

Abstract
Major depressive disorder (MDD) is the single largest contributor to global disability and up to 20–30% of patients do not respond to at least two antidepressants (treatment-resistant depression, TRD). This study leveraged imputed gene expression in TRD to perform a drug repurposing analysis. Among those with MDD, we defined TRD as having at least two antidepressant switches according to primary care records in UK Biobank (UKB). We performed a transcriptome-wide association study (TWAS) of TRD (n = 2165) vs healthy controls (n = 11,188) using FUSION and gene expression levels from 21 tissues. We identified compounds with opposite gene expression signatures (ConnectivityMap data) compared to our TWAS results using the Kolmogorov-Smirnov test, Spearman and Pearson correlation. As symptom patterns are routinely assessed in clinical practice and could be used to provide targeted treatments, we identified MDD subtypes associated with TRD in UKB and analysed them using the same pipeline described for TRD. Anxious MDD (n = 14,954) and MDD with weight gain (n = 4697) were associated with TRD. In the TWAS, two genes were significantly dysregulated (TMEM106B and ATP2A1 for anxious and weight gain MDD, respectively). A muscarinic receptor antagonist was identified as top candidate for repurposing in TRD; inhibition of heat shock protein 90 was the main mechanism of action identified for anxious MDD, while modulators of metabolism such as troglitazone showed promising results for MDD with weight gain. This was the first TWAS of TRD and associated MDD subtypes. Our results shed light on possible pharmacological approaches in individuals with difficult-to-treat depression.
Subject terms: Gene expression, Predictive markers
Introduction
Major depressive disorder (MDD) is the single largest contributor to global disability and the main contributor to suicide deaths, which number close to 800,000 per year [1]. Antidepressants are the first line treatment for moderate–severe MDD and their efficacy compared to placebo has been demonstrated by large meta-analyses;[2] however, up to 20–30% of patients develop treatment-resistant depression (TRD), i.e. their depressive symptoms do not sufficiently improve after two or more antidepressant treatments [3, 4]. TRD is associated with recurrent depression, frequent hospitalizations, negative repercussions on occupational/social functioning, decline of physical health and increased all cause-mortality [5]. A longitudinal study of TRD showed that 40% of patients had persistent depression or subsyndromal symptoms after 3 years, demonstrating the chronic course and long-term impairment associated with TRD [6].
MDD and antidepressant response have a heritable component [7, 8], the study of which can be useful to better understand the inter-individual differences in the clinical course of the disease and treatment response. Genetic studies have indeed suggested that patients with TRD have distinctive biological characteristics compared to responders [9] and the clinical heterogeneity of MDD at least partly reflects the involvement of specific genetic factors [10]. Therefore, genetic data are a precious resource to disentangle the mechanisms responsible for the heterogeneous manifestations of MDD and develop more effective treatments, i.e. treatments that target the specific biological dysfunctions found in certain groups of patients with MDD. Certain subtypes of MDD have been associated with the risk of TRD or with a chronic course and higher disability, particularly anxious, melancholic and atypical depression [11–13]. These findings confirm the hypothesis that clinical manifestations may be connected to specific biological mechanisms and to the risk of TRD. According to this hypothesis, clinical symptoms can be used not only to predict the risk of TRD, but also to guide the prescription of targeted treatments based on patterns that are easily recognizable at the first clinical assessment.
Drug repurposing guided by genetic findings is a promising approach to identify new candidate compounds for TRD and depression subtypes of interest, as selecting genetically supported targets could double the success rate in clinical development [14]. Previous studies leveraged genetic findings to identify possible drugs for repurposing in MDD (e.g. [15, 16]), while only one examined specifically TRD to the best of our knowledge and it represented a first step to the identification of drugs with genetic support of efficacy in this group of patients [17]. It used an approach based on the enrichment of drug gene targets in TRD-associated genes, therefore it could not discriminate the direction of the pharmacological effect on the disease (therapeutic or detrimental) and the results needed interpretation based on the known pathogenetic mechanisms of TRD [17]. This limitation is common also to other drug repurposing studies [18] and a possible method to address it is to impute gene expression from genome-wide trait-associated variants, then to compare imputed gene expression profiles to drug-induced gene expression profiles [16].
Therefore, in the present study we analysed UK Biobank (UKB) data with the aim to: (1) identify gene expression changes associated with TRD and depression subtypes associated with TRD risk; (2) compare the disease-associated gene expression profiles with drug-induced gene expression to select compounds that show opposite patterns of expression and could counteract the pathogenetic alterations of TRD.
Material and methods
Selection of depression phenotypes
We selected MDD subtypes associated with TRD and TRD itself to prioritize the identification of compounds potentially effective in cases of MDD poorly responsive to the available antidepressants and because previous studies have focused on MDD as a whole group [15, 16, 19].
TRD was defined using electronic health records (EHR) of primary care events in UKB as participants with MDD having at least two switches between different antidepressant drugs (independently from the class) satisfying the following criteria [20]:
Each drug was prescribed for at least six consecutive weeks (noting that adequate duration for efficacy is four weeks, and our conservative threshold should reduce the risk that drug switch was due to side effects);
The time interval between the prescription of two consecutive drugs was no longer than 14 weeks (to ensure that treatment had not been suspended).
Secondly, we tested if any MDD subtype was associated with an increased risk of TRD, considering MDD with typical and atypical neurovegetative symptoms, MDD with weight gain, anxious MDD, psychotic, seasonal, peripartum, stress-related or reactive MDD and endogenous MDD. These depression subtypes were defined using primary care EHR of diagnostic codes and/or symptoms reported as part of the mental health questionnaire (MHQ), particularly those assessed by the Composite International Diagnostic Interview Short Form (CIDI-SF) [21]; cases of bipolar, psychotic or substance use disorders were excluded and further details are available as Supplementary Methods. We decided to examine also MDD with weight gain because the subtype with atypical neurovegetative symptoms had relatively small sample size in UKB [22] and previous studies suggested that weight increase during depression drives relevant biological characteristics of this group [10]. The investigated subtypes include those previously associated with worse treatment response and worse prognosis (see “Introduction”), and the subtypes that reflect the current nosology and therefore are usually routinely assessed in the clinical practice [23]. Though reactive and endogenous depression are no longer part of the current nosology, they were present among primary care EHR as these mostly started in the 1990s, and endogenous MDD was the equivalent of melancholic depression [24]. Reactive or stress-related depression was considered because recent studies suggested it may show distinctive genetic factors compared to other MDD cases [25].
