

Summary
Phosphatidylserine (PS) is an integral component of eukaryotic cell membranes and organelles. The Drosophila genome contains a single PS synthase (PSS)-encoding gene (Pss) homologous to mammalian PSSs. Flies with Pss loss-of-function alleles show a reduced life span, increased bang sensitivity, locomotor defects, and vacuolated brain, which are the signs associated with neurodegeneration. We observed defective mitochondria in mutant adult brain, as well as elevated production of reactive oxygen species, and an increase in autophagy and apoptotic cell death. Intriguingly, glial-specific knockdown or overexpression of Pss alters synaptogenesis and axonal growth in the larval stage, causes developmental arrest in pupal stages, and neurodegeneration in adults. This is not observed with pan-neuronal up- or down-regulation. These findings suggest that precisely regulated expression of Pss in glia is essential for the development and maintenance of brain function. We propose a mechanism that underlies these neurodegenerative phenotypes triggered by defective PS metabolism.
Subject areas: Developmental neuroscience, Cell biology, Developmental biology
Graphical abstract
Highlights
-
•
Loss of Pss leads to developmental defects and neurodegeneration
-
•
Loss of Pss causes a mitochondrial defect, elevated ROS, and secondary necrosis
-
•
Pss functions in glia are essential for synaptogenesis and neuronal maintenance
-
•
Glial Pss expression level must be tightly regulated to maintain a healthy nervous system
Developmental neuroscience; Cell biology; Developmental biology
Introduction
Neurodegenerative diseases (NDs), like Alzheimer's disease (AD), Parkinson's disease (PD), Huntington's disease, and frontotemporal dementia, are a major threat to healthy aging as life expectancy has increased (Heemels, 2016). The best-characterized cause of ND is associated with protein aggregates, such as amyloid-β plaques and Tau-tangles in AD and α-synuclein in PD (Irvine et al., 2008; Ross and Poirier, 2004; Serpell and Smith, 2000; Sharon et al., 2003). Interestingly, some studies have shown changes in the phospholipid composition of neural tissue, including in patients with AD and PD (Buccoliero et al., 2004; Li et al., 2015; Pettegrew et al., 2001; Wang et al., 2014). Defective phospholipid biosynthetic enzymes are a likely cause of changes in phospholipid composition in ND models (Kretzschmar et al., 1997; Pavlidis et al., 1994). These studies speak to the important role of phospholipids in neural function. However, it is unclear whether changes in phospholipid composition are a consequence or a cause of ND.
Phosphatidylserine (PS) is one of the major phospholipids in cellular membrane and performs a variety of functions, including the regulation of intracellular signaling proteins such as protein kinase C (PKC), Akt, and Raf-1 (Kim et al., 2014). It also plays a role in the fusion of exocytotic vesicles with the plasma membrane (Williams et al., 2009) and acts as an “eat-me” signal for phagocytic clearance of apoptotic cells (Fadok et al., 1992; Leventis and Grinstein, 2010; Segawa and Nagata, 2015). Some clinical studies have shown that PS consumption significantly improves cognitive function and memory in patients with AD (Crook et al., 1991, 1992; Heiss et al., 1994). Furthermore, dietary intake of PS suppresses circadian and sleep disorders in PD models (Valadas et al., 2018). These results suggest a potentially important role for PS in the maintenance and/or function of neural tissues.
In mammals, PS is synthesized from phosphatidylcholine (PC) and phosphatidylethanolamine (PE) through enzymatic replacement of the head group with serine by two PS synthases, PSS1 for PC and PSS2 for PE (Vance and Steenbergen, 2005). Both enzymes are present in the endoplasmic reticulum (ER) and the mitochondria-associated membrane (MAM) (Stone and Vance, 2000). PS synthesized by PSS1 is transferred to the mitochondria, where it is converted into PE by PS decarboxylase (Choi et al., 2005) (Figure 1A). PE and PS also migrate to the Golgi apparatus and then to the plasma membrane using vesicular transport mode (Leventis and Grinstein, 2010). Mice that carry a loss-of-function mutation for either PSS1 (Pss1−/−) or PSS2 (Pss2−/−) are viable, but double knockout mice (Pss1−/−; Pss2−/−) are prenatally lethal, indicating that PSS1 and PSS2 have a redundant function (Arikketh et al., 2008; Bergo et al., 2002). Mutations in human PSS1 (PTDSS1) cause Lenz-Majewski hyperostotic dwarfism (LMHD), a rare disease characterized by multiple congenital anomalies featuring generalized craniotubular hyperostosis, developmental defects, and intellectual disability (Sousa et al., 2014). However, the mechanisms underlying the association between PTDSS1 function and the neural defects seen in LMHD are not known.
Figure 1.

The fly Pss gene: structure and expression pattern
(A) PS synthesis in mammalian cell and Drosophila. Drosophila PSS is the only PS synthase in Drosophila.
(B) Homology between Drosophila PSS and human PSS1 and PSS2.
(C) Pss mutant alleles. Three P element insertions are clustered in a small region of the 5′ UTR. PssΔ1 is a deletion mutation in the second exon, which causes a frameshift in the ORF.
(D) Complementation tests. The trans-heterozygous mutants are in the same w+ background as we used the Pss32 line to collect virgins for crosses. With the ubiquitous overexpression of Pss, the viabilities of trans-heterozygous mutants were increased up to 139% of expected flies. Numbers indicate the ratio of observed/expected flies.
(E) Identification of cell types expressing Pss in the larval CNS and adult brain. mCherry expression by Pss-GAL4 (Pss>mCherry.nls) in the third instar larval (a', c') ventral nerve cord (VNC) and (b', d') brain, and (e', f') adult brain. (a", b", e") Glial cells were labeled by anti-Repo and (c", d", f") neuronal cells by anti-Elav. (a-f) Merged image of Pss>mCherry.nls with either anti-Repo or anti-Elav. Scale bar, 25 μm in a-d, 100 μm in e-f.
In this study, we report that loss of Pss causes neurodegenerative features, including reduced life span, increased bang sensitivity, locomotor defects, and increased formation of vacuoles in the brain observed. In addition, we document that glial-specific knockdown or overexpression of Pss caused altered synaptogenesis and axonal growth in the larval stage, developmental arrest in pupae, and neurodegeneration in the adults, indicating that PSS plays a critical role in glia.
Results
Pss encodes a Drosophila homolog of mammalian PSS genes
The Drosophila genome contains a single PSS-encoding gene (Pss) (Figure 1A) (Midorikawa et al., 2010; Vance and Steenbergen, 2005; Vance and Tasseva, 2013). It is the homolog to both human PSS1 and PSS2 (Figure 1B). Drosophila PSS exhibits 53% homology to human PSS1 across the entire sequence, and PSS and PSS1 share a well-conserved PSS domain with 61% homology (DIOPT score 15 of 15) (Hu et al., 2011). Drosophila PSS shares 35% homology across the entire PSS2 protein and 40% homology in the PSS domain (DIOPT score 3 of 15), indicating that Drosophila PSS is more closely related to PSS1 than PSS2 (Figure S1A).
To delve into a function of Pss in vivo, we characterized three different P element insertion lines (Pss15, Pss32, PssGAL4) inserted in the 5′ UTR of the Pss gene (Figures 1C and S1B). We also generated a deletion mutation (PssΔ1) by removing 14 bp in the second exon using the CRISPR/Cas9 strategy (Kondo and Ueda, 2013). This deletion causes a frameshift in the ORF of Pss, suggesting it is a null-allele. These alleles (Pss15, Pss32, PssGAL4, PssΔ1) are all homozygous embryonic lethal, and PssGAL4/PssGAL4 animals can be rescued to pharate adults with UAS-Pss (Figure S1C). Complementation tests show that some alleles partially complement each other and produce trans-heterozygous viable adults (Figure 1D). As the Pss15, PssGAL4, and PssΔ1 are not in the w+ background, we used the Pss32 (P{PZ}Pss04521 ry506/TM3, ryRK Sb1 Ser1) as female parent to make the trans-heterozygous mutants with the same w+ background. When we cross two different Pss heterozygous mutant flies to generate trans-heterozygous progenies, we also get heterozygous progenies of two different genotypes. We counted the number of all heterozygous progenies, and half of them would be the theoretically expected number of trans-heterozygotes. The reduced viability of these trans-heterozygous animals can be rescued by actin-GAL4 or Pss-GAL4>UAS-Pss showing that they indeed affect Pss (Figure 1D) and result in partial loss of function of PSS (Figures 2A and S1D). To assess the level of Pss transcripts in Pss mutant flies, we used quantitative RT-PCR (qRT-PCR), which showed that trans-heterozygous flies have an 84–88% reduction in Pss expression compared with wild-type flies (Canton-S), while heterozygous flies showed an approximately 65% reduction when compared with wild-type flies (Figures 2A and S1D). We, therefore, performed the experiments using trans-heterozygous mutant flies to examine the function of Pss in larvae and adults. We selected the weakest allelic combination Pss32/Pss15 for most experiments.
Figure 2.

Pss mutants exhibit neurodegenerative phenotypes
(A) qRT-PCR showing Pss expression levels in Pss32/15 flies and Act>Pss; Pss32/15 flies compared with the wild type (WT). Bars represent max and min values.
(B) Survival curves at 29°C. WT n = 200, Pss32/15 n = 199, Act>Pss; Pss32/15 n = 172 (Log rank test: p < 0.0001).
(C and D) Bang-sensitive phenotypes of 3-day-old flies. (C)Trans-heterozygous Pss mutants were more sensitive to shock than wild-type flies and rescue flies. (D) Recovery time for paralyzed flies. Pss mutant flies took longer to recover (WT 2.9 sec, Pss32/15 42.5 sec, Act>Pss; Pss32/15 7.1 sec). Bars represent mean ± SEM. ∗∗∗p < 0.001; n.s., not significant. Numbers within the bars indicate mean values. WT n = 18, Pss32/15 n = 18, Act>Pss; Pss32/15 n = 14.
(E) Pss mutation reduces the flies' climbing ability. Climbing assays were analyzed in 3-day-old flies of the indicated genotypes. Bars represent mean ± SEM. ∗∗∗p < 0.001. The numbers above the bars indicate mean values. WT n = 100, Pss32/15 n = 100, Act>Pss; Pss32/15 n = 100.
(F) Head sections showing vacuoles in the brains of 5-day-old flies. Vacuoles were significantly more abundant in Pss32/15 mutant brains. Some of the vacuoles are indicated by black arrowheads or white arrows. (a, b) WT. (c-h)Trans-heterozygous Pss mutants. Scale bar, 50 μm. (i) quantification of vacuoles. Pss32/15 flies with Actin-GAL4 and UAS-Pss partially rescued the vacuolization. Bars represent mean ± SEM. ∗∗∗p < 0.001. Numbers above the bars indicate mean values. WT n = 13, Pss32/15 n = 11, Act>Pss; Pss32/15 n = 14.
The loss of one copy of Pss does not exhibit any obvious behavior or morphological defects right after eclosion, but trans-heterozygous escapers (Pss32/15, Pss32/GAL4, Pss32/Δ1) exhibit severe movement defects and are barely mobile. To assess if PSS is expressed in the central nervous system (CNS), we crossed PssGAL4 with UAS-mCherry.nls or UAS-GFP.nls to mark the nuclei of cells expressing Pss (Figure 1E). Interestingly, mCherry-expressing cells were co-labeled mostly with subsets of Repo-positive cells (Figure 1E a, b, e) and few Elav-positive cells (Figure 1E c, d, f), indicating that Pss is highly enriched in subsets of glia in the CNS. In addition, Pss is also expressed in some other tissues such as salivary gland (sg), fat body (fb), gut (g), trachea (tc), and oenocytes (oc) besides the nervous system (Figure S1E).
