

Abstract
Polycomb group (PcG) proteins are epigenetic regulators that facilitate both embryonic development and cancer progression. PcG proteins form Polycomb repressive complexes 1 and 2 (PRC1 and PRC2). PRC2 trimethylates histone H3 lysine 27 (H3K27me3), a histone mark recognized by the N-terminal chromodomain (ChD) of the CBX subunit of canonical PRC1. There are five PcG CBX paralogs in humans. CBX2 in particular is upregulated in a variety of cancers, particularly in advanced prostate cancers. Using CBX2 inhibitors to understand and target CBX2 in prostate cancer is highly desirable; however, high structural similarity among the CBX ChDs has been challenging for developing selective CBX ChD inhibitors. Here, we utilize selections of focused DNA encoded libraries (DELs) for the discovery of a selective CBX2 chromodomain probe, SW2_152F. SW2_152F binds to CBX2 ChD with a Kd of 80 nM and displays 24–1000-fold selectivity for CBX2 ChD over other CBX paralogs in vitro. SW2_152F is cell permeable, selectively inhibits CBX2 chromatin binding in cells, and blocks neuroendocrine differentiation of prostate cancer cell lines in response to androgen deprivation.
Keywords: CBX2, DNA-Encoded Library, Neuroendocrine Differentiation, Prostate Cancer, Chromodomain Inhibitor
Graphical Abstract
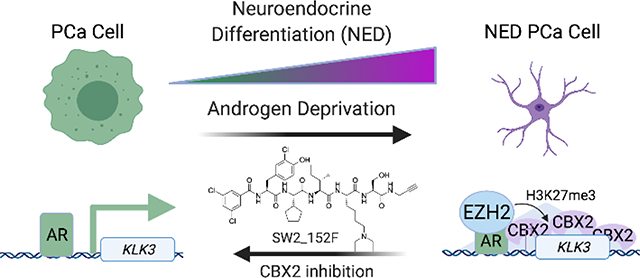
Prostate cancers survive androgen deprivation by going through a process called neuroendocrine differentiation (NED), which requires gene repression by CBX2, a histone methyllysine reader protein. Selective inhibition of the CBX2 chromodomain using a cell permeable ligand blocks NED and promotes prostate cancer cell death.
Introduction
Chromatin regulators with fundamental functions in embryonic development are often also involved in cancer progression,[1,2] maintaining stem cell-like properties, enhancing metastatic potential, and promoting resistance to therapy.[3,4,5] Polycomb group (PcG) proteins are one such set of chromatin regulators that repress the transcription of differentiation genes in both progenitor cells and cancers. PcG proteins combinatorially assemble into two distinct complexes, Polycomb repressive complex 1 (PRC1) and Polycomb repressive complex 2 (PRC2) (Fig 1A).[6] Polycomb-mediated repression is initiated by non-canonical PRC1 and PRC2 that establish sites of Polycomb activity through H2AK119 ubiquitination and H3K27me3, respectively.[7,8] Canonical PcG-mediated compaction of these regions begins with allosteric activation of canonical PRC2 by H3K27me3 association with EED,[9] leading to the spread of H3K27me3 marks to nearby nucleosomes.[10,11] H3K27me3 is recognized by the chromodomain (ChD) of the CBX (chromobox homolog) subunit of PRC1, which compacts chromatin to promote transcriptional repression at target loci.[12,13] PcG CBX is represented by five paralogs (CBX2, CBX4, CBX6, CBX7, CBX8), which share a conserved N-terminal ChD that recognizes H3K27me3 and a conserved C-terminal domain Pc-box for incorporation into PRC1 complexes.[14]
Figure 1.
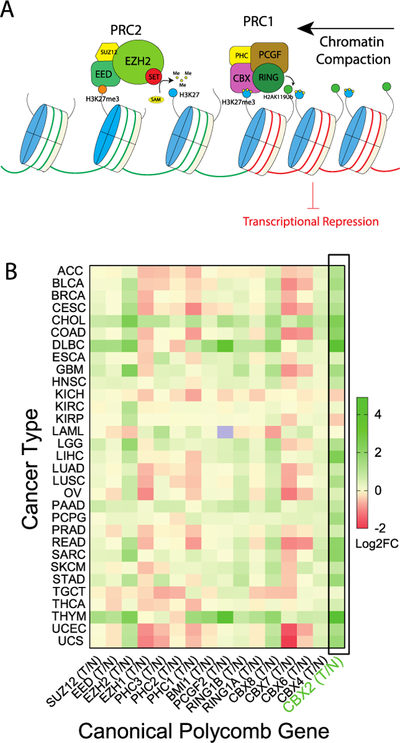
A). Canonical Polycomb-mediated transcriptional repression. EZH2 catalyzes the transfer of methyl group on H3K27, which can be recognized by CBX chromodomain, followed up with ubiquitylation on H2A119 by RING and downstream transcriptional repression. B). Polycomb group gene expression changes in tumor normalized to normal tissue. Using GEPIA, the gene expression fold changes across all tumor samples and paired normal tissues (T/N) were plotted as Log2 fold change in heatmap.
EZH2, a subunit of PRC2, is frequently upregulated during cancer progression and metastasis,[15,16] and EZH2 inhibitors are currently under development in preclinical studies and phase 1/2 clinical trials.[17–19] Due to an essential role for EZH2/H3K27me3 mark in maintaining certain normal cell populations,[20] targeting downstream canonical PRC1 (cPRC1) complexes in cancer is a desirable alternative for drug development. In particular, emerging evidence indicates a critical function for individual CBX paralogs in cancer initiation[21,22,23,24,25] and progression.[26,27]
Selective inhibitors of individual CBX proteins would be immensely helpful in understanding paralog-specific function in cancer. The high flexibility and structural similarities (>67% conserved residues) of the PcG ChDs, as well as the shallow, extended binding site for “induced-fit” binding of H3K27me3, present significant challenges for the development of potent and selective inhibitors to individual CBX ChDs.[28,29] Small molecule inhibitors with weak affinity (Kd ~20 μM) have been developed for the CBX7 ChD,[30,31] and peptidomimetic ligands (5–6-mers) with higher affinity (Kd <1 μM) have been developed with selectivity for CBX4/CBX7,[28,32] CBX6,[32] and CBX8[33] ChDs; however, no ligand has been developed to date with selectivity for the CBX2 ChD.
