

Abstract
Myosin 18Aα is a myosin 2-like protein containing unique N- and C-terminal protein interaction domains that co-assembles with myosin 2. One protein known to bind to myosin 18Aα is β-Pix, a guanine nucleotide exchange factor (GEF) for Rac1 and Cdc42 that has been shown to promote dendritic spine maturation by activating the assembly of actin and myosin filaments in spines. Here we show that myosin 18A⍺ concentrates in the spines of cerebellar Purkinje neurons via co-assembly with myosin 2 and through an actin binding site in its N-terminal extension. miRNA-mediated knockdown of myosin 18A⍺ results in a significant defect in spine maturation that is rescued by an RNAi-immune version of myosin 18A⍺. Importantly, β-Pix co-localizes with myosin 18A⍺ in spines, and its spine localization is lost upon myosin 18A⍺ knockdown or when its myosin 18A⍺ binding site is deleted. Finally, we show that the spines of myosin 18A⍺ knockdown Purkinje neurons contain significantly less F-actin and myosin 2. Together, these data argue that mixed filaments of myosin 2 and myosin 18A⍺ form a complex with β-Pix in Purkinje neuron spines that promotes spine maturation by enhancing the assembly of actin and myosin filaments downstream of β-Pix’s GEF activity.
Keywords: F-actin, myosin 2, spine development
INTRODUCTION
Dendritic spines are small protrusions on the surface of neuronal dendrites that form the postsynaptic component of most excitatory synapses in the brain, function as signaling microcompartments, and serve as the primary site of memory formation (1–13). Actin filaments comprise the major structural element of spines, and changes in spine size and shape that occur upon synapse usage are thought to result largely from changes in spine actin content, organization and dynamics (2, 6–10, 14, 15). Moreover, these actin-dependent changes in spine morphology (structural plasticity) are thought to promote the long-lasting changes in synaptic strength (functional plasticity) that underlie learning and memory (3–5, 11–13). Consistently, many learning and memory disorders (e.g. autism, psychosis) are associated with actin-related defects in dendritic spine morphology and dynamics (16, 17). Defining the molecular basis of learning and memory in both health and disease will require, therefore, a thorough understanding of the origin, organization and dynamics of actin in spines.
While several models have been proposed for how dendritic spines are created, the prevailing model is that they begin life as dendritic filopodia, and that these filopodia then mature into spines (15, 18–21). Like conventional filopodia, dendritic filipodia contain long, unbranched actin filaments (22) created by a formin (in this case mDia2 downstream of the Rho-related GTPase Rif) (23). Unlike conventional filopodia, dendritic filopodia also contain branched actin filaments (22). Whether these branched filaments are created by the Arp2/3 complex is not entirely clear (22, 24). What is much clearer, however, is that the maturation of dendritic filopodia into dendritic spines involves a large increase in the creation of branched actin filaments by the Arp2/3 complex (24–28). This increase drives the expansion of the maturing spine head and coincides with shortening of the filopodia precursor and constriction at the base of the maturing spine. These changes yield the prototypical mature, mushroom-shaped spine possessing an enlarged head filled with branched actin that is connected to the dendrite by a narrow spine neck containing both branched and unbranched actin filaments.
Guanine nucleotide exchange factors (GEFs) for Rho-related GTPases likely play key roles in the regulation of actin dynamics underlying spine formation, spine maturation, and spine structural plasticity, as these proteins link signal transduction pathways to the activation of factors required for actin assembly and turnover (29–34). One GEF that has been implicated repeatedly in driving spine maturation is the Rac1 and Cdc42 GEF β-Pix (30, 35–39). The activation of Rac1 and Cdc42 within spines by β-Pix is thought to drive spine maturation via several distinct pathways. First, GTP-bound Rac1 and GTP-bound Cdc42 can activate two nucleation promoting factors (NPFs) for the Arp2/3 complex (WAVE in the case of Rac1-GTP, WASp in the case of Cdc42-GTP), resulting in the activation of branched actin filament assembly (40). Several published studies are consistent with the idea that β-Pix promotes spine maturation via this pathway (24–26, 28, 32, 33, 41). Second, both GTP-bound Rac1 and GTP-bound Cdc42 can activate PAK kinase, leading to the activation of myosin 2 filament assembly and contractility within spines via the PAK-dependent phosphorylation of the myosin’s regulatory light chain (RLC) (42). Direct evidence that this β-Pix-dependent pathway promotes spine maturation has been presented (35, 36). Active PAK also appears to promote spine maturation by phosphorylating/activating LIM-kinase (LIM-K), as this leads to the stabilization of spine actin via the LIM-K-dependent inhibition cofilin-driven actin filament turnover (43, 44). Finally, the morphogenesis of spines that occurs in response to NMDA receptor activation has been shown to be mediated by a signaling complex containing two related calcium-calmodulin-dependent protein kinases, β-Pix, and Rac1 (38, 39).
In most cellular contexts where F-actin plays a critical role, myosin 2 also plays a critical role. Consistent with this paradigm, myosin 2B is enriched along with F-actin in spines, and attenuating the myosin’s function in developing neurons either pharmacologically or by knockdown results in a strong block in spine maturation (45–54). Moreover, blocking myosin 2B function in fully-developed neurons shows that it also plays a critical role in the activity-dependent changes in spine actin morphology underlying learning and memory (48, 50, 52–54). While myosin 2B is present in dendritic filopodia as well as in mature spines (22), its function in mature spines has been studied most. Myosin 2B resides primarily at the base of these structures (i.e. in the spine neck and base of the head), most likely in the form of bipolar filaments (22). The contractile activity of these bipolar filaments is thought to drive the shortening of immature spines during spine maturation, the constriction of the spine neck, the tip-to-base flow of a dynamic spine actin pool, the cross-linking of a stable spine actin pool, and possibly global spine actin dynamics by catalyzing actin filament turnover downstream of myosin-dependent actin filament breakage (52). Interestingly, Purkinje neurons express an alternatively spliced version of myosin 2B known as myosin 2B-B2 in addition to regular myosin 2B (47). Mice lacking this splice variant exhibit aberrant Purkinje neuron development (reduced numbers of dendritic spines and branches) and impaired motor coordination (47).
The focus of this study is on the function of the myosin 2-like protein myosin 18Aα (55, 56), one of three alternatively spliced isoforms generated from the mouse M18A gene. Like myosin 2, all three myosin 18A isoforms possess a globular head domain followed by a rod-like coiled-coil tail domain. Unlike myosin 2, all three isoforms possess shared and unique N- and C-terminal extensions that harbor both recognizable (e.g. PDZ domain, SH3 domain binding sites) and uncharacterized protein: protein interaction domains (55, 56). Also unlike myosin 2, two of these three isoforms (myosin 18Aα and myosin 18Aβ) have been shown to lack motor activity and to be incapable of self-assembling into bipolar filaments, the working form of myosin 2 (55, 57, 58). Despite these peculiarities, and the fact that myosin 18A isoforms are present in cells in amounts significantly sub-stochiometric to myosin 2 (about 1 myosin 18Aα or myosin 18Aβ for every 10 to 50 myosin 2s) (55), the knockout of myosin 18A results in embryonic lethality in both flies (59) and mice (56). Moreover, at least one spliced isoform of myosin 18A appears to be expressed in every differentiated mammalian cell type based on expression profiling.
A major breakthrough in resolving the enigma of myosin 18A came from biochemical data showing that both myosin 18Aα and myosin 18Aβ co-assemble with myosin 2 in vitro to make mixed filaments, and that such mixed filaments can be seen in living cells using super-resolution light microscopy (55). Importantly, co-assembly provided explanations for myosin 18A’s unusual properties: it does not need to self-assemble because it can co-assemble with myosin 2, it does not need to be a motor because myosin 2 in the mixed filament will provide that function, and its low stoichiometry ensures that mixed filaments contain mostly myosin 2. Moreover, co-assembly suggested a clear functional role for myosin 18A: display of its N- and C-terminal protein interaction domains on the surface of mixed filaments could serve to recruit proteins to mixed filaments and/or attach them to cellular structures. Implicit in this hypothesis is that myosin 18A function is inextricably linked to myosin 2 function. Given that myosin 18A is ubiquitously expressed, it may be the case that most past studies of myosin 2 function in organisms ranging from fly to man were actually interrogating the function of mixed filaments of myosin 2 and myosin 18A.
One bona fide myosin 18A binding partner is β-Pix, which binds via 12 residues near its C-terminus to myosin 18A’s ~110-residue non-helical tailpiece (60, 61). This interaction has been shown to regulate cell migration and focal adhesion dynamics in mesenchymal cells (60, 61). A second binding partner is the myosin 2 regulatory light chain kinase MRCK, which binds to myosin 18A’s N-terminal PDZ domain through the adaptor protein LRAP (62). This interaction has been shown to mediate myosin 18A’s role in regulating the dynamics of actomyosin 2 networks in mesenchymal cells by phosphorylating and activating myosin 2 (62). Relevant to this, β-Pix is typically found in a complex with PAK kinase (and the Arf GTPase activating protein GIT1), so the interaction of myosin 18A with β-Pix could conceivably lead to myosin 2 activation downstream of PAK (36, 37).
Given that both myosin 2 and β-Pix play important roles in spine maturation and function, that myosin 18A co-assembles with myosin 2 and binds β-Pix, and that myosin 18A is highly expressed throughout the brain (e.g. in pyramidal neurons in the hippocampus and cortex, in cerebellar Purkinje neurons), we decided to investigate myosin 18A function in a neuronal cell type. With regard to which cell type to study, previous work from our lab showed that myosin Va transports tubules of smooth endoplasmic reticulum into Purkinje neuron spines to support synaptic plasticity and motor learning, thereby explaining the ataxia exhibited by dilute/myosin Va null mice (63). Our subsequent efforts have resulted in improvements in culturing and transfecting Purkinje neurons, in the development of a Purkinje neuron-specific miRNA-mediated knockdown system (64), and in creating Purkinje neurons from embryonic stem cells (65). For these reasons we decided to investigate myosin 18A function in Purkinje neurons. Here we show that myosin 18A targets β-Pix to Purkinje neuron spines and promotes spine maturation.
METHODS
Primary cerebellar cultures and mice
Timed-pregnant C57BL/6 females of gestation day 17–18 were purchased from Charles Rivers Laboratories. E17-E18 embryos were harvested and mixed cerebellar cultures were prepared as described previously (64). While a detailed description of the generation of the myosin 18A conditional knockout mouse will be presented elsewhere, the mouse was created using a targeting vector containing a Neomycin resistance cassette flanked by Flp sites and exons 11 to 13 of myosin 18A, all of which was bounded by LoxP sites. Deletion of exons 11 to 13 deletes residues 685 to 835 within the myosin’s motor domain, introduces a frame shift, and should block expression of all known myosin 18A spliced isoforms. This vector was electroporated into C57BL/6 ES cells, Neomycin-resistant ES cells were selected, and targeted clones were confirmed by Southern blotting. Confirmed clones were microinjected into wild type albino (C57BL/6 BrdCrtHsd-Tyrc) blastocysts and transferred to pseudo-pregnant females. Chimeric male mice were bred with wild type albino females and heterozygous myosin 18A Neo-cKO offspring were identified by PCR genotyping. After crossing to homozygosity, the Neo cassette was excised by crossing with a FLP mouse. Finally, homozygous myosin 18A cKO mice were back crossed ten times to C57BL/6. Efforts were made to limit the number of animals used in this study. All animal experiments were approved by the Institutional Animal Care and Use Committee of the National Heart, Lung, and Blood Institute in accordance with the National Institutes of Health guidelines.
Immunofluorescence
Primary cerebellar cultures plated on 35mm round dishes with coverslip bottoms were fixed for 15 minutes at room temperature in 4% paraformaldehyde (PFA) in Cytoskeleton Fixation Buffer (10 mM PIPES (pH 6.8), 100 mM NaCl, 10% (v/v) sucrose, 3 mM MgCl2, 1 mM EGTA) by diluting a 16% PFA stock solution (Electron Microscope Sciences, 15710). Samples were then washed three times with PBS (Gibco, 70011044), permeabilized in PERM buffer (300 mM Glycine, 0.5% Triton X-100 in PBS (pH 7.4)) for 10 minutes, washed once with PBS, and blocked for 30 minutes in IF Blocking Buffer (3% (w/v) BSA (Sigma A8412), 0.1 % (v/v) Tween 20 in PBS (pH 7.4)). Purkinje neurons were identified using anti-Calbindin D28K antibody (guinea pig, 1:500, Synaptic Systems, 214 004) or anti-mCherry antibody (mouse, 1:500, MyBioSource, MBS555186). Depending on the experiment, cultures were also stained with the following primary antibodies at the indicated dilutions: an anti-myosin 18Aα raised against the C-terminal 18 residues of myosin 18Aα/β (rabbit, 1:50) (55, 66), anti-Bassoon (rabbit, 1:200, Synaptic Systems, 141 003), anti-myosin 2B (rabbit, 1:200, ThermoFisher, PA517026), or anti-β-Pix (mouse, 1:200, BD Biosciences, 611648) in 200 μl of IF Blocking Solution for 45 minutes at room temperature. Following three 5-minute washes in PBS, samples were incubated for 45 minutes at room temperature with 1:500 dilutions of the appropriate labeled secondary antibody (Alexa Fluor 546 goat anti-guinea pig, Thermo Fisher, A11074; Alexa Fluor 546 goat anti-mouse, Thermo Fisher, A11003; Alexa Fluor 488 goat anti-rabbit, Thermo Fisher, A11008). Samples were then washed three times with PBS. To visualize F-actin, cells were incubated during the final wash step with Alexa Fluor 647 labelled Phalloidin for 5 minutes (1:100, Thermo Fisher Scientific, A12379). Following all wash steps, cells were overlaid with 200 μl of Fluromount-G mounting medium (Electron Microscope Sciences, 179840–25), covered with a 25 mm circular coverslip (Electron Microscope Sciences, 72223–01), and sealed with slide sealant. Images were obtained using a Zeiss LSM 780 laser scanning confocal microscope equipped with a Zeiss 63 X, 1.4 NA oil objective. Perfusion fixed sagittal cerebellar slices were a kind gift of Dr. Herbert Geller (NHLBI/NIH) and prepared as previously described (67).
Cloning
All oligonucleotides used in this study were purchased from Eurofins Genomics at 50 nM scale and purified by standard desalting. Table 1 shows the oligonucleotides used to generate the pL7-based (68), Purkinje neuron-specific expression plasmids for myosin 18Aα, myosin 18Aβ, myosin 2B, and β-Pix using the following DNA templates: mouse myosin 18A (Riken clone F730020C19), mouse myosin 2B-B2 (47), and mouse β-Pix (Mammalian Gene Collection clone MMM1013–202702676). DNA fragments containing 21 base pair overhangs homologous to the vector backbone were generated by PCR using Phusion High-Fidelity DNA polymerase (NEB, M0530), purified, and inserted into Bgl II/Sal I-linearized pL7 mGFP or pL7 mCherry (68) using an In-Fusion cloning kit (Takara, 638920). The specific fragments generated using the appropriate template and indicated primers were: myosin 18Aα (oM18_1 and oM18_2), myosin 18Aβ (oM18_3 and oM18_4), myosin 2B-B2 (oM18_5 and oM18_6), myosin 18Aα-ΔNHT (lacking C-terminal residues 1940–2050) (oM18_7 and oM18_8), myosin 18Aα/β-CC (containing residues 1241–2035 of myosin 18Aα, which are common to myosin 18Aα and myosin 18Aβ) (oM18_9 and oM18_10), and β-Pix (oM18_19 and oM18_20). pL7 mGFP myosin 18Aα-ABmut in which residues V117 and L118 were both changed to A (69) was generated by site directed mutagenesis using primers oM18_17 and oM18_18 (Table 1) and a Quikchange II Site-directed Mutagenesis kit (Agilent, 200523). The PCR product was digested with DpnI enzyme to remove template vector and transformed into Stbl2 chemically competent cells (Thermo Fisher, 10268019). β-Pix lacking its myosin 18A binding site (residues 639–647) was synthesized by Gene Universal and cloned into pL7 mGFP as a Bgl II/Sal I fragment. The myosin 18Aα N-terminal extension (NT) constructs were generated using traditional cloning methods. Specifically, the wild type N-terminal extension (NT; residues 1–334), the N-terminus lacking the KE-rich region (NT-ΔKE; residues 1–29 deleted), the N-terminus containing the PDZ domain mutation (NT-PDZmut; residue G232 changed to P (70)), and the N-terminus containing the actin binding site mutation (NT-ABmut; residues V117 and L118 changed to A) were amplified by PCR with Bgl II/Sal I ends using the appropriate template and primers indicated in Table 1, or they were synthesized by Gene Universal. The purified NT fragments were digested with Bgl II/Sal I and ligated into Bgl II/Sal I-linearized pL7 mGFP. To generate the E. coli expression plasmids NT (residues 1–334) and NT-ABmut (containing the VLΔAA point mutations), fragments with Nco I/Sal I ends were PCR amplified using the appropriate pL7 template and primers oM18_21 and oM18_22. The PCR products were purified, digested with Nco I/Sal I, and ligated into the GST expression plasmid pGEX-Parallel 2 (71) linearized with Nco I/Sal I to generate GST fusion proteins NT and NT-ABmut. All plasmids generated in this study were confirmed by sequencing.
