

Abstract
Second trimester foetal human amniotic fluid‐derived stem cells (hAFS) have been shown to possess remarkable cardioprotective paracrine potential in different preclinical models of myocardial injury and drug‐induced cardiotoxicity. The hAFS secretome, namely the total soluble factors released by cells in their conditioned medium (hAFS‐CM), can also strongly sustain in vivo angiogenesis in a murine model of acute myocardial infarction (MI) and stimulates human endothelial colony‐forming cells (ECFCs), the only truly recognized endothelial progenitor, to form capillary‐like structures in vitro. Preliminary work demonstrated that the hypoxic hAFS secretome (hAFS‐CMHypo) triggers intracellular Ca2+ oscillations in human ECFCs, but the underlying mechanisms and the downstream Ca2+‐dependent effectors remain elusive. Herein, we found that the secretome obtained by hAFS undergoing hypoxic preconditioning induced intracellular Ca2+ oscillations by promoting extracellular Ca2+ entry through Transient Receptor Potential Vanilloid 4 (TRPV4). TRPV4‐mediated Ca2+ entry, in turn, promoted the concerted interplay between inositol‐1,4,5‐trisphosphate‐ and nicotinic acid adenine dinucleotide phosphate‐induced endogenous Ca2+ release and store‐operated Ca2+ entry (SOCE). hAFS‐CMHypo‐induced intracellular Ca2+ oscillations resulted in the nuclear translocation of the Ca2+‐sensitive transcription factor p65 NF‐κB. Finally, inhibition of either intracellular Ca2+ oscillations or NF‐κB activity prevented hAFS‐CMHypo‐induced ECFC tube formation. These data shed novel light on the molecular mechanisms whereby hAFS‐CMHypo induces angiogenesis, thus providing useful insights for future therapeutic strategies against ischaemic‐related myocardial injury.
Keywords: angiogenesis, Ca2+ signalling, endothelial colony‐forming cells, human amniotic fluid stem cell secretome, InsP3Rs, NAADP, NF‐κB, paracrine therapy, TRPV4, tubulogenesis
1. INTRODUCTION
Endothelial colony‐forming cells (ECFCs) represent vasculogenic cells that are truly committed to the endothelial lineage and, throughout postnatal life, are mobilized upon ischaemic injury to restore the damaged vascular network.1, 2 ECFCs possess a high clonogenic potential, form capillary‐like structures in in vitro Matrigel tubulogenesis assays, integrate into pre‐existing vasculature and rescue local blood perfusion in murine models of ischaemia.2, 3 Therefore, ECFCs hold remarkable promise as the most suitable cellular substrate to induce therapeutic angiogenesis in ischaemic disorders, such as acute myocardial infarction (AMI), peripheral artery disease, and stroke.2, 3, 4 Genetic manipulation and pharmacological conditioning could be exploited to improve ECFCs’ angiogenic activity and/or to improve their survival/engraftment within the harsh microenvironment of the ischaemic tissue.2, 5 Mesenchymal stromal progenitors obtained from leftover samples of second trimester amniotic fluid for prenatal diagnosis have been lately described as appealing therapeutics in several preclinical models of disease. In particular, human amniotic fluid‐derived stem cells (hAFS) have been shown to express a remarkable pro‐angiogenic paracrine potential.6, 7 A more recent investigation showed that the hAFS secretome collected under hypoxic conditions stimulated circulating ECFCs to assemble into bidimensional capillary‐like networks in vitro through an oscillatory increase in intracellular Ca2+ concentration ([Ca2+]i)8, 9; furthermore, it actively induced local angiogenesis and promoted cardiac repair in a murine model of AMI.8, 9 Therefore, the local injection of the hypoxic hAFS secretome could represent an alternative strategy to recruit circulating ECFCs towards the damaged myocardium and induce therapeutic angiogenesis.2 The paracrine therapy of AMI would be promptly accessible to the patients and could overcome many of the drawbacks associated with cell‐based therapy, including the time‐consuming procedure and the requirement for huge amounts of cells.10, 11 Clinical translation of this approach would benefit from the elucidation of the signalling pathways whereby the hAFS secretome triggers the pro‐angiogenic Ca2+ response in ECFCs.
Intracellular Ca2+ oscillations drive vascular endothelial growth factor (VEGF)‐induced proliferation and tube formation in both circulating12 and umbilical cord blood (UCB)‐derived ECFCs.13 The oscillatory Ca2+ signal elicited by VEGF in ECFCs is shaped by rhythmical inositol‐1,4,5‐trisphosphate (InsP3)‐induced Ca2+ release from the endoplasmic reticulum (ER) and sustained over time by store‐operated Ca2+ entry (SOCE).12 Furthermore, nicotinic acid adenine dinucleotide phosphate (NAADP)‐induced Ca2+ mobilization from endolysosomal (EL) vesicles through two‐pore channel 1 (TPC1) may contribute to pattern the spiking response.14 VEGF‐induced intracellular Ca2+ oscillations, in turn, stimulate angiogenesis by inducing the nuclear translocation of the Ca2+‐sensitive transcription factor, nuclear factor κB (NF‐κB).12 The intracellular Ca2+ oscillations induced by the hypoxic hAFS secretome in circulating ECFCs strongly resemble those elicited by VEGF.9 However, the same pattern of intracellular Ca2+ signalling could be underlain by diverse mechanisms,15, 16, 17 as previously reported for VEGF in different types of ECFCs. For instance, the dynamic interplay between InsP3 receptors (InsP3Rs) and SOCE is triggered by extracellular Ca2+ entry through Transient Receptor Potential Channel 3 (TRPC3) in UCB‐derived, but not circulating, ECFCs.12, 13 Moreover, the Ca2+‐dependent molecular decoder which translates hAFS secretome‐induced intracellular Ca2+ oscillations into a pro‐angiogenic output remains elusive. Therefore, in the present investigation, we have characterized for the first time the mechanisms whereby hypoxic hAFS secretome induces pro‐angiogenic intracellular Ca2+ oscillations and one of the downstream Ca2+‐dependent decoders, that is NF‐κB, in circulating ECFCs.
2. MATERIALS AND METHODS
2.1. Isolation and cultivation of ECFCs
Blood samples (40 mL) collected in EDTA (ethylenediaminetetraacetic acid)‐containing tubes were obtained from healthy human volunteers aged from 22 to 28 years. The Institution Review Board at “Istituto di Ricovero e Cura a Carattere Scientifico Policlinico San Matteo Foundation” in Pavia approved all the protocols. Informed written consent was obtained according to the Declaration of Helsinki of 1975 as revised in 2008. ECFCs were isolated from circulating mononuclear cells, as described in Supplementary Information and in REF.18, 19
2.2. Isolation and preconditioning of hAFS
Human amniotic fluid‐derived stem cells were isolated from leftover samples of second trimester amniotic fluid obtained by prenatal screening from the Prenatal Diagnosis and Perinatal Medicine Unit, IRCCS San Martino Hospital, and the Fetal and Perinatal Medical and Surgery Unit and Human Genetics Laboratory at IRCCS Istituto Gaslini hospital (Genova, Italy). Informed written consent was obtained from all donors, according to local ethical committee authorization (protocol PR 428REG2015) and in compliance with Helsinki Declaration guidelines. After obtaining adherent amniotic fluid mesenchymal cells, hAFS were isolated by immunomagnetic sorting for c‐KIT expression (CD117 MicroBead Kit, Miltenyi Biotechnology), as previously defined8, 9, 11, 20 (PMID: 17206138). c‐KIT+ hAFS were then cultured in Minimal Essential Medium (MEM)‐alpha with 15% FBS (Gibco—Thermo Fisher Scientific), 18% Chang B and 2% Chang C Medium (Irvine Scientific) with 1% L‐glutamine and 1% penicillin/streptomycin (Gibco—Thermo Fisher Scientific), at 75% confluency before being used to isolate their secretome.
In order to trigger the hAFS paracrine potential and to enrich their secretome with trophic soluble factors, cells were primed in vitro for 24 hours in serum‐free medium (high glucose Dulbecco's Modified Eagle's Medium, DMEM, with 1% L‐glutamine and 1% penicillin/streptomycin, Gibco—Thermo Fisher Scientific) under normoxic (20% O2 and 5% CO2 at 37°C) or stimulatory hypoxic (1% O2 and 5% CO2 at 37°C in a Galaxy® 48 R CO2 incubators; Eppendorf) conditions.8, 9, 11, 20
2.3. hAFS secretome separation and concentration
The total cell secretome, as represented by the cell‐conditioned medium (hAFS‐CM) from either hAFS in control normoxic condition (hAFS‐CMNormo) or hypoxic preconditioning (hAFS‐CMHypo), was collected as previously described.8, 9, 20 Briefly, hAFS‐CM formulations were centrifuged to remove cell debris and further concentrated using ultrafiltration membranes with a 3 kDa selective cut‐off (Amicon Ultra‐15; Millipore) at 4°C first at 3000 ×g for 90’ and then at 3000 ×g for additional 30’. hAFS‐CM protein concentration was assessed by BiCinchoninic Acid (BCA) assay (Gibco—Thermo Fisher Scientific). hAFS‐CM was used for in vitro experiments as 80 mg/mL solution to be added to the cell culture medium as from previous studies.8, 9
2.3.1. Solutions
Physiological salt solution (PSS) had the following composition (in mmol/L): 150 NaCl, 6 KCl, 1.5 CaCl2, 1 MgCl2, 10 Glucose and 10 Hepes. In Ca2+‐free solution (0Ca2+), Ca2+ was substituted with 2 mmol/L NaCl, and 0.5 mmol/L EGTA was added. Solutions were titrated to pH 7.4 with NaOH. The osmolality of PSS as measured with an osmometer (Wescor 5500) was 338 mmol/kg.
