

Graphical abstract

Keywords: Pulsed ultrasound, Continuous reactive crystallization, MSMPR, Lithium carbonate
Highlights
-
•
Pulsed ultrasound was used to facilitate continuous reactive crystallization.
-
•
Experimental analyses of the stirred tank MSMPR cascade were performed.
-
•
A population balance model was developed and applied.
-
•
A three-stage MSMPR continuous reactive crystallization was successfully designed.
Abstract
In this work, pulsed ultrasound was used to facilitate steady-state reactive crystallization and increase the final yield and productivity of lithium carbonate in continuously operated single and multistage mixed suspension mixed product removal (MSMPR) crystallizers. Experimental analyses of the stirred tank MSMPR cascade were performed to investigate the effects of ultrasound field, residence time and temperature which contributed to the steady-state yield, crystal size distribution and crystal morphology. The results show that pulsed ultrasound can not only significantly enhance the reaction rate, but also help to improve the particle size distribution and the crystal habit. Subsequently, a population balance model was developed and applied to estimate the final yield of the continuous process of the lithium bicarbonate thermal decomposition reaction coupling lithium carbonate crystallization. The consistency of the final yield between the experiments and the simulations proved the reliability of the established model. Through the experimental and simulation analyses, it is demonstrated that the use of pulsed ultrasound, higher final stage temperature, MSMPR cascade design and appropriate residence time help to achieve higher yield and productivity. Furtherly, based on the conclusion drawn, pulsed ultrasound enhanced three-stage MSMPR cascaded lithium carbonate continuous crystallization processes were designed, and the maximum productivity of 44.0 g/h was obtained experimentally.
1. Introduction
Crystallization, a typical solid–liquid separation unit operation that can be employed to purify a great diversity of chemical compounds, is widely applied in the production of solid-state intermediates or final products [1], [2]. Traditionally, for a simplification of crystallization equipment and manual operation, chemical enterprises predominantly conducted batch methods in industrial crystallization procedures. However, the existence of the inevitable batch-to-batch variations lead to fluctuations in product quality, eventually resulting in declination of the reliability of the crystallization process, and other circumstances, such as the existence of dead-flow zones in the crystallizer, would make it worse [3]. In comparison with batch crystallization, continuous crystallization has the advantages of taking a short development period to reach pilot-scale and commercial production, improving product robustness, increasing product consistency, and realizing a superior control of product attributes. Hence, it attracted an increasing attention in recent years [4].
Mixed-suspension mixed-product-removal (MSMPR) crystallizers are considered as the most commonly used continuous crystallizers because they are easier to convert from batch-to-batch operation and lower maintenance cost [5], [6]. More importantly, single stage and multistage MSMPR crystallizers can provide sufficient time for nucleation and growth, which is appropriate for processes with slower conversion, such as reactive crystallization [7]. In recent years, researchers have tried to investigate the relationship in operating variables and particle size distribution, purity and yield in the continuous reaction crystallization process through experiments or simulations. Quon et al. [8] described a two-stage MSMPR continuous reactive crystallization. Since the reaction rate, solubility and production efficiency were all affected by the temperature, the researchers controlled the yield and purity of the product by means of a reasonable selection of the temperature of the two-stage MSMPR cascade, and mathematical descriptions of the continuous crystallization process were established. McDonald et al. [9], [10] have done works in continuous reactive crystallization. They developed a model for the simulation of continuous reactive crystallization of β-lactam antibiotics catalyzed by penicillin G acylase. By calculating a Pareto optimal surface for productivity, fractional yield and conversion, the 6-APA conversion was increased up to 87%, both productivity and fractional yield decrease. Different from continuous cooling or anti-solvent crystallization, continuous reactive crystallization relies on the reaction to provide the driving force for crystallization, which will lead to rapid accumulation of local supersaturation. This circumstance will not only lead to the product quality decline, but also cause the issues of fouling, encrustation and blockages. Implementing process intensification strategies to improve mixing in the crystallizer are the best choices to avoid uneven distribution of supersaturation.
Ultrasound is a feasible process intensification technique which can be used to improve mixing. It is generally believed that ultrasound irradiation of a solution results in pressure waves in the liquid which cause compression and rarefaction of liquid molecules, and eventually mixing is improved [11]. When a large amount of acoustic energy is introduced into a solvent mediated crystallization process, cavitation bubbles, which is considered as the most important effect of ultrasound, are generated in the liquid, which have internal high temperatures and pressures. When the cavitation bubbles implode, they create jets of fluid and shockwaves that yield stronger implosions, which dominates and improves local mixing much better. Cavitation could also induce heterogeneous primary nucleation due to provide new crystal nucleation sites causing by phase interface under certain conditions. Although the exact effect of cavitation may be multifactor and difficult to determine, there is consensus that it could use to intensify chemical processes including reaction and crystallization [12]. Several papers have reported on the effects of ultrasound on different classes of nucleation and growth. It has been proved that ultrasound could decrease the induction time, nucleation at lower supersaturation or a reduction in the Metastable Zone Width (MZW) for primary homogeneous and heterogeneous nucleation [13], [14], [15], [16]. The cavitation bubbles implode can lead to the breakage of existing crystals, which promote subsequent induction of secondary nucleation [17], [18]. In addition, ultrasound is also known to reduce particle agglomeration and clogging in tube, which could beneficial to scale up and industrial application [12], [19]. In spite of these promising advantages, the extra energy requirement of ultrasound often inhibits scale up and industrial application [20], [21]. Pulsed ultrasound, which operated as periodically turning the sound field on and off, is considered as an interesting approach to solving this restriction [22], [23]. Moreover, an enhanced crystallization effect could be achieved by using pulsed ultrasound in previous studies [24], [25]. This low energy consumption and high process efficiency method provide the possibility of using ultrasonic fields in continuous crystallization. Nevertheless, there are few reports on the application of pulsed ultrasound to continuous reactive crystallization.
Herein this work focuses mainly on developing an ultrasound enhanced continuous reactive crystallization technique adjusted to the production of lithium carbonate (Li2CO3, LCB). The workflow of this work is available in Fig. 1. LCB, one of the most important lithium compounds, has been widely applied to lithium-ion batteries [26], medicines [27], ceramics [28], etc [29]. This article deals with the development of the crystallization step in manufacturing, from which the LCB is isolated by continuous reactive crystallization from the lithium bicarbonate (LiHCO3, LBCB) thermal decomposition. In order to improve the efficiency and product quality of continuous manufacturing, pulsed ultrasound is used in different stages of MSMPR crystallizers. Additionally, the process parameter dependence of the yield, the product quality, and also the crystal morphology had to be examined in detail. A population balance equation based on coupled kinetics functions was established to verify and predict the final yield at different operation conditions. Finally, a three-stage MSMPR crystallizer was designed and optimized by simulation, which was also verified experimentally.
Fig. 1.

The workflow of this work.
2. Materials and methods
2.1. Materials
99% pure LCB was purchased from Tianqi Lithium, China. Distilled deionized water (conductivity b 0.5 μS cm−1) prepared in our laboratory was used throughout the entire reaction and crystallization process. High purity carbon dioxide (CO2, >99% pure), purchased from Tianjin Liufang Industrial Gas Distribution Co., Ltd, China, was used as the reactant to produce LBCB.