In this study, we identify the CBX2-specific chromodomain inhibitor SW2_152F from DNA-encoded libraries (DELs) of peptidic compounds.[33] This inhibitor exhibits high affinity (Kd ~80 nM), and selectivity (24 to 1000-fold over other paralogs) in vitro, and high selectivity for CBX2 in cells. Using this inhibitor, we demonstrate a role for the CBX2 ChD in prostate cancer neuroendocrine differentiation (NED), and define the utility of using CBX2 inhibitors in the treatment of therapy-resistant prostate cancer.
Results
Bioinformatic Analysis of Canonical PRC Subunits in Cancer
Using Gene Expression Profiling Interactive Analysis (GEPIA),[34] and data from The Cancer Genome Atlas (TCGA), we determined the change in canonical PcG gene expression in tumors compared to matched normal tissue. In addition to EZH2, both CBX2 and CBX8 are upregulated in multiple cancers, while CBX7 is generally downregulated (Fig 1B). CBX2 is the most highly and commonly upregulated CBX paralog in cancer and has recently emerged as a potential oncogenic target in multiple malignancies.[35,36,37,38] For example, CBX2 is upregulated in metastatic castration resistant prostate cancer (CRPC)[39] as well as neuroendocrine prostate cancer (NEPC), a clinically advanced, highly treatment-resistant subtype.[40] Although NEPC can occur de novo, the majority of the NEPC patients arise from castration-resistant prostate cancer (CRPC) patients treated with hormone therapy, radiotherapy or chemotherapy. Long term treatment results in the trans-differentiation of adenocarcinoma cells into treatment-induced neuroendocrine prostate cancer (NEPC).[41,42] NEPC is characterized by decreased expression of androgen receptor (AR) and AR target genes, and increased expression of neuroendocrine markers synaptophysin (SYP), chromogranin A (CHGA) and neuron-specific enolase (NSE).[1,41,43] Neuroendocrine differentiation requires multiple epigenetic regulators, [41–44] including EZH2,[1,45] which cooperates with the oncogenic regulator N-Myc (MYCN)[43,46] to repress AR target genes during neuroendocrine differentiation (NED).[40,43,47]
Bioinformatic Analysis of PcG Genes in Neuroendocrine Prostate Cancer (NEPC)
Using publicly available clinical datasets, we confirmed a decrease in AR and AR-target genes (KLK3, TMPRSS2) in castration-resistant prostate cancer CRPC with neuroendocrine characteristics (CRPC-NE), compared to CRPC with intact AR signaling (CRPC-Adeno) (Fig 2A).[44] Similar to EZH2,[44] CBX2 expression is upregulated in prostate cancer adenocarcinoma (PRAD) compared with normal tissue (Fig 1B). In fact, CBX2, is the most highly upregulated CBX subunit in NEPC compared to prostate cancer as a whole, using the clinical NEPC/PCa cohort and PDX samples (Fig 2B).[40,48] Across all TCGA prostate cancer samples, higher expression of EZH2 and CBX2 both correlate with lower rates of disease-free survival (Fig 2C) and correlate with each other (Fig 2D), providing initial evidence for CBX2 as the primary “reader” of EZH2-mediated H3K27me3 in prostate cancer.
Figure 2: CBX2 expression in patient samples.

A) AR and AR target gene expression in castration resistant prostate cancer (CRPC)-Adeno and CRPC-NE using clinical patient data.[42]B) Average expression fold changes of CBX paralogs and EZH2 in clinical/PDX NEPC cells compared to localized prostate cancer.[38,46] C). Disease-free survival analysis of EZH2 and CBX2 expression in PRAD (GEPIA). D) Correlation analysis of EZH2 with CBX2 in PRAD (GEPIA).
Discovery of CBX2 Chromodomain Ligands via Focused DNA-Encoded Libraries
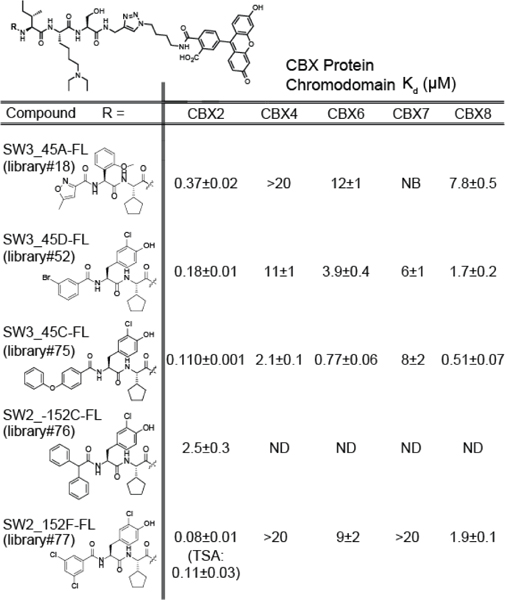
We recently reported the use of an on-DNA medicinal chemistry approach for the optimization of ligands to the CBX8 ChD. In this work, selections of focused DNA-encoded libraries against all five ChDs of the Polycomb CBX paralogs and the ChD of CBX5, a heterochromatin protein (HP1) were conducted to develop ligands with improved affinity and selectivity (Fig 3A).[33,49] While CBX2 was not our intended target, we observed several molecules with increased affinity and selectivity to CBX2 (relative to a parental ligand) from the DNA sequencing data (Fig 3B). These analogs have differing substitution at position [−3] (library member 18) or position [−4] (library members 52, 75, 76 and 77) (Fig 3B).