2.3.2. [Ca2+]i measurements
Endothelial colony‐forming cells were loaded with 4 µmol/L fura‐2 acetoxymethyl ester (fura‐2/AM; 1 mmol/L stock in dimethyl sulfoxide) in PSS for 1 hour at room temperature. The details of the Ca2+ recording set‐up have been described in REF9, 14 and are reported in the Supplementary Information. All the experiments were performed at room temperature. All the data have been collected from ECFCs isolated from peripheral blood of at least three healthy volunteers.
2.3.3. Immunofluorescence
Twenty‐four hours before treatment with hAFS‐CMHypo and the specific blockers of intracellular Ca2+ signalling, 6 × 104 ECFCs were plated onto 13 mm coverslips in 24‐well plates. ECFCs were fixed in 4% formaldehyde in PBS for 15 minutes at room temperature, permeabilized for 7 minutes in PBS with 0.1% Triton X‐100 and blocked for 30 minutes in 2% gelatin. Then, primary (incubated for 1 hour at 37°C) and secondary (incubated for 1 hour at room temperature) antibodies were applied in PBS with 2% gelatin. The primary anti‐p65 (NF‐κB subunit) antibody specific for immunocytochemistry (Santa Cruz Biotechnology, catalog no. Sc‐372) was used at 1:50 dilution, whereas the AlexaFluor 488 secondary antibody from Invitrogen (catalog no. A‐21441) was used at 1:200. After washing (3 times for 5 minutes each), nuclei were stained with 40,6‐diamidino‐2‐phenylindole dihydrochloride (DAPI) for 15 minutes at room temperature. Fluorescence images were acquired using a Leica epifluorescence microscope equipped with S Fluor X40/1.3 objective using MetaMorph software.
2.3.4. In vitro tube formation assay
Early passage (P2‐P3) ECFCs were cultured in basal medium EBM‐2 supplemented with 2% FBS in Cultrex (Trevigen)‐coated 96‐well plates, in the absence or in the presence of hAFS‐CMHypo for 24 hours. Capillary network formation was assessed starting from 4 up to 24 hours later. The angiogenic response was measured by evaluating both dimensional and topological parameters. The length of endothelial tube‐like structures (tubules or TLS), number of polygon structures established by TLS, referred to as meshes and indicative of endothelial cell migration, and number of master junctions were measured from acquired bright‐field pictures by using the Angiogenesis Analyzer plugin of ImageJ (Gilles Carpentier, Faculte’ des Sciences et Technologie, Universite’ Paris Est, Creteil Val de Marne, France).21, 22 Micrographs were captured by using an Olympus IX71‐inverted microscope (Olympus Europa GmbH) equipped with a CPlan F1 10 ×/0.30 objective. Three different sets of experiments, each performed in duplicate, were carried out. To evaluate the effect of Ca2+ signalling, the same protocol was repeated by priming ECFC with hAFS‐CMHypo in the presence of RN‐1734 (20 μmol/L), a selective blocker of transient receptor potential vanilloid 4 (TRPV4),22, 23 or thymoquinone (25 μmol/L), a specific NF‐κB inhibitor.12
2.3.5. Chemicals
Fura‐2/AM was obtained from Molecular Probes (Molecular Probes Europe BV). YM‐58483/BTP‐2 (BTP‐2; 4‐methyl‐4'‐[3,5‐bis(trifluoromethyl)‐1H‐pyrazol‐1‐yl]‐1,2,3‐thiadiazole‐5‐carboxanilide) was purchased from Tocris Bioscience. Glycyl‐l‐phenylalanine 2‐naphthylamide (GPN) was obtained from Santa Cruz Biotechnology. All the chemicals were of analytical grade and obtained from Sigma Chemical Co.
2.3.6. Statistics
All the data have been collected from ECFCs deriving from at least three distinct donors. Pooled data are given as mean ± SE and statistical significance (P < .05) was evaluated by Student's t test or one‐way ANOVA followed by the post hoc Dunnett's test as appropriate. Data relative to Ca2+ signals are presented as mean ± SE, while the number of cells analysed is indicated in the corresponding bar histograms.
3. RESULTS
3.1. Extracellular Ca2+ entry triggers hAFS‐CMHypo‐induced intracellular Ca2+ oscillations in circulating ECFCs
Human amniotic fluid‐derived stem cells medium conditioned under hypoxia (hAFS‐CMHypo) (Figure 1A), but not under normoxia (hAFS‐CMNormo) (Figure 1B), immediately induced repetitive oscillations in [Ca2+]i in circulating ECFCs loaded with the Ca2+‐sensitive dye Fura‐2/AM (4 µmol/L), thereby confirming the findings recently reported in.8, 9 Furthermore, the percentage of ECFCs displaying a Ca2+ signal was significantly (P < .05) larger when hAFS‐CMHypo was administered (Figure 1C,D). The frequency of the intracellular Ca2+ spikes arising during 1‐hour recording ranged between 5 and 11 oscillations/hour and averaged 8.8 ± 1.1 oscillations/hour (n = 101). The endothelial Ca2+ response to extracellular stimuli impinges on two Ca2+ sources: the extracellular milieu and the endogenous Ca2+ stores located within ER cisternae and EL vesicles.24, 25 hAFS‐CMHypo induced intracellular Ca2+ oscillations in the presence (Figure 2A), but not in the absence (Figure 2B), of extracellular Ca2+. However, the spiking Ca2+ signal promptly resumed upon Ca2+ restitution to the recording solution (Figure 2B). Therefore, extracellular Ca2+ entry was required to trigger hAFS‐CMHypo‐induced intracellular Ca2+ oscillations. SOCE, which can be recruited by a spatially restricted InsP3‐induced ER Ca2+ pulse undetectable by epifluorescence imaging,26, 27 represents the main Ca2+‐entry pathway in circulating ECFCs.28 The pyrazole derivative BTP‐2 has been shown to specifically inhibit SOCE in ECFCs.29, 30 Pre‐treating the cells with BTP‐2 (10 µmol/L, 20 minutes) did not prevent the onset of the Ca2+ response to hAFS‐CMHypo (Figure 2C,D), but significantly (P < .05) reduced the percentage of oscillating cells (Figure 2E) and the amplitude of the 1st Ca2+ spike (Figure 2F). Furthermore, BTP‐2 curtailed the frequency of the Ca2+ transients to 1‐2 oscillations/h (Figure 2G). These data demonstrate that SOCE is required to maintain the oscillations over time but is not responsible for the onset of the Ca2+ response to hAFS‐CMHypo. Therefore, a store‐independent Ca2+‐permeable route initiates the oscillatory signal recorded in the presence of extracellular Ca2+, as previously shown in UCB‐derived ECFCs stimulated with VEGF.13
FIGURE 1.
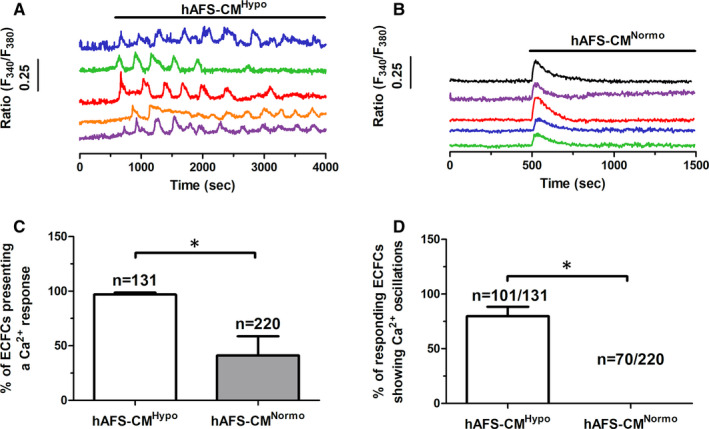
The hypoxic human amniotic fluid‐derived stem cell (hAFS) secretome triggers intracellular Ca2+ oscillations in ECFCs. A, intracellular Ca2+ oscillations induced by hypoxic hAFS secretome (hAFS‐CMHypo) in circulating endothelial colony‐forming cells (ECFCs) from the same coverslip. B, transient intracellular Ca2+ signals evoked by normoxic hAFS secretome (hAFS‐CMNormo) in the same population of circulating ECFCs, plated on a different coverslip. The horizontal bar above the Ca2+ tracings indicates when Hypo (A) and Normo (B) hAFS secretomes were applied. C, mean ± SE of the percentage of cells displaying a Ca2+ response to the different treatments. D, mean ± SE of the percentage of responding cells displaying intracellular Ca2+ oscillations (ie more than one Ca2+ transient) in response to the different treatments. * indicates P < .05 (Student's t test)
FIGURE 2.