Statistical analysis
Transcriptome-wide association study
We performed a genome-wide association study (GWAS) of depression subtypes found to be associated with TRD using BGENIE v1.2 and imputed genotype dosages [26]; phenotypes were residualized for six genetic ancestry principal components, assessment centre and batch effects. Sex and age were not considered as covariates as they are not risk factors for TRD according to the literature [11]. A higher average body mass index (BMI) was found in MDD cases vs healthy controls (28.34 [SD = 5.43] vs 26.43 [SD = 4.15], p < 5e-324), particularly in cases who reported weight gain during their worst depressive episode (30.19 [SD = 5.56]). We did not adjust the analyses for BMI, because shared genetic factors and a bi-directional relationship have been reported between MDD and overweight/obesity [27]. We included participants of European ancestry; details on quality control of genotypes are in the Supplementary Methods. Healthy controls were selected from those who completed the MHQ and/or had primary care records available and no psychiatric disorder (n = 54,974 after quality control). GWAS summary statistics of TRD vs healthy controls (n = 2,165 vs n = 11,188) were generated as part of a previous study [20].
SNP weights from distinct tissues and samples (of European ancestry) were used, applying the same procedure described in a recent study [28]. As in this previous transcriptome-wide association study (TWAS) of MDD, FUSION SNP-weights from postmortem brain tissue, whole blood, peripheral blood, adrenal, pituitary and thyroid glands were obtained (http://gusevlab.org/projects/fusion/#reference-functional-data). We chose to use FUSION software because it compares several models when deriving SNP-weights to account for different architecture and it has in-built functionality for conditional and colocalization analyses. We did not focus our analyses exclusively on brain tissues because: (1) a high correlation between cis-eQTLs effects in blood and brain tissues has been demonstrated, as well as a gain of power in gene discovery for brain-related phenotypes using blood cis-eQTL data with large sample sizes [29]; (2) MDD is considered a systemic disease (e.g. increased systemic inflammation, metabolic and endocrine dysfunctions) [30]; (3) Cmap mostly includes data on cancer cell lines that are not neuronal in origin [31]. The weights pertained to the following RNA reference samples: NTR (Netherlands Twin Register) and YFS (Young Finns Study), both of which provide information on blood tissue gene expression; CMC (CommonMind Consortium) and PsychENCODE Consortium, both of which assessed the dorsolateral prefrontal cortex (DLPFC); and the GTEx Consortium, which measured expression in multiple brain and peripheral tissues [32–34].
A TWAS was performed for each phenotype of interest using FUSION, setting the transcriptome-wide significance at p = 1.37 × 10−6 in line with a previous study [28]; colocalization, conditional analysis and fine mapping of significant signals were performed according to [28] and are described in the Supplementary Methods.
Screening of compounds for drug repurposing
As the high number of compound signatures (>130 K) available in ConnectivityMap (Cmap) and the lack of information to select a priori certain experimental conditions (e.g. cell line, dose, time of exposure), we used the Cmap Query tool (https://clue.io/query) to screen the available signatures and identify those to include in the following step (see next paragraph). To the best of our knowledge, Cmap represents the largest repository of compound-induced gene expression profiles in terms of number of available compounds and type of experimental conditions (cell lines, doses, time of exposure).
We selected the compound signatures generating gene expression profiles most dissimilar to our top TWAS results, given the hypothesis that these could counteract the alterations observed in TRD and MDD subgroups of interest [16]. Cmap catalogs transcriptional responses of human cells to chemical and genetic perturbation; >1 M replicate-collapsed signatures are available in the 2020 version of the database (Expanded CMap LINCS Resource 2020) [31]. We used the top 25, 50, 100, 150 and 250 genes dysregulated in each TWAS (with no pre-specified p value threshold) to identify candidate signatures generating opposite transcriptional responses, since we do not know how many genes underlie TRD or depression subtypes [16]. This method was applied in a previous study [16], although we used 25 top genes instead of 500, as we preferred to be more conservative given the relatively small size of our samples. We used the R package “biomaRt” to convert gene symbols to Entrez ID which is the nomenclature used by Cmap. When multiple features were available for the same gene (i.e. weights for multiple tissues), we selected the feature with the highest cross-validation coefficient of determination (CV R2) [35]. Cmap Query provides a measure of similarity of the provided up- or downregulated genes to those induced by perturbagens in the database, namely the connectivity score. The connectivity score represents a non-parametric similarity measure based on the weighted Kolmogorov-Smirnov (KS) enrichment statistic and it is positive for signatures that are positively related and negative for those that are inversely related [36]. To allow for comparison of connectivity scores across cell types and perturbation types, the scores are normalized by dividing a raw positive connectivity score by the mean of positive connectivity scores and a raw negative connectivity score by the mean of negative connectivity scores. A p value is calculated by comparing the similarity between the query and reference signature (KS test) to a null distribution of random queries and then it is adjusted for multiple testing (false discovery rate, FDR) [36].
Ranking of top compounds for repurposing
Selected signatures from the previous step were then examined using the approach described in a previous study that includes the R code made available by the authors [16], in order to identify those with the strongest dissimilarity to the genes dysregulated in our TWAS. In detail, the selected signatures were investigated using a combination of different methods (KS test, Spearman and Pearson correlation with all or with the top 25, 50, 100, 150 and 250 differentially expressed genes). For each compound, we calculated the average rank within each method across the examined sets of genes; then we computed the average rank across the different methods [16]. To assess the significance of the ranks, we performed permutations by shuffling the disease-expression z-scores and repeating the same procedure above to determine average ranks. We performed 100 permutations for each signature–phenotype pair and combined the distribution of ranks under the null across all signature–phenotype pairs; the p value was calculated by comparing the distribution of the permuted ranks to the observed ranks.
For these analyses we downloaded the Expanded CMap LINCS Resource 2020 level 5 data which includes replicate-collapsed z-score vectors representing gene expression levels [31]. We applied the following selection criteria to identify signatures of interest: (1) a negative connectivity score to our TWAS dysregulated genes; (2) FDR p < 0.10 (liberal threshold was used for inclusion in subsequent analyses); (3) the signature corresponded to a compound (and not the genetic loss/gain of function perturbagens included in Cmap); (4) the corresponding compound was not an antidepressant or antipsychotic medication and did not have known toxicity or relevant side effects (e.g. chemotherapy, immunosuppressant activity); (5) the signature satisfied Cmap quality control criteria [36]. If a signature satisfied all criteria but not (5), we selected an alternative signature for the same compound passing quality control and having the most similar characteristics in terms of cell line, dose of the compound and time of exposure; when more than one alternative signature having overlapping characteristics was available, we prioritized those labelled as high quality if any, otherwise we included all the available alternatives.