Figure 3:

A) Preparation and selection of DNA-encoded chemical libraries for CBX ChD ligands. Positional scanning libraries (PSL) were prepared by parallel chemical modification of an amine-modified oligonucleotide immobilized on a solid support. Subsequent encoding was performed by parallel PCRs with unique templates. Libraries were pooled and selected against immobilized CBX ChDs. Enriched libraries were then additionally barcoded with index sequences, pooled, and sequenced. B) Representative monomers with increased affinity or selectivity are depicted and enrichment heat map from previous selections were illustrated. Enrichment was normalized to a non-ligand control. C). In vitro affinity determination of optimal CBX2 ligand SW2_152F using fluorescence polarization assay with fluorescein-conjugated ligand. Selectivity was calculated from Kd values (Table 1).
Off-DNA CBX2 Hit Synthesis and Validation
To validate potential CBX2 inhibitors, the five enriched ligands were synthesized off-DNA using solid phase synthesis (Table 1). Instead of the DNA barcodes, an alkyne was incorporated at the C-terminus for conjugation chemistry. In addition, a diethyllysine was incorporated in the place of trimethyllysine to improve cellular activity.[28] Previous studies find that substituting the trimethyllysine with diethyllysine does not change the affinity of peptidic ligands to CBX chromodomains.[28] To measure Kd values, fluorescein conjugates were synthesized and used in fluorescence polarization (FP) assays with all five Polycomb CBX ChDs. Except for SW2_152C_FL, all the ligands bound with submicromolar affinity to CBX2 ChD and exhibited moderate to high selectivity for CBX2 ChD over the other paralogs (Table 1, SI1A). We selected SW2_152F, which displayed the highest affinity to CBX2 (~Kd = 80 nM) and highest selectivity over the other paralogs (at least 24-fold) (Fig 3C) for follow up studies. To verify that the fluorophore in SW2_152F_FL does not contribute to CBX2 binding affinity, we performed a competitive FP assay with unlabeled SW2_152F. The IC50 was 1.08 μM, which corresponds to an approximate Ki of 89 nM (SI1C).[50] In addition, thermal shift analysis with unlabeled SW2_152F resulted in a similar Kd of 110 nM (SI1C). We also performed the competitive FP assay with the trimethyllysine variant, SW2_152F_Kme3, and obtained an IC50 of 2.07 μM (Ki = 228 nM) (SI1D). This indicates that neither the addition of the fluorophore nor the substitution of the trimethyllysine with diethyllysine significantly alters binding affinity to CBX2 ChD.
Table 1:
Kd values of fluorescein-conjugated ligands to PcG CBX ChDs.
|
Evaluation of Cytosolic Access using ChloroAlkane Penetration Assay (CAPA)
To quantify the cell permeability of SW2_152F, we used the chloroalkane penetration assay (CAPA).[51] This assay utilizes a HeLa cell line stably transfected with HaloTag protein for a pulse-chase experiment. Cells are first incubated with chloroalkane (CA) conjugated ligands (pulse), which covalently react with the HaloTag protein when/if the ligand reaches the cytoplasm. The cells are then treated with excess chloroalkane-TAMRA (chase), which labels any remaining, unblocked HaloTag resulting in fluorescence in the cell. This fluorescence is inversely proportional to the CA-molecule cytosolic concentration and is quantified using flow cytometry. SW2_152F was conjugated to the chloroalkane via its alkyne (denoted SW2_152F-CA) and CAPA was performed. In comparison to the CP50 value of 26 ± 1.0 μM for our previously reported chloroalkane-conjugated CBX8 ligand SW2_110A-CA,[33] SW2_152F has increased permeability, with CP50 value of 6.2 ± 1.0 μM (Fig 4A).
Figure 4: Cellular Activity of CBX2 ChD Ligand.

A) Relative cytosolic access of SW2_152F conjugated to a chloroalkane (SW2_152F-CA) was evaluated using chloroalkane penetration assay (CAPA). The half maximal cell penetration value (CP50) of SW2_152F-CA was determined for SW2_152F-CA and CBX8 inhibitor SW2_110A-CA. B) Chemoprecipitations from HEK293T nuclear lysates using biotinylated SW2_152F (SW2_152F-B) were analyzed using immunoblot analysis. C) Analysis and quantification of SW2_152F in abrogating bulk binding of endogenous CBX proteins to chromatin in HEK293T cells by Sequential Salt Extraction. D). Chromatin immunoprecipitation (ChIP) from K562 cells using antibodies against IgG, H3K27me3, CBX2 and CBX8 was followed by quantitative PCR of genomic regions near Tm9SF4 (negative locus), TCF21, Fyn-2, and PAX7. Cells were treated with 10 μM SW2_152F for 4h prior to harvest. E) Chromatin immunoprecipitation (ChIP) in Hs68 fibroblast cells using antibodies against IgG, CBX7, and CBX8 followed by quantitative PCR of genomic regions at LMNB2 (negative locus), CCND2, and RUNX3. Cells were treated with 100 μM SW2_152F for 4h prior to harvest. For all qPCR, error bars represent SEM n=3 biological replicates, p-values were calculated using two-tailed Student’s t-test, * = p < 0.05, ** = p <0.01, *** = p < 0.001, **** = p <0.0001.
