

Abstract
As part of the adaptive immune system, T cells are vital for the eradication of infected and malignantly transformed cells. To perform their protective function, T cells produce effector molecules that are either directly cytotoxic, such as granzymes, perforin, interferon‐γ and tumour necrosis factor α, or attract and stimulate (immune) cells, such as interleukin‐2. As these molecules can also induce immunopathology, tight control of their production is required. Indeed, inflammatory cytokine production is regulated on multiple levels. Firstly, locus accessibility and transcription factor availability and activity determine the amount of mRNA produced. Secondly, post‐transcriptional mechanisms, influencing mRNA splicing/codon usage, stability, decay, localization and translation rate subsequently determine the amount of protein that is produced. In the immune suppressive environments of tumours, T cells gradually lose the capacity to produce effector molecules, resulting in tumour immune escape. Recently, the role of post‐transcriptional regulation in fine‐tuning T‐cell effector function has become more appreciated. Furthermore, several groups have shown that exhausted or dysfunctional T cells from cancer patients or murine models possess mRNA for inflammatory mediators, but fail to produce effector molecules, hinting that post‐transcriptional events also play a role in hampering tumour‐infiltrating lymphocyte effector function. Here, the post‐transcriptional regulatory events governing T‐cell cytokine production are reviewed, with a specific focus on the importance of post‐transcriptional regulation in anti‐tumour responses. Furthermore, potential approaches to circumvent tumour‐mediated dampening of T‐cell effector function through the (dis)engagement of post‐transcriptional events are explored, such as CRISPR/Cas9‐mediated genome editing or chimeric antigen receptors.
Keywords: AU‐rich elements, cancer, effector function, post‐transcriptional regulation, T cells
Post‐transcriptional control of T‐cell responses ensures the correct timing and magnitude of T‐cell cytokine production. Here, the post‐transcriptional events governing T‐cell effector function are discussed, with a specific focus on the implications thereof for cancer immunotherapy.

Abbreviations
- ARE
adenylate/uridylate‐rich element
- AREbp
AU‐rich element binding protein
- CDE
constitutive decay element
- CTLA‐4
cytotoxic T‐lymphocyte associated protein 4
- GAIT
gamma‐interferon‐activated inhibitor of translation
- hnRNP
heterogeneous nuclear ribonucleoprotein
- IFN
interferon
- IL
interleukin
- JRE
c‐Jun NH2‐terminal protein kinase‐responsive element
- PD‐1
programmed cell death 1
- PD‐L1
programmed death‐ligand 1
- SHP‐2
Src homology region 2 domain‐containing phosphatase‐2
- TCR
T‐cell receptor
- TLR
Toll‐like receptor
- TNF
tumour necrosis factor
- UTR
untranslated region
INTRODUCTION
T cells form an integral part of the immune system and are critical to combat (viral) infections and cancer cells. While patrolling the body, T cells eradicate infected and malignant cells through the production of effector molecules. These include granzymes, perforin and cytokines, such as the three key cytokines interferon (IFN)‐γ, tumour necrosis factor (TNF)‐α and interleukin (IL)‐2 [1, 2].
Yet, the production thereof must be tightly regulated, as excessive cytokine production can play a role in diverse autoimmune diseases, such as inflammatory bowel disease [3, 4, 5, 6], multiple sclerosis [7] and rheumatoid arthritis [8]. Furthermore, disproportionate cytokine production can also lead to immunopathology [9, 10, 11, 12, 13]. However, lack of cytokine production by T cells or defects in downstream signalling pathways also pose risks for the host. For instance, patients with defects in (one of) the IFNG receptors are highly susceptible to mycobacterial infections [14, 15]. Furthermore, mice lacking the Ifng gene, or Stat1, a signalling molecule downstream of the IFN‐γ receptors, spontaneously develop tumours [16, 17]. Lastly, copy‐number variations of IFNG pathway genes in cancer patients correlate with poor outcome in cancer immunotherapy [18].
Therefore, tight control of cytokine production by T cells is required to maintain the precarious balance between protective immunity and immunopathology. Several (molecular) mechanisms are in place not only to limit cytokine production to the right time and magnitude, but also to ensure the proper resolution thereof. Firstly, once the invading pathogen is eradicated, pathogen‐derived antigen will no longer be loaded into major histocompatibility complex, or MHC, molecules. As a result, T‐cell receptor (TCR) signalling ceases, and T cells become quiescent. Secondly, in response to T‐cell activation, T cells upregulate inhibitory receptors, such as programmed cell death 1 (PD‐1) and cytotoxic T‐lymphocyte associated protein 4 (CTLA‐4) [19]. While CTLA‐4 directly interferes with costimulatory signalling by competing with CD28 for the binding of CD80/CD86 on antigen‐presenting cells [20, 21], binding of PD‐1 to its ligands programmed death‐ligand 1 (PD‐L1) and PD‐L2 rather supplies inhibitory signals to T cells [22]. Recent work has shown that PD‐1 triggering recruits the phosphatase Src homology region 2 domain‐containing phosphatase‐2 (SHP‐2) to inhibit CD28 signalling [23]. This negative feedback of blocking cytokine production also has a downside. In fact, inhibitory signalling is exploited by some pathogens. For example, viruses such as hepatitis C virus and human immunodeficiency virus 1 induce the expression of inhibitory receptor ligands such as PD‐L1 on infected cells to evade the immune system [24].
Also tumours exploit inhibitory signalling to dampen CD8+ T‐cell responses [25, 26]. Indeed, in the immune suppressive tumour microenvironment, T cells gradually lose the capacity to produce effector molecules [27, 28], resulting in tumour immune escape. Tumours employ several well‐known strategies to hamper T‐cell responses. By modulating antigen presentation and increasing the uptake of nutrients by tumour cells, antigen‐specific T cells can no longer be triggered through their TCR, and the lack of nutrients results in cellular starvation [29, 30, 31]. As discussed, tumour cells also exploit the mechanisms that are in place to prevent excessive T‐cell function by upregulating inhibitory receptor ligands, for example PD‐1 and CTLA‐4 [25, 26], resulting in the inhibition of T‐cell effector function.
The production of inflammatory cytokines, such as IFN‐γ, is regulated on multiple levels (Figure 1). Gene locus accessibility and the presence and activation status of transcription factors determine the amount of mRNA that is produced [32, 33, 34, 35, 36]. Post‐transcriptional mechanisms, influencing mRNA codon usage through splicing, stability, decay, localization and translation rate, subsequently determine the amount of protein that is produced [37, 38, 39, 40]. Recently, the role of post‐transcriptional regulation in fine‐tuning T‐cell effector function has become more appreciated [6, 50]. Furthermore, several groups have described that exhausted or dysfunctional T cells from cancer patients or murine cancer models possess mRNA of inflammatory mediators such as IFN‐γ, but fail to produce effector molecules [51, 52, 53]. This implies that post‐transcriptional events also play a role in hampering the effector function of tumour‐infiltrating lymphocytes (TILs). Indeed, upon uncoupling part of the post‐transcriptional regulatory events that govern IFN‐γ production in a murine melanoma model, TILs were able to sustain IFN‐γ production, resulting in prolonged survival [52]. Understanding how tumour cells exploit post‐transcriptional regulatory mechanisms is paramount to enhance T‐cell‐based therapies or boost naturally occurring anti‐tumour T‐cell responses.
FIGURE 1.
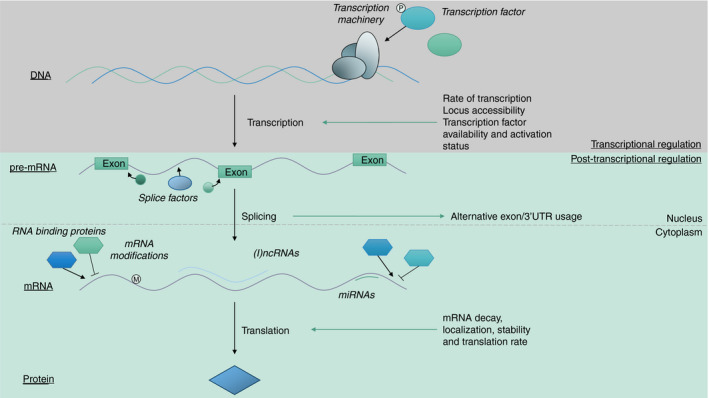
Transcriptional and post‐transcriptional regulatory events together shape protein production. Firstly, transcription factor availability and activation status, locus accessibility and cellular transcription rates determine the amount of pre‐mRNA that is produced. Secondly, post‐transcriptional splicing affects the inclusion of alternative exons and determines the usage of alternative 3’UTRs. Thirdly, post‐transcriptional regulatory events, mediated by mRNA modifications, and binding by (long) non‐coding RNAs, miRNAs and RBPs influence mRNA decay, localization, stability and translational rate, and thus determine the amount of protein that is produced from a single mRNA molecule
Here, the current knowledge regarding the role of post‐transcriptional regulation of T‐cell cytokine production is reviewed. Furthermore, the importance of post‐transcriptional regulation in anti‐tumour responses is discussed. Lastly, potential approaches to circumvent tumour‐mediated dampening of T‐cell effector function through the engagement of post‐transcriptional events are explored.
T‐CELL EFFECTOR FUNCTION IS GOVERNED BY POST‐TRANSCRIPTIONAL REGULATION OF T‐CELL EFFECTOR FUNCTION
As discussed, several intracellular molecular layers of regulation fine‐tune the production of T‐cell effector molecules [54]. These regulatory mechanisms can be subdivided into two categories: transcriptional and post‐transcriptional regulation (Figure 1). Transcriptional regulation, enforced by epigenetic markers present on DNA and histones, and transcription factor availability, localization and activation status determine the amount of mRNA that is produced from a single gene [32, 33, 36]. The role of transcriptional regulation in T‐cell differentiation and effector function has been extensively reviewed elsewhere [55, 56, 57, 58].
Once gene transcription has occurred, another regulatory pathway dictates the amount of protein that is formed from one mRNA molecule. Indeed, post‐transcriptional events dictate mRNA maturation through RNA splicing, and mRNA location, stability and translational rate [38, 39, 59, 60]. Firstly, the RNA molecule produced upon gene transcription requires splicing to excise non‐coding introns. Through alternative splicing, the specific inclusion or exclusion of coding exons results in the production of different proteins from the same gene [61, 62]. Furthermore, through the use of alternative polyadenylation sites, transcripts can contain alternative 3’‐untranslated regions (UTRs), while mRNA modifications such as 5’ capping or methylation can influence mRNA localization, decay, stability and translation [63, 64, 65]. Lastly, cis‐elements present in the 5’ and 3’UTRs of mRNA transcripts, functioning as binding hubs for miRNAs and RNA binding proteins (RBPs), can also alter mRNA localization, stability and translation. As such, the amount of effector molecule mRNA and the amount of effector molecules produced by T cells do not always correlate at a 1:1 ratio [66, 67, 68, 69] and depend on cellular activation status. Here, the current knowledge regarding the contribution of (alternative) splicing, RNA cis‐elements and RBP binding on T‐cell cytokine production is discussed.