These analyses were performed using the R code made available by [16] in R version 4.0.3 and the R package “cmapR”.
Results
We identified three subtypes of MDD that were associated with increased TRD risk after Bonferroni correction, namely MDD with weight gain (n total = 5826 and 4697 after quality control), anxious MDD (n total = 18,034 and 14,954 after quality control) and endogenous MDD (n total 1014 and 860 after quality control) (Fig. 1). Interestingly, MDD with typical neurovegetative symptoms was associated with reduced risk of TRD. An overview of the number of cases for each subtype and their association with TRD is in Supplementary Table 1.
Fig. 1. OR and 95% confidence intervals for treatment-resistant depression (TRD) in each of the examined depression subtypes.
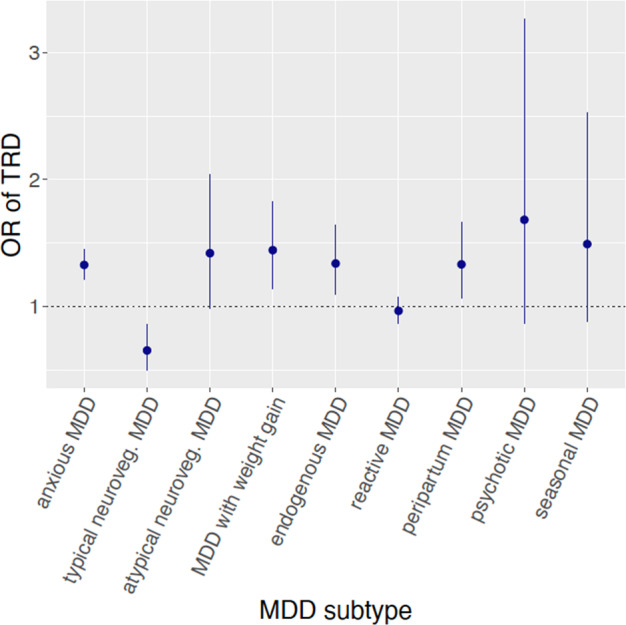
The analysed MDD subtypes were selected to reflect those with higher risk of TRD according to the previous literature and those part of the psychiatric nosology. MDD = major depressive disorder.
As the endogenous MDD subtype had a very limited sample size for further analyses, we decided to include anxious MDD, MDD with weight gain and TRD in the TWAS and subsequent analyses.
TWAS results
We identified two transcriptome-wide significant signals: TMEM106B for depression with anxiety (panel CMC DLPFC splicing, p = 1.23 × 10−6) and ATP2A1 for depression with weight gain (panel PsychENCODE, p = 1.34 × 10−6); no significant features were identified for TRD. The top features (p < 5e-5) for each phenotype are described in Supplementary Table 2 and z-scores across tissues in Fig. 2; QQ plots and Manhattan plots are in Supplementary Fig. 1.
Fig. 2. Z-scores across SNP-weight sets for genes with p < 5e-5.

Comparisons of z-scores across SNP-weight sets for genes with p < 5e-5 for anxious depression (A), depression with weight gain (B) and treatment-resistant depression (TRD) (C) (described in Supplementary Table 2). White spaces correspond to genes that were not tested in the TWAS due to their not significant heritability. Blue shades indicate downregulation while red ones represent upregulation of gene expression. Black vertical lines indicate where the major histocompatibility complex region starts and ends. Transcriptome-wide significant genes are underlined in red. ACC = anterior cingulate cortex; CMC = CommonMind Consortium; DLPFC = dorsolateral prefrontal cortex; GTEx = genotype tissue expression; NTR = Netherlands Twins Register; YFS = Young Finns Study.
For both TMEM106B and ATP2A1, the colocalization analysis showed that the same causal SNP was likely affecting both the risk of anxious or weight gain depression and transcription (Table 1). FOCUS suggested that both features were in the 90% credible set, though it could not identify any feature in the corresponding regions that was more likely to be causal than others (Table 1 and Supplementary Table 2). The results of the conditional analysis showed that the identified features were independent (jointly significant) (Table 1, Supplementary Table 2 and Supplementary Fig. 2).
Table 1.
Transcriptome-wide significant genes in at least one tissue; we reported a comparison with the latest TWAS of major depressive disorder (MDD) (Dall’Aglio et al., 2021 [28]) and with previous GWASs of neuropsychiatric and cardio-metabolic traits (according to GWAS Catalog: https://www.ebi.ac.uk/, accessed on 4th May 2021; GWAS Catalog annotations are based on the last Ensembl release, genes in which a variant maps are reported or the closest upstream or downstream gene within 50 kb).
| Phenotype | Gene | Panel | z-score | p value | PP3 | PP4 | In FOCUS credibility set | Significant in previous MDD TWAS | Traits with significant associations in previous GWASs |
|---|---|---|---|---|---|---|---|---|---|
| Anxious MDD | TMEM106B | CMC DLPFC splicing | −4.85 | 1.23e-6a | 0.022 | 0.972 | Yes | No | Neuroticism, feeling tense, mood swings, depressive symptoms, depression, frontotemporal dementia, aging, help-seeking from a GP, years of education, well-being spectrum, anxiety disorders, cardio-metabolic traits |
| GTEx adrenal gland | 4.79 | 1.65e-6 | 0.013 | 0.978 | Yes | Yes | |||
| YFS blood | 4.67 | 3.02e-6 | 0.011 | 0.986 | Yes | Yes | |||
| PsychENCODE | −4.63 | 3.58e-6 | 0.062 | 0.934 | Yes | Yes | |||
| GTEx whole blood | 4.05 | 5.11e-5 | – | – | No | Yes | |||
| GTEx cerebellar hemisphere | 0.47 | 0.64 | – | – | No | No | |||
| MDD with weight gain | ATP2A1 | PsychENCODE | 4.83 | 1.34e-6a | 0.084 | 0.875 | Yes | No | Cannabis use, risk taking behaviour, bipolar disorder, schizophrenia, intelligence, years of education, cardio-metabolic traits |
| GTEx thyroid | 3.69 | 2.23e-4 | – | – | Yes | No | |||
| GTEx pituitary | 3.57 | 3.64e-4 | – | – | Yes | No | |||
| GTEx cerebellar hemisphere | 2.68 | 7.45e-3 | – | – | No | No |
Posterior probability PP3 = probability that two causal SNPs in linkage disequilibrium affect transcription and the phenotype separately; posterior probability PP4 = probability that the same causal SNP affects both transcription and the phenotype. CMC CommonMind Consortium, DLPFC dorsolateral prefrontal cortex, GTEx genotype tissue expression, YFS Young Finns Study, GP general practitioner.
aTranscriptome-wide significant results.