Cellular Selectivity and Activity Studies
Chemoprecipitations
A biotinylated derivative of CBX2 inhibitor SW2_152F, SW2_152F-B, was used to evaluate endogenous CBX protein enrichment from HEK293T nuclear lysates (Fig 4B). SW2_152F-B robustly enriched CBX2 and CBX8 with limited enrichment of other paralogs compared to streptavidin beads alone. In addition, RING1B, a subunit of PRC1 that interacts with the C-terminus of CBX, was also enriched, indicating that the compound binds to full-length CBX2 within PRC1 complexes. Furthermore, enrichment of CBX2, CBX8 and RING1B was reduced in the presence of excess SW2_152F added to the lysate, confirming that enrichment is a result of specific binding of CBX chromodomains to SW2_152F-B. As a non-specific control, BRG1 was not significantly enriched compared to input or beads alone. CBX6 and CBX7 were similarly not enriched, consistent with the in vitro FP assay results with recombinant chromodomains (Table 1).
Sequential Salt Extraction
Since SW2_152F binds to the ChD of CBX2, we next verified if it can disrupt CBX2 binding to chromatin. We adapted a sequential salt extraction (SSE) assay for evaluating the relative binding properties of chromatin-bound proteins with and without inhibitor treatment.[52,53] In this assay, bulk chromatin is sequentially resuspended in increasing concentrations of sodium chloride, and the proteins eluted with each wash are quantified using immunoblotting. From quantification of CBX paralogs in each fraction as a percent of total CBX protein amount (SI 2A), we confirmed that 10 μM SW2_152F dramatically abrogated CBX2 binding to chromatin, while only a modest change was observed with the closest paralog CBX8. No effect was seen with CBX7 or the non-specific control, BAF155 (Fig 4C, SI 2A).
ChIP-qPCR
After validating selectivity using lysate and bulk chromatin, we wanted to determine whether the inhibitor can disrupt CBX2 binding to chromatin in live cells. We used chromatin immunoprecipitation (ChIP) followed by quantitative PCR (ChIP-qPCR) of sites with CBX2, CBX8 and H3K27me3 enrichment in K562 ChIP-Seq datasets in ENCODE.[54] We evaluated effects of compound treatment at both 10 μM and 100 μM SW2_152F. Treatment of K562 cells with 10 μM SW2_152F resulted in a significant reduction of CBX2 binding, but not CBX8 binding, (or H3K27me3 enrichment) at these sites (Fig 4D). Incubation of cells with 100 μM SW2_152F resulted in a significant reduction of both CBX2 and CBX8 binding at these sites, in agreement with the in vitro affinity to CBX8 (SI 2B). In order to evaluate SW2_152F activity against a paralog with no binding to SW2_152F in vitro, CBX7 ChIP-qPCR was performed in Hs68 fibroblast cells, which have no detectable CBX2 expression.[55] Incubation of Hs68 cells with 100 μM SW2_152F resulted in a significant reduction of CBX8 binding, but not CBX7 binding, to shared genomic target loci (Fig 4E), consistent with the in vitro binding profiles.
Cellular Activity of CBX2 Inhibitor in Neuroendocrine Differentiation of Prostate Cancer
Generation of a Prostate Cancer Neuroendocrine Differentiation (NED) System
To generate an in vitro model for NED, we cultured the androgen-sensitive prostate cancer cell line LNCaP in charcoal-stripped serum (CSS), which lacks androgens as well as other steroids. Upon treatment with CSS media, LNCaP cells adopted morphological characteristics of neuroendocrine-like cells (LNCaP_NED), which include dominant nucleus, limited cytoplasm, dendrites (Fig 5A) and a decreased proliferation rate. To monitor transcriptional changes during androgen deprivation, we quantified the changes in expression of two NED markers (ENO2 and CHGA) and two AR target genes (KLK3 and TMPRSS2) by RT-qPCR during 15 days of androgen deprivation (SI 3A). After 6 days of androgen deprivation, the expression of AR-target genes significantly decreased, and the expression of NED markers significantly increased (Fig 5B). To verify that LNCaP_NED cells become resistant to AR antagonists, we tested the effects of the AR antagonist enzalutamide (ENZA) on cell proliferation. LNCaP_NED showed no further decrease in proliferation with enzalutamide treatment, while LNCaP cells demonstrated highly decreased proliferation in response to ENZA treatment (SI 3B).
Figure 5: Targeting the CBX2 chromodomain in Neuroendocrine Prostate Cancer.

A). Neuroendocrine differentiation (NED) of prostate cancer induced by androgen deprivation. Representative pictures of LNCaP cells treated with normal media or charcoal-stripped serum (CSS) media for 6 days to induce neuroendocrine differentiation (LNCaP_NED). B). Transcriptional fold changes of KLK3/TMPRSS2/ENO2/CHGA were quantified normalized to control gene YWHAZ upon CSS media treatment for 6 or 9 days. C). Western blotting of CBX paralogs in LNCaP and LNCaP_NED cells generated from 14-day CSS treatment. D). Cell proliferation of LNCaP_NED following 5 days incubation with 2 or 10 μM SW2_152F. DMSO was used as negative control. E). LNCaP_NED cells were treated with 10 μM SW2_152F for 24h-96h. Cell morphology pictures were captured starting from 24h. Average cell size were plotted at 24h or 48h for both DMSO group and compound treated group. F). LNCaP_NED cells were treated with 10 μM or 50 μM SW2_152F for 48 hours. Transcriptional fold change of genes (KLK3/TMPRSS2/AR/CBX2) were normalized to YWHAZ in LNCaP_NED cells with SW2_152F treatment or DMSO. G). LNCaP cells were treated with DMSO. LNCaP_NED cells were treated with DMSO, 10 μM GSK343 or 10 μM SW2_152F for 48 hours. Whole cell lysates were extracted for western blotting. H). LNCaP_NED cells were treated with DMSO or 10 μM SW2_152F for 4 hours before harvest. Binding of H3K27me3 and CBX2 at specific genomic sites (KLK3/TMPRSS2, e: enhancer, p: promoter) were evaluated using chromatin immunoprecipitation-qPCR. For all experiments in the figure, error bars represent SEM n=3 biological replicates, p-values were calculated using two-tailed Student’s t-test, * = p < 0.05, ** = p <0.01, *** = p < 0.001, **** = p <0.0001.