Pss mutants exhibit neurodegenerative phenotypes
The life span of Pss32/15 flies is significantly reduced, and most flies die within 12 days (Figure 2B). The mean T50 (time at 50% survival) of Pss32/15 flies was 9.6 days, whereas the T50 for wild-type flies was 36 days. The Pss32/15 flies that contain Actin-GAL4>UAS-Pss showed a partially rescued life span with a T50 of 22.8 days. The Pss32/+ and PssΔ1/+ flies showed no reduction in life span compared with wild-type flies (Figure S1F).
We also examined Pss32/15 flies for bang sensitivity and climbing ability. Bang sensitivity assay can induce seizures in flies (Kuebler and Tanouye, 2000; Parker et al., 2011). Upon vortexing the flies for 5 sec, wild-type flies right themselves within a few seconds, but flies that are sensitive show stereotypical cycles of shaking and paralysis for many seconds and up to minutes. We used 3-day-old adults. Pss32/15 flies showed significant shock-induced paralysis (100%) when compared with wild-type flies (28%). Ubiquitous expression of Pss (Act>UAS-Pss) rescues (36%) the Bang sensitivity of Pss32/15 flies (Figure 2C). We also measured recovery time from shock-induced paralysis, and Pss32/15 flies took significantly longer to recover when compared with wild-type flies and rescue flies (2.9 sec on average for wild-type flies, 7.1 sec for rescue flies vs. 42.5 sec for Pss32/15 mutants) (Figure 2D).
We also performed climbing test in 3-day-old wild-type flies climbed above 18 cm on average in 10 sec, whereas Pss32/15 mutants managed only 4.47 cm (Figure 2E). Ubiquitous expression of Pss (Act>UAS-Pss) partially rescues the climbing defects of Pss32/15 flies (Figure 2E). We further noticed that heterozygous Pss (Pss32/+ and PssΔ1/+) flies exhibit a progressive decline in locomotion (Figure S1G). These results suggest that Pss heterozygosity does not affect longevity but does affect certain behaviors with age, such as climbing ability. In summary, a severe loss of Pss causes significant defects in longevity and climbing ability and causes bang sensitivity.
Vacuolization in Pss mutant brains
Vacuoles are frequently considered a histological hallmark of neurodegeneration in Drosophila (Fergestad et al., 2008; Palladino et al., 2002). To examine whether the Pss mutant's phenotypes stem from neurodegeneration in the CNS, we examined vacuole formation in 5-day-old brains of Pss32/15 and wild-type flies. Pss32/15 mutant brains contain many vacuoles (black arrowheads in Figure 2F a-h). After measuring the diameter of each vacuole by ImageJ, the total numbers of vacuoles with diameter from 3 μm to 6 μm and over 6μm in each fly brain were divided by the total number of slices of each fly brain to obtain the average number of vacuoles per plane (Figure 2F i). We frequently observed very large vacuoles in mutant flies (white arrows in Figure 2F c-g) but not in the wild-type flies (Figure 2F a-b). Pss32/15 flies with Actin-GAL4 and UAS-Pss are partially rescued as the number of vacuoles decreased significantly (Figure 2F i). On the basis of these observations, we conclude that Pss mutant brains exhibit hallmarks of neurodegeneration, which may underlie the behavioral phenotypes.
Defective mitochondria in Pss mutant flies
Mammalian PSS1 is commonly found in the membrane of the ER and is enriched in the MAM of ER contacts with the mitochondria (Stone and Vance, 2000). PS synthesized by PSS1 is translocated to the mitochondria, where it is converted into PE (Vance and Tasseva, 2013). Because PE is an important phospholipid for maintaining the stability of the mitochondrial inner membrane in mammals (Schenkel and Bakovic, 2014; Tamura et al., 2012; Vance, 2014), we asked whether the mitochondrial inner membrane is compromised when Pss expression is reduced. We used transmission electron microscopy (TEM) to examine mitochondrial morphology in the brain and retina of 5-day-old Pss32/15 mutants and wild-type flies. Mitochondria in the wild-type flies were, in general, elliptically shaped with orderly, tightly packed cristae (Figures 3A a, a', a'', and S2A a, a', a''). In contrast, the majority of mitochondria in Pss32/15 flies were round in shape and appeared swollen or deformed (Figures 3A b-c, b'-c', b''-c'', d, and S2A b-c, b'-c', b''-c'', d). They also displayed irregular cristae structures and ruptured membranes (white arrows in Figures 3A b', b'', c'', and S2A b'-c', b''-c''). Such structural deformities suggest that reduced PS production due to reduced Pss expression undermines the stability of the mitochondrial inner membrane, leading to disorganized cristae, swelling, and rupture.
Figure 3.

Pss mutation results in abnormal mitochondrial morphology and increased ROS production in the brain
(A) TEM images of mitochondria in the brain of 5-day-old (a) WT and (b-c) Pss32/15 adults. (a-c) Areas indicated by red boxes in the upper panels are shown at higher magnification in the lower panels (a'-c', a''-c''). (a', a'') WT mitochondria were elliptical with orderly cristae structures. Pss32/15 mitochondria have abnormal shapes (non-elliptical), sizes, and cristae patterns (defective mitochondria are indicated by yellow stars). (b', b'', c'') Occasionally, ruptured mitochondria are observed as indicated by white arrows, and (c') swollen mitochondria are also detected. Scale bar, 1 μm. (e) Quantification of abnormal mitochondria. Data points indicate the ratio of the 'number of abnormal mitochondria/number of total mitochondria' observed from the different TEM figures. ∗∗∗p < 0.001.
(B) Detection of ROS in the brain with dihydroethidium (DHE). (a, a') Pharate adult WT. (b, b') Pharate adult Pss32/15. Enhanced ROS production was found in the brain of Pss mutants. (a, b) Images are shown in 'fire' mode for better contrast. (c) Boxplots showing quantification of the DHE intensities. At least five specimens were measured for quantification. Scale bar, 100 μm. ∗p < 0.05.
(C) Detection of lipid peroxidation in the brains by C11-BODIPY(581/591). (a, a') 3-day-old WT. (b, b') 3-day-old Pss32/15. (a, b) Images of the green fluorescent signals were converted to 'fire' modes for better contrast. (c) Boxplots showing quantified fluorescence intensities. At least five specimens were used for the quantification. Scale bar, 100 μm. ∗p < 0.05.
Elevation of ROS in Pss mutant flies
Reactive oxygen species (ROS) are produced mostly by complex I in mitochondria, and healthy mitochondria contain enzymatic mechanisms to lower ROS levels (Starkov, 2008). Since elevated levels of oxidative stress are known to be a major cause of neurodegeneration (Sies, 2017), we hypothesized that the abnormal mitochondrial structure in Pss mutants may be associated with a systemic increase in ROS levels in the brain. To determine whether this is the case, we measured ROS levels in the brains of both Pss mutant and wild-type flies using red fluorescence emitted by dihydroethidium (DHE) in response to ROS (Owusu-Ansah et al., 2008).
Since Pss32/15 mutants showed behavioral defects in young adults (Figures 2C–2E), we measured ROS levels in late pupal brains. Pharate adult brains of Pss32/15 mutants exhibit significantly higher levels of red fluorescence than wild-type brains (Figure 3B). Because the ND phenotype presents later in heterozygotes than in the trans-heterozygotes, we examined the aging effect in the heterozygotes by measuring ROS levels in brains of older Pss32/+ flies. 5-day-old Pss32/+ flies had higher levels of red fluorescence than 5-day-old wild-type flies (Figure S2B). These results suggest that reduced Pss expression increases ROS levels.
Lipids, especially polyunsaturated fatty acid (PUFA), are more susceptible to oxidation in the presence of ROS. Thus, lipid peroxidation products can be used as a biomarker for in vivo oxidative stress (Abuja and Albertini, 2001). We measured lipid peroxidation by staining brains with C11-BODIPY(581/591) in 3-day-old wild-type and Pss32/15 flies. As shown in Figure 3C, lipid peroxidation was significantly higher in mutant brains than in wild-type brains. We also found increased lipid peroxidation in the brains of 10-day-old Pss32/+ flies relative to wild-type flies (Figure S2C). On the basis of these data, we suggest that mitochondrial dysfunction in Pss mutant flies is associated with increased ROS production in the brain.
Increased autophagic activity in the brain and retina of Pss mutants
The data presented above show that the majority of mitochondria in the brain and retina of Pss32/15 mutants are abnormally shaped (Figures 3A and S2A). Since autophagy is an important mechanism for removing dysfunctional organelles (Mizushima, 2007), we wondered whether the autophagic activity was also affected by Pss mutation. Acridine orange (AO) is a lysotropic dye that is protonated at acidic pH and trapped in acidic vesicular organelles such as autophagosomes/autolysosomes. Hence, red fluorescence of 579–620 nm emitted from AO with excitation at 561 nm can be used to detect cellular autophagic activity (Thome et al., 2016). We stained 1-day-old brains from Pss32/15, and wild-type flies with AO. Brains from Pss32/15 flies showed strong fluorescent signals (Figure 4A). We also examined autophagic activities in Pss32/+ flies to assess whether aging also affects autophagic activity. Notably, in contrast to the Pss32/15 flies, there was no significant difference in fluorescence between the 1-day-old Pss32/+ and wild-type flies; however, 10-day-old Pss32/+ flies showed high levels of red fluorescence (Figure S2D). These results suggest that even heterozygous Pss32/+ flies display higher levels of autophagic activity upon aging.
Figure 4.

Increased autophagy, apoptosis, and secondary necrosis in Pss mutants
(A) Autophagy in the brain detected by acridine orange (AO) (excitation/emission: 561/579–620). (a, a') 1-day-old WT. (b, b') 1-day-old Pss32/15. (a, b) Brain images are shown in 'fire' mode for better contrast. (c) Boxplots of quantified fluorescence intensities. At least five specimens were used for the quantification. Scale bar, 100 μm. ∗p < 0.05.
(B) Dying cells detected by AO in the brains. AO binding with ssDNA emits fluorescence at a longer wavelength than 650 nm (excitation: 488 nm). (a, a') 5-day-old WT. (b, b') 5-day-old Pss32/15. (a, b) Brain images are shown in 'fire' mode for better contrast. (c) Boxplots of quantified fluorescence intensities. At least five specimens were used for the quantification. Scale bar, 100 μm. ∗p < 0.05.
(C) TEM images of ommatidia of (a) 5-day-old WT and (b) Pss32/15 flies. The inter-ommatidial area that is composed of pigment cells is greatly expanded in Pss32/15 flies. Red boxes in a, b are magnified in a', b'. R: rhabdomeres, m: mitochondria. Autophagic bodies are indicated by asterisks (∗). (c) A bar graph showing numbers of autophagosomes/autolysosomes per ommatidium. Pss32/15 flies display about 4.2 times more autophagosomes/autolysosomes per ommatidium than WT flies. Twenty specimens were used for the quantification. The error bars represent mean ± SD (∗∗∗p < 0.001). The numbers above the error bars indicate mean values. Scale bar, 5 μm.
(D) Detection of apoptotic cells with anti-cleaved caspase 3 antibody. (a, d) WT. (b, e) Pss32/+. (c) Boxplots of quantified immunofluorescent puncta per 104μm2 of 1-day-old flies' central brains ((total puncta number/area)x104). (f) Boxplots of quantified immunosignal area in cortex of 5-day-old flies' brains ((signal positive area/total cortex area)x100). AL: antennal lobe, CB: central brain, CT: cortex, OL: optic lobe. Scale bar, 50 μm.
(E) Secondary necrosis in Pss mutants. WT and Pss32/15 brains were stained with a necrosis detector. (a) While only faint signals appeared in the WT brains, (b) strong and widespread signals appeared in the mutant brains. (c) Boxplots of quantified fluorescence intensities. At least four specimens were used for the quantification. Scale bar, 100 μm. ∗p < 0.05.
