soror and modern omeiensis, respectively. This latter conclusion is also supported by
) (Fig. 6; Supplementary Table S5 available on Dryad). Significant gene flow was also detected between valentini and omeiensis or soror with low D-statistic values when whistleri was set as P1, valentini was set as P2, and omeiensis or soror were set as P3 (D-statistic values ranging from 0.025 to 0.037). We also detected significant gene flow between intermedius or poliogenys and all four species in clade I, with low D-statistic values (ranging from 0.022 to 0.032) (Fig. 6; Supplementary Table S5 available on Dryad). Therefore, according to the parsimony principle described above, three gene flow events were inferred: one between omeiensis and the ancestral lineage of whistleri and valentini; one between the ancestral lineage of all four species in clade I and the ancestral lineage of intermedius and poliogenys; and one between valentini and the ancestral lineage of soror and omeiensis. In consideration of the recent divergence between whistleri and valentini and the much older divergence between soror and omeiensis (see Fig. 9), the third of these gene flow events is not likely, instead suggesting that the D-statistic values reflect a less parsimonious interpretation of two independent gene flow events from valentini into modern soror and modern omeiensis, respectively. This latter conclusion is also supported by  a
a i results (see below).
i results (see below).
Figure 6.
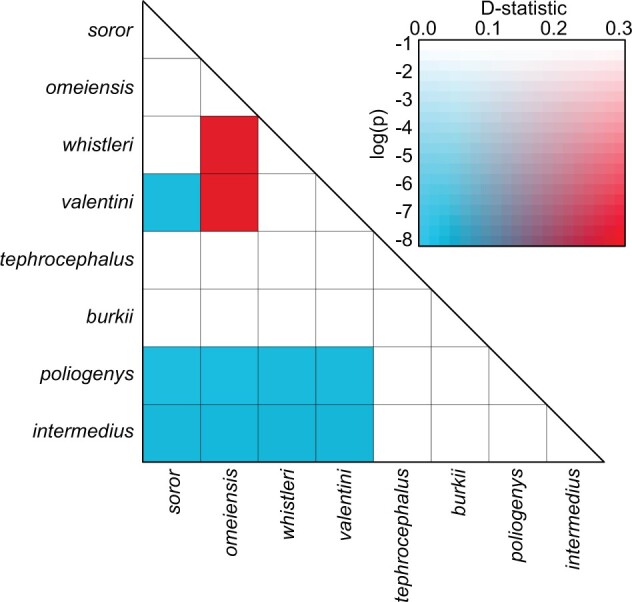
Heatmap of D-statistic values. Only combinations with unadjusted  values
values  are shown here. A color legend is shown in the right panel: the darker the color, the smaller the p value (Dsuite uses a standard block-jackknife procedure to estimate
are shown here. A color legend is shown in the right panel: the darker the color, the smaller the p value (Dsuite uses a standard block-jackknife procedure to estimate 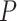 values) and the higher (red) or lower (blue) the absolute D-statistic value.
values) and the higher (red) or lower (blue) the absolute D-statistic value.
Figure 9.
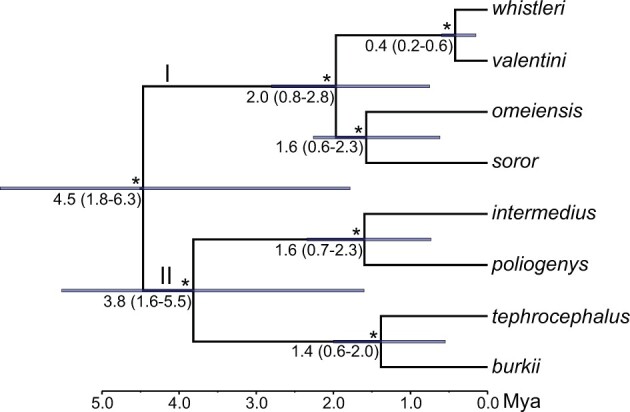
Species tree estimated using SNAPP based on 4444 SNVs which were least affected by ancient gene flow between omeiensis and whistleri/valentini. Asterisks represent posterior probabilities of 1.00, and divergence times with 95% highest posterior density (HPD) are shown below these nodes (95% HPD also indicated by blue bars). Time scale in millions of years.
The conclusions of  a
a i analysis were in broad agreement with D-statistics. With respect to gene flow (or lack thereof) between valentini and omeiensis, an ancient asymmetric gene flow model exhibited a much better fit than the remaining models; for valentini and soror, all three models incorporating gene flow displayed similar likelihood values although isolation with bidirectional gene flow achieved the best fit (Supplementary Table S4 available on Dryad). As for the four sister species pairs, models incorporating gene flow exhibited a much better fit than strict isolation without gene flow (Supplementary Table S4 available on Dryad).
i analysis were in broad agreement with D-statistics. With respect to gene flow (or lack thereof) between valentini and omeiensis, an ancient asymmetric gene flow model exhibited a much better fit than the remaining models; for valentini and soror, all three models incorporating gene flow displayed similar likelihood values although isolation with bidirectional gene flow achieved the best fit (Supplementary Table S4 available on Dryad). As for the four sister species pairs, models incorporating gene flow exhibited a much better fit than strict isolation without gene flow (Supplementary Table S4 available on Dryad).
