

Abstract
It has been reported that paclitaxel activates cell cycle arrest and increases caspase protein expression to induce apoptosis in head and neck squamous cell carcinoma (HNSCC) cell lines. However, the potential signaling pathway regulating this apoptotic phenomenon remains unclear. The present study used OEC-M1 cells to investigate the underlying molecular mechanism of paclitaxel-induced apoptosis. Following treatment with paclitaxel, cell viability was assessed via the MTT assay. Necrosis, apoptosis, cell cycle and mitochondrial membrane potential (∆Ψm) were analyzed via flow cytometric analyses, respectively. Western blot analysis was performed to detect the expression levels of proteins associated with the MAPK and caspase signaling pathways. The results demonstrated that low-dose paclitaxel (50 nM) induced apoptosis but not necrosis in HNSCC cells. In addition, paclitaxel activated the c-Jun N-terminal kinase (JNK), but not extracellular signal-regulated kinase or p38 mitogen-activated protein kinase. The paclitaxel-activated JNK contributed to paclitaxel-induced apoptosis, activation of caspase-3, −6, −7, −8 and −9, and reduction of ∆Ψm. In addition, caspase-8 and −9 inhibitors, respectively, significantly decreased paclitaxel-induced apoptosis. Notably, Bid was truncated following treatment with paclitaxel. Taken together, the results of the present study suggest that paclitaxel-activated JNK is required for caspase activation and loss of ∆Ψm, which results in apoptosis of HNSCC cells. These results may provide mechanistic basis for designing more effective paclitaxel-combining regimens to treat HNSCC.
Keywords: paclitaxel, HNSCC, apoptosis, JNK, caspase
Introduction
The main treatment of early stage head and neck squamous cell carcinoma (HNSCC) is surgery and/or radiotherapy (1). Chemotherapy is often used in various combinations with surgery and radiotherapy for patients in late stage HNSCC to improve poor survival rate or increase organ preservation (1–4). Increasing evidence suggest that cisplatin/5-fluorouracil (PF)-based regimens are useful in improving the clinical outcomes of patients in late stage HNSCC; however, they are far from satisfactory (5–8). For example, the 5-year survival rate for patients with oral cancer remains at ~60% over the last few decades (9).
Paclitaxel (also known as taxol), a natural product extracted from the bark of Pacific yew Taxus brevifolia, can promote tubulin polymerization and inhibit microtubules disassembly, causing cell death by disrupting the microtubule dynamics required for cell division and vital interphase process (10). Paclitaxel and docetaxel are the prototypes of microtubule-targeting taxane drugs, and are currently used as active chemotherapeutic agents against different types of human cancer, including HNSCC (3,5,11,12). Recent studies have demonstrated that taxane-containing triplets (taxane/cisplatin/5 fluorouracil) are superior as an induction regimen compared with the standard cisplatin/5 fluorouracil regimen for locally advanced HNSCC, and may be superior as an induction regimen followed by chemo-radiation compared with chemo-radiation alone (13,14).
Although previous studies have investigated the molecular mechanism of taxanes (15–18), only a few have focused on paclitaxel-induced cell death in HNSCC (19–21). Given that taxane-induced cell death signaling pathways may be dependent on the genotype of cancer cells and may be cell-type specific (22,23), understanding paclitaxel-induced HNSCC cell death may be useful in designing effective taxane-based regimens against HNSCC. It has been reported that paclitaxel can significantly induce apoptosis in most HNSCC cell lines, including FaDu, OC3 and OEC-M1 cells (24). In addition, activation of initiator caspases (caspase-8 and −9), downstream effector caspases (caspase-3, −6 and −7), and poly-ADP-ribose polymerase cleavage were also observed in these HNSCC cell lines (24), suggesting that activation of both death receptors and mitochondria apoptotic pathways is a common phenomenon in paclitaxel-treated HNSCC cell death.
The mitogen-activated protein kinase (MAPK) superfamily is composed of extracellular signal-regulated kinases (ERKs), c-Jun N-terminal kinases (JNKs) and p38 MAPKs (25). ERK, JNK and p38 MAPK have been reported to play important roles in promoting the activation of pro-apoptotic proteins (26–29). For example, ERK is involved in promoting caspase-3 activation in cisplatin-induced apoptosis (26). Additionally, JNK has been reported to be involved in promoting caspase-9 and caspase-3 activation induced by gemcitabine (27), and it also contributes to Bax activation, a pro-apoptotic Bcl-2 protein, following treatment with sunitinib (28). In addition, p38 MAPK is associated with caspase-8 activation in TGFβ-mediated apoptosis (29).
It has been reported that treatment of cancer cells with the anticancer drugs decreased the mitochondrial membrane potential (∆Ψm) (30), a phenomenon reflecting that mitochondrial outer membrane permeabilization (MOMP) is induced. Activation of several pro-apoptotic proteins, such as Bax, Bak and Bid, has been demonstrated to contribute to MOMP induction (31,32). Once MOMP is induced, cytochrome c is released into the cytosol, which activates caspase-9 (33,34). This in turn activates the downstream effector caspases, such as caspase-3 or caspase-7, resulting in apoptosis (35). Thus, ∆Ψm may serve as an indicator of apoptosis.
Although our previous study demonstrated that paclitaxel can activate caspases and induce apoptosis in HNSCC cells (24), the pivotal signaling pathway for driving caspase activation and apoptosis induced by paclitaxel remains unclear. Thus, the present study aimed to investigate the underlying mechanism of paclitaxel-induced apoptosis in HNSCC cells.
Materials and methods
Reagents and antibodies
Paclitaxel (Sigma-Aldrich; Merck KGaA) was solubilized in dimethyl sulfoxide (DMSO), at a final concentration of 1 mM. SP600125 was dissolved in DMSO as a 100 mM stock solution and stored at −20°C. RNase A, propidium iodide (PI), BSA, DMSO, penicillin/streptomycin, phenylmethylsulfonyl fluoride (PMSF), dithiothreitol, copper sulfate, K+-Na+-tartrate and MTT reagent were purchased from Sigma-Aldrich; Merck KGaA). Fetal bovine serum (FBS), RPMI-1640 medium and lyophilized trypsin-EDTA were purchased from Gibco (Thermo Fisher Scientific, Inc.). Tween-20, sodium hydroxide and hydrochloric acid were purchased from Merck KGaA. SDS, acrylamide and Tris-base were purchased from J.T. Baker (Avantor, Inc.). PD184352 (Enzo Life Sciences, Inc.) was dissolved in DMSO as a 5 mM stock solution and stored at −20°C. SB202190 (Merck KGaA) was dissolved in DMSO as a 10 mM stock solution and stored at −20°C.
Antibodies against cleaved caspase-3, cleaved caspase-6, cleaved caspase-7, cleaved caspase-8, cleaved caspase-9, phospho-JNK, JNK, phospho-ERK, ERK, phospho-p38, p38, β-actin and Bid were purchased from Cell Signaling Technology, Inc. Caspase-8 inhibitor (Z-IETD-FMK) and caspase-9 inhibitor (Z-LEHD-FMK) were purchased from R&D Systems, Inc. HEPES was purchased from Avantor, Inc. Sodium bicarbonate, sodium carbonate and sodium chloride were purchased from Riedel-de Haen (Honeywell International, Inc.).