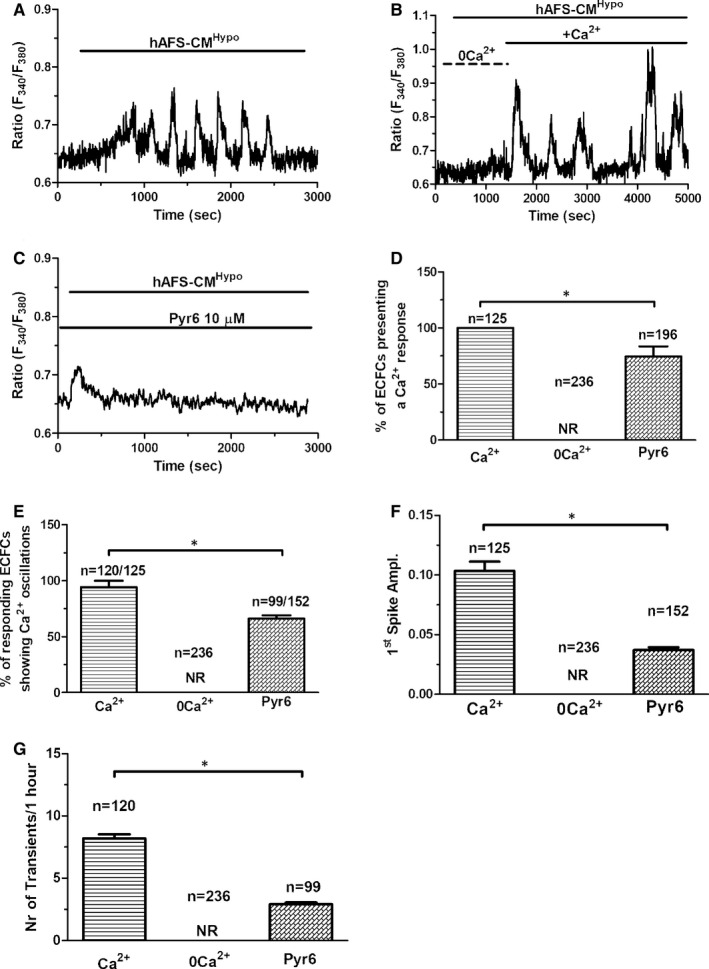
Extracellular Ca2+ entry triggers hypoxic human amniotic fluid‐derived stem cell (hAFS) secretome‐induced intracellular Ca2+ oscillations in circulating endothelial colony‐forming cells (ECFCs). A, intracellular Ca2+ oscillations induced by hypoxic hAFS secretome (hAFS‐CMHypo) in the presence of external Ca2+. The horizontal bar above the Ca2+ tracings indicates the application period of hAFS‐CMHypo. B, extracellular Ca2+ was removed (0 Ca2+) at 200 s, and hAFS‐CMHypo was applied at 300 s from the beginning of the recording. Intracellular Ca2+ oscillations were not recorded under 0Ca2+ conditions. Extracellular Ca2+ was restored at 1500 s, thereby resuming the spiking Ca2+ response. Horizontal bars above the Ca2+ tracings indicate the application period of hAFS‐CMHypo and of physiological salt solution (PSS) supplemented (Ca2+) or not (0 Ca2+) with Ca2+. C, 10‐min pre‐incubation with Pyr6 (10 μmol/L), a selective inhibitor of store‐operated Ca2+ entry (SOCE), curtailed, but did not prevent, the onset of the Ca2+ response to hAFS‐CMHypo. The horizontal bars above the Ca2+ tracings indicate the application period of hAFS‐CMHypo and Pyr6. D, mean ± SE of the percentage of cells displaying a Ca2+ response to hAFS‐CMHypo under the designated treatments. E, mean ± SE of the percentage of cells displaying intracellular Ca2+ oscillations (ie more than one Ca2+ transient) in response to hAFS‐CMHypo under the designated treatments. F, mean ± SE of the amplitude of the 1st Ca2+ spike elicited by hAFS‐CMHypo under the designated treatments. G, mean ± SE of the intracellular Ca2+ transients elicited by hAFS‐CMHypo under the designated treatments. * indicates P < .05 (Student's t test). NR, no response
3.2. hAFS‐CMHypo‐induced intracellular Ca2+ oscillations are triggered by TRP Vanilloid 4 (TRPV4) and shaped by InsP3 receptors (InsP3Rs) and TPC1
Phospholipase C (PLC) plays a crucial role in the onset of the Ca2+ response to chemical stimulation in circulating ECFCs and vascular endothelial cells.28 PLC cleaves the minor membrane phospholipid, phosphatidylinositol 4,5‐bisphosphate (PIP2), to generate InsP3 and DAG. InsP3 induces ER Ca2+ release by priming InsP3Rs to be activated by cytosolic Ca2+,12, 19 whereas DAG may be converted by DAG lipase into arachidonic acid (AA), which in turn activates TRPV4.31 The pharmacological blockade of PLC with the aminosteroid U73122 (10 µmol/L, 20 minutes) abrogated hAFS‐CMHypo‐induced intracellular Ca2+ oscillations (Figure 3A,B). The same inhibitory effect was achieved by Xestospongin C (XeC; 1 µmol/L, 10 minutes) (Figure 3B), a selective blocker of InsP3Rs,32 thereby confirming that InsP3 contributes to shape the spiking Ca2+ signal. The involvement of DAG was assessed by measuring the Ca2+ response to hAFS‐CMHypo in the presence of RHC‐80267 (50 µmol/L, 10 minutes) or RN‐1734 (20 µmol/L, 10 minutes), which, respectively, inhibit DAG lipase33 and TRPV4.23 Both drugs suppressed hAFS‐CMHypo‐induced intracellular Ca2+ oscillations in circulating ECFCs (Figure 3C,D). Similarly, the spiking Ca2+ signal was prevented by pre‐treating the cells with ruthenium red (10 µmol/L, 10 minutes) (Figure 3E), a less specific TRPV4 inhibitor.34 Collectively, these data provide the evidence that, upon PLC engagement, DAG gates TRPV4 to mediate the influx of extracellular Ca2+ that triggers the rhythmical Ca2+‐dependent recruitment of InsP3Rs. To further corroborate this model, hAFS‐CMHypo was administered to circulating ECFCs exposed for 30 minutes to cyclopiazonic acid (CPA; 10 µmol/L), which selectively blocks Sarco‐Endoplasmic Reticulum Ca2+‐ATPase (SERCA) activity, in the presence of extracellular Ca2+ to deplete the ER Ca2+ store and full activate SOCE.13, 31 Therefore, under these conditions, only second messengers‐operated channels may be recruited by extracellular stimuli to increase the [Ca2+]i. When hAFS‐CMHypo was delivered upon 20‐minutes exposure to CPA, it induced a sustained elevation in [Ca2+]i (Figure 2F) that was abrogated in the absence of extracellular Ca2+ and in the presence of RN‐1734 (Figure 2G). Interestingly, addition of the hAFS‐CMHypo in the presence of external Ca2+ caused a transient reduction in [Ca2+]i, which preceded the gradual re‐emergence of extracellular Ca2+ entry and reflects AA‐dependent inhibition of SOCE.31, 35 Taken together, these findings endorse the view that TRPV4‐mediated extracellular Ca2+ entry is required to initiate InsP3‐dependent intracellular Ca2+ oscillations. A recent report showed that NAADP‐induced EL Ca2+ release through TPC1 supports oscillations in [Ca2+]i mediated by InsP3Rs in ECFCs.14 In agreement with these observations, hAFS‐CMHypo failed to trigger cytosolic Ca2+ signals in the presence of the lysosomotropic compound, GPN (200 µmol/L, 30 minutes),36, 37 and of NED‐19 (100 µmol/L, 30 minutes), a selective TPC antagonist.38, 39 The statistical analysis of pharmacological manipulation of hAFS‐CMHypo‐induced intracellular Ca2+ oscillations has been reported in Figure 4.
FIGURE 3.