Cellular Activity of CBX2i during Neuroendocrine Differentiation of Prostate Cancer
To confirm that the CBX expression changes observed in NEPC patients also occurs in LNCaP_NED cells, we performed immunoblotting before and after 14 days of androgen deprivation. CBX2 and CBX8, but not CBX7, were upregulated in LNCaP_NED cells, in line with analysis of patient tumors (Fig 5C). Further, knockdown of CBX2 in LNCaP_NED cells using lentiviral-mediated shRNA resulted in a dramatic increase in the expression of AR target genes and a decrease in the expression of NED marker ENO2 and (SI 3C).
While SW2_152F treatment did not affect the viability of non-transformed prostate epithelial RWPE-1 cells or HEK293T cells (SI 3D), it significantly inhibited LNCaP_NED cell proliferation (Fig 5D). In addition, SW2_152F treatment significantly decreased average cell size and reduced dendrites in LNCaP_NED cells (Fig 5E), consistent with a loss in the neuroendocrine phenotype, and similar to reported effects of an EZH2 inhibitor.[56]
To assess how CBX2 inhibition modulates transcription during NED, LNCaP_NED cells were treated with SW2_152F at two different doses for 48 hours. Expression of AR and AR-target genes (TMPRSS2 and KLK3) was significantly increased, while CBX2 expression remained unchanged (Fig 5F). To determine whether CBX2 inhibition can re-sensitize LNCAP_NED cells to ENZA treatment, LNCaP_NED cells were treated with SW2_152F and/or ENZA for 4 days. Cell viability was significantly decreased in cells treated with both compounds compared to SW2_152F or ENZA treatment alone (SI 3E). This indicates that CBX2 inhibition can re-sensitize LNCaP_NED cells to AR signaling inhibition by enzalutamide.
In another androgen-sensitive prostate cancer cell line, VCaP, 6 days of androgen deprivation decreases AR protein expression and increases ENO2, N-Myc, H3K27me3 and CBX2 protein expression (Fig 5G), similar to published findings.[40,47,56] Using this NED cell line (VCaP_NED), we tested how addition of EZH2 inhibitor GSK343 or CBX2 inhibitor SW2_152F affects protein expression changes during NED. Consistent with the different mechanisms of inhibition, GSK343 reduces H3K27me3 levels while SW2_152F does not. Both inhibitors, however, similarly increase AR and decrease ENO2 and N-Myc expression in NED cells, compared to DMSO treatment alone. An unexpected effect was a decrease in CBX2 expression in VCaP_NED cells treated with GSK343 (Fig 5G). This could mean that CBX2 expression is dependent on EZH2 activity, or it could mean that the subset of cells with high CBX2 expression are selectively killed upon EZH2 inhibition. The high correlation between EZH2 and CBX2 expression could reflect either possibility.
To determine whether SW2_152F inhibits CBX2 binding at AR target genes repressed during NED, we used ChIP-qPCR in LNCaP_NED cells. Addition of SW2_152F abrogates CBX2 binding at the KLK3 (PSA)/TMPRSS2 enhancers and promoters. Interestingly, SW2_152F also reduces H3K27me3 enrichment specifically at these sites (Fig 5H), without affecting global levels of H3K27me3 in VCaP cells (Fig 5G), or H3K27me3 levels at CBX2 binding sites in K562 cells (Fig 4D). Similar to the decrease in CBX2 with EZH2 inhibition this could reflect a feedback loop between PRC1 and PRC2 during the repression of AR target genes in NEPC[57] or reflect that the subset of cells with high H3K27me3 at AR target genes are dependent on CBX2 for viability.
Discussion and Conclusions
Previous studies have identified increased CBX2 in NEPC and implicated CBX2 as a potential target in therapy-resistant prostate cancers.[39,40] This evidence, confirmed with our own bioinformatic analysis, implicates CBX2 as the primary CBX paralog involved in polycomb-mediated transition of prostate cancer adenocarcinoma to neuroendocrine prostate cancer.[39,40,47,58] We developed the first CBX2-selective ChD ligand, and used it to confirm that the CBX2 ChD is critical for mediating CBX2 binding at AR target genes during NED. Treatment with the inhibitor also prevents CBX2-mediated repression of AR-target genes, and the subsequent induction of NE genes. As a result, CBX2 ChD inhibition increases cell death during androgen deprivation and restores prostate cancer sensitivity to AR antagonists, suggesting CBX2 inhibition as a promising strategy to both treat and prevent NEPC.
In addition to establishing CBX2 as a potential therapeutic target in NEPC, the development of our CBX2 inhibitor will facilitate in-depth understanding of how epigenetic regulators facilitate prostate cancer transition to a therapy-resistant state. While CBX2 is clearly important for NED, mechanistically it is still unclear whether CBX2 inhibitors revert neuroendocrine-like cells to an androgen sensitive state, or if they selectively inhibit survival of the subset of cells undergoing neuroendocrine differentiation at that time. The temporal control CBX2 inhibitors provide will allow for mechanistic investigation of CBX2 function along specific time points during the differentiation process.