Table 1.
Oligonucleotides used in this study
| Oligonucleotide | Oligonucleotide full name | Sequence (5’ to 3’) |
|---|---|---|
| oM18_1 | pL7_Myo18Aa_F | GTCCGGACTCAGATCTATGTTTAACCTCATGAAGAAAGATAAGGA |
| oM18_2 | pL7_Myo18Aa_R | AGCAGGATCCGTCGACCTATGCACTGGTCTCTGTCAGCTT |
| oM18_3 | pL7_Myo18Ab_F | GTCCGGACTCAGATCTATGGCCTTTTCAACG |
| oM18_4 | pL7_Myo18Aa_R | CCGCGGTACCGTCGACCAGTGGTGCCCCTCTATCCT |
| oM18_5 | pL7_M2B2_R | GTCCGGACTCAGATCTATGGCGCAGAGAACTG |
| oM18_6 | pL7_M2B2_F | AGCAGGATCCGTCGACCAACTTTACTCTGACTGGGG |
| oM18_7 | pL7_Myo18AaΔNHT_R | GTCCGGACTCAGATCTATCTAAATGCGTTTGAACGCCAGC |
| oM18_8 | pL7_Myo18AaΔNHT_F | AGCAGGATCCGTCGATGTTTAACCTCATGAAGAAAGATAAGGA |
| oM18_9 | pL7_Myo18Aa_CC_F | GTCCGGACTCAGATCTCCCCTCATCCAAGTTCAGCTG |
| oM18_10 | pL7_Myo18Aa_CC_R | CCGCGGTACCGTCGACCTATCCACTGGTCTCTGTCAG |
| oM18_11 | pL7_NT1_F | CTCAGATCTATGTCTGCGGCAGAACTG |
| oM18_12 | pL7_NT1_R | TCCGTCGACCTAGCGCTCGC |
| oM18_13 | pL7_NT1_ΔKE_F | CTCAGATCTATGTTTAACCTCATG |
| oM18_14 | pL7_NT1_ΔKE_R | TCCGTCGACCTAGCGCTCGC |
| oM18_15 | pL7_NT1_PDZmut_F | CCCACAGGAGACTTTCCCTTCTCACTGCGGC |
| oM18_16 | pL7_NT1_PDZmut_R | GCCGCAGTGAGAAGGGAAAGTCTCCTGTGGG |
| oM18_17 | pL7_M18Aa_ABMut_F | CCGGGGCTCTGCGGCGCAGCGGGCAGCC |
| oM18_18 | pL7_M18Aa_ABMut_R | GGCTGCCCGCTGCGCCGCAGAGCCCCGG |
| oM18_19 | pL7_βPIX_F | CAAGTCCGGACTCAGATCTACTGATAACACCAACAGCC |
| oM18_20 | pL7_βPIX_R | CAGCAGGATCCGTCGACCTATAGATTGGTCTCATCCCA |
| oM18_21 | pGST_NT1_F | ATTGCCATGGGCATGACTGATAACAC |
| oM18_22 | pGST_NT1_R | TAATGTCGACTATAGATTGGTCTC |
miRNA-mediated knockdown
Purkinje neuron-specific, miRNA-mediated protein knockdown was performed as described previously (64) using the BLOCK-iT Inducible Pol II miR RNAi Expression Vector Kit (Life Technologies K4939–00). Briefly, complementary oligonucleotides for generating miRNAs directed against the 3’ UTR of target proteins were designed using the Thermo Fisher BLOCK-iT RNAi Design Tool (https://rnaidesigner.thermofisher.com/rnaexpress/) and NM_001291213 (mouse myosin 18A) and NM_001346804 (mouse β-PIX) as reference sequences. The oligonucleotides were ligated into the pcDNA6.2/emGFP plasmid provided with the kit and the resulting plasmids used as templates to PCR amplify complete miRNA cassettes with Bgl II and Sal I ends for ligation into Bgl II/Sal I cut pL7 mCherry (64). The final constructs were sequence verified.
Western Blotting
Cerebella from E17 embryos and adult (~8 weeks old) mice were harvested and lysed in RIPA buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 2 mM EDTA, 0.5% (w/v) Na-deoxycholate, 1% (v/v) NP-40, and 0.1% (v/v) SDS) supplemented with Complete Protease Inhibitor Cocktail (Roche, 11697498001)). Whole cell extracts of Jurkat cells, a human leukemic T cell line (ATCC, TIB-152), were prepared as described previously (72). Protein concentrations were determined using a BCA Protein Assay (Thermo Fisher, 23225). Equal amounts of protein were run on 6% SDS-PAGE gels and blotted onto nitrocellulose using a semi-dry Trans-Blot (Bio-Rad, 1703940). Blots were incubated overnight at 4°C with a rabbit anti-myosin 18A polyclonal antibody that recognizes the C-terminal 18 residues of both myosin 18Aα and 18Aβ (66) diluted 1:1000 in TRIS-buffered saline containing 0.05% (v/v) Tween-20 (TBS-T) and supplemented with 5% (w/v) dry nonfat milk. Blots were washed extensively with TBS-T and then incubated with anti-rabbit HRP secondary antibody (GE Lifesciences, NA934) diluted 1:5000 in TBS-T supplemented with 5% (w/v) dry nonfat milk. Bands were detected using the SuperSignal West Pico PLUS Chemiluminescent detection system (Thermo Fisher, 34577). The blot in Figure S1 also contained an extract of adult mouse cardiac muscle, and was probed with a rabbit anti-myosin 18A polyclonal antibody raised against the C-terminal 300 residues of myosin 18Aα and myosin 18β (66) (this antibody should also see myosin 18Aγ because the immunogen overlaps with 252 residues of myosin 18Aγ). The extent of miRNA-mediated knockdown of myosin 18A was estimated using NIH 3T3 mouse embryonic fibroblasts (ATCC CRL-1658) cultured in DMEM medium containing 4 mM Glutamax (Thermo Fisher, 10566) and 10% (v/v) FBS. Cells were grown to ~70% confluency, harvested with TripLE dissociation reagent (Thermo Fisher, 12604013) and counted. Each miRNA, under the control of the CMV promoter, was introduced into cells by nucleofection using an Amaxa Nucleofector (Lonza). Briefly, 1 × 106 cells were collected and resuspended in 100 ul of Nucleofection Solution (5 mM KCl, 15 mM MgCl2, 120 mM Na2HPO4/NaH2PO4 (pH 7.2), 50 mM Mannitol) containing 2 ug of plasmid DNA. Nucleofection was carried out using program U-(0)03. Cells were immediately transferred to one well of a 6-well plate containing 3 ml of growth medium, cultured for 36 h, harvested, lysed in SDS-PAGE loading buffer, and subjected to SDS-PAGE. Proteins were transferred to nitrocellulose, probed with the rabbit anti-myosin 18A polyclonal antibody directed against the C-terminal 18 residues of myosin 18Aα/β (1:1000, cite JB Cytoskeleton 2017) and with a mouse anti-RACK1 monoclonal antibody (1:1000, Santa Cruz, sc-17754) as the loading control, and developed using an anti-rabbit HRP secondary antibody (1:5000, GE Lifesciences, NA931) and an anti-mouse HRP secondary antibody (1:10,000, GE Healthcare, NA935). Blots were imaged using an Amersham Imager 600 (GE Healthcare Life Sciences) and the extent of knockdown was estimated by densitometry using Fiji (73). For the Western blot in Figure S5, Panel D, mouse embryo fibroblasts (MEFs) from the myosin 18A cKO mouse were prepared and cultured as described previously (74), transfected with a puromycin resistance plasmid that expresses Cre recombinase (Addgene, 17408), grown for 48 hours and then selected for using 2.5 μg/ml puromycin (Invivogen, ant-pr), and processed as for the miRNA-treated 3T3 fibroblasts except that a rabbit polyclonal against the heavy chain of clathrin (1:1000, Abcam, ab21679) was used as the loading control.
F-actin co-sedimentation assay
GST-NT and GST-NT-ABmut were expressed in BL21-CodonPlus (DE3)-RIPL cells (Agilent, 230280), purified as described previously (75), and dialyzed into KMEI buffer (50 mM KCl, 1 mM MgCl2, 1 mM EGTA and 10 mM imidazole (pH 7.0)). Actin was purified as described previously (76) and dialyzed into G-buffer (2 mM TRIS (pH 8.0), 0.2 mM ATP, 0.5 mM DTT, 0.1 mM CaCl2 and 1 mM NaN3). The affinities of NT and NT-ABmut for F-actin were determined by high speed co-sedimentation assays essentially as described previously (75, 76). Briefly, 1 μM of NT or NT-ABmut were incubated for 60 min at room temperature with increasing molar ratios of F-actin (1:0, 1:1, 1:2.5, 1:3.5 or 1:5 for NT; 1:0, 1:10, 1:15, 1:25 or 1:40 for NT-ABmut), centrifuged at 100,000 x g for 1 h at room temperature, and the supernatant (S) and pellet (P) fractions resolved by SDS-PAGE. The amount of NT fusion protein in each sample was determined by densitometry using Fiji (73). The data were fit to a one-site binding hyperbola to obtain Kd values.
Image analyses
Stained cells were imaged using a Zeiss LSM 780 confocal microscope equipped with 63 X Plan Apo objectives (NA 1.4). All morphological measurements of Purkinje neurons were performed using Fiji Image Software (73). To measure dendritic spine length, serial 0.4 μm confocal sections that encompass the entire spine (identified using the cell volume marker mCherry in the 546 nm channel) together with the adjacent dendrite were summed using the Fiji Z projection function. Using the line tool, individual dendritic spines were masked by drawing a line from the tip of the spine head through to the dendrite at the base of the spine. Spine length was then determined by measuring the length of each corresponding line. To determine dendritic spine density, four serial 0.4 μm confocal sections (a total of 1.2 μm) were summed using the Fiji Z projection function. The number of dendritic spines in 10 μm lengths of dendrite were counted and used to determine the average number of spines per μm2. The enrichment of proteins in dendritic spines relative to dendrites was determined by ratio imaging as described previously (77). Briefly, cells were co-transfected with a GFP-tagged protein of interest and mCherry as a volume marker. Serial 0.4 μm confocal sections that encompass an entire spine (identified using the mCherry volume marker in the 546 nm channel) together with the adjacent dendrite were summed using the Fiji Z projection function. The total fluorescence for the GFP signal (488 nm channel) in the spine and in an equal-sized area of the dendrite immediately under the spine were then measured. To correct for differences in volume between these two regions, the total fluorescence for the mCherry volume marker (546 nm channel) was also measured in both regions. The apparent fold-enrichment of the GFP-tagged protein in the spine relative to the subjacent dendrite region was then corrected for any difference in volume between these two regions to obtain an accurate value for the fold-enrichment of the GFP-tagged protein in the spine. To determine the content of F-actin per spine, cells were fixed and stained with a rabbit anti-mCherry antibody (1:200, MyBioSource, MBS9400754) followed by an Alexa 546-conjugated goat anti-rabbit secondary antibody (1:500, Thermo Fisher, A11035) (this staining serves to both identify myosin 18Aα miRNA-expressing cells and mark their volume), and with Alexa 647-conjugated Phalloidin to label F-actin. Control cells were fixed and stained with Alexa 647-conjugated Phalloidin and with anti-Calbindin D28K followed by Alexa 546-conjugated anti-Guinea Pig secondary antibody to mark their volume. Serial 0.4 μm confocal sections that encompass the entire dendrite were collected and summed using the Fiji Z projection function. The mCherry or anti-Calbindin volume signals were then thresholded to determine the outline of the dendrite and dendritic spines, and this outline was then used to mask any Phalloidin signal in the 647 channel outside the Purkinje neuron volume. The total Phalloidin fluorescence per spine was then measured. To determine the content of myosin 2B per spine, cells were fixed and stained with the anti-mCherry antibody as described above to identify cells expressing the myosin 18Aα or Scrambled miRNA and to mark their volume, and with the rabbit anti-myosin 2B antibody (1:200, ThermoFisher, PA517026) to label the myosin. The content of myosin 2B per spine was determined as for F-actin. As discussed in the relevant Figure legends, we also provided estimates of the reductions in spine F-actin and myosin 2B content in knockdown cells that included corrections for differences between control and knockdown cells in spine volume.
Statistical Analyses
Statistics for all experiments were performed using Graphpad Prism v7 software (Mac Version), and all figures were prepared using Inkscape, Inkscape Project v0.92.3 (Mac Version). Statistical details, including N values and what these values represent, are included in the appropriate figures, figure legends and text. For comparison of two groups, a parametric Student’s t-test with Welch’s correction was performed. For the Pearson’s correlation coefficient data in Figure S2, the signals in the 594 channel (Calbindin D28K) and 488 channel (myosin 18Aα) were measured using the Fiji co-localization tool. As a control, the 488 channel (myosin 18Aα) was rotated 90 degrees and the Pearson’s correlation coefficient was re-calculated. Three equal-sized regions of cerebellar slices were analyzed. All of the statistical data is presented as means with standard deviations. Significance values, which are included in figures, figure legends, and text, are defined as follows: * p=<0.05, ** p=<0.01, *** p=<0.001, **** p=<0.0001.
Immunoelectron microscopy
Immunoelectron microscopy was performed using a pre-embedding immunoperoxidase method followed by silver/gold enhancement as described previously (78, 79). Briefly, two 5-week old Sprague-Dawley rats were anesthetized with Ketamine/xylazine and perfused with 4% paraformaldehyde in PBS. Fifty micrometer brain sections in 30% sucrose/PBS were frozen using acetone in dry ice and stored at −80°C. Thawed sections were washed in PBS and incubated for 1 hour in 10% (v/v) normal goat serum diluted in PBS, and then in a 1:2000 dilution of the rabbit C-terminal myosin 18Aα primary antibody in PBS at 4°C overnight. Samples were processed the following day using a Vectastain kit (biotinylated goat-anti-rabbit IgG secondary antibody and avidin-biotin-peroxidase complex) and ImpacDAB (Vector Laboratories). Control sections lacking the primary antibody were unlabeled. Subsequent silver/gold toning was performed in cacodylate buffer, including incubation for 10 minutes at 60°C in 0.2% (w/v) silver nitrate, 0.2% (w/v) sodium borate and 2.6% (v/v) hexamethylenetetramine, followed by 2 minutes each with 0.05% (w/v) gold chloride and 3% (w/v) sodium thiosulfate. Finally, sections were fixed in 2% (w/v) glutaraldehyde and then 1% (w/v) osmium tetroxide, dehydrated in alcohols and propylene oxide, and embedded in Epon. Thin sections from the 2 rats were produced on a Leica Ultracut ultramicrotome and were examined in a JEOL JSM-1010 transmission electron microscope.