The top 250 genes for each phenotype available in Cmap are in Supplementary Table 3.
Candidate compounds for repurposing
The total number of examined signatures of compound perturbagens was 136,460, which corresponded to 29,679 unique compounds. A total of 76, 41 and 21 compounds showed a negative connectivity score and FDR p < 0.10 for TRD, depression with anxiety and depression with weight gain, respectively (Supplementary Table 4). They included four approved psychotropic medications (one antidepressant, two antipsychotics and one antiepileptic).
For TRD, the most common mechanisms of action of the identified compounds were modulation of cell survival–proliferation–differentiation (13%) and monoaminergic neurotransmission (7%). For anxious depression, modulation of cell survival–proliferation–differentiation was again the most common mechanism of action (15%), but interestingly inhibition of heat shock proteins (HSP) was the second one (10%). Candidate compounds for depression with weight gain included mostly modulators of metabolism (14%), such as peroxisome proliferator-activated receptor (PPAR) agonism. A relatively high proportion of the identified compounds had no reported mechanisms of action in the Cmap database (53%, 39% and 67% for TRD, depression with anxiety and depression with weight gain, respectively).
Ranking of candidate compounds for repurposing
For TRD, anxious and weight gain MDD, respectively, a total of 58, 35 and 18 compounds had at least one available expression signature passing quality control (Supplementary Table 4). For TRD we identified three compounds with permuted p < 0.05, which included zamifenacin (a muscarinic M3 and M5 receptor antagonist) and two molecules with unknown mechanism of action.
Four compounds showed permuted p < 0.05 for anxious depression: two were HSP90 inhibitors, one was a miR122 inhibitor and one was a modulator of cell cytoskeleton. Finally, for MDD with weight gain, only one signature corresponding to a molecule with unknown mechanism of action had significant permuted (BRD-K60636255). The top results for each group are summarized in Table 2.
Table 2.
Compounds showing an oppositive transcriptomic profile to the expression profiles imputed in TRD, anxious MDD and MDD with weight gain vs healthy controls.
| Phenotype | Compound | Signature ID | p value | MOA | Previous findings |
|---|---|---|---|---|---|
| TRD | Zamifenacin | ERG021_PC3_24H.BRD.K80451230.3.33 | 0.002 | Acetylcholine M3 and M5 receptor antagonist | Scopolamine has AD effects in TRD [46] but antagonisms on M2 seems to be responsible for the AD effects, though more data are needed to clarify the role of other muscarinic receptor subtypes [48] |
| BRD-K92033419 | CPC010_HT29_6H.BRD.K92033419.001.06.7.10 | 0.004 | Unknown | / | |
| BRD-K95664364 | DOS005_VCAP_6H.BRD.K95664364.001.01.2.4.79 | 0.039 | Unknown | / | |
| Dantrolene | CPC017_ASC_24H.BRD.K81272440.236.06.9.10 | 0.061 | Calcium channel blocker | It shows also inhibition of monoamine oxidase B and acetylcholinesterase, it has neuroprotective effects [57] | |
| Zardaverine | CPC016_HEPG2_6H.BRD.K37561857.001.06.4.10 | 0.065 | PDE4 and PDE3 inhibitor | PDE4 and PDE3 inhibition has AD-like effects [58] | |
| BRD-K09661167 | CPC012_ASC_24H.BRD.K09661167.001.06.1.10 | 0.065 | RAR-related orphan receptor gamma inhibitor, Bcl2-A1 Inhibitor, NOD1 inhibitor | / | |
| BRD-K39284479 | DOSBIO001_PC3_24H.BRD.K39284479.10.0519 | 0.070 | Unknown | / | |
| Zebularine | CPC006_HEPG2_6H.BRD.A01145011.001.01.4.11.1 | 0.073 | DNA methylation inhibitor | It reverses the behavioural deficits induced by chronic stress [59] | |
| Anxious MDD | Tanespimycin | MUC.CP007_P1A82_24H.G17 | 0.009 | HSP90 inhibitor | HSP90 inhibition increases lifespan and health in animal models [50]. FKBP51, acting via HSP90, is a pathogenetic mechanism in MDD [51] |
| SNX-2112 | REP.B022_HA1E_24H.I01 | 0.013 | HSP90 inhibitor | See tanespimycin | |
| Cytochalasin-d | CPC013_HCC515_6H.BRD.K25504083.001.03.1.10 | 0.016 | Tubulin inhibitor | It acts as inhibitor of the G-actin–cofilin interaction [60] that has a detrimental role in neurodegenerative disorders as it promotes neuronal cell death [55]. Cytochalasin-d increases the clearance of neuron-released α-synuclein by microglia [61] | |
| BRD-K85392418 | MUC.CP007_P1A82_24H.G17 | 0.026 | miR122 inhibitor | miR122 activates HSP70 and NF-κB pathway; NF-κB signalling activated by stress has a role in the pathogenesis of depressive behaviours [54] | |
| Azithromycin | REP.B024_HA1E_24H.B22 | 0.062 | Bacterial 50 S ribosomal subunit inhibitor | β-lactam antibiotics were found to promote the expression of the glutamate transporter GLT1 and have a neuroprotective role [62] | |
| MDD with weight gain | BRD-K60636255 | DOSBIO001_NPC_24H.BRD.K60636255.10.0166 | 0.025 | Unknown | Unknown |
Ranking was obtained by averaging the results of a KS test, Spearman correlation with all or with the most differentially expressed genes and Pearson correlation with all or with the most differentially expressed genes; 100 permutations were performed to assess the significance of the ranks (permuted p values < 0.10 are reported). MOA mechanisms of action, AD antidepressant.
Discussion
This study leveraged imputed gene expression profiles of TRD and associated depression subtypes to identify compounds showing opposite transcriptomic changes, which may have therapeutic effects in these groups as they show poor response to standard treatments.
Our TWAS findings for anxious MDD found that TMEM106B expression was downregulated in brain tissues, in line with the results of a recent TWAS of MDD [28]; in total, ten of our top findings for anxious MDD (p < 5e-5) were significantly dysregulated in the same study (see Supplementary Table 2 for a comparison of our results with the previous MDD TWAS [28]). In previous GWASs, SNPs in TMEM106B have been associated with MDD (e.g. the intronic SNP rs10950398 [7]), anxiety disorders (2 kb upstream variant rs3807866 [37]) and other traits (Table 1). The intronic rs5011432 SNP in TMEM106B has been associated with MDD in a previous GWAS [38], and it was the strongest eQTL for the TMEM106B TWAS association with anxious MDD (YFS BLOOD).