CBX2-selective inhibitors can also facilitate better understanding of CBX2 dependency in other cancers,[59,60] and in development, specifically sex-determination.[61] While there is substantial evidence that individual CBX paralogs have unique and non-overlapping functions in development and disease,[21,23] it is unclear what biochemical functions are important for CBX2’s unique roles. Considering the high homology between the chromodomain and Pc-box of CBX paralogs, the non-homologous sequence in between is likely responsible for non-redundant functions among paralogs.[62] CBX2 has a unique serine-rich patch region which contributes to nucleosome binding and phase separation.[63,64] Additionally, CBX2 is unique in having a short chromodomain-containing variant missing the Pc box[65] indicating that CBX2 can function independently of PRC1. CBX2-specific probes will be instrumental in dissecting the specific biochemical role for CBX2 in different cell types.
Lastly, this result demonstrates the potential in using DNA-encoding for the optimization of ligands in medicinal chemistry. In our prior efforts in developing CBX8 ChD ligands,[33] we showed how the DNA-encoded ligands can be used to greatly facilitate the testing of directed libraries in parallel against a panel of protein targets, which allowed for a concurrent optimization of both potency and selectivity. This yielded not only a potent and selective ligand to the intended target but also a wealth of structure activity relationship data useful for ligand development with off-target proteins. Additionally, this discovery of a potent, selective, and cellularly active CBX2 ChD inhibitor using DNA-encoded libraries provides further evidence that the development of effective and selective probes for all five CBX paralogs may be possible despite the high homology between the ChDs. Further optimization of ligands with DNA-encoded libraries will facilitate the discovery of improved CBX2 ChD inhibitors, as well as expedite the discovery of new inhibitors for the many unexplored chromatin binding domains in the human proteome.
Supplementary Material
Figure 6: Overview of CBX2 inhibitor function during prostate cancer NED.

A) Androgen deprivation initiates the gradual repression of AR target genes by EZH2 and CBX2, resulting in the emergence of a neuroendocrine phenotype that is resistant to anti-androgen therapy. B) Inhibition of CBX2 during androgen deprivation prevents neuroendocrine differentiation and maintains prostate cancer cell susceptibility to anti-androgen therapy.
Acknowledgements
We thank Dr. Josh Kritzer for providing Halo-GFP-Mito Hela cell lines. Support for this research was provided by the Purdue Center for Cancer Research, NIH grant P30 CA023168 in the form of the Genomics Core and the Pilot Grant Program. S.W. is partially supported by Lilly Endowment Gift Graduate Research Award. C.J.K. is supported by NIH grant (R35GM128894). E.C.D. is supported by NIH grant (U01CA207532). Figures created with BioRender.com
Footnotes
Conflict of Interest
The authors declare no conflict of interest.
Supporting information for this article is given via a link at the end of the document.
References
- [1].Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, Ghosh D, Pienta KJ, Sewalt RGAB, Rubin MA, Chinnaiyan AM, Nature 2002, 419, 624–629. [DOI] [PubMed] [Google Scholar]
- [2].Jueng SY, Jae KK, Seo DW, Jae HP, Jong WP, Jae CL, Yae JJ, Eun JC, Han JW, Cancer Res 2009, 69, 5716–5725. [DOI] [PubMed] [Google Scholar]
- [3].McDonald OG, Wu H, Timp W, Doi A, Feinberg AP, Nat. Struct. Mol. Biol 2011, 18, 867–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Timp W, Feinberg AP, Nat. Rev. Cancer 2013, 13, 497–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Sparmann A, Van Lohuizen M, Nat. Rev. Cancer 2006, 6, 846–856. [DOI] [PubMed] [Google Scholar]
- [6].Di Croce L, Helin K, Nat. Struct. Mol. Biol 2013, 20, 1147–1155. [DOI] [PubMed] [Google Scholar]
- [7].Blackledge NP, Farcas AM, Kondo T, King HW, McGouran JF, Hanssen LLP, Ito S, Cooper S, Kondo K, Koseki Y, Ishikura T, Long HK, Sheahan TW, Brockdorff N, Kessler BM, Koseki H, Klose RJ, Cell 2014, 157, 1445–1459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Zepeda-Martinez JA, Pribitzer C, Wang J, Bsteh D, Golumbeanu S, Zhao Q, Burkard TR, Reichholf B, Rhie SK, Jude J, Moussa HF, Zuber J, Bell O, Sci. Adv 2020, 6, eaax5692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Margueron R, Justin N, Ohno K, Sharpe ML, Son J, Drury WJ, Voigt P, Martin SR, Taylor WR, De Marco V, Pirrotta V, Reinberg D, Gamblin SJ, Nature 2009, 461, 762–767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Leicher R, Ge EJ, Lin X, Reynolds MJ, Xie W, Walz T, Zhang B, Muir TW, Liu S, Proc. Natl. Acad. Sci. U. S. A 2020, 117, 30465–30475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Poepsel S, Kasinath V, Nogales E, Nat. Struct. Mol. Biol 2018, 25, 154–162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Francis NJ, Kingston RE, Woodcock CL, Science 2004, 306, 1574–7. [DOI] [PubMed] [Google Scholar]