RESULTS
Purkinje neurons express myosin 18Aα
As discussed briefly in the Introduction, myosin 18A mRNA (detected using a probe that should recognize all spliced isoforms) is present at moderate to high levels throughout the central nervous system, including in cerebellar Purkinje neurons (80). Three myosin 18A splice isoforms have been described to date: 18Aα, 18Aβ, and 18Aγ (55, 56). These three isoforms share head and coiled-coil domains but differ in their N- and C-terminal extensions, with myosin 18Aβ lacking myosin 18Aα’s N-terminal extension, and myosin 18Aγ having large, unique N- and C-terminal domains (Figure 1A). A Western blot of embryonic and adult mouse cerebellar extracts (Figure 1B, lanes 2 and 3, respectively) probed with an antibody that recognizes the C-terminal 18 residues of both myosin 18Aα and 18Aβ (66) detected only the ~235 kDa myosin 18Aα isoform in both cerebellar extracts (lane 1 shows the strong signal for myosin 18Aβ at ~200 kDa in a Jurkat T cell whole cell extract; (72)). A second Western blot of extracts from Jurkat T cells, adult mouse cerebellum, and adult mouse cardiac muscle probed with an antibody to the C-terminal 300 residues of myosin 18Aα/myosin 18Aβ, which overlap with 252 residues within the tail domain of myosin 18Ay, again detected only myosin 18Aβ in T cells and myosin 18Aα in the cerebellar extract (Figure S1, lanes 1 and 2, respectively), but also a ~290 kDa isoform specific to the cardiac muscle extract (Figure S1, lane 3). This ~290 kDa band probably corresponds to myosin 18Aƴ given that its expression is reported to be restricted to cardiac muscle ((56); our RT PCR data also shows that myosin 18Aƴ mRNA is not detectable in adult mouse brain (data not shown)). Based on all of these observations, we conclude that Purkinje neurons express myosin 18A, and that they express predominantly if not exclusively the 18Aα isoform.
Figure 1. Endogenous myosin 18Aα localizes to the dendritic spines of Purkinje neurons.
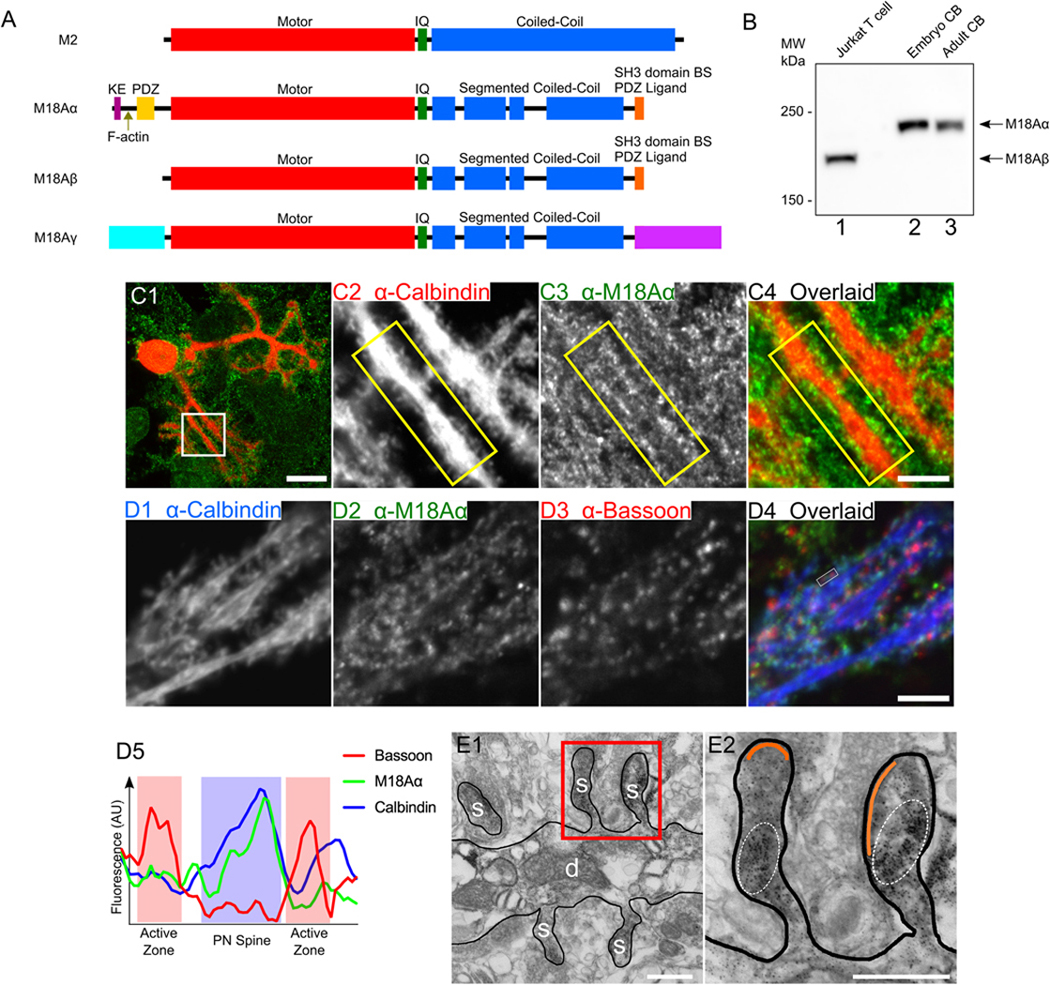
(A) Domain architecture for myosin 2 (M2) and the three known isoforms of myosin 18A, myosin 18Aα (M18Aα), myosin 18Aβ (M18Aβ), and myosin 18Aγ (M18Aγ). Domains discussed in the text are labeled. (B) Representative Western blot performed on a human Jurkat T cell extract (lane 1), a whole mouse embryonic cerebellum extract (lane 2), and a whole mouse adult cerebellum extract (lane 3), and probed with an anti-myosin 18A antibody against the C-terminal 18 residues of myosin 18Aα/myosin 18Aβ. The expected positions for the heavy chains of myosin 18Aα and myosin 18Aβ are indicated. (C1-C4) Panel C1 shows a representative cultured Purkinje neuron (DIV 18) that was double-stained for Calbindin D28K in red and myosin 18Aα in green, while Panels C2-C4 show enlargements of the boxed region in Panel C1. The yellow boxes highlight the localization of endogenous myosin 18Aα in dendritic spines. (D1-D5) Panels D1-D4 show a portion of a dendrite from a cultured Purkinje neuron (DIV 18) that was tripled-stained for Calbindin D28K in blue (D1), myosin 18Aα in green (D2), and the granule neuron pre-synaptic marker Bassoon in red (D3) (D4, Overlaid image), while Panel D5 shows a line scan across a representative spine bounded by closely-associated Bassoon signals (Calbindin signal, blue line; myosin 18Aα signal, green line; Bassoon signal, red line; Purkinje neuron spine, blue shading; presynaptic active zones, red shading). (E1 and E2) Panel E1 shows a representative electron micrograph taken of a cerebellar section from a five-week old Sprague-Dawley rat that was perfusion-fixed and labeled with a pre-embedding immunoperoxidase method for myosin 18Aα, followed by silver/gold enhancement. The dendrite (d) and several spines (s) are labeled. Panel E2 shows an enlargement of the two spines present within the boxed region in Panel E1. The area within each spine that is enriched for myosin 18Aα immunogold labeling (black dots) is encircled by a dashed line, while the PSD in each spine is pseudo-colored orange. The image in E1 is representative of over 200 images taken of several sections obtained from two rats. Scale bars: 20 μm (C1 and D1), 5 μm (C4 and D5), and 500 nm (E1 and E2).
Endogenous myosin 18Aα localizes to Purkinje neuron spines
To demonstrate more directly that Purkinje neurons express myosin 18Aα, and to begin to define its intracellular localization, we stained primary cerebellar cultures with an antibody to the Purkinje neuron-specific marker Calbindin D28K in red to identify Purkinje neurons (this staining also acts as a fill marker to reveal dendritic spines) (65, 68, 77, 81, 82), and with the antibody to the C-terminal 18 residues of myosin 18Aα in green. Figure 1, Panel C1, shows a representative stained Purkinje neuron, while Panels C2 through C4 show enlarged images of the individual channels and the overlaid image for the boxed region in Panel C1. Careful inspection reveals that the edges of the dendrites present within this boxed region are studded with Calbindin-positive spines that contain the majority of the signal for endogenous myosin 18Aα (compare the positions of the two signals within the yellow boxes in Panels C2 through C4). Similar results were obtained using frozen sections of cerebellar tissue that were fixed and stained for Calbindin and myosin 18Aα (Figure S2; see the arrowheads). Together, these initial observations argue that Purkinje neurons do indeed express myosin 18Aα, and that this myosin localizes primarily to dendritic spines.
Cultured Purkinje neurons make extensive synaptic contacts with granule neurons that are also present in primary cultures (77, 83, 84). To address the possibility that the spine-associated signals described above might actually be on the presynaptic side, we stained primary Purkinje neuron cultures for Calbindin in blue, myosin 18Aα in green, and the presynaptic marker Bassoon in red (see Panels D1-D3, respectively, in Figure 1). Inspection of the overlaid image in Panel D4 shows that while the green signals for myosin 18Aα and the red signals for Bassoon are clearly quite close, they appear to be largely non-overlapping. Line scans bore this out, as shown for a representative spine in Panel D5, where the signals for myosin 18Aα (green line) and Calbindin (blue line) marking the spine (blue shading), and the signal for Bassoon (red line) marking two adjacent presynaptic active zones (red shading), are largely non-overlapping.
To demonstrate conclusively that myosin 18Aα is postsynaptic, rat cerebellar tissue that had been perfusion-fixed was labeled with a pre-embedding immunoperoxidase method using the C-terminal myosin 18Aα antibody, followed by silver/gold enhancement. Figure 1, Panel E1, and the enlarged inset in Panel E2, show that the myosin 18Aα signal, represented by the mottled black precipitate encircled with dashed lines, lies within the Purkinje neuron spine and well below the post synaptic density (PSD, pseudo-colored in orange). Together, these observations indicate that myosin 18Aα is expressed by Purkinje neurons and that endogenous myosin 18Aα localizes predominantly to Purkinje neuron spines.
Expressed, tagged myosin 18Aα localizes to and is enriched in Purkinje neuron spines along with F-actin
Efforts to define the function of myosin 18Aα in Purkinje neurons would be facilitated if expressed versions of the myosin localize within dendritic spines like the endogenous protein. To test this, we co-transfected Purkinje neurons with mCherry as a fill marker and myosin 18Aα tagged at its N-terminus with GFP. Figure 2, Panel A1 through A3 show a representative transfected Purkinje neuron while Panels A4 through A6 show enlarged images of the individual channels and the overlaid image for the boxed region in Panel A3. What is immediately apparent is that expressed, GFP-tagged myosin 18Aα also localizes dramatically to spines. Previous work from our lab using F-Tractin, a live-cell, indirect reporter for F-actin, showed that spines are the major actin-rich compartment in Purkinje neurons (63). Consistently, Purkinje neurons transfected with GFP-tagged F-Tractin and mCherry-tagged myosin 18Aα show robust co-localization of the two signals within spines (Figure 2; Panels B1 through B3 show the individual channels and overlaid image for a portion of a Purkinje neuron dendritic arbor, Panels B4 through B6 show enlarged images of the boxed region in Panel B3, and Panels B7 through B9 show enlarged images of the boxed region in Panel B6). To determine the fold-enrichment of myosin 18Aα and F-Tractin in spines over dendrites, GFP-tagged myosin 18Aα and GFP-tagged F-Tractin were separately co-transfected with mCherry as a volume marker and ratio imaging was performed to determine the actual difference in the concentrations of each protein in spines relative to the dendrite proper by correcting for differences in the volumes imaged (see Methods). Consistent with the images in Figure 2, myosin 18Aα and F-Tractin are concentrated 3.47 ± 1.11 fold and 3.67 ± 0.99 fold in spines relative to dendrites, respectively (see Figure 4; Panels B1 through B3 and C1 through C3 show representative images for the mCherry/GFP-F-Tractin pair and the mCherry/GFP-myosin 18Aα pair, respectively; Panels A1 through A3 show a representative image of the free mCherry/free GFP control pair; Panel H shows the cumulative results for these three samples). We conclude, therefore, that expressed, tagged myosin 18Aα can be used to interrogate the function of this myosin within Purkinje neuron spines, and that both myosin 18Aα and F-actin are concentered ~3.5 fold in spines relative to dendrites.
Figure 2. Exogenously expressed myosin 18Aα localizes to dendritic spines together with F-actin.
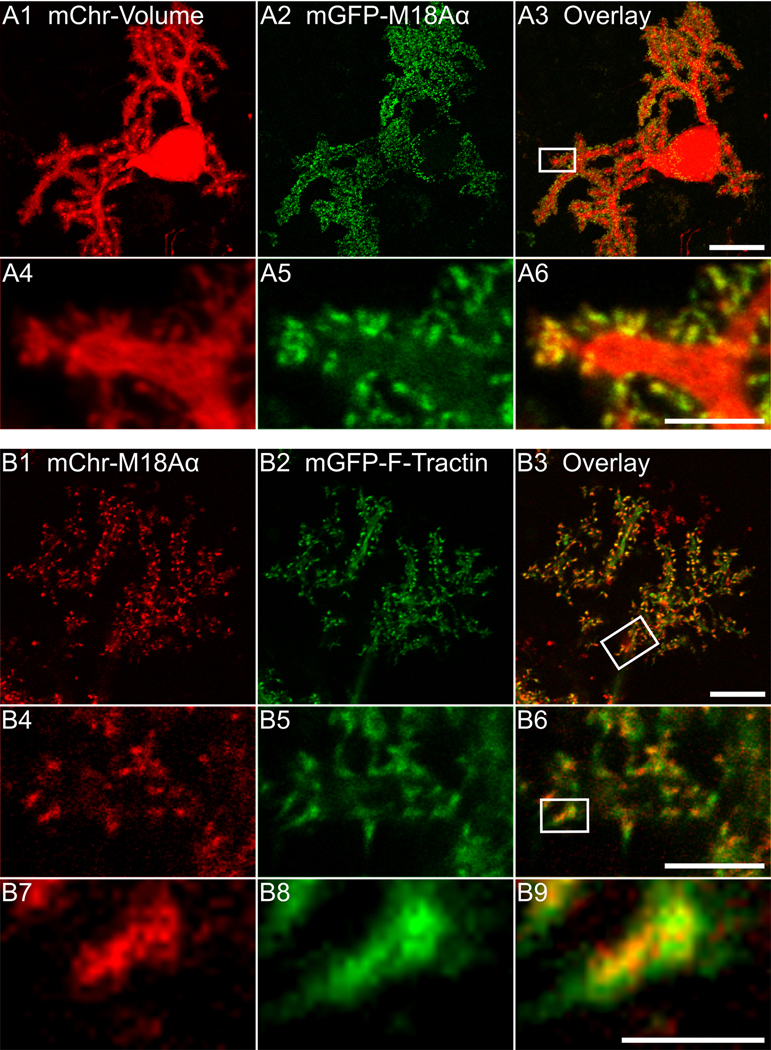
(A1-A6) Panels A1-A3 show a representative cultured Purkinje neuron (DIV 18) that was expressing mCherry as a volume marker (A1) and mGFP-myosin 18α (A2); the overlaid image is shown in Panel A3. Panels A4-A6 show enlargements of the boxed region in Panel A3. (B1-B9) Panels B1-B3 show a portion of the dendritic arbor of a representative cultured Purkinje neuron (DIV 18) that was expressing mCherry-myosin 18Aα (B1) and mGFP-F-Tractin (B2); the overlaid image is shown in (B3). Panels B4-B6 show enlargements of the boxed region in Panel B3. Panels B7-B9 show enlargements of the boxed region in Panel B6. Scale bars: 20 μm (A3), 10 μm (B3), 5 μm (A6 and B6) and 1 μm (B9).
Figure 4. Myosin 18Aα is targeted to spines through a combination of its N-terminal F-actin binding site and co-assembly with myosin 2.
