

Abstract
The utility of circulating tumor DNA (ctDNA) as a biomarker in patients with advanced cancers receiving immunotherapy is uncertain. We therefore analyzed pretreatment (n = 978) and on-treatment (n = 171) ctDNA samples across 16 advanced-stage tumor types from three phase I/II trials of durvalumab (± the anti-CTLA4 therapy tremelimumab). Higher pretreatment variant allele frequencies (VAF) were associated with poorer overall survival (OS) and other known prognostic factors, but not objective response, suggesting a prognostic role for patient outcomes. On-treatment reductions in VAF and lower on-treatment VAF were independently associated with longer progression-free survival and OS and increased objective response rate, but not prognostic variables, suggesting that on-treatment ctDNA dynamics are predictive of benefit from immune checkpoint blockade. Accordingly, we propose a concept of “molecular response” using ctDNA, incorporating both pretreatment and on-treatment VAF, that predicted long-term survival similarly to initial radiologic response while also permitting early differentiation of responders among patients with initially radiologically stable disease.
Introduction:
Immune checkpoint inhibitors improve survival across a wide range of solid tumors (1) and are now a routine component of treatment for many cancer types (2–17). Despite their wide use, a minority of patients experience long-term benefit, highlighting the need for tools to guide treatment to those most likely to respond. There are few biomarkers for immune checkpoint inhibitor response, particularly ones that can be applied across cancer types (18–20). Cell-free circulating tumor DNA (ctDNA) analysis is a noninvasive tool with potentially broad use (21–29), including emerging applications in the context of immunotherapy (30). For example, we have previously reported that early decreases in ctDNA were associated with improved survival in patients receiving PD-L1 inhibitors (31).
Ongoing challenges to routine use of ctDNA in clinical practice include clarification of the prognostic and/or predictive associations with immune checkpoint therapy, validation of results in larger patient cohorts, and demonstration of added clinical utility beyond routine radiologic assessment. Here, we present a comprehensive analysis of ctDNA data from patients with 16 different solid tumor types across three phase I/II trials of durvalumab (alone or in combination with the CTLA4 inhibitor tremelimumab). This represents the largest patient cohort receiving immune checkpoint blockade with ctDNA and clinical outcome data. We characterized the prognostic and predictive impact of pretreatment and on-treatment ctDNA. Using training and validation sets, we propose an approach to interrogate “molecular response” using ctDNA, which enables the prediction of long-term survival and adjudicates which patients with initial radiologically stable disease (SD) will ultimately respond to immunotherapy.
Results:
Detectability of ctDNA Variants in 16 Advanced-Stage Tumor Types
Among the three cohorts included here (Study 1108, ATLANTIC, and Study 10), we detected ctDNA variants in the pretreatment samples of 814 (83.2%) of 978 patients across 16 tumor types. The detection rate varied from 21.4% for patients with glioblastoma multiforme (GBM) to >95% for patients with small-cell lung cancer (SCLC) and naso-pharyngeal carcinoma (Fig. 1A). Fifteen of the 16 tumor types had a mean pretreatment variant allele frequency (VAF) <10% (Supplementary Fig. S1A), and the median VAF among all the pretreatment samples was 2.4% (Supplementary Fig. S1B). ctDNA was more likely to be detected in patients with higher tumor burden, defined as the sum of the diameters of the target lesions (Supplementary Fig. S1C). There was no association between detectability and PD-L1 expression in tumor cells or other clinical features (Supplementary Fig. S1D; Supplementary Table S1).
Figure 1.