2.2. Experimental setup
Multistage continuous crystallization experiments were carried out in a one to three stage continuous crystallizer under 101.325 kPa. Each was operated as a mixed suspension mixed product removal (MSMPR) crystallizer. The experiment system consisted of one jacketed glass feed tank (3 L) and three jacketed glass crystallizers (500 mL each) with independent mechanical stirring and temperature control (Julabo, CF41). Series insulated polyethylene tubing, wrapping with flexible polyurethane foam for realizing temperature stability during suspension transportation, were used to connect the feed tank and crystallizers. An approximately 15 cm height drop was adopted among crystallizers to avoid blockage in tube. An ultrasonic transducer (JY92-IIDN, Scientz, China) was bolted to the crystallizer. The frequency was maintained to be constant at 20 kHz. A schematic drawing of the complete setup is given in Fig. 2.
Fig. 2.

Schematic image of a three-stage continuous reactive crystallization experiment setup.
The reaction probed in this study was the thermal decomposition reaction of LBCB to LCB shown in Eq.(1). The mechanism and kinetics of this reaction were reported in Fig. S1 and Table S1 [30], [31].
| (1) |
For all continuous experiments, the pure solution of LBCB in the feed tank was prepared by the reaction of LCB solution and CO2 (gas). CO2 (gas) was continuously injected into LCB suspension solution over 24 h with stirring to completely convert LCB into LBCB at room temperature. When the preparation steps of LBCB solution were completed, the carbon dioxide gas valve was switched off. Peristaltic pumps (Longer Pump, China) were used for solution and slurry transfer. Thermal decomposition reaction and nucleation were induced spontaneously by high temperature and stirring in each stage. In order to prevent the peristaltic pump pipeline from being affected by the disturbance of the internal flow field to reach a steady state, the peristaltic pump inlet pipeline was fixed at about 4/5 of the bottom of the crystallizer to ensure the stability and repeatability of each experiment. The slurry removed from the last crystallizer was transferred to a volumetric flask (500 mL) maintained at a controlled temperature.
2.3. Analyses of crystallization
2.3.1. Experiment procedure of batch crystallization
A batch operated reactive crystallization of LCB was carried out in a 500 mL crystallizer. A clear solution of LBCB at room temperature was heated to 363.15 K and maintained at this temperature for 2 h. In order to better understand the reactive crystallization behavior affected by thermal decomposition of LBCB, particle video microscope (PVM, Mettler-Toledo, Switzerland) was used to observe the batch operated reactive crystallization process online. The solid phase sample was taken at the end of the process.
2.3.2. Experiment procedure of MSMPR crystallization
Reactive crystallization experiments were carried out by using the continuous crystallization apparatus described above. For each initial configuration, a mass ratio of 3.2 g LCB/100 g H2O was weighed and added to a tank at room temperature. Then, high purity CO2(gas) was continuously injected into solution. Under the conditions of continuous reaction and 500 rpm mechanical stirring, the clarification of the suspension means that LCB is completely converted to LBCB. The extra 24 h is necessary to ensure that small particles in the solution that cannot be detected by the naked eye can be completely dissolved and reacted. Hence, pure LBCB solution was prepared and used as the feed solution. The suspension was agitated with 400 rpm speed to enhance the degree of micromixing and avoid sedimentation of the crystals. The jacket temperature was set at 333.15 K, 343.15 K or 363.15 K at each stage that did not change during the whole process. For all experiments setup, the flow rate for these experiments was set to 8.33–33.33 mL/min, so that the corresponding residence time (RT) of the solution in each stage was 15 min to 60 min. Experiments lasted for 12 RTs, as this can provide enough time for the system to reach a steady-state condition. Therefore, the duration of experiments was 3, 6, or 12 h depending on the RT. In order to increase the yield of the process, a continuous reactive crystallization experiment with ultrasonic intensification was carried out. Ultrasonic field is used to intensify the reaction and crystallization process. In experiments with ultrasound, it was noticed that the heat generation due to and the cavitation led to evaporation caused by local overheating within the crystallizers. Therefore, a low-intensity pulsed ultrasound was used to tackle this problem, and yields at different operation conditions were compared. The pulse-on time was kept constant at 1s, and the pulse-off time was set to 5s.
All experimental designs are shown in Table 1, and were all repeated three times. The design of the experiments was guided by the following considerations. First, all experimental designs have experienced 363.15 K, so that the relatively fast reaction and crystallization rate at high temperature can provide a higher steady-state yield for the process. Second, under different experimental designs, pulsed ultrasound was added to the first or second stage to compare the enhancement degree of the reaction and crystallization in the continuous process by the ultrasonic field. Third, the total experimental time was set as 3 h, 6 h, and 12 h respectively (corresponding RT to 0.25 h, 0.5 h, and 1 h) to compare the influence of operating conditions on product quality and steady-state yield at the same time. Fourth, the concentration of LBCB solution, volume of crystallizers and stirring speed are not considered in the current study.
Table 1.
Experimental Conditions of Continuous Reactive crystallization.
| EXP. ID | Stage number | Ultrasonic field | Temperature (K) | RT (h) | Overall time of experiments (h) |
|---|---|---|---|---|---|
| 1 | one | Without | 363.15 | 0.5 | 6 |
| 2 | one | With | 363.15 | 0.5 | 6 |
| 3 | one | With | 363.15 | 0.25 | 3 |
| 4 | one | Without | 363.15 | 1 | 12 |
| 5 | two | Without | 333.15–363.15 | 0.5 | 6 |
| 6 | two | Without | 363.15–333.15 | 0.5 | 6 |
| 7 | two | With | 333.15–363.15 | 0.5 | 6 |
| 8 | two | With | 363.15–333.15 | 0.5 | 6 |
| 9 | two | With | 333.15–363.15 | 1 | 12 |
| 10 | two | Without | 333.15–363.15 | 1 | 12 |
| 11 | two | Without | 363.15–353.15 | 1 | 12 |
| 12 | two | Without | 353.15–363.15 | 1 | 12 |
2.3.3. Liquid and solid-state characterizations
In the continuous crystallization process, the quality of LCB inside the crystallizer varies with time and is determined by offline sampling by the previous method [32]. This is applied to determine whether the process has achieved a steady state inside the crystallizer. At approximately every 30 min, about 4 mL of solution in crystallizer was withdrawn by the pre-heated and pre-weight injector with a water syringe filter (0.22 μm) and moved into a pre-weighted glass beaker. The weight difference can be used to determine the LCB weight at a certain time by the balance (type AB204-N, Mettler-Toledo, Switzerland) with a precision of ±0.0001 g. Then the LCB weight obtained by offline sampling can be used to calculate the product yield at certain experimental moment.