Interestingly, we found that ATP2A1 showed significant transcriptomic-wide association with MDD with weight gain, and this gene was implicated in GWASs of anthropometric traits such as BMI, type 2 diabetes, cognitive abilities, cannabis use, schizophrenia and bipolar disorder, therefore it probably represents a pleiotropic genetic factor regulating both the risk of metabolic and psychiatric disorders [39–43] (Table 1). This study was the first to determine the significance of ATP2A1 on a psychiatric trait using a TWAS to the best of our knowledge. The strongest eQTL for ATP2A1 was associated with cannabis use in a previous GWAS [41] (rs10499, GTEx pituitary).
Our screening of molecules for drug repurposing in TRD identified 76 compounds, one of which was shared with anxious MDD, two were antipsychotics, and six were identified also in a previous drug repurposing study for TRD [17], though the latter applied a different methodology and compared TRD vs antidepressant responders. However, the distribution of the mechanisms of action between the two studies was similar, with a prevalence of compounds modulating cell survival, proliferation, differentiation, monoaminergic neurotransmission and inflammation. One of the identified drugs, aprepitant, is a tachykinin antagonist and it failed phase III clinical trials for depression, but it has been suggested that insufficient doses were employed based on PET occupancy data [44]. Other identified compounds have known antidepressant-like or neuroprotective effects, such as vincamine, bergenin, zebularine, zardaverine, dantrolene, clenbuterol and stiripentol (Supplementary Table 4). The ranking of compounds combining the connectivity score, Spearman and Pearson correlation, identified three molecules with significant permuted p value, zamifenacin (a muscarinic M3 and M5 receptor antagonist) and two compounds with still unknown activity. Some antidepressants and atypical antipsychotics show M3 receptor antagonism, and scopolamine, a non-selective antimuscarinic drug, shows rapid antidepressant effects in TRD [45, 46]. However, the positive effects on mood are mostly attributed to antagonism on the M2 receptor subtype [47], but the knowledge about the role of M3 and M5 receptor subtypes is still limited [48]. Dantrolene (a calcium channel blocker) and zebularine (a modulator of DNA methylation) were very close to the significance threshold and have previous evidence of involvement in pathways relevant to MDD (Table 2).
For anxious depression, one of the 41 compounds identified in the screening phase was an approved antidepressant (agomelatine) and two were identified in the previously cited TRD repurposing study [17]. The modulation of cell survival/proliferation was again the most common known effect of the identified molecules, but another relatively common mechanism was the inhibition of HSP90, and two compounds with this effect were among the top ranking drugs in the following step of the analysis (SNX-2112 and tanespimycin). HSP expression is associated with stress response and HSP effects are complex: they can be pro-cell survival or pro-death depending on the molecules they interact with, on the tissue, on the cell type [49]. Interestingly, HSP90 inhibition (including by tanespimycin) increases lifespan and health in animal models [50]. The available studies suggest that although increased HSP levels may be beneficial for acute conditions, such increases can be detrimental for chronic conditions, as exemplified by acute and chronic heart conditions [51]. In regard to MDD, the co-chaperone FKBP51, acting via HSP90, is an established pathogenetic mechanism, through the induction of glucocorticoid resistance and a poor stress coping phenotype [51]. BRD-K85392418 was another of the identified compounds for anxious MDD and it was reported to act as miR122 inhibitor. This micro-RNA was found to be altered in MDD [52] and it increases the activity of HSP70 and the NF-κB pathway, which have protective effects during acute stress conditions [53], and also in this case their activity has likely detrimental effects in the long term as they upregulate proinflammatory cytokine expression and cause impaired neurogenesis [54]. The last significant compound for repurposing in anxious MDD was cytochalasin-d, which acts as inhibitor of the G-actin–cofilin interaction, a process that has been implicated in neurodegenerative disorders as it promotes neuronal cell death [55].
Among the 20 molecules identified in the screening step for MDD with weight gain, one was also significant in our previous study of drug repurposing in TRD [17]. This was troglitazone, a proliferator-activated receptor (PPAR) agonist which was among the most promising results since PPAR agonists showed antidepressant effects in four open-label studies and in three out of four RCTs in patients with major depression [56]. However, troglitazone did not show significant permuted p value in the drug ranking using the connectivity score, Spearman and Pearson correlation, and this analysis identified only one molecule with unknown mechanism of action (Table 2).
Our results should be interpreted in the light of the limitations of this study. The sample sizes included in the TWAS were relatively limited, as they were constrained to the number of cases available for each MDD subtype and TRD, and to the number of participants of European ancestry. The weights used in the TWAS were calculated based on existing eQTL datasets, which were often obtained in small samples, particularly for brain tissues in GTEx, though PsychENCODE is larger. On the other hand, the use of imputed gene expression from GWAS is much more feasible on a large scale than the direct measurement of gene expression levels. The tested tissues were selected based on the previous literature, however, the inclusion of 21 distinct SNP-weight sets from different tissues might capture noncausal genes [28]. The restriction of the analysis to brain tissues could be an alternative option, particularly on large datasets; however, Cmap expression signatures were mostly obtained in non-neural cell lines. Our TWAS approach solely assessed the cis-genetic component of gene expression, while it could not capture trans-eQTL effects. Finally, the approach used for drug repurposing was based on the dissimilarity of gene expression profiles in the TWAS and Cmap compound-induced expression profiles; however, the latter were determined in vitro and under heterogenous experimental conditions (different cell lines, different drug dose and time of exposure), and we did not know which ones were more similar to in vivo gene expression changes.
In conclusion, our study identified two genes showing transcriptomic alterations in depression subtypes associated with TRD: TMEM106B in anxious MDD and ATP2A1 in MDD with weight gain; both genes had previous evidence of involvement in psychiatric traits and also metabolic traits in the case of ATP2A1. Our drug repurposing analyses suggested that the inhibition of HSP90 and the modulation of cell cytoskeleton may represent alternative strategies for the treatment of anxious MDD, while drugs modulating metabolism may be beneficial in MDD with weight gain. For the treatment of TRD, potentially useful pharmacological mechanisms included the antagonism of muscarinic receptors, the modulation of DNA methylation and calcium channels.