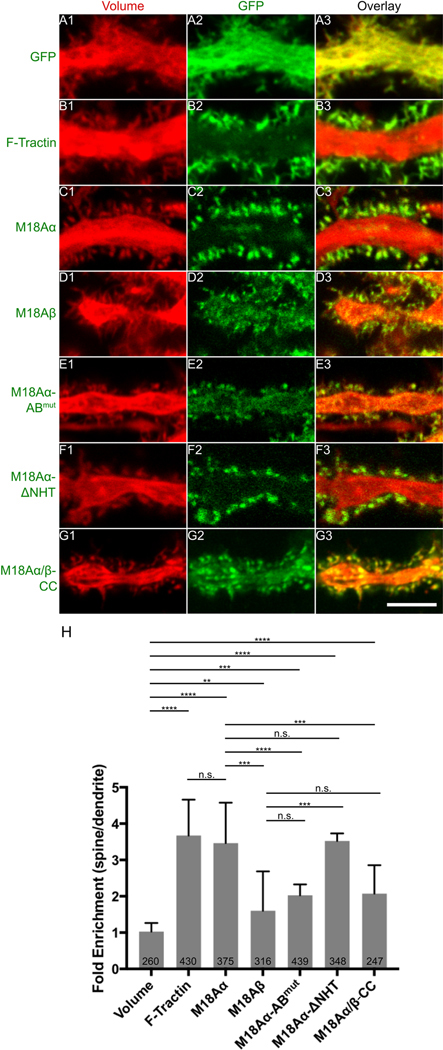
Shown are portions of dendrites from representative cultured Purkinje neurons (DIV 18) that were expressing mCherry as a volume marker (A1-G1) and one of the following mGFP-tagged proteins: mGFP alone (A2), F-Tractin (B2), myosin 18Aα (C2), myosin 18Aβ (D2), myosin 18 Aα-ABmut (N-terminal actin binding site mutated) (E2), myosin 18 Aα-ΔNHT (missing non-helical tailpiece) (F2), or myosin 18 Aα/β-CC (coiled coil domain only) (G2). Panels A3-G3 show the overlaid images. (H) Shown are the fold-enrichments in the spine over the adjacent dendrite for each GFP construct, as determined by ratio imaging. The number of spines analyzed for each construct, which were derived from three independent experiments, is indicated within each bar. Scale bar: 5 μm (G3). ** p<0.01; *** p<0.001; **** p<0.0001; n.s., not significant.
Myosin 18Aα is enriched towards the base of spines along with myosin 2
To look for evidence that myosin 18Aα might be enriched within a specific region of the spine, we transfected neurons with mCherry-tagged myosin 18Aα and with GFP-tagged PSD93 to mark post synaptic densities. Figure 3, Panels A1 through A3, show the individual channels and overlaid image for a portion of a representative Purkinje neuron dendritic arbor, while Panels A4 through A6 show enlarged images of the boxed region in Panel A3. What is evident, especially from the enlarged images, is that these two signals are largely non-overlapping within spines. Consistently, line scans of fluorescent intensities taken from the base to the tip of spines shows that myosin 18Aα is enriched towards the spine base (Figure 3, Panel D, red curve), while PSD93 is enriched towards the spine tip (Figure 3, Panel D, orange curve). Notably, lines scans of F-Tractin intensities made using images like those in Figure 4, Panels B1 through B3, indicate that F-actin is also enriched towards the spine base, although it usually extends further towards the spine tip than myosin 18Aα (Figure 3, Panel D, blue curve). Together, these light microscopic images argue that myosin 18Aα is enriched towards the base of spines, consistent with the EM images in Figure 1.
Figure 3. Myosin 18Aα localizes towards the base of the dendritic spine along with myosin 2B.
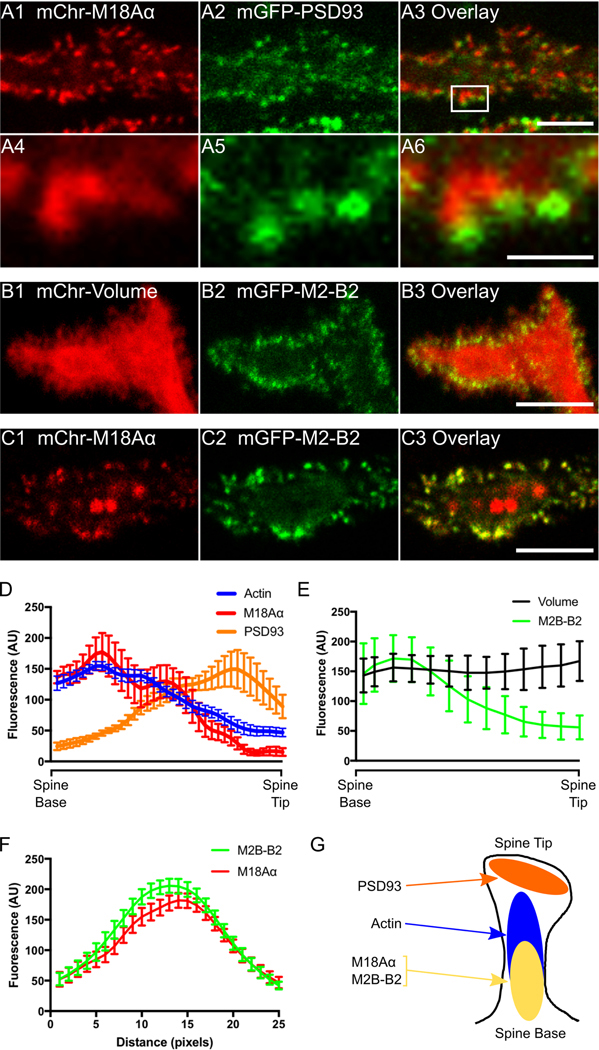
(A1-A6) Panels A1-A3 show a portion of a dendrite from a representative cultured Purkinje neuron (DIV 18) that was expressing mCherry-myosin 18Aα (A1) and mGFP-PSD93 (A2); the overlaid image is shown in (A3). Panels A4-A6 show enlargements of the boxed region in Panel A3. (B1-B3) Shown is a portion of a dendrite from a representative cultured Purkinje neuron (DIV 18) that was expressing mCherry as a volume marker (B1) and mGFP-myosin 2B-B2 (B2); the overlaid image is shown in (B3). (C1-C3) Shown is a portion of a dendrite from a representative cultured Purkinje neuron (DIV 18) that was expressing mCherry-myosin 18Aα (C1) and mGFP-myosin 2B-B2 (C2); the overlaid image is shown in (C3). (D) Shown are the fluorescence intensities (in arbitrary units; AU) from spine base to spine tip for F-actin (blue; determined using mGFP-F-Tractin), myosin 18Aα (red; determined using mCherry-myosin 18Aα), and PSD-93 (orange; determined using mGFP-PSD93) (each plot represents data from at least 20 spines). (E) Shown are the fluorescence intensities (in arbitrary units; AU) from spine base to spine tip for volume (black; determined using an mCherry fill marker) and myosin 2B-B2 (green; determined using mGFP-myosin B2–2B) (each plot represents data from at least 20 spines). (F) Shown are the fluorescence intensities for myosin 2B-B2 (green; determined using mGFP-myosin B2–2B) and myosin 18Aα (red; determined using mCherry-myosin 18Aα) from line scans performed along the long axis of spines (each plot represents data from at least 20 spines). (G) Cartoon depicting the distribution within a typical spine of PSD93 (orange), F-actin (blue), myosin 2B-B2 (yellow), and myosin 18Aα (yellow). Scale bars: 5 μm (A3), 1 μm (A6), 5 μm (B3 and C3).
Previous studies have shown that myosin 2B is enriched towards the base of Hippocampal neuron spines (22, 45, 51). A similar localization for myosin 2B in Purkinje neuron spines would be consistent with the general thinking that actomyosin contractile activity localizes towards the base of spines, and more specifically with the fact that myosin 18Aα co-assembles with myosin 2 (55). To address this, we co-transfected Purkinje neurons with mCherry as a volume marker and the B2-spliced isoform of myosin 2B tagged at its N-terminus with GFP (this myosin 2B isoform is Purkinje neuron-specific; (47)). Figure 3, Panels B1 through B3, show the individual channels and overlaid image for a portion of a representative Purkinje neuron dendritic arbor. As with myosin 18Aα and F-Tractin, myosin 2B-B2 is clearly enriched in spines. Consistently, ratio imaging indicated that it is concentrated 2.7 ± 1.2 fold in spines over dendrites (Figure 4, Panel H). Moreover, lines scans show that myosin 2B-B2 is enriched towards the base of spines like myosin 18Aα (Figure 3, Panel E, green line). Finally, Purkinje neurons expressing both mCherry-tagged myosin 18Aα and GFP-tagged myosin 2B-B2 show strong co-localization of the two myosins within spines (Figure 3, Panels C1 through C3) that is borne out using line scans (Figure 3, Panel F). We conclude, therefore, that myosin 2B-B2 and myosin 18Aα colocalize towards the base of Purkinje neuron spines where they likely co-assemble to form mixed bipolar filaments (Figure 3, Panel G) (55).
Myosin 18Aα is targeted to spines by a combination the F-actin binding domain present in its N-terminal extension and its ability to co-assemble with myosin 2
To define the motifs within myosin 18Aα responsible for its targeting to spines, we first looked at the targeting of the myosin 18Aβ isoform to spines as a proxy for the role of myosin 18Aα’s N-terminal extension because myosin 18Aβ differs from myosin 18Aα only in lacking this extension (Figure 1A). For this and all subsequent efforts, we performed ratio imaging using a volume marker to obtain values for the concentration of the protein in the spine relative to the dendrite that was corrected for differences in the volumes imaged. Panels D1-D3 and Panel H in Figure 4 together show that while myosin 18Aβ is concentrated to a significant extent in spines, its targeting is less robust than that of myosin 18Aα (1.60 ± 1.10 fold versus 3.47 ± 1.11 fold for myosin 18Aα). This result argues that myosin 18Aα’s N-terminal extension is contributing significantly to spine targeting. To gain further support for this conclusion, we measured the targeting of myosin 18Aα’s isolated N-terminal extension, referred to here as NT. Panels A1 through A3 and Panel E in Figure S3 together show that NT is indeed enriched in spines 3.26 ± 1.44 fold over dendrites.
The NT sequence contains three recognizable domains: a KE-rich region, a nucleotide-independent F-actin binding site (69), and a PDZ domain (70) (Figure 1A). To define the relative importance of these three domains for spine targeting, we measured the targeting of NT constructs in which (i) the N-terminal, 29-residue KE-rich region was deleted (NT-ΔKE), (ii) a function blocking point mutation (70) was introduced into the PDZ domain (NT-PDZmut), or (iii) two point mutations were introduced into the F-actin binding site that together reduce its affinity for F-actin in vitro by ~10-fold (NT-ABmut; see Figure S4 for the F-actin binding assays that were performed using NT and NT-ABmut). Panels B1 through B3, C1 through C3, D1 through D3, and Panel E in Figure S3 together show that only the mutation of the F-actin binding site in NT significantly reduced its spine targeting (from 3.26 ± 1.44 fold for NT to 1.71 ± 0.81 fold for NT-ABmut). Consistent with this observation, Panels E1 through E3 and Panel H in Figure 4 together show that full length myosin 18Aα containing this F-actin binding site mutation exhibited a significant reduction in spine targeting (2.03 ± 0.35 fold versus 3.47 ± 1.11 fold for wild type myosin 18Aα).
While the preceding results indicate that the nucleotide-independent F-actin binding site present within M18Aα’s N-terminal extension contributes to its spine targeting, the spine targeting that persists when this binding site is impaired or absent indicates that another motif(s) within myosin 18Aα must be contributing significantly to targeting. The two most obvious remaining candidates are the PDZ ligand comprising the C-terminal 7 residues of myosin 18Aα, and myosin 18Aα’s coiled-coil domain, which drives its co-assembly with myosin 2 (55). Panels F1 through F3 and Panel H in Figure 4 together show that deletion of myosin 18Aα’s 110-residue non-helical tailpiece (ΔNHT), which contains the PDZ ligand, does not have any adverse effect on the targeting of myosin 18Aα to spines. In contrast, Panels G1 though G3 and Panel H in Figure 4 show that myosin 18Aα’s coiled-coil domain exhibits significant spine targeting (2.07 ± 0.78 fold spine enrichment versus 3.47 ± 1.11 fold for full length myosin 18Aα). Based on all of these results, we conclude that the targeting of myosin 18Aα to Purkinje neuron spines is driven by a combination of the F-actin binding site present within its N-terminal extension and its ability to co-assemble with myosin 2 via its coiled-coil domain (55).
Myosin 18A knockdown results in increases in spine length and density consistent with an impairment in spine maturation
Having established that myosin 18Aα targets robustly to Purkinje neuron spines, we next asked what effect the loss of this myosin might have on spine development. To address this question, we made use of a Purkinje neuron-specific miRNA-mediated knockdown system we recently developed (64). Like the custom plasmids we use to express genes specifically within Purkinje neurons (63, 68), this knockdown system makes use of the Purkinje neuron-specific promoter L7/Pcp2 to drive miRNA expression. Validation of this system was obtained previously (64) by showing that wild type Purkinje neurons expressing a miRNA directed against myosin Va exhibit the same phenotype as Purkinje neurons isolated from mice homozygous for a myosin Va functional null allele (loss of inheritance of endoplasmic reticulum into spines) (63). Because the efficiency of knockdown within cultured Purkinje neurons cannot be accessed by Western blotting (as they represent a very minor fraction of the cells present in mixed primary cultures), we first measured the extent of myosin 18Aα knockdown in NIH/3T3 fibroblasts using two candidate miRNAs directed against the 3’ untranslated region of myosin 18Aα. Both miRNAs yielded ~80% knockdown of myosin 18Aα (Figure S5, Panel A, lanes 1–3). Given this, we introduced miRNA #2 into wild type Purkinje neurons at day 0 using our Purkinje neuron-specific knockdown plasmid. Of note, this plasmid also drives the expression of free mCherry, which serves to identify Purkinje neurons expressing the miRNA and to mark the cell’s volume. As a control, we used a scrambled miRNA (64). Consistent with penetrant knockdown, antibody staining of wild type Purkinje neurons expressing the scrambled miRNA (Figure S5, Panels B1-B3) side by side with Purkinje neurons expressing miRNA #2 (Figure S5, Panels C1-C3) after 18 days in culture revealed a large decease in the signal for myosin 18Aα within the spines of the neurons expressing miRNA #2.
Live-cell imaging of control Purkinje neurons expressing the scrambled miRNA (Figure 5, Panels A1 and A2) and Purkinje neurons expressing myosin 18Aα miRNA #2 (Figure 5, Panels B1 and B2) after 18 days in culture revealed an apparent difference between them in terms of spine length, with the knockdown neurons appearing to exhibit longer spines (see also representative higher magnification images in Panel E (scrambled miRNA) and Panel F (myosin 18Aα miRNA)). Indeed, quantitation (Figure 5, Panel I) showed that myosin 18Aα knockdown Purkinje neurons exhibit about a 30% increase in spine length relative to control neurons (1.32 ± 0.39 μm for knockdown neurons versus 1.03 ± 0.29 μm for control neurons; note that this later value matches values reported previously (85) for the length of spines on mature wild type Purkinje neurons present in primary culture). More generally, the spines on the myosin 18Aα knockdown Purkinje neurons appear more filopodial in nature (Figure 5, Panels E and F), as if the maturation of filopodial precursors into mature spines (15, 18–21, 86–88) is impaired by the knockdown of myosin 18Aα. Quantitation (Figure 5, Panel J) also showed that myosin 18Aα knockdown Purkinje neurons exhibit about a 1.7-fold increase in the density of spines relative to control neurons (3.43 ± 1.33 spines per μm2 for knockdown neurons versus 2.05 ± 1.1 spines per μm2 for control neurons). This phenotype may also reflect an impairment in spine maturation, given that the spines of myosin 18Aα knockdown Purkinje neurons contain less myosin 2B ((49); see also below and Discussion). Importantly, these defects in spine length and density were both completely rescued by reintroduction of wild type, miRNA-resistant myosin 18Aα into myosin 18Aα knockdown Purkinje neurons (see Figure 5, Panels C1 though C4, for a representative rescued Purkinje neuron, Panel G for a representative higher magnification image of a dendrite from one such neuron, and Panels I and J for quantitation of spine length and density; spine length: 1.00 ± 0.28 μm; spine density: 2.05 ± 1.10 spines per μm2). This result argues that the apparent defect in spine maturation exhibited by Purkinje neurons expressing the myosin 18Aα miRNA is due to the knockdown of myosin 18Aα and not to possible off-target effects. We also note that 68.2 ± 8.8% of spines on myosin 18Aα knockdown Purkinje neurons had a closely associated Bassoon-positive puncta, which is not significantly different from the value for control neurons (73.7 ± 3.3%). This observation argues that the defect in spine maturation exhibited by myosin 18Aα knockdown Purkinje neurons is not caused by a reduction in the fraction of spines that have formed a synapse.
Figure 5. The knockdown of myosin 18Aα in Purkinje neurons results in an increase in spine length and density, consistent with a defect in spine maturation.