Samples used for crystallinity, size, and crystal habit analysis were collected at the outlet after immediate vacuum filtration (0.22 μm filter) so as to avoid any growth after the crystallizer. Twelve samples obtained from different continuous reactive crystallization were analyzed. Samples were air dried for at least 36 h. Powder X-ray diffractometer was employed to determine the crystallinity of LCB samples. The X-ray diffraction measurement was performed on Rigaku D/max-2500 with Cu Kα radiation (1.5405 Å). The powder X-ray diffraction (PXRD, Rigaku, Japan) pattern was collected over the diffraction angle range from 2 to 40° at a scanning rate of 8°/min. Particle size measurements were performed in a Malvern Mastersizer 3000 (Malvern Panalytical, England) Hydro MV laser diffractometer. The dispersant used was ethanol. Before sample analysis, the background (ethanol – 99.9%) was analyzed. LCB was then added to the dispersant until the desired obscuration was reached. Each sample was measured 5 times as the mean of these values is presented. Among samples, at least 3 cleaning cycles were conducted. The volumetric distribution values Dv10, Dv50, and Dv90 were applied to characterize the crystal size distribution (CSD) of the products. The CSD plots represent volume-based distributions, where crystal size intervals were plotted as a function of the volume fraction (%). The volume fraction (%) of crystal size interval represents the volume percent of these crystals relative to the total crystal fraction. The crystal habit of the products was monitored by using an optical inverted microscope (Eclipse E200, Nikon). The analyses of product quality and total yield in this work all comes from the products of the last stage crystallizer of each experiment. The yield at steady state was calculated based on the total dissolved and reacted weight of LCB, which could be referred as:
| (2) |
3. Results & discussion
3.1. Evaluation of experiments in batch
The batch experiment was carried out under a constant 363.15 K with mechanical agitation. PVM was also used to observe the thermal decomposition process of LBCB. At the beginning of the reaction, CO2 bubbles were gradually generated in clear solution. As the reaction proceeded, the number and size of bubbles increased (Fig. 3, A-B), which also led to the accumulation of supersaturation in solution. When the supersaturation of LCB reached a certain degree, the primary nucleation occurred spontaneously (approximately after 60 min). With the continuous progress of the reaction, the number of LCB particles increased significantly. Meanwhile, CO2 bubbles could also be observed (Fig. 3, D-E), representing the thermal decomposition reaction was still going on after about 2 h. An obvious conclusion is that the reaction rate of the thermal decomposition of LBCB is low, and the effect of reaction kinetics on the crystallization process needs to be considered in reactive crystallization process design and optimization. In addition, it can be seen from Fig. 3F that due to the non-ideal local mixing, a high degree of partial supersaturation could increase the primary nucleation, which could eventually lead to the agglomeration of LCB crystals. Therefore, the application of pulsed ultrasound in such system has the following purposes: 1). Speeding up the process of reaction and promoting the release of CO2 bubbles; 2). Using the ultrasonic field to enhance mixing to avoid high local supersaturation; 3). Bursting of the cavitation bubbles leading to the LCB agglomerates breaking into fragments. These fragments acted as new crystal nucleation sites and hence induced secondary nucleation.
Fig. 3.

PVM images depicted (a, b, c, d, e, and f) time lapse images of the thermal decomposition reaction of LBCB resulting in the generation of CO2 bubbles, and also accompanied by the crystallization of LCB.
3.2. Evaluation of experiments with/without pulsed ultrasound.
3.2.1. The effect pulsed ultrasound on steady state.
Beginning with an initial concentration of LBCB of 29.4 g/kg, it could be observed that there was nucleation in all 12 experiments. Crystal size distribution and morphology were analyzed by detecting the LCB crystals after exiting the crystallizer (in the collection beaker) to determine a steady-state condition. Besides, whether achieving a steady-state condition in crystallizers were investigated by offline sampling mentioned in section 2.3.3. Multiple experimental conditions were tested by varying the temperature, stage number and ultrasonic field, details are given in Table 1.
The yield determined by offline sampling of MSMPR_1, MSMPR_3, MSMPR_6, and MSMPR_9 advancing with time were shown in Fig. 4. It was found that the yield gradually increased with the advancing of time, and the final yield tended to be stable. The yield gradually increased during the start-up period, and stabilized after a certain period of time. The explanation for this phenomenon is that the reactive crystallization process needs a longer start-up period, since the supersaturation is generated by the reaction which is slower than that in cooling or anti-solvent crystallization. The yields in crystallizers after achieving a stable condition were almost numerically equal to the final yield. The steady-state condition was obtained in continuous reactive crystallization could be confirmed. To illustrate the main tendency in crystal product quality changes during continuous reactive crystallization, Dv10, Dv50, Dv90, and D[4,3]-D[3,2] values of the samples, and waterfall plots were selected and shown in Table 2, Table 3.
Fig. 4.

The last stage yield during crystallization process of MSMPR_1, MSMPR_3, MSMPR_6, and MSMPR_9.
Table 2.
Comparison of MSMPR_1, MSMPR_2, and MSMPR_3 experiment regarding the crystal size of the last stage alteration during the crystallization process.
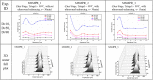 |
Table 3.
Comparison of MSMPR_5, MSMPR_6, MSMPR_7, and MSMPR_8 experiment regarding the crystal size of the last stage alteration during the crystallization process.
 |
In Table 2, three selected single-stage MSMPR crystallizations were described, and shown a similar process in which the crystal sizes varied from fine to large, then fell after rising, and eventually remained stable. Because seeds with a mean crystal size of 10 µm were added to the crystallizer (with the ratio of 2 g/kg), the increase in crystal size was the result of the seed crystal growth at a lower supersaturation. Since the LBCB solution was continuously fed into the suspension, as the supersaturation accumulated to a higher degree, a secondary nucleation process occurred spontaneously. The nucleation led to the generation of a large number of small particles, which mainly grew within 1-2 RT. Finally, as the supersaturation in the crystallizer stabilized, the crystal size only fluctuated in an insignificant range. A similar tendency has been observed in two-stage MSMPR crystallization. This tendency is not common in continuous cooling crystallization and continuous anti-solvent crystallization. Because in these two crystallization modes, supersaturation generates almost instantaneously and then reaches the maximum value, which is obviously different from the slow generation of supersaturation in continuous reaction crystallization. The start-up period of single-stage MSMPR is five to seven RTs. But when applying pulsed ultrasound, steady state conditions can be reached faster (MSMPR_2 and MSMPR_3). The presence of acoustic streaming can break the bubbles and accelerate the release of the carbon dioxide gas, forcing the reaction proceed to become faster in the direction of obtaining lithium carbonate. Simultaneously, the local temperature rise due to pulsed ultrasound can also lead to increasing the reaction rate and reducing the carbon dioxide solubility. In addition, the continuous process with an RT of 15 min reached the steady state faster than that of 30 min. This is due to the fact that the crystal does not achieve sufficient nucleation and growth within a shorter RT, which also results in a decrease in the final yield. According to the experimental results of batch operation in section 3.1, when pulsed ultrasound was applied to the system within a short period of time, the weight loss in the system was almost the same as the weight difference when carbon dioxide was completely discharged in 10 min. This represents that within one random RT, the applied pulsed ultrasound can fully convert LBCB into LCB, so that supersaturation can be quickly generated and provided to the continuous crystallization process. In Table 3, compared with the low (333.15 K)-high (363.15 K) temperature design, when the temperature was designed as high (363.15 K)-low (333.15 K), the maximum crystal sizes were achieved earlier (less than 2 RT). This is because the rapid reaction speed at high temperature, leading to a more rapid accumulation of supersaturation, which promoted crystal growth and secondary nucleation. A comparison of MSMPR_11 and MSMPR_12 experiment regarding crystal size alteration during the crystallization process is available in Table S2.