To gain further histological evidence for enhanced autophagy in Pss mutants, we processed sections of the compound eyes for TEM to examine autophagosome/autolysosome structures in retinal pigment cells, a type of glial cell (Edwards and Meinertzhagen, 2010). To our surprise, the inter-ommatidial area where retinal pigment cells are located was atypically broad in Pss32/15 flies (Figure 4C a vs. b). Moreover, we observed numerous dark and white bodies enclosed by a double lipid bilayer, characteristic of autophagosomes/autolysosomes (Eskelinen et al., 2011) and Pss mutants contained more autophagosomes than wild-type flies (Figure 4C). These results provide further support for increased autophagic activity in Pss mutant flies, likely to remove dysfunctional mitochondria.
Loss of Pss leads to enhanced cell death and secondary necrosis in the CNS
We wondered whether vacuole formation in the brain of Pss32/15 mutants was due to excessive cell death caused by the increased number of dysfunctional mitochondria and/or by elevated oxidative stress induced by high levels of ROS. Both have been implicated in cell death (Gaschler and Stockwell, 2017). Deep red fluorescent signals (longer than 650 nm) from AO after excitation at 488 nm can be used to detect dying cells (Sarkissian et al., 2014). Hence, we examined deep red fluorescent signals in AO-treated brain tissue from 5-day-old Pss32/15 and wild-type flies. We observed weak fluorescent signals in various parts of the wild-type brain (Figures 4B a, a'). By comparison, the signals were much more intense in Pss32/15 mutants (Figures 4B b, b', c). Similar results were obtained for 20-day-old Pss32/+ (Figure S2E) flies. Together, these results show enhanced cell death in Pss mutant brains.
To determine if cell death in Pss mutants is caused by caspase-mediated apoptosis, brain samples were stained for caspase 3 (anti-cleaved caspase 3). Since this antibody reacts selectively with activated effector caspases in Drosophila, it is commonly used to detect apoptotic cells. We detected immunofluorescent puncta throughout the central brain of 1-day-old Pss32/15 flies, more than in 1-day-old wild-type flies (Figure 4D a-c). Notably, more prominent immunosignal was detected in 5-day-old Pss32/15 brains (Figure 4D e). These signals appeared to be more concentrated in the cortex (CT), whereas such signals were rarely observed in similarly aged wild-type flies (Figure 4D d-f). We propose that enhanced autophagy and apoptosis are both causally associated with the neurodegenerative phenotype observed in Pss mutant brains.
Necrotic cell death is also associated with PS. PS is involved in the clearance of apoptotic cells by acting as an eat-me signal that is recognized by scavenging cells (Segawa and Nagata, 2015). The absence or reduction of phagocytic clearance due to reduced PS signals results in the autolysis of apoptotic cells termed secondary necrosis (Silva, 2010). Cytoplasmic contents, including lysosomal enzymes and ROS that are released from autolyzed apoptotic cells, further damage surrounding cells, eventually killing them (Muñoz et al., 2010). Because Pss mutants contain a reduced amount of PS, we hypothesized that secondary necrosis might occur more frequently in mutants than in wild-type flies. To test this hypothesis, wild-type and Pss32/15 brains were stained with a necrosis detector, which emits green fluorescence only when it enters necrotic cells. Indeed, we detected either no signals or very faint signals in wild-type brains at both 5 days (Figure 4E a) and 10 days old (Figure S2F a). However, 5-day-old Pss32/15 mutants showed strong, widespread signals (Figures 4E b, c) that increased further in 10-day-old flies (Figure S2F b). These results indicate that Pss mutation led to secondary necrosis in the brain tissue, which is accelerated with aging.
Glia-specific function of Pss
To further investigate Pss's function, we employed an RNAi-mediated silencing tool (Pss-RNAi). Ubiquitous knockdown of Pss expression by da-GAL4 caused developmental arrest at early larval stages (Figure 5A). da>Pss RNAi embryos hatched into first instar larvae but died before reaching the third instar larval stage. Pss knockdown using Pss-GAL4 showed the same results as da-GAL4. The larval lethality phenotype, when compared with the embryonic lethality of homozygous Pss mutants, likely reflects the hypomorphic nature of RNAi-mediated knockdown. Nevertheless, these results further indicate that Pss plays a vital role in both embryonic and larval development.
Figure 5.

Glia-specific functions of Pss
(A) Pss RNAi expression with Dcr2 by da-GAL4, Pss-GAL4, or repo-GAL4 caused lethality, whereas knockdown by neuron-specific elav-GAL4 produced viable adults without any noticeable defects.
(B and C) Escapers of Pan-glial Pss knockdown without Dcr2 (+; repo>Pss RNAi) produced a significant reduction in life span and climbing ability. (B) life span, control n = 190, +; repo>Pss RNAi n = 96 (Log rank test: p < 0.0001). (C) Climbing behavior was analyzed in 10-day-old flies. control n = 77, +; repo>Pss RNAi n = 52. Bars represent the mean ± SEM. ∗∗∗p < 0.001. Numbers within bars indicate mean values.
(D and E) Pan-neuronal Pss knockdown (Dcr2; elav>Pss RNAi) does not obviously affect development, longevity, and climbing ability. (D) life span, control n = 189, Dcr2; elav>Pss RNAi n = 180 (Log rank test: p = 0.7291). (E) Climbing behavior was analyzed in 10-day-old flies. control n = 40, Dcr2; elav>Pss RNAi n = 40. Bars represent the mean ± SEM. n.s., not significant. Numbers within bars indicate mean values.
(F and G) Pan-glial Pss overexpression (repo>Pss) does not affect development but leads to a significant reduction in life span and climbing ability. (F) life span, control n = 212, repo>Pss n = 200 (Log rank test: p < 0.0001). (G) Climbing behavior was analyzed in 10- and 20-day-old flies. WT n = 100, repo>Pss n = 100. Bars represent the mean ± SEM. ∗∗∗p < 0.001. Numbers within bars indicate mean values.
(H and I) Pan-neuronal Pss overexpression did not affect development, longevity, and climbing ability. (H) life span, control n = 160, Dcr2; elav>Pss RNAi n = 366 (Log rank test: p = 0.0157). (I) Climbing behavior was analyzed in 10-day-old flies. WT n = 100, +; repo>Pss RNAi n = 100. Bars represent the mean ± SEM. n.s., not significant. Numbers within bars indicate mean values.
(J) Lethality caused by glia subtype-specific Pss knockdown.
(K) Survival rate of cortex glia-specific Pss knockdown (Dcr2; NP2222>Pss RNAi) mutants. Most died within 48 hr at 29°C. control n = 150, Dcr2; NP2222>Pss RNAi n = 140 (Log rank test: p < 0.0001).
(L and M) Bang-sensitive phenotypes of 1-day-old flies. (L) Cortex glia Pss knockdown (Dcr2; NP2222>PssRNAi) mutants (n = 94) were much more sensitive to shock than the control flies (n = 122). (M) Recovery time for paralyzed flies. Pss mutant flies took longer to recover. Bars represent the mean ± SEM. ∗∗∗p < 0.001.
(N) Relative Pss expression level in the head of cortex glia-specific knockdown flies and control flies. Bars represent max and min values.
Reporter expression driven by Pss-GAL4 was highly enriched in subsets of the glial population in the CNS (Figure 1E). We found that the pan-glial knockdown of Pss (repo>Pss RNAi) resulted in pupal lethality (Figure 5A). However, the pan-neuronal knockdown of Pss (elav>Pss RNAi) did not produce any noticeable defects either during development or in adults (Figures 5A, 5D, and 5E). Together, these results suggest that glial-specific Pss expression appears to be essential for pupal development.
As we did not observe any adult flies from repo>Pss RNAi developed in 25°C, we generated milder knockdowns in flies by raising the flies expressing repo>Pss RNAi without Dcr2 at 22°C during development. The escapers (+; repo>Pss RNAi) showed a dramatically decreased life span (T50, 15 days) compared with control flies (T50, 37.2 days) (Figure 5B). They also exhibit climbing defects at day 10 when compared with the control flies (Figure 5C).
Furthermore, to eliminate the effects of Pss knockdown during development and to assess if pan-glial knockdown of Pss after eclosion can also cause neurodegeneration, we used the temperature-sensitive GAL80ts, which provides a temporal regulation of UAS transgene by preventing GAL4-dependent transcription (Duffy, 2002). As a result, +; tubGAL80; repo>Pss RNAi flies (T50, 33.5 days) and Dcr2; tubGAL80; repo>Pss RNAi flies (T50, 28 days) have a decreased life span compared with the control flies (T50, 42.5 days) (Figure S3A). In the climbing assay, both mutant flies did not show any significant difference compared with the control flies at day 10 (Figure S3B). However, mutant flies showed severe climbing defects when aged for 30 days (Figure S3C). These results strongly support that glial-specific Pss is also essential for the maintenance of the nervous system in adults.
Given Act>Pss; Pss32/15 flies partially rescued the Pss32/15 but still displayed a relatively shorter life span than wild-type flies (Figure 2B), we examined whether overexpression of Pss has any deleterious effects. Adult flies carrying ectopic Pss expression in pan-neuronal cells (elav>Pss) developed normally and did not show any noticeable changes in life span or climbing, suggesting that neuronal cells were not affected by the elevated Pss expression (Figures 5H and 5I). Moreover, Pan-glial overexpression of Pss (repo>Pss) did not grossly alter either embryonic or postembryonic development. However, these adults had a significantly reduced life span (T50, 19.8 days) compared with controls (T50, 31.8 days) (Figure 5F) and exhibited reduced climbing ability: a 42% reduction at 10 days old and an 80% reduction at 20 days (Figure 5G). Even wild-type flies showed an approximately 2-fold reduction in climbing height when aged 10 days old or more, suggesting that aging affects this behavior. Nonetheless, our results show that climbing performance deteriorated more precipitously with aging in repo>Pss flies. On the basis of these results, we speculated that pan-glial overexpression of Pss accelerates aging-associated neurodegeneration. In line with this, we found more vacuoles in the brains of repo>Pss flies, whereas wild-type brains were relatively well-shaped with fewer vacuoles (Figure S3D).
To assess if repo>Pss also increases apoptosis of glial cells, we co-labeled the brains of 15-day-old repo>Pss flies with anti-cleaved caspase 3 and anti-Repo. Although controls (repo-GAL4/+) showed little caspase 3 immunoreactivity, repo>Pss flies had higher levels of immunoreactivity throughout the brain, as well as dense label in some small areas (Figure S3E a vs. b), anti-Repo signals are limited to the nucleus, and anti-cleaved caspase 3 immunoreactive signals are seen mainly in the cytoplasm. As shown in magnified images (Figure S3E b'), we observed high levels of anti-cleaved caspase 3 immunosignals surrounding Repo-expressing nuclei, as well as in some cells lacking Repo expression, suggesting that cell death events were taking place in glial cells, as well as in neuronal cells. Since both pan-glial knockdown and overexpression of Pss led to defects indicative of neurodegeneration, we speculate that glial Pss expression must be tightly regulated to maintain the adult CNS.
To determine the glial subtype(s) that require Pss function, GAL4 drivers specific to each glial subtype were used to knockdown Pss expression: NP2222-GAL4 (cortex glia), NP6293-GAL4 (perineurial glia), NP2276-GAL4 (subperineurial glia), Alrm-GAL4 (astrocyte-like glia), and Mz97-GAL4 (wrapping glia). Each GAL4 line was crossed to UAS-RNAi-Pss expressing Dcr2, and the offspring were examined for developmental defects (Figure 5J). Interestingly, knockdown of Pss in the cortex glial subtype (Dcr2; NP2222>Pss-RNAi) produced semi-lethality during pupal development and only 37.4% of expected flies eclosed. Knockdown of Pss in perineurial glia (Dcr2; NP6293>Pss-RNAi) produced delayed development. Pss knockdown in the other glial subtypes did not produce any noticeable defects. From these results, we infer that both cortex and perineurial glia require Pss and that Pss expression in these glial subtypes is required for normal development.