Supplementary information
Supplementary Methods-Table S1 - Figures.
Acknowledgements
The authors acknowledge use of the research computing facility at King’s College London, Rosalind (https://rosalind.kcl.ac.uk), which is delivered in partnership with the NIHR Biomedical Research Centres at South London & Maudsley and Guy’s & St. Thomas’ NHS Foundation Trusts, and part-funded by capital equipment grants from the Maudsley Charity (award 980) and Guy’s & St. Thomas’ Charity (TR130505). This research has been conducted using the UK Biobank Resource under Application Number 56514 “Stratification of health outcomes in mood disorders”. UK Biobank is an open access resource for bona fide researchers (https://www.ukbiobank.ac.uk/enable-your-research).
Author contributions
C.F. designed the study and performed the analyses using a pipeline developed by O.P. (available at: https://opain.github.io/MDD-TWAS/MDD_TWAS.html#222_twas_analysis). S.P.H. contributed to the creation of the depression phenotypes in UK Biobank, A.S. and C.M.L. supervised the work and revised the final draft of the manuscript.
Funding
This research was supported by the UK Medical Research Council (Saskia Hagenaars: MR/S0151132) and by the National Institute for Health Research (NIHR) Biomedical Research Centre at South London and Maudsley NHS Foundation Trust and King’s College London. The views expressed are those of the authors and not necessarily those of the NHS, the NIHR or the Department of Health and Social Care. Chiara Fabbri was supported by Fondazione Umberto Veronesi (https://www.fondazioneveronesi.it).
Competing interests
C.M.L. is a member of the Scientific Advisory Board of Myriad Neurosciences. A.S. is or has been consultant/speaker for: Abbott, Abbvie, Angelini, Astra Zeneca, Clinical Data, Boheringer, Bristol Myers Squibb, Eli Lilly, GlaxoSmithKline, Innovapharma, Italfarmaco, Janssen, Lundbeck, Naurex, Pfizer, Polifarma, Sanofi, Servier. The other authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
Supplementary information
The online version contains supplementary material available at 10.1038/s41386-021-01059-6.
References
- 1.World Health Organization. Depression and Other Common Mental Disorders: Global Health Estimates. 2017. https://apps.who.int/iris/bitstream/handle/10665/254610/WHO-MSD-MER-2017.2-eng.pdf;jsessionid=2A52412EC4FD7E2707B9228C4ED61604?sequence=1. Accessed 20 July 2020.
- 2.Cipriani A, Furukawa TA, Salanti G, Chaimani A, Atkinson LZ, Ogawa Y, et al. Comparative efficacy and acceptability of 21 antidepressant drugs for the acute treatment of adults with major depressive disorder: a systematic review and network meta-analysis. Lancet. 2018;391:1357–66. doi: 10.1016/S0140-6736(17)32802-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rush AJ, Trivedi MH, Wisniewski SR, Nierenberg AA, Stewart JW, Warden D, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–17. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- 4.Rizvi SJ, Grima E, Tan M, Rotzinger S, Lin P, Mcintyre RS, et al. Treatment-resistant depression in primary care across Canada. Can J Psychiatry. 2014;59:349–57. doi: 10.1177/070674371405900702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li G, Fife D, Wang G, Sheehan JJ, Bodén R, Brandt L, et al. All-cause mortality in patients with treatment-resistant depression: a cohort study in the US population. Ann Gen Psychiatry. 2019;18:23. doi: 10.1186/s12991-019-0248-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Fekadu A, Rane LJ, Wooderson SC, Markopoulou K, Poon L, Cleare AJ. Prediction of longer-term outcome of treatment-resistant depression in tertiary care. Br J Psychiatry. 2012;201:369–75. doi: 10.1192/bjp.bp.111.102665. [DOI] [PubMed] [Google Scholar]
- 7.Wray NR, Ripke S, Mattheisen M, Trzaskowski M, Byrne EM, Abdellaoui A, et al. Genome-wide association analyses identify 44 risk variants and refine the genetic architecture of major depression. Nat Genet. 2018;50:668–81. doi: 10.1038/s41588-018-0090-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Pain O, Hodgson K, Trubetskoy V, Ripke S, Marshe V, Adams M, et al. Antidepressant response in major depressive disorder: a genome-wide association study. 2020. Preprint at https://www.medrxiv.org/content/10.1101/2020.12.11.20245035v1.
- 9.Fabbri C, Kasper S, Kautzky A, Zohar J, Montgomery S, Albani D, et al. A polygenic predictor of treatment-resistant depression using whole exome sequencing and genome-wide genotyping. Transl Psychiatry. 2020;10:50. doi: 10.1038/s41398-020-0738-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milaneschi Y, Lamers F, Peyrot WJ, Baune BT, Breen G, Dehghan A, et al. Genetic association of major depression with atypical features and obesity-related immunometabolic dysregulations. JAMA Psychiatry. 2017;74:1214–25. doi: 10.1001/jamapsychiatry.2017.3016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bartova L, Dold M, Kautzky A, Fabbri C, Spies M, Serretti A, et al. Results of the European Group for the Study of Resistant Depression (GSRD) - basis for further research and clinical practice. World J Biol Psychiatry. 2019;20:427–48. doi: 10.1080/15622975.2019.1635270. [DOI] [PubMed] [Google Scholar]
- 12.Wu Z, Chen J, Yuan C, Hong W, Peng D, Zhang C, et al. Difference in remission in a Chinese population with anxious versus nonanxious treatment-resistant depression: a report of OPERATION study. J Affect Disord. 2013;150:834–9. doi: 10.1016/j.jad.2013.03.012. [DOI] [PubMed] [Google Scholar]
- 13.Matza LS, Revicki DA, Davidson JR, Stewart JW. Depression with atypical features in the national comorbidity survey: classification, description, and consequences. Arch Gen Psychiatry. 2003;60:817. doi: 10.1001/archpsyc.60.8.817. [DOI] [PubMed] [Google Scholar]
- 14.Nelson MR, Tipney H, Painter JL, Shen J, Nicoletti P, Shen Y, et al. The support of human genetic evidence for approved drug indications. Nat Genet. 2015;47:856–60. doi: 10.1038/ng.3314. [DOI] [PubMed] [Google Scholar]