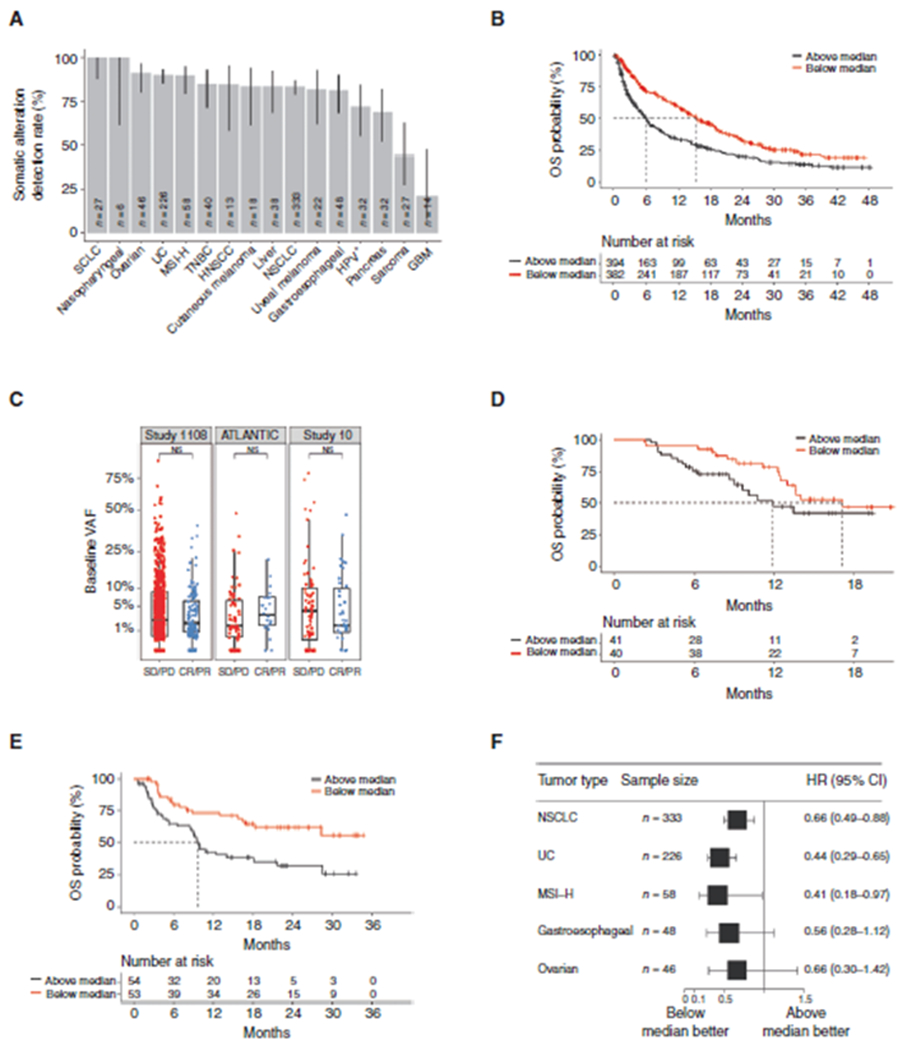
A, Somatic alteration detection rate by tumor type. Detection rate varied from 21.4% for patients with GBM to >95% for patients with SCLC and nasopharyngeal carcinoma. The error bars denote the 95% CI of detection rate for each tumor type given the sample sizes. B, Kaplan–Meier analysis of OS in the discovery cohort stratified by ≥median versus <median pretreatment VAF. For OS, unadjusted HR, 0.58; 95% CI, 0.49–0.69; P < 0.0001 and adjusted HR, 0.58; 95% CI, 0.48–0.7; P < 0.0001. C, Pretreatment VAF stratified by objective response (complete and partial responses vs. SD and progressive disease) in the discovery and validation cohorts. Pretreatment VAF was not significantly associated with response in any cohort. The horizontal bar represents the mean, the box represents the 25th and 75th percentiles, and whiskers ± 1.5 × interquartile range. Kaplan–Meier analysis of OS in the validation cohorts. D, Pretreatment VAF was not significantly associated with OS in ATLANTIC (HR, 0.58; 95% CI, 0.3–1.16; P = 0.12). E, Pretreatment VAF was significantly associated with OS in Study 10 (HR, 0.42; 95% CI, 0.24–0.74; P = 0.0019). F, Forest plot for OS by tumor type stratified by median pretreatment VAF using pooled data from the discovery cohort and both validation cohorts. CR, complete response; HNSCC, head and neck squamous cell carcinoma; HPV, human papillomavirus; PD, progressive disease; PR, partial response; TNBC, triple-negative breast cancer; UC, urothelial cancer.
Mean VAF of variants detected in ctDNA correlated with several patient characteristics, including Eastern Cooperative Oncology Group (ECOG) performance status (P = 0.0004; Supplementary Fig. S1E), presence of liver metastases (P < 0.0001; Supplementary Fig. S1F), and tumor burden (P < 0.0001; Supplementary Fig. S1G), but did not correlate with lymph node metastases, lines of prior therapy, or smoking status [assessed only in patients with non–small cell lung cancer (NSCLC); Supplementary Fig. S1H–S1J]. PD-L1 expression on either tumor or immune cells was not correlated with mean VAF (Supplementary Fig. S1K–S1N), suggesting that intratumoral inflammation (reflected by PD-L1 level; refs. 32, 33) is not associated with changes in ctDNA levels.
Pretreatment ctDNA VAF Is Prognostic
As pretreatment mean VAF correlated with several known prognostic factors, including ECOG performance status and the presence of liver metastases, we hypothesized that pretreatment ctDNA might itself be prognostic. To explore this hypothesis, Study 1108 (n = 790) was first used as the discovery cohort to understand the relationship of pretreatment ctDNA levels with outcomes. A significant association was found between pretreatment mean VAF and overall survival [OS; stratified by the median; unadjusted HR, 0.58; 95% confidence interval (CI), 0.49–0.69; P < 0.0001; Fig. 1B] and progression-free survival (PFS; Supplementary Fig. S2A). In addition, pretreatment VAF was not simply a surrogate of tumor burden or other prognostic variables (ECOG performance status, baseline liver metastases, baseline lymph node metastases, smoking status, tumor burden, and tumor PD-L1 score), as the association with OS remained after adjustment for these features (adjusted HR, 0.58; 95% CI, 0.48–0.7; P < 0.0001) and with varying VAF cutoff points (Supplementary Fig. S2B). Furthermore, pretreatment VAF did not correlate with objective response rate (ORR; Fig. 1C). Use of maximum VAF (instead of mean) yielded similar associations (Supplementary Fig. S3A–S3C). Taken together, the correlation with known prognostic features, the lack of association with ORR, and the observed association with long-term survival suggest that pretreatment ctDNA VAF levels may be prognostic, rather than specifically predictive, of response to treatment with immune checkpoint inhibitors.