Because of the observed developmental defect, we investigated the phenotype of Dcr2; NP2222>Pss-RNAi further. Intriguingly, most of the adults carrying Dcr2; NP2222>Pss-RNAi died within 48 hr when grown at 29°C (Figure 5K). They also showed significantly increased bang sensitivity: shock-induced paralysis was observed in 93.1% of 1-day-old flies, but only 33.4% of control flies (Figure 5L). These flies also took 101.2 s on average to recover from shock-induced paralysis, whereas control flies took 9.9 s (Figure 5M). We performed qRT-PCR using heads extracts and observed a 50% reduction in Pss transcripts in the cortex glia specific knockdown flies (Figure 5N). This is a significant change considering that the heads extracts include numerous other cell types, and cortex glia only correspond to about to a few % of the number of neurons and glia in adult brains. These results suggest that Pss expression in cortex glia is essential for pupal development and adult CNS function.
Glia-specific Pss functions are important for the synaptogenesis of larval motoneurons
Glial cells play an essential role in synaptic formation and differentiation (Ullian et al., 2004). For example, glial cells are known to regulate the growth of synapses in the larval neuromuscular junction (NMJ) (Brink et al., 2012). Because the previous data suggest an essential role of proper Pss expression in glial cells, we investigated whether Pss is also important for NMJ synapse development in larvae. NMJs were stained with anti-HRP antibody (red) to label the neuronal membrane and anti-CSP antibody (green) to visualize pre-synaptic vesicles (Brent et al., 2009). We counted synaptic boutons and branches, and measured the NMJ length for muscles 6/7 of the third abdominal segment (Figure 6A). Glia-specific knockdown (Dcr2; repo>RNAi-Pss) resulted in a significant reduction in the number of boutons and branches and NMJ length when compared with controls (Figure 6A a, c, and g–i). Conversely, ectopic Pss expression in glial cells (repo>Pss) enhanced synaptogenesis, as indicated by increased numbers of boutons and branches (Figures 6A a, b, and g–i). By contrast, neither knockdown nor overexpression of Pss in the larval motoneurons using D42-GAL4 (Dcr2; D42>RNAi-Pss and D42>Pss) had any significant effect on synaptogenesis (Figures 6A d–f, and g–i). These results further suggest that glia-specific Pss functions are required for optimal synaptogenesis at the larval NMJ.
Figure 6.

Pss expression alters synaptogenesis and axonal development in larval motoneurons and larval behavior
NMJ morphology was assessed in the sixth and seventh muscles in the third abdominal segment of third-instar larvae.
(A) NMJ structures affected by mutations and overexpression of Pss in a glial-specific manner. (a-f) Merged images of HRP immunostaining for the neuronal membrane (red) and CSP immunostaining for pre-synaptic vesicles (green) are shown for the genotypes indicated in each panel. Overexpression and knockdown of Pss were achieved by repo-GAL4 in glial cells and by D42-GAL4 in larval motoneurons. Scale bar, 50 μm. (g-i) Quantitative analysis of (g) bouton numbers, (h) axonal branches, and (i) NMJ length. Each genotype was color-coded, as indicated below the graphs. At least 17 specimens were assessed for each genotype. ∗p < 0.05; ∗∗p < 0.01; n.s., not significant.
(B) Effect of pan-glial overexpression (repo>Pss) and knockdown of Pss (Dcr2; repo>Pss RNAi) on the growth of glial processes and NMJ length. repo>mCD8.GFP was used as a marker of the glial cell membrane. (a-c) HRP immunostaining (red), (a'-c') mCD8.GFP expression (green), and (a"-c") merged pictures of both labelings. Scale bar, 50 μm. (d) A histogram showing the average lengths of NMJs and glial processes affected by glial-specific overexpression and knockdown of Pss. At least 14 specimens were observed for each genotype. Bars represent the mean ± SEM values. ∗p < 0.05; ∗∗p < 0.01.
(C) Larval roll-over assay. Larvae carrying glial-specific knockdown or overexpression of Pss take significantly longer to roll over. Bar colors for the genotypes are the same as in (A). At least 20 larvae were tested for each genotype. Bars represent the mean ± SEM values. ∗p < 0.05; ∗∗p < 0.01; ∗∗∗p < 0.001; n.s., not significant.
To assess if the development of glial is affected by abnormal levels of Pss expression, we measured the lengths of the NMJs and glial processes (Figure 6B). Glial processes were labeled with membrane-tethered mCD8.GFP driven by repo-GAL4, and motoneuronal axon membranes were counterstained with anti-HRP. We found that both glial process length and distal axon length (also termed the NMJ length) were indeed correlated with Pss levels, whereas repo>Pss produced an increase in the length of both NMJs and glial processes when compared with repo-GAL4/+ control (Figure 6B a, b, and d), and the opposite was observed with Pss knockdown (Figure 6B c, and d). From these data, we conclude that glia-specific Pss functions are required for the optimal growth of glial processes that guide axonal growth and synaptogenesis of motor neurons.
It is possible that both subnormal and excessive synaptogenesis of motoneurons affect larval motor skills. To test this, we performed roll-over assays in which we inverted a larva so that the ventral side is facing up and measured the time the larva to recover to their normal posture. Indeed, larvae with either glial knockdown (Dcr2; repo>Pss-RNAi) or overexpression (repo>Pss) of Pss took significantly longer to recover than control larvae (Figure 6C). Meanwhile, the same transgenic manipulations in motoneurons driven by D42-GAL4 produced no significant deficits in the roll-over assay (Figure 6C). These data suggest that abnormal synaptogenesis at NMJs resulting from altered expression of Pss in glial cells impairs the function of larval motoneurons. These results further support the hypothesis that the expression levels of Pss in glial cells must be tightly regulated.
Discussion
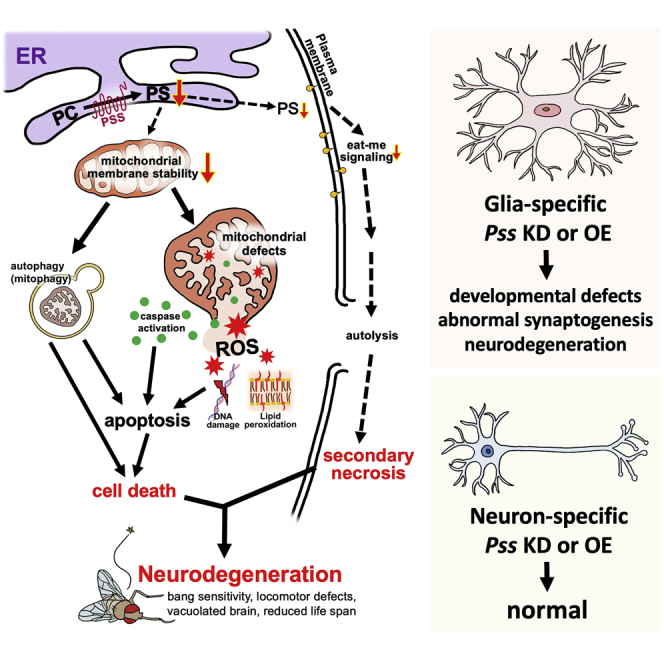
Changes in phospholipid levels have been reported in the brains of patients with neurodegeneration, such as AD and PD (Li et al., 2015; Pettegrew et al., 2001). However, it has not yet been determined whether changes in phospholipid levels are sufficient to lead to NDs. In this study, we demonstrate that loss of Pss and overexpression of the Pss gene encoding a Drosophila homolog of mammalian PSS1 and 2 cause severe neurodegenerative phenotypes. These flies display defects in longevity, climbing ability, and bang sensitivity. We further document the formation of numerous vacuoles in brains with reduced Pss. Finally, we showed that either glia-specific knockdown or overexpression of Pss is sufficient to induce neurodegenerative features (Figure 5) in aging flies. We argue that the level of PSS is critical and needs to be properly controlled.
PS is one of the major substrates for the production of PE, which is critical for the stability of the mitochondrial inner membrane (Schenkel and Bakovic, 2014; Tamura et al., 2012; Vance, 2014). A decrease in mitochondrial PE impairs cell growth, respiratory capacity, and ATP production and profoundly alters mitochondrial morphology in mammalian cells (Tasseva et al., 2013). Also, reduction of PE production in mitochondria causes fragmented, misshapen mitochondria in mice (Steenbergen et al., 2005). A recent study has shown that the level of PS in salivary gland is dramatically reduced in salivary gland-specific Pss RNAi flies to around 20% of control flies (Yang et al., 2019). Hence, reduced PS synthesis in our Pss mutants may affect PE production in mitochondria, which in turn affects mitochondrial structure and function. This is consistent with our observation of a large population of swollen and ruptured mitochondria with disorganized cristae in Pss mutant. Abnormal and dysfunctional mitochondria are removed by autophagy because ruptured mitochondria release excessive ROS to the cytoplasm (Thomas and Gustafsson, 2013). Increases in either cytoplasmic ROS levels or the number of dysfunctional mitochondria will eventually kill affected cells (Bhat et al., 2015; Jena, 2012). Indeed, Pss mutants had a large population of misshapen and ruptured mitochondria, higher ROS levels, and increased cell death by both autophagy and apoptosis. Therefore, the neurodegeneration observed in Pss mutants may be a consequence of mitochondrial dysfunction.
PS has also been well recognized as an “eat-me” signal for phagocytic clearance of apoptotic cells (Fadok et al., 1992; Leventis and Grinstein, 2010; Segawa and Nagata, 2015), and PS exposure is necessary for detection by phagocytes (Borisenko et al., 2003; Fadok et al., 2001). Phagocytosis is a vital component in the development and homeostasis of neural tissue, and the abnormal phagocytic ability has been linked to many diseases (Napoli and Neumann, 2009; Poon et al., 2014; Ravichandran, 2011; Salter and Stevens, 2017; Von Bernhardi et al., 2015; White and Gallin, 1986). Also, the absence of the phagocytic receptor Draper (drpr) in glia leads to a pronounced accumulation of apoptotic neurons in the brain of Drosophila (Etchegaray et al., 2016). Pss mutants are expected to have lower external PS levels but increased cell death events. Recent studies have shown that if apoptotic cells are not properly removed, the cells will eventually autolyze (secondary necrosis), releasing cytotoxic substances and killing exposed neighboring cells (Muñoz et al., 2010). Therefore, increased cell death in the Pss mutant brain may be due to secondary necrosis. Indeed, Pss mutants showed a strong increase in necrotic signals. Therefore, the primary apoptosis can be initiated by mitochondrial defect, but the reduction of phagocytic clearance and the secondary necrosis should accelerate the neurodegeneration in the Pss mutants and may even have a primary role.
Glia are non-neuronal cells that play an essential role in the trophic support of neurons, regulation of neuronal morphology, extracellular homeostasis, establishment of the blood-brain barrier, myelination, and axon growth and guidance (Chotard and Salecker, 2007; Liu et al., 2015), and abnormal glial function is associated with many NDs (Chung et al., 2020; Lee and Sun, 2015). Interestingly, we found that Pss is mostly expressed in a subset of glia in the CNS, and glia-specific knockdown or overexpression of Pss altered synaptogenesis and axonal growth in larval motoneurons. Moreover, glia-specific knockdown of Pss caused reduced life span and climbing defect. Importantly, the glial knockdown of Pss specifically after eclosion using the Gal80 system induces neurodegeneration as the flies aged. These results indicate that Pss is essential for glia not only during developmental stages but also for the maintenance of the nervous system in the adult. Furthermore, glia-specific overexpression of Pss also resulted in neurodegenerative phenotypes in the adult brain, and trans-heterozygous mutants in which UAS-Pss is expressed also showed reduction of life span and climbing ability compared with the wild-type flies due to the excessive expression of Pss, suggesting that Pss expression levels must be tightly regulated to maintain a healthy nervous system. In a recent study, increased expression of phagocytic receptors, SIMU or Drpr, in glia causes neuronal loss, motor defects, and early onset death, and these phenotypes are reversed by driving MFG-E8, which specifically binds PS and prevent the interaction between PS and the receptors (Hakim-Mishnaevski et al., 2019). In this context, overexpression of Pss is expected to increase the level and exposure of the PS in the plasma membrane, which may lead to excessive phagocytosis, resulting in neurodegeneration.