(ALTERNATIVE) SPLICING AS A MEANS TO MODULATE T‐CELL CYTOKINE PRODUCTION
Upon locus accessibility and sufficient intracellular signalling, the concerted actions of transcription factors and the transcriptional machinery result in gene transcription. This pre‐mRNA, however, contains both introns and exons and first requires splicing before translation can occur. Splicing is mediated by the spliceosome, which is comprised out of several subunits: 5 small nuclear ribonucleoproteins (or snRNPs) in complex with 6 small nuclear RNAs named U1‐U6 [70]. Several subsequent steps are required for the assembly of the active spliceosome (Figure 2a). In their inactive state, subunits U4 and U6 are bound together. For splicing to occur, SR proteins first need to recognize an intronic splicing site (ISS). Next, U1 and U2 bind to the RNA at the 5’ and 3’ splice sites (complex A). Then, the interaction between U4 and U6 is disrupted, U5 is recruited to the splice site (complex B, inactive spliceosome), and U4 and U1 are released. This allows for the interaction between U2 and U6, forming the catalytically active spliceosome (complex C) [71]. Subsequently, introns are excised by the catalytically active spliceosome. After splicing has been completed, as well as 5’ capping and 3’ polyadenylation, the formation of a mature mRNA molecule is complete.
FIGURE 2.
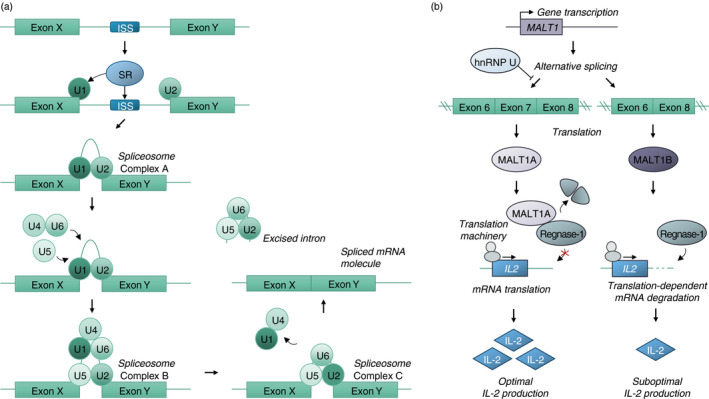
Splicing of pre‐mRNA and the impact of alternative splicing on IL‐2 production. (a) Intronic splicing relies on the recognition of the internal splicing site (ISS) by SR proteins. Subsequently, splicing factors U1 and U2 bind the 5’ and 3’ splicing sites, forming spliceosome complex A. U5 and U4‐U6 are recruited, together forming spliceosome complex B. Through splicing factor rearrangement, U1 and U4 are released, and the active spliceosome is formed, which is catalytically active and able to excise the intron. (b) Alternative splicing of MALT1 under the influence of several RBPs, including hnRNP U, results in the expression of MALT1 isoforms MALT1A and MALT1B. MALT1A is capable of cleaving Regnase‐1, an RNAse involved in the translation‐dependent breakdown of IL2 mRNA. As a result, cells expressing MALT1A are superior IL‐2‐producing cells
However, splicing events do not always result in the same mRNA molecule from one particular pre‐mRNA gene product. Indeed, alternative splicing occurs, in which exons are selectively included or excised, resulting in transcript variety [72]. As a result, one pre‐mRNA can encode for different protein isoforms, or contain alternative UTRs, resulting in altered engagement of the post‐transcriptional machinery governing mRNA decay, localization, stability and translational rate. These splicing events can be cell‐type‐specific, resulting in differential protein expression by for instance B and T cells [73], or rather result in heterogeneous expression patterns [74].
For instance, one well‐studied example of alternative splicing is the expression of different CD45 isoforms by B and T cells. Through the divergent expression of the RBP heterogeneous nuclear ribonucleoprotein (hnRNP) LL, CD45 pre‐mRNA is differentially spliced in B and T cells, resulting in the expression of different isoforms of the CD45 molecule [61, 62]. Also, the role of splicing in immune responses has become more appreciated in recent years [37]. Indeed, in both naïve T cells and during T‐cell activation, alternative splicing actively occurs [75, 76, 77, 78], resulting in ~10‐15% of transcripts containing alternative exons in activated T cells compared with resting T cells [75]. Similarly, also during T‐cell proliferation, a significant amount of transcripts display alternative exon usage [79]. However, splicing and the splicing machinery have been mainly investigated in cell lines such as Jurkat cells [75]. As much, little is known about the role of (alternative) splicing in T‐cell cytokine production. Nontheless, recently, several reports have been published investigating this phenomenon.
One of the crucial proteins downstream of TCR and CD28 costimulatory signalling required for T‐cell activation is MALT1, or paracaspase [80]. MALT1 exists in 2 isoforms, MALT1A and MALT1B, depending on the inclusion of a 11‐amino acid exon (exon 7, included in isoform MALT1A). Splicing factors hnRNP R, SRSF3 and SRSF9 favour MALT1A splicing, while hnRNP U inhibits MALT1A splicing [81] (Figure 2b). Through reconstitution studies of MALT1 knockout Jurkat T cells, it was shown that MALT1A+ Jurkat T cells were superior IL‐2 producers compared with MALT1B+ Jurkat T cells [81]. However, while MALT1 is required for the production of both IFN‐γ and IL‐17 by murine T cells, both MALT1 isoforms induced TH1 and TH17 differentiation to a similar extent [81]. Furthermore, upon T‐cell activation, the proportion of MALT1 proteins containing exon 7 (i.e. MALT1A) increases, and the expression of the MALT1A isoform was shown to be required for optimal TCR signalling and IL‐2 production [81]. Subsequent work has shown that the increased IL‐2 production by CD4+ T cells was mediated through MALT1‐induced cleavage of the RNAse MCPIP1/Regnase‐1/ZC3H12A [82]. Interestingly, TCR stimulation upregulates the expression of splicing factors favouring both MALT1A (hnRNP R, SRSF3 and SRSF9) and MALT1B (hnRNP U) splicing [81]. The exact mechanism behind how TCR stimulation results in increased MALT1A over MALT1B expression despite upregulating factors that favour splicing towards both MALT1 isoforms remains to be determined.
In summary, while recent years have seen increasing interest in mRNA splicing in T cells, little is known regarding the role of splicing‐mediated regulation of T‐cell effector function. Nonetheless, the alternative splicing mechanism through which MALT1 mediates IL‐2 production by T cells shows that post‐transcriptional regulation through alternative splicing contributes, at least indirectly, to the regulation of cytokine production. Further research is needed to investigate whether splicing events are directly regulating T‐cell effector function as well. For this, data on a single‐cell level would be of extreme importance. To this end, potentially, single‐cell RNA sequencing data, either de novo‐generated or obtained from previously deposited data sets, could be utilized in conjunction with recently described methodology [74], which again confirmed splicing heterogeneity in diverse CD4+ and CD8+ T‐cell subsets.
REGULATING CYTOKINE PRODUCTION THROUGH MODULATION OF mRNA DECAY, STABILITY AND TRANSLATIONAL RATE
While (alternative) pre‐mRNA splicing is important for the generation of mature mRNA molecules and the variation in composition therein, subsequent post‐transcriptional regulatory events are important for determining protein output. As such, it is not surprising that the timing and magnitude of antigen‐experienced T‐cell responses are regulated on a post‐transcriptional level and mediated through the complex interplay between multiple mediators [38, 41, 42, 46, 47, 54] (summarized in Figure 3). Indeed, all T‐cell effector molecules have their own distinct pattern of production due to the engagement of different post‐transcriptional mechanisms. For instance, the production of TNF‐α by antigen‐experienced T cells primarily relies on increased translation of pre‐formed Tnfa mRNA that is already present in resting memory T cells [47]. Interestingly, T‐cell activation does not lead to stabilization of Tnfa mRNA [47]. In contrast, antigen‐experienced T cells do not contain pre‐formed Il2 mRNA [47]. The onset of IL‐2 protein production by antigen‐experienced T cells fully relies on de novo Il2 mRNA transcription, while the magnitude and duration of IL‐2 production are dependent on transient mRNA stabilization [47]. Lastly, the rapid and initial production of IFN‐γ protein by antigen‐experienced T cells depends on translation of pre‐formed mRNA [47], similar to TNF‐α. However, IFN‐γ protein production at later time points depends on TCR signal strength, de novo transcription and mRNA stabilization, together defining the magnitude and duration of IFN‐γ production [47].
FIGURE 3.
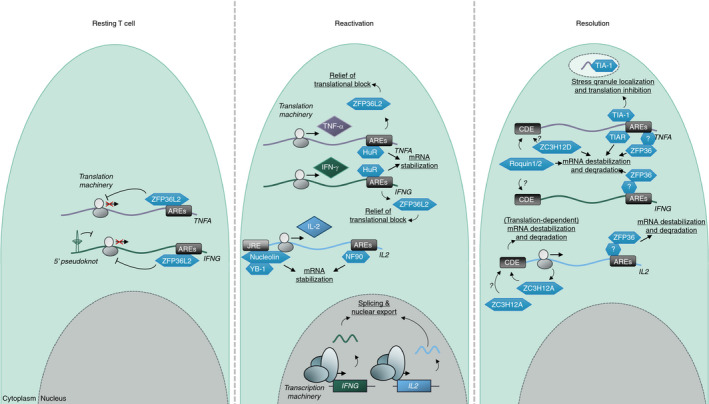
Post‐transcriptional regulatory processes governing T‐cell cytokine production. Left, antigen‐experienced T cells possess IFNG and TNFA mRNA that is kept translationally silent through the binding of their respective 3’UTR AREs by ZFP36L2. Middle, upon T‐cell activation, ZFP36L2 dissociates from IFNG and TNFA mRNA, enabling translation. HuR transiently binds the 3’UTR AREs in IFNG and TNFA mRNA, stabilizing the transcripts. De novo transcription of IFNG and IL2 mRNA results in increasing mRNA levels. IL2 mRNA is stabilized through the binding of Nucleolin, YB‐1 and NF‐90. Right, upon resolution of the stimulatory signal, cytokine mRNA is destabilized and degraded through the binding of multiple RBPs (IFNG: ZFP36, Roquin1/2; IL2: ZC3H12A, ZC3H12D, ZFP36; TNFA: Roquin1/2, TIA‐1, TIAR, ZC3H12D)
Other reports confirmed that antigen‐experienced T cells retain mRNA not only of IFN‐γ and TNF‐α, but also of other effector molecules, including granzyme B and RANTES/CCL5 [83, 84]. However, due to post‐transcriptional regulatory events, this mRNA is kept translationally silent [83]. This stored mRNA is rapidly translated into protein upon antigen re‐encounter [83], allowing for the rapid secondary T‐cell responses observed upon reinfection.
The engagement of post‐transcriptional regulatory processes is dependent on the type of stimulus T cells receive [41, 47]. T cells can be (re)activated through the triggering of myriad receptors. Besides classical antigen‐dependent TCR‐mediated T‐cell activation, T cells also produce effector molecules upon the sensing of (pro‐inflammatory) molecules such as type I IFNs, IL‐2, IL‐12, IL‐15 or IL‐18, or a combination thereof [85, 86, 87]. Recently, it has become more evident that T cells can also sense pathogens in a more innate fashion through the triggering of pattern recognition receptors such as Toll‐like receptors (TLRs) or stimulator of interferon genes, or STING [41, 45, 88, 89], and that these pathogen‐derived molecules can provide costimulatory signals [41, 90]. The triggering of multiple (co)stimulatory receptors results in the optimal induction of T‐cell effector function. One of the most studied costimulatory molecules is CD28. Besides the well‐known effects on the T‐cell transcriptional landscape [91], CD28 costimulation also stabilizes cytokine mRNA [92]. Similarly, also other costimulatory signals, such as LFA‐1 and TLR2 costimulation, engage post‐transcriptional mechanisms that result in mRNA stabilization and increased cytokine production by T cells [41, 93].