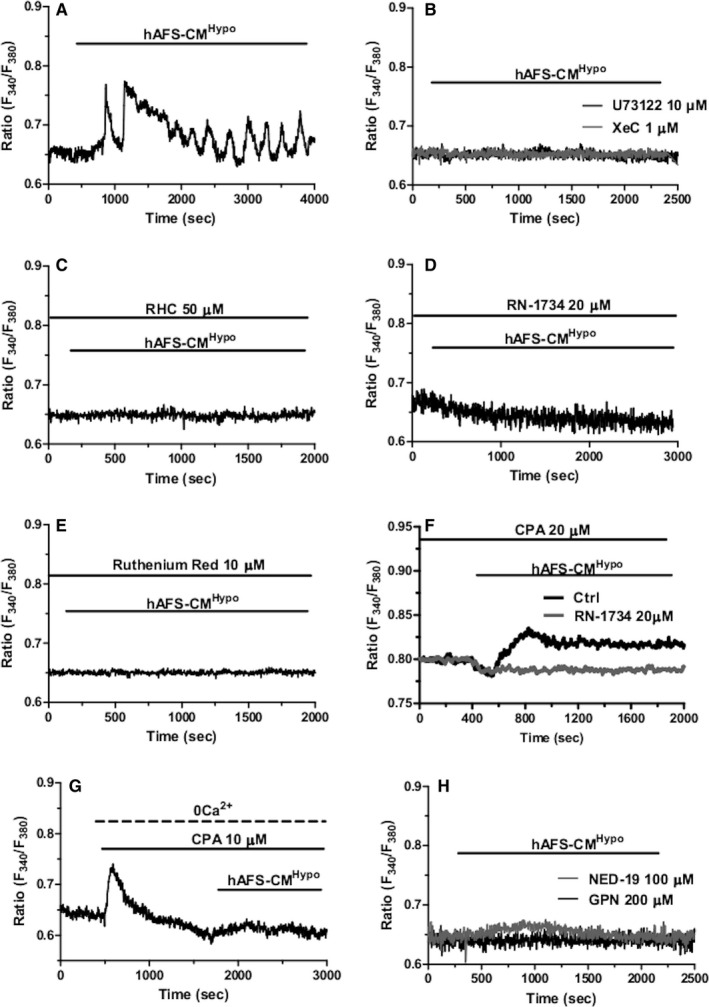
The role of transient receptor potential vanilloid 4 (TRPV4), InsP3 receptors (InsP3Rs) and two‐pore channel 1 (TPC1) in hypoxic human amniotic fluid‐derived stem cell (hAFS) (hAFS) secretome‐induced intracellular Ca2+ oscillations in circulating endothelial colony‐forming cells (ECFCs). A, intracellular Ca2+ oscillations induced by the hypoxic hAFS secretome (hAFS‐CMHypo) in circulating ECFCs under control conditions, that is in the presence of extracellular Ca2+ and in the absence of inhibitors of the Ca2+ signalosome. The horizontal bar above the Ca2+ tracings indicates the application period of hAFS‐CMHypo. B, 30‐min pre‐incubation with U73122 (10 μmol/L), an antagonist of phospholipase C (PLC), and 10‐min pre‐incubation with Xestospongin C (XeC; 1 μmol/L, 10 min), a blocker of InsP3Rs, prevented the oscillatory response to hAFS secretome. The horizontal bar above the Ca2+ tracings indicates the application period of hAFS‐CMHypo. C, 10‐min pre‐incubation with RHC‐80267 (RHC; 50 μmol/L), which selectively interferes with diacylglycerol (DAG) lipase activity, suppressed hAFS‐CMHypo‐induced intracellular Ca2+ oscillations in circulating ECFCs. The horizontal bars above the Ca2+ tracings indicate the application period of hAFS‐CMHypo and RHC. D, 30‐min pre‐incubation with RN‐1734 (20 μmol/L), a selective TRPV4 blocker, prevented the oscillatory Ca2+ response to hAFS‐CMHypo in circulating ECFCs. The horizontal bars above the Ca2+ tracings indicate the application period of hAFS‐CMHypo and RN‐1734. E, 10‐min pre‐incubation with ruthenium red (10 μmol/L), a pan‐specific inhibitor of TRPV channels, also suppressed hAFS‐CMHypo‐induced intracellular Ca2+ oscillations in circulating ECFCs. The horizontal bars above the Ca2+ tracings indicate the application period of hAFS‐CMHypo and ruthenium red. F, ECFCs were pretreated for 30 min with cyclopiazonic acid (CPA; 10 μmol/L) to fully deplete the endoplasmic reticulum (ER) Ca2+ reservoir and activate store‐operated Ca2+ entry (SOCE). Thereafter, hAFS‐CMHypo was added and caused a transient reduction in intracellular Ca2+ levels followed by a sustained increase in [Ca2+]i. 30‐min pre‐incubation with RN‐1734 (20 μmol/L) to block TRPV4 prevented this [Ca2+]i rise and unmasked the progressive decrease in Fura‐2 fluorescence, which reflects AA‐dependent SOCE inhibition (please, see the text for a wider explanation). The horizontal bars above the Ca2+ tracings indicate the application period of hAFS‐CMHypo and CPA. G, 30‐min pre‐incubation with NED‐19 (100 µmol/L), a selective two‐pore channel (TPC) blocker, and 30‐min pre‐incubation with the lysosomotropic agent, glycyl‐l‐phenylalanine 2‐naphthylamide (GPN; 200 µmol/L), inhibited the oscillatory Ca2+ response to hAFS‐CMHypo. The horizontal bar above the Ca2+ tracings indicates the application period of hAFS‐CMHypo
FIGURE 4.
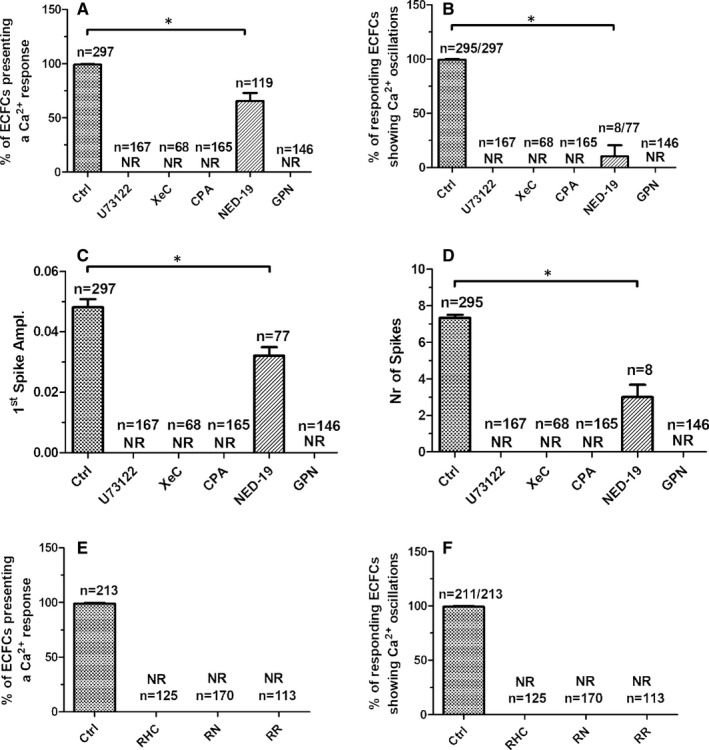
Statistical analysis of phospholipase C (PLC) and nicotinic acid adenine dinucleotide phosphate (NAADP) signalling. Mean ± SE of the percentage of endothelial colony‐forming cells (ECFCs) presenting a Ca2+ response (A) and, among these, of ECFCs presenting intracellular Ca2+ oscillations (ie more than one Ca2+ transient) (B) upon exposure to the hypoxic human amniotic fluid‐derived stem cell (hAFS) secretome (hAFS‐CMHypo) under the designated treatments. Mean ± SE of the amplitude of the 1st Ca2+ spike (C) and of the intracellular Ca2+ transients (D) elicited by hAFS‐CMHypo under the designated treatments. Mean ± SE of the percentage of ECFCs presenting a Ca2+ response (E) and, among these, of ECFCs presenting intracellular Ca2+ oscillations (ie more than one Ca2+ transient) (F) when exposed to hAFS‐CMHypo in the absence (Ctrl) and presence of blockers of transient receptor potential vanilloid 4 (TRPV4) signalling. * indicates P < .05 (Student's t test). NR, no response
3.3. hAFS‐CMHypo induces the nuclear translocation of NF‐κB in a Ca2+‐dependent manner
The transcription factor NF‐κB has long been known to translate intracellular Ca2+ signals in a pro‐angiogenic output in ECFCs.12, 22, 40 In quiescent cells, the p65 NF‐κB subunit is retained in the cytoplasm by the physical association with the inhibitory IκB protein, but is released from inhibition and primed to translocate into the nucleus by an oscillatory increase in [Ca2+]i.15, 40 Immunofluorescence revealed that p65 NF‐κB displayed a cytosolic distribution in non‐stimulated ECFCs (Ctrl; Figure 5A,B), whereas it was mainly accumulated in the nucleus upon exposure to hAFS‐CMHypo (Figure 5A,B). Conversely, pre‐treating the cells with RN‐1734 (20 µmol/L, 10 minutes), which suppresses the Ca2+ spikes, or BAPTA (30 µmol/L, 2 hours), a membrane‐permeable Ca2+ buffer,9, 12 significantly (P < .05) inhibited hAFS‐CMHypo‐induced nuclear translocation of p65 NF‐κB (Figure 5A,B). These data, therefore, demonstrate that intracellular Ca2+ oscillations drive hAFS‐CMHypo‐induced p65 NF‐κB translocation into the nucleus in circulating ECFCs.
FIGURE 5.