Recent studies emphasize the role of the lipid metabolism enzymes in glial cells (Chung et al., 2020; Griffin and Ackerman, 2020). One of the enzymes of the very-long-chain fatty acid (VLCFA) β-oxidation pathway in peroxisomes, ACOX1 (acyl-CoA oxidase 1), is mostly expressed and required in glia and both loss and gain of ACOX1 lead to glial degeneration. Moreover, another study indicates that elevated VLCFA levels can induce PS exposure on cell surfaces (Gong et al., 2017). Therefore, it will be interesting to test whether the PS containing VLCFA may also have important roles in glial cells.
There are six glial subtypes in Drosophila — cortex glia, ensheathing glia, astrocyte, perineurial glia, subperineurial glia, and wrapping glia — characterized in large part by their morphology and association with neurons (Freeman, 2015). Our genetic data suggest that Pss functions in cortex glia. Specific knockdown of Pss in cortex glia led to semi-lethality, a severely reduced life span as well as a significant increase in bang sensitivity, suggesting a crucial role for Pss in cortex glia. Cortex glia surrounds and supports neuronal cell bodies and the proximal regions of neurites as they extend toward the neuropil (Awasaki and Lee, 2011). Despite their notable morphology, cortex glial cells are the least well-characterized glial subtypes of the Drosophila CNS (Freeman, 2015), and further studies will be needed to clarify the functional relationships between Pss and cortex glia.
PSS seems to be essential not only for the maintenance of the nervous system but also for overall development. In humans, mutations in PTDSS1 cause LMHD (MIM 151050), a rare disease which shows developmental defects, severe growth retardation, and intellectual disability. Overexpression of mutant PTDSS1 RNA in zebrafish embryos also disrupts normal development (Sousa et al., 2014). PSS1 and PSS2 double knockout mice are prenatally lethal (Arikketh et al., 2008). These results and our observations suggest that Phosphatidylserine synthase plays a vital role in development, and further studies are necessary in order to uncover the specific role of these proteins in developmental processes.
In summary, our studies in Drosophila show that PSS is required for neuronal maintenance, and its loss or gain may cause NDs triggered by defective PS metabolism. Abnormal PS production in glial cells may have deleterious effects on the nervous system, including its development, maintenance, and/or function. We propose that changes in phospholipid composition, due mainly to abnormal levels of PS, are a cause of neurodegeneration rather than merely a consequence of disease.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse anti-GFP | DSHB | Cat# GFP-G1, RRID: AB_2619561 |
| Rabbit anti-GFP | Abcam | Cat# ab183734, RRID:AB_2732027 |
| Mouse anti-Repo | DSHB | Cat# 8D12 anti-Repo, RRID:AB_528448 |
| Mouse anti-Elav | DSHB | Cat# ELAV 9F8A9, RRID:AB_2314364 |
| Mouse anti-CSP | DSHB | Cat# DCSP-1 (ab49), RRID:AB_2307345 |
| Rabbit anti-cleaved caspase 3 | Cell signal | Cat# 9661, RRID:AB_2341188 |
| Goat anti-Mouse Alexa 488 | Invitrogen | Cat# A-11001, RRID:AB_2534069 |
| Goat anti-Mouse Alexa 594 | Invitrogen | Cat# A-11005, RRID:AB_2534073 |
| Goat anti-Rabbit Alexa 488 | Invitrogen | Cat# A21222, RRID:AB_10373853 |
| Goat anti-Rabbit Alexa 594 | Invitrogen | Cat# A-11072, RRID:AB_2534116 |
| Alexa 594 AffiniPure Goat Anti-HRP | Jackson ImmunoResearch Labs | Cat# 123-585-021, RRID: AB_2338966 |
| Bacterial and virus strains | ||
| DH5α Chemically Competent E. coli | enzynomics | Cat# CP010 |
| Chemicals, peptides, and recombinant proteins | ||
| 4% paraformaldehyde | Biosesang | Cat# PC2031-050-00 |
| 20X PBS | Biosesang | Cat# PR2007-100-00 |
| Triton X-100 | Sigma-Aldrich | Cat# X100 |
| Fluoromount-G | SouthernBiotech | Cat# 0100-01 |
| Schneider's Drosophila Medium (SDM) | Gibco | Cat# 21720024 |
| Dihydroethidium (DHE) | Invitrogen | Cat# D11347 |
| C11-BODIPY 581/591 | Invitrogen | Cat# D3861 |
| Acridine Orange (AO) | Invitrogen | Cat# A1301 |
| RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay | Promega | Cat# JA1011 |
| Ethyl alcohol, anhydrous, 99.9% | Samchun | Cat# E0243 |
| Chloroform, 99.5% | Samchun | Cat# C0584 |
| Acetic acid (glacial) 100% | Merck | Cat# 100063 |
| Paraplast | Leica | Cat# 39601006 |
| Paraplast plus | Sigma-Aldrich | Cat# P3683 |
| Hematoxylin | Sigma-Aldrich | Cat# 105175 |
| Eosin Y | Sigma-Aldrich | Cat# 318906 |
| Xylene, 99.9% | Samchun | Cat# X0097 |
| 5X All-In-One RT MasterMix (with AccuRT Genomic DNA Removal Kit) | Abm | Cat# G492 |
| TOPreal qPCR 2X PreMix (SYBR green with low ROX) | Enzynomics | Cat# RT500S |
| iTaq Universal SYBR Green Supermix | Bio-Rad | Cat# 1725120 |
| 4% glutaraldehyde | Ted Pella | Cat# 18427 |
| 0.1 M cacodylate buffer | Ted Pella | Cat# 18851 |
| 100% propylene | EMS | Cat# 20401 |
| eponate 812 resin | Ted Pella | Cat# 18005 |
| Uranyl acetate | Ted Pella | Cat# 19481 |
| Lead citrate | EMS | Cat# 17800 |
| Critical commercial assays | ||
| RNeasy Plus Mini Kit | Qiagen | Cat# 74134 |
| Deposited data | ||
| FlyBase | - | http://flybase.org |
| Experimental models: Organisms/strains | ||
| Pss15 | BDSC | RRID: BDSC_2215 |
| Pss32 | BDSC | RRID: BDSC_11632 |
| PssGAL4 | Kyoto Stock Center | RRID: Kyoto Stock Center_104172 |
| PssΔ1 | Jeon lab (This paper) | N/A |
| Df(3L)BSC448 | BDSC | RRID: BDSC_24952 |
| da-GAL4 | BDSC | RRID: BDSC_55851 |
| Act-GAL4 | BDSC | RRID: BDSC_4414 |
| repo-GAL4 | BDSC | RRID: BDSC_7415 |
| elav-GAL4 | BDSC | RRID: BDSC_8765 |
| D42-GAL4 | BDSC | RRID: BDSC_8816 |
| alrm-GAL4 | BDSC | RRID: BDSC_67031 |
| Mz97-GAL4 | BDSC | RRID: BDSC_9488 |
| UAS-mCherry.nls | BDSC | RRID: BDSC_38424 |
| UAS-GFP.nls | BDSC | RRID: BDSC_4776 |
| UAS-mCD8.GFP | BDSC | RRID: BDSC_32186 |
| NP2222-GAL4 | Kyoto Stock Center | RRID: Kyoto Stock Center_112830 |
| NP6293-GAL4 | Kyoto Stock Center | RRID: Kyoto Stock Center_105188 |
| NP2276-GAL4 | Kyoto Stock Center | RRID: Kyoto Stock Center_112853 |
| UAS-Pss RNAi | VDRC | RRID: VDRC_V5391 |
| y1v1nos-phiC31; attP40 | NIG-fly | RRID: SHIGEN TBX-0002 |
| y2cho2v1 | NIG-fly | RRID: SHIGEN TBX-0004 |
| y2cho2v1; Sp/CyO | NIG-fly | RRID: SHIGEN TBX-0009 |
| y2cho2v1; attP40{nos-Cas9}/CyO | NIG-fly | RRID: SHIGEN CAS-0001 |
| y2cho2v1; Pr Dr/TM6C, Sb Tb | NIG-fly | RRID: SHIGEN TBX-0010 |
| UAS-Pss | Jeon lab (This paper) | N/A |
| Oligonucleotides | ||
|
PssΔ1 gRNA top oligo: CTTCGAACGTGGATGAGAA TCTCT |
Bioneer | N/A |
|
PssΔ1 gRNA bottom oligo: AAACAGAGATTCTCAT CCACGTTC |
Bioneer | N/A |
| qRT-PCR primer for Rp49 forward oligo: FW 5'-ACA GGCCCAAGATCGTGAAGA-3' |
Bioneer | N/A |
| qRT-PCR primer for Rp49 reverse oligo: RV 5’-CGCA CTCTGTTGTCGATACCCT-3′ |
Bioneer | N/A |
| qRT-PCR primer for Pss forward oligo: FW 5'-ACAGG CCCAAGATCGTGAAGA-3' |
Bioneer | N/A |
| qRT-PCR primer for Pss reverse oligo: RV 5’-CGCACTC TGTTGTCGATACCCT-3′ |
Bioneer | N/A |
| Software and algorithms | ||
| BLAST | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| PRALINE | (Simossis and Heringa, 2005) https://doi.org/10.1093/nar/gki390 | https://www.ibi.vu.nl/programs/pralinewww/ |
| Rotor-Gene Q Software | Qiagen | https://www.qiagen.com/kr/resources/resourcedetail?id=8435805b-2c5d-4fa9-948c-a43de75a7ee1&lang=en |
| CFX Manger Software | Bio-Rad | https://www.bio-rad.com/en-kr/sku/1845000-cfx-manager-software?ID=1845000 |
| QuantStudio 5 Real-Time PCR System Software | ThermoFisher | https://www.thermofisher.com/kr/en/home/global/forms/life-science/quantstudio-3-5-software.html |
| ImageJ | Version 2.1.0 | https://imagej.nih.gov/ij/ |
| Zen Black | Zeiss | https://www.zeiss.com/microscopy/us/products/microscope-software/zen-lite.html |
| LAS X | Leica | https://www.leica-microsystems.com/products/microscope-software/p/leica-las-x-ls/ |
| MetaMorph | Version 7.8.6.0 | https://www.moleculardevices.com/products/cellular-imaging-systems/acquisition-and-analysis-software/metamorph-microscopy#gref |
| Tracker | Version 5.1.5 | https://tracker.physlets.org/ |
Resource availability
Lead contact and materials availability
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Sang-Hak Jeon (jeonsh@snu.ac.kr). All fly lines and plasmids generated in this study are available from the Lead Contact without restriction.