Cell line and cell culture
OEC-M1 is a cell line derived from a surgical specimen of buccal mucosa squamous carcinoma from a Taiwanese, a unique oral cancer indigenous in Taiwan, which was generously gifted by Professor Kuo-Wei Chang at National Yang-Ming University (Taipei, Taiwan) (36). Cells were maintained and serially passaged in RPMI-1640 medium supplemented with 10% FBS, 24 mM NaHCO3, 25 mM HEPES, 100 U/ml penicillin and 100 µg/ml streptomycin, at pH 7.4, 37°C and in a humidified atmosphere containing 95% air and 5% CO2.
MTT assay
Cell viability was assessed via the MTT assay, as previously described (20). Briefly, cells (1×104 cells/well) were seeded into a 96-well plate. Following incubation for 24 h at 37°C with 5% CO2, cells were treated with different concentrations of paclitaxel for 48 h. Subsequently, MTT reagent was added to each well (0.5 mg/ml final concentration). Following incubation for 4 h, the medium was removed and the precipitate in each well was dissolved in DMSO. The optical density (OD) values were measured at a wavelength of 590 nm, using an ELISA reader (Dynex Opsys MR; Aspect Scientific Ltd.). Each experiment was performed in triplicate.
Flow cytometry
To demonstrate necrotic fractions of paclitaxel-treated cells, cells (6×105) were seeded into 6-cm dishes and treated with different concentrations of paclitaxel for 24 h. Following treatment, cells were trypsinized, washed with PBS and stained with Annexin V for 15 min at room temperature in the dark, then resuspended in staining solution containing RNase A (100 µg/ml in PBS) and PI (40 µg/ml in PBS). Stained cells were analyzed using a FACScan flow cytometer (Becton-Dickinson and Companu), using CellQuest software (version 5.1) (Becton-Dickinson and Company). Cells that were positively stained with Annexin V and PI were considered necrotic cells (37).
Cell cycle analysis and DNA fragmentation were assessed via flow cytometry using PI staining (38). For experiments involving JNK inhibition, cells were treated with 50 nM paclitaxel for 24 h, in the presence or absence of different concentrations of a specific JNK inhibitor, SP600125. For experiments involving caspase-8 or caspase-9 inhibition, cells were treated with caspase-8 inhibitor (Z-IETD-FMK) 100 µM or caspase-9 inhibitor (Z-LEHD-FMK) 100 µM in the presence of 50 nM paclitaxel for 24 h, respectively. Following treatment, cells were trypsinized, washed with PBS and fixed in 70% ethanol for 20 min at room temperature. The fixed cells were then re-washed with PBS and resuspended in staining solution containing RNase A (100 µg/ml in PBS) and PI (40 µg/ml in PBS) for 1 h at room temperature. The fractions of cells in subG1, G0/G1 and G2/M phases were analyzed using a FACScan flow cytometer (Becton-Dickinson and Company), using CellQuest software (Becton-Dickinson and Company). To assess paclitaxel-induced ∆Ψm changes in OEC-M1 cells, a fluorochrome (DiOC6, 3, 3-dihexyloxacarbocyanine) was used, which exclusively emits with the spectrum of green light and accumulates in the mitochondrial matrix under the influence of ∆Ψm (39). Briefly, near-confluent cells were treated with or without 50 nM paclitaxel in culture media for 12, 24, 36 or 48 h, collected and subsequently incubated with 40 nM DiOC6 at 37°C for 20 min. ∆Ψm-related DiOC6 fluorescence was subsequently recorded using an FL1 photomultiplier tube via a FACScan flow cytometer (Becton-Dickinson and Company). Data from 10,000 cells were acquired for each sample. Analysis was performed using CellQuest software (Becton-Dickinson and Company). All experiments were performed at least three times independently.
Western blotting
Cell lysates were harvested from adherent and floating cells following drug treatment and lysed in lysis buffer (50 mM Tris-base, 150 mM NaCl, 1% NP-40, 0.1% SDS, 0.5% deoxychloride acid and 1 mM PMSF). Protein concentration was determined via the Lowry assay (40), using BSA as standard. Proteins from each sample (40 µg/lane) were separated via SDS-PAGE on a 12% gel for caspases-8, −9, phospho-JNK, JNK, phospho-ERK, ERK, phospho-p38 MAPK, p38 MAPK and Bid, or on a 15% gel for caspase-3, −6 and −7. Standard SDS-PAGE running buffer (24 mM Tris/HCl, 0.19 M glycine, 0.5% SDS, pH 8.3) was used as electrophoresis buffer to resolve proteins, which were subsequently transferred onto polyvinylidene difluoride membranes at 80 mA for 1.5 h in transfer buffer (20 mM Tris/HCl, 150 mM glycine, 10% methanol, 0.01% SDS). The membranes were blocked with 5% non-fat milk for 1 h at room temperature and subsequently incubated with the following primary antibodies (all from Cell Signaling Technology, Inc.) for 16–18 h at 4°C: Anti-caspase-3 (cat. no. 9661; 1:1,000), anti-cleaved caspase-6 (cat. no. 9761; 1:1,000), anti-cleaved caspase-7 (cat. no. 8438; 1:1,000), anti-cleaved caspase-8 (cat. no. 9429; 1:1,000), anti-cleaved caspase-9 (cat. no. 9509; 1:1,000), anti-phospho-JNK (cat. no. 9251; 1:4,000), anti-JNK (cat. no. 9252; 1:1,000), anti-phospho-ERK (cat. no. 9101; 1:4,000), anti-ERK (cat. no. 9102; 1:4,000), anti-phospho-p38 (cat. no. 9215; 1:1,000), p38 (cat. no. 9212; 1:4,000), anti-β-actin (cat. No. 58169; 1:5,000), and anti-Bid (cat. no. 8762; 1:1,000). Following the primary incubation, membranes were washed with TBS with Tween 20 (0.15 M NaCl, 0.050 M Tris/HCl, 0.1% Tween-20, pH 7.6), and then incubated with horseradish peroxidase (HRP)-conjugated goat anti-rabbit IgG (cat. no. 111-035-144; 1:5,000; Jackson ImmunoResearch, Inc.) or HRP-conjugated goat anti-mouse IgG (cat. no. 111-035-146; 1:5,000; Jackson ImmunoResearch, Inc.) secondary antibodies for 1 h at room temperature. Protein bands were visualized using the enhanced chemiluminescence detection kit (Amersham; Cytiva), and optical densities were quantitated using a Quantity One (ProVision Diagnostics, Inc.) computer-assisted image analysis system. All experiments were performed at least three times independently.
ELISA
Cytokeratin 18 concentration in the cell culture supernatants was determined using the SimpleStep ELISA kit (cat. no. ab227896; Abcam), according to the manufacturer's instructions.
Statistical analysis
All experiments were performed in triplicate and data are presented as the mean ± standard error of the mean. The statistical analysis was performed using SPSS software version 17.0 (SPSS, Inc.). Unpaired Student's t-test was used to compare differences between two groups, while one-way ANOVA and Tukey's post-hoc test were used to compare differences between multiple groups. P<0.05 was considered to indicate a statistically significant difference.
Results
Paclitaxel (50 nM) induces apoptosis but not necrosis in OEC-M1 cells
It has been reported that low-dose paclitaxel can induce apoptosis, whereas high-dose paclitaxel induces necrosis (41). To determine the optimal experimental concentration of paclitaxel used in the present study, OEC-M1 cells were treated with different concentrations of paclitaxel for 48 h and the MTT assay was performed to assess cell viability. As presented in Fig. 1A, different doses of paclitaxel (50 nM to 1 µM) significantly inhibited cell viability (P<0.05). The necrosis effect of these doses was analyzed via flow cytometry. As presented in Fig. 1B, the necrotic fraction of OEC-M1 cells did not significantly increase following treatment with 50 nM paclitaxel. However, a dose-dependent increase of necrotic cell death was observed following treatment with 250 nM or higher concentrations of paclitaxel (P<0.05). Given that 50 nM paclitaxel induced apoptosis in OEC-M1 cells in a time-dependent manner (P<0.05; Fig. 1C) and did not significantly increase necrosis in cells (Fig. 1B), 50 nM paclitaxel was used as the standard concentration to assess the apoptosis-inducing effect of paclitaxel treatment on OEC-M1 cells.