Phylogenetic Networks
In the PhyloNet analyses, when reticulations were set to 1, 2, and 3, all the corresponding optimal networks showed that omeiensis was sister to soror with significant gene flow between omeiensis and the ancestral lineage of whistleri/valentini (Fig. 7), consistent with hypothesis H2 and the main gene flow event of the Dsuite analysis. The ancestral lineage of whistleri/valentini was connected to omeiensis by an inheritance probability of 42%, 32%, and 69%, respectively, under the three different reticulation scenarios. This result is consistent with the extremely high D-statistic values in the Dsuite analysis. When reticulations were set to 2 and 3, the corresponding optimal networks also recovered gene flow between the ancestral lineage of all four species in clade I and the ancestral lineage of intermedius and poliogenys with relatively low inheritance probabilities, consistent with the second described gene flow event and the low D-statistic values in Dsuite analysis.
Figure 7.

Phylogenetic network analysis using PhyloNet. a) Reticulation 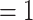 ; b) reticulation
; b) reticulation 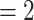 ; c) reticulation
; c) reticulation 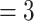 . Numerical values indicate inheritance probabilities.
. Numerical values indicate inheritance probabilities.
The Genomic Distribution of Sequence Windows Supporting Different Phylogenetic Topologies
The distribution of sequence windows supporting tree1 and tree2, respectively, was highly interleaved (Fig. 8a). These two tree topologies were supported by a total of 88.9% of the genome, with overall proportions of support for tree1 and tree2 at 55.4% and 33.5%, respectively. D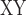 decrease, D-statistic, and F
decrease, D-statistic, and F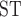 between omeiensis and whistleri were significantly higher in sequence windows supporting tree1 than in those supporting tree2. Nucleotide diversity of omeiensis and whistleri and the population recombination rate were significantly lower in sequence windows supporting tree1 than in those supporting tree2 (Wilcoxon rank sum test, Fig. 8b). In general, tree1 was enriched in genomic regions with higher D
between omeiensis and whistleri were significantly higher in sequence windows supporting tree1 than in those supporting tree2. Nucleotide diversity of omeiensis and whistleri and the population recombination rate were significantly lower in sequence windows supporting tree1 than in those supporting tree2 (Wilcoxon rank sum test, Fig. 8b). In general, tree1 was enriched in genomic regions with higher D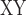 -decrease, D-statistic, and F
-decrease, D-statistic, and F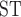 , and lower recombination and nucleotide diversity (Fig. 8b).
, and lower recombination and nucleotide diversity (Fig. 8b).
Figure 8.

The distribution patterns of sequence windows supporting different topologies and their population genomic parameters. a) Distribution of 50 Kb sequence windows in support of different topologies. Orange, blue, and black indicate windows in support of tree1, tree2, and other tree topologies, respectively. b) Tree-wise comparisons (between tree1 and tree2) of D-statistic, D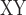 decrease, F
decrease, F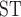 between omeiensis and whistleri, nucleotide diversities (
between omeiensis and whistleri, nucleotide diversities (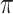 ) of omeiensis and whistleri, and population recombination rate (Rho);
) of omeiensis and whistleri, and population recombination rate (Rho); 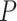 values estimated by Wilcoxon rank sum test.
values estimated by Wilcoxon rank sum test.
Time Calibrated Tree
Using a small data set of 4444 SNVs which were least affected by ancient gene flow between omeiensis and the ancestral lineage of whistleri and valentini, we inferred a bifurcating species tree for the eight species. The root divergence time was estimated at 4.5 Ma (95% highest posterior density [HPD]: 1.8–6.3 Ma), and the basal divergence time of clade I and clade II were estimated at 2.0 Ma (95% HPD: 0.8–2.8 Ma) and 3.8 Ma (95% HPD: 1.6–5.5 Ma), respectively (Fig. 9). The divergence time between whistleri and valentini (0.4 Ma) was much younger than between members of the other three species pairs (1.4–1.6 Ma) (Fig. 9).
Discussion
Bifurcation with Reticulation
Using genome-wide variants and sequences, we recovered two primary clades in the radiation comprising the eight Phylloscopus species under study (Figs. 3 and 4), and several main gene flow events were identified on the basis of Dsuite, PhyloNet, fastsimcaol2, and 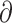 a
a i analyses (Figs. 6 and 7; Supplementary Tables S2 and S4 available on Dryad). Not all gene flow events had an equally disturbing effect on accurate phylogenetic reconstruction. For example, the gene flow event between omeiensis and the ancestral lineage of whistleri/valentini has profoundly misled phylogenetic reconstruction due to ancient gene flow affecting large parts of the genome, as suggested by both high D-statistic values (Fig. 6; Supplementary Table S5 available on Dryad) and high inheritance probabilities (Fig. 7). On the other hand, the gene flow event between the ancestral lineage of all four species in clade I and the ancestral lineage of intermedius and poliogenys generated limited bias in the phylogenetic reconstruction using genome-wide data, probably as a result of its lesser magnitude as suggested by the low D-statistic values (Fig. 6; Supplementary Table S5 available on Dryad) and low inheritance probabilities (Fig. 7).
i analyses (Figs. 6 and 7; Supplementary Tables S2 and S4 available on Dryad). Not all gene flow events had an equally disturbing effect on accurate phylogenetic reconstruction. For example, the gene flow event between omeiensis and the ancestral lineage of whistleri/valentini has profoundly misled phylogenetic reconstruction due to ancient gene flow affecting large parts of the genome, as suggested by both high D-statistic values (Fig. 6; Supplementary Table S5 available on Dryad) and high inheritance probabilities (Fig. 7). On the other hand, the gene flow event between the ancestral lineage of all four species in clade I and the ancestral lineage of intermedius and poliogenys generated limited bias in the phylogenetic reconstruction using genome-wide data, probably as a result of its lesser magnitude as suggested by the low D-statistic values (Fig. 6; Supplementary Table S5 available on Dryad) and low inheritance probabilities (Fig. 7).