Altogether, cytokine production by T cells is governed by post‐transcriptional regulatory mechanisms. Through understanding which post‐transcriptional events result in optimal T‐cell responses, harnessing and amplifying these pathways could be useful in therapeutic settings.
SEQUENCE ELEMENTS AND RNA BINDING PROTEINS MEDIATING POST‐TRANSCRIPTIONAL CONTROL OF CYTOKINE PRODUCTION
Post‐transcriptional events at the mRNA level are mediated by mRNA modifications and the binding of trans‐factors, for example, of miRNAs and (long) non‐coding RNAs, and RBPs to cis‐elements located in mRNA transcripts [44, 54, 94, 95, 96]. Often, these cis‐elements are located in the 5’ or 3’UTR [54, 97]. Regulatory cis‐elements can exert their function by either forming a physical structure, such as 5’ hairpins and pseudoknots [94, 98, 99, 100], or by forming specific or repetitive sequences, such as microRNA seed regions [59, 101], adenylate/uridylate‐rich elements (AREs) [102, 103, 104], constitutive decay elements (CDEs) [102, 105], gamma‐interferon‐activated inhibitor of translation (GAIT) elements [106] and c‐Jun NH2‐terminal protein kinase‐responsive elements (JREs) [107, 108]. Many T‐cell effector molecules contain at least one cis‐regulatory element [54]. An overview of the regulatory elements present in the three key cytokines produced by T cells in the context of anti‐tumour effector function (IFN‐γ, TNF‐α and IL‐2) and the regulators thereof can be found in Table 1. While miRNA transcript binding and mRNA modifications can significantly alter lymphocyte (cytokine) protein production [54, 109, 110], here, only post‐transcriptional regulation through (repetitive) sequence elements and the role RBPs play therein are discussed (summarized in Figure 3), as these processes have recently been explored as potential therapeutic targets through genome editing with, for example, CRISPR/Cas9 [42, 111].
TABLE 1.
Cis‐elements and trans‐factors involved in fine‐tuning anti‐tumour cytokine production by T cells through post‐transcriptional regulation
| mRNA | Pre‐formed mRNA in memory T cells | Expressed by dysfunctional TILs | 5’UTR cis‐element(s) | 3’UTR cis‐element(s) | trans‐factor(s) | Mode(s) of action | References |
|---|---|---|---|---|---|---|---|
| IFN‐γ | + | + | Pseudoknot | ARE, MRE | HuR, Roquin, TTP/ZFP36, ZFP36L2, miR29, miR181a | mRNA stability, translational block | [47, 103, 124, 125, 172, 173, 174] |
| IL−2 | − | n.d. | JRE | ARE, CDE, MRE | NF−90, Nucleolin, YB−1, ZC3H12A, ZC3H12D, TTP/ZFP36, miR181c | mRNA stability, translational block | [47, 82, 107, 108, 118, 123, 132, 133] |
| TNF‐α | ++ | n.d. | n.d. | ARE, CDE, MRE | HuR, TIA−1, TIAR, ZC3H12A, Roquin, TTP/ZFP36, ZFP36L2, miR16, miR181, miR221 | mRNA stability, translational block | (6, 178) |
Abbreviations: −, not present; +, present; ++, present in large amounts; n.d., not determined.
Amongst the most studied mRNA cis‐elements are AREs. AREs are encoded in the 3’UTR of mRNA transcripts [112, 113], and it has been estimated that 8 to 16 percent of mammalian genes contain AREs [112, 113]. In fact, most of the genes encoding cytokines contain AREs in their 3’UTR [54, 60, 104]. This includes not only the three key T‐cell cytokines for effective anti‐tumour responses (IFN‐γ, TNF‐α and IL‐2), but also type 2 cytokines, such as IL‐4, IL‐10 and IL‐13, have been described to contain AREs [104].
Several classes of AREs have been identified [60, 114, 115]. Class I AREs are defined as multimeric AUUUA pentamers [60] and are present in, for example, the 3’UTR of the murine and the human IFNG gene [49]. Class II AREs possess 2 overlapping UUAUUUA(U/A)(U/A) nonamers and are, for instance, present in the 3’UTR of the IL2 and TNFA genes [115, 116]. Lastly, class III AREs are less well characterized, but have been previously defined as U‐rich regions [115, 117].
These AU‐rich sequence elements facilitate the binding of a specialized subset of RBPs, also known as AU‐rich element binding proteins (AREbps), resulting in modulation of mRNA half‐life, localization and translational rate [49, 93, 115, 118]. As the different ARE classes each have unique sequence identities, the AREbps or AREbp complexes interacting with the different classes of AREs are also profoundly different [115]. Therefore, unsurprisingly, it has been speculated that the number and class of AREs present in the 3’UTR, the secondary structure of the RNA molecule and the surrounding sequences determine which AREbps can access the binding site of a specific transcript [60].
As previously discussed, it has been shown that memory T cells express pre‐formed Ifng and Tnfa mRNA, but IFN‐γ and TNF‐α proteins are only produced from this pre‐formed mRNA upon T‐cell reactivation [47, 84] (Figure 3). This block in Ifng and Tnfa mRNA translation is ARE‐dependent [6, 49]. Indeed, memory T cells from mice with a germline deletion of the 3’UTR AREs present in the Ifng gene spontaneously produce IFN‐γ in the absence of stimulation [49], exhibit an increased half‐life of Ifng mRNA in after in vitro culture [49] and produce more IFN‐γ protein upon stimulation compared with wild‐type T cells [68]. Also, more human T cells in which the 3’UTR AREs have been removed from the IFNG gene via CRISPR/Cas9‐genome editing produce IFN‐γ compared with wild‐type T cells due to increased IFNG mRNA stability [42]. Similarly, the AREs in the Tnfa gene are also crucial to keep TNF‐α production contained to the right time and location. ARE deletion in the Tnfa gene results in immunopathology in mice through continuous TNF‐α production [6].
For Ifng, this ARE‐mediated block in protein production was shown to be facilitated through the binding of the AREbp ZFP36L2 [49]. Upon T‐cell activation, ZFP36L2 rapidly dissociates from Ifng mRNA, allowing for the recruitment of the Ifng mRNA into ribosomes to initiate mRNA translation [49]. Of note, mice with a T‐cell conditional ZFP36L2 deletion also exhibited enhanced TNF‐α production through increased Tnfa mRNA translation [49]. Whether ZFP36L2 binds Tnfa mRNA directly through the AREs present in the 3’UTR of Tnfa, binds to other elements present in the 3’UTR of Tnfa, or is indirectly involved through the regulation of other regulatory proteins remains to be determined. However, considering the RNA‐binding moieties present in ZFP36L2, it seems highly likely that also for TNF‐α, the ZFP36L2‐mediated block in translation is mediated through 3’UTR AREs. Of note, it is currently unclear what role, if any, ZFP36L2 also plays later on in cytotoxic T‐cell activation.
T cells express many other RBPs that can also influence T‐cell cytokine production [38, 43, 48, 49, 69]. Another AREbp that has been shown to be important in regulating T‐cell effector function is human antigen R (HuR) or ELAVL1 [93, 119]. Upon T‐cell costimulation with LFA‐1, HuR translocates from the nucleus to the cytoplasm and stabilizes IFNG and TNFA mRNA by binding to the AREs present in their respective 3’UTRs [93]. In contrast, conditional HuR knockout increases Il2 mRNA stability without altering Il2 mRNA translational rate [120]. However, whether this is a direct effect or rather through deregulation of other factors that influence mRNA stability remains to be determined. For instance, the RBP NF‐90 is essential for the ARE‐mediated stabilization of IL2 mRNA observed upon T‐cell activation [118, 121, 122]. In contrast, ZFP36, also known as TTP, has been shown to destabilize IFN‐γ, TNF‐α and IL‐2 mRNA in T cells, inhibiting effective T‐cell responses [123, 124, 125]. This was especially evident in an infection model, where ZFP36 knockout mice were able to more rapidly clear an LCMV infection [125]. While previous reports indicate ARE‐mediated binding for ZFP36 is important for the regulation of IFN‐γ and IL‐2 [123, 124], more recent CLIP data indicate indirect binding of ZFP36 to the 3’UTRs and thus rather argue for the importance of ZFP36 through indirect binding via the formation of RBP complexes [125]. Of note, mice with a germline TTP deletion develop a complex autoimmune disorder that is related to the continuous production of TNF‐α [126], highlighting again the importance of proper control of T‐cell cytokine production. Similarly, both TIA‐1 and TIAR bind the AREs in the 3’UTR of the TNFA gene, resulting in mRNA destabilization [116, 127, 128]. mRNA binding by TIA‐1 regulates subcellular localization [129]. Specifically, TIA‐1 binding to TNFA results in mRNA inclusion in stress granules [130].
However, while the 3’ UTRs of IFNG, TNFA and IL2 all contain AREs [102, 103, 104], other cis‐elements are also present (Table 1). For example, IL2 and TNFA contain a CDE, and TNFA, IFNG and IL2 each contain transcript‐specific miRNA seeding sites [54]. For instance, the RNAse MCPIP1/Regnase‐1/ZC3H12A was shown to influence IL‐2 expression in an ARE‐independent fashion [131]. ZC3H12A binds CDE stem–loop structures in the 3’UTR of IL‐2 in a translation‐dependent fashion, inducing mRNA degradation [82, 132, 133]. As discussed, this process is inhibited by MALT1‐mediated cleavage of ZC3H12A [81]. ZC3H12A has also been suggested to play a role in the regulation of IFN‐γ production [134, 135]. However, this was rather due to dysregulation of the Roquin family of proteins [134], which themselves negatively impact cytokine production by T cells by destabilizing Ifng and Tnfa mRNA [105, 136, 137] and, similar to ZC3H12A [81], are also targets of MALT1 [138]. Similar to ZC3H12A [133], post‐transcriptional regulation via Roquin family proteins occurs through binding to 3’UTR CDE stem–loop structures [105, 139], which was confirmed for TNF‐α [105]. The exact mechanism behind how Roquin proteins influence IFN‐γ mRNA stability remains to be determined.
ZC3H12D, a family member of ZC3H12A, was also shown to be crucial for the regulation of T‐cell cytokine production. Upon ZC3H12D knockout, murine T cells produced higher amounts of IL‐2 due to increased Il2 mRNA stability [140]. ZC3H12D overexpression in Jurkat cells also reduced expression of luciferase reporter constructs fused to the 3’UTRs of Il2, Il6, Il17a and Tnfa, but not Ifng or Il10 [140].
Of note, also the 5’UTR is important for the post‐transcriptional regulation of T‐cell cytokines (Figure 3). For instance, it was shown that for IL2 mRNA stabilization, binding of Nucleolin and YB‐1 to the IL‐2 5’UTR JRE is crucial [108]. Furthermore, the 5’UTR of IFN‐γ contains repetitive sequences that fold into a 5’ pseudoknot [98]. Unfolding of this pseudoknot is required for translation initiation [98].