3.2.2. The effect of pulsed ultrasound on product quality.
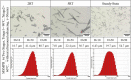
To monitor the crystal morphology in MSMPR crystallizations, four processes were demonstrated. For MSMPR_5 (Table 4), because the supersaturation was generated slowly by the reaction within 2 RT, the crystals mainly grew under such a low supersaturation condition. The crystal size of obtained crystals was in the mean range of 11.0 μm (Dv 10)–77.3 μm (Dv 90). As the supersaturation rose, the secondary nucleation led to the generation of small crystals, which eventually resulted in a smaller crystal size of 6.96 μm (Dv 10)–61.8 μm (Dv 90) at steady state. In MSMPR_6, the influence of temperature on the continuous reactive crystallization process has been evaluated by switching the temperature design between the crystallizers. Under the same energy consumption situation, the crystal size was smaller and in a narrower distribution of 6.43 μm (Dv 10)–54.7 μm (Dv 90) (Table 5), which was because the high temperature at the first-stage accelerated the decomposition reaction and produced a higher local supersaturation faster. Besides, a faster reaction rate would result in faster CO2 releasing, and the gas–liquid interface provided by more small bubbles was easier to promote the nucleation of solute [12]. The above two reasons together lead to the formation of fines. When the RT was extended to 60 min (MSMPR_11, Table S3), the crystal had more sufficient time to grow, so a wider crystal size distribution was obtained.
Table 4.
Microscopic pictures, Dv values, and CSD plots of the MSMPR_5 samples (2 RT, 6 RT, and from steady-state of the experiment).
 |
Table 5.
Microscopic pictures, Dv values, and CSD plots of the MSMPR_6 samples (2 RT, 5 RT, and from steady-state of the experiment).
 |
To investigate the enhancement of the reaction and crystallization process by ultrasonic field in MSMPR_5 in Table 5, the ultrasonic probe was inserted into the first stage of the MSMPR cascade. The outflowing slurry of the first stage was transferred to the second stage at 363.15 K. The second stage outflowing slurry was used for morphology and particle size analysis after being filtered, washed and dried. Comparing the needle-like crystals produced within 2 RT, shorter and thicker LCB particles upon sonication were found. This phenomenon can be explained as in stage one, ultrasonic energy (power) accelerates effectively the reaction and mass transfer in the mixture and thus enhances the driving force of crystal growth [33]. The speed of insonated molecules is fast enough for them to approach each side of the crystal to compensate partly for differences in the growth rate of each side in conventional crystallization. Thus, it is reasonable that crystal grows uniformly into shorter and thicker. Although the crystal habit showed a significant difference, the mean sizes of MSMPR_5 (2 RT, Table 5) and MSMPR_7 (2 RT, Table 6), measured by the Mastersizer, were very similar, because the instrument seemed unable to detect differences in the shapes. With the increase of crystal size, when the crystals flow through the peristaltic pump, a part of the large crystals will inevitably go through the breakage process. Secondary nucleation can occur by the crystals fragmenting to produce more nucleation sites, leading to an increase in small crystals [34]. Besides, the growth of crystals is also accompanied by an increase in the risk of breakage due to mechanical agitation. Since the nucleation and growth of the LCB crystals were not affected by pulsed ultrasound in the second-stage MSMPR, the steady-state crystal size did not show an obvious difference with MSMPR_5. The effects of the ultrasound field on the crystallization process can be investigated by inserting the ultrasonic probe into the second stage. In MSMPR_8 (Table 7), the reaction proceeded more completely at the 363.15 K in the first stage. Pulsed ultrasound maintained reasonably uniform supersaturation conditions throughout the second stage crystallizer. The microscopic observation showed that more uniform rod-like crystals were obtained. Such crystals have the advantages of flowability and filterability compared with needle-like crystals, which are commonly seen in other LCB continuous reactive crystallization experiments. Offline microscope images in Table 7 confirm some crystals shows obvious breakage, this was caused by the effects of particle-shockwave interactions and the mechanical force during the slurry transformation.
Table 6.
Microscopic pictures, Dv values, and CSD plots of the MSMPR_7 samples (2 RT, 5 RT, and from steady-state of the experiment).
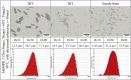 |
Table 7.
Microscopic pictures, Dv Values, and CSD plots of the MSMPR_8 samples (2 RT, 6 RT, and from steady-state of the experiment).
 |
The steady-state crystal habit, crystal size, and CSD of MSMPR_6 - MSMPR_9 can be referred to in Table 8. Although MSMPR_8 showed the narrowest CSD among four experiments, the mean crystal size was a little bit larger than MSMPR_6. This indicated that particle-shockwave interactions induced sonofragmentation was not the main reason for the decrease of the crystal size. As the RT expended to 60 min, a larger mean crystal size was obtained. This is because the higher supersaturation and the longer RT could generate sufficient growth of the LCB crystals when applied the ultrasonic field, which led to a wider CSD. Regardless of whichever stage of crystallizers the ultrasonic probe inserted into, the effect was all to continuously and uniformly distribute the supersaturation, and to improve the reaction rate by enhanced molecular thermal motion and accelerating the CO2 bubbles overflowing. In summary, only for optimizing the crystal morphology and CSD, the advantageous design strategy is to apply high temperature in the first stage to provide a higher reaction rate and a larger supersaturation, followed by applying pulsed ultrasound at a lower temperature in the second stage to enhance local-mixing and to induce nucleation and crystal growth uniformly.
Table 8.
Summary of the MSMPR crystallization experiments regarding crystal habit, crystal size, and crystal size distribution of the experiments under steady-state operation.