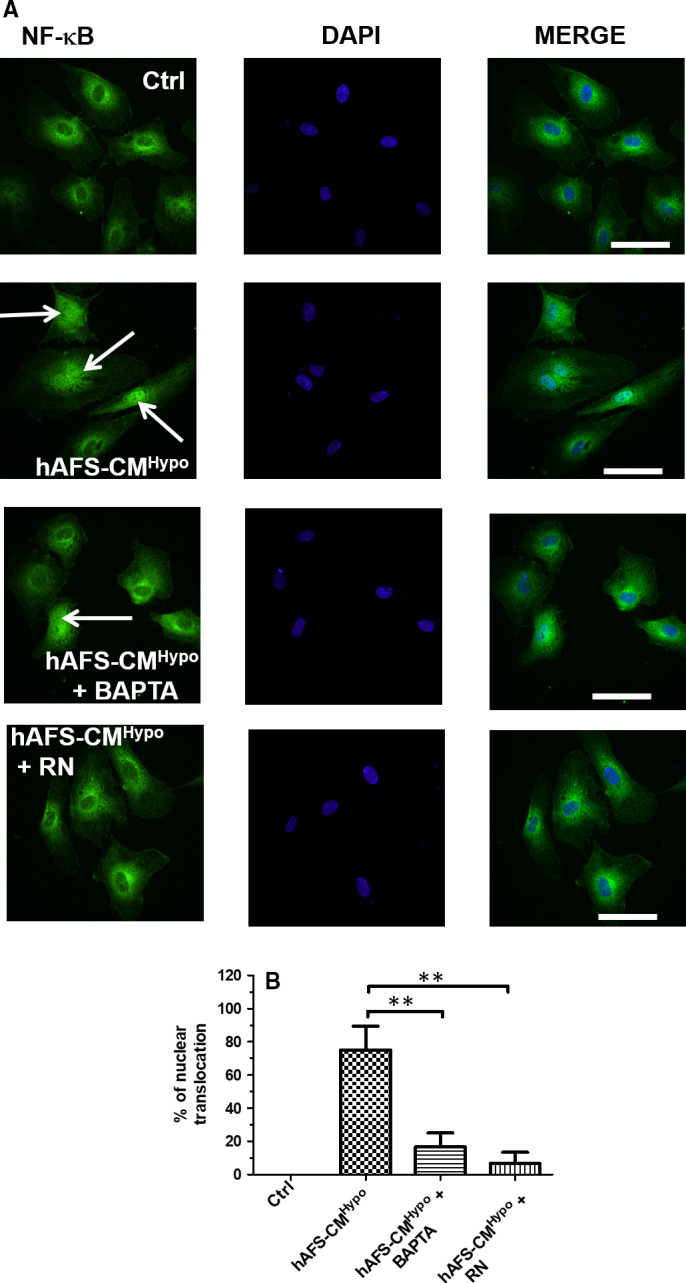
Hypoxic human amniotic fluid‐derived stem cell (hAFS) secretome induces Ca2+‐dependent p65 NF‐κB nuclear translocation in circulating endothelial colony‐forming cells (ECFCs). A, immunofluorescence analysis of p65 NF‐κB nuclear translocation in ECFCs untreated (Ctrl) and treated with hypoxic hAFS secretome (hAFS‐CMHypo) for 2 h under control conditions and in the presence of BAPTA (30 µmol/L, 2‐h pre‐incubation before stimulation with hAFS‐CMHypo) or RN‐1734 (RN; 20 μmol/L, 30‐min pre‐incubation before stimulation with hAFS‐CMHypo). The first column shows the fluorescent p65 signal, the second column the nuclei coloured by DAPI (2‐[4‐(Aminoiminomethyl)phenyl]‐1H‐Indole‐6‐carboximidamide hydrochloride), and the third one the merge. B, quantification of nuclear staining of p65 NF‐κB from three coverslips from three independent experiments. Data were shown as mean ± SE. ** indicates P < .01 (one‐way ANOVA followed by the post hoc Dunnett's test)
3.4. NF‐κB drives hAFS‐CMHypo‐induced ECFC tubulogenesis
A recent report from our group demonstrated that hAFS‐CM collected under hypoxic conditions specifically induced ECFC tubulogenesis in vitro.8, 9 Unlike other in vitro assays, for example migration and invasion, the Matrigel‐based tube formation assay involves all the main physiological steps of the angiogenic process, including endothelial cell proliferation, adhesion, migration and differentiation.41 We, therefore, evaluated both topologic (number of meshes and junctions per picture) and dimensional (total number of tubules per picture) of the capillary‐like networks formed by circulating ECFCs placed in a Matrigel scaffold, as shown elsewhere.19, 21 Preventing the oscillatory increase in [Ca2+]i with BAPTA has previously been shown to interfere with hAFS‐CMHypo‐induced ECFC assembly in a bidimensional tubular network.8, 9 Likewise, ECFCs did not originate capillary tube‐like structures when stimulated with hAFS‐CMHypo in the presence of RN‐1734 (20 µmol/L, 10 minutes), which prevents the onset of the intracellular Ca2+ oscillations, and of thymoquinone (25 µmol/L, 10 minutes) (Figure 6), a selective NF‐κB blocker.12, 42 Collectively, these data show that hAFS‐CMHypo requires TRPV4‐mediated extracellular Ca2+ entry to trigger the nuclear translocation of p65 NF‐κB and promote ECFC tubulogenesis.
FIGURE 6.
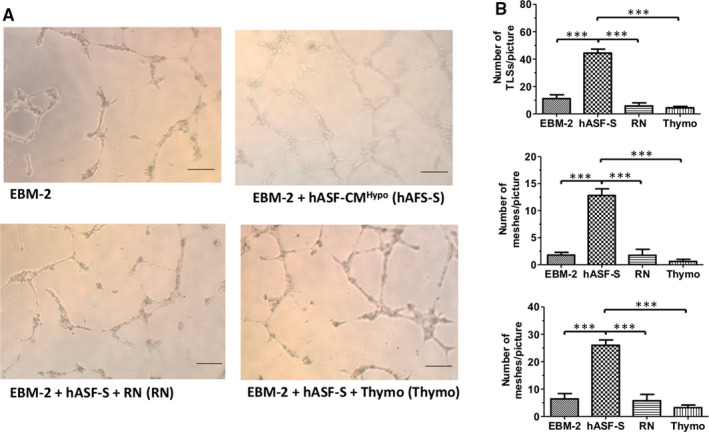
Hypoxic human amniotic fluid‐derived stem cell (hAFS) secretome induces endothelial colony‐forming cell (ECFC) tubulogenesis in Ca2+‐ and NF‐κB‐dependent manner. Tubulogenesis assay on ECFCs plated in the presence of EBM‐2 supplemented with 2% foetal bovine serum (FBS) and treated with or without 80 µg/mL of the secretome obtained from hAFS after hypoxic conditioning (hAFS‐CMHypo) supplemented or not with RN‐1734 (RN; 20 μmol/L, 30‐min pre‐incubation before stimulation) or thymoquinone (25 µmol/L, 10‐min pre‐incubation before stimulation). Digital images of endothelial tubes were obtained by bright‐field light microscopy 10 h after plating cells on Matrigel‐coated wells; scale bar: 50 µm. B, mean ± SE of the following parameters evaluated from digital images: number of master tubules (TLSs)/picture (Ba), number of meshes/picture (Bb), number of master junctions/picture (Bc). *** indicate P < .0001 (one‐way ANOVA followed by the post hoc Dunnett's test). hAFS‐CMHypo, RN and thymoquinone were maintained during the tubulogenic assay
4. DISCUSSION
Paracrine therapy through stem cells‐secreted mediators is emerging as an alternative, promising strategy to treat AMI by instructing resident cells, for example cardiac stromal cells, cardiomyocytes and endothelial cells, to optimize endogenous mechanism of cardiac repair.10, 43 Local delivery of different cell‐conditioned media halted maladaptive remodelling and improved cardiac performance in murine models of AMI. Paracrine signalling was shown to act by restoring cell‐cycle activity or by inhibiting apoptosis/senescence in adult cardiomyocytes and by stimulating angiogenesis in coronary microvascular endothelial cells.20, 44, 45 A recent study by Balbi et al provided a paracrine therapy proof‐of‐principle by demonstrating that single administration via intramyocardial injection of the hAFS secretome in the form of the cell‐conditioned medium reduced infarct area, increased left ventricular ejection fraction, stimulated local angiogenesis and supported cell‐cycle re‐entry of resident surviving cardiomyocytes.8, 9 In vitro analysis further showed that the hAFS‐CMHypo stimulated circulating human ECFCs to form capillary‐like networks through an oscillatory increase in [Ca2+]i.9 However, the mechanisms whereby paracrine signalling induces repetitive Ca2+ spikes were not dissected. Understanding how the hAFS‐CMHypo elicits intracellular Ca2+ oscillations in ECFCs will provide the biological bases required to decipher the components of the Ca2+ toolkit that could be specifically targeted to induce vascular regrowth in infarcted hearts.
4.1. hAFS‐CMHypo induces intracellular Ca2+ oscillations by activating TRPV4
Human amniotic fluid‐derived stem cells ‐conditioned medium obtained from human amniotic fluid stem cells maintained under hypoxic over control normoxic conditions induced intracellular Ca2+ oscillations in circulating ECFCs, thereby confirming previous results from our group.9 Paracrine medium secreted by bone marrow‐derived mesenchymal stem cells cultured under hypoxia was previously shown to dampen mitochondrial Ca2+ overload during ischaemia/reperfusion injury.46 Thus, paracrine mediators released from stem cells could vary depending on their source and/or their microenvironment and could activate different steps of the complex process of cardiac repair.43 The intracellular Ca2+ oscillations induced by hAFS‐CMHypo in circulating ECFCs strongly resembled those induced by VEGF.12, 19, 21 However, hAFS‐CMHypo does not contain VEGF, whereas it is enriched with multiple cytokines and chemokines, including interleukin 8 (IL‐8), angiogenin, Extracellular matrix metalloproteinase inducer and monocyte chemoattractant protein‐1 (MCP‐1).47 Interestingly, an increase in [Ca2+]i can be evoked by IL‐8 in mouse lymphokine‐activated killer (LAK) cells48 and by MCP‐1 in human monocytes,49 although these Ca2+ signals do not adopt an oscillatory pattern.
Extracellular stimuli cause an increase in endothelial [Ca2+]i, as well as in circulating ECFCs,28 by promoting extracellular Ca2+ entry and/or endogenous Ca2+ release.24, 25, 50, 51 Removal of extracellular Ca2+ prevented the onset of hAFS‐CMHypo‐induced intracellular Ca2+ oscillations, which promptly resumed upon restitution of external Ca2+. This finding further supports the notion that VEGF is not involved in the spiking Ca2+ signal. Indeed, VEGF is still able to trigger 1‐4 intracellular Ca2+ spikes in the absence of extracellular Ca2+ entry in circulating ECFCs.12 Nonetheless, early reports demonstrated that extracellular Ca2+ entry through two distinct DAG‐sensitive pathways, that is TRPC1 and TRPC3, induced intracellular Ca2+ oscillations, respectively, in primary myelofibrosis‐derived ECFCs21 and UCB‐derived ECFCs.13 The circulating ECFCs employed in the present investigation do not express TRPC3.13 Furthermore, in these cells, TRPC1 is not sensitive to DAG,21 but is part of a super‐molecular complex including also Orai1 and STIM1, which is assembled upon depletion of the ER Ca2+ store.28, 52 However, DAG may be converted by DAG lipase in AA,53 which selectively gates extracellular Ca2+ entry through TRPV4 in circulating ECFCs31, 54 and vascular endothelial cells.23, 25 The following pieces of evidence demonstrate that TRPV4‐mediated extracellular Ca2+ entry triggers hAFS‐CMHypo‐induced intracellular Ca2+ oscillations in circulating ECFCs. First, the pharmacological blockade of TRPV4 with two structurally distinct inhibitors, RN‐1734 and ruthenium red, suppressed the onset of the oscillatory signal. RN‐1734 is a selective TRPV4 antagonist,34 and it does not inhibit the other TRPV isoform expressed in circulating ECFCs, that is TRPV1.22, 54 Second, U73122 and RHC‐80267, which, respectively, inhibit PLC activity12 and DAG lipase,53 also prevented the oscillatory Ca2+ response to hAFS‐CMHypo. Interestingly, the intracellular Ca2+ signals evoked by MCP‐1, which is quite abundant in hAFS‐CM,8 also require DAG metabolism by DAG lipase in human monocytes.49 Third, the pharmacological blockade of Orai1, which contributes to SOCE,28, 52 curtailed, but did not abrogate, the number of Ca2+ transients evoked by hAFS‐CMHypo. Therefore, while SOCE is required to maintain the intracellular Ca2+ oscillations by reloading the ER with Ca2+ in a SERCA‐dependent manner,12, 15 it does not ignite the oscillatory Ca2+ response.