Data and code availability
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
Experimental model and subject details
Drosophila melanogaster strains
Pss mutant alleles used for this experiment were designated according to the last two digits taken from the stock number: Pss15 (BL22115, w1118; P{EPg}PssHP31723), Pss32 (BL11632, P{PZ}Pss04521 ry506 /TM3, ryRK Sb1 Ser1), and PssGAL4 (#104172, w∗; P{GawB}PssNP2363 / TM3, Sb1 Ser1) (Bellen et al., 2004, 2011; Hayashi et al., 2002). We used the Pss32 line to collect virgins for crosses to generate trans-heterozygous mutants (Pss32/15, Pss32/GAL4, Pss32/Δ1, Pss32/Df), and used Canton-S as female parent to generate heterozygous mutants (Pss32/+, Pss Δ1/+) to make their backgrounds same in w+. Canton-S was used as a control. For the transgenic manipulations, the GAL4-UAS system was employed (Brand and Perrimon, 1993). We obtained Pss15 (BL22115), Pss32 (BL11632), PssGAL4 (BL104172), Df(3L)BSC448 (BL24952), da-GAL4 (BL55851), Act-GAL4 (BL4414), repo-GAL4 (BL7415), elav-GAL4 (BL8765), D42-GAL4 (BL8816), alrm-GAL4 (BL67031), Mz97-GAL4 (BL9488), UAS-mCherry.nls (BL38424), UAS-GFP.nls (BL4776) and UAS-mCD8.GFP (BL32186) from the Bloomington Drosophila Stock Center (BDSC); PssGAL4, NP2222-GAL4 (#112830), NP6293-GAL4 (#105188), and NP2276-GAL4 (#112853) from the Kyoto Stock Center; and UAS-Pss RNAi (V5391) from the Vienna Drosophila Resource Center (VDRC). The V5391 was already validated in a recent study (Yang et al., 2019) as it dramatically reduced the expression level of Pss in salivary gland (ppl>Pss RNAi, almost 90% reduced). Flies were raised in vials containing cornmeal–yeast–agar medium at 22°C or 25°C during development and at 29°C after eclosion, with 50% relative humidity and a 12 h:12 h light:dark cycle. Except for the complementary test, we used only male progenies for all the other assays on adult flies.
Method details
Generation of PssΔ1 and UAS-Pss
We employed the CRISPR/Cas9 system to generate a small deletion in the 2nd exon of the Pss gene, as described previously (Kondo and Ueda, 2013). To construct gRNA, two complementary oligonucleotides targeting a region in the 2nd exon, 'CTTCGAACGTGGATGAGAATCTCT' and 'AAACAGAGATTCTCATCCACGTTC', were annealed and cloned into the pBFv-U6.2 vector (NIG-fly) at the BbsI site, and the resulting recombinant plasmids were injected into y1 v1 nos-phiC31; attP40 host embryos. Subsequent methods for producing a gRNA-mediated deletion line followed the previously described procedures (Kondo and Ueda, 2013). G0 males that survived to adulthood were individually mated with y2 cho2 v1 (NIG-fly). Male offspring showing wild-type eye color (i.e., transformants) were crossed with y2 cho2 v1; Sp/CyO (NIG-fly) virgins, and then the transgenic line was balanced and established as a stock (U6-Pss-gRNA). Male flies of this line were crossed with y2 cho2 v1; attP40{nos-Cas9}/CyO (NIG-Fly), which express the Cas9 protein specifically in the germ line. The male founders containing both U6-Pss-gRNA and nos-Cas9 transgenes were mated with y2 cho 2 v1; Pr Dr/TM6C, Sb Tb flies. Offspring balanced with TM6C, Sb Tb were collected to establish a stock and used for molecular characterization. To generate UAS-Pss transgenic lines, a full-length cDNA was cloned into a pUAST vector, and the resulting plasmid was used for conventional germline transformation.
Protein homology and alignment of Pss
Pss gene sequence and protein sequence information were obtained from the FlyBase (http://flybase.org). Protein BLAST was conducted using web-based software (https://blast.ncbi.nlm.nih.gov/Blast.cgi) to determine the similarities between fly PSS and human PSSs. Amino acid sequence alignment was performed with PRALINE (Simossis and Heringa, 2005).
Quantitative real-time PCR
Total RNA was extracted from pupae using RNeasy Plus Mini Kit (Qiagen, #74134), and then ∼1.0 μg RNA was reverse transcribed to synthesize cDNA using a 5X All-In-One RT MasterMix with AccuRT Genomic DNA Removal Kit (Abm, G492). qPCR was performed with TOPreal qPCR 2X PreMix (SYBR green with low ROX, enzynomics) and Rotor-Gene Q (QIAGEN) or with iTaq Universal SYBR Green Supermix (BIO-RAD) and CFX96 Touch Real-Time PCR Detection System (BIO-RAD) or QuantStudio™ 5 System (Thermo Fisher Scientific). Rp49 was used for normalization. The following primers were used for qPCR:
Rp49-for (5'-ACAGGCCCAAGATCGTGAAGA-3')
Rp49-rev (5’-CGCACTCTGTTGTCGATACCCT-3′)
Pss-for (5'-ATGAAGAAGCGCACTAATTCACG-3′)
Pss-rev (5′-CCTGATTTGTAGGCGGGATG-3′)
Longevity, climbing ability, and bang sensitivity
For all assays, newly eclosed flies were collected and kept at 29°C until used. For the longevity assay, each genotype was collected at eclosion, divided into vials (up to 20 flies per vial), and then incubated at 29°C. Flies were transferred to fresh vials every 3 days, and the number of dead flies was counted. Survival rates were calculated from the total population. To test climbing ability, we made a climbing kit with 5 vials (diameter: 2.6cm, height: 27.7cm) and a frame that enables us to compare up to 5 alleles at once. 3, 10, 20, or 30-day-old flies were transferred to an empty vial (each containing up to 20 flies). The vials in the frame were gently tapped to force all flies to the bottom and then videotaped for 10 s. This was repeated ten times for each vial, and this was considered as one set. 3-5 sets were analyzed by Tracker software (Brown, 2008) (https://tracker.physlets.org/) for final data. Error bars for the climbing assay are SEM. For the bang sensitivity test, 3-day-old flies were transferred to an empty vial and vortexed for 5 s at maximum speed (3000 rpm). For each individual fly, we measured the time from the end of the impact to the moment that flies right themselves. At least 14 flies were tested for each genotype.
Detection of vacuoles in the brain
To detect vacuoles in the brain, fly heads were fixed in fresh Carnoy's solution (ethanol:chloroform:acetic acid at 6:3:1) at 4°C for at least 24 h, washed, serially dehydrated with ethanol, and paraffin-embedded using standard histological procedures (Palladino et al., 2002). Serial 5 μm sections were stained with hematoxylin and eosin and examined under a light microscope. Images were taken with a DE/Axio Imager A1 microscope (Carl Zeiss) equipped with a DE/Axiocam HR. The diameter of the vacuoles was measured by ImageJ. The total numbers of vacuoles with diameter from 3μm to 6μm and over 6μm in each fly brain were divided by the total number of slices of each fly brain to obtain the average number of vacuoles per slice. For each genotype, at least 11 heads were used.
NMJ analyses and larval roll-over assay
NMJs were dissected from wandering third instar larvae, as described previously (Smith and Taylor, 2011). The NMJs between the 6/7 muscles of the A3 segment were stained with anti-HRP and anti-CSP. An Intravital multi-photon confocal laser scanning microscope (IMP-LSM; LSM 780 NLO, Carl Zeiss) was used for observing and photographing NMJs. ImageJ was used for the quantification of boutons, branches, and NMJ length. Synaptic branches with two or more synaptic boutons were considered branches, according to (Miller et al., 2012).
For the larval roll-over assay, a third instar larva was placed on an apple juice plate. After a brief adjustment period, the larva was gently inverted (ventral side up). The time taken for the original posture (dorsal side up) to be restored was measured. For each genotype, at least 20 larvae were tested.
Immunohistochemistry
Mouse anti-GFP (G1-S, 1:25, Developmental Studies Hybridoma Bank (DSHB)), rabbit anti-GFP (1:25, Abcam), mouse anti-Repo (8D12, 1:20, DSHB), mouse anti-Elav (9F8A9, 1:40, DSHB), and mouse anti-CSP (ab49, 1/20, DSHB) were used as primary antibodies. For secondary antibodies, we used goat anti-mouse-IgG and anti-rabbit-IgG conjugated with Alexa Fluor 488 or Alexa Fluor 594 (1:1000 dilution, Invitrogen) and Alexa Fluor 594-conjugated AffiniPure Goat Anti-Horseradish Peroxidase (1:400, Jackson ImmunoResearch).
Tissue was fixed with 4% paraformaldehyde for 30 min, washed in PT (1X PBS, 0.1% Triton X-100), incubated with primary antibodies overnight at 4°C, and then washed again before incubation with secondary antibodies for 1–2 h at room temperature. Tissue was thoroughly rinsed in PT and mounted with Fluoromount-G for imaging.
Detection of ROS in brain tissue
Fly brains were dissected in Schneider's medium (SDM). After brief washing with SDM, the brains were incubated for 5 min either with 150 μL DHE (final concentration 30 μM) to detect ROS or with C11-BODIPY(581/591) (final concentration 2 μM) to measure lipid peroxidation. After washing three times in SDM, brains were mounted with Fluoromount-G and then observed immediately with an Intravital multi-photon confocal laser scanning microscope (IMP-LSM; LSM 780 NLO, Carl Zeiss). Images were obtained as Z series with the same interval for the whole brain. Z series images were merged by ImageJ (Image-Stacks-Z projection-SUM slices), and then the fluorescence intensity was measured. To compare the fluorescence intensity, a ratio was calculated by dividing the fluorescence value from the mutant brain by the fluorescence value from the wild-type brain obtained immediately after the mutant.
Detection of cell death in brain tissue
Acridine orange (AO) staining and anti-caspase 3 immunohistochemistry were used to detect dying cells in adult brains. The chemicals were prepared as follows: 1.3 μL of 1 mM AO (Invitrogen, A1301) in 1 mL of 1X PBS; rabbit anti-cleaved caspase 3 antibody (#9661, 1:100, Cell signal); and Alexa Fluor 594 conjugated goat anti-rabbit-IgG (1:1000, Invitrogen).
For AO staining, fly brains were dissected in PBS, washed with PBS, and then incubated in 150 μL AO for 10 min. The tissue was washed three times with PBS, mounted in Fluoromount-G, and observed immediately with an Intravital multi-photon confocal laser scanning microscope (IMP-LSM; LSM 780 NLO, Carl Zeiss). Each observation was conducted under the following conditions: single-strand DNA and RNA (excitation: 488 nm, emission: 650 nm); lysosome (excitation: 561 nm, emission: 579–620 nm). Images were analyzed in the same way as described above in ROS detection.
Transmission electron microscopy
Fly heads without the proboscis were fixed with 2% glutaraldehyde and 2% paraformaldehyde in phosphate buffer (pH 7.4) for 1h at 4°C and then postfixed osmium tetroxide for 40 min at 4°C. The samples were dehydrated in a graded series of ethanol. The samples were treated with graded propylene oxide series and embedded into Epon. The sections were then ultra-thin sectioned in 80-nm and placed on copper grid. The final samples were stained with uranyl acetate and lead citrate. After the observed using a transmission electron microscope (JEOL-2100F, USA, 200kV) at the Korea Basic Science Institute, Chuncheon Center.
Detection of necrosis
We used the necrosis detection reagent (1000X) from the RealTime-Glo™ Annexin V Apoptosis and Necrosis Assay (Promega, JA1011). Reagents were diluted to 1X in SDM immediately before use. Fly heads without the proboscis were stained in 1X necrosis detection reagent for 2 h at room temperature. Then, the tissue was washed three times with SDM. The cuticles were removed gently and quickly, and the brains were mounted in Fluoromount-G. Images were obtained with an Olympus BX51 microscope, a Q Imaging optiMOS camera, and MetaMorph software.
Quantification and statistical analysis
Quantified data were analyzed statistically with the Excel program's data analysis tools for the f-test and the t-test. Log-rank test was used to compare survival (lifespan) curves. Statistical significance is shown as follows: ∗p < 0.05, ∗∗p < 0.01, ∗∗∗p < 0.001. p > 0.05 was considered not significant (n.s.).