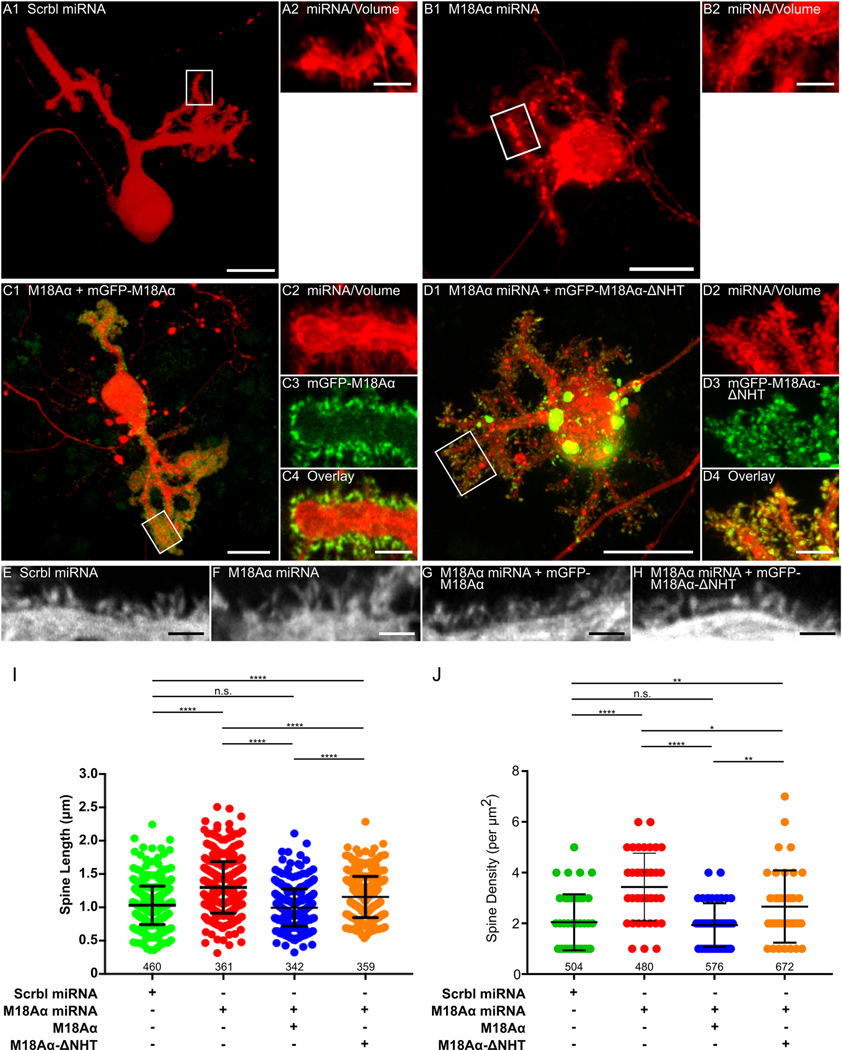
(A1 and A2) Panel A1 shows a representative cultured Purkinje neuron (DIV 18) that was expressing a scrambled miRNA (indicated by the presence of the mCherry volume marker). Panel A2 shows an enlargement of the boxed region in Panel A1. (B1 and B2) As in A1 and A2, except the Purkinje neuron was expressing myosin 18 Aα miRNA #2. (C1-C4) Panel C1 shows a representative cultured Purkinje neuron (DIV 18) that was expressing myosin 18 Aα miRNA #2 (indicated by the presence of the mCherry volume marker) and mGFP-myosin 18 Aα. Panels C2-C4 show enlargements of the boxed region in Panel C1. (D1-D4) Panel D1 shows a representative cultured Purkinje neuron (DIV 18) that was expressing myosin 18 Aα miRNA #2 (indicated by the presence of the mCherry volume marker) and mGFP-myosin 18 Aα-ΔNHT. Panels D2-D4 show enlargements of the boxed region in Panel D1. Panels E-H show representative, high-magnification images of spines on Purkinje neurons treated as in Panels A1, B1, C1, and D1, respectively. (I) Measurements of spine length (in μm) on DIV 18 Purkinje neurons treated as indicated (green, scrambled miRNA; red, myosin 18Aα miRNA; blue, myosin 18Aα miRNA plus GFP-myosin 18Aα; orange, myosin 18Aα miRNA plus GFP-myosin 18Aα-ΔNHT). The N values, which correspond to the total number of spines scored using at least 10 neurons across three independent experiments, are indicated below each measurement. (J) Measurements of spine density (in numbers per μm2) on DIV 18 Purkinje neurons treated as indicated. The N values, which correspond to the total area scored using at least 10 neurons from three independent experiments, are indicated below each measurement. Scale bars: 20 μm (A1, B1, C1 and D1), 5 μm (A2, B2, C4 and D4), 1 μm (E-H). * p<0.05; ** p<0.01; **** p<0.0001; n.s., not significant.
To gain further support for the results obtained using miRNA-mediated myosin 18Aα knockdown, we examined Purkinje neurons isolated from a myosin 18A conditional knockout (cKO) mouse following the delivery of Cre recombinase into these neurons using TAT-Cre. While the creation and characterization of this mouse will be described elsewhere, we show in Figure S5 (Panel D) that MEFs isolated from this mouse and transfected with a plasmid expressing Cre recombinase lose essentially all of the Western blot signals for both myosin 18Aα and myosin 18Aβ, indicating that the conditional allele we created is functional. Figure S6 shows representative images at day 18 in culture (with enlarged insets) of a cKO Purkinje neuron that was not treated with TAT-Cre (Panel A), a cKO Purkinje neuron that was treated TAT-Cre at day 5 (Panel B), and a wild type Purkinje neuron that was treated with TAT-Cre at day 5 (Panel C). Quantitation (Figure S6, Panels D and E) shows that cKO Purkinje neurons treated with TAT-Cre exhibit significant increases in both spine length (0.92 ± 0.22 μm for cKO neurons without TAT-Cre treatment versus 1.28 ± 0.35 μm for cKO Purkinje neurons with TAT-Cre treatment) and spine density (2.95 ± 1.38 spines per μm2 for cKO neurons without TAT-Cre treatment versus 4.08 ± 2.14 spines per μm2 for cKO Purkinje neurons with TAT-Cre treatment). Importantly, the length and density of spines on wild type Purkinje neurons treated at day 5 with TAT-Cre (Figure S6, Panel C) was not significantly different from cKO Purkinje neurons not treated with TAT-Cre (Figure S6, Panels D and E; spine length: 0.93 ± 0.28 μm; spine density: 2.51 ± 1.24 spines per μm2). This indicates that the effect of TAT-Cre on spine length and density in cKO Purkinje neurons is likely due to the loss of myosin 18Aα and not to some non-specific effect caused by the introduction of Cre recombinase. Together, these results, like the knockdown results described above, argue that myosin 18Aα plays a significant role in the maturation of Purkinje neuron spines.
An additional phenotype arising from the knockdown/knockout of myosin 18Aα that is seen in all of the related images discussed above (compare, for example, Panel B1 to Panel A1 in Figure 5) is an apparent defect in cell polarity manifested as an increase in the number of primary dendrites in myosin 18Aα knockdown/knockout neurons. Consistently, Figure S7 shows that while wild type Purkinje neurons, control Purkinje neurons expressing the scrambled miRNA, cKO Purkinje neurons not treated with TAT-Cre, and wild type Purkinje neurons treated with TAT-Cre all exhibit about two primary dendrites per neuron (2.0 ± 0.9, 1.9 ± 0.7, 2.50 ± 1.0, and 1.7 ± 0.8, respectively), myosin 18Aα knockdown Purkinje neurons and cKO Purkinje neurons treated with TAT-Cre exhibit significantly higher numbers of primary dendrites per neuron (5.6 ± 2.2 and 4.00 ± 1.7 primary dendrites per neuron, respectively). Importantly, myosin 18Aα knockdown Purkinje neurons in which wild type myosin 18Aα had been reintroduced exhibit normal numbers of dendrites per cell (2.1 ± 0.8 primary dendrites per neuron; Figure S7), arguing that the polarity defect is due to the loss of myosin 18Aα. While we did not pursue the underlying mechanism of this defect in dendrite morphogenesis, it may be related to the reported interaction of myosin 18A with Golgi membranes ((89); also see Discussion).
Myosin 18Aα targets the Rac/Cdc42 GEF β-Pix to Purkinje neuron spines
We next sought to gain insight into the mechanism by which myosin 18Aα contributes to spine maturation. As discussed in the Introduction, myosin 18A isoforms probably contribute to cell function primarily by co-assembling with myosin 2, where they then recruit proteins that regulate the myosin filament, influence the local environment around the filament, and/or attach the filament to cellular structures. One protein known to interact with myosin 18Aα is the Rac and Cdc42 GEF β-Pix, which binds to the myosin’s C-terminal non-helical tailpiece (60, 61). This interaction could be particularly relevant here, as β-Pix has been implicated in promoting spine maturation via several distinct pathways involving actin assembly and myosin assembly/contractility (see Introduction). Given this, we asked if myosin 18Aα’s contribution to spine maturation is mediated at least in part by its interaction with β-Pix.
We first asked if β-Pix is targeted to Purkinje neuron spines. Figure 6, Panels A1 through A3, show that endogenous β-Pix appears enriched in spines (see arrowheads). Consistently, ratio imaging indicated that it is concentrated 3.51 ± 1.27 fold in spines over dendrites (Figure 6, Panel I). Figure 6, Panels B1 through B3, show that GFP-tagged β-Pix is also enriched in spines, with ratio imaging indicating that it is concentrated 3.14 ± 0.79 fold in spines over dendrites (Figure 6, Panel I). Moreover, line scans of individual spines showed that, like myosin 18Aα and myosin 2, β-Pix is enriched towards the base of the spine (Figure 6, Panel G). Consistently, mCherry-tagged myosin 18Aα and mGFP-tagged β-Pix colocalize dramatically in spines (Figure 6, Panels C1 through C3, and Panel H). Together, these results show that β-Pix is targeted to Purkinje neuron spines and colocalizes with myosin 18Aα.
Figure 6. Myosin 18Aα targets β-Pix to Purkinje neuron spines.
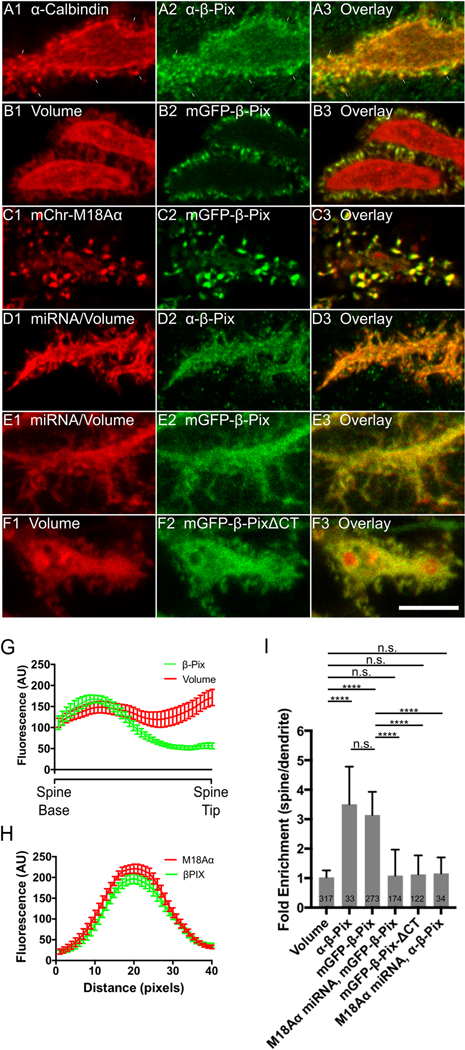
Shown are portions of dendrites from representative cultured Purkinje neurons (DIV 18) treated as follows. (A1-A3) Fixed and stained for Calbindin (A1) and endogenous β-Pix (A2); Overlay (A3). (B1-B3) Expressing mCherry fill marker (B1) and mGFP- β-Pix (B2); Overlay (B3). (C1-C3) Expressing mCherry-myosin 18Aα (C1) and mGFP-β-Pix (C2); Overlay (C3). (D1-D3) Expressing myosin 18Aα miRNA #2 (D1; associated mCherry fill marker shown) and fixed/stained for endogenous β-Pix (D2); Overlay (D3). (E1-E3) Expressing myosin 18 Aα miRNA #2 (E1; associated mCherry fill marker shown) and mGFP-β-Pix (E2); Overlay (E3). (F1-F3) Expressing mCherry fill marker (F1) and mGFP-β-PixΔCT (F2); Overlay (F3). (G) Shown are the fluorescence intensities (in arbitrary units; AU) from spine base to spine tip for volume (red; determined using an mCherry fill marker) and β-Pix (green; determined using mGFP-β-Pix) (each plot represents data from at least 20 spines). (H) Shown are the fluorescence intensities for myosin 18Aα (red; determined using mCherry-myosin 18Aα) and β-Pix (green; determined using mGFP-β-Pix) from line scans performed along the long axis of spines (each plot represents data from at least 20 spines). (I) Shown are the fold-enrichments in the spine over the adjacent dendrite for each GFP construct or stained protein, as determined by ratio imaging. The number of spines analyzed for each construct or stained protein is indicated within each bar. A total of at least 6 cells from three independent experiments were analyzed for each GFP construct. Scale bar: 5 μm (F3). **** p<0.0001; n.s. not significant.
We next asked if the targeting of β-Pix to spines depends on myosin 18Aα. Figure 6, Panels D1 through D3, show that the enrichment of endogenous β-Pix in spines appears to be lost upon myosin 18Aα knockdown (and, as predicted, the spines appear more filopodia-like). Consistently, ratio imaging indicated that endogenous β-Pix is indeed no longer concentrated in spines relative to dendrites following myosin 18Aα knockdown (Figure 6, Panel I). Similar results were obtained using GFP-tagged β-Pix, which is also no longer concentrated in spines relative to dendrites following myosin 18Aα knockdown (Figure 6, Panels E1 through E3, and Panel I). Finally, a GFP-tagged version of β-Pix lacking C-terminal residues 639–647 (β-Pix-ΔCT), which is no longer able to bind to myosin 18Aα (60, 61), does not exhibit significant spine enrichment (Figure 6, Panels F1 through F3, and Panel I). Together, these results argue that the recruitment of β-Pix to Purkinje neuron spines depends on myosin 18Aα.
The spines of myosin 18Aα knockdown neurons exhibit significant reductions in the content of both F-actin and myosin 2
The above results are consistent with the idea that the defect in spine maturation exhibited by Purkinje neurons lacking myosin 18Aα might be caused, at least in part, by the loss of β-Pix targeting to spines. Based on previous studies (see Introduction), β-Pix can promote spine maturation downstream of its GEF activity towards Rac1 and Cdc42 via at least two pathways: increased Arp2/3 complex-dependent branched actin filament nucleation and increased myosin 2 filament assembly/contractility following the PAK-dependent phosphorylation of the myosin’s RLC. In our particular case, this latter pathway could be driven by the myosin 18Aα-dependent recruitment of PAK to spines (as it is commonly in a complex with β-Pix and GIT1; see Discussion), as well as by the activation of PAK downstream of β-Pix’s GEF activity. To look for evidence that the defect in spine maturation exhibited by Purkinje neurons lacking myosin 18Aα is due at least in part to defects in these two pathways, we measured the spine content of F-actin and myosin 2 in control and myosin 18Aα knockdown neurons by staining cells with either Phalloidin or an antibody to myosin 2B, respectively. Figure 7, Panels A1 through A3, show a representative example of a dendrite from a control Purkinje neuron (DIV 18) that was fixed and stained for Calbindin and F-actin, while Panels B1 through B3 show a representative example of a dendrite from a Purkinje neuron that was treated with myosin 18Aα miRNA #2 (DIV 18) and then fixed and stained for F-actin. Comparison of the images in Panels A2 and B2 suggested that myosin 18Aα knockdown results in a significant reduction in Phalloidin staining/F-actin content per spine. Consistently, quantitation showed that knockdown neurons exhibit on average a ~35% reduction in total F-actin content per spine relative to control neurons (Figure 7, Panel C).
Figure 7. Myosin 18Aα knockdown neurons exhibit reduced F-actin content per spine.