Figure 1.

Low-dose paclitaxel (50 nM) induces apoptosis but not necrosis in OEC-M1 cells. Cells were treated without or with different concentrations of paclitaxel (5, 50, 250, 500 nM and 1 µM) for 48 h, and the percentage of (A) cell viability and (B) necrotic cell death were determined via the MTT assay and FACScan analysis following Annexin V and PI staining, respectively. (C) Cells were treated with or without 50 nM paclitaxel for 12, 24, 36 and 48 h. Following PI staining, cell cycle events were measured via FACScan analysis. Fractions of subG1 phase (apoptosis) were quantified using CellQuest software. *P<0.05 vs. control. PI, propidium iodide.
JNK is activated in OEC-M1 cells treated with low-dose paclitaxel
The MAPK superfamily governs the ERK, JNK and p38 MAPK pathways, which are distinctly modulated by microtubule-targeting agents, such as paclitaxel (42). Thus, the present study investigated which MAPK kinase pathway is more important in paclitaxel-induced apoptosis of OEC-M1 cells. The JNK pathway was activated (phosphorylated) following treatment with 50 nM paclitaxel at 12 and 24 h (P<0.05; Fig. 2A and B); the ERK pathway was stimulated (phosphorylated) following treatment with 50 nM paclitaxel at 6, 12, 24 and 36 h (P<0.05; Fig. 2A and C); and the p38 pathway was induced (phosphorylated) following treatment with 50 nM paclitaxel at 24 h (P<0.05; Fig. 2A and D) in OEC-M1 cells, respectively.
Figure 2.

Activation of the MAPK pathway in low-dose paclitaxel-treated OEC-M1 cells. Cells were treated with control or 50 nM paclitaxel at different time points. (A) Western blot analysis was performed to detect the protein expression levels of JNK, ERK and p38 MAPK pathways in OEC-M1 cells. Activation of (B) JNK, (C) ERK and (D) p38 were quantified using Quantity One image analysis system. OEC-M1 cells were treated with control or 50 nM paclitaxel, without or with 10 µM SP, 5 µM PD and 5 µM SB, respectively, and (E) the expression levels of cytokeratin 18 were determined via ELISA. *P<0.05 vs. control; #P<0.05 vs. paclitaxel alone treatment. MAPK, mitogen-activated protein kinase; JNK, c-Jun N-terminal kinase; ERK, extracellular signal-regulated kinase; C, control, P, paclitaxel; SP, JNK inhibitor-SP600125; PD, ERK inhibitor-PD184352; SB, p38 inhibitor-SB202190.
Caspase-cleaved cytokeratin 18, a biomarker of apoptotic cell death, is released from epithelial cells during apoptosis (43). The results of the present study demonstrated that paclitaxel significantly promoted the release of cytokeratin 18 from OEC-M1 cells compared with the control group (P<0.05; Fig. 2E). Treatment with JNK inhibitor, but not ERK inhibitor or p38 inhibitor, remarkably inhibited the paclitaxel-induced release of cytokeratin 18 (P<0.05; Fig. 2E), suggesting that the JNK pathway is the candidate MAPK pathway mediating paclitaxel-induced apoptosis in OEC-M1 cells.
Inactivation of JNK effectively prevents paclitaxel-induced apoptosis and caspase activation in OEC-M1 cells
Our previous study demonstrated that paclitaxel can induce apoptosis by activating caspases in the two HNSCC cell lines, OEC-M1 and OC3, through the activation of initiator caspases (caspase-8 and −9) and effector caspases (caspase-3, −6 and −7) (24). The results of the present study demonstrated that paclitaxel-activated JNK was important for apoptosis in OEC-M1 cells (Fig. 2). To determine the role of JNK activation in paclitaxel-induced apoptosis, OEC-M1 cells were treated with 50 nM paclitaxel, with or without different concentrations of JNK inhibitor, SP600125, for 24 h. Apoptosis was subsequently quantified by measuring the subG1 fraction via flow cytometric analysis. As presented in Fig. 3A, 10 µM SP600125 abolished JNK activation and cleaved caspase-3 expression to near background level, respectively. The expression levels of cleaved caspase-6 and −7 were also blocked following treatment with 10 µM SP600125, respectively (Fig. 3B). Inhibition of JNK activation with 10 µM SP600125 effectively rescued paclitaxel-induced apoptosis in OEC-M1 cells (P<0.05; Fig. 3C and D), suggesting that JNK activation plays a central role in low-dose paclitaxel-induced apoptosis of OEC-M1 cells. JNK inhibition also significantly decreased the G0/G1 fraction (P<0.05; Fig. 3C and E), and notably increased the G2/M fraction (P<0.05; Fig. 3C and F) in paclitaxel-treated OEC-M1 cells.
Figure 3.

JNK inhibitor (SP600125) inhibits paclitaxel-induced apoptosis and caspases effectors in OEC-M1 cells. JNK inhibitor, SP600125, (0.1, 1, 10 and 20 µM) inhibited paclitaxel-induced phosphorylation of (A) JNK and cleaved caspase-3, and (B) cleaved caspase-6 and cleaved caspase-7. Cells (6×105) were subsequently treated with 50 nM paclitaxel, with or without different concentrations of SP600125 (10 and 20 µM) for 24 h and fixed in 70% alcohol. (C) Following PI staining, cell cycle events were measured via FACScan analysis. Fractions of (D) subG1 phase (apoptosis), (E) G0/G1 phase and (F) G2/M phase were quantified using CellQuest software. In (C) red and blue dotted lines were plotted to illustrate the changes of subG1 (left to red line), G0/G1 (between red and blue lines) and G2/M phases (right to blue line) in the different treatment groups. *P<0.05 vs. control; #P<0.05 vs. paclitaxel alone treatment. JNK, c-Jun N-terminal kinase; PI, propidium iodide.
As presented in Fig. 4, paclitaxel-induced activation of initiator caspase-8 and −9 was significantly inhibited to near background level with JNK inhibitors, respectively (P<0.05; Fig. 4A and B), suggesting that low-dose paclitaxel-induced activation of caspases is mediated via the JNK pathway.
Figure 4.

JNK inhibition prevents paclitaxel-induced activation of caspase-8 and −9 initiators in OEC-M1 cells. Cells were treated with 50 nM paclitaxel, with or without different concentrations of SP600125 (0.1, 1, 10 and 20 µM) for 24 h. Cleavage of initiator (A) caspase-8 and (B) caspase-9 were detected via western blotting and quantified, respectively. *P<0.05 vs. control; #P<0.05 vs. paclitaxel alone treatment. JNK, c-Jun N-terminal kinase.