The magnitude of gene flow required to mislead phylogenetic reconstruction is not clear. Our fastsimcoal2 analysis (Supplementary Table S3 available on Dryad) indicated a lower level of gene flow between omeiensis and the ancestral lineage of whistleri/valentini than shown by D-statistics and PhyloNet (Figs. 6 and 7), suggesting that even smaller amounts of gene flow may be capable of affecting large parts of the genome. Regardless of how much gene flow actually occurred, all divergence models incorporating gene flow generally exhibited a better fit than the strict isolation divergence models for the four sister species pairs tested in  a
a i (Supplementary Table S4 available on Dryad). This outcome highlights the pervasiveness of gene flow at early stages of divergence (Rheindt and Edwards 2011), in contrast to the strong premating reproductive isolating barriers that presently keep these leaf warbler species separate (Alström and Olsson 1999; Price 2010). We conclude that ancient gene flow between omeiensis and the ancestral lineage of whistleri/valentini has resulted in a situation where phylogenetic analyses of genomic data in most cases will infer a tree that is incongruent with the true bifurcating species phylogeny (Fig. 9). In other words, our hypothesis H2 is supported, but H1 and H3 (Fig. 2) are rejected.
i (Supplementary Table S4 available on Dryad). This outcome highlights the pervasiveness of gene flow at early stages of divergence (Rheindt and Edwards 2011), in contrast to the strong premating reproductive isolating barriers that presently keep these leaf warbler species separate (Alström and Olsson 1999; Price 2010). We conclude that ancient gene flow between omeiensis and the ancestral lineage of whistleri/valentini has resulted in a situation where phylogenetic analyses of genomic data in most cases will infer a tree that is incongruent with the true bifurcating species phylogeny (Fig. 9). In other words, our hypothesis H2 is supported, but H1 and H3 (Fig. 2) are rejected.
The placement of omeiensis as sister to whistleri/valentini was supported by data sets with a much higher number of SNVs (Figs. 3a,b and 4a) as well as data sets with a considerably higher number of 50-Kb sequence windows (Fig. 8a) than data sets supporting a sister relationship between omeiensis and soror. In addition, a sister relationship between omeiensis and whistleri/valentini were six times more frequently recovered in trees based on 100 concatenated 10-Kb windows than in analyses of only 10 concatenated 10-Kb windows. In contrast, a sister position between omeiensis and soror was supported by analyses based on a relatively smaller set of SNVs (Figs. 3c and 4b) or sequence windows (Fig. 8a). Although 50-Kb sequence windows in support of tree1 outnumbered those in support of tree2 by about 1.7 times, our hypothesis-exclusion trials (Fig. 5), phylogenetic network analysis (Fig. 7) and demographic inference in fastsimcoal2 (Supplementary Table S2 available on Dryad) all revealed that tree2 should be the most likely species tree topology, suggesting that the higher proportion of loci in support of tree1 was caused by ancient gene flow between omeiensis and the ancestral lineage of whistleri/valentini across large parts of the genome.
This result is consistent with a number of other recent comparative genomic studies, including on an Anopheles mosquito species complex (Fontaine et al. 2015) and Lonchura munias (Faust Stryjewski and Sorenson 2017), in which only a small fraction of the genome was shielded from gene flow, thereby reflecting the true phylogeny against a backdrop of pervasive gene flow. The synthesis of these important novel genomic results advocates Maddison’s (1997) early assertion that phylogenetic history is not a “winner-takes-all democracy” (Maddison 1997). If gene flow affected the vast majority of the genome, the whole-genomic or “democratic majority” trees would only reflect reticulation history but not bifurcation history (Mallet et al. 2016). Under such circumstances, species relationships cannot be resolved simply by relying on additional data (Degnan and Rosenberg 2009) because the majority gene tree topology does not necessarily reflect the true species phylogeny (Degnan and Rosenberg 2006). Although larger data sets might alleviate problems associated with lineage sorting, interspecific gene flow, which has been shown to be pervasive in nature (Mallet 2005; Rheindt and Edwards 2011; Payseur and Rieseberg 2016; Ottenburghs et al. 2017), can only be accounted for by harnessing the phylogenetic signal of those parts of the genome that are most shielded from gene flow.
Interestingly, the true bifurcation tree of the eight Phylloscopus species (Fig. 9) was also recovered by using only a small number of genes (Alström et al. 2018), which is consistent with the conclusions of the present study that smaller sets of genomic loci can sometimes avoid the effects of gene flow due to stochastic sampling. The hidden reticulation histories are likely to emerge preferentially from the use of larger data sets, especially whole-genome data sets. With an increasing availability of genomic data, phylogenomic trees should be interpreted with caution especially for closely related species because the whole-genomic tree will not always represent bifurcation history (Fontaine et al. 2015; Mallet et al. 2016; Edelman et al. 2019). Whole-genomic data may further inflate branch support values, which can additionally mislead interspecific relationships as indicated in this study. Only when both bifurcation and reticulation are taken into account can the true tree of life be unveiled.