- [13].Lavigne M, Francis NJ, King IFG, Kingston RE, Mol. Cell 2004, 13, 415–25. [DOI] [PubMed] [Google Scholar]
- [14].Kaustov L, Ouyang H, Amaya M, Lemak A, Nady N, Duan S, Wasney GA, Li Z, Vedadi M, Schapira M, Min J, Arrowsmith CH, J. Biol. Chem 2011, 286, 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Bracken AP, Helin K, Nat. Rev. Cancer 2009, 9, 773–784. [DOI] [PubMed] [Google Scholar]
- [16].Tan J, Yang X, Zhuang L, Jiang X, Chen W, Puay LL, Karuturi RKM, Tan PBO, Liu ET, Yu Q, Genes Dev 2007, 21, 1050–1063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Bradley WD, Arora S, Busby J, Balasubramanian S, Gehling VS, Nasveschuk CG, Vaswani RG, Yuan C-C, Hatton C, Zhao F, Williamson KE, Iyer P, Mé J, Campbell R, Cantone N, Garapaty-Rao S, Audia JE, Cook AS, Dakin LA, Albrecht BK, Harmange J-C, Daniels DL, Cummings RT, Bryant BM, Normant E, Trojer P, Chem. Biol 2014, 21, 1463–1475. [DOI] [PubMed] [Google Scholar]
- [18].Garapaty-Rao S, Nasveschuk C, Gagnon A, Chan EY, Sandy P, Busby J, Balasubramanian S, Campbell R, Zhao F, Bergeron L, Audia JE, Albrecht BK, Harmange J-C, Cummings R, Trojer P, Chem. Biol 2013, 20, 1329–1339. [DOI] [PubMed] [Google Scholar]
- [19].Campbell JE, Kuntz KW, Knutson SK, Warholic NM, Keilhack H, Wigle TJ, Raimondi A, Klaus CR, Rioux N, Yokoi A, Kawano S, Minoshima Y, Choi HW, Porter Scott M, Waters NJ, Smith JJ, Chesworth R, Moyer MP, Copeland RA, ACS Med. Chem. Lett 2015, 6, 491–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Chou RH, Yu YL, Hung MC, Am. J. Transl. Res 2011, 3, 243–250. [PMC free article] [PubMed] [Google Scholar]
- [21].Klauke K, Radulović V, Broekhuis M, Weersing E, Zwart E, Olthof S, Ritsema M, Bruggeman S, Wu X, Helin K, Bystrykh L, De Haan G, Nat. Cell Biol 2013, 15, 353–362 [DOI] [PubMed] [Google Scholar]
- [22].Kloet SL, Makowski MM, Baymaz HI, Van Voorthuijsen L, Karemaker ID, Santanach A, Jansen PWTC, Di Croce L, Vermeulen M, Nat. Struct. Mol. Biol 2016, 23, 682–690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Morey L, Pascual G, Cozzuto L, Roma G, Wutz A, Benitah SA, Di Croce L, Cell Stem Cell 2012, 10, 47–62. [DOI] [PubMed] [Google Scholar]
- [24].Morey L, Santanach A, Blanco E, Aloia L, Nora EP, Bruneau BG, Di Croce L, Cell Stem Cell 2015, 17, 300–15. [DOI] [PubMed] [Google Scholar]
- [25].O’Loghlen A, Muñoz-Cabello AM, Gaspar-Maia A, Wu HA, Banito A, Kunowska N, Racek T, Pemberton HN, Beolchi P, Lavial F, Masui O, Vermeulen M, Carroll T, Graumann J, Heard E, Dillon N, Azuara V, Snijders AP, Peters G, Bernstein E, Gil J, Cell Stem Cell 2012, 10, 33–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Mills AA, Nat. Rev. Cancer 2010, 10, 669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Koppens M, Van Lohuizen M, Oncogene 2016, 35, 1341–1352. [DOI] [PubMed] [Google Scholar]
- [28].Stuckey JI, Dickson BM, Cheng N, Liu Y, Norris JL, Cholensky SH, Tempel W, Qin S, Huber KG, Sagum C, Black K, Li F, Huang X-P, Roth BL, Baughman BM, Senisterra G, Pattenden SG, Vedadi M, Brown PJ, Bedford MT, Min J, Arrowsmith CH, James LI, V Frye S, Nat. Chem. Biol 2016, 12, 180–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Kaustov L, Ouyang H, Amaya M, Lemak A, Nady N, Duan S, Wasney G, Li Z, Vedadi M, Schapira M, Min J, Arrowsmith CH, J. Biol. Chem 2011, 286, 521–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Ren C, Morohashi K, Plotnikov AN, Jakoncic J, Smith SG, Li J, Zeng L, Rodriguez Y, Stojanoff V, Walsh M, Zhou MM, Chem. Biol 2015, 22, 161–168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Ren C, Smith SG, Yap K, Li S, Li J, Mezei M, Rodriguez Y, Vincek A, Aguilo F, Walsh MJ, Zhou MM, ACS Med. Chem. Lett 2016, 7, 601–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Milosevich N, Gignac MC, McFarlane J, Simhadri C, Horvath S, Daze KD, Croft CS, Dheri A, Quon TTH, Douglas SF, Wulff JE, Paci I, Hof F, ACS Med. Chem. Lett 2016, 7, 139–144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Wang S, Denton KE, Hobbs KF, Weaver T, McFarlane JMB, Connelly KE, Gignac MC, Milosevich N, Hof F, Paci I, Musselman CA, Dykhuizen EC, Krusemark CJ, ACS Chem. Biol 2020, 15, 112–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Tang Z, Li C, Kang B, Gao G, Li C, Zhang Z, Nucleic Acids Res 2017, 45, W98–W102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Mao J, Tian Y, Wang C, Jiang K, Li R, Yao Y, Zhang R, Sun D, Liang R, Gao Z, Wang Q, Wang L, J. Cancer 2019, 10, 2706–2719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Zheng S, Lv P, Su J, Miao K, Xu H, Li M, Am. J. Transl. Res 2019, 11, 1668–1682. [PMC free article] [PubMed] [Google Scholar]