As feedback loops, transcriptional and post‐transcriptional events and translational rates play a role in determining protein output, it is not surprising that the regulation of IFN‐γ, IL‐2 and TNF‐α protein production is profoundly different [47]. Cytokine‐specific post‐transcriptional regulatory events dictate the timing and magnitude of protein production to prevent the unwanted production of cytokines by memory T cells [49, 52], while also facilitating the storage of pre‐formed mRNA by memory T cells to allow for immediate responses upon antigen sensing. Post‐transcriptional regulatory events thus directly contribute to effective recall responses. Recent work has identified several RBPs that play a crucial role here. However, how these identified RBPs interact is currently unknown. Furthermore, many other RBPs can play a role, either through direct binding or through the formation of RBP complexes. Therefore, unravelling the cytokine mRNA interactome, that is all RBPs and binding partners in RBP complexes able to bind cytokine mRNA species of interest (e.g. IFN‐γ, IL‐2 and TNF‐α), would provide novel insights into the networks of molecular pathways regulating cytokine production.
THE IMPORTANCE OF RBPS IN T‐CELL FORMATION, PROLIFERATION AND DIFFERENTIATION
RBPs are not only instrumental for maintaining immune balance and fine‐tuning immune responses. Also for the generation of a fully functional immune system, the differentiation of naïve T cells after first antigen encounter, and immune homeostasis, the timely expression and concerted action of RBPs are of paramount importance. For instance, it has been shown that dual ZFP36L1 and ZFP36L2 knockout in CD2‐Cre mice impairs thymic T‐cell maturation at the double‐negative stage and results in lethal lymphoma [141], while Roquin knockout mice exhibit skewed CD4+ TH17 differentiation and gastritis [138]. Other RBP knockouts were shown to be (partially) lethal. For instance, NF‐90‐deficient mice exhibit perinatal lethality [142]. Similarly, TIA‐1 knockout is partially embryonically lethal [127], while HuR knockout is embryonically lethal [143]. Of note, HuR knockout in late‐stage thymocytes does not impair thymic regression or seeding of peripheral tissues by T cells [144]. Furthermore, homozygous ZC3H12A knockout mice die within 12 weeks after birth due to a complex immune disorder stemming mostly from hyperimmunoglobulinaemia and granuloma formation due to defects in lymphocyte formation and differentiation [134]. Interestingly, this phenotype was recapitulated in mice with a T‐cell conditional ZC3H12A knockout [82]. In contrast to ZFP36L1/L2 knockout mice, thymic development and regulatory T‐cell formation were not impaired in ZC3H12A conditional knockout mice. Rather, upon TCR‐mediated CD4+ T‐cell activation, immunopathology was observed due to non‐restrained effector molecule production of IL‐2 and IL‐6 [82]. In contrast, ZC3H12D knockout mice do not show developmental defects [140].
In short, post‐transcriptional regulation is not only instrumental for the concerted production of T‐cell effector molecules. Rather, RBP‐mediated post‐transcriptional events are of great importance not only for embryonic maturation and perinatal development, but also for T‐cell generation, differentiation and peripheral tissue seeding.
FINE‐TUNING TILS: POST‐TRANSCRIPTIONAL CONTROL OF ANTI‐TUMOUR RESPONSES
In the tumour immune suppressive microenvironment, T cells gradually lose the capacity to perform their effector function and to produce cytokines [27, 28]. It has been shown that TILs derived from lung cancer resections can still benefit from costimulatory signals from, for example, CD27 and/or CD28 triggering [145]. However, in tumours, TILs are rather exposed to inhibitory signals instead. One of the most studied mechanisms tumours employ is the exploitation of inhibitory receptors expressed by T cells, such as PD‐1. Indeed, targeting the PD‐1/PD‐L1 axis via antibody‐mediated cancer therapy resulted in a remarkable 50% survival rate in melanoma patients [146] and is currently being investigated for other cancer types [147]. Furthermore, also genetic ablation of PD‐1 in T cells from cancer patients through CRISPR/Cas9‐mediated genome editing has resulted in significant increases in T‐cell cytokine production in vitro and in xenograft models [148, 149]. Of note, recently, several investigational trials have published (preliminary) results of TIL therapy with TILs that have been genetically engineered to lack PD‐1 expression [150, 151, 152]. Currently, several other clinical trials investigating the benefit of PD‐1 knockout in other cancer types are still planned or in progress.
Another cellular therapy that exploits the modulation of T‐cell effector function is CAR T‐cell therapy. Chimeric antigen receptors, or CARs, are comprised of the variable region of an antibody with a very high affinity to a defined tumour antigen such as CD19, fused to one or more signalling modules, often incorporating at least CD3 and CD28 domains [153, 154]. This type of therapy is also being combined with PD‐1 ablation [155], with the first (partial) results of several trials showing therapeutic promise [156, 157], Furthermore, while one of the main concerns for CAR T therapy are the diverse pathologies resulting from the abundant release of cytokines by CAR T cells, also known as cytokine release syndrome (CRS) [158], this was not observed in one of the ongoing clinical trials’ interim report [156].
The inhibitory function of PD‐1 has been attributed to the PD‐1‐mediated recruitment and phosphorylation of SHP‐2 [23, 159, 160]. However, recently, it was shown that PD‐1 triggering also affects the post‐transcriptional regulation of T‐cell effector function. While blocking the engagement of PD‐1 through anti‐PD‐L1 therapy, and triggering of CD28, resulted in similar amounts of IFN‐γ‐producing T cells in an in vitro B16‐OVA melanoma model [52], it was shown that anti‐PD‐1 and CD28 stimulation differently modulate T‐cell cytokine protein production. As previously discussed, CD28 costimulation enhanced IFN‐γ production through the stabilization of Ifng mRNA [52, 92]. In contrast, PD‐1 blockade resulted in similar Ifng mRNA levels and mRNA stability compared with non‐treated cells [52]. Therefore, PD‐1 blockade enhances the translation efficiency of Ifng mRNA. Of note, the expression of PD‐L1 by tumour cells is also mediated by post‐transcriptional regulatory mechanisms [161, 162]. Indeed, it was shown that ARE‐mediated binding by ZFP36 destabilizes PD‐L1 mRNA [161]. This again highlights the importance of investigating post‐transcriptional networks in the tumour context.
Which mediators downstream of PD‐1 are responsible for the observed block in Ifng mRNA translation is yet unknown; however, it was recently shown that dysfunctional T cells from melanoma patients express high levels of ZFP36L1 and ZFP36L2 [53]. As ZFP36L2 keeps Ifng mRNA translationally quiescent in memory T cells [49], and exhausted or dysfunctional T cells from cancer patients or murine models possess mRNA for inflammatory mediators such as IFN‐γ, but fail to produce effector molecules [51, 52, 53], it is tempting to speculate that ZFP36L2 might play a role in TILs [125]. However, whether ZFP36L2, or one of the other ZFP36 family members that have been shown to modulate IFN‐γ production by T cells [125], plays a role in dampening TIL function remains to be determined. Of note, as discussed, a myriad of other RBPs is also involved in governing the post‐transcriptional regulation of cytokine mRNA. Therefore, identifying the cytokine mRNA interactome in TILs, especially upon PD‐1 engagement, is of utmost importance.
Other external signals that engage post‐transcriptional mechanisms to influence the anti‐tumour effector function of T cells have also been described [41]. For instance, TLR2 costimulation enhances the production of effector cytokines in an in vitro B16‐OVA melanoma model [41], and has also enhanced tumour‐specific T‐cell responses in weakly immunogenic tumour models [163]. Furthermore, TLR2 costimulation prolonged the survival of mice in a B16‐OVA melanoma model [164] and resulted in increased IFN‐γ production by human T cells in vitro [41]. Interestingly, TLR7 ligand costimulation only enhanced IFN‐γ production by murine T cells [41]. However, in contrast to TLR2 triggering, TLR7 triggering did not enhance Ifng mRNA stability, but rather induced only increased Ifng mRNA transcription [41]. Therefore, the exploitation of TLR2, rather than TLR7, triggering and/or downstream signalling to enhance T‐cell effector function in the context of anti‐tumour responses might be more beneficial [165]. Of note, TLR2 signalling has been employed in a CAR T‐cell therapeutic setting. CAR T cells equipped with a CD28 and a TLR2 signalling module produced more IFN‐γ and IL‐2 compared with T cells with only a CD28 signalling module [166]. Whether this was due to altered post‐transcriptional regulatory events is unclear, but treatment of cancer patients with the TLR2 signalling module containing CAR resulted in complete remission in 4 out of 5 patients [167]. Nonetheless, which downstream mediators are involved in the altered engagement of post‐transcriptional mechanisms when comparing TLR2 versus TLR7 triggering remains unknown and warrants further investigation.
Interestingly, one of the characteristics TILs share with memory T cells is the presence of translationally silent IFNG mRNA. As discussed, several groups have shown that dysfunctional T cells from cancer patients or murine models express IFNG mRNA, but fail to translate it into IFN‐γ protein [51, 52, 53]. These reports show that post‐transcriptional regulatory mechanisms silence TIL effector function and highlight the importance of assessing mRNA and protein simultaneously in TILs [168]. One of the cis‐elements that are involved in blocking cytokine production in TILs is AREs. As discussed, the AREs in the 3’UTR of the IFNG gene are essential to block the production of IFN‐γ in resting murine T cells and to limit IFN‐γ production in human T cells upon antigen withdrawal [42, 52]. The 3’UTR AREs in the Ifng gene are also critical for hampering T‐cell responses in a murine melanoma model [52]. Deletion of the 3’UTR AREs in the IFN‐γ gene (ARE‐del) increases the stability of IFN‐γ mRNA in murine and human T cells [49, 52]. This enhanced mRNA stability leads to prolonged IFN‐γ production [52], a feature that is critical for anti‐tumour responses. Indeed, ARE‐Del T‐cell therapy significantly delayed B16‐OVA tumour outgrowth in a murine model compared with mice treated with wild‐type T‐cell therapy [52]. Upon ex vivo examination, ARE‐Del TILs derived from melanoma tumours exhibited enhanced IFN‐γ production capacity compared with wild‐type TILs [52]. This increased IFN‐γ produced by ARE‐Del TILs acted directly on the tumour, as the survival benefit observed was directly linked to the sensing of IFN‐γ by the tumour cells [52]. Upon treatment of IFN‐γR−/− B16 tumour‐bearing mice with ARE‐Del T cells, the survival benefit was lost [52]. Similar to their murine counterparts, also human ARE‐Del T cells genetically engineered to express a MART‐1 TCR possessed superior IFN‐γ production in an in vitro coculture model with patient‐derived melanoma cells compared with wild‐type T cells [42].
As discussed, also the CDE stem–loop structures present in many cytokine mRNAs are important for the regulation of T‐cell effector function. Recently, it was shown that they also play a role in anti‐tumour T‐cell responses. Indeed, B16‐OVA melanoma‐bearing mice treated with murine ZC3H12A knockout T cells generated via CRISPR/Cas9‐technology showed an increased survival compared with control T‐cell therapy‐treated mice [135]. Furthermore, ZC3H12A deficient TILs produced more IL‐2, IFN‐γ, TNF‐α and granzyme B, while displaying a long‐lived effector phenotype, as characterized by the increased expression of the transcription factor TCF‐1 [135].
Of note, recently, it was also shown that TILs from liver tumours display a high degree of splicing heterogeneity [74]. The authors highlighted the differential splicing of WARS, which allowed differentiation between exhausted/dysfunctional T cells and other T cells [74]. Alternative WARS splicing also correlated negatively with survival [74], albeit marginally. While the implications of the observed heterogeneous splicing in TILs is still unclear, this phenomenon warrants further investigation, as it might shed light on deregulated splicing pathways in TILs.