True Species Tree Topology Tends to be Supported by Genomic Regions with Higher Recombination Rate and Nucleotide Diversity
Genomic regions of low recombination rate show low local nucleotide diversity mainly due to the effects of linked selection (Charlesworth et al. 1993; Charlesworth 2012). At the same time, regions of low recombination are more likely to be linked with deleterious loci because recombination effectively breaks the linkage between neutral and deleterious loci, which may further reduce gene flow in regions of low recombination (Edelman et al. 2019; Li et al. 2019; Martin et al. 2019). Therefore, regions of low recombination have been suggested to be effective markers in reconstructing the true species tree in the presence of gene flow (Pease and Hahn 2013; Li et al. 2019). On the other hand, the influence of selection may reduce the ability of these regions to produce accurate species tree reconstructions (Nater et al. 2015). In our Phylloscopus radiation, recombination rate and nucleotide diversity were lower in genomic regions supporting tree1 than in those supporting tree2, indicating higher levels of gene flow in genomic regions with low recombination rates (Fig. 8b). Genomic regions supporting tree1 also exhibited higher F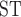 between whistleri and omeiensis than those supporting tree2 (Fig. 8b). This outcome is not in line with empirical studies suggesting that gene flow is lower in regions of low recombination (Edelman et al. 2019; Li et al. 2019; Martin et al. 2019). However, gene flow can be pronounced in highly differentiated genomic regions (“differentiation islands”) compared to their genomic backgrounds (Bay and Ruegg 2017; Zhang et al. 2017), and “differentiation islands” are usually characterized by low recombination and nucleotide diversity (Kawakami et al. 2014; Burri et al. 2015), which may account for the pattern we have uncovered.
between whistleri and omeiensis than those supporting tree2 (Fig. 8b). This outcome is not in line with empirical studies suggesting that gene flow is lower in regions of low recombination (Edelman et al. 2019; Li et al. 2019; Martin et al. 2019). However, gene flow can be pronounced in highly differentiated genomic regions (“differentiation islands”) compared to their genomic backgrounds (Bay and Ruegg 2017; Zhang et al. 2017), and “differentiation islands” are usually characterized by low recombination and nucleotide diversity (Kawakami et al. 2014; Burri et al. 2015), which may account for the pattern we have uncovered.
When two species with reasonably large population sizes hybridize and alleles of one species infiltrate the other, genetic drift constitutes a formidable barrier to the gene flow of neutral alleles from the donor species. Instead, alleles under positive selection will likely form a large proportion of loci that lastingly establish themselves in the genome of the recipient species. Our results strongly support this model, indicating that introgressed loci in the genome of omeiensis exhibited lower recombination and the hallmarks of high selection (Fig. 8), allowing them to occupy substantial portions of the recipient’s genome. However, high selection may not be enough to affect large parts of the genome. High-frequency gene flow may be a necessary requirement. Importantly, our study confirms that low recombination and nucleotide diversity may not be a good indicator of genomic regions suitable for inferring the true phylogeny in the context of ancient gene flow.
Acknowledgements
We thank the editors and three anonymous reviewers for the constructive comments, which improve this manuscript substantially. We thank Zuohua Yin and Xiaobing Li for sample collections. We also thank the National Zoological Museum of China for the assistance of specimen examination.
Supplementary Material
Data available from the Dryad Digital Repository: https://doi.org/10.5061/dryad.fbg79cnsz.
Funding
This research was funded by the National Science Foundation of China [31900320] and China Postdoctoral Science Foundation to D.Z., the Second Tibetan Plateau Scientific Expedition and Research (STEP) program [2019QZKK0501, 2019QZKK0304 to F.L. and D.Z.]; the National Science and Technology Basic Resources Survey Program of China [2019FY100204 to F.L.]; the Swedish National Science Foundation [2019-04486], Jornvall Foundation and Mark and Mo Constantine (to P.A) and the National Science Foundation of China [32070434] to G.S.
Data Accessiblity
Whole-genome resequencing data and short and long read data for de novo assembly produced in this study were deposited at the NCBI Sequence Read Archive (SRA) under Bioproject PRJNA646997. The genome assembly was deposited at NCBI under GenBank assembly accession GCA 017589585.1.
017589585.1.