Inhibition of caspase-8 and −9 activation partially rescues paclitaxel-induced apoptosis in OEC-M1 cells
It has been demonstrated that microtubule-targeting agents (MTA) preferentially activate different caspases in different types of cancer (42,44–46). Thus, the present study investigated whether caspase-8 and −9 activation is responsible for the apoptosis-inducing effect caused by low-dose paclitaxel treatment in OEC-M1 cells. Notably, addition of either caspase-8 inhibitor (Z-IETD-FMK) or caspase-9 inhibitor (Z-LEHD-FMK) rescued about half of the apoptotic cells following treatment with paclitaxel in OEC-M1 cells (P<0.05; Fig. 5A and B). Neither caspase inhibitors affected the G0/G1 fraction of paclitaxel-treated OEC-M1 cells (P<0.05; Fig. 5C). However, both caspase inhibitors increased the G2/M fraction of paclitaxel-treated cells (P<0.05; Fig. 5D), suggesting that a significant proportion of paclitaxel-treated cells are arrested in G2/M phase undergoing apoptosis through activation of initiator caspase-8 and −9. In addition, a less than additive effect was observed when the two caspase inhibitors were used in combination (Fig. 5E and F). Taken together, these results suggest that other caspases, or caspase-independent apoptotic mechanism, may also be involved in paclitaxel-induced apoptosis in OEC-M1 cells.
Figure 5.

Caspase inhibitors partially rescue paclitaxel-induced apoptosis in OEC-M1 cells. (A) Cells were treated with 50 nM paclitaxel with either caspase-8 (Z-IETD-FMK) and/or caspase-9 (Z-LEHD-FMK) inhibitor (100 µM) for 24 h, and cell cycle events were measured via FACScan analysis following PI staining. Fractions of (B) subG1 phase, (C) G0/G1 phase and (D) G2/M phase were quantified using CellQuest software. (E) Combined usage of these two caspase inhibitors, with or without treatment with 50 nM paclitaxel for 24 h, and cell cycle events were measured via FACScan analysis following PI staining. (F) Fractions of subG1 phase were quantified using CellQuest software. In (A) and (E) red and blue dotted lines were plotted to illustrate the changes of subG1 (left to red line), G0/G1 (between red and blue lines) and G2/M phases (right to blue line) in the different treatment groups. *P<0.05 vs. control; #P<0.05 vs. paclitaxel alone treatment. PI, propidium iodide.
Inactivation of JNK inhibits late-phase (48 h) ∆Ψm loss in OEC-M1 cells
The effectiveness of MTAs is a consequence of caspase activation through the intrinsic mitochondrial apoptotic pathway (42), which is closely associated with ∆Ψm collapse (33). To investigate the association between JNK activation and ∆Ψm collapse, the preset study investigated whether low-dose paclitaxel can affect ∆Ψm in OEC-M1 cells. As presented in Fig. 6A, 50 nM paclitaxel significantly induced depolarization of ∆Ψm of OEC-M1 cells, in a time-dependent manner (P<0.05). The present study also investigated whether the ∆Ψm loss is associated with JNK activation. Notably, inactivation of JNK failed to reverse the early-phase (24 h) ∆Ψm loss of OEC-M1 cells, suggesting that JNK activation is dispensable for the early-phase ∆Ψm loss (Fig. 6B). However, inactivation of JNK effectively prevented paclitaxel-induced late-phase (48 h) ∆Ψm loss (P<0.05; Fig. 6B), suggesting that JNK activation is responsible for the late-phase (48 h) ∆Ψm collapse. The JNK-dependent, late-phase ∆Ψm loss may, at least partially, be explained by caspase-8-mediated truncation of Bid occurring at 36 and 48 h following treatment with low-dose paclitaxel (P<0.05; Fig. 6C).
Figure 6.

Inactivation of JNK inhibits late-phase (48 h) ∆Ψm loss in OEC-M1 cells. (A) Paclitaxel induced time-dependent ∆Ψm loss in OEC-M1 cells. (B) Inactivation of JNK with specific JNK inhibitor (SP600125, 10 µM) only significantly rescued the late-phase (48 h) ∆Ψm loss. (C) Bid truncation occurred at 36 and 48 h following treatment with paclitaxel. *P<0.05 vs. control; #P<0.05 vs. paclitaxel alone treatment. JNK, c-Jun N-terminal kinase; ∆Ψm, mitochondrial membrane potential; C, control, P, paclitaxel.
Schematic chart of paclitaxel action on the apoptosis of OEC-M1 cells
The potential signaling pathway of paclitaxel-induced apoptosis in OEC-M1 cells is illustrated in Fig. 7, which shows that JNK activated by paclitaxel contributes to caspase-8 activation and ∆Ψm reduction. Activation of caspase-8 results in Bid cleavage, which may contribute to ∆Ψm loss (47). The reduction of ∆Ψm results in activation of caspase-9 (33,34). Activation of both caspase-8 and −9 promote activation of the downstream caspases (caspase-3, −6 and −7) (48), resulting in apoptosis of OEC-M1 cells.
Figure 7.
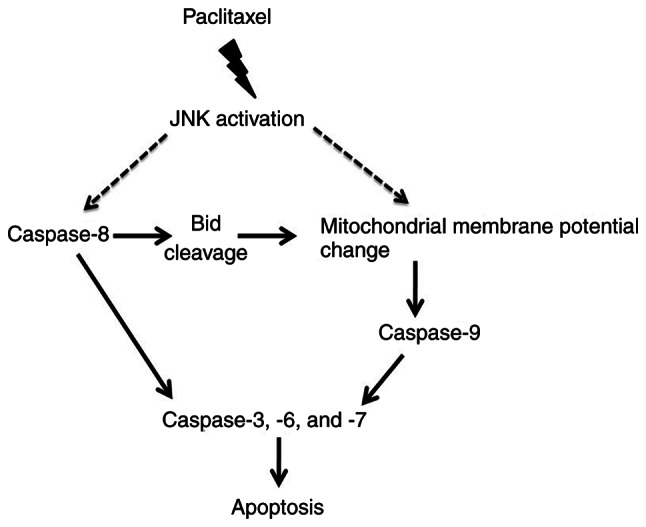
Potential signaling pathways of paclitaxel-induced apoptosis in OEC-M1 cells. Paclitaxel-induced JNK activation plays an important role in promoting loss of ∆Ψm and activation of pro-apoptotic proteins, resulting in apoptosis of OEC-M1 cells. JNK, c-Jun N-terminal kinase; ∆Ψm, mitochondrial membrane potential.
Discussion
The results of the present study demonstrated that most of the cytotoxic effect of paclitaxel was achieved at a relatively low concentration (50 nM) compared with high concentration (1 µM) of paclitaxel in OEC-M1 cells. A similar cytotoxic plateau has been reported in breast cancer cells and, less remarkably, in OEC-M1 cells (41,49). The cytotoxic plateau phenomenon observed in the present study may largely be due to the apoptotic plateau in OEC-M1 cells treated with different concentrations of paclitaxel (50 nM to 1 µM; data not shown). Similar to the results of present study, an apoptotic plateau has also been reported in paclitaxel-treated breast cancer cells (41), as well as in paclitaxel-treated HNSCC histocultures (46). These results suggest that most of the apoptotic-inducing effect in HNSCC cells can be achieved with a relative low dose of paclitaxel. In addition, the results of the present study demonstrated that in OEC-M1 cells, high concentrations of paclitaxel (250 and 500 nM and 1 µM) induced necrosis, an unprogrammed form of cell death (50). It is well-known that necrotic cells are able to release cellular cytoplasmic contents into extracellular space, which evoke inflammatory reactions and contribute to tumor progression (51). Given that high concentrations of paclitaxel can induce necrosis and prolonged exposure to paclitaxel concentrations exceeding the thresholds of 0.05 or 0.1 µM induce side effects in patients with cancer, such as neutropenia and peripheral neuropathy (52), in terms of clinical usage, lower doses of paclitaxel are recommended to minimize serious side effects while retaining most of the apoptosis-inducing effect of this drug. This concept is supported by a recent report demonstrating that, in treating patients with metastatic breast cancer, low-dose weekly docetaxel/paclitaxel infusion schedules can achieve overall response rates comparable to several high dose infusion schedules, with fewer grade 3–4 toxicities (53).