 |
3.2.3. The effect of pulsed ultrasound on crystal size distribution.
In Fig. 4 to Fig. 7, the volume density distributions of the output are presented to illustrate the effect of the pulsed ultrasound, temperatures and RTs on CSD. Multiple works have shown a decrease in particle size with ultrasound [13], [35], but the opposite result was obtained in single-stage MSMPR. When pulsed ultrasound was applied to continuous crystallization systems under different operating conditions, increasing or constant average particle size was observed. Three factors were thought to explain this behavior. One possible explanation for large crystal formation is that using pulsed ultrasound increase the collision frequency between crystals, which helps inclusion of microcrystal to larger crystals [36]. Because the thermal decomposition reaction generates CO2 bubbles, pulsed ultrasonic waves and mechanical stirring cause the CO2 bubbles to burst rapidly, which increases the degree of turbulence in the crystallizer, leading to the increased chance of direct collision of crystals. The second reason is that, as already established, a few large isometric crystals were identified at the beginning of the process. These crystals could break into fragments induced by both mechanical and ultrasound effects, which could boost secondary nucleation further. Besides, ultrasound can create sonofragmentation in both supersaturated and undersaturated conditions. When this fragmentation occurs in a supersaturated solution, the fragments can act as nucleation sites for new nuclei. When the system reaches a steady state, the supersaturation in the crystallizer in the presence of pulsed ultrasound is higher than that under traditional conditions. Therefore, fines generated by secondary nucleation have a larger growth rate, resulting in continuously producing larger crystals. Thirdly, during the expansion phase of the cavitation bubble, evaporation occurs from the surface of the cavitation bubble to the bubble interior [12]. As a result, the surface is cooled and it is suggested that this cooling causes the decrease of local supersaturation and hence dissolution (because the solubility of lithium carbonate decreases with increasing temperature). The dissolution of small-sized crystals near the gas–liquid interface also be one of the reasons why the average particle size does not decrease significantly in the presence of an ultrasonic field. When the RT was limited to 15 min, time for crystal growth was shortened under the ultrasonic field, which led to a smaller crystal size of MSMPR_3 shown in Fig. 5. In consideration of the two-stage MSMPR, different from MSMPR_10 in Fig. 6, smaller crystals were obtained in MSMPR_9. This is because pulsed ultrasound was applied to the first stage (333.15 K), which caused the thermal decomposition reaction to being enhanced. More uniform LCB crystals were contained in the mother liquor, and these crystals represented a smaller mean crystal size and a narrow CSD under the low supersaturation of the second-stage MSMPR. The above analyses prove that under the same energy consumption conditions, designing the continuous process of experiencing high temperature first and then experiencing low temperature will help to obtain the LCB crystals with narrower particle size distribution and smaller particle size. The conclusion was also supported by MSMPR_11 and MSMPR_12 (Fig. S2). An interesting phenomenon is that when ultrasound was applied to the low-temperature section of the high-low-temperature design (the ultrasonic probe was inserted into the second-stage crystallizer), the mean crystal size did not change significantly (Fig. 7). As mentioned earlier, higher supersaturation is one of the reasons that promote the growth of the first-stage MSMPR crystal. In the high-low temperature design, the high supersaturation generated at high temperature has been consumed by the nucleation and growth of LCB crystals in the first-stage crystallizer, and the supersaturation in the second-stage crystallizer was relatively low. Therefore, it is acceptable that there is no significant crystal growth in such conditions. Thus, one could expect that in pulsed ultrasound enhanced reaction continuous crystallization process, crystal sizes would be larger at relatively high supersaturation but that does not seem to be the case. It must be mentioned that crystal size would also be influenced by cavitation bubbles and the CO2 bubble sizes and numbers, which maybe a reason for the absence of a specific trend in crystal sizes at steady-state conditions.
Fig. 7.

Particle size distributions for MSMPR_6(Two Stages, Tstage1 = 363.15 K, Tstage2 = 333.15 K, without ultrasound enhancing, τ = 30 min), and MSMPR_8(Two Stages, Tstage1 = 363.15 K, Tstage2 = 333.15 K, with ultrasound enhancing, τ = 30 min) at steady state.
Fig. 5.

Particle size distributions for MSMPR_1(One Stage, Tstage1 = 363.15 K, without ultrasound enhancing, τ = 30 min), MSMPR_2(One Stage, Tstage1 = 363.15 K, with ultrasound enhancing, τ = 30 min), and MSMPR_3 (One Stage, Tstage1 = 363.15 K, with ultrasound enhancing, τ = 15 min) at steady state.
Fig. 6.

Particle size distributions for MSMPR_5(Two Stages, Tstage1 = 333.15 K, Tstage2 = 363.15 K, without ultrasound enhancing, τ = 30 min), MSMPR_7(Two Stages, Tstage1 = 333.15 K, Tstage2 = 363.15 K, with ultrasound enhancing, τ = 30 min), MSMPR_9(Two Stages, Tstage1 = 333.15 K, Tstage2 = 363.15 K, with ultrasound enhancing, τ = 60 min), and MSMPR_10(Two Stages, Tstage1 = 333.15 K, Tstage2 = 363.15 K, without ultrasound enhancing, τ = 60 min) at steady state.
3.3. Yield assessment of crystallization with/without ultrasound
3.3.1. Mathematical modeling and parameter estimation of multistage MSMPR
The process in this work was developed for obtaining crystalline solids of the LCB. Therefore, yield is the most important process and crystal property that should be emphasized. A model of the continuous reactive crystallization process based on the simultaneous solution of the population balance equation (PBE) and mass balance was developed. The thermal decomposition reaction kinetics, the crystal growth rate, and nucleation rate are all established and used in the model so that yields in the multistage continuous crystallizer can be optimized around the desired product quality. Yields determined by experiments at steady state were also used to evaluate the reliability of the constructed model.
The multistage continuous crystallization system can be described as a series of MSMPR crystallizers, which can be modeled with PBEs. Assuming a continuous steady state operation with negligible agglomeration or breakage, where the crystal size distribution of the product is the same as that found within the crystallizer and the feed stream is free of suspended solids in the first stage, the governing equations at steady state for stage i are:
| (3) |
| (4) |
Here, Vi is the volume of MSMPR i, Fi-1 and Fi are the volumetric flow rates of streams entering and leaving MSMPR i, respectively, N is the number of crystallizers in the cascade, ni is the crystal population density at stage i, and L is the characteristic length of the crystal. The system of ordinary differential equations (ODEs) formed by the population balance equations is satisfied by the boundary condition, ni0 = ni (L = 0), corresponding to the population density of nuclei. The ODEs were firstly analytically solved by Randolph and Larson [37], [38], which are:
| (5) |
| (6) |
| (7) |
At steady state, there is no accumulation of the mass in each stage. Thus, the rate at which solute is lost from the solution phase is equal to the rate at which mass is gained by the solid phase. A mass balance on the solute crystallized for stage i gives:
| (8) |
| (9) |
Fi and Ci are the fresh volumetric flow rate and mother liquor LCB concentration of the i stage crystallizer, respectively. Crea,i is the LCB generated by the decomposition reaction in the i stage crystallizer. MTi is the suspension density at stage i, which is calculated from the population density function as follows:
| (10) |
| (11) |
| (12) |
where kv is the crystal volume shape factor, and ρ is the LCB crystal density.
The thermal decomposition reaction kinetics can be expressed as a function of the LBCB concentration shown as eq (13), where k0 is the pre-exponential factor, and Ea is the activation energy. Details can be found in the supporting information and Fig. S3. The crystal growth rate and nucleation rate can be expressed as a function of supersaturation in the form of empirical power-law eqs (14), 15, and 16, where Gi and Bi are the crystal growth rate and nucleation rate at stage i, respectively, Si is the steady state supersaturation at stage i, Ea,g and Ea,b are the energy barrier for growth and nucleation. T is the temperature, and R is the gas constant (8.314 J/(mol K)), while kg,0, kb,0, g, b, and m are model parameters [39], [40].
| (13) |
| (14) |
| (15) |
| (16) |
The homogenous ordinary equation system coupled with mass balance and kinetics were solved numerically. The results of the parameter estimation and a comparison between the calculated and experimental are shown in Table 9 and Fig. S3.
Table 9.