References
- Alström P., Olsson U. 1999. The Golden-spectacled Warbler: a complex of sibling species, including a previously undescribed species. Ibis 141:545–568. [Google Scholar]
- Alström P., Rheindt F.E., Zhang R., Zhao M., Wang J., Zhu X., Gwee C.Y., Hao Y., Ohlson J., Jia C., Prawiradilaga D.M., Ericson P.G.P., Lei F., Olsson U. 2018. Complete species-level phylogeny of the leaf warbler (Aves: Phylloscopidae) radiation. Mol. Phylogenet. Evol. 126:141–152. [DOI] [PubMed] [Google Scholar]
- Avise J.C., Robinson T.J. 2008. Hemiplasy: a new term in the lexicon of phylogenetics. Syst. Biol. 57:503–507. [DOI] [PubMed] [Google Scholar]
- Bay R.A., Ruegg K. 2017. Genomic islands of divergence or opportunities for introgression? Proc. Biol. Sci. 284:20162414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bensch S., Andersson T., Åkesson S. 1999. Morphological and molecular variation across a migratory divide in willow warblers, Phylloscopus trochilus. Evolution 53:1925–1935. [DOI] [PubMed] [Google Scholar]
- Bouckaert R., Heled J., Kuhnert D., Vaughan T., Wu C.H., Xie D., Suchard M.A., Rambaut A., Drummond A.J. 2014. BEAST 2: a software platform for Bayesian evolutionary analysis. PLoS Comput. Biol. 10:e1003537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bouckaert R.R. 2010. DensiTree: making sense of sets of phylogenetic trees. Bioinformatics 26:1372–1373. [DOI] [PubMed] [Google Scholar]
- Bryant D., Bouckaert R., Felsenstein J., Rosenberg N.A., RoyChoudhury A. 2012. Inferring species trees directly from biallelic genetic markers: bypassing gene trees in a full coalescent analysis. Mol. Biol. Evol. 29:1917–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burbrink F.T., Gehara M. 2018. The biogeography of deep time phylogenetic reticulation. Syst. Biol. 67:743–744. [DOI] [PubMed] [Google Scholar]
- Burri R., Nater A., Kawakami T., Mugal C.F., Olason P.I., Smeds L., Suh A., Dutoit L., Bures S., Garamszegi L.Z., Hogner S., Moreno J., Qvarnstrom A., Ruzic M., Saether S.A., Saetre G.P., Torok J., Ellegren H. 2015. Linked selection and recombination rate variation drive the evolution of the genomic landscape of differentiation across the speciation continuum of Ficedula flycatchers. Genome Res. 25:1656–1665. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B. 2012. The effects of deleterious mutations on evolution at linked sites. Genetics 190:5–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Charlesworth B., Morgan M.T., Charlesworth D. 1993. The effect of deleterious mutations on neutral molecular variation. Genetics 134:1289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S., Zhou Y., Chen Y., Gu J. 2018. fastp: an ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 34:i884-i890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danecek P., Auton A., Abecasis G., Albers C.A., Banks E., DePristo M.A., Handsaker R.E., Lunter G., Marth G.T., Sherry S.T., McVean G., Durbin R., Genomes Project Analysis G. 2011. The variant call format and VCFtools. Bioinformatics 27:2156–2158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H., Rosenberg N.A. 2006. Discordance of species trees with their most likely gene trees. PLoS Genet. 2:e68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Degnan J.H., Rosenberg N.A. 2009. Gene tree discordance, phylogenetic inference and the multispecies coalescent. Trends Ecol. Evol. 24:332–340. [DOI] [PubMed] [Google Scholar]
- Durand E.Y., Patterson N., Reich D., Slatkin M. 2011. Testing for ancient admixture between closely related populations. Mol. Biol. Evol. 28:2239–2252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelman N.B., Frandsen P.B., Miyagi M., Clavijo B., Davey J., Dikow R.B., García-Accinelli G., Van Belleghem S.M., Patterson N., Neafsey D.E., Challis R., Kumar S., Moreira G.R.P., Salazar C., Chouteau M., Counterman B.A., Papa R., Blaxter M., Reed R.D., Dasmahapatra K.K., Kronforst M., Joron M., Jiggins C.D., McMillan W.O., Di Palma F., Blumberg A.J., Wakeley J., Jaffe D., Mallet J. 2019. Genomic architecture and introgression shape a butterfly radiation. Science 366:594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards S.V. 2009. Is a new and general theory of molecular systematics emerging? Evolution 63:1–19. [DOI] [PubMed] [Google Scholar]
- Edwards S.V., Xi Z., Janke A., Faircloth B.C., McCormack J.E., Glenn T.C., Zhong B., Wu S., Lemmon E.M., Lemmon A.R., Leache A.D., Liu L., Davis C.C. 2016. Implementing and testing the multispecies coalescent model: a valuable paradigm for phylogenomics. Mol. Phylogenet. Evol. 94:447–462. [DOI] [PubMed] [Google Scholar]
- Excoffier L., Dupanloup I., Huerta-Sanchez E., Sousa V.C., Foll M. 2013. Robust demographic inference from genomic and SNP data. PLoS Genet. 9:e1003905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faust Stryjewski K., Sorenson M.D. 2017. Mosaic genome evolution in a recent and rapid avian radiation. Nat. Ecol. Evol. 1:1912–1922. [DOI] [PubMed] [Google Scholar]
- Feder J.L., Egan S.P., Nosil P. 2012. The genomics of speciation-with-gene-flow. Trends Genet. 28:342–350. [DOI] [PubMed] [Google Scholar]