Together, these data show that combining genome editing to enhance TIL effector function through modulation of post‐transcriptional events and TCR engineering for integration of antigen specificity could further advance cellular therapies. CRISPR/Cas9‐mediated genome editing can also be combined with CARs to maximize T‐cell effector function [111]. However, current CAR T‐cell therapies already pose significant threats to the host. As mentioned, one side effect of CAR administration is CRS, where the potent cytokine production of the CARs results in, for example, immune activation, neurotoxicity and anaphylactic shock [158]. These potentially deadly complications can be ameliorated by incorporating either a ‘death’ switch or ‘on/off’ switch into the CAR [158]. These measures have increased the on‐target CAR T‐cell effector function [158] and could provide a safer option for patients undergoing CAR T‐cell treatment. The increased effector function of CAR T cells could be further optimized by modulating post‐transcriptional responses, for instance by combining CAR integration with a deletion of RBPs that hamper T‐cell effector function, for example, TTP/ZFP36 or ZFP36L2. However, deleting RBPs would not only affect cytokine production, but rather have an impact on cellular functioning. For instance, the ZFP36 family has been shown to be instrumental for early T‐cell development [141], while the continuous TNF‐α production observed upon TTP knockout resulted in a complex autoimmune disorder [126]. However, knockout of ZC3H12A resulted in polyfunctional TILs with a long‐lived phenotype in murine models. Lastly, a more targeted approach, for example, IFNG 3’UTR ARE deletion, might also be of interest to enhance TIL effector function. However, much research into the long‐term effects on TIL survival, feasibility and patient safety is required to determine whether these approaches could be employed to increase the therapeutic window of TIL therapy in the future.
CONCLUSION AND OUTLOOK
As T‐cell therapy takes a more prominent spot in the fight against cancer, understanding the mechanisms governing the production of effector molecules is paramount. In recent years, much information has been gained regarding how post‐transcriptional regulatory processes influence T‐cell effector function, and it has become evident that also in TILs, post‐transcriptional regulation plays a major role in determining effector function. Recently, several new techniques have allowed for identifying not only RBPs binding to specific sequences [49, 105] and probing RBP complex formation [169, 170], but also all cellular protein interaction with RNA [171]. Together with older techniques, that is, PAR‐CLIP, these types of approaches have identified proteins previously not identified as RBPs/part of RBP complexes, thereby expanding the pool of potential therapeutic targets. Translating these methodologies to T cells and unravelling the RNA interactome in TILs would allow for the identification of deregulated (post‐transcriptional) regulatory pathways. Similarly, identifying the RBPs and RBP complexes specifically regulating cytokine production in TILs is paramount for the therapeutic fine‐tuning of TIL effector function. By making use of novel technologies, such as CRISPR/Cas9‐mediated genome editing to eliminate inhibitory factors, or CARs that integrate signals that exploit post‐transcriptional regulation, post‐transcriptional regulatory events can in future be exploited to further enhance TIL therapy.
DISCLOSURE
The author has no competing interests to declare.
CONSENT FOR PUBLICATION
Not applicable.
ACKNOWLEDGEMENTS
Nothing to declare.
REFERENCES
- 1.Pennock ND, White JT, Cross EW, Cheney EE, Tamburini BA, Kedl RM. T cell responses: naïve to memory and everything in between. Adv Physiology Educ. 2013;37:273–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Zhang B, Karrison T, Rowley DA, Schreiber H. IFN‐γ‐ and TNF‐dependent bystander eradication of antigen‐loss variants in established mouse cancers. J Clin Invest. 2008;118:1398–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bisping G, Lugering N, Lutke‐Brintrup S, Pauels HG, Schurmann G, Domschke W, et al. Patients with inflammatory bowel disease (IBD) reveal increased induction capacity of intracellular interferon‐gamma (IFN‐g) in peripheral CD8+ lymphocytes co‐cultured with intestinal epithelial cells. Clin Exp Immunol. 2001;123:15–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ito R, Shin‐Ya M, Kishida T, Urano A, Takada R, Sakagami J, et al. Interferon‐γ is causatively involved in experimental inflammatory bowel disease in mice. Clin Exp Immunol. 2006;146:330–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Rafa H, Amri M, Saoula H, Belkhelfa M, Medjeber O, Boutaleb A. Involvement of Interferon‐γ in bowel disease pathogenesis by nitric oxide pathway: A study in algerian patients. J Interf Cytokine Res. 2010;30:691–7. [DOI] [PubMed] [Google Scholar]
- 6.Kontoyiannis D, Pasparakis M, Pizarro TT, Cominelli F, Kollias G. Impaired On/Off Regulation of TNF Biosynthesis in Mice Lacking TNF AU‐Rich Elements: Implications for Joint and Gut‐Associated Immunopathologies. Immunity. 1999;10:387–98. [DOI] [PubMed] [Google Scholar]
- 7.Arellano G, Ottum PA, Reyes LI, Burgos PI. Stage‐specific role of interferon‐γ in experimental autoimmune encephalomyelitis and multiple sclerosis. Front Immunol. 2015;6:492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Mori L, Iselin S, De Libero G, Lesslauer W. Attenuation of collagen‐induced arthritis in 55‐kDa TNF receptor type 1 (TNFR1)‐IgG1‐treated and TNFR1‐deficient mice. J Immunol. 1996;157:3178–82. [PubMed] [Google Scholar]
- 9.Yoshinaga M, Takeuchi O. RNA binding proteins in the control of autoimmune diseases. Immunol Med. 2019;42:53–64. [DOI] [PubMed] [Google Scholar]
- 10.De Bruin AM, Voermans C, Nolte MA. Impact of interferon‐γ on hematopoiesis. Blood. 2014;124:2479–86. [DOI] [PubMed] [Google Scholar]
- 11.Goedhart M, Cornelissen AS, Kuijk C, Geerman S, Kleijer M, Van Buul JD, et al. Interferon‐gamma impairs maintenance and alters hematopoietic support of bone marrow mesenchymal stromal cells. Stem Cells Dev. 2018;27:579–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.de Bruin AM, Libregts SF, Valkhof M, Boon L, Touw IP, Nolte MA. IFN‐γ induces monopoiesis and inhibits neutrophil development during inflammation. Blood. 2012;119:1543–55. [DOI] [PubMed] [Google Scholar]
- 13.Libregts SF, Gutie L, De BAM, Wensveen FM, Papadopoulos P, Van IW, et al. Chronic IFN‐γ production in mice induces anemia by reducing erythrocyte life span and inhibiting erythropoiesis through an IRF‐1/PU.1 axis. Blood. 2011;118:2578–89. [DOI] [PubMed] [Google Scholar]
- 14.Newport M, Huxley C, Huston S, Hawrylowicz C, Oostra B, Williamson R, et al. A Mutation in the interferon‐g‐receptor gene and susceptibility to mycobacterial infection. N Engl J Med. 1996;355:1941–9. [DOI] [PubMed] [Google Scholar]
- 15.Dorman SE, Holland SM. Mutation in the signal‐transducing chain of the interferon‐γ receptor and susceptibility to mycobacterial infection. J Clin Invest. 1998;101:2364–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kaplan DH, Shankaran V, Dighe AS, Stockert E, Aguet M, Old LJ, et al. Demonstration of an interferon‐gamma dependent tumor surveillance system in immunocompetent mice. Proc Natl Acad Sci. 1998;95:7556–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Shankaran V, Ikeda H, Bruce AT, White JM, Swanson PE, Old LJ, et al. IFN‐g and lymphocytes prevent primary tumour development and shape tumour immunogenicity. Nature. 2001;410:1107–11. [DOI] [PubMed] [Google Scholar]
- 18.Gao J, Shi LZ, Zhao H, Chen J, Xiong L, He Q, et al. Loss of IFN‐γ pathway genes in tumor cells as a mechanism of resistance to anti‐CTLA‐4 therapy. Cell. 2016;167:397–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Turnis ME, Andrews LP, Vignali DAA. Inhibitory receptors as targets for cancer immunotherapy. Eur J Immunol. 2015;45:1892–905. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Krummel M, Allison J. CD28 and CTLA‐4 have opposing effects on the response of T cells to stimulation. J Exp Med. 1995;182:459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov. 2018;8:1069–86. [DOI] [PubMed] [Google Scholar]
- 22.Nishimura H, Nose M, Hiai H, Minato N, Honjo T. Development of lupus‐like autoimmune diseases by disruption of the PD‐1 gene encoding an ITIM motif‐carrying immunoreceptor. Immunity. 1999;11:141–51. [DOI] [PubMed] [Google Scholar]
- 23.Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD‐1 inhibits T‐cell receptor induced phosphorylation of the ZAP70/CD3ζ signalosome and downstream signaling to PKCθ. FEBS Lett. 2004;574:37–41. [DOI] [PubMed] [Google Scholar]
- 24.Ong EZ, Chan KR, Ooi EE. Viral manipulation of host inhibitory receptor signaling for immune evasion. PLoS Pathog. 2016;12:e1005776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Seidel JA, Otsuka A, Kabashima K. Anti‐PD‐1 and Anti‐CTLA‐4 therapies in cancer: mechanisms of action, efficacy, and limitations. Front Oncol. 2018;8:86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Tang F, Zheng P. Tumor cells versus host immune cells: whose PD‐L1 contributes to PD‐1/PD‐L1 blockade mediated cancer immunotherapy? Cell Biosci. 2018;8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wherry EJ, Kurachi M. Molecular and cellular insights into T cell exhaustion. Nat Rev Immunol. 2015;15:486–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ando M, Ito M, Srirat T, Kondo T, Yoshimura A. Memory T cell, exhaustion, and tumor immunity. Immunol Med. 2020;43:1–9. [DOI] [PubMed] [Google Scholar]
- 29.Garrido F, Aptsiauri N, Doorduijn EM, Lora AMG, Van HT. The urgent need to recover MHC class I in cancers for effective immunotherapy. Curr Opin Immunol. 2016;39:44–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Molon B, Calì B. T Cells and Cancer: How Metabolism Shapes Immunity. Front Immunol. 2016;7:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Morrison BJ, Steel JC, Morris JC. Reduction of MHC‐I expression limits T‐lymphocyte‐mediated killing of Cancer‐initiating cells. BMC Cancer. 2018;18:469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lugo‐Villarino G, Maldonado‐López R, Possemato R, Peñaranda C, Glimcher LH. T‐bet is required for optimal production of IFN‐γ and antigen‐specific T cell activation by dendritic cells. Proc Natl Acad Sci U S A. 2003;100:7749–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pearce EL, Mullen AC, Martins GA, Krawczyk CM, Hutchins AS, Zediak VP, et al. Control of effector CD8+ T cell function by the transcription factor eomesodermin. Science (80‐). 2003;302:1041–3. [DOI] [PubMed] [Google Scholar]