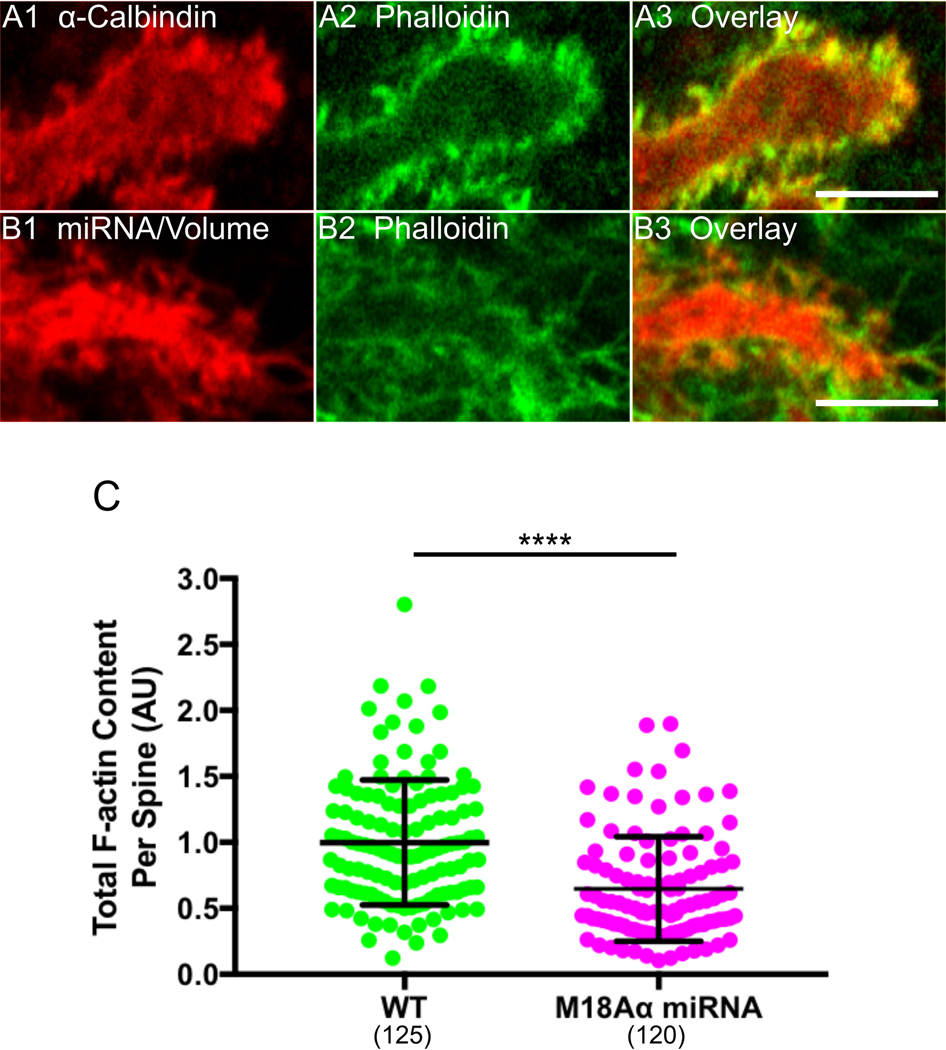
(A1-A3) Shown is a portion of a dendrite from a representative cultured control Purkinje neuron (DIV 18) that was fixed and stained for Calbindin (A1) and F-actin using Phalloidin (A2); Overlay (A3). (B1-B3) Shown is a portion of a dendrite from representative cultured Purkinje neuron (DIV 18) that was expressing myosin 18 Aα miRNA #2 (indicated by the presence of the mCherry volume marker) (B1) and then fixed and stained for F-actin using Phalloidin (B2); Overlay (B3). (C) Phalloidin fluorescence/F-actin content per spine in arbitrary units (AU) (WT, green, n = 125 spines measured; myosin 18Aα miRNA, magenta, n = 120 spines measured; the results are from two independent experiments). The fluorescence values were normalized such that the mean intensity in WT spines equals 1.0. Of note, the difference in spine F-actin content between WT and knockdown neurons was maintained when the two samples were normalized for spine volume by ratio imaging (34.6% reduction in F-actin content per unit spine volume versus 35.2% reduction in total F-actin content per spine). Scale bar: 5 μm (A3 and B3). **** p<0.0001.
With regard to the content of myosin 2B in spines, Figure 8, Panels A1 through A3 (and the corresponding insets in Panels A4 through A6), show a representative example of a dendrite from a control Purkinje neuron (DIV 18) that was treated with a scrambled miRNA and then fixed and stained for myosin 2B, while Panels B1 through B6 show the same except that the Purkinje neuron was treated with myosin 18Aα miRNA #2. Comparison of the images in Panels A2/A5 with those in Panels B2/B5 suggested that myosin 18Aα knockdown results in a significant reduction in myosin 2B content per spine. Consistently, quantitation showed that knockdown neurons exhibit on average a ~39% reduction in total myosin 2B content per spine relative to control neurons (Figure 8, Panel C). These results, together with those in Figure 7, argue that the defect in spine maturation caused by the loss of myosin 18Aα may be due, at least in part, to an attenuation of the two β-Pix-driven maturation pathways discussed above.
Figure 8. Myosin 18Aα knockdown neurons exhibit reduced myosin 2B content per spine.
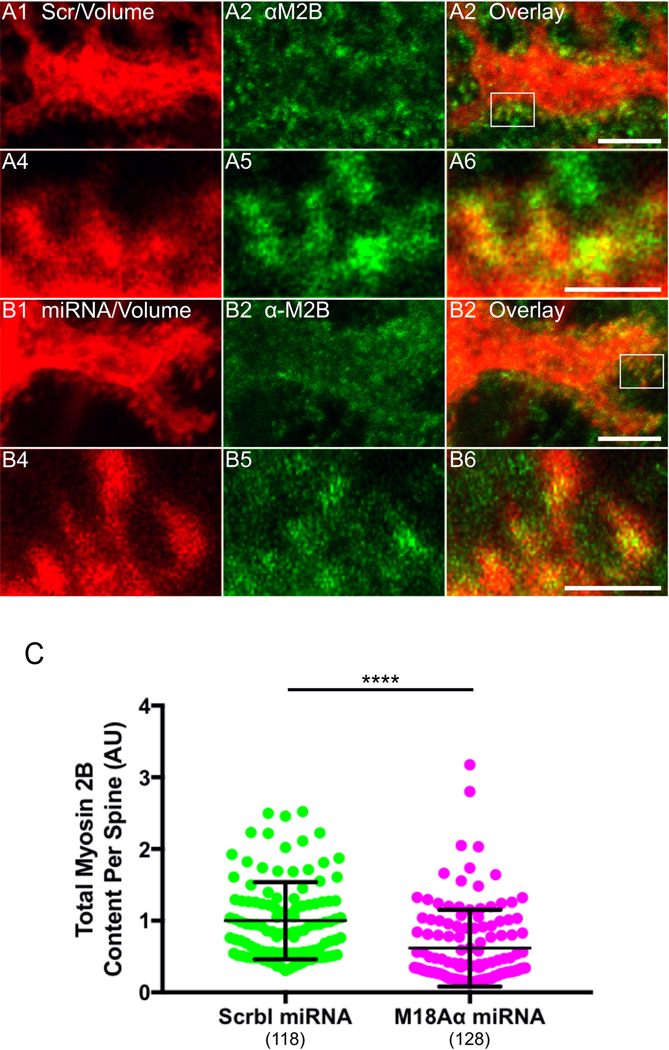
(A1-A6) Panels A1-A3 show a portion of a dendrite from a representative cultured control Purkinje neuron (DIV 18) that was expressing a scrambled miRNA (indicated by the presence of the mCherry volume marker) (A1) and then fixed and stained for myosin 2B (A2); Overlay (A3). Panels A4-A6 show enlargements of the boxed region in Panel A3. (B1-B6) Panels B1-B3 show a portion of a dendrite from a representative cultured Purkinje neuron (DIV 18) that was expressing myosin 18Aα miRNA #2 (indicated by the presence of the mCherry volume marker) (B1) and then fixed and stained for myosin 2B (B2); Overlay (B3). Panels B4-B6 show enlargements of the boxed region in Panel B3. (C) Myosin 2B fluorescence/content per spine in arbitrary units (AU) (Control, green, n = 118 spines measured; myosin 18Aα knockdown, magenta, n = 128 spines measured; the results are from three independent experiments). The fluorescence values were normalized such that the mean intensity in control spines equals 1.0. Of note, the difference in spine myosin 2B content between control and knockdown neurons was maintained when the two samples were normalized for spine volume by ratio imaging (34.6% reduction in myosin 2B content per unit spine volume versus 39.3% reduction in total myosin 2B content per spine). Scale bar: 5 μm (A3 and B3) and 1 μm (A6 and B6). **** p<0.0001.
Myosin 18Aα’s contribution to spine maturation depends significantly on its interaction with β-Pix but extends beyond it as well
If myosin 18Aα’s contribution to spine maturation is due entirely to its ability to recruit β-Pix to spines, then a version of myosin 18Aα lacking its C-terminal non-helical tailpiece (M18Aα ΔNHT), which can no longer bind to β-Pix (60, 61), should not rescue knockdown cells. Somewhat surprisingly, Figure 5, Panels D1 through D4, together with Panels H, I and J, show that this construct results in a significant degree of rescue of both spine length and spine density. That said, the values for spine length and density using this construct are also significantly different from the values for control cells. In other words, a version of myosin 18Aα that cannot target β-Pix to spines yields a partial rescue of knockdown cells in which the values for spine length and density are in between, and significantly different from, the values for both control and knockdown cells. The simplest interpretation of this result is that while the interaction of myosin 18Aα with β-Pix plays a significant role in driving spine maturation, there must be at least one other pathway by which myosin 18Aα contributes significantly to spine maturation (see Discussion).
DISCUSSION
Here we show that myosin 18A⍺ recruits the Rac1 and Cdc42 GEF β-Pix to the spines of cerebellar Purkinje neurons and promotes spine maturation. With regard to the underlying mechanism of this effect, our demonstration that the spines of myosin 18A⍺ knockdown Purkinje neurons contain significantly less F-actin and myosin 2B argues that the myosin 18A⍺-dependent recruitment of β-Pix serves to enhance the assembly of actin and myosin filaments in spines. This conclusion is consistent with previous studies showing that β-Pix promotes spine maturation in hippocampal neurons downstream of its Rac1 and Cdc42 GEF activity by increasing Arp2/3 complex-dependent branched actin filament nucleation (via activation of the NPFs WAVE and WASp) (24–26, 28, 32, 33, 41), and by increasing myosin 2 filament assembly/contractility (via the activation of PAK-dependent myosin RLC phosphorylation) (35, 36). Together, our results add additional support to the emerging view that myosin 18A isoforms confer via their N- and C-terminal protein interaction domains novel functions upon myosin 2 by co-assembling with it (55, 60–62). Our results also underscore the need to interpret studies of myosin 2 function within dendritic spines in the context of myosin 18Aα function.
One result that surprised us was that myosin 18Aα lacking its non-helical tailpiece where β-Pix binds was able to partially rescue knock down cells with regard to spine length and spine density. This result argues that the interaction of myosin 18Aα with β-Pix, while important, is not the whole story, and that there must be at least one other pathway by which myosin 18Aα contributes significantly to spine maturation. While we did not attempt to define this pathway, a number of candidates come to mind that could guide future work. First, myosin 18Aα could promote spine maturation by stabilizing myosin 2 filaments (55). Second, myosin 18Aα could promote spine maturation by recruiting the myosin 2 regulatory light chain kinase MRCK via its N-terminal PDZ domain (and through the adaptor protein LRAP; (62)). Third, other yet-to-be identified proteins that interact with myosin 18Aα’s N-terminal extension could promote spine maturation by promoting actin and/or myosin assembly. Future work should address these and other possible mechanisms by which myosin 18Aα promotes spine maturation beyond its role in recruiting β-Pix to spines.
Given that β-Pix promotes the maturation of hippocampal neuron spines via its GEF activity (30, 35–39), it seems likely that the portion of myosin 18Aα’s contribution to spine maturation due to its targeting of β-Pix to spines requires β-Pix’s GEF activity. Efforts to prove this by simultaneously knocking down β-Pix and replacing it with wild type and GEF-dead versions of the protein were unfortunately unsuccessful (no transfected Purkinje neurons were observed on three attempts). Future efforts should seek to overcome this hurdle so as to cement the link between the myosin 18Aα-dependent targeting of β-Pix to spines, β-Pix’s GEF activity, and myosin 18Aα-dependent spine maturation.
The only morphological indicator of a defect in spine maturation that we provided at the individual spine level was increased spine length. This is in contrast to studies in cultured hippocampal neurons, where reduced spine head size is usually included along with increased spine length as an indicator of a defect in spine maturation (90–94). Unlike hippocampal neurons in situ, where ~70% of mature spines exhibit an enlarged spine head, only ~10% of mature spines on Purkinje neurons in situ exhibit an enlarged spine head (95). Consistently, we rarely see spines with enlarged heads in cultured Purkinje neurons at DIV18. It is for this reason that our only measure of a defect in spine maturation at the individual spine level was increased spine length.
The other spine parameter we measured was spine density, which increased upon myosin 18Aα knockdown and knockout. This change could reflect an attenuation of spine pruning (96), as this complex process often parallels spine maturation, although we have no direct evidence that spine pruning slows in knockdown/knockout neurons. A more likely explanation, however, has to do with our observation that the spines of myosin 18Aα knockdown Purkinje neurons contain significantly less myosin 2B, as attenuation of myosin 2B function in hippocampal neurons leads to an increase in the density of immature, filopodial-like spine precursors (49). If this is the case, then the increase in spine density exhibited by myosin 18Aα knockdown and knockout Purkinje neurons represents a second morphological indicator of a defect in spine maturation. Finally, these defects in spine maturation are most likely not secondary to a reduction in synapse formation, as the percentage of Purkinje neuron spines with closely-associated Bassoon-positive puncta was very similar for control and myosin 18Aα knockdown Purkinje neurons (~70%). Moreover, studies of Weaver mice, Reeler mice, and gamma-irradiated neonatal mice all show that Purkinje neurons in situ develop morphologically normal dendritic spines at roughly normal densities in the almost complete absence of innervation, i.e. that spinogenesis in this particular neuron is largely intrinsic (reviewed in (15)).
We presented two supplementary figures containing data obtained using Purkinje neurons isolated from a myosin 18Aα cKO mouse. The availability of this mouse opens the door to the characterization of myosin 18Aα function at the level of the cerebellum (e.g. its role in synaptic plasticity) and the animal (e.g. its role in coordination and motor learning). That said, when we crossed our myosin 18Aα cKO mouse with the L7/Pcp2 Cre driver mouse (97), only a small fraction of Purkinje neurons in 3 month-old animals appeared to have lost myosin 18Aα expression based on tissue staining (data not shown). This may be due to issues with the access of Cre to the LoxP sites, although the ability to delete the gene in cKO MEFs (Figure S5) would appear to argue against this. A second issue may be the relative weakness of the L7/Pcp2 promoter in vivo (98). Available alternatives are a L7/Pcp2 Cre driver mouse created using BAC transgene technology (99), or stereotaxic injection of an adenovirus harboring Cre driven by an improved version of the L7/Pcp2 promoter (100).
Our β-Pix targeting data indicates that the bulk of the protein is present in the central and lower regions of Purkinje neuron spines. This differs from older reports in hippocampal neurons that β-Pix, as well as other GEFs (and GAPs) for Rho-related GTPases, are targeted to the PSD (30, 101). Indeed, even key effectors of these signaling molecules like the NPFs for the Arp2/3 complex were reported to be localized at the PSD. While our results for β-Pix may differ because we used Purkinje neurons, it is important to note that more recent studies in hippocampal neurons have provided clear evidence that Rho GTPases, as well as their upstream regulators and downstream effectors, localize away from the PSD as well (31, 101). This is consistent with the fact that many of the pathways driven by these signaling and effector molecules (e.g. actin assembly and myosin contractility) are operating at considerable distances from the PSD (e.g. in the middle and at the base of spines). Future efforts should seek to determine if GIT1 and PAK, which are usually in a complex with β-Pix (30, 35–37), are also recruited to the center and base of spines by myosin 18Aα.