- Felsenstein J. 2004. Inferring phylogenies. Sunderland, MA: Sinauer Associates. [Google Scholar]
- Flouri T., Jiao X., Rannala B., Yang Z. 2020. A Bayesian implementation of the multispecies coalescent model with introgression for phylogenomic analysis. Mol. Biol. Evol. 37:1211–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fontaine M.C., Pease J.B., Steele A., Waterhouse R.M., Neafsey D.E., Sharakhov I.V., Jiang X., Hall A.B., Catteruccia F., Kakani E., Mitchell S.N., Wu Y.-C., Smith H.A., Love R.R., Lawniczak M.K., Slotman M.A., Emrich S.J., Hahn M.W., Besansky N.J. 2015. Extensive introgression in a malaria vector species complex revealed by phylogenomics. Science 347:1258524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao F., Ming C., Hu W., Li H. 2016. New software for the fast estimation of population recombination rates (FastEPRR) in the genomic era. G3-Genes Genom. Genet. 6:1563–1571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill F., Donsker D., Rasmussen P. 2020. IOC World Bird List (v10.2). 10.14344/IOC.ML.10.2. [DOI]
- Green R.E., Krause J., Briggs A.W., Maricic T., Stenzel U., Kircher M., Patterson N., Li H., Zhai W., Fritz M.H., Hansen N.F., Durand E.Y., Malaspinas A.S., Jensen J.D., Marques-Bonet T., Alkan C., Prufer K., Meyer M., Burbano H.A., Good J.M., Schultz R., Aximu-Petri A., Butthof A., Hober B., Hoffner B., Siegemund M., Weihmann A., Nusbaum C., Lander E.S., Russ C., Novod N., Affourtit J., Egholm M., Verna C., Rudan P., Brajkovic D., Kucan Z., Gusic I., Doronichev V.B., Golovanova L.V., Lalueza-Fox C., de la Rasilla M., Fortea J., Rosas A., Schmitz R.W., Johnson P.L.F., Eichler E.E., Falush D., Birney E., Mullikin J.C., Slatkin M., Nielsen R., Kelso J., Lachmann M., Reich D., Paabo S. 2010. A draft sequence of the Neandertal genome. Science 328:710–722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gutenkunst R.N., Hernandez R.D., Williamson S.H., Bustamante C.D. 2009. Inferring the joint demographic history of multiple populations from multidimensional SNP frequency data. PLoS Genet. 5:e1000695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harris R.B., Alström P., Odeen A., Leache A.D. 2018. Discordance between genomic divergence and phenotypic variation in a rapidly evolving avian genus (Motacilla). Mol. Phylogenet. Evol. 120:183–195. [DOI] [PubMed] [Google Scholar]
- Huson D.H., Richter D.C., Rausch C., Dezulian T., Franz M., Rupp R. 2007. Dendroscope: an interactive viewer for large phylogenetic trees. BMC Bioinformatics 8:460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin D.E. 2020. Assortative mating in hybrid zones is remarkably ineffective in promoting speciation. Am. Nat. 195:E150-E167. [DOI] [PubMed] [Google Scholar]
- Johansson U.S., Alström P., Olsson U., Ericson P.G.P., Sundberg P., Price T.D. 2007. Build-up of the Himalayan avifauna through immigration: a biogeographical analysis of the Phylloscopus and Seicercus warblers. Evolution 61:324–333. [DOI] [PubMed] [Google Scholar]
- Kawakami T., Backström N., Burri R., Husby A., Olason P., Rice A.M., Ålund M., Qvarnström A., Ellegren H. 2014. Estimation of linkage disequilibrium and interspecific gene flow in Ficedula flycatchers by a newly developed 50k single-nucleotide polymorphism array. Mol. Ecol. Resour. 14:1248–1260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koren S., Walenz B.P., Berlin K., Miller J.R., Bergman N.H., Phillippy A.M. 2017. Canu: scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 27:722–736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubatko L.S., Degnan J.H. 2007. Inconsistency of phylogenetic estimates from concatenated data under coalescence. Syst. Biol. 56:17–24. [DOI] [PubMed] [Google Scholar]
- Laine V.N., Gossmann T.I., Schachtschneider K.M., Garroway C.J., Madsen O., Verhoeven K.J., de Jager V., Megens H.J., Warren W.C., Minx P., Crooijmans R.P., Corcoran P., Great Tit HapMap C., Sheldon B.C., Slate J., Zeng K., van Oers K., Visser M.E., Groenen M.A. 2016. Evolutionary signals of selection on cognition from the great tit genome and methylome. Nat. Commun. 7:10474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Durbin R. 2009. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 25:1754–1760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H., Handsaker B., Wysoker A., Fennell T., Ruan J., Homer N., Marth G., Abecasis G., Durbin R., Genome Project Data Processing Subgroup. 2019. The Sequence Alignment/Map format and SAMtools. Bioinformatics 25:2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Davis B.W., Eizirik E., Murphy W.J. 2016. Phylogenomic evidence for ancient hybridization in the genomes of living cats (Felidae). Genome Res. 26:1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li G., Figueiro H.V., Eizirik E., Murphy W.J. 2019. Recombination-aware phylogenomics reveals the structured genomic landscape of hybridizing cat species. Mol. Biol. Evol. 36:2111–2126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu L., Anderson C., Pearl D., Edwards S.V. 2019. Modern phylogenomics: building phylogenetic trees using the multispecies coalescent model. In: Anisimova M, editor. Evolutionary genomics: statistical and computational methods. New York, NY: Springer New York. p. 211–239. [DOI] [PubMed] [Google Scholar]