Reaction and crystallization kinetic parameters for the reactive crystallization of LCB.
| Parameter | Value | Units |
|---|---|---|
| k0 | 1.911010 | mol0.5 min−1 |
| Ea | 7.30104 | J mol−1 |
| kg0 | 3.0510-4 | m min−1 |
| Eag/R | 1732.02 | K |
| g | 1.02 | dimensionless |
| kb0 | 2.781011 | # crystals m−3 min−1 |
| Eab/R | 4.25103 | K |
| b | 1.3 | dimensionless |
| m | 2/3 | dimensionless |
| kv | 0.6 | dimensionless |
3.3.2. Yield assessment
The continuous reactive crystallization experiments were characterized by yield and productivity. As can be seen in Table 10, application pulsed ultrasound resulted in a significant increase in yield, while using pulsed ultrasound with a longer RT had no remarkable effects. In most experiments, the standard deviation of yield remained under 5%. The highest standard deviation regarding yield was 5.3% in the MSMPR_3 experiment.
Table 10.
Summary of the MSMPR crystallization experiments including process parameters, yield, and productivity.
| ID of the experiment | Temperature (K) | RT (h) | Ultrasonic field | Experimental Resultsa |
Simulation Results |
||
|---|---|---|---|---|---|---|---|
| yield (%) | productivity (g/h) | yield (%) | productivity (g/h) | ||||
| MSMPR_1 | 363.15 | 0.5 | Without | 33.7 ± 2.7 | 10.1 ± 0.8 | 27.0 | 8.1 |
| MSMPR_2 | 363.15 | 0.5 | With | 63.8 ± 2.4 | 19.2 ± 0.7 | – | – |
| MSMPR_3 | 363.15 | 0.25 | With | 43.0 ± 5.3 | 25.9 ± 3.2 | – | – |
| MSMPR_4 | 363.15 | 1 | Without | 62.1 ± 1.8 | 9.3 ± 0.3 | 64.2 | 9.7 |
| MSMPR_5 | 333.15–363.15 | 0.5 | Without | 31.2 ± 3.3 | 9.4 ± 1.0 | 36.7 | 11.0 |
| MSMPR_6 | 363.15–333.15 | 0.5 | Without | 46.9 ± 1.3 | 14.1 ± 0.4 | 48.0 | 14.4 |
| MSMPR_7 | 333.15–363.15 | 0.5 | With | 74.1 ± 2.4 | 22.3 ± 0.7 | 77.0 | 23.2 |
| MSMPR_8 | 363.15–333.15 | 0.5 | With | 70.3 ± 4.8 | 21.1 ± 1.4 | – | – |
| MSMPR_9 | 333.15–363.15 | 1 | With | 73.9 ± 3.6 | 10.3 ± 0.5 | – | – |
| MSMPR_10 | 333.15–363.15 | 1 | Without | 63.9 ± 3.7 | 9.6 ± 0.6 | 66.5 | 10.0 |
| MSMPR_11 | 363.15–353.15 | 1 | Without | 68.3 ± 5.2 | 10.3 ± 0.8 | 76.9 | 11.6 |
| MSMPR_12 | 353.15–363.15 | 1 | Without | 70.2 ± 2.7 | 10.6 ± 0.4 | 76.7 | 11.5 |
The experimental results are reported as the mean ± standard deviation of three parallel experiments.
The yield was found to increase with the application of sonication to the continuous system by up to 23.4%-42.9% at the same RT. In single stage MSMPR, because of the application of pulsed ultrasound, the experiment with shorter RT (15 min) can reach almost the same yield of a longer residence (30 min) time experiment (MSMPR_2/4). Also, in two stage MSMPR cascade, the experiments with lower heating duty and shorter RT can even obtain an extra 3.9 % yield (MSMPR_7/12). It can be concluded that after using pulsed ultrasound, almost the same yield can be obtained within a shorter RT and a lower operating temperature. This conclusion is undoubtedly very attractive, because the pursuit of minimum investment while ensuring the yield is the eternal goal pursued by industrial crystallization.
With the same heating duty input, MSMPR_7/8, and MSMPR_11/12 showed that the final stage temperature controlled the final yield [37]. But in MSMPR_5/6, an opposite conclusion has been drawn because without the use of ultrasound field, a higher temperature in stage one has benefits to improve the reaction rate, and then provided a higher supersaturation of LCB. Under such a condition, the time for secondary nucleation and growth of the LCB crystals is more sufficient. The productivity of the experiments varied in the range of 9.3–25.9 g/h. A shorter RT and using pulsed ultrasound favor an increase in productivity. Hence, the highest productivity could be obtained when the RT is shorter (15 min), but the yield is relatively low even with the use of pulsed ultrasound (43.0 ± 5.3%).
As already mentioned in section 3.2.1, the thermal decomposition reaction can be completed within one RT after being enhanced by pulsed ultrasound, which provided the possibility for the yield simulation of MSMPR_7. In the eight experiments shown in Table 10 and Fig. 8, the results of the experiment and the simulation are in good agreement, and the difference between the two ranges 1.1%-8.6%. This proves that the established secondary nucleation and crystal growth kinetics are reliable. It is worth mentioning that several reasons could contribute to a difference in the yield between experiment and simulation. These reasons are as follows: ①The first is the deposition and attachment of the crystals on the crystallizer wall and slurry transfer tube wall, which is the main reason that experimental yield is lower than simulation; ② The continuously transferred slurry would be affected by the flow field in the crystallizer, resulting in an unstable obtained weight per unit time, which caused the yield to fluctuate during any steady-state RT. Such fluctuations are very common in laboratory-scale experiments [7], [41]; ③ Although pulsed ultrasound or high temperature greatly increased the reaction rate, there were still situations where carbon dioxide bubbles were not completely released in the system. This would cause the dissolved LCB to react with the residual carbon dioxide to form LBCB, and eventually lead to a decrease in final yield. It is worth mentioning that because the LCB solubility decreases with increasing temperature, the local temperature rise caused by ultrasound would not cause the particle dissolution behavior. In fact, based on the LCB solubility curve, the effect of temperature changes caused by pulsed ultrasound on the LCB solubility can be negligible. Besides, reasons like the position of feeding tubes, the selection of crystal size due to the inappropriate tube wide, had been experienced during the process development and efforts have been made to eliminate all these obstacles in the presented results. In this work, the chemical reaction limits the attainable crystallization yields for a constant feed concentration. From the perspective of achieving higher yield and higher productivity, the adoption of multi-stage MSMPR cascades, the design of highest temperature at the last stage, and the process intensification by pulsed ultrasound should be considered when the design strategy of continuous reactive crystallization is determined.
Fig. 8.

A comparison of yield between the simulation results and the experimental results for eight experiments.
3.4. Design of three stages MSMPR reactive crystallization of LCB
Based on above mentioned investigations, a clear conclusion is that using pulsed ultrasound and the multi-stage cascade help to improve the yield of continuous reactive crystallization. At the same time, higher yield and shorter RT have the benefit to obtain higher productivity. This inspired the idea of designing a three-stage MSMPR cascade. The two MSMPR cascades were designed as: MSMPR_13 and 14 with/without the ultrasonic probe inserted. The RT was set to 15 min, and pulsed ultrasound was added to the first-stage MSMPR to meet the requirements of productivity. The final yield simulated by established model were Y(MSMPR_13) = 76.9% and productivity = 46.3 [g/h], and Y(MSMPR_14) = 35.2% and productivity = 21.2 [g/h]. Furthermore, two experiments verified the reliability of the yield and productivity obtained by the simulation (Y(MSMPR_13) = 73.19% and productivity = 44.0 [g/h], and Y(MSMPR_14) = 40.9% and productivity = 24.6 [g/h]). A microscope photo of the product is available in Fig. 9.