One additional phenotype we observed upon myosin 18Aα knockdown and knockout was a significant increase in the number of primary dendrites. This phenotype could be quite interesting to pursue for two related reasons. The first has to do with the growing evidence that acentrosomal Golgi elements like Golgi outposts and Golgi satellites, which reside in primary dendrites and at dendritic branch points (outposts), as well as throughout dendritic arbors (satellites), play key roles in the morphogenesis of neuronal dendrites (102–107). In addition to possessing a variety of Golgi processing enzymes, these dendritic Golgi elements commonly serve as sites of microtubule nucleation, allowing the creation of acentrosomal microtubule arrays that support their role in the local processing of ER-derived secretory cargo like transmembrane proteins (108). Consistently, perturbations of either the localization or microtubule nucleating activity of Golgi outposts result in alterations in dendrite morphology. These alternations range from reduced dendritic arborization in Drosophila dopaminergic neurons (108) to excessive numbers of primary dendrites in zebrafish Purkinje neurons (109) (matching the phenotype we observed here in myosin 18Aα knockdown/knockout Purkinje neurons).
The second and related reason to pursue the defect in dendrite morphology exhibited by myosin 18Aα knockdown/knockout Purkinje neurons is the published evidence that myosin 18A interacts with the resident Golgi protein GOLPH3 to promote the ribbon-like morphology of the Golgi apparatus in tissue culture cells (89). Adding to the intrigue is the recent report that endogenous myosin 18A is present on Golgi outposts in oligodendrocytes (110). Together, these observations suggest that the defect in primary dendrite number exhibited by Purkinje neurons lacking myosin 18Aα, as well as other possible defects in dendrite morphology that might be revealed upon more extensive analyses, could be due to a defect in the localization and/or function of Golgi outposts and satellites. Interestingly, myosin 18Aα lacking its non-helical tailpiece was incapable of rescuing the defect in primary dendrite number (Figure S7), arguing that β-Pix may play a critical role in the underlying process of primary dendrite specification. Consistently, multiple studies have demonstrated that Cdc42 (one target of β-Pix’s GEF activity) plays a central role in Golgi outpost formation and trafficking (111–114). Future studies should seek to elaborate upon these apparent connections between myosin 18Aα, β-Pix and dendritic Golgi elements in regulating dendrite morphogenesis.
Supplementary Material
ACKNOWLEDGMENTS
This work was supported in part by the Intramural Research Program of the National Heart, Lung, and Blood Institute (NHLBI) (ZIAHL006123 to JAH) and in part by the Intramural Research Program of the National Institute on Deafness and Other Communication Disorders (NIDCD) (DC000039 to TBF; Advanced Imaging Core DC000081 to RSP). The authors thank Dr. Xufeng Wu (NHLBI Light Microscopy Core) for imaging support.
NON-STANDARD ABREVIATIONS
- ABmut
Actin binding site mutant
- CT
C-terminus
- DIV
Days in vitro
- KE
rich region: Lysine, Glutamate rich region
- M18Aα
Myosin 18A alpha
- M18Aβ
Myosin 18A beta
- M18Aγ
Myosin 18A gamma
- NHT
non-helical tailpiece
- NT
N-terminus
- PDZmut
PDZ domain mutant
REFERENCES
- 1.Yuste R and Bonhoeffer T. (2001) Morphological Changes in Dendritic Spines Associated with Long-Term Synaptic Plasticity. Annu. Rev. Neurosci 24, 1071–1089 [DOI] [PubMed] [Google Scholar]
- 2.Okamoto KI, Nagai T, Miyawaki A, and Hayashi Y. (2004) Rapid and persistent modulation of actin dynamics regulates postsynaptic reorganization underlying bidirectional plasticity. Nat. Neurosci 7, 1104–1112 [DOI] [PubMed] [Google Scholar]
- 3.Hlushchenko I, Koskinen M, and Hotulainen P. (2016) Dendritic spine actin dynamics in neuronal maturation and synaptic plasticity. Cytoskeleton 73, 435–441 [DOI] [PubMed] [Google Scholar]
- 4.Basu S and Lamprecht R. (2018) The role of actin cytoskeleton in dendritic spines in the maintenance of long-term memory. Front. Mol. Neurosci 11, 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Nakahata Y and Yasuda R. (2018) Plasticity of spine structure: Local signaling, translation and cytoskeletal reorganization. Front. Synaptic Neurosci 10, 29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Lamprecht R and LeDoux J. (2004) Structural plasticity and memory. Nat. Rev. Neurosci 5, 45–54 [DOI] [PubMed] [Google Scholar]
- 7.Matsuzaki M, Honkura N, Ellis-Davies GCR, and Kasai H. (2004) Structural basis of long-term potentiation in single dendritic spines. Nature 429, 761–766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Bourne JN and Harris KM (2008) Balancing Structure and Function at Hippocampal Dendritic Spines. Annu. Rev. Neurosci 31, 47–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Holtmaat A and Svoboda K. (2009) Experience-dependent structural synaptic plasticity in the mammalian brain. Nat. Rev. Neurosci 10, 647–658 [DOI] [PubMed] [Google Scholar]
- 10.Newpher TM and Ehlers MD (2009) Spine microdomains for postsynaptic signaling and plasticity. Trends Cell Biol. 19, 218–227 [DOI] [PubMed] [Google Scholar]
- 11.Roberts TF, Tschida KA, Klein ME, and Mooney R. (2010) Rapid spine stabilization and synaptic enhancement at the onset of behavioural learning. Nature 463, 948–952 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kasai H, Fukuda M, Watanabe S, Hayashi-Takagi A, and Noguchi J. (2010) Structural dynamics of dendritic spines in memory and cognition. Trends Neurosci. 33, 121–129 [DOI] [PubMed] [Google Scholar]
- 13.Fortin DA, Srivastava T, and Soderling TR (2012) Structural modulation of dendritic spines during synaptic plasticity. Neuroscientist 18, 326–341 [DOI] [PubMed] [Google Scholar]
- 14.Cingolani LA and Goda Y. (2008) Actin in action: The interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci 9, 344–356 [DOI] [PubMed] [Google Scholar]
- 15.Yuste R and Bonhoeffer T. (2004) Genesis of dendritic spines: Insights from ultrastructural and imaging studies. Nat. Rev. Neurosci 5, 24–34 [DOI] [PubMed] [Google Scholar]
- 16.Yan Z, Kim E, Datta D, Lewis DA, and Soderling SH (2016) Synaptic actin dysregulation, a convergent mechanism of mental disorders? J. Neurosci 36, 11411–11417 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lima Caldeira G, Peça J, and Carvalho AL (2019) New insights on synaptic dysfunction in neuropsychiatric disorders. Curr. Opin. Neurobiol 57, 62–70 [DOI] [PubMed] [Google Scholar]
- 18.Ethell IM and Pasquale EB (2005) Molecular mechanisms of dendritic spine development and remodeling. Prog. Neurobiol 75, 161–205 [DOI] [PubMed] [Google Scholar]
- 19.Tada T and Sheng M. (2006) Molecular mechanisms of dendritic spine morphogenesis. Curr. Opin. Neurobiol 16, 95–101 [DOI] [PubMed] [Google Scholar]
- 20.Yoshihara Y, De Roo M, and Muller D. (2009) Dendritic spine formation and stabilization. Curr. Opin. Neurobiol 19, 146–153 [DOI] [PubMed] [Google Scholar]
- 21.Kanjhan R, Noakes PG, and Bellingham MC (2016) Emerging roles of filopodia and dendritic spines in motoneuron plasticity during development and disease. Neural Plast. 2016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Korobova F and Svitkina TM (2010) Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol. Biol. Cell 21, 165–176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hotulainen P, Llano O, Smirnov S, Tanhuanpää K, Faix J, Rivera C, and Lappalainen P. (2009) Defning mechanisms of actin polymerization and depolymerization during Dendritic spine morphogenesis. J. Cell Biol 185, 323–339 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Spence EF, Kanak DJ, Carlson BR, and Soderling SH (2016) The Arp2/3 complex is essential for distinct stages of spine synapse maturation, including synapse unsilencing. J. Neurosci 36, 9696–9709 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Kim Y, Sung JY, Ceglia I, Lee KW, Ahn JH, Halford JM, Kim AM, Kwak SP, Park JB, Ho Ryu S, Schenck A, Bardoni B, Scott JD, Nairn AC, and Greengard P. (2006) Phosphorylation of WAVE1 regulates actin polymerization and dendritic spine morphology. Nature 442, 814–817 [DOI] [PubMed] [Google Scholar]
- 26.Soderling SH, Guire ES, Kaech S, White J, Zhang F, Schutz K, Langeberg LK, Banker G, Raber J, and Scott JD (2007) A WAVE-1 and WRP signaling complex regulates spine density, synaptic plasticity, and memory. J. Neurosci 27, 355–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Haeckel A, Ahuja R, Gundelfinger ED, Qualmann B, and Kessels MM (2008) The actin-binding protein Abp1 controls dendritic spine morphology and is important for spine head and synapse formation. J. Neurosci 28, 10031–10044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wegner AM, Nebhan CA, Hu L, Majumdar D, Meier KM, Weaver AM, and Webb DJ (2008) N-WASP and the Arp2/3 complex are critical regulators of actin in the development of dendritic spines and synapses. J. Biol. Chem 283, 15912–15920 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Newey SE, Velamoor V, Govek E-E, and Van Aelst L. (2005) Rho GTPases, dendritic structure, and mental retardation. J. Neurobiol 64, 58–74 [DOI] [PubMed] [Google Scholar]
- 30.Kiraly DD, Eipper-Mains JE, Mains RE, and Eipper BA (2010) Synaptic plasticity, a symphony in GEF. ACS Chem. Neurosci 1, 348–365 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Penzes P and Cahill ME (2012) Deconstructing signal transduction pathways that regulate the actin cytoskeleton in dendritic spines. Cytoskeleton 69, 426–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen C, Wirth A, and Ponimaskin E. (2012) Cdc42: An important regulator of neuronal morphology. Int. J. Biochem. Cell Biol 44, 447–451 [DOI] [PubMed] [Google Scholar]
- 33.Tejada-Simon MV (2015) Modulation of actin dynamics by Rac1 to target cognitive function. J. Neurochem 133, 767–779 [DOI] [PubMed] [Google Scholar]
- 34.Woolfrey KM and Srivastava DP (2016) Control of Dendritic Spine Morphological and Functional Plasticity by Small GTPases. Neural Plast. 2016:30259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Zhang H, Webb DJ, Asmussen H, and Horwitz AF (2003) Synapse formation is regulated by the signaling adaptor GIT1. J. Cell Biol 161, 131–142 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang H, Webb DJ, Asmussen H, Niu S, and Horwitz AF (2005) A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J Neurosci 25, 3379–3388 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Za L, Albertinazzi C, Paris S, Gagliani M, Tacchetti C, and de Curtis I. (2006) βPIX controls cell motility and neurite extension by regulating the distribution of GIT1. J. Cell Sci 119, 2654–2666 [DOI] [PubMed] [Google Scholar]
- 38.Saneyoshi T, Wayman G, Fortin D, Davare M, Hoshi N, Nozaki N, Natsume T, and Soderling TR (2008) Activity-Dependent Synaptogenesis: Regulation by a CaM-Kinase Kinase/CaM-Kinase I/βPIX Signaling Complex. Neuron 57, 94–107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Penzes P, Cahill ME, Jones KA, and Srivastava DP (2008) Convergent CaMK and RacGEF signals control dendritic structure and function. Trends Cell Biol. 18, 405–413 [DOI] [PubMed] [Google Scholar]
- 40.Rotty JD, Wu C, and Bear JE (2013) New insights into the regulation and cellular functions of the ARP2/3 complex. Nat. Rev. Mol. Cell Biol 14, 7–12 [DOI] [PubMed] [Google Scholar]
- 41.Rácz B and Weinberg RJ (2008) Organization of the Arp2/3 complex in hippocampal spines. J. Neurosci 28, 5654–5659 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Beach JR and Hammer JA (2015) Myosin II isoform co-assembly and differential regulation in mammalian systems. Exp. Cell Res 334, 2–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Meng Y, Zhang Y, Tregoubov V, Janus C, Cruz L, Jackson M, Lu WY, MacDonald JF, Wang JY, Falls DL, and Jia Z. (2002) Abnormal spine morphology and enhanced LTP in LIMK-1 knockout mice. Neuron 35, 121–133 [DOI] [PubMed] [Google Scholar]
- 44.Chen LY, Rex CS, Casale MS, Gall CM, and Lynch G. (2007) Changes in synaptic morphology accompany actin signaling during LTP. J. Neurosci 27, 5363–5372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Morales M and Fifková E. (1989) In situ localization of myosin and actin in dendritic spines with the immunogold technique. J. Comp. Neurol 279, 666–674 [DOI] [PubMed] [Google Scholar]
- 46.Ryu J, Liu L, Wong TP, Wu DC, Burette A, Weinberg R, Wang YT, and Sheng M. (2006) A critical role for myosin IIB in dendritic spine morphology and synaptic function. Neuron 49, 175–182 [DOI] [PubMed] [Google Scholar]
- 47.Ma X, Kawamoto S, Uribe J, and Adelstein RS (2006) Function of the neuron-specific alternatively spliced isoforms of nonmuscle myosin II-B during mouse brain development. Mol. Biol. Cell 17, 2138–2149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Rex CS, Gavin CF, Rubio MD, Kramar EA, Chen LY, Jia Y, Huganir RL, Muzyczka N, Gall CM, Miller CA, Lynch G, and Rumbaugh G. (2010) Myosin IIb Regulates actin dynamics during synaptic plasticity and memory formation. Neuron 67, 603–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hodges JL, Newell-Litwa K, Asmussen H, Vicente-Manzanares M, and Horwitz AR (2011) Myosin IIB activity and phosphorylation status determines dendritic spine and post-synaptic density morphology. PLoS One 6, e24149 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Rubio MD, Johnson R, Miller CA, Huganir RL, and Rumbaugh G. (2011) Regulation of synapse structure and function by distinct myosin II motors. J. Neurosci 31, 1448–1460 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Koskinen M, Bertling E, Hotulainen R, Tanhuanpää K, and Hotulainen P. (2014) Myosin IIb controls actin dynamics underlying the dendritic spine maturation. Mol. Cell. Neurosci 61, 56–64 [DOI] [PubMed] [Google Scholar]
- 52.Ozkan ED, Aceti M, Creson TK, Rojas CS, Hubbs CR, McGuire MN, Kakad PP, Miller CA, and Rumbaugh G. (2015) Input-specific regulation of hippocampal circuit maturation by non-muscle myosin IIB. J. Neurochem 134, 429–444 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Briggs SB, Hafenbreidel M, Young EJ, Rumbaugh G, and Miller CA. (2018) The role of nonmuscle myosin II in polydrug memories and memory reconsolidation. Learn. Mem 25, 391–398 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Gavin CF, Rubio MD, Young E, Miller C, and Rumbaugh G. (2012) Myosin II motor activity in the lateral amygdala is required for fear memory consolidation. Learn. Mem 19, 9–14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Billington N, Beach JR, Heissler SM, Remmert K, Guzik-Lendrum S, Nagy A, Takagi Y, Shao L, Li D, Yang Y, Zhang Y, Barzik M, Betzig E, Hammer JA, and Sellers JR (2015) Myosin 18A coassembles with nonmuscle myosin 2 to form mixed bipolar filaments. Curr. Biol 25, 942–948 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Horsthemke M, Nutter LMJ, Bachg AC, Skryabin BV, Honnert U, Zobel T, Bogdan S, Stoll M, Seidl MD, Müller FU, Ravens U, Unger A, Linke WA, Van Gorp PRR, De Vries AAF, Bähler M, and Hanley PJ (2019) A novel isoform of myosin 18A (Myo18Aγ) is an essential sarcomeric protein in mouse heart. J. Biol. Chem 294, 7202–7218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Guzik-Lendrum S, Nagy A, Takagi Y, Houdusse A, and Sellers JR (2011) Drosophila melanogaster myosin-18 represents a highly divergent motor with actin tethering properties. J. Biol. Chem 286, 21755–21766 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Guzik-Lendrum S, Heissler SM, Billington N, Takagi Y, Yang Y, Knight PJ, Homsher E, and Sellers JR (2013) Mammalian myosin-18A, a highly divergent myosin. J. Biol. Chem 288, 9532–9548 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bonn BR, Rudolf A, Hornbruch-Freitag C, Daum G, Kuckwa J, Kastl L, Buttgereit D, and Renkawitz-Pohl R. (2013) Myosin heavy chain-like localizes at cell contact sites during Drosophila myoblast fusion and interacts in vitro with Rolling pebbles 7. Exp. Cell Res 319, 402–416 [DOI] [PubMed] [Google Scholar]