- MacGuigan D.J., Near T.J. 2019. Phylogenomic signatures of ancient introgression in a rogue lineage of darters (Teleostei: Percidae). Syst. Biol. 68:329–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maddison W.P. 1997. Gene trees in species trees. Syst. Biol. 46:523–536. [Google Scholar]
- Malinsky M., Matschiner M., Svardal H. 2020. Dsuite-fast D-statistics and related admixture evidence from VCF files. Mol. Ecol. Resour. doi: 10.1111/1755-0998.13265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mallet J. 2005. Hybridization as an invasion of the genome. Trends Ecol. Evol. 20:229–237. [DOI] [PubMed] [Google Scholar]
- Mallet J., Besansky N., Hahn M.W. 2016. How reticulated are species? Bioessays 38:140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.H., Davey J.W., Jiggins C.D. 2015. Evaluating the use of ABBA-BABA statistics to locate introgressed loci. Mol. Biol. Evol. 32:244–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin S.H., Davey J.W., Salazar C., Jiggins C.D. 2019. Recombination rate variation shapes barriers to introgression across butterfly genomes. PLoS Biol. 17:e2006288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meier J.I., Marques D.A., Mwaiko S., Wagner C.E., Excoffier L., Seehausen O. 2017. Ancient hybridization fuels rapid cichlid fish adaptive radiations. Nat. Commun. 8:14363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nachman M.W., Payseur B.A. 2012. Recombination rate variation and speciation: theoretical predictions and empirical results from rabbits and mice. Philos. Trans. R. Soc. Lond. B Biol. Sci. 367:409–421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakamura T., Yamada K.D., Tomii K., Katoh K. 2018. Parallelization of MAFFT for large-scale multiple sequence alignments. Bioinformatics 34:2490–2492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nater A., Burri R., Kawakami T., Smeds L., Ellegren H. 2015. Resolving evolutionary relationships in closely related species with whole-genome sequencing data. Syst. Biol. 64:1000–1017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen L.T., Schmidt H.A., von Haeseler A., Minh B.Q. 2015. IQ-TREE: a fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 32:268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nichols R. 2001. Gene trees and species trees are not the same. Trends Ecol. Evol. 16:358–364. [DOI] [PubMed] [Google Scholar]
- Olsson U., Alström P., Ericson P.G.P., Sundberg P. 2005. Non-monophyletic taxa and cryptic species—evidence from a molecular phylogeny of leaf-warblers (Phylloscopus, Aves). Mol. Phylogenet. Evol. 36:261–276. [DOI] [PubMed] [Google Scholar]
- Olsson U., Alström P., Sundberg P. 2004. Non-monophyly of the avian genus Seicercus (Aves: Sylviidae) revealed by mitochondrial DNA. Zool. Scr. 33:501–510. [Google Scholar]
- Ottenburghs J., Kraus R.H.S., van Hooft P., van Wieren S.E., Ydenberg R.C., Prins H.H.T. 2017. Avian introgression in the genomic era. Avian Res. 8:30. [Google Scholar]
- Päckert M., Martens J., Sun Y., Severinghaus L.L., Nazarenko A.A., Ting J., Töpfer T., Tietze D.T. 2012. Horizontal and elevational phylogeographic patterns of Himalayan and Southeast Asian forest passerines (Aves: Passeriformes). J. Biogeogr. 39:556–573. [Google Scholar]
- Päckert M., Martens J., Sun Y.-H., Veith M. 2004. The radiation of the Seicercus burkii complex and its congeners (Aves: Sylviidae): molecular genetics and bioacoustics. Org. Divers. Evol. 4:341–364. [Google Scholar]
- Pamilo P., Nei M. 1988. Relationships between gene trees and species trees. Mol. Biol. Evol. 5:568–583. [DOI] [PubMed] [Google Scholar]
- Payseur B.A., Rieseberg L.H. 2016. A genomic perspective on hybridization and speciation. Mol. Ecol. 25:2337–2360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pease J.B., Hahn M.W. 2013. More accurate phylogenies inferred from low-recombination regions in the presence of incomplete lineage sorting. Evolution 67:2376–2384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price T.D. 2010. The roles of time and ecology in the continental radiation of the Old World leaf warblers (Phylloscopus and Seicercus). Philos. Trans. R. Soc. Lond. B Biol. Sci. 365:1749–1762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pryszcz L.P., Gabaldon T. 2016. Redundans: an assembly pipeline for highly heterozygous genomes. Nucleic Acids Res. 44:e113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pulido-Santacruz P., Aleixo A., Weir J.T. 2020. Genomic data reveal a protracted window of introgression during the diversification of a neotropical woodcreeper radiation. Evolution 74:842–858. [DOI] [PubMed] [Google Scholar]
- Rancilhac L., Irisarri I., Angelini C., Arntzen J.W., Babik W., Bossuyt F., Künzel S., Lüddecke T., Pasmans F., Sanchez E., Weisrock D., Veith M., Wielstra B., Steinfartz S., Hofreiter M., Philippe H., Vences M. 2021. Phylotranscriptomic evidence for pervasive ancient hybridization among Old World salamanders. Mol. Phylogenet. Evol. 155:106967. [DOI] [PubMed] [Google Scholar]