- 15.Gaspar HA, Gerring Z, Hübel C, Major Depressive Disorder Working Group of the Psychiatric Genomics Consortium. Middeldorp CM, Derks EM, et al. Using genetic drug-target networks to develop new drug hypotheses for major depressive disorder. Transl Psychiatry. 2019;9:117. doi: 10.1038/s41398-019-0451-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.So H-C, Chau CK-L, Chiu W-T, Ho K-S, Lo C-P, Yim SH-Y, et al. Analysis of genome-wide association data highlights candidates for drug repositioning in psychiatry. Nat Neurosci. 2017;20:1342–9. doi: 10.1038/nn.4618. [DOI] [PubMed] [Google Scholar]
- 17.Fabbri C, Kasper S, Zohar J, Souery D, Montgomery S, Albani D, et al. Drug repositioning for treatment-resistant depression: hypotheses from a pharmacogenomic study. Prog Neuropsychopharmacol Biol Psychiatry. 2021;104:110050. doi: 10.1016/j.pnpbp.2020.110050. [DOI] [PubMed] [Google Scholar]
- 18.Pritchard J-LE, O’Mara TA, Glubb DM. Enhancing the promise of drug repositioning through genetics. Front Pharmacol. 2017;8:896. doi: 10.3389/fphar.2017.00896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.So H-C, Chau CK-L, Lau A, Wong S-Y, Zhao K. Translating GWAS findings into therapies for depression and anxiety disorders: gene-set analyses reveal enrichment of psychiatric drug classes and implications for drug repositioning. Psychol Med. 2019;49:2692–708. doi: 10.1017/S0033291718003641. [DOI] [PubMed] [Google Scholar]
- 20.Fabbri C, Hagenaars SP, John C, Williams AT, Shrine N, Moles L, et al. Genetic and clinical characteristics of treatment-resistant depression using primary care records in two UK cohorts. Mol Psychiatry. 2021. 10.1038/s41380-021-01062-9 [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 21.Davis KAS, Coleman JRI, Adams M, Allen N, Breen G, Cullen B, et al. Mental health in UK Biobank - development, implementation and results from an online questionnaire completed by 157 366 participants: a reanalysis. BJPsych Open. 2020;6:e18. doi: 10.1192/bjo.2019.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Badini I, Coleman JRI, Hagenaars SP, Hotopf M, Breen G, Lewis CM, et al. Depression with atypical neurovegetative symptoms shares genetic predisposition with immuno-metabolic traits and alcohol consumption. Psychol Med. 2020:1–11. [Epub ahead of print]. [DOI] [PMC free article] [PubMed]
- 23.American Psychiatric Association. The diagnostic and statistical manual of mental disorders (DSM–5) 5th ed. Washington, DC: American Psychiatric Publishing; 2013. [Google Scholar]
- 24.Shorter E. The doctrine of the two depressions in historical perspective. Acta Psychiatr Scand Suppl. 2007;433:5–13. [DOI] [PMC free article] [PubMed]
- 25.Dahl A, Cai N, Ko A, Laakso M, Pajukanta P, Flint J, et al. Reverse GWAS: using genetics to identify and model phenotypic subtypes. PLoS Genet. 2019;15:e1008009. doi: 10.1371/journal.pgen.1008009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Bycroft C, Freeman C, Petkova D, Band G, Elliott LT, Sharp K, et al. The UK Biobank resource with deep phenotyping and genomic data. Nature. 2018;562:203–9. doi: 10.1038/s41586-018-0579-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Milaneschi Y, Simmons WK, van Rossum EFC, Penninx BW. Depression and obesity: evidence of shared biological mechanisms. Mol Psychiatry. 2019;24:18–33. doi: 10.1038/s41380-018-0017-5. [DOI] [PubMed] [Google Scholar]
- 28.Dall’Aglio L, Lewis CM, Pain O. Delineating the genetic component of gene expression in major depression. Biol Psychiatry. 2021;89:627–36. doi: 10.1016/j.biopsych.2020.09.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Qi T, Wu Y, Zeng J, Zhang F, Xue A, Jiang L, et al. Identifying gene targets for brain-related traits using transcriptomic and methylomic data from blood. Nat Commun. 2018;9:2282. doi: 10.1038/s41467-018-04558-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zheng X, Zhang X, Wang G, Hao H. Treat the brain and treat the periphery: toward a holistic approach to major depressive disorder. Drug Discov Today. 2015;20:562–8. doi: 10.1016/j.drudis.2015.03.015. [DOI] [PubMed] [Google Scholar]
- 31.Broad Institute. Connectivity Map. 2020. https://clue.io. Accessed 15 December 2020.
- 32.Gusev A, Ko A, Shi H, Bhatia G, Chung W, Penninx BWJH, et al. Integrative approaches for large-scale transcriptome-wide association studies. Nat Genet. 2016;48:245–52. doi: 10.1038/ng.3506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gusev A, Mancuso N, Won H, Kousi M, Finucane HK, Reshef Y, et al. Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nat Genet. 2018;50:538–48. doi: 10.1038/s41588-018-0092-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.GTEx Consortium. Human genomics. The Genotype-Tissue Expression (GTEx) pilot analysis: multitissue gene regulation in humans. Science. 2015;348:648–60. doi: 10.1126/science.1262110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Pain O, Pocklington AJ, Holmans PA, Bray NJ, O’Brien HE, Hall LS, et al. Novel insight into the etiology of autism spectrum disorder gained by integrating expression data with genome-wide association statistics. Biol Psychiatry. 2019;86:265–73. doi: 10.1016/j.biopsych.2019.04.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Subramanian A, Narayan R, Corsello SM, Peck DD, Natoli TE, Lu X, et al. A next generation connectivity map: L1000 platform and the first 1,000,000 profiles. Cell. 2017;171:1437–52.e17. doi: 10.1016/j.cell.2017.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Purves KL, Coleman JRI, Meier SM, Rayner C, Davis KAS, Cheesman R, et al. A major role for common genetic variation in anxiety disorders. Mol Psychiatry. 2020;25:3292–303. doi: 10.1038/s41380-019-0559-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Howard DM, Adams MJ, Shirali M, Clarke T-K, Marioni RE, Davies G, et al. Genome-wide association study of depression phenotypes in UK Biobank identifies variants in excitatory synaptic pathways. Nat Commun. 2018;9:1470. doi: 10.1038/s41467-018-03819-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Winkler TW, Justice AE, Graff M, Barata L, Feitosa MF, Chu S, et al. The influence of age and sex on genetic associations with adult body size and shape: a large-scale genome-wide interaction study. PLoS Genet. 