Acknowledgments
This work was supported by the National Research Foundation of Korea Grant (NRF-2015R1D1A1A01059861, NRF-2019R1F1A1061772) to S.H.J. H.J.B. is an Investigator of the Howard Hughes Medical Institute. H.-L.C. is supported by the Warren Alpert Foundation. We appreciate the technical support of the Korea Basic Science Institute (KBSI), Chuncheon Center, and Seoul Center.
Author contributions
Y.-J.P., S.K., H.-P.S., H.-L.C., H.J.B., and S.-H.J. designed the experiments and wrote the manuscript. Y.-J.P., S.K., H.-P.S., T.-Y.K., M.-C.J., M.L, S.L., and J.Y. conducted experiments. A.-Y.K. generated the CRISPR/Cas9 mutation. J.H.P. and G.L. produced the UAS-Pss lines and wrote the manuscript. S.-H.K. conducted the analysis of confocal microscopy images.
Declaration of interests
The authors declare no competing interests.
Published: August 20, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2021.102899.
Supplemental information
References
- Abuja P.M., Albertini R. Methods for monitoring oxidative stress, lipid peroxidation and oxidation resistance of lipoproteins. Clinica Chim. Acta Int. J. Clin. Chem. 2001;306:1–17. doi: 10.1016/s0009-8981(01)00393-x. [DOI] [PubMed] [Google Scholar]
- Arikketh D., Nelson R., Vance J.E. Defining the importance of phosphatidylserine synthase-1 (PSS1): unexpected viability of PSS1-deficient mice. J. Biol. Chem. 2008;283:12888–12897. doi: 10.1074/jbc.M800714200. [DOI] [PubMed] [Google Scholar]
- Awasaki T., Lee T. New tools for the analysis of glial cell biology in Drosophila. Glia. 2011;59:1377–1386. doi: 10.1002/glia.21133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellen H.J., Levis R.W., He Y., Carlson J.W., Evans-Holm M., Bae E., Kim J., Metaxakis A., Savakis C., Schulze K.L. The <em>Drosophila</em> gene disruption project: progress using transposons with distinctive site specificities. Genetics. 2011;188:731. doi: 10.1534/genetics.111.126995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bellen H.J., Levis R.W., Liao G., He Y., Carlson J.W., Tsang G., Evans-Holm M., Hiesinger P.R., Schulze K.L., Rubin G.M. The BDGP gene disruption project. Genetics. 2004;167:761. doi: 10.1534/genetics.104.026427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergo M.O., Gavino B.J., Steenbergen R., Sturbois B., Parlow A.F., Sanan D.A., Skarnes W.C., Vance J.E., Young S.G. Defining the importance of phosphatidylserine synthase 2 in mice. J. Biol. Chem. 2002;277:47701–47708. doi: 10.1074/jbc.M207734200. [DOI] [PubMed] [Google Scholar]
- Bhat A.H., Dar K.B., Anees S., Zargar M.A., Masood A., Sofi M.A., Ganie S.A. Oxidative stress, mitochondrial dysfunction and neurodegenerative diseases; a mechanistic insight. Biomed. Pharmacother. 2015;74:101–110. doi: 10.1016/j.biopha.2015.07.025. [DOI] [PubMed] [Google Scholar]
- Borisenko G.G., Matsura T., Liu S.X., Tyurin V.A., Jianfei J., Serinkan F.B., Kagan V.E. Macrophage recognition of externalized phosphatidylserine and phagocytosis of apoptotic Jurkat cells--existence of a threshold. Arch. Biochem. Biophys. 2003;413:41–52. doi: 10.1016/s0003-9861(03)00083-3. [DOI] [PubMed] [Google Scholar]
- Brand A.H., Perrimon N. Targeted gene expression as a means of altering cell fates and generating dominant phenotypes. Development. 1993;118:401–415. doi: 10.1242/dev.118.2.401. [DOI] [PubMed] [Google Scholar]
- Brent J., Werner K., McCabe B.D. Drosophila larval NMJ immunohistochemistry. J. Vis. Exp. 2009:1108. doi: 10.3791/1108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brink D.L., Gilbert M., Xie X., Petley-Ragan L., Auld V.J. Glial processes at the Drosophila larval neuromuscular junction match synaptic growth. PLoS One. 2012;7:e37876. doi: 10.1371/journal.pone.0037876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown D. 2008. Video Modeling: Combining Dynamic Model Simulations with Traditional Video Analysis. [Google Scholar]
- Buccoliero R., Bodennec J., Van Echten-Deckert G., Sandhoff K., Futerman A.H. Phospholipid synthesis is decreased in neuronal tissue in a mouse model of Sandhoff disease. J. Neurochem. 2004;90:80–88. doi: 10.1111/j.1471-4159.2004.02457.x. [DOI] [PubMed] [Google Scholar]
- Choi J.Y., Wu W.I., Voelker D.R. Phosphatidylserine decarboxylases as genetic and biochemical tools for studying phospholipid traffic. Anal. Biochem. 2005;347:165–175. doi: 10.1016/j.ab.2005.03.017. [DOI] [PubMed] [Google Scholar]
- Chotard C., Salecker I. Glial cell development and function in the Drosophila visual system. Neuron Glia Biol. 2007;3:17–25. doi: 10.1017/S1740925X07000592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H.L., Wangler M.F., Marcogliese P.C., Jo J., Ravenscroft T.A., Zuo Z., Duraine L., Sadeghzadeh S., Li-Kroeger D., Schmidt R.E. Loss- or gain-of-function mutations in ACOX1 cause axonal loss via different mechanisms. Neuron. 2020;106:589–606.e586. doi: 10.1016/j.neuron.2020.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crook T., Petrie W., Wells C., Massari D.C. Effects of phosphatidylserine in Alzheimer's disease. Psychopharmacol. Bull. 1992;28:61–66. [PubMed] [Google Scholar]
- Crook T.H., Tinklenberg J., Yesavage J., Petrie W., Nunzi M.G., Massari D.C. Effects of phosphatidylserine in age-associated memory impairment. Neurology. 1991;41:644–649. doi: 10.1212/wnl.41.5.644. [DOI] [PubMed] [Google Scholar]
- Duffy J.B. GAL4 system in drosophila: a fly geneticist's swiss army knife. genesis. 2002;34:1–15. doi: 10.1002/gene.10150. [DOI] [PubMed] [Google Scholar]
- Edwards T.N., Meinertzhagen I.A. The functional organisation of glia in the adult brain of Drosophila and other insects. Prog. Neurobiol. 2010;90:471–497. doi: 10.1016/j.pneurobio.2010.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eskelinen E.L., Reggiori F., Baba M., Kovacs A.L., Seglen P.O. Seeing is believing: the impact of electron microscopy on autophagy research. Autophagy. 2011;7:935–956. doi: 10.4161/auto.7.9.15760. [DOI] [PubMed] [Google Scholar]
- Etchegaray J.I., Elguero E.J., Tran J.A., Sinatra V., Feany M.B., McCall K. Defective phagocytic corpse processing results in neurodegeneration and can Be rescued by TORC1 activation. J. Neurosci. 2016;36:3170–3183. doi: 10.1523/JNEUROSCI.1912-15.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fadok V.A., de Cathelineau A., Daleke D.L., Henson P.M., Bratton D.L. Loss of phospholipid asymmetry and surface exposure of phosphatidylserine is required for phagocytosis of apoptotic cells by macrophages and fibroblasts. J. Biol. Chem. 2001;276:1071–1077. doi: 10.1074/jbc.M003649200. [DOI] [PubMed] [Google Scholar]
- Fadok V.A., Voelker D.R., Campbell P.A., Cohen J.J., Bratton D.L., Henson P.M. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J. Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- Fergestad T., Olson L., Patel K.P., Miller R., Palladino M.J., Ganetzky B. Neuropathology in Drosophila mutants with increased seizure susceptibility. Genetics. 2008;178:947–956. doi: 10.1534/genetics.107.082115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Freeman M.R. Drosophila central nervous system glia. Cold Spring Harb. Perspect. Biol. 2015;7:a020552. doi: 10.1101/cshperspect.a020552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaschler M.M., Stockwell B.R. Lipid peroxidation in cell death. Biochem. Biophys. Res. Commun. 2017;482:419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong Y., Sasidharan N., Laheji F., Frosch M., Musolino P., Tanzi R., Kim D.Y., Biffi A., El Khoury J., Eichler F. Microglial dysfunction as a key pathological change in adrenomyeloneuropathy. Ann. Neurol. 2017;82:813–827. doi: 10.1002/ana.25085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffin E.N., Ackerman S.L. Lipid metabolism and axon degeneration: an ACOX1 balancing act. Neuron. 2020;106:551–553. doi: 10.1016/j.neuron.2020.04.030. [DOI] [PubMed] [Google Scholar]
- Hakim-Mishnaevski K., Flint-Brodsly N., Shklyar B., Levy-Adam F., Kurant E. Glial phagocytic receptors promote neuronal loss in adult Drosophila brain. Cell Rep. 2019;29:1438–1448.e1433. doi: 10.1016/j.celrep.2019.09.086. [DOI] [PubMed] [Google Scholar]
- Hayashi S., Ito K., Sado Y., Taniguchi M., Akimoto A., Takeuchi H., Aigaki T., Matsuzaki F., Nakagoshi H., Tanimura T. GETDB, a database compiling expression patterns and molecular locations of a collection of gal4 enhancer traps. Genesis. 2002;34:58–61. doi: 10.1002/gene.10137. [DOI] [PubMed] [Google Scholar]
- Heemels M.-T. Neurodegenerative diseases. Nature. 2016;539:179. doi: 10.1038/539179a. [DOI] [PubMed] [Google Scholar]
- Heiss W.D., Kessler J., Mielke R., Szelies B., Herholz K. Long-term effects of phosphatidylserine, pyritinol, and cognitive training in Alzheimer's disease. A neuropsychological, EEG, and PET investigation. Dementia. 1994;5:88–98. doi: 10.1159/000106702. [DOI] [PubMed] [Google Scholar]
- Hu Y., Flockhart I., Vinayagam A., Bergwitz C., Berger B., Perrimon N., Mohr S.E. An integrative approach to ortholog prediction for disease-focused and other functional studies. BMC Bioinformatics. 2011;12:357. doi: 10.1186/1471-2105-12-357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irvine G.B., El-Agnaf O.M., Shankar G.M., Walsh D.M. Protein aggregation in the brain: the molecular basis for Alzheimer's and Parkinson's diseases. Mol. Med. 2008;14:451–464. doi: 10.2119/2007-00100.Irvine. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jena N.R. DNA damage by reactive species: mechanisms, mutation and repair. J. Biosci. 2012;37:503–517. doi: 10.1007/s12038-012-9218-2. [DOI] [PubMed] [Google Scholar]
- Kim H.Y., Huang B.X., Spector A.A. Phosphatidylserine in the brain: metabolism and function. Prog. lipid Res. 2014;56:1–18. doi: 10.1016/j.plipres.2014.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo S., Ueda R. Highly improved gene targeting by germline-specific Cas9 expression in Drosophila. Genetics. 2013;195:715–721. doi: 10.1534/genetics.113.156737. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kretzschmar D., Hasan G., Sharma S., Heisenberg M., Benzer S. The swiss cheese mutant causes glial hyperwrapping and brain degeneration in Drosophila. J. Neurosci. 1997;17:7425–7432. doi: 10.1523/JNEUROSCI.17-19-07425.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuebler D., Tanouye M.A. Modifications of seizure susceptibility inDrosophila. J. Neurophysiol. 2000;83:998–1009. doi: 10.1152/jn.2000.83.2.998. [DOI] [PubMed] [Google Scholar]
- Lee Y.M., Sun Y.H. Drosophila as a model to study the role of glia in neurodegeneration. J. Neurogenet. 2015;29:69–79. doi: 10.3109/01677063.2015.1076816. [DOI] [PubMed] [Google Scholar]