- 34.Jones B, Chen J. Inhibition of IFN‐gamma transcription by site‐specific methylation during T helper cell development. EMBO J. 2006;25:2443–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Intlekofer AM, Takemoto N, Wherry EJ, Longworth SA, Northrup JT, Palanivel VR, et al. Effector and memory CD8+ T cell fate coupled by T‐bet and eomesodermin. Nat Immunol. 2005;6:1236–44. [DOI] [PubMed] [Google Scholar]
- 36.Gray SM, Kaech SM, Staron MM. The interface between transcriptional and epigenetic control of effector and memory. Immunol Rev. 2014;261:157–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schaub A, Glasmacher E. Splicing in immune cells—mechanistic insights and emerging topics. Int Immunol. 2017;29:173–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turner M, Díaz‐Muñoz MD. RNA‐binding proteins control gene expression and cell fate in the immune system. Nat Immunol. 2018;19:120–9. [DOI] [PubMed] [Google Scholar]
- 39.Salerno F, Turner M, Wolkers MC. Dynamic post‐transcriptional events governing CD8+ T cell homeostasis and effector function. Trends Immunol. 2020;41:240–54. [DOI] [PubMed] [Google Scholar]
- 40.Wolf T, Jin W, Zoppi G, Vogel IA, Akhmedov M, Bleck CKE, et al. Dynamics in protein translation sustaining T cell preparedness. Nat Immunol. 2020;21:927–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salerno F, Freen‐van Heeren JJ, Guislain A, Nicolet BP, Wolkers MC. Costimulation through TLR2 Drives Polyfunctional CD8+ T Cell Responses. J Immunol. 2019;202:714–23. [DOI] [PubMed] [Google Scholar]
- 42.Freen‐van Heeren JJ, Popović B, Guislain A, Monika C. Human T cells employ conserved AU‐rich elements to fine‐tune IFN‐γ production. Eur J Immunol. 2020;50:949–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Hoefig KP, Reim A, Gallus C, Wong EH, Behrens G, Conrad C, et al. Defining the RBPome of T helper cells to study higher order post‐transcriptional gene regulation. bioRxiv. 2020. 10.1101/2020.08.20.259234 [DOI] [Google Scholar]
- 44.Kafasla P, Skliris A, Kontoyiannis DL. Post‐transcriptional coordination of immunological responses by RNA‐binding proteins. Nat Immunol. 2014;15:492–502. [DOI] [PubMed] [Google Scholar]
- 45.Salerno F, Guislain A, Cansever D, Wolkers MC. TLR‐mediated innate production of IFN‐γ by CD8+ T cells is independent of glycolysis. J Immunol. 2016;196:3695–705. [DOI] [PubMed] [Google Scholar]
- 46.Ganguly K, Giddaluru J, August A, Khan N. Post‐transcriptional Regulation of Immunological Responses through Riboclustering. Front Immunol. 2016;7:161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Salerno F, Paolini NA, Stark R, von Lindern M, Wolkers MC. Distinct PKC‐mediated posttranscriptional events set cytokine production kinetics in CD8+ T cells. Proc Natl Acad Sci. 2017;114:9677–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hoefig KP, Heissmeyer V. Posttranscriptional regulation of T helper cell fate decisions. J Cell Biol. 2018;217:2615–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Salerno F, Engels S, van den Biggelaar M, Van AFPJ, Guislain A, Zhao W, et al. Translational repression of pre‐formed cytokine‐encoding mRNA prevents chronic activation of memory T cells. Nat Immunol. 2018;19:828–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Gagnon JD, Kageyama R, Shehata HM, Fassett MS, Mar DJ, Wigton EJ, et al. miR‐15/16 restrain memory T cell differentiation, cell cycle, and survival. Cell Rep. 2019;28:2169–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Baitsch L, Baumgaertner P, Devevre E, Raghav SK, Legat A, Barba L, et al. Exhaustion of tumour‐specific CD8+ T cells in metastases from melanoma patients. J Clin Invest. 2011;121:2350–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Salerno F, Guislain A, Freen‐van Heeren JJ, Benoit P, Young HA, Wolkers MC. Critical role of post‐transcriptional regulation for IFN‐γ in tumor‐infiltrating T cells. Oncoimmunology. 2019;8:e1532762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Li H, van der Leun AM, Yofe I, Lubling Y, Gelbard‐Solodkin D, van Akkooi ACJ, et al. Dysfunctional CD8 T cells form a proliferative, dynamically regulated compartment within human melanoma. Cell. 2019;176:775–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Salerno F, Wolkers MC. T‐cells require post‐transcriptional regulation for accurate immune responses. Biochem Soc Trans. 2015;43:1201–7. [DOI] [PubMed] [Google Scholar]
- 55.Kuo CT, Leiden JM. Transcriptional regulation of t lymphocyte development and function. Annu Rev Immunol. 1999;17:149–87. [DOI] [PubMed] [Google Scholar]
- 56.Chang JT, Wherry EJ, Goldrath AW. Molecular regulation of effector and memory T cell differentiation. Nat Immunol. 2014;15:1104–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Conley JM, Gallagher MP, Berg LJ. T cells and gene regulation: The switching on and turning up of genes after T cell receptor stimulation in CD8 T cells. Front Immunol. 2016;7:76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Chen Y, Zander R, Khatun A, Schauder DM, Cui W. Transcriptional and epigenetic regulation of effector and memory CD8 T cell differentiation. Front Immunol. 2018;9:2826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Asirvatham AJ, Magner WJ, Tomasi TB. miRNA regulation of cytokine genes. Cytokine. 2009;45:58–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Beisang D, Bohjanen P. Perspectives on the ARE as it turns 25 years old. Wiley Interdiscip Rev RNA. 2012;3:719–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Oberdoerffer S, Moita LF, Neems D, Freitas RP, Hacohen N, Rao A. Regulation of CD45 alternative splicing by heterogeneous ribonucleoprotein, hnRNPLL. Science. 2008;321:686–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Topp JD, Jackson J, Melton AA, Lynch KW. A cell‐based screen for splicing regulators identifies hnRNP LL as a distinct signal‐induced repressor of CD45 variable exon 4. RNA. 2008;14:2038–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mauer J, Luo X, Blanjoie A, Jiao X, Grozhik AV, Patil DP, et al. Reversible methylation of m6 Am in the 5′ cap controls mRNA stability. Nature. 2017;541:371–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jiao X, Doamekpor SK, Bird JG, Nickels BE, Tong L, Hart RP, et al. 5′ End nicotinamide adenine dinucleotide cap in human cells promotes RNA decay through DXO‐mediated deNADding. Cell. 2017;168:1015–1027.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Galloway A, Cowling VH. mRNA cap regulation in mammalian cell function and fate. Biochimica et Biophysica Acta (BBA) ‐ Gene Regulatory Mechanisms. 2019;1862:270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nicolet BP, Guislain A, Wolkers MC. Combined single‐cell measurement of cytokine mRNA and protein identifies T cells with persistent effector function. J Immunol. 2017;198:962–70. [DOI] [PubMed] [Google Scholar]
- 67.Freen‐van Heeren JJ, Nicolet BP, Wolkers MC. Measuring T cell responses by flow cytometry‐based fluorescence in situ hybridization. Crit Rev Immunol. 2018;38:131–43. [DOI] [PubMed] [Google Scholar]
- 68.Freen‐van Heeren JJ, Nicolet BP, Wolkers MC. Combined Single‐Cell Measurement of Cytokine mRNA and Protein in Immune Cells. Methods Mol Biol. 2020;2108:259–71. [DOI] [PubMed] [Google Scholar]
- 69.Nicolet B, Wolkers M. Limited but gene‐class specific correlation of mRNA and protein expression in human CD8+ T cells. bioRxiv. 2020. 10.1101/2020.04.21.053884 [DOI] [Google Scholar]
- 70.Matera AG, Wang Z. A day in the life of the spliceosome. Nat Rev Mol Cell Biol. 2014;15:108–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Murray HL, Jarrell KA. Flipping the Switch to an Active Spliceosome. Cell. 1999;96:599–602. [DOI] [PubMed] [Google Scholar]
- 72.Wang ET, Sandberg R, Luo S, Khrebtukova I, Zhang L, Mayr C, et al. Alternative isoform regulation in human tissue transcriptomes. Nature. 2008;456:470–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Ergun A, Doran G, Costello JC, Paik HH, Collins JJ, Mathis D, et al. Differential splicing across immune system lineages. Proc Natl Acad Sci U S A. 2013;110:14324–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Liu S, Zhou B, Wu L, Sun Y, Chen J, Liu S. Single‐cell differential splicing analysis reveals high heterogeneity of liver tumor‐infiltrating T cells. Sci Rep. 2021;11:5325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Ip JY, Tong A, Pan Q, Topp JD, Blencowe BJ, Lynch KW. Global analysis of alternative splicing during T‐cell activation. RNA. 2007;13:563–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Martinez NM, Pan Q, Cole BS, Yarosh CA, Babcock GA, Heyd F, et al. Alternative splicing networks regulated by signaling in human T cells. RNA. 2012;18:1029–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.da Glória VG, Martins de Araújo M, Mafalda Santos A, Leal R, de Almeida SF, Carmo AM, et al. T cell activation regulates CD6 alternative splicing by transcription dynamics and SRSF1. J Immunol. 2014;193:391–9. [DOI] [PubMed] [Google Scholar]
- 78.Liu X, Andrews MV, Skinner JP, Johanson TM, Chong MMW. A comparison of alternative mRNA splicing in the CD4 and CD8 T cell lineages. Mol Immunol. 2021;133:53–62. [DOI] [PubMed] [Google Scholar]
- 79.Whistler T, Chiang CF, Lonergan W, Hollier M, Unger ER. Implementation of exon arrays: Alternative splicing during T‐cell proliferation as determined by whole genome analysis. BMC Genom. 2010;14:496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Ruland J, Duncan GS, Wakeham A, Mak TW. Differential requirement for Malt1 in T and B cell antigen receptor signaling. Immunity. 2003;19:749–58. [DOI] [PubMed] [Google Scholar]
- 81.Meininger I, Griesbach RA, Hu D, Gehring T, Seeholzer T, Bertossi A, et al. Alternative splicing of MALT1 controls signalling and activation of CD4 + T cells. Nat Commun. 2016;12:11292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Uehata T, Iwasaki H, Vandenbon A, Matsushita K, Hernandez‐Cuellar E, Kuniyoshi K, et al. Malt1‐induced cleavage of regnase‐1 in CD4+ helper T cells regulates immune activation. Cell. 2013;153:1036. [DOI] [PubMed] [Google Scholar]
- 83.Swanson BJ, Murakami M, Mitchell TC, Kappler J, Marrack P. RANTES production by memory phenotype T cells is controlled by a posttranscriptional, TCR‐dependent process. Immunity. 2002;17:605–15. [DOI] [PubMed] [Google Scholar]
- 84.Kaech SM, Hemby S, Kersh E, Ahmed R. Molecular and functional profiling of memory CD8 T cell differentiation. Cell. 2002;111:837–51. [DOI] [PubMed] [Google Scholar]