- 60.Hsu RM, Tsai MH, Hsieh YJ, Lyu PC, and Yu JS (2010) Identification of MYO18A as a novel interacting partner of the PAK2/βPIX/GIT1 complex and its potential function in modulating epithelial cell migration. Mol. Biol. Cell 21, 287–301 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Hsu RM, Hsieh YJ, Yang TH, Chiang YC, Kan CY, Lin YT, Chen JT, and Yu JS (2014) Binding of the extreme carboxyl-terminus of PAK-interacting exchange factor β (βPIX) to myosin 18A (MYO18A) is required for epithelial cell migration. Biochim. Biophys. Acta - Mol. Cell Res 1843, 2513–2527 [DOI] [PubMed] [Google Scholar]
- 62.Tan I, Yong J, Dong JM, Lim L, and Leung T. (2008) A tripartite complex containing MRCK modulates lamellar actomyosin retrograde flow. Cell 135, 123–136 [DOI] [PubMed] [Google Scholar]
- 63.Wagner W, Brenowitz SD, and Hammer JA (2011) Myosin-Va transports the endoplasmic reticulum into the dendritic spines of Purkinje neurons. Nat. Cell Biol 13, 40–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Alexander CJ and Hammer JA (2016) Optimization of cerebellar purkinje neuron cultures and development of a plasmid-based method for purkinje neuron-specific, miRNA-mediated protein knockdown. Methods Cell Biol. 131, 177–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Alexander CJ and Hammer JA (2019) An Improved Method for Differentiating Mouse Embryonic Stem Cells into Cerebellar Purkinje Neurons. Cerebellum 18, 406–421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bruun K, Beach JR, Heissler SM, Remmert K, Sellers JR, and Hammer JA (2017) Re-evaluating the roles of myosin 18Aα and F-actin in determining Golgi morphology. Cytoskeleton 74, 205–218 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yi JH, Katagiri Y, Susarla B, Figge D, Symes AJ, and Geller HM (2012) Alterations in sulfated chondroitin glycosaminoglycans following controlled cortical impact injury in mice. J. Comp. Neurol 520, 3295–3313 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wagner W, McCroskery S, and Hammer JA (2011) An efficient method for the long-term and specific expression of exogenous cDNAs in cultured Purkinje neurons. J. Neurosci. Methods 200, 95–105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Taft MH, Behrmann E, Munske-Weidemann LC, Thiel C, Raunser S, and Manstein DJ (2013) Functional characterization of human myosin-18A and its interaction with F-actin and GOLPH3. J. Biol. Chem 288, 30029–30041 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Tonikian R, Zhang Y, Sazinsky SL, Currell B, Yeh JH, Reva B, Held HA, Appleton BA, Evangelista M, Wu Y, Xin X, Chan AC, Seshagiri S, Lasky LA, Sander C, Boone C, Bader GD, and Sidhu SS (2008) A specificity map for the PDZ domain family. PLoS Biol. 6, 2043–2059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sheffield P, Garrard S, and Derewenda Z. (1999) Overcoming expression and purification problems of RhoGDI using a family of “parallel” expression vectors. Protein Expr. Purif 15, 34–39 [DOI] [PubMed] [Google Scholar]
- 72.Murugesan S, Hong J, Yi J, Li D, Beach JR, Shao L, Meinhardt J, Madison G, Wu X, Betzig E, and Hammer JA (2016) Formin-generated actomyosin arcs propel T cell receptor microcluster movement at the immune synapse. J Cell Biol 215, 383–399 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Rueden CT, Schindelin J, Hiner MC, DeZonia BE, Walter AE, Arena ET, and Eliceiri KW (2017) ImageJ2: ImageJ for the next generation of scientific image data. BMC Bioinformatics 18, Article: 529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Beach JR, Bruun KS, Shao L, Li D, Swider Z, Remmert K, Zhang Y, Conti MA, Adelstein RS, Rusan NM, Betzig E, and Hammer JA (2017) Actin dynamics and competition for myosin monomer govern the sequential amplification of myosin filaments. Nat. Cell Biol 19, 85–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fujiwara I, Remmert K, Piszczek G, and Hammer JA (2014) Capping protein regulatory cycle driven by CARMIL and V-1 may promote actin network assembly at protruding edges. Proc. Natl. Acad. Sci. U. S. A 111, E1970–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Fujiwara I, Remmert K, and Hammer JA (2010) Direct observation of the uncapping of capping protein-capped actin filaments by CARMIL homology domain 3. J. Biol. Chem 285, 2707–2720 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Alexander CJ, Wagner W, Copeland NG, Jenkins NA, and Hammer JA (2018) Creation of a myosin Va-TAP-tagged mouse and identification of potential myosin Va-interacting proteins in the cerebellum. Cytoskeleton 75, 395–409 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Petralia RS, Wang YX, Hua F, Yi Z, Zhou A, Ge L, Stephenson FA, and Wenthold RJ (2010) Organization of NMDA receptors at extrasynaptic locations. Neuroscience 167, 68–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.He S, Wang YX, Petralia RS, and Brenowitz SD (2014) Cholinergic modulation of large-conductance calcium- activated potassium channels regulates synaptic strength and spine calcium in cartwheel cells of the dorsal cochlear nucleus. J. Neurosci 34, 5261–5272 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Lein ES, Hawrylycz MJ, Ao N, Ayres M, Bensinger A, Bernard A, Boe AF, Boguski MS, Brockway KS, Byrnes EJ, Chen L, Chen L, Chen TM, Chin MC, Chong J, Crook BE, Czaplinska A, Dang CN, Datta S, Dee NR, Desaki AL, Desta T, Diep E, Dolbeare TA, Donelan MJ, Dong HW, Dougherty JG, Duncan BJ, Ebbert AJ, Eichele G, Estin LK, Faber C, Facer BA, Fields R, Fischer SR, Fliss TP, Frensley C, Gates SN, Glattfelder KJ, Halverson KR, Hart MR, Hohmann JG, Howell MP, Jeung DP, Johnson RA, Karr PT, Kawal R, Kidney JM, Knapik RH, Kuan CL, Lake JH, Laramee AR, Larsen KD, Lau C, Lemon TA, Liang AJ, Liu Y, Luong LT, Michaels J, Morgan JJ, Morgan RJ, Mortrud MT, Mosqueda NF, Ng LL, Ng R, Orta GJ, Overly CC, Pak TH, Parry SE, Pathak SD, Pearson OC, Puchalski RB, Riley ZL, Rockett HR, Rowland SA, Royall JJ, Ruiz MJ, Sarno NR, Schaffnit K, Shapovalova NV, Sivisay T, Slaughterbeck CR, Smith SC, Smith KA, Smith BI, Sodt AJ, Stewart NN, Stumpf KR, Sunkin SM, Sutram M, Tam A, Teemer CD, Thaller C, Thompson CL, Varnam LR, Visel A, Whitlock RM, Wohnoutka PE, Wolkey CK, Wong VY, Wood M, Yaylaoglu MB, Young RC, Youngstrom BL, Yuan XF, Zhang B, Zwingman TA, and Jones AR (2007) Genome-wide atlas of gene expression in the adult mouse brain. Nature 445, 168–176 [DOI] [PubMed] [Google Scholar]
- 81.Laure-Kamionowska M and Málińska D. (2009) Calbindin positive Purkinje cells in the pathology of human cerebellum occurring at the time of its development. Folia Neuropathol. 47, 300–305 [PubMed] [Google Scholar]
- 82.Yang Q, Hashizume Y, Yoshida M, Wang Y, Goto Y, Mitsuma N, Ishikawa K, and Mizusawa H. (2000) Morphological Purkinje cell changes in spinocerebellar ataxia type 6. Acta Neuropathol. 100, 371–376 [DOI] [PubMed] [Google Scholar]
- 83.Seil FJ (2014) The changeable nervous system: Studies on neuroplasticity in cerebellar cultures. Neurosci. Biobehav. Rev 45, 212–232 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Richter K, Langnaese K, Kreutz MR, Olias G, Zhai R, Scheich H, Garner CC, and Gundelfinger ED (1999) Presynaptic cytomatrix protein bassoon is localized at both excitatory and inhibitory synapses of rat brain. J. Comp. Neurol 408, 437–448 [DOI] [PubMed] [Google Scholar]
- 85.Heintz TG, Eva R, and Fawcett JW (2016) Regional regulation of purkinje cell dendritic spines by integrins and Eph/ephrins. PLoS One 11, e0158558–e0158558 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Gao Y, Perkins EM, Clarkson YL, Tobia S, Lyndon AR, Jackson M, and Rothstein JD (2011) β-III spectrin is critical for development of Purkinje cell dendritic tree and spine morphogenesis. J. Neurosci 31, 16581–16590 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Velázquez-Zamora DA, Martínez-Degollado M, and González-Burgos I. (2011) Morphological development of dendritic spines on rat cerebellar Purkinje cells. Int. J. Dev. Neurosci 29, 515–520 [DOI] [PubMed] [Google Scholar]
- 88.Lee KJ, Jung JG, Arii T, Imoto K, and Rhyu IJ (2007) Morphological changes in dendritic spines of Purkinje cells associated with motor learning. Neurobiol. Learn. Mem 88, 445–450 [DOI] [PubMed] [Google Scholar]
- 89.Dippold HC, Ng MM, Farber-Katz SE, Lee SK, Kerr ML, Peterman MC, Sim R, Wiharto PA, Galbraith KA, Madhavarapu S, Fuchs GJ, Meerloo T, Farquhar MG, Zhou H, and Field SJ (2009) GOLPH3 Bridges Phosphatidylinositol-4- Phosphate and Actomyosin to Stretch and Shape the Golgi to Promote Budding. Cell 139, 337–351 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Harris KM and Stevens JK (1989) Dendritic spines of CA1 pyramidal cells in the rat hippocampus: Serial electron microscopy with reference to their biophysical characteristics. J. Neurosci 9, 2982–2997 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Harris KM, Jensen FE, and Tsao B. (1992) Three-dimensional structure of dendritic spines and synapses in rat hippocampus (CA 1) at postnatal day 15 and adult ages: Implications for the maturation of synaptic physiology and long-term potentiation. J. Neurosci 12, 2685–2705 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Fiala JC, Feinberg M, Popov V, and Harris KM (1998) Synaptogenesis via dendritic filopodia in developing hippocampal area CA1. J. Neurosci 18, 8900–8911 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Oray S, Majewska A, and Sur M. (2005) Effects of Synaptic Activity on Dendritic Spine Motility of Developing Cortical Layer V Pyramidal Neurons. Cereb. Cortex 16, 730–741 [DOI] [PubMed] [Google Scholar]
- 94.Takahashi H, Sekino Y, Tanaka S, Mizui T, Kishi S, and Shirao T. (2003) Drebrin-dependent actin clustering in dendritic filopodia governs synaptic targeting of postsynaptic density-95 and dendritic spine morphogenesis. J. Neurosci 23, 6586–6595 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Lee KJ, Kim H, and Rhyu IJ (2005) The roles of dendritic spine shapes in Purkinje cells. Cerebellum 4, 97–104 [DOI] [PubMed] [Google Scholar]
- 96.Stein IS and Zito K. (2018) Dendritic Spine Elimination: Molecular Mechanisms and Implications. Neurosci. 25, 27–47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Barski JJ, Dethleffsen K, and Meyer M. (2000) Cre recombinase expression in cerebellar Purkinje cells. Genesis 28, 93–98 [PubMed] [Google Scholar]
- 98.Sugawara T, Hisatsune C, Le TD, Hashikawa T, Hirono M, Hattori M, Nagao S, and Mikoshiba K. (2013) Type 1 inositol trisphosphate receptor regulates cerebellar circuits by maintaining the spine morphology of purkinje cells in adult mice. J. Neurosci 33, 12186–12196 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang XM, Ng AHL, Tanner JA, Wu WT, Copeland NG, Jenkins NA, and Huang JD (2004) Highly restricted expression of Cre recombinase in cerebellar Purkinje cells. Genesis 40, 45–51 [DOI] [PubMed] [Google Scholar]
- 100.Nitta K, Matsuzaki Y, Konno A, and Hirai H. (2017) Minimal Purkinje Cell-Specific PCP2/L7 Promoter Virally Available for Rodents and Non-human Primates. Mol. Ther. - Methods Clin. Dev 6, 159–170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Chazeau A and Giannone G. (2016) Organization and dynamics of the actin cytoskeleton during dendritic spine morphological remodeling. Cell. Mol. Life Sci 73, 3053–3073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Jan YN and Jan LY (2010) Branching out: Mechanisms of dendritic arborization. Nat. Rev. Neurosci 11, 316–328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lewis TL and Polleux F. (2012) Neuronal Morphogenesis: Golgi Outposts, Acentrosomal Microtubule Nucleation, and Dendritic Branching. Neuron 76, 862–864 [DOI] [PubMed] [Google Scholar]
- 104.Valenzuela JI and Perez F. (2015) Diversifying the secretory routes in neurons. Front. Neurosci 9: 358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hanus C and Ehlers MD (2016) Specialization of biosynthetic membrane trafficking for neuronal form and function. Curr. Opin. Neurobiol 39, 8–16 [DOI] [PubMed] [Google Scholar]
- 106.Kennedy MJ and Hanus C. (2019) Architecture and Dynamics of the Neuronal Secretory Network. Annu. Rev. Cell Dev. Biol 35, 543–566 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mikhaylova M, Bera S, Kobler O, Frischknecht R, and Kreutz MR (2016) A Dendritic Golgi Satellite between ERGIC and Retromer. Cell Rep. 14, 189–199 [DOI] [PubMed] [Google Scholar]
- 108.Ori-McKenney KM, Jan LY, and Jan YN (2012) Golgi Outposts Shape Dendrite Morphology by Functioning as Sites of Acentrosomal Microtubule Nucleation in Neurons. Neuron 76, 921–930 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Tanabe K, Kani S, Shimizu T, Bae Y-K, Abe T, and Hibi M. (2010) Atypical Protein Kinase C Regulates Primary Dendrite Specification of Cerebellar Purkinje Cells by Localizing Golgi Apparatus. J. Neurosci 30, 16983 LP – 16992 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Fu M. meng, McAlear TS, Nguyen H, Oses-Prieto JA, Valenzuela A, Shi RD, Perrino JJ, Huang TT, Burlingame AL, Bechstedt S, and Barres BA (2019) The Golgi Outpost Protein TPPP Nucleates Microtubules and Is Critical for Myelination. Cell 179, 132–146.e14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Ramakers GJA, Wolfer D, Rosenberger G, Kuchenbecker K, Kreienkamp H-J, Prange-Kiel J, Rune G, Richter K, Langnaese K, Masneuf S, Bösl MR, Fischer K-D, Krugers HJ, Lipp H-P, van Galen E, and Kutsche K. (2011) Dysregulation of Rho GTPases in the αPix/Arhgef6 mouse model of X-linked intellectual disability is paralleled by impaired structural and synaptic plasticity and cognitive deficits. Hum. Mol. Genet 21, 268–286 [DOI] [PubMed] [Google Scholar]
- 112.Meseke M, Rosenberger G, and Förster E. (2013) Reelin and the Cdc42/Rac1 guanine nucleotide exchange factor αPIX/Arhgef6 promote dendritic Golgi translocation in hippocampal neurons. Eur. J. Neurosci 37, 1404–1412 [DOI] [PubMed] [Google Scholar]
- 113.Long M and Simpson JC (2017) Rho GTPases operating at the Golgi complex: Implications for membrane traffic and cancer biology. Tissue Cell 49, 163–169 [DOI] [PubMed] [Google Scholar]
- 114.Friesland A, Zhao Y, Chen YH, Wang L, Zhou H, and Lu Q. (2013) Small molecule targeting Cdc42-intersectin interaction disrupts Golgi organization and suppresses cell motility. Proc. Natl. Acad. Sci. U. S. A 110, 1261–1266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Hirai H and Launey T. (2000) The regulatory connection between the activity of granule cell NMDA receptors and dendritic differentiation of cerebellar Purkinje cells. J. Neurosci 20, 5217–5224 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Tanaka M, Yanagawa Y, and Hirashima N. (2009) Transfer of small interfering RNA by single-cell electroporation in cerebellar cell cultures. J. Neurosci. Methods 178, 80–86 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.