- Leventis P.A., Grinstein S. The distribution and function of phosphatidylserine in cellular membranes. Annu. Rev. Biophys. 2010;39:407–427. doi: 10.1146/annurev.biophys.093008.131234. [DOI] [PubMed] [Google Scholar]
- Li Z., Zhang J., Sun H. Increased plasma levels of phospholipid in Parkinson's disease with mild cognitive impairment. J. Clin. Neurosci. 2015;22:1268–1271. doi: 10.1016/j.jocn.2015.02.013. [DOI] [PubMed] [Google Scholar]
- Liu L., Zhang K., Sandoval H., Yamamoto S., Jaiswal M., Sanz E., Li Z., Hui J., Graham B.H., Quintana A. Glial lipid droplets and ROS induced by mitochondrial defects promote neurodegeneration. Cell. 2015;160:177–190. doi: 10.1016/j.cell.2014.12.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Midorikawa R., Yamamoto-Hino M., Awano W., Hinohara Y., Suzuki E., Ueda R., Goto S. Autophagy-dependent rhodopsin degradation prevents retinal degeneration in Drosophila. J. Neurosci. 2010;30:10703–10719. doi: 10.1523/JNEUROSCI.2061-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller D.L., Ballard S.L., Ganetzky B. Analysis of synaptic growth and function in Drosophila with an extended larval stage. J. Neurosci. 2012;32:13776–13786. doi: 10.1523/JNEUROSCI.0508-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizushima N. Autophagy: process and function. Genes Development. 2007;21:2861–2873. doi: 10.1101/gad.1599207. [DOI] [PubMed] [Google Scholar]
- Muñoz L.E., Lauber K., Schiller M., Manfredi A.A., Herrmann M. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat. Rev. Rheumatol. 2010;6:280. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- Napoli I., Neumann H. Microglial clearance function in health and disease. Neuroscience. 2009;158:1030–1038. doi: 10.1016/j.neuroscience.2008.06.046. [DOI] [PubMed] [Google Scholar]
- Owusu-Ansah E., Yavari A., Banerjee U. Protocol Exchange; 2008. A Protocol for in Vivo Detection of Reactive Oxygen Species. [Google Scholar]
- Palladino M.J., Hadley T.J., Ganetzky B. Temperature-sensitive paralytic mutants are enriched for those causing neurodegeneration in Drosophila. Genetics. 2002;161:1197–1208. doi: 10.1093/genetics/161.3.1197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker L., Howlett I.C., Rusan Z.M., Tanouye M.A. Seizure and epilepsy: studies of seizure disorders in Drosophila. In: Atkinson N., editor. International Review of Neurobiology. Academic Press; 2011. pp. 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlidis P., Ramaswami M., Tanouye M.A. The Drosophila easily shocked gene: a mutation in a phospholipid synthetic pathway causes seizure, neuronal failure, and paralysis. Cell. 1994;79:23–33. doi: 10.1016/0092-8674(94)90397-2. [DOI] [PubMed] [Google Scholar]
- Pettegrew J.W., Panchalingam K., Hamilton R.L., McClure R.J. Brain membrane phospholipid alterations in Alzheimer's disease. Neurochem. Res. 2001;26:771–782. doi: 10.1023/a:1011603916962. [DOI] [PubMed] [Google Scholar]
- Poon I.K.H., Lucas C.D., Rossi A.G., Ravichandran K.S. Apoptotic cell clearance: basic biology and therapeutic potential. Nat. Rev. Immunol. 2014;14:166–180. doi: 10.1038/nri3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ravichandran Kodi S. Beginnings of a good apoptotic meal: the find-me and eat-me signaling pathways. Immunity. 2011;35:445–455. doi: 10.1016/j.immuni.2011.09.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ross C.A., Poirier M.A. Protein aggregation and neurodegenerative disease. Nat. Med. 2004;10:S10–S17. doi: 10.1038/nm1066. [DOI] [PubMed] [Google Scholar]
- Salter M.W., Stevens B. Microglia emerge as central players in brain disease. Nat. Med. 2017;23:1018–1027. doi: 10.1038/nm.4397. [DOI] [PubMed] [Google Scholar]
- Sarkissian T., Timmons A., Arya R., Abdelwahid E., White K. Detecting apoptosis in Drosophila tissues and cells. Methods. 2014;68:89–96. doi: 10.1016/j.ymeth.2014.02.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel L.C., Bakovic M. Formation and regulation of mitochondrial membranes. Int. J. Cell Biol. 2014;2014:709828. doi: 10.1155/2014/709828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Segawa K., Nagata S. An apoptotic 'Eat Me' signal: phosphatidylserine exposure. Trends Cell Biol. 2015;25:639–650. doi: 10.1016/j.tcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- Serpell L.C., Smith J.M. Direct visualisation of the beta-sheet structure of synthetic Alzheimer's amyloid. J. Mol. Biol. 2000;299:225–231. doi: 10.1006/jmbi.2000.3650. [DOI] [PubMed] [Google Scholar]
- Sharon R., Bar-Joseph I., Frosch M.P., Walsh D.M., Hamilton J.A., Selkoe D.J. The formation of highly soluble oligomers of alpha-synuclein is regulated by fatty acids and enhanced in Parkinson's disease. Neuron. 2003;37:583–595. doi: 10.1016/s0896-6273(03)00024-2. [DOI] [PubMed] [Google Scholar]
- Sies H. Hydrogen peroxide as a central redox signaling molecule in physiological oxidative stress: oxidative eustress. Redox Biol. 2017;11:613–619. doi: 10.1016/j.redox.2016.12.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva M.T. Secondary necrosis: the natural outcome of the complete apoptotic program. FEBS Lett. 2010;584:4491–4499. doi: 10.1016/j.febslet.2010.10.046. [DOI] [PubMed] [Google Scholar]
- Simossis V.A., Heringa J. PRALINE: a multiple sequence alignment toolbox that integrates homology-extended and secondary structure information. Nucleic Acids Res. 2005;33:W289–W294. doi: 10.1093/nar/gki390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith R., Taylor J.P. Dissection and imaging of active zones in the Drosophila neuromuscular junction. J. Vis. Exp. 2011:2676. doi: 10.3791/2676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa S.B., Jenkins D., Chanudet E., Tasseva G., Ishida M., Anderson G., Docker J., Ryten M., Sa J., Saraiva J.M. Gain-of-function mutations in the phosphatidylserine synthase 1 (PTDSS1) gene cause Lenz-Majewski syndrome. Nat. Genet. 2014;46:70–76. doi: 10.1038/ng.2829. [DOI] [PubMed] [Google Scholar]
- Starkov A.A. The role of mitochondria in reactive oxygen species metabolism and signaling. Ann. N. Y. Acad. Sci. 2008;1147:37–52. doi: 10.1196/annals.1427.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steenbergen R., Nanowski T.S., Beigneux A., Kulinski A., Young S.G., Vance J.E. Disruption of the phosphatidylserine decarboxylase gene in mice causes embryonic lethality and mitochondrial defects. J. Biol. Chem. 2005;280:40032–40040. doi: 10.1074/jbc.M506510200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stone S.J., Vance J.E. Phosphatidylserine synthase-1 and -2 are localized to mitochondria-associated membranes. J. Biol. Chem. 2000;275:34534–34540. doi: 10.1074/jbc.M002865200. [DOI] [PubMed] [Google Scholar]
- Tamura Y., Onguka O., Itoh K., Endo T., Iijima M., Claypool S.M., Sesaki H. Phosphatidylethanolamine biosynthesis in mitochondria: phosphatidylserine (PS) trafficking is independent of a PS decarboxylase and intermembrane space proteins UPS1P and UPS2P. J. Biol. Chem. 2012;287:43961–43971. doi: 10.1074/jbc.M112.390997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasseva G., Bai H.D., Davidescu M., Haromy A., Michelakis E., Vance J.E. Phosphatidylethanolamine deficiency in Mammalian mitochondria impairs oxidative phosphorylation and alters mitochondrial morphology. J. Biol. Chem. 2013;288:4158–4173. doi: 10.1074/jbc.M112.434183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas R.L., Gustafsson A.B. Mitochondrial autophagy--an essential quality control mechanism for myocardial homeostasis. Circ. J. 2013;77:2449–2454. doi: 10.1253/circj.cj-13-0835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thome M.P., Filippi-Chiela E.C., Villodre E.S., Migliavaca C.B., Onzi G.R., Felipe K.B., Lenz G. Ratiometric analysis of Acridine Orange staining in the study of acidic organelles and autophagy. J. Cell Sci. 2016;129:4622–4632. doi: 10.1242/jcs.195057. [DOI] [PubMed] [Google Scholar]
- Ullian E.M., Christopherson K.S., Barres B.A. Role for glia in synaptogenesis. Glia. 2004;47:209–216. doi: 10.1002/glia.20082. [DOI] [PubMed] [Google Scholar]
- Valadas J.S., Esposito G., Vandekerkhove D., Miskiewicz K., Deaulmerie L., Raitano S., Seibler P., Klein C., Verstreken P. ER lipid defects in neuropeptidergic neurons impair sleep patterns in Parkinson's disease. Neuron. 2018;98:1155–1169.e1156. doi: 10.1016/j.neuron.2018.05.022. [DOI] [PubMed] [Google Scholar]
- Vance J.E. MAM (mitochondria-associated membranes) in mammalian cells: lipids and beyond. Biochim. Biophys. Acta. 2014;1841:595–609. doi: 10.1016/j.bbalip.2013.11.014. [DOI] [PubMed] [Google Scholar]
- Vance J.E., Steenbergen R. Metabolism and functions of phosphatidylserine. Prog. lipid Res. 2005;44:207–234. doi: 10.1016/j.plipres.2005.05.001. [DOI] [PubMed] [Google Scholar]
- Vance J.E., Tasseva G. Formation and function of phosphatidylserine and phosphatidylethanolamine in mammalian cells. Biochim. Biophys. Acta. 2013;1831:543–554. doi: 10.1016/j.bbalip.2012.08.016. [DOI] [PubMed] [Google Scholar]
- Von Bernhardi R., Eugenin-von Bernhardi L., Eugenin J. Microglial cell dysregulation in brain aging and neurodegeneration. Front. Aging Neurosci. 2015;7:124. doi: 10.3389/fnagi.2015.00124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S., Zhang S., Liou L.C., Ren Q., Zhang Z., Caldwell G.A., Caldwell K.A., Witt S.N. Phosphatidylethanolamine deficiency disrupts alpha-synuclein homeostasis in yeast and worm models of Parkinson disease. Proc. Natl. Acad. Sci. U S A. 2014;111:E3976–E3985. doi: 10.1073/pnas.1411694111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White C.J., Gallin J.I. Phagocyte defects. Clin. Immunol. 1986;40:50–61. doi: 10.1016/0090-1229(86)90068-1. [DOI] [PubMed] [Google Scholar]
- Williams D., Vicogne J., Zaitseva I., McLaughlin S., Pessin J.E. Evidence that electrostatic interactions between vesicle-associated membrane protein 2 and acidic phospholipids may modulate the fusion of transport vesicles with the plasma membrane. Mol. Biol. Cell. 2009;20:4910–4919. doi: 10.1091/mbc.E09-04-0284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang X., Liang J., Ding L., Li X., Lam S.-M., Shui G., Ding M., Huang X. Phosphatidylserine synthase regulates cellular homeostasis through distinct metabolic mechanisms. PLoS Genet. 2019;15:e1008548. doi: 10.1371/journal.pgen.1008548. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data reported in this paper will be shared by the lead contact upon request. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.