- 85.Kohlmeier JE, Cookenham T, Roberts AD, Miller SC, David L. Type I interferons regulate cytolytic activity of memory CD8+ T cells in the lung airways during respiratory virus challenge. Immunity. 2010;33:96–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Berg RE, Cordes CJ, Forman J. Contribution of CD8+ T cells to innate immunity: IFN‐γ secretion induced by IL‐12 and IL‐18. Eur J Immunol. 2002;32:2807–16. [DOI] [PubMed] [Google Scholar]
- 87.Berg RE, Crossley E, Murray S, Forman J. Memory CD8+ T cells provide innate immune protection against listeria monocytogenes in the absence of cognate antigen. J Exp Med. 2003;198:1583–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Cottalorda A, Verschelde C, Marçais A, Tomkowiak M, Musette P, Uematsu S, et al. TLR2 engagement on CD8 T cells lowers the thresholdfor optimal antigen‐induced T cell activation. Eur J Immunol. 2006;36:1684–93. [DOI] [PubMed] [Google Scholar]
- 89.Imanishi T, Unno M, Kobayashi W, Yoneda N, Matsuda S, Ikeda K, et al. Reciprocal regulation of STING and TCR signaling by mTORC1 for T‐cell activation and function. Life Sci Alliance. 2019;2:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Larkin B, Ilyukha V, Sorokin M, Buzdin A, Vannier E, Poltorak A. Cutting Edge: Activation of STING in T Cells Induces Type I IFN Responses and Cell Death. J Immunol. 2017;199:397–402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Diehn M, Alizadeh A, Rando O, Liu C, Stankunas K, Botstein D, et al. Genomic expression programs and the integration of the CD28 costimulatory signal in T cell activation. Proc Natl Acad Sci. 2002;99:11796–801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lindstein T, June CH, Ledbetter JA, Stella G, Thompson CB. Regulation of Lymphokine Messenger RNA Stability by a Surface‐Mediated T cell Activation Pathway. Science (80‐). 1989;244:339–43. [DOI] [PubMed] [Google Scholar]
- 93.Wang JG, Collinge M, Ramgolam V, Fan XC, Pardi R, Jeffrey R, et al. LFA‐1‐dependent HuR nuclear export and cytokine mRNA stabilization in T cell activation. J Immunol. 2006;176:2105–13. [DOI] [PubMed] [Google Scholar]
- 94.Cohen‐Chalamish S, Hasson A, Weinberg D, Namer LS, Banai Y, Osman F, et al. Dynamic refolding of IFN‐gamma mRNA enables it to function as PKR activator and translation template. Nat Chem Biol. 2009;5:896–903. [DOI] [PubMed] [Google Scholar]
- 95.Turner M, Galloway A, Vigorito E. Noncoding RNA and its associated proteins as regulatory elements of the immune system. Nat Immunol. 2014;15:484–91. [DOI] [PubMed] [Google Scholar]
- 96.Yue Y, Liu J, He C. RNA N6‐methyladenosine methylation in post‐transcriptional gene expression regulation. Genes Dev. 2015;29:1343–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Moore KS, Von LM. RNA Binding Proteins and Regulation of mRNA Translation in Erythropoiesis. Front Physiol. 2018;9:910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Ben‐Asouli Y, Banai Y, Pel‐Or Y, Shir A, Kaempfer R. Human interferon‐gamma mRNA autoregulates its translation through a pseudoknot that activates the interferon‐inducible protein kinase PKR. Cell. 2002;108:221–32. [DOI] [PubMed] [Google Scholar]
- 99.Staple DW, Butcher SE. Pseudoknots: RNA structures with diverse functions. PLoS Biol. 2005;3:e213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Babendure JR, Babendure JL, Ding J‐H, Tsien RY. Control of mammalian translation by mRNA structure near caps. RNA. 2006;12:851–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rodríguez‐Galán A, Fernández‐Messina L, Sánchez‐Madrid F. Control of Immunoregulatory Molecules by miRNAs in T Cell Activation. Front Immunol. 2018;9:2148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Stoecklin G, Lu M, Rattenbacher B, Moroni C. A constitutive decay element promotes tumor necrosis factor alpha mRNA degradation via an AU‐rich element‐independent pathway. Mol Cell Biol. 2003;23:3506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hodge DL, Berthet C, Coppola V, Kastenmüller W, Buschman MD, Schaughency PM, et al. IFN‐gamma AU‐rich element removal promotes chronic IFN‐gamma expression and autoimmunity in mice. J Autoimmun. 2014;53:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vlasova‐St. Louis I, Bohjanen PR. Post‐transcriptional regulation of cytokine signaling by AU‐rich and GU‐rich elements. J Interf Cytokine Res. 2014;34:233–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Leppek K, Schott J, Reitter S, Poetz F, Hammond MC, Stoecklin G. Roquin promotes constitutive mRNA decay via a conserved class of stem‐loop recognition motifs. Cell. 2013;153:869–81. [DOI] [PubMed] [Google Scholar]
- 106.Arif A, Chatterjee P, Moodt RA, Fox PL. Heterotrimeric GAIT complex drives transcript‐selective translation inhibition in murine macrophages. Mol Cell Biol. 2012;32:5046–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Chen C. Stabilization of interleukin‐2 mRNA by the c‐Jun NH2‐terminal kinase pathway. Science. 1998;280:1945–9. [DOI] [PubMed] [Google Scholar]
- 108.Chen CY, Gherzi R, Andersen JS, Gaietta G, Jürchott K, Royer HD, et al. Nucleolin and YB‐1 are required for JNK‐mediated interleukin‐2 mRNA stabilization during T‐cell activation. Genes Dev. 2000;14:1236–48. [PMC free article] [PubMed] [Google Scholar]
- 109.Grenov AC, Moss L, Edelheit S, Cordiner R, Schmiedel D, Biram A, et al. B Cell division capacity in germinal centers depends on Myc Transcript stabilization through m6A mRNA methylation and IGF2BP3 functions. bioRxiv. 2020;2020.09.08.287433. [Google Scholar]
- 110.Rodríguez‐Galán A, Dosil SG, José Gómez M, Fernández‐Delgado I, Sánchez‐Cabo F, Sánchez‐Madrid F. Impaired miRNA degradation by post‐transcriptional addition of 3’ cytosine and adenine in T cell activation. bioRxiv. 2020. 10.1101/2020.08.19.257816 [DOI] [Google Scholar]
- 111.Freen‐van Heeren JJ. Using CRISPR to enhance T cell effector function for therapeutic applications. Cytokine X. 2021;3:100049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Bakheet T, Frevel M, Williams BR, Greer W, Khabar KS. ARED: human AU‐rich element‐containing mRNA database reveals an unexpectedly diverse functional repertoire of encoded proteins. Nucleic Acids Res. 2001;29:246–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Gruber AR, Fallmann J, Kratochvill F, Kovarik P, Hofacker IL. AREsite: a database for the comprehensive investigation of AU‐rich elements. Nucleic Acids Res. 2011;39(Database):D66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Xu N, Chen C‐YA, Shyu A‐B. Modulation of the fate of cytoplasmic mRNA by AU‐rich elements: Key sequence features controlling mRNA deadenylation and decay. Mol Cell Biol. 1997;17:4611–4621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Barreau C, Paillard L, Osborne HB. AU‐rich elements and associated factors: are there unifying principles? Nucleic Acids Res. 2005;33:7138–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gueydan C, Droogmans L, Chalon P, Huez G, Caput D, Kruys V. Identification of TIAR as a protein binding to the translational regulatory AU‐rich element of tumor necrosis factor α mRNA. J Biol Chem. 1999;274:2322–6. [DOI] [PubMed] [Google Scholar]
- 117.Peng SS, Chen CY, Shyu AB. Functional characterization of a non‐AUUUA AU‐rich element from the c‐jun proto‐oncogene mRNA: evidence for a novel class of AU‐rich elements. Mol Cell Biol. 1996;16:1490–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Shim J, Lim H, Yates JR, Karin M. Nuclear export of NF90 is required for interleukin‐2 mRNA stabilization. Mol Cell. 2002;10:1331–44. [DOI] [PubMed] [Google Scholar]
- 119.Dean JLE, Wait R, Mahtani KR, Clark AR, Saklatvala J, Sully G. The 3 ′Untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA‐stabilizing factor HuR The 3J untranslated region of tumor necrosis factor alpha mRNA is a target of the mRNA‐stabilizing factor HuR. Mol Cell Biol. 2001;21:721–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Gubin MM, Techasintana P, Magee JD, Dahm GM, Calaluce R, Martindale JL, et al. Conditional knockout of the RNA‐binding protein HuR in CD4+ T cells reveals a gene dosage effect on cytokine production. Mol Med. 2014;20:93–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Shi L, Godfrey WR, Lin J, Zhao G, Kao PN. NF90 regulates inducible IL‐2 gene expression in T cells. J Exp Med. 2007;204:971–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pei Y, Zhu P, Dang Y, Wu J, Yang X, Wan B, et al. Nuclear Export of NF90 to stabilize IL‐2 mRNA is mediated by AKT‐dependent phosphorylation at Ser 647 in response to CD28 costimulation. J Immunol. 2008;180:222–9. [DOI] [PubMed] [Google Scholar]
- 123.Ogilvie RL, Abelson M, Hau HH, Vlasova I, Blackshear PJ, Bohjanen PR. Tristetraprolin down‐regulates IL‐2 gene expression through AU‐rich element‐mediated mRNA decay. J Immunol. 2005;174:953–61. [DOI] [PubMed] [Google Scholar]
- 124.Ogilvie RL, John JRS, Rattenbacher B, Vlasova IA, Williams DA, Hau HH, et al. Tristetraprolin mediates interferon‐γ mRNA decay. J Biol Chem. 2009;284:11216–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Moore MJ, Blachere NE, Fak JJ, Park CY, Sawicka K, Parveen S, et al. ZFP36 RNA‐binding proteins restrain T cell activation and anti‐viral immunity. Elife. 2018;7:e33057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Taylor GA, Carballo E, Lee DM, Lai WS, Thompson MJ, Patel DD, et al. A pathogenetic role for TNFα in the syndrome of cachexia, arthritis, and autoimmunity resulting from tristetraprolin (TTP) deficiency. Immunity. 1996;4:445–54. [DOI] [PubMed] [Google Scholar]
- 127.Piecyk M, Wax S, Beck AR, Kedersha N, Gupta M, Maritim B, et al. TIA‐1 is a translational silencer that selectively regulates the expression of TNF‐alpha. EMBO J. 2000;19:4154–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Yamasaki S, Stoecklin G, Kedersha N, Simarro M, Anderson P. T‐cell intracellular antigen‐1 (TIA‐1)‐induced translational silencing promotes the decay of selected mRNAs. J Biol Chem. 2007;282:30070–7. [DOI] [PubMed] [Google Scholar]
- 129.Díaz‐Muñoz MD, Kiselev VY, Le NN, Curk T, Ule J, Turner M. Tia1 dependent regulation of mRNA subcellular location and translation controls p53 expression in B cells. Nat Commun. 2017;8:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kedersha N, Cho MR, Li W, Yacono PW, Chen S, Gilks N, et al. Dynamic shuttling of TIA‐1 accompanies the recruitment of mRNA to mammalian stress granules. J Cell Biol. 2000;151:1257–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Li M, Cao W, Liu H, Zhang W, Liu X, Cai Z, et al. MCPIP1 Down‐regulates IL‐2 expression through an ARE‐independent pathway. PLoS One. 2012;7:e49841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Mino T, Murakawa Y, Fukao A, Vandenbon A, Wessels HH, Ori D, et al. Regnase‐1 and roquin regulate a common element in inflammatory mRNAs by spatiotemporally distinct mechanisms. Cell. 2015;161:1058–73. [DOI] [PubMed] [Google Scholar]