4.2. InsP3Rs and TPC1 mediate intracellular Ca2+ release during hAFS‐CMHypo‐induced intracellular Ca2+ oscillations
InsP3Rs provide the main ER Ca2+‐releasing pathway that is periodically activated to support intracellular Ca2+ oscillations.15, 55 InsP3 is synthesized in response to extracellular stimuli recruiting PLC and sensitizes InsP3Rs towards feedback activation by cytosolic Ca2+, thereby producing brief Ca2+ transients.13, 52 ECFCs express all the three known InsP3R sub‐types, that is InsP3R1‐3.12 InsP3R1 and InsP3R2 are especially suitable to support intracellular Ca2+ oscillations as they have shown a “bell‐shaped” dependence on surrounding Ca2+ 28, 55: a relatively small increase in local Ca2+ concentration activates the InsP3‐primed receptors, while the subsequent increase in cytosolic Ca2+ (>1 µmol/L) inhibits further Ca2+ release. That InsP3Rs are required to support hAFS‐CMHypo‐induced intracellular Ca2+ oscillations is indicated by the inhibitory effect of U73122 and XeC, which, respectively, target PLC and InsP3Rs. Furthermore, depletion of the ER Ca2+ store with CPA transformed the repetitive Ca2+ transients into a sustained elevation in [Ca2+]i, which was caused by TRPV4 activation. Therefore, we propose that TRPV4‐mediated extracellular Ca2+ entry provides the source of Ca2+ that is required to induce the Ca2+‐dependent recruitment of InsP3Rs by hAFS‐CMHypo. The subsequent InsP3‐induced depletion of the ER Ca2+ store can, in turn, engage STIM1, a sensor of ER Ca2+ concentration, to bind to and gate Orai1 and TRPC1 to mediate SOCE.28 NAADP‐induced EL Ca2+ release through TPCs may cooperate with InsP3Rs to trigger endothelial Ca2+ waves.56, 57 For instance, TPC1 activation is required to trigger VEGF‐induced repetitive Ca2+ spikes in circulating ECFCs.14 Herein, we provided the evidence that interfering with EL Ca2+ release with either GPN or NED‐19, which, respectively, disrupt the EL Ca2+ store and inhibit TPC1, also impairs hAFS‐CMHypo‐induced intracellular Ca2+ oscillations. Therefore, NAADP‐induced Ca2+ release is likely to contribute with TRPV4 to ignite periodical InsP3‐induced ER Ca2+ release under these circumstances. Interestingly, IL‐8, which is also enriched in hAFS‐CM,8 was recently found to stimulate endosomal CD38 to produce NAADP in LAK cells.48
4.3. hAFS‐CMHypo promotes in vitro tubulogenesis by inducing the nuclear translocation of NF‐κB
Endothelial Ca2+ oscillations support in vitro tubulogenesis and neovessel formation in vivo.24 Moreover, we recently demonstrated that preventing the oscillatory increase in [Ca2+]i with BAPTA‐AM impaired hAFS‐CMHypo‐induced ECFC tubulogenesis in Matrigel scaffolds. The Ca2+‐sensitive transcription factor, NF‐κB, may translate intracellular Ca2+ signals into a pro‐angiogenic input in circulating ECFCs. For instance, NF‐κB decodes VEGF‐induced intracellular Ca2+ oscillations in ECFCs12 and is activated by TRPV1‐mediated extracellular Ca2+ entry to induce proliferation and tube formation upon optical excitation of the photosensitive conjugated polymer, P3HT.22, 50 Furthermore, hAFS‐CMHypo promotes the nuclear translocation of NF‐κB to counteract doxorubicin‐induced cardiotoxicity in murine neonatal ventricular myocytes.20 In agreement with these observations, immunofluorescence revealed that nuclear accumulation of the p65 subunit of the NF‐κB complex was impaired by suppressing intracellular Ca2+ oscillations with either RN‐1734 (to block the triggering TRPV4‐mediated Ca2+ signal) or BAPTA‐AM (to buffer intracellular Ca2+ levels). Of note, repetitive increases in [Ca2+]i represent the most suitable Ca2+ waveform to recruit NF‐κB.15, 58 Furthermore, the frequency of hAFS‐CMHypo‐induced intracellular Ca2+ oscillations, that is ~9 oscillations/hour or 2.5 mHz, is within the same range as that required to efficiently activate NF‐κB, that is 0.56‐10 mHz.58 Of note, preventing the intracellular Ca2+ oscillations with RN‐1734 and the downstream recruitment of NF‐κB with thymoquinone potently inhibited hAFS‐CMHypo‐induced ECFC tube formation. This finding is strongly supported by the evidence that multiple pro‐angiogenic genes, for example intercellular adhesion molecule 1, selectin E and various matrix metalloproteinases, are expressed upon the Ca2+‐dependent activation of NF‐κB in circulating ECFCs.22, 59 Furthermore, NF‐κB regulates the expression of a large array of pro‐angiogenic genes, for example those encoding for growth factors (eg VEGF), chemokines and cell adhesion molecules.59 Thus, the Ca2+‐dependent engagement of NF‐κB is likely to play a crucial role in hAFS‐CMHypo‐induced revascularization in murine models of AMI8 and hindlimb ischaemia.11 The involvement of other pro‐angiogenic signalling pathways, such as phosphoinositide 3‐kinase/Akt, which can also be activated by hAFS‐CMHypo 20 and is sensitive to Ca2+ in ECFCs,18 cannot be ruled out.
5. CONCLUSIONS
The present investigation reveals for the first time the signalling pathways whereby hAFS‐CMHypo induces pro‐angiogenic Ca2+ oscillations in circulating ECFCs, which represent the most suitable cellular substrate to achieve therapeutic angiogenesis in ischaemic disorders. hAFS‐CMHypo promotes TRPV4‐mediated extracellular Ca2+ entry, which thereby results in InsP3‐dependent periodical ER Ca2+ release accompanied by SOCE activation (Figure 7). Intracellular Ca2+ transients are also supported by NAADP‐induced EL Ca2+ release through TPC1, which favours the Ca2+‐dependent recruitment of InsP3Rs (Figure 7). These findings endorse the emerging view that TRPV4, InsP3Rs, TPC1 and SOCE may be targeted through genetic or pharmacological manipulation to enhance the therapeutic outcome of ECFCs‐based therapy.2, 60 For instance, autologous ECFCs could be genetically manipulated to overexpress TRPV4, thereby boosting their vasoreparative potential in ischaemic disorders. Alternately, specific TRPV4 agonists, such as GSK1016790A,23 could be injected into the infarcted myocardium, to boost proliferation and tube formation in ECFCs recruited towards the damaged tissue by the ischaemic insult. Furthermore, they extend at molecular level our knowledge of the mechanisms whereby paracrine therapy through hAFS secretome formulations may induce significant vascular regrowth in widespread ischaemic disorders, such as AMI and hindlimb ischaemia. In the light of such evidence, profiling the components of the hAFS‐CMHypo that induce these pro‐angiogenic Ca2+ oscillations could lead to the formulation of a more efficient cocktail of bioactive factors, which bear the potential to be locally delivered to the ischaemic heart, thereby replacing the time‐consuming and costly cell‐based therapy. This task is currently under way in our laboratories. Due to their ability to induce intracellular Ca2+ signals in other cell types, we are assessing whether IL‐848 and MCP‐149 elicit intracellular Ca2+ oscillations in ECFCs either alone or in combination. If effective at inducing pro‐angiogenic Ca2+ signals, these mediators could be directly injected into the infarcted myocardium to boost ECFCs’ vasoreparative activity.
FIGURE 7.