Fig. 9.

Microscope images of the LCB crystals at steady-state condition of a) MSMPR_13 and b) MSMPR_14.
It can be concluded that for the continuous multi-stage reactive crystallization process, applying pulsed ultrasound, using the three-stage MSMPR cascade, and setting 363.15 K at the final stage crystallizer can help to obtain a higher yield under lower heating duty. Try to extend this conclusion to other continuous reactive crystallization systems. In fact, for small molecule drugs, although the higher temperature will bring about a higher reaction rate, it is unavoidable that the increase in solubility caused by the increase in temperature leads to a decrease in the highest theoretical yield that can be achieved. At the same time, although the multi-stage MSMPR cascade has obvious productivity advantages and lower operating expenditures (mainly reflected in lower energy consumption), the capital expenditures are relatively high. These factors also need to be considered in the actual design of an industrial crystallization process.
4. Conclusion
Pulsed ultrasound enhanced continuous reactive crystallization in single and multistage MSMPR crystallizers were studied in this work. Experiments were carried out in stirred tank crystallizers with or without an ultrasonic probe inserted to investigate the effect of the operation conditions on the entire process and the final LCB product. The thermal decomposition reaction kinetics was probed by the offline gravimetric method. Furthermore, mass balance and population balance equation were coupled and established as the population balance model to verify and predict the steady state yield of the continuous process. The final yield obtained by the simulation was consistent with the experimental results. According to the experimental analyses and simulation, pulsed ultrasound enhanced three-stage MSMPR processes were designed and experimentally verified. The highest productivity was achieved as 44.0 g/h.
Only relying on temperature changes to develop continuous reactive crystallization processes could take a longer time to reach the steady-state condition, and higher yield and productivity can be obtained only through the multi-stage cascade and high-temperature design. Applying pulsed ultrasound in the MSMPR crystallizer setup accompanied with different temperature gradient is a suitable design to overcome the mentioned issues. When the ultrasonic probe is inserted into the front crystallizer, the reaction process is greatly enhanced, which leads to an increase in the steady-state yield. When the ultrasonic probe is inserted into the last-stage crystallizer, well-dispersion crystalline and products with narrow crystal size distribution can be obtained. This can meet the demands of flexibility for products.
With the development of continuous crystallization processes in the industry crystallization process, the limitations of traditional continuous crystallization design have attracted increasing concern. The pulsed ultrasound based-process intensification technique developed in this work demonstrates a way to improve the performance at steady state and is expected to provide a new perspective for the industry in moving from batch to continuous manufacturing.
CRediT authorship contribution statement
Yiming Ma: Conceptualization, Methodology, Validation, Formal analysis, Investigation, Data curation, Writing – original draft, Writing - review & editing, Visualization. Zhixu Li: Data curation, Writing - review & editing, Validation, Investigation. Peng Shi: Validation, Investigation, Resources. Jiawei Lin: Validation, Investigation. Zhenguo Gao: Writing - review & editing. Menghui Yao: Writing - review & editing. Mingyang Chen: Writing - review & editing. Jingkang Wang: Funding acquisition, Writing - review & editing. Songgu Wu: Writing - review & editing. Junbo Gong: Conceptualization, Resources, Writing - review & editing, Supervision, Project administration, Funding acquisition.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors are grateful to the financial support of National Natural Science Foundation of China (Nos. NNSFC 21938009 and NNSFC 22078234).
Footnotes
Supplementary data to this article can be found online at https://doi.org/10.1016/j.ultsonch.2021.105698.
Appendix A. Supplementary data
The following are the Supplementary data to this article:
References
- 1.Ma Y., Wu S., Macaringue E.G.J., Zhang T., Gong J., Wang J. Recent progress in continuous crystallization of pharmaceutical products: precise preparation and control. Org. Process Res. Dev. 2020;24(10):1785–1801. [Google Scholar]
- 2.Wood B., Girard K.P., Polster C.S., Croker D.M. Progress to date in the design and operation of continuous crystallization processes for pharmaceutical applications. Org. Process Res. Dev. 2019;23(2):122–144. [Google Scholar]
- 3.Zhang D., Xu S., Du S., Wang J., Gong J. Progress of pharmaceutical continuous crystallization. Engineering. 2017;3(3):354–364. [Google Scholar]
- 4.Chen J., Sarma B., Evans J.M.B., Myerson A.S. Pharmaceutical crystallization. Cryst. Growth Des. 2011;11(4):887–895. [Google Scholar]
- 5.Lührmann M.-C., Timmermann J., Schembecker G., Wohlgemuth K. Enhanced product quality control through separation of crystallization phenomena in a four-stage MSMPR cascade. Cryst. Growth Des. 2018;18(12):7323–7334. [Google Scholar]
- 6.Ferguson S., Morris G., Hao H., Barrett M., Glennon B. Characterization of the anti-solvent batch, plug flow and MSMPR crystallization of benzoic acid. Chem. Eng. Sci. 2013;104:44–54. [Google Scholar]
- 7.Tacsi K., Pataki H., Domokos A., Nagy B., Csontos I., Markovits I., Farkas F., Nagy Z.K., Marosi G. Direct processing of a flow reaction mixture using continuous mixed suspension mixed product removal crystallizer. Cryst. Growth Des. 2020;20(7):4433–4442. [Google Scholar]
- 8.Quon J.L., Zhang H., Alvarez A., Evans J., Myerson A.S., Trout B.L. Continuous crystallization of aliskiren hemifumarate. Cryst. Growth Des. 2012;12(6):3036–3044. [Google Scholar]
- 9.McDonald M.A., Bommarius A.S., Rousseau R.W., Grover M.A. Continuous reactive crystallization of β-lactam antibiotics catalyzed by penicillin G acylase. Part I: model development. Comput. Chem. Eng. 2019;123:331–343. [Google Scholar]
- 10.McDonald M.A., Bommarius A.S., Grover M.A., Rousseau R.W. Continuous reactive crystallization of β-lactam antibiotics catalyzed by penicillin G acylase. Part II: case study on ampicillin and product purity. Comput. Chem. Eng. 2019;126:332–341. [Google Scholar]