- Rannala B., Yang Z. 2003. Bayes Estimation of species divergence times and ancestral population sizes using DNA sequences from multiple loci. Genetics 164:1645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rheindt F.E., Edwards S.V. 2011. Genetic introgression: an integral but neglected component of speciation in birds. Auk 128:620–632. [Google Scholar]
- Simao F.A., Waterhouse R.M., Ioannidis P., Kriventseva E.V., Zdobnov E.M. 2015. BUSCO: assessing genome assembly and annotation completeness with single-copy orthologs. Bioinformatics 31:3210–3212. [DOI] [PubMed] [Google Scholar]
- Stamatakis A. 2014. RAxML version 8: a tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 30:1312–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stange M., Sanchez-Villagra M.R., Salzburger W., Matschiner M. 2018. Bayesian divergence-time estimation with genome-wide single-nucleotide polymorphism data of sea catfishes (Ariidae) supports Miocene closure of the Panamanian Isthmus. Syst. Biol. 67:681–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thawornwattana Y., Dalquen D., Yang Z. 2018. Coalescent analysis of phylogenomic data confidently resolves the species relationships in the Anopheles gambiae species complex. Mol. Biol. Evol. 35:2512–2527. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thom G., Amaral F.R.D., Hickerson M.J., Aleixo A., Araujo-Silva L.E., Ribas C.C., Choueri E., Miyaki C.Y. 2018. Phenotypic and genetic structure support gene flow generating gene tree discordances in an amazonian floodplain endemic species. Syst. Biol. 67:700–718. [DOI] [PubMed] [Google Scholar]
- Van Doren B.M., Campagna L., Helm B., Illera J.C., Lovette I.J., Liedvogel M. 2017. Correlated patterns of genetic diversity and differentiation across an avian family. Mol. Ecol. 26:3982–3997. [DOI] [PubMed] [Google Scholar]
- Vijay N., Weissensteiner M., Burri R., Kawakami T., Ellegren H., Wolf J.B.W. 2017. Genomewide patterns of variation in genetic diversity are shared among populations, species and higher-order taxa. Mol. Ecol. 26:4284–4295. [DOI] [PubMed] [Google Scholar]
- Vilella A.J., Severin J., Ureta-Vidal A., Heng L., Durbin R., Birney E. 2009. EnsemblCompara GeneTrees: complete, duplication-aware phylogenetic trees in vertebrates. Genome Res. 19:327–335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang J., Walker B.J., Abeel T., Shea T., Priest M., Abouelliel A., Sakthikumar S., Cuomo C.A., Zeng Q., Wortman J., Young S.K., Earl A.M. 2014. Pilon: an integrated tool for comprehensive microbial variant detection and genome assembly improvement. PLoS One 9:e112963. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D., Yu Y., Hahn M.W., Nakhleh L. 2016. Reticulate evolutionary history and extensive introgression in mosquito species revealed by phylogenetic network analysis. Mol. Ecol. 25:2361–2372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wen D., Yu Y., Zhu J., Nakhleh L., Posada D. 2018. Inferring phylogenetic networks using PhyloNet. Syst. Biol. 67:735–740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarza E., Faircloth B.C., Tsai W.L., Bryson R.W. Jr., Klicka J., McCormack J.E. 2016. Hidden histories of gene flow in highland birds revealed with genomic markers. Mol. Ecol. 25:5144–5157. [DOI] [PubMed] [Google Scholar]
- Zhang D., Song G., Gao B., Cheng Y., Qu Y., Wu S., Shao S., Wu Y., Alström P., Lei F. 2017. Genomic differentiation and patterns of gene flow between two long-tailed tit species (Aegithalos). Mol. Ecol. 26:6654–6665. [DOI] [PubMed] [Google Scholar]
- Zhang D., Tang L., Cheng Y., Hao Y., Xiong Y., Song G., Qu Y., Rheindt F.E., Alström P., Jia C., Lei F. 2019. “Ghost Introgression” as a cause of deep mitochondrial divergence in a bird species complex. Mol. Biol. Evol. 36:2375–2386. [DOI] [PubMed] [Google Scholar]
- Zhang G., Li C., Li Q., Li B., Larkin D.M., Lee C., Storz J.F., Antunes A., Greenwold M.J., Meredith R.W., Ödeen A., Cui J., Zhou Q., Xu L., Pan H., Wang Z., Jin L., Zhang P., Hu H., Yang W., Hu J., Xiao J., Yang Z., Liu Y., Xie Q., Yu H., Lian J., Wen P., Zhang F., Li H., Zeng Y., Xiong Z., Liu S., Zhou L., Huang Z., An N., Wang J., Zheng Q., Xiong Y., Wang G., Wang B., Wang J., Fan Y., da Fonseca R.R., Alfaro-Núñez A., Schubert M., Orlando L., Mourier T., Howard J.T., Ganapathy G., Pfenning A., Whitney O., Rivas M.V., Hara E., Smith J., Farré M., Narayan J., Slavov G., Romanov M.N., Borges R., Machado J.P., Khan I., Springer M.S., Gatesy J., Hoffmann F.G., Opazo J.C., Håstad O., Sawyer R.H., Kim H., Kim K.-W., Kim H.J., Cho S., Li N., Huang Y., Bruford M.W., Zhan X., Dixon A., Bertelsen M.F., Derryberry E., Warren W., Wilson R.K., Li S., Ray D.A., Green R.E., O’Brien S.J., Griffin D., Johnson W.E., Haussler D., Ryder O.A., Willerslev E., Graves G.R., Alström P., Fjeldså J., Mindell D.P., Edwards S.V., Braun E.L., Rahbek C., Burt D.W., Houde P., Zhang Y., Yang H., Wang J., Jarvis E.D., Gilbert M.T.P., Wang J. 2014. Comparative genomics reveals insights into avian genome evolution and adaptation. Science 346:1311–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]