It has been reported that paclitaxel not only causes JNK activation but also causes ERK inactivation and a reduction of basal p38 MAPK activity concomitantly in KB-3 human epidermoid carcinoma cells (54). However, in the present study, JNK, ERK and p38 MAPK were activated by paclitaxel. This difference may be caused by the high paclitaxel concentration used in the previous study (500 nM) compared with the present study (50 nM). Furthermore, activation of specific MAPK pathways may be cell-type specific.
The results of the present study demonstrated that paclitaxel-induced JNK activation was responsible for most of the apoptosis-inducing effect in OEC-M1 cells. JNK activation was also responsible for paclitaxel-induced caspase activation. However, the effect of inactivation of caspase-8 and/or caspase-9 in rescuing paclitaxel-induced apoptosis was inferior compared with the effect of JNK inhibition, suggesting that other caspase, such as caspase-10 (55), or caspase-independent apoptosis-like programmed cell death (56,57), may also be involved in JNK-mediated, paclitaxel-induced apoptosis of HNSCC cells.
Failure of JNK inactivation to prevent early-phase (24 h) depolarization of OEC-M1 cells suggests that early-phase ∆Ψm loss is not caused by JNK activation. The JNK-dependent, late-phase (48 h) ∆Ψm loss can be explained, at least partially, by caspase-8-induced truncation of Bid (a process of Bid activation), which occurred at 36 and 48 h following treatment with paclitaxel. Whether paclitaxel-induced JNK activation also causes activation of other pro-apoptotic ‘BH-3 only’ proteins (58,59) and contributes to the late-phase (48 h) ∆Ψm loss remain unknown.
The upstream events leading to JNK activation by low-dose paclitaxel treatment are yet to be investigated. Reactive oxygen species (ROS) production has been reported to be an early and crucial step for paclitaxel-induced lung cancer cell death (60). ROS production has also been reported to mediate docetaxel-induced apoptosis of HNSCC cells (61), and ROS is associated with ASK1 activation, an upstream kinase of JNK (62). On the other hand, paclitaxel treatment may also induce ceramide formation (63,64), which can subsequently lead to both JNK activation (65) and ∆Ψm collapse (66,67). It is speculated that paclitaxel may induce activation of JNK via the ROS-ASK1 axis and ceramide formation. However, further studies are required to verify these speculations.
p53 is able to upregulate several pro-apoptosis related proteins, such as Bax and PUMA (68,69), or promote activation of Bid and caspases (70,71). Thus, p53 plays an important role in regulating apoptosis (72). It has been reported that JNK can promote apoptosis by activating p53 (73). OEC-M1 is a cell line with p53 mutation, and our previous study demonstrated that paclitaxel can induce apoptosis and activation of caspases in OEC-M1 cells (24), suggesting that p53 is not involved in paclitaxel-induced apoptosis. Apoptosis can be divided into two pathways, death receptor apoptotic pathway (the extrinsic pathway) and mitochondrial apoptotic pathway (the intrinsic pathway) (48). The former, activated by binding of Fas ligand to its receptor, Fas, leads to formation of death-inducing signaling complex, which promotes activation of initiator caspase-8 (48). The latter, activated by activation of several pro-apoptotic Bcl-2 proteins, such as Bim and Bad, results in formation of apoptosome, which promotes initiator caspase-9 activation (48). In addition, the extrinsic pathway is involved in activating the intrinsic pathway through activation of the caspase-8-Bid axis (48). The results of the present study demonstrated that paclitaxel activated caspase-8, Bid and caspase-9, and caused ∆Ψm loss, suggesting that both pathways are activated in paclitaxel-induced apoptosis. Given that JNK can upregulate Fas ligand (74) and promote the activation of Bax and Bim (58,75), it was speculated that during the progression of paclitaxel-induced apoptosis, JNK may trigger activation of the extrinsic and the intrinsic pathways to activate the initiator caspases (caspase-8 and −9) in OEC-M1 cells, which may contribute to activation of downstream caspases (caspase-3, −6 and −7). Prospective studies are required to investigate whether paclitaxel can upregulate Fas ligand and activate Bax and Bim.
In conclusion, the results of the present study demonstrated that paclitaxel-activated JNK is required for caspase activation and loss of ∆Ψm to induce apoptosis of OEC-M1 cells. To the best of our knowledge, the present study was the first to demonstrate that JNK can activate caspases and reduce ∆Ψm, which is important for paclitaxel-induced apoptosis of OEC-M1 cells. These results may be applied to improve or enhance the therapeutic efficacy of paclitaxel as a chemotherapeutic drug.
Acknowledgements
Not applicable.
Glossary
Abbreviations
- ERK
extracellular signal-regulated kinase
- HNSCC
head and neck squamous cell carcinoma
- JNK
c-Jun N-terminal kinase
- MAPK
mitogen-activated protein kinase
- MOMP
mitochondrial outer membrane permeabilization
- MTA
microtubule-targeting agents
- ROS
reactive oxygen species
- ∆Ψm
mitochondrial membrane potential
Funding Statement
The present study was supported by the Ministry of Science and Technology, Taiwan (grant no. MOST-107-2320-B-471-001 to YYL; grant no. MOST-109-2314-B-218-001 to HYC; and grant nos. MOST-105-2320-B-006-028-MY3 and MOST-106-2320-B-006-001-MY3 to BMH).
Funding
The present study was supported by the Ministry of Science and Technology, Taiwan (grant no. MOST-107-2320-B-471-001 to YYL; grant no. MOST-109-2314-B-218-001 to HYC; and grant nos. MOST-105-2320-B-006-028-MY3 and MOST-106-2320-B-006-001-MY3 to BMH).
Availability of data and materials
The datasets used and analyzed in the present study are available from the corresponding authors upon reasonable request.
Authors' contributions
YYL, YHC, CL and BMH designed the present study and analyzed the data. YYL, YHC, KLT, YTW and SCL performed the experiments. YCC, CHW and HYC interpreted the results. YYL, YHC, CL and YCC drafted the initial manuscript. YCC, CHW, HYC and BMH revised the manuscript for important intellectual content. YYL, YCC and BMH confirm the authenticity of all the raw data. All authors read and approved the final manuscript and agreed to be accountable for all aspects of the research in ensuring that the accuracy or integrity of any part of the work are appropriately investigated and resolved.
Ethics approval and consent to participate
Not applicable.
Patient consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing interests.