- 11.Eder R.J.P., Schrank S., Besenhard M.O., Roblegg E., Gruber-Woelfler H., Khinast J.G. Continuous sonocrystallization of acetylsalicylic acid (ASA): control of crystal size. Cryst. Growth Des. 2012;12(10):4733–4738. [Google Scholar]
- 12.Jordens J., Gielen B., Xiouras C., Hussain M.N., Stefanidis G.D., Thomassen L.C.J., Braeken L., Van Gerven T. Sonocrystallisation: observations, theories and guidelines. Chem. Eng. Process. 2019;139:130–154. [Google Scholar]
- 13.Mao Y., Li F., Wang T., Cheng X., Li G., Li D., Zhang X., Hao H. Enhancement of lysozyme crystallization under ultrasound field. Ultrason. Sonochem. 2020;63 doi: 10.1016/j.ultsonch.2020.104975. [DOI] [PubMed] [Google Scholar]
- 14.Fang C., Tang W., Wu S., Wang J., Gao Z., Gong J. Ultrasound-assisted intensified crystallization of L-glutamic acid: crystal nucleation and polymorph transformation. Ultrason. Sonochem. 2020;68:105227. doi: 10.1016/j.ultsonch.2020.105227. [DOI] [PubMed] [Google Scholar]
- 15.Sayan P., Sargut S.T., Kiran B. Effect of ultrasonic irradiation on crystallization kinetics of potassium dihydrogen phosphate. Ultrason. Sonochem. 2011;18(3):795–800. doi: 10.1016/j.ultsonch.2010.11.003. [DOI] [PubMed] [Google Scholar]
- 16.Hazi Mastan T., Lenka M., Sarkar D. Nucleation kinetics from metastable zone widths for sonocrystallization of l-phenylalanine. Ultrason. Sonochem. 2017;36:497–506. doi: 10.1016/j.ultsonch.2016.12.017. [DOI] [PubMed] [Google Scholar]
- 17.Chow R., Blindt R., Chivers R., Povey M. A study on the primary and secondary nucleation of ice by power ultrasound. Ultrasonics. 2005;43(4):227–230. doi: 10.1016/j.ultras.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 18.Bari A.H., Chawla A., Pandit A.B. Sono-crystallization kinetics of K2SO4: Estimation of nucleation, growth, breakage and agglomeration kinetics. Ultrason. Sonochem. 2017;35:196–203. doi: 10.1016/j.ultsonch.2016.09.018. [DOI] [PubMed] [Google Scholar]
- 19.Gajendragadkar C.N., Gogate P.R. Ultrasound assisted acid catalyzed lactose hydrolysis: understanding into effect of operating parameters and scale up studies. Ultrason. Sonochem. 2017;37:9–15. doi: 10.1016/j.ultsonch.2016.12.029. [DOI] [PubMed] [Google Scholar]
- 20.Gogate P.R., Shaha S., Csoka L. Intensification of cavitational activity in the sonochemical reactors using gaseous additives. Chem. Eng. J. 2014;239:364–372. [Google Scholar]
- 21.Gogate P.R., Sutkar V.S., Pandit A.B. Sonochemical reactors: important design and scale up considerations with a special emphasis on heterogeneous systems. Chem. Eng. J. 2011;166:1066–1082. [Google Scholar]
- 22.Gielen B., Kusters P., Jordens J., Thomassen L.C.J., Van Gerven T., Braeken L. Energy efficient crystallization of paracetamol using pulsed ultrasound. Chem. Eng. Process. 2017;114:55–66. [Google Scholar]
- 23.McCausland L.J., Cains P.W. Power ultrasound – a means to promote and control crystallization in biotechnology. Biotechnol. Genet. Eng. 2004;21(1):3–10. doi: 10.1080/02648725.2004.10648048. [DOI] [PubMed] [Google Scholar]
- 24.Ciaravino V., Flynn H.G., Miller M.W. Pulsed enhancement of acoustic cavitation. Ultrasound Med. Biol. 1981;7:403–404. doi: 10.1016/0301-5629(81)90005-3. [DOI] [PubMed] [Google Scholar]
- 25.Choi P.-K., Kaneko Y., Meguro T. Enhancement of sonoluminescence and bubble dynamics using pulsed ultrasound at 103 kHz. Jpn. J. Appl. Phys. 2008;47(5):4111–4114. [Google Scholar]
- 26.Chi S.S., Liu Y., Song W.L., Fan L.Z., Zhang Q. Prestoring lithium into stable 3D nickel foam host as dendrite-free lithium metal anode. Adv. Funct. Mater. 2017;27:24. [Google Scholar]
- 27.Xiao J.-L., Sun S.-Y., Song X., Li P., Yu J.-G. Lithium ion recovery from brine using granulated polyacrylamide–MnO2 ion-sieve. Chem. Eng. J. 2015;279:659–666. [Google Scholar]
- 28.Swain B. Recovery and recycling of lithium: a review. Sep. Purif. Technol. 2017;172:388–403. [Google Scholar]
- 29.Lv W., Wang Z., Cao H., Sun Y., Zhang Y.i., Sun Z. A critical review and analysis on the recycling of spent lithium-ion batteries. Acs Sustain. Chem. Eng. 2018;6(2):1504–1521. [Google Scholar]
- 30.Zhou Z., Liang F., Qin W., Fei W. Coupled reaction and solvent extraction process to form Li2CO3: mechanism and product characterization. AIChE J. 2014;60:282–288. [Google Scholar]
- 31.Sun Y.-Z., Song X.-F., Jin M.-M., Jin W., Yu J.-G. Gas–liquid reactive crystallization of lithium carbonate by a falling film column. Ind. Eng. Chem. Res. 2013;52(49):17598–17606. [Google Scholar]
- 32.Ma Y., Cao Y., Yang Y., Li W., Shi P., Wang S., Tang W. Thermodynamic analysis and molecular dynamic simulation of the solubility of vortioxetine hydrobromide in three binary solvent mixtures. J. Mol. Liq. 2018;272:676–688. [Google Scholar]
- 33.Hong Li J.W., Bao Ying, Guo Zhichao, Zhang Muyan. Rapid sonocrystallization in the salting-out process. J. Cryst. Growth. 2003;247:192–198. [Google Scholar]
- 34.Dalvi-Isfahan M., Hamdami N., Xanthakis E., Le-Bail A. Review on the control of ice nucleation by ultrasound waves, electric and magnetic fields. J. Food Eng. 2017;195:222–234. [Google Scholar]
- 35.Jordens J., Canini E., Gielen B., Van Gerven T., Braeken L. Ultrasound assisted particle size control by continuous seed generation and batch growth. Crystals. 2017;7:195. [Google Scholar]
- 36.Nii S., Takayanagi S. Growth and size control in anti-solvent crystallization of glycine with high frequency ultrasound. Ultrason. Sonochem. 2014;21(3):1182–1186. doi: 10.1016/j.ultsonch.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 37.Alvarez A.J., Singh A., Myerson A.S. Crystallization of cyclosporine in a multistage continuous MSMPR crystallizer. Cryst. Growth Des. 2011;11(10):4392–4400. [Google Scholar]
- 38.Power G., Hou G., Kamaraju V.K., Morris G., Zhao Y., Glennon B. Design and optimization of a multistage continuous cooling mixed suspension, mixed product removal crystallizer. Chem. Eng. Sci. 2015;133:125–139. [Google Scholar]
- 39.Zhao S., Gao J., Ma S., Li C., Ma Y., He Y., Gong J., Zhou F., Zhang B., Tang W. Mechanism and modelling of reactive crystallization process of lithium carbonate. Processes. 2019;7:248. [Google Scholar]
- 40.Liu H., Azimi G. Process analysis and study of factors affecting the lithium carbonate crystallization from sulfate media during lithium extraction. Hydrometallurgy. 2021;199 [Google Scholar]
- 41.Li J., Lai T.-t.C., Trout B.L., Myerson A.S. Continuous crystallization of cyclosporine: effect of operating conditions on yield and purity. Cryst. Growth Des. 2017;17:1000–1007. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.