- 133.Mino T, Iwai N, Endo M, Inoue K, Akaki K, Hia F, et al. Translation‐dependent unwinding of stem‐loops by UPF1 licenses Regnase‐1 to degrade inflammatory mRNAs. Nucleic Acids Res. 2019;47:8838–59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Matsushita K, Takeuchi O, Standley DM, Kumagai Y, Kawagoe T, Miyake T, et al. Zc3h12a is an RNase essential for controlling immune responses by regulating mRNA decay. Nature. 2009;458:1185–90. [DOI] [PubMed] [Google Scholar]
- 135.Wei J, Long L, Zheng W, Dhungana Y, Lim SA, Guy C, et al. Targeting REGNASE‐1 programs long‐lived effector T cells for cancer therapy. Nature. 2019;576:471–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chang P‐P, Lee SK, Hu X, Davey G, Duan G, Cho J‐H, et al. Breakdown in repression of IFN‐γ mRNA leads to accumulation of self‐reactive effector CD8+ T Cells. J Immunol. 2012;189:701–10. [DOI] [PubMed] [Google Scholar]
- 137.Lee SK, Silva DG, Martin JL, Pratama A, Hu X, Chang PP, et al. Interferon‐γ excess leads to pathogenic accumulation of follicular helper T cells and germinal centers. Immunity. 2012;37:880–92. [DOI] [PubMed] [Google Scholar]
- 138.Jeltsch KM, Hu D, Brenner S, Zöller J, Heinz GA, Nagel D, et al. Cleavage of roquin and regnase‐1 by the paracaspase MALT1 releases their cooperatively repressed targets to promote TH17 differentiation. Nat Immunol. 2014;15:1079–89. [DOI] [PubMed] [Google Scholar]
- 139.Essig K, Kronbeck N, Guimaraes JC, Lohs C, Schlundt A, Hoffmann A, et al. Roquin targets mRNAs in a 3′‐UTR‐specific manner by different modes of regulation. Nat Commun. 2018;9:3810. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Minagawa K, Wakahashi K, Kawano H, Nishikawa S, Fukui C, Kawano Y, et al. Posttranscriptional modulation of cytokine production in T cells for the regulation of excessive inflammation by TFL. J Immunol. 2014;192:1512–24. [DOI] [PubMed] [Google Scholar]
- 141.Hodson DJ, Janas ML, Galloway A, Bell SE, Andrews S, Li CM, et al. Deletion of the RNA‐binding proteins ZFP36L1 and ZFP36L2 leads to perturbed thymic development and T lymphoblastic leukemia. Nat Immunol. 2010;11:717–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Shi L, Zhao G, Qiu D, Godfrey WR, Vogel H, Rando TA, et al. NF90 Regulates cell cycle exit and terminal myogenic differentiation by direct binding to the 3’‐untranslated region of MyoD and p21WAF1/CIP1 mRNAs. J Biol Chem. 2005;280:18981–9. [DOI] [PubMed] [Google Scholar]
- 143.Katsanou V, Milatos S, Yiakouvaki A, Sgantzis N, Kotsoni A, Alexiou M, et al. The RNA‐binding protein Elavl1/HuR is essential for placental branching morphogenesis and embryonic development. Mol Cell Biol. 2009;29:2762–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Techasintana P, Ellis JS, Glascock J, Gubin MM, Ridenhour SE, Magee JD, et al. The RNA‐binding protein HuR posttranscriptionally regulates IL‐2 homeostasis and CD4 + Th2 differentiation. ImmunoHorizons. 2017;1:109–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Oja AE, Piet B, van der Zwan D, Blaauwgeers H, Mensink M, de Kivit S, et al. Functional heterogeneity of CD4+ tumor‐infiltrating lymphocytes with a resident memory phenotype in NSCLC. Front Immunol. 2018;9:2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Wolchok JD, Chiarion‐Sileni V, Gonzalez R, Rutkowski P, Grob JJ, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med. 2017;377:1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Kagamu H, Kaira K. Efficacy of PD‐1 blockade therapy and T cell immunity in lung cancer patients. Immunol Med. 2020;43:10–5. [DOI] [PubMed] [Google Scholar]
- 148.Su S, Zou Z, Chen F, Ding N, Du J, Shao J, et al. CRISPR‐cas9‐mediated disruption of PD‐1 on human T cells for adoptive cellular therapies of EBV positive gastric cancer. Oncoimmunology. 2017;6:e1249558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Zhao Z, Shi L, Zhang W, Han J, Zhang S, Fu Z, et al. CRISPR knock out of programmed cell death protein 1 enhances anti‐tumor activity of cytotoxic T lymphocytes. Oncotarget. 2018;9:5208–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lu Y, Huang M, Deng T, Zhou X, Yu K, Liang M, et al. A phase I trial of PD‐1 deficient engineered T cells with CRISPR/Cas9 in patients with advanced non‐small cell lung cancer with PD‐L1 expression. Cancer Res. 2018;78(13 Suppl):CT133. [Google Scholar]
- 151.Lu Y, Xue J, Deng T, Zhou X, Yu K, Deng L, et al. Safety and feasibility of CRISPR‐edited T cells in patients with refractory non‐small‐cell lung cancer. Nat Med. 2020;26:732–40. [DOI] [PubMed] [Google Scholar]
- 152.Stadtmauer EA, Fraietta JA, Davis MM, Cohen AD, Weber KL, Lancaster E, et al. CRISPR‐engineered T cells in patients with refractory cancer. Science. 2020;367:eaba7365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Pettitt D, Arshad Z, Smith J, Stanic T, Holländer G, Brindley D. CAR‐T cells : A systematic review and mixed methods analysis of the clinical trial landscape. Mol Ther. 2018;26:342–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Weinkove R, George P, Dasyam N, McLellan AD. Selecting costimulatory domains for chimeric antigen receptors: functional and clinical considerations. Clin Transl Immunol. 2019;8:e1049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.McGowan E, Lin Q, Ma G, Yin H, Chen S, Lin Y. PD‐1 disrupted CAR‐T cells in the treatment of solid tumors: Promises and challenges. Biomed Pharmacother. 2019;2020:109625. [DOI] [PubMed] [Google Scholar]
- 156.Chen S, Lin Y, Zhong S, An H, Lu Y, Yin M, et al. Anti‐MUC1 CAR‐T cells combined with PD‐1 knockout engineered T cells for patients with non‐small cell lung cancer (NSCLC): A pilot study. Ann Oncol. 2018;29(Suppl 10):x11. [Google Scholar]
- 157.Jing Z, Zhang N, Ding L, Wang X, Hua Y, Jiang M, et al. Safety and activity of programmed cell death‐1 gene knockout engineered t cells in patients with previously treated advanced esophageal squamous cell carcinoma: An open‐label, single‐arm phase I study. J Clin Oncol. 2018;36(15 Suppl):3054. [Google Scholar]
- 158.Bonifant CL, Jackson HJ, Brentjens RJ, Curran KJ. Toxicity and management in CAR T‐cell therapy. Mol Ther ‐ Oncolytics. 2016;3:16011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a primary target for PD‐1–mediated inhibition. Science (80‐). 2017;355:1428–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells by PD‐1–targeted therapies is CD28‐dependent. Science (80‐.). 2017;355:1423–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Coelho MA, de Carné TS, Rana S, Zecchin D, Moore C, Molina‐Arcas M, et al. Oncogenic RAS signaling promotes tumor immunoresistance by stabilizing PD‐L1 mRNA. Immunity. 2017;47:1083–1099.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Papalexi E, Mimitou E, Butler A, Foster S, Bracken B, Mauck W III, et al. Characterizing the molecular regulation of inhibitory immune checkpoints with multi‐modal single‐ cell screens. Nat Genet. 2021;53:322–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Geng D, Zheng L, Srivastava R, Velasco‐Gonzales C, Riker A, Markovic SN, et al. Amplifying TLR‐MyD88 signals within tumor‐specific T‐cells enhances antitumor activity to suboptimal levels of weakly‐ immunogenic tumor‐antigens. Cancer Res. 2010;70:7442–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Asprodites N, Zheng L, Geng D, Velasco‐Gonzalez C, Sanchez‐Perez L, Davila E. Engagement of Toll‐like receptor‐2 on cytotoxic T‐lymphocytes occurs in vivo and augments antitumor activity. FASEB J. 2008;22:3628–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Freen‐van Heeren JJ. TLR‐2/7‐mediated T cell activation: an innate potential to augment CD8+ T cell cytokine production. Scand J Immunol. 2021;93:e13019. [DOI] [PubMed] [Google Scholar]
- 166.Lai Y, Weng J, Wei X, Qin L, Lai P, Zhao R, et al. Toll‐like receptor 2 costimulation potentiates the antitumor efficacy of CAR T Cells. Leukemia. 2018;32:801–8. [DOI] [PubMed] [Google Scholar]
- 167.Weng J, Lai P, Qin L, Lai Y, Jiang Z, Luo C, et al. A novel generation 1928zT2 CAR T cells induce remission in extramedullary relapse of acute lymphoblastic leukemia. J Hematol Oncol. 2018;11:25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Freen‐van Heeren JJ. Harnessing high‐throughput flow cytometric protein and RNA detection to enhance TIL therapy. Cytom Part A. 2021. 10.1002/cyto.a.24323 [DOI] [PubMed] [Google Scholar]
- 169.Castello A, Horos R, Strein C, Fischer B, Eichelbaum K, Steinmetz LM, et al. Comprehensive identification of RNA‐binding proteins by RNA interactome capture. In: Dassi E, eds. Post‐Transcriptional Gene Regulation. New York: Humana Press Inc.; 2015. p. 131–9. [Google Scholar]
- 170.Whisenant TC, Peralta ER, Aarreberg LD, Gao NJ, Head SR, Ordoukhanian P, et al. The activation‐induced assembly of an RNA/Protein interactome centered on the splicing factor U2AF2 regulates gene expression in human CD4 T Cells. PLoS One. 2015;10:e0144409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Shchepachev V, Bresson S, Spanos C, Petfalski E, Fischer L, Rappsilber J, et al. Defining the RNA interactome by total RNA ‐associated protein purification. Mol Syst Biol. 2019;15:e8689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Ma F, Xu S, Liu X, Zhang Q, Xu X, Liu M, et al. The microRNA miR‐29 controls innate and adaptive immune responses to intracellular bacterial infection by targeting interferon‐γ. Nat Immunol. 2011;12:861–9. [DOI] [PubMed] [Google Scholar]
- 173.Amado T, Amorim A, Enguita FJ, Romero PV, Inácio D, de Miranda MP, et al. MicroRNA‐181a regulates IFN‐γ expression in effector CD8+ T cell differentiation. J Mol Med. 2020;98:309–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Chang P‐P, Lee SK, Hu X, Davey G, Duan G, Cho J‐H, et al. Breakdown in repression of IFN‐γ mRNA leads to accumulation of self‐reactive effector CD8+ T cells. J Immunol. 2012;189:701–10. [DOI] [PubMed] [Google Scholar]
- 175.Dan C, Jinjun B, Zi‐Chun H, Lin M, Wei C, Xu Z, et al. Modulation of TNF‐α mRNA stability by human antigen R and miR181s in sepsis‐induced immunoparalysis. EMBO Mol Med. 2015;7:140–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Jing Q, Huang S, Guth S, Zarubin T, Motoyama A, Chen J, et al. Involvement of MicroRNA in AU‐rich element‐mediated mRNA instability. Cell. 2005;120:623–34. [DOI] [PubMed] [Google Scholar]
- 177.Katsanou V, Papadaki O, Milatos S, Blackshear PJ, Anderson P, Kollias G, et al. HuR as a negative posttranscriptional modulator in inflammation. Mol Cell. 2005;19:777–89. [DOI] [PubMed] [Google Scholar]
- 178.El Gazzar M, McCall CE. MicroRNAs distinguish translational from transcriptional silencing during endotoxin tolerance. J Biol Chem. 2010;285:20940–51. [DOI] [PMC free article] [PubMed] [Google Scholar]