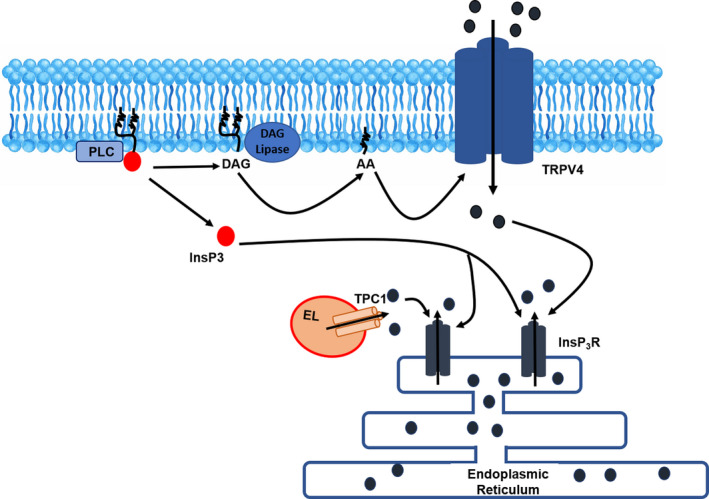
The mechanisms leading to the onset of hypoxic human amniotic fluid‐derived stem cell (hAFS) secretome‐induced intracellular Ca2+ oscillations in endothelial colony‐forming cells (ECFCs). Exposure of circulating ECFCs to hypoxic hAFS secretome (hAFS‐CMHypo) results in phospholipase C (PLC) engagement, followed by production of diacylglycerol (DAG) and inositol‐1,4,5‐trisphosphate (InsP3). DAG is converted by DAG lipase into arachidonic acid (AA), which gates transient receptor potential vanilloid 4 (TRPV4) to mediate extracellular Ca2+ entry through the plasma membrane. InsP3 primes ER‐embedded to be activated by the incoming Ca2+. Nicotinic acid adenine dinucleotide phosphate (NAADP)‐induced endolysosomal (EL) Ca2+ release mediated by two‐pore channel 1 (TPC1) is also likely to contribute to the Ca2+‐dependent recruitment of InsP3 receptors (InsP3Rs). Endoplasmic reticulum (ER) Ca2+ depletion, in turn, leads to store‐operated Ca2+ entry (SOCE) activation and maintenance of intracellular Ca2+ oscillations over time (not shown)
CONFLICT OF INTEREST
The authors confirm that there are no conflicts of interest.
AUTHOR CONTRIBUTIONS
Valentina Balducci: Formal analysis (lead); Investigation (lead); Methodology (equal); Writing‐review & editing (supporting). Pawan Faris: Formal analysis (supporting). Carolina Balbi: Investigation (equal). Ambra Costa: Investigation (equal). Sharon Negri: Formal analysis (equal). Vittorio Rosti: Conceptualization (equal); Investigation (equal); Validation (equal); Writing‐review & editing (equal). Sveva Bollini: Conceptualization (equal); Funding acquisition (equal); Investigation (equal); Supervision (equal); Validation (equal); Writing‐review & editing (equal). Francesco Moccia: Conceptualization (lead); Funding acquisition (lead); Project administration (lead); Supervision (lead); Validation (lead); Writing‐original draft (lead).
Supporting information
Supplementary Information
ACKNOWLEDGEMENTS
The authors would like to acknowledge Dr Pierangela De Biasio, from IRCCS San Martino Hospital and Dr Dario Paladini and Dr Domenico Coviello from IRCCS Istituto Gaslini in Genova, Italy, for their assistance in providing the leftover samples of human amniotic fluid.
This research was funded by the following: Italian Ministry of Education, University and Research (MIUR): Dipartimenti di Eccellenza Program (2018–2022)—Dept. of Biology and Biotechnology "L. Spallanzani," University of Pavia (FM); Fondo Ricerca Giovani from the University of Pavia (FM); Programma Giovani Ricercatori “Rita Levi Montalcini” 2012 from MIUR—Italian Ministry of Education and Research; and University of Genova, Genova, Italy, “Curiosity Driven” grant (SB). This study contributes to the aims of the Horizon 2020 COST Action CA17116 SPRINT—International Network for Translating Research on Perinatal Derivatives into Therapeutic Approaches (SB and CB).
Balducci V, Faris P, Balbi C, et al. The human amniotic fluid stem cell secretome triggers intracellular Ca2+ oscillations, NF‐κB nuclear translocation and tube formation in human endothelial colony‐forming cells. J Cell Mol Med. 2021;25:8074–8086. 10.1111/jcmm.16739
DATA AVAILABILITY STATEMENT
All the data are fully available upon reasonable request.
REFERENCES
- 1.Medina RJ, Barber CL, Sabatier F, et al. Endothelial progenitors: a consensus statement on nomenclature. Stem Cells Transl Med. 2017;6(5):1316‐1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Faris P, Negri S, Perna A, Rosti V, Guerra G, Moccia F. Therapeutic potential of endothelial colony‐forming cells in ischemic disease: strategies to improve their regenerative efficacy. Int J Mol Sci. 2020;21(19):7406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.O'Neill CL, McLoughlin KJ, Chambers SEJ, Guduric‐Fuchs J, Stitt AW, Medina RJ. The vasoreparative potential of endothelial colony forming cells: a journey through pre‐clinical studies. Front Med. 2018;5:273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Paschalaki KE, Randi AM. Recent advances in endothelial colony forming cells toward their use in clinical translation. Front Med. 2018;5:295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Tasev D, Koolwijk P, van Hinsbergh VW . Therapeutic potential of human‐derived endothelial colony‐forming cells in animal models. Tissue Eng Part B Rev. 2016;22(5):371‐382. [DOI] [PubMed] [Google Scholar]
- 6.Mellows B, Mitchell R, Antonioli M, et al. Protein and molecular characterization of a clinically compliant amniotic fluid stem cell‐derived extracellular vesicle fraction capable of accelerating muscle regeneration through enhancement of angiogenesis. Stem Cells Dev. 2017;26(18):1316‐1333. [DOI] [PubMed] [Google Scholar]
- 7.Mirabella T, Hartinger J, Lorandi C, Gentili C, van Griensven M , Cancedda R. Proangiogenic soluble factors from amniotic fluid stem cells mediate the recruitment of endothelial progenitors in a model of ischemic fasciocutaneous flap. Stem Cells Dev. 2012;21(12):2179‐2188. [DOI] [PubMed] [Google Scholar]
- 8.Balbi C, Lodder K, Costa A, et al. Reactivating endogenous mechanisms of cardiac regeneration via paracrine boosting using the human amniotic fluid stem cell secretome. Int J Cardiol. 2019;287:87‐95. [DOI] [PubMed] [Google Scholar]
- 9.Balbi C, Lodder K, Costa A, et al. Supporting data on in vitro cardioprotective and proliferative paracrine effects by the human amniotic fluid stem cell secretome. Data Brief. 2019;25:104324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Maghin E, Garbati P, Quarto R, Piccoli M, Bollini S. Young at heart: combining strategies to rejuvenate endogenous mechanisms of cardiac repair. Front Bioeng Biotechnol. 2020;8:447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Balbi C, Piccoli M, Barile L, et al. First characterization of human amniotic fluid stem cell extracellular vesicles as a powerful paracrine tool endowed with regenerative potential. Stem Cells Transl Med. 2017;6(5):1340‐1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Dragoni S, Laforenza U, Bonetti E, et al. Vascular endothelial growth factor stimulates endothelial colony forming cells proliferation and tubulogenesis by inducing oscillations in intracellular Ca2+ concentration. Stem Cells. 2011;29(11):1898‐1907. [DOI] [PubMed] [Google Scholar]
- 13.Dragoni S, Laforenza U, Bonetti E, et al. Canonical transient receptor potential 3 channel triggers vascular endothelial growth factor‐induced intracellular Ca2+ oscillations in endothelial progenitor cells isolated from umbilical cord blood. Stem Cells Dev. 2013;22(19):2561‐2580. [DOI] [PubMed] [Google Scholar]
- 14.Moccia F, Zuccolo E, Di Nezza F, et al. Nicotinic acid adenine dinucleotide phosphate activates two‐pore channel TPC1 to mediate lysosomal Ca(2+) release in endothelial colony‐forming cells. J Cell Physiol. 2020;236(1):688‐705. [DOI] [PubMed] [Google Scholar]
- 15.Parekh AB. Decoding cytosolic Ca2+ oscillations [Review]. Trends Biochem Sci. 2011;36(2):78‐87. [DOI] [PubMed] [Google Scholar]
- 16.Di Capite J, Ng SW, Parekh AB. Decoding of cytoplasmic Ca(2+) oscillations through the spatial signature drives gene expression. Curr Biol. 2009;19(10):853‐858. [DOI] [PubMed] [Google Scholar]
- 17.Kar P, Parekh AB. Distinct spatial Ca2+ signatures selectively activate different NFAT transcription factor isoforms. Mol Cell. 2015;58(2):232‐243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Zuccolo E, Di Buduo C, Lodola F, et al. Stromal cell‐derived factor‐1alpha promotes endothelial colony‐forming cell migration through the Ca(2+)‐dependent activation of the extracellular signal‐regulated kinase 1/2 and phosphoinositide 3‐Kinase/AKT pathways. Stem Cells Dev. 2018;27(1):23‐34. [DOI] [PubMed] [Google Scholar]
- 19.Lodola F, Laforenza U, Cattaneo F, et al. VEGF‐induced intracellular Ca2+ oscillations are down‐regulated and do not stimulate angiogenesis in breast cancer‐derived endothelial colony forming cells. Oncotarget. 2017;8:95223‐95246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazzarini E, Balbi C, Altieri P, et al. The human amniotic fluid stem cell secretome effectively counteracts doxorubicin‐induced cardiotoxicity. Sci Rep. 2016;6:29994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dragoni S, Reforgiato M, Zuccolo E, et al. Dysregulation of VEGF‐induced proangiogenic Ca2+ oscillations in primary myelofibrosis‐derived endothelial colony‐forming cells. Exp Hematol. 2015;43(12):1019‐1030.e3. [DOI] [PubMed] [Google Scholar]