2015;11:e1005378. doi: 10.1371/journal.pgen.1005378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Vujkovic M, Keaton JM, Lynch JA, Miller DR, Zhou J, Tcheandjieu C, et al. Discovery of 318 new risk loci for type 2 diabetes and related vascular outcomes among 1.4 million participants in a multi-ancestry meta-analysis. Nat Genet. 2020;52:680–91. doi: 10.1038/s41588-020-0637-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Pasman JA, Verweij KJH, Gerring Z, Stringer S, Sanchez-Roige S, Treur JL, et al. GWAS of lifetime cannabis use reveals new risk loci, genetic overlap with psychiatric traits, and a causal influence of schizophrenia. Nat Neurosci. 2018;21:1161–70. doi: 10.1038/s41593-018-0206-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lam M, Hill WD, Trampush JW, Yu J, Knowles E, Davies G, et al. Pleiotropic meta-analysis of cognition, education, and schizophrenia differentiates roles of early neurodevelopmental and adult synaptic pathways. Am J Hum Genet. 2019;105:334–50. doi: 10.1016/j.ajhg.2019.06.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Pisanu C, Williams MJ, Ciuculete DM, Olivo G, Del Zompo M, Squassina A, et al. Evidence that genes involved in hedgehog signaling are associated with both bipolar disorder and high BMI. Transl Psychiatry. 2019;9:315. doi: 10.1038/s41398-019-0652-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Rupniak NMJ, Kramer MS. NK1 receptor antagonists for depression: Why a validated concept was abandoned. J Affect Disord. 2017;223:121–5. doi: 10.1016/j.jad.2017.07.042. [DOI] [PubMed] [Google Scholar]
- 45.Tran Y-H, Schuiling-Veninga CCM, Bergman JEH, Groen H, Wilffert B. Impact of muscarinic M3 receptor antagonism on the risk of type 2 diabetes in antidepressant-treated patients: a case-controlled study. CNS Drugs. 2017;31:483–93. doi: 10.1007/s40263-017-0436-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ionescu DF, Rosenbaum JF, Alpert JE. Pharmacological approaches to the challenge of treatment-resistant depression. Dialogues Clin Neurosci. 2015;17:111–26. doi: 10.31887/DCNS.2015.17.2/dionescu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Jeon WJ, Dean B, Scarr E, Gibbons A. The role of muscarinic receptors in the pathophysiology of mood disorders: a potential novel treatment? Curr Neuropharmacol. 2015;13:739–49. doi: 10.2174/1570159X13666150612230045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Dulawa SC, Janowsky DS. Cholinergic regulation of mood: from basic and clinical studies to emerging therapeutics. Mol Psychiatry. 2019;24:694–709. doi: 10.1038/s41380-018-0219-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Campanella C, Pace A, Caruso Bavisotto C, Marzullo P, Marino Gammazza A, Buscemi S, et al. Heat shock proteins in Alzheimer’s disease: role and targeting. Int J Mol Sci. 2018;19:2603. doi: 10.3390/ijms19092603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Janssens GE, Lin X-X, Millan-Ariño L, Kavšek A, Sen I, Seinstra RI, et al. Transcriptomics-based screening identifies pharmacological inhibition of Hsp90 as a means to defer aging. Cell Rep. 2019;27:467–80.e6. doi: 10.1016/j.celrep.2019.03.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Edkins AL, Price JT, Pockley AG, Blatch GL. Heat shock proteins as modulators and therapeutic targets of chronic disease: an integrated perspective. Philos Trans R Soc Lond B. 2018;373:20160521. doi: 10.1098/rstb.2016.0521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Garbett KA, Vereczkei A, Kálmán S, Brown JA, Taylor WD, Faludi G, et al. Coordinated messenger RNA/microRNA changes in fibroblasts of patients with major depression. Biol Psychiatry. 2015;77:256–65. doi: 10.1016/j.biopsych.2014.05.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Guo D, Ma J, Li T, Yan L. Up-regulation of miR-122 protects against neuronal cell death in ischemic stroke through the heat shock protein 70-dependent NF-κB pathway by targeting FOXO3. Exp Cell Res. 2018;369:34–42. doi: 10.1016/j.yexcr.2018.04.027. [DOI] [PubMed] [Google Scholar]
- 54.Koo JW, Russo SJ, Ferguson D, Nestler EJ, Duman RS. Nuclear factor-kappaB is a critical mediator of stress-impaired neurogenesis and depressive behavior. Proc Natl Acad Sci USA. 2010;107:2669–74. doi: 10.1073/pnas.0910658107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hoffmann L, Rust MB, Culmsee C. Actin(g) on mitochondria - a role for cofilin1 in neuronal cell death pathways. Biol Chem. 2019;400:1089–97. doi: 10.1515/hsz-2019-0120. [DOI] [PubMed] [Google Scholar]
- 56.Colle R, de Larminat D, Rotenberg S, Hozer F, Hardy P, Verstuyft C, et al. PPAR-γ agonists for the treatment of major depression: a review. Pharmacopsychiatry. 2017;50:49–55. doi: 10.1055/s-0042-120120. [DOI] [PubMed] [Google Scholar]
- 57.Bolognino I, Giangregorio N, Pisani L, de Candia M, Purgatorio R, Tonazzi A, et al. A prospective repurposing of dantrolene as a multitarget agent for Alzheimer’s disease. Molecules. 2019;24:4298. doi: 10.3390/molecules24234298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Murthy VS, Mangot AG. Psychiatric aspects of phosphodiesterases: an overview. Indian J Pharmacol. 2015;47:594–9. doi: 10.4103/0253-7613.169593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Vialou V, Feng J, Robison AJ, Nestler EJ. Epigenetic mechanisms of depression and antidepressant action. Annu Rev Pharmacol Toxicol. 2013;53:59–87. doi: 10.1146/annurev-pharmtox-010611-134540. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Shoji K, Ohashi K, Sampei K, Oikawa M, Mizuno K. Cytochalasin D acts as an inhibitor of the actin-cofilin interaction. Biochem Biophys Res Commun. 2012;424:52–7. doi: 10.1016/j.bbrc.2012.06.063. [DOI] [PubMed] [Google Scholar]
- 61.Choi I, Zhang Y, Seegobin SP, Pruvost M, Wang Q, Purtell K, et al. Microglia clear neuron-released α-synuclein via selective autophagy and prevent neurodegeneration. Nat Commun. 2020;11:1386. doi: 10.1038/s41467-020-15119-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Obregon D, Parker-Athill EC, Tan J, Murphy T. Psychotropic effects of antimicrobials and immune modulation by psychotropics: implications for neuroimmune disorders. Neuropsychiatry. 2012;2:331–43. doi: 10.2217/npy.12.41. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Methods-Table S1 - Figures.