References
- 1.Rades D, Seidl D, Wollenberg B, Schild SE, Hakim SG. Radiochemotherapy with paclitaxel for recurrent previously irradiated squamous cell carcinoma of the head and neck. Anticancer Res. 2016;36:5463–5468. doi: 10.21873/anticanres.11126. [DOI] [PubMed] [Google Scholar]
- 2.Forastiere AA, Goepfert H, Maor M, Pajak TF, Weber R, Morrison W, Glisson B, Trotti A, Ridge JA, Chao C, et al. Concurrent chemotherapy and radiotherapy for organ preservation in advanced laryngeal cancer. N Engl J Med. 2003;349:2091–2098. doi: 10.1056/NEJMoa031317. [DOI] [PubMed] [Google Scholar]
- 3.Psyrri A, Kwong M, DiStasio S, Lekakis L, Kassar M, Sasaki C, Wilson LD, Haffty BG, Son YH, Ross DA, et al. Cisplatin, fluorouracil, and leucovorin induction chemotherapy followed by concurrent cisplatin chemoradiotherapy for organ preservation and cure in patients with advanced head and neck cancer: Long-term follow-up. J Clin Oncol. 2004;22:3061–3069. doi: 10.1200/JCO.2004.01.108. [DOI] [PubMed] [Google Scholar]
- 4.Lo Nigro C, Denaro N, Merlotti A, Merlano M. Head and neck cancer: Improving outcomes with a multidisciplinary approach. Cancer Manag Res. 2017;9:363–371. doi: 10.2147/CMAR.S115761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Adamo V, Ferraro G, Pergolizzi S, Sergi C, Laudani A, Settineri N, Alafaci E, Scimone A, Spano F, Spitaleri G. Paclitaxel and cisplatin in patients with recurrent and metastatic head and neck squamous cell carcinoma. Oral Oncol. 2004;40:525–531. doi: 10.1016/j.oraloncology.2003.10.010. [DOI] [PubMed] [Google Scholar]
- 6.Monnerat C, Faivre S, Temam S, Bourhis J, Raymond E. End points for new agents in induction chemotherapy for locally advanced head and neck cancers. Ann Oncol. 2002;13:995–1006. doi: 10.1093/annonc/mdf172. [DOI] [PubMed] [Google Scholar]
- 7.Pignon JP, Bourhis J, Domenge C, Designe L. Chemotherapy added to locoregional treatment for head and neck squamous-cell carcinoma: Three meta-analyses of updated individual data. MACH-NC Collaborative Group. Meta-analysis of chemotherapy on head and neck cancer. Lancet. 2000;355:949–955. doi: 10.1016/S0140-6736(00)90011-4. [DOI] [PubMed] [Google Scholar]
- 8.Pendleton KP, Grandis JR. Cisplatin-based chemotherapy options for recurrent and/or metastatic squamous cell cancer of the head and neck. Clin Med Insights Ther. 2013 doi: 10.4137/CMT.S10409. 10.4137/CMT.S10409, 2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Miller KD, Siegel RL, Lin CC, Mariotto AB, Kramer JL, Rowland JH, Stein KD, Alteri R, Jemal A. Cancer treatment and survivorship statistics, 2016. CA Cancer J Clin. 2016;66:271–289. doi: 10.3322/caac.21349. [DOI] [PubMed] [Google Scholar]
- 10.Rowinsky EK, Donehower RC. Paclitaxel (taxol) N Engl J Med. 1995;332:1004–1014. doi: 10.1056/NEJM199504133321507. [DOI] [PubMed] [Google Scholar]
- 11.Abu Samaan TM, Samec M, Liskova A, Kubatka P, Busselberg D. Paclitaxel's mechanistic and clinical effects on breast cancer. Biomolecules. 2019;9:789. doi: 10.3390/biom9120789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Habib S, Delourme J, Dhalluin X, Petyt G, Tacelli N, Scherpereel A, Lafitte JJ, Cortot AB. Bevacizumab and weekly paclitaxel for non-squamous non small cell lung cancer patients: A retrospective study. Lung Cancer. 2013;80:197–202. doi: 10.1016/j.lungcan.2013.01.015. [DOI] [PubMed] [Google Scholar]
- 13.Specenier P, Vermorken JB. The role of taxanes and targeted therapies in locally advanced head and neck cancer. Curr Opin Oncol. 2007;19:195–201. doi: 10.1097/CCO.0b013e3280f00fe7. [DOI] [PubMed] [Google Scholar]
- 14.Ferrari D, Ghi MG, Franzese C, Codeca C, Gau M, Fayette J. The slippery role of induction chemotherapy in head and neck cancer: Myth and reality. Front Oncol. 2020;10:7. doi: 10.3389/fonc.2020.00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Xiao P, Ma T, Zhou C, Xu Y, Liu Y, Zhang H. Anticancer effect of docetaxel induces apoptosis of prostate cancer via the cofilin-1 and paxillin signaling pathway. Mol Med Rep. 2016;13:4079–4084. doi: 10.3892/mmr.2016.5000. [DOI] [PubMed] [Google Scholar]
- 16.Miller AV, Hicks MA, Nakajima W, Richardson AC, Windle JJ, Harada H. Paclitaxel-induced apoptosis is BAK-dependent, but BAX and BIM-independent in breast tumor. PLoS One. 2013;8:e60685. doi: 10.1371/journal.pone.0060685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Han TD, Shang DH, Tian Y. Docetaxel enhances apoptosis and G2/M cell cycle arrest by suppressing mitogen-activated protein kinase signaling in human renal clear cell carcinoma. Genet Mol Res. 2016;15 doi: 10.4238/gmr.15017321. [DOI] [PubMed] [Google Scholar]
- 18.Pan Z, Avila A, Gollahon L. Paclitaxel induces apoptosis in breast cancer cells through different calcium-regulating mechanisms depending on external calcium conditions. Int J Mol Sci. 2014;15:2672–2694. doi: 10.3390/ijms15022672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hu J, Zhang NA, Wang R, Huang F, Li G. Paclitaxel induces apoptosis and reduces proliferation by targeting epidermal growth factor receptor signaling pathway in oral cavity squamous cell carcinoma. Oncol Lett. 2015;10:2378–2384. doi: 10.3892/ol.2015.3499. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 20.Nonaka M, Ikeda H, Fujisawa A, Uehara M, Inokuchi T. Induction of apoptosis by paclitaxel in human oral carcinoma cells. Int J Oral Maxillofac Surg. 2006;35:649–652. doi: 10.1016/j.ijom.2006.01.011. [DOI] [PubMed] [Google Scholar]
- 21.Holsinger FC, Doan DD, Jasser SA, Swan EA, Greenberg JS, Schiff BA, Bekele BN, Younes MN, Bucana CD, Fidler IJ, Myers JN. Epidermal growth factor receptor blockade potentiates apoptosis mediated by Paclitaxel and leads to prolonged survival in a murine model of oral cancer. Clin Cancer Res. 2003;9:3183–3189. [PubMed] [Google Scholar]
- 22.Ganansia-Leymarie V, Bischoff P, Bergerat JP, Holl V. Signal transduction pathways of taxanes-induced apoptosis. Curr Med Chem Anticancer Agents. 2003;3:291–306. doi: 10.2174/1568011033482422. [DOI] [PubMed] [Google Scholar]
- 23.Huang CY, Ju DT, Chang CF, Muralidhar Reddy P, Velmurugan BK. A review on the effects of current chemotherapy drugs and natural agents in treating non-small cell lung cancer. Biomedicine (Taipei) 2017;7:23. doi: 10.1051/bmdcn/2017070423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hsiao JR, Leu SF, Huang BM. Apoptotic mechanism of paclitaxel-induced cell death in human head and neck tumor cell lines. J Oral Pathol Med. 2009;38:188–197. doi: 10.1111/j.1600-0714.2008.00732.x. [DOI] [PubMed] [Google Scholar]