- [37].Wheeler LJ, Watson ZL, Qamar L, Yamamoto TM, Post MD, Berning AA, Spillman MA, Behbakht K, Bitler BG, Oncogenesis 2018, 7, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Clermont PL, Sun L, Crea F, Thu KL, Zhang A, Parolia A, Lam WL, Helgason CD, Br. J. Cancer 2014, 111, 1663–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Clermont PL, Crea F, Chiang YT, Lin D, Zhang A, Wang JZL, Parolia A, Wu R, Xue H, Wang Y, Ding J, Thu KL, Lam WL, Shah SP, Collins CC, Wang Y, Helgason CD, Clin. Epigenetics 2016, 8, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Clermont PL, Lin D, Crea F, Wu R, Xue H, Wang Y, Thu KL, Lam WL, Collins CC, Wang Y, Helgason CD, Clin. Epigenetics 2015, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Rubin MA, Bristow RG, Thienger PD, Dive C, Imielinski M, Mol. Cell 2020, 80, 562–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Beltran H, Jendrisak A, Landers M, Mosquera JM, Kossai M, Louw J, Krupa R, Graf RP, Schreiber NA, Nanus DM, Tagawa ST, Marrinucci D, Dittamore R, Scher HI, Clin. Cancer Res 2016, 22, 1510–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Vlachostergios PJ, Puca L, Beltran H, Curr. Oncol. Rep 2017, 19, 32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BVSK, Varambally S, Tomlins SA, Nanus DM, Tagawa ST, Van Allen EM, Elemento O, Sboner A, Garraway LA, Rubin MA, Demichelis F, Nat. Med 2016, 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Crea F, Hurt EM, Mathews LA, Cabarcas SM, Sun L, Marquez VE, Danesi R, Farrar WL, Mol. Cancer 2011, 10, 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Lee JK, Phillips JW, Smith BA, Park JW, Stoyanova T, McCaffrey EF, Baertsch R, Sokolov A, Meyerowitz JG, Mathis C, Cheng D, Stuart JM, Shokat KM, Gustafson WC, Huang J, Witte ON, Cancer Cell 2016, 29, 536–547. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Dardenne E, Beltran H, Benelli M, Gayvert K, Berger A, Puca L, Cyrta J, Sboner A, Noorzad Z, MacDonald T, Cheung C, Yuen KS, Gao D, Chen Y, Eilers M, Mosquera JM, Robinson BD, Elemento O, Rubin MA, Demichelis F, Rickman DS, Cancer Cell 2016, 30, 563–577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Beltran H, Rickman DS, Park K, Chae SS, Sboner A, MacDonald TY, Wang Y, Sheikh KL, Terry S, Tagawa ST, Dhir R, Nelson JB, de la Taille A, Allory Y, Gerstein MB, Perner S, Pienta KJ, Chinnaiyan AM, Wang Y, Collins CC, Gleave ME, Demichelis F, Nanus DM, Rubin MA, Cancer Discov 2011, 1, 487–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Denton KE, Wang S, Gignac MC, Milosevich N, Hof F, Dykhuizen EC, Krusemark CJ, SLAS Discov 2018, 23, 417–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Huang X, J. Biomol. Screen 2003, 8, 34–38. [DOI] [PubMed] [Google Scholar]
- [51].Peraro L, Zou Z, Makwana KM, Cummings AE, Ball HL, Yu H, Lin YS, Levine B, Kritzer JA, J. Am. Chem. Soc 2017, 139, 7792–7802. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Porter EG, Connelly KE, Dykhuizen EC, J. Vis. Exp 2017, 55369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Porter EG, Dykhuizen EC, J. Biol. Chem 2017, 292, 2601–2610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Azkanaz M, López AR, De Boer B, Huiting W, Angrand PO, Vellenga E, Kampinga HH, Bergink S, Martens JHA, Schuringa JJ, ven den Boom V, Elife 2019, 8, e45205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Pemberton H, Anderton E, Patel H, Brookes S, Chandler H, Palermo R, Stock J, Rodriguez-Niedenführ M, Racek T, de Breed L, Stewart A, Matthews N, Peters G, Genome Biol 2014, 15, R23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Zhang Y, Zheng D, Zhou T, Song H, Hulsurkar M, Su N, Liu Y, Wang Z, Shao L, Ittmann M, Gleave M, Han H, Xu F, Liao W, Wang H, Li W, Nat. Commun 2018, 9, 4080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kalb R, Latwiel S, Baymaz HI, Jansen PWTC, Müller CW, Vermeulen M, Müller J, Nat. Struct. Mol. Biol 2014, 21, 569–571. [DOI] [PubMed] [Google Scholar]
- [58].Gu X, Wang X, Su D, Su X, Lin L, Li S, Wu Q, Liu S, Zhang P, Zhu X, Jiang X, Front. Mol. Neurosci 2018, 11, 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Béguelin W, Teater M, Gearhart MD, Calvo Fernández MT, Goldstein RL, Cárdenas MG, Hatzi K, Rosen M, Shen H, Corcoran CM, Hamline MY, Gascoyne RD, Levine RL, Abdel-Wahab O, Licht JD, Shaknovich R, Elemento O, Bardwell VJ, Melnick AM, Cancer Cell 2016, 30, 197–213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Chung CY, Sun Z, Mullokandov G, Bosch A, Qadeer ZA, Cihan E, Rapp Z, Parsons R, Aguirre-Ghiso JA, Farias EF, Brown BD, Gaspar-Maia A, Bernstein E, Cell Rep 2016, 16, 472–486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Tan J, Jones M, Koseki H, Nakayama M, Muntean AG, Maillard I, Hess JL, Cancer Cell 2011, 20, 563–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Vincenz C, Kerppola TK, Proc. Natl. Acad. Sci. U. S. A 2008, 105, 16572–16577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Tatavosian R, Kent S, Brown K, Yao T, Duc HN, Huynh TN, Zhen CY, Ma B, Wang H, Ren X, J. Biol. Chem 2019, 294, 1451–1463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Plys AJ, Davis CP, Kim J, Rizki G, Keenen MM, Marr SK, Kingston RE, Genes Dev 2019, 33, 799–813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Völkel P, le Faou P, Vandamme J, Pira D, Angrand PO, Epigenetics 2012, 7, 482–491. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.