- 22.Lodola F, Rosti V, Tullii G, et al. Conjugated polymers optically regulate the fate of endothelial colony‐forming cells. Sci Advan. 2019;5(9):eaav4620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Berra‐Romani R, Faris P, Negri S, Botta L, Genova T, Moccia F. Arachidonic acid evokes an increase in intracellular Ca(2+) concentration and nitric oxide production in endothelial cells from human brain microcirculation. Cells. 2019;8(7):689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Moccia F, Negri S, Shekha M, Faris P, Guerra G. Endothelial Ca(2+) signaling, angiogenesis and vasculogenesis: just what it takes to make a blood vessel. Int J Mol Sci. 2019;20(16):3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Negri S, Faris P, Berra‐Romani R, Guerra G, Moccia F. Endothelial transient receptor potential channels and vascular remodeling: extracellular Ca(2 +) entry for angiogenesis, arteriogenesis and vasculogenesis. Front Physiol. 2019;10:1618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zuccolo E, Laforenza U, Ferulli F, et al. Stim and Orai mediate constitutive Ca(2+) entry and control endoplasmic reticulum Ca(2+) refilling in primary cultures of colorectal carcinoma cells. Oncotarget. 2018;9(57):31098‐31119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chang WC, Di Capite J, Nelson C, Parekh AB. All‐or‐none activation of CRAC channels by agonist elicits graded responses in populations of mast cells. J Immunol. 2007;179(8):5255‐5263. [DOI] [PubMed] [Google Scholar]
- 28.Moccia F, Guerra G. Ca(2+) signalling in endothelial progenitor cells: friend or foe? J Cell Physiol. 2016;231(2):314‐327. [DOI] [PubMed] [Google Scholar]
- 29.Sanchez‐Hernandez Y, Laforenza U, Bonetti E, et al. Store‐operated Ca(2+) entry is expressed in human endothelial progenitor cells. Stem Cells Dev. 2010;19(12):1967‐1981. [DOI] [PubMed] [Google Scholar]
- 30.Moccia F, Dragoni S, Lodola F, et al. Store‐dependent Ca(2+) entry in endothelial progenitor cells as a perspective tool to enhance cell‐based therapy and adverse tumour vascularization. Curr Med Chem. 2012;19(34):5802‐5818. [DOI] [PubMed] [Google Scholar]
- 31.Zuccolo E, Dragoni S, Poletto V, et al. Arachidonic acid‐evoked Ca2+ signals promote nitric oxide release and proliferation in human endothelial colony forming cells. Vascul Pharmacol. 2016;87:159‐171. [DOI] [PubMed] [Google Scholar]
- 32.Mikoshiba K. IP3 receptor/Ca2+ channel: from discovery to new signaling concepts. J Neurochem. 2007;102(5):1426‐1446. [DOI] [PubMed] [Google Scholar]
- 33.Wasilewski A, Krajewska U, Owczarek K, Lewandowska U, Fichna J. Fatty acid amide hydrolase (FAAH) inhibitor PF‐3845 reduces viability, migration and invasiveness of human colon adenocarcinoma Colo‐205 cell line: an in vitro study. Acta Biochim Pol. 2017;64(3):519‐525. [DOI] [PubMed] [Google Scholar]
- 34.White JP, Cibelli M, Urban L, Nilius B, McGeown JG, Nagy I. TRPV4: molecular conductor of a diverse orchestra. Physiol Rev. 2016;96(3):911‐973. [DOI] [PubMed] [Google Scholar]
- 35.Peppiatt CM, Holmes AM, Seo JT, et al. Calmidazolium and arachidonate activate a calcium entry pathway that is distinct from store‐operated calcium influx in HeLa cells. Biochem J. 2004;381(Pt 3):929‐939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kilpatrick BS, Eden ER, Schapira AH, Futter CE, Patel S. Direct mobilisation of lysosomal Ca2+ triggers complex Ca2+ signals. J Cell Sci. 2013;126(Pt 1):60‐66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Faris P, Pellavio G, Ferulli F, et al. Nicotinic acid adenine dinucleotide phosphate (NAADP) induces intracellular Ca(2+) release through the two‐pore channel TPC1 in metastatic colorectal cancer cells. Cancers. 2019;11(4):542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Di Nezza F, Zuccolo E, Poletto V, et al. Liposomes as a putative tool to investigate NAADP signaling in vasculogenesis. J Cell Biochem. 2017;118(11):3722‐3729. [DOI] [PubMed] [Google Scholar]
- 39.Moccia F, Zuccolo E, Di Nezza F, et al. Nicotinic acid adenine dinucleotide phosphate activates two‐pore channel TPC1 to mediate lysosomal Ca(2+) release in endothelial colony‐forming cells. J Cell Physiol. 2021;236(1):688‐705. [DOI] [PubMed] [Google Scholar]
- 40.Song S, Li J, Zhu L, et al. Irregular Ca(2+) oscillations regulate transcription via cumulative spike duration and spike amplitude. J Biol Chem. 2012;287(48):40246‐40255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arnaoutova I, Kleinman HK. In vitro angiogenesis: endothelial cell tube formation on gelled basement membrane extract. Nat Protoc. 2010;5(4):628‐635. [DOI] [PubMed] [Google Scholar]
- 42.Sethi G, Ahn KS, Aggarwal BB. Targeting nuclear factor‐kappa B activation pathway by thymoquinone: role in suppression of antiapoptotic gene products and enhancement of apoptosis. Mol Cancer Res. 2008;6(6):1059‐1070. [DOI] [PubMed] [Google Scholar]
- 43.Hodgkinson CP, Bareja A, Gomez JA, Dzau VJ. Emerging concepts in paracrine mechanisms in regenerative cardiovascular medicine and biology. Circ Res. 2016;118(1):95‐107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Barile L, Lionetti V, Cervio E, et al. Extracellular vesicles from human cardiac progenitor cells inhibit cardiomyocyte apoptosis and improve cardiac function after myocardial infarction. Cardiovasc Res. 2014;103(4):530‐541. [DOI] [PubMed] [Google Scholar]
- 45.Barile L, Cervio E, Lionetti V, et al. Cardioprotection by cardiac progenitor cell‐secreted exosomes: role of pregnancy‐associated plasma protein‐A. Cardiovasc Res. 2018;114(7):992‐1005. [DOI] [PubMed] [Google Scholar]
- 46.DeSantiago J, Bare DJ, Banach K. Ischemia/Reperfusion injury protection by mesenchymal stem cell derived antioxidant capacity. Stem Cells Dev. 2013;22(18):2497‐2507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Costa A, Ceresa D, De Palma A, et al. Comprehensive profiling of secretome formulations from fetal‐ and perinatal human amniotic fluid stem cells. Int J Mol Sci. 2021;22(7):3713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Nam TS, Park DR, Rah SY, et al. Interleukin‐8 drives CD38 to form NAADP from NADP(+) and NAAD in the endolysosomes to mobilize Ca(2+) and effect cell migration. FASEB J. 2020;34(9):12565‐12576. [DOI] [PubMed] [Google Scholar]
- 49.Day P, Burrows L, Richards D, Fountain SJ. Inhibitors of DAG metabolism suppress CCR2 signalling in human monocytes. Br J Pharmacol. 2019;176(15):2736‐2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Negri S, Faris P, Rosti V, Antognazza MR, Lodola F, Moccia F. Endothelial TRPV1 as an emerging molecular target to promote therapeutic angiogenesis. Cells. 2020;9(6):1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Moccia F, Negri S, Faris P, Berra‐Romani R. Targeting the endothelial Ca 2+ tool kit to rescue endothelial dysfunction in obesity associated‐hypertension. Curr Med Chem. 2020;27(2):240‐257. [DOI] [PubMed] [Google Scholar]
- 52.Lodola F, Laforenza U, Bonetti E, et al. Store‐operated Ca2+ entry is remodelled and controls in vitro angiogenesis in endothelial progenitor cells isolated from tumoral patients. PLoS One. 2012;7(9):e42541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Antoniotti S, Fiorio Pla A, Pregnolato S, Mottola A, Lovisolo D, Munaron L. Control of endothelial cell proliferation by calcium influx and arachidonic acid metabolism: a pharmacological approach. J Cell Physiol. 2003;197(3):370‐378. [DOI] [PubMed] [Google Scholar]
- 54.Dragoni S, Guerra G, Fiorio Pla A, et al. A functional transient receptor potential vanilloid 4 (TRPV4) channel is expressed in human endothelial progenitor cells. J Cell Physiol. 2015;230(1):95‐104. [DOI] [PubMed] [Google Scholar]
- 55.Berridge MJ. The inositol trisphosphate/calcium signaling pathway in health and disease. Physiol Rev. 2016;96(4):1261‐1296. [DOI] [PubMed] [Google Scholar]
- 56.Berra‐Romani R, Faris P, Pellavio G, et al. Histamine induces intracellular Ca(2+) oscillations and nitric oxide release in endothelial cells from brain microvascular circulation. J Cell Physiol. 2020;235(2):1515‐1530. [DOI] [PubMed] [Google Scholar]
- 57.Negri S, Faris P, Pellavio G, et al. Group 1 metabotropic glutamate receptors trigger glutamate‐induced intracellular Ca(2+) signals and nitric oxide release in human brain microvascular endothelial cells. Cell Mol Life Sci. 2020;77(11):2235‐2253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Smedler E, Uhlen P. Frequency decoding of calcium oscillations. Biochim Biophys Acta. 2014;1840(3):964‐969. [DOI] [PubMed] [Google Scholar]
- 59.Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF‐kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. Oct‐Dec. 2010;1799(10–12):775‐787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Moccia F, Berra‐Romani R, Rosti V. Manipulating intracellular Ca2+ signals to stimulate therapeutic angiogenesis in cardiovascular disorders. Curr Pharm Biotechnol. 2018;19(9):686‐699. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Information
Data Availability Statement
All the data are fully available upon reasonable request.