- 25.Cargnello M, Roux PP. Activation and function of the MAPKs and their substrates, the MAPK-activated protein kinases. Microbiol Mol Biol Rev. 2011;75:50–83. doi: 10.1128/MMBR.00031-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Wang X, Martindale JL, Holbrook NJ. Requirement for ERK activation in cisplatin-induced apoptosis. J Biol Chem. 2000;275:39435–39443. doi: 10.1074/jbc.M004583200. [DOI] [PubMed] [Google Scholar]
- 27.Teraishi F, Zhang L, Guo W, Dong F, Davis JJ, Lin A, Fang B. Activation of c-Jun NH2-terminal kinase is required for gemcitabine's cytotoxic effect in human lung cancer H1299 cells. FEBS Lett. 2005;579:6681–6687. doi: 10.1016/j.febslet.2005.10.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Uzu M, Sato H, Shimizu A, Shibata Y, Ueno K, Hisaka A. Connexin 43 enhances Bax activation via JNK activation in sunitinib-induced apoptosis in mesothelioma cells. J Pharmacol Sci. 2017;134:101–107. doi: 10.1016/j.jphs.2017.05.005. [DOI] [PubMed] [Google Scholar]
- 29.Schrantz N, Bourgeade MF, Mouhamad S, Leca G, Sharma S, Vazquez A. p38-mediated regulation of an Fas-associated death domain protein-independent pathway leading to caspase-8 activation during TGFbeta-induced apoptosis in human Burkitt lymphoma B cells BL41. Mol Biol Cell. 2001;12:3139–3151. doi: 10.1091/mbc.12.10.3139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhao H, Wu S, Li H, Duan Q, Zhang Z, Shen Q, Wang C, Yin T. ROS/KRAS/AMPK signaling contributes to gemcitabine-induced stem-like cell properties in pancreatic cancer. Mol Ther Oncolytics. 2019;14:299–312. doi: 10.1016/j.omto.2019.07.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.James D, Parone PA, Terradillos O, Lucken-Ardjomande S, Montessuit S, Martinou JC. Mechanisms of mitochondrial outer membrane permeabilization. Novartis Found Symp. 2007;287:170–176. discussion 176-182. [PubMed] [Google Scholar]
- 32.Gahl RF, Dwivedi P, Tjandra N. Bcl-2 proteins bid and bax form a network to permeabilize the mitochondria at the onset of apoptosis. Cell Death Dis. 2016;7:e2424. doi: 10.1038/cddis.2016.320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Tait SW, Green DR. Mitochondrial regulation of cell death. Cold Spring Harb Perspect Biol. 2013;5:a008706. doi: 10.1101/cshperspect.a008706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li P, Zhou L, Zhao T, Liu X, Zhang P, Liu Y, Zheng X, Li Q. Caspase-9: Structure, mechanisms and clinical application. Oncotarget. 2017;8:23996–24008. doi: 10.18632/oncotarget.15098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McIlwain DR, Berger T, Mak TW. Caspase functions in cell death and disease. Cold Spring Harb Perspect Biol. 2015;7:a026716. doi: 10.1101/cshperspect.a026716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Foo NP, Ko CL, Chu CY, Wang CY, So EC, Huang BM. Arsenic compounds activate the MAPK and caspase pathways to induce apoptosis in OEC-M1 gingival epidermal carcinoma. Oncol Rep. 2020;44:2701–2714. doi: 10.3892/or.2020.7793. [DOI] [PubMed] [Google Scholar]
- 37.Krysko DV, Vanden Berghe T, D'Herde K, Vandenabeele P. Apoptosis and necrosis: Detection, discrimination and phagocytosis. Methods. 2008;44:205–221. doi: 10.1016/j.ymeth.2007.12.001. [DOI] [PubMed] [Google Scholar]
- 38.Nicoletti I, Migliorati G, Pagliacci MC, Grignani F, Riccardi C. A rapid and simple method for measuring thymocyte apoptosis by propidium iodide staining and flow cytometry. J Immunol Methods. 1991;139:271–279. doi: 10.1016/0022-1759(91)90198-O. [DOI] [PubMed] [Google Scholar]
- 39.Galluzzi L, Zamzami N, de La Motte Rouge T, Lemaire C, Brenner C, Kroemer G. Methods for the assessment of mitochondrial membrane permeabilization in apoptosis. Apoptosis. 2007;12:803–813. doi: 10.1007/s10495-007-0720-1. [DOI] [PubMed] [Google Scholar]
- 40.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. doi: 10.1016/S0021-9258(19)52451-6. [DOI] [PubMed] [Google Scholar]
- 41.Yeung TK, Germond C, Chen X, Wang Z. The mode of action of taxol: Apoptosis at low concentration and necrosis at high concentration. Biochem Biophys Res Commun. 1999;263:398–404. doi: 10.1006/bbrc.1999.1375. [DOI] [PubMed] [Google Scholar]
- 42.Esteve MA, Carre M, Braguer D. Microtubules in apoptosis induction: Are they necessary? Curr Cancer Drug Targets. 2007;7:713–729. doi: 10.2174/156800907783220480. [DOI] [PubMed] [Google Scholar]
- 43.Ulukaya E, Karaagac E, Ari F, Oral AY, Adim SB, Tokullugil AH, Evrensel T. Chemotherapy increases caspase-cleaved cytokeratin 18 in the serum of breast cancer patients. Radiol Oncol. 2011;45:116–122. doi: 10.2478/v10019-011-0006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lu KH, Lue KH, Chou MC, Chung JG. Paclitaxel induces apoptosis via caspase-3 activation in human osteogenic sarcoma cells (U-2 OS) J Orthop Res. 2005;23:988–994. doi: 10.1016/j.orthres.2005.01.018. [DOI] [PubMed] [Google Scholar]
- 45.Mhaidat NM, Wang Y, Kiejda KA, Zhang XD, Hersey P. Docetaxel-induced apoptosis in melanoma cells is dependent on activation of caspase-2. Mol Cancer Ther. 2007;6:752–761. doi: 10.1158/1535-7163.MCT-06-0564. [DOI] [PubMed] [Google Scholar]
- 46.Janssen K, Pohlmann S, Janicke RU, Schulze-Osthoff K, Fischer U. Apaf-1 and caspase-9 deficiency prevents apoptosis in a Bax-controlled pathway and promotes clonogenic survival during paclitaxel treatment. Blood. 2007;110:3662–3672. doi: 10.1182/blood-2007-02-073213. [DOI] [PubMed] [Google Scholar]
- 47.Li H, Zhu H, Xu CJ, Yuan J. Cleavage of BID by caspase 8 mediates the mitochondrial damage in the Fas pathway of apoptosis. Cell. 1998;94:491–501. doi: 10.1016/S0092-8674(00)81590-1. [DOI] [PubMed] [Google Scholar]
- 48.Baig S, Seevasant I, Mohamad J, Mukheem A, Huri HZ, Kamarul T. Potential of apoptotic pathway-targeted cancer therapeutic research: Where do we stand? Cell Death Dis. 2016;7:e2058. doi: 10.1038/cddis.2015.275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Huang GC, Liu SY, Lin MH, Kuo YY, Liu YC. The synergistic cytotoxicity of cisplatin and taxol in killing oral squamous cell carcinoma. Jpn J Clin Oncol. 2004;34:499–504. doi: 10.1093/jjco/hyh091. [DOI] [PubMed] [Google Scholar]
- 50.Dhuriya YK, Sharma D. Necroptosis: A regulated inflammatory mode of cell death. J Neuroinflammation. 2018;15:199. doi: 10.1186/s12974-018-1235-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lee SY, Ju MK, Jeon HM, Jeong EK, Lee YJ, Kim CH, Park HG, Han SI, Kang HS. Regulation of tumor progression by programmed necrosis. Oxid Med Cell Longev. 2018;2018:3537471. doi: 10.1155/2018/3537471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Mielke S. Individualized pharmacotherapy with paclitaxel. Curr Opin Oncol. 2007;19:586–589. doi: 10.1097/CCO.0b013e3282a08f8c. [DOI] [PubMed] [Google Scholar]
- 53.Eniu A, Palmieri FM, Perez EA. Weekly administration of docetaxel and paclitaxel in metastatic or advanced breast cancer. Oncologist. 2005;10:665–685. doi: 10.1634/theoncologist.10-9-665. [DOI] [PubMed] [Google Scholar]
- 54.Stone AA, Chambers TC. Microtubule inhibitors elicit differential effects on MAP kinase (JNK, ERK, and p38) signaling pathways in human KB-3 carcinoma cells. Exp Cell Res. 2000;254:110–119. doi: 10.1006/excr.1999.4731. [DOI] [PubMed] [Google Scholar]
- 55.Park SJ, Wu CH, Gordon JD, Zhong X, Emami A, Safa AR. Taxol induces caspase-10-dependent apoptosis. J Biol Chem. 2004;279:51057–51067. doi: 10.1074/jbc.M406543200. [DOI] [PubMed] [Google Scholar]
- 56.Broker LE, Kruyt FA, Giaccone G. Cell death independent of caspases: A review. Clin Cancer Res. 2005;11:3155–3162. doi: 10.1158/1078-0432.CCR-04-2223. [DOI] [PubMed] [Google Scholar]
- 57.Constantinou C, Papas KA, Constantinou AI. Caspase-independent pathways of programmed cell death: The unraveling of new targets of cancer therapy? Curr Cancer Drug Targets. 2009;9:717–728. doi: 10.2174/156800909789271512. [DOI] [PubMed] [Google Scholar]
- 58.Qu L, Liu FX, Cao XC, Xiao Q, Yang X, Ren KQ. Activation of the apoptosis signal-regulating kinase 1/c-Jun N-terminal kinase pathway is involved in the casticin-induced apoptosis of colon cancer cells. Exp Ther Med. 2014;8:1494–1500. doi: 10.3892/etm.2014.1934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Brockmann A, Bluwstein A, Kogel A, May S, Marx A, Tschan MP, Brunner T. Thiazolides promote apoptosis in colorectal tumor cells via MAP kinase-induced Bim and Puma activation. Cell Death Dis. 2015;6:e1778. doi: 10.1038/cddis.2015.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alexandre J, Batteux F, Nicco C, Chéreau C, Laurent A, Guillevin L, Weill B, Goldwasser F. Accumulation of hydrogen peroxide is an early and crucial step for paclitaxel-induced cancer cell death both in vitro and in vivo. Int J Cancer. 2006;119:41–48. doi: 10.1002/ijc.21685. [DOI] [PubMed] [Google Scholar]
- 61.Taniguchi T, Takahashi M, Shinohara F, Sato T, Echigo S, Rikiishi H. Involvement of NF-kappaB and mitochondrial pathways in docetaxel-induced apoptosis of human oral squamous cell carcinoma. Int J Mol Med. 2005;15:667–673. [PubMed] [Google Scholar]
- 62.Zhao Q, Liu Y, Zhong J, Bi Y, Liu Y, Ren Z, Li X, Jia J, Yu M, Yu X. Pristimerin induces apoptosis and autophagy via activation of ROS/ASK1/JNK pathway in human breast cancer in vitro and in vivo. Cell Death Discov. 2019;5:125. doi: 10.1038/s41420-019-0208-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Asakuma J, Sumitomo M, Asano T, Asano T, Hayakawa M. Selective Akt inactivation and tumor necrosis actor-related apoptosis-inducing ligand sensitization of renal cancer cells by low concentrations of paclitaxel. Cancer Res. 2003;63:1365–1370. [PubMed] [Google Scholar]
- 64.Swanton C, Marani M, Pardo O, Warne PH, Kelly G, Sahai E, Elustondo F, Chang J, Temple J, Ahmed AA, et al. Regulators of mitotic arrest and ceramide metabolism are determinants of sensitivity to paclitaxel and other chemotherapeutic drugs. Cancer Cell. 2007;11:498–512. doi: 10.1016/j.ccr.2007.04.011. [DOI] [PubMed] [Google Scholar]
- 65.Lu S, Natarajan SK, Mott JL, Kharbanda KK, Harrison-Findik DD. Ceramide induces human hepcidin gene transcription through JAK/STAT3 pathway. PLoS One. 2016;11:e0147474. doi: 10.1371/journal.pone.0147474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Villena J, Henriquez M, Torres V, Moraga F, Díaz-Elizondo J, Arredondo C, Chiong M, Olea-Azar C, Stutzin A, Lavandero S, Quest AF. Ceramide-induced formation of ROS and ATP depletion trigger necrosis in lymphoid cells. Free Radic Biol Med. 2008;44:1146–1160. doi: 10.1016/j.freeradbiomed.2007.12.017. [DOI] [PubMed] [Google Scholar]
- 67.Kogot-Levin A, Saada A. Ceramide and the mitochondrial respiratory chain. Biochimie. 2014;100:88–94. doi: 10.1016/j.biochi.2013.07.027. [DOI] [PubMed] [Google Scholar]
- 68.Yamaguchi H, Chen J, Bhalla K, Wang HG. Regulation of Bax activation and apoptotic response to microtubule-damaging agents by p53 transcription-dependent and -independent pathways. J Biol Chem. 2004;279:39431–39437. doi: 10.1074/jbc.M401530200. [DOI] [PubMed] [Google Scholar]
- 69.Lee WT, Chang CW. Bax is upregulated by p53 signal pathway in the SPE B-induced apoptosis. Mol Cell Biochem. 2010;343:271–279. doi: 10.1007/s11010-010-0522-6. [DOI] [PubMed] [Google Scholar]
- 70.Henry RE, Andrysik Z, Paris R, Galbraith MD, Espinosa JM. A DR4:tBID axis drives the p53 apoptotic response by promoting oligomerization of poised BAX. EMBO J. 2012;31:1266–1278. doi: 10.1038/emboj.2011.498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Shen YH, Utama B, Wang J, Raveendran M, Senthil D, Waldman WJ, Belcher JD, Vercellotti G, Martin D, Mitchelle BM, Wang XL. Human cytomegalovirus causes endothelial injury through the ataxia telangiectasia mutant and p53 DNA damage signaling pathways. Circ Res. 2004;94:1310–1317. doi: 10.1161/01.RES.0000129180.13992.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Aubrey BJ, Kelly GL, Janic A, Herold MJ, Strasser A. How does p53 induce apoptosis and how does this relate to p53-mediated tumour suppression? Cell Death Differ. 2018;25:104–113. doi: 10.1038/cdd.2017.169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Buschmann T, Potapova O, Bar-Shira A, Ivanov VN, Fuchs SY, Henderson S, Fried VA, Minamoto T, Alarcon-Vargas D, Pincus MR, et al. Jun NH2-terminal kinase phosphorylation of p53 on Thr-81 is important for p53 stabilization and transcriptional activities in response to stress. Mol Cell Biol. 2001;21:2743–2754. doi: 10.1128/MCB.21.8.2743-2754.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Dong Y, Shen X, He M, Wu Z, Zheng Q, Wang Y, Chen Y, Wu S, Cui J, Zeng Z. Activation of the JNK-c-Jun pathway in response to irradiation facilitates Fas ligand secretion in hepatoma cells and increases hepatocyte injury. J Exp Clin Cancer Res. 2016;35:114. doi: 10.1186/s13046-016-0394-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chu R, Upreti M, Ding WX, Yin XM, Chambers TC. Regulation of Bax by c-Jun NH2-terminal kinase and Bcl-xL in vinblastine-induced apoptosis. Biochem Pharmacol. 2009;78:241–248. doi: 10.1016/j.bcp.2009.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The datasets used and analyzed in the present study are available from the corresponding authors upon reasonable request.