We then explored the relationship between pretreatment ctDNA and outcomes in two independent, smaller cohorts, ATLANTIC (n = 81) and Study 10 (n = 107). In both studies, low levels of mean pretreatment VAF were also associated with OS (stratified by median), although not reaching statistical significance in the smaller of the two cohorts (Fig. 1D and E). Pretreatment VAF showed no significant associations with ORR or PFS in ATLANTIC or Study 10 (Fig. 1C; Supplementary Fig. S3D and S3E).
Consistent with the overall analysis, within subgroups of the five most prevalent tumor types [NSCLC (n = 333), urothelial cancer (UC; n = 226), microsatellite instability– high (MSI-H) cancers (n = 58), gastroesophageal cancer (n = 48), and ovarian cancer (n = 46)], pooled across the three trials, pretreatment VAF had a significant inverse relationship with OS, to a similar degree in each tumor type (Fig. 1F), highlighting the potential applicability of pretreatment ctDNA as an independent, prognostic biomarker across tumors.
On-Treatment ctDNA Dynamics Are Predictive of Immunotherapy Benefit
In a subset of patients with NSCLC and UC, we previously showed that reduction in ctDNA mean VAF at 6 weeks after treatment initiation was associated with better response and survival (31). Here, the cohort was expanded to include an additional 48 patients. Overall paired on-treatment ctDNA samples were available for a subset of 171 (17.5%) patients in the three cohorts; their clinical characteristics were generally consistent with those of the full cohort (Supplementary Table S2). Similar to our prior study, a lower on-treatment VAF was associated with longer PFS and OS (Fig. 2A).
Figure 2.
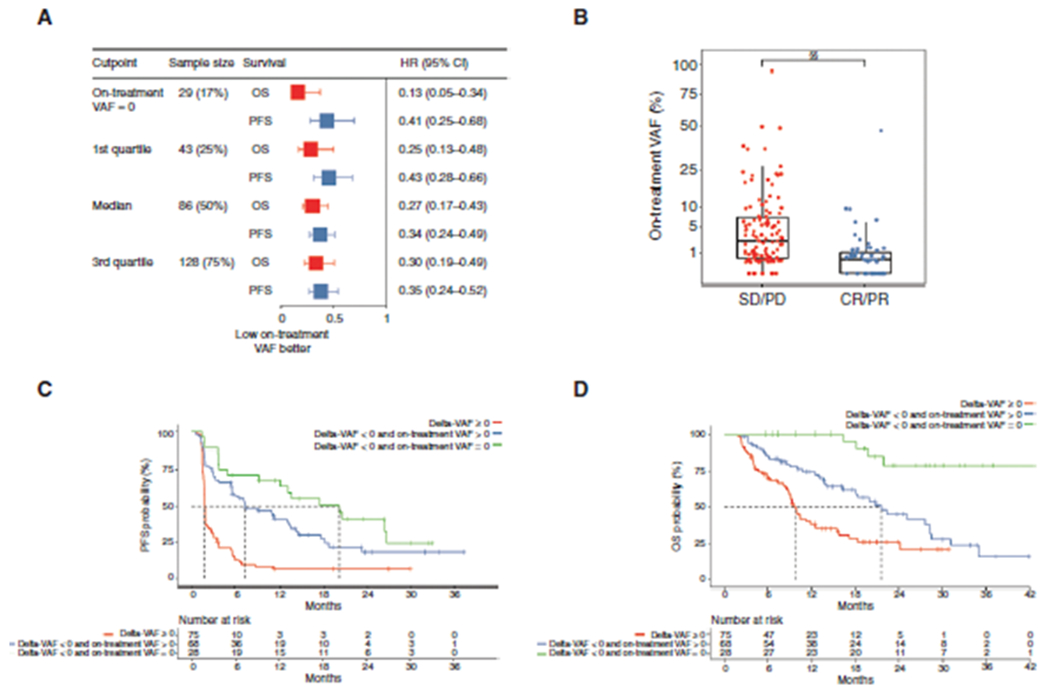
A, Forest plot of PFS and OS by on-treatment VAF. The best OS was observed in patients with complete ctDNA clearance on treatment. B, RECIST responders and nonresponders had significantly different on-treatment VAF (P < 0.0001). C and D, Kaplan–Meier analysis of PFS (C) and OS (D) in the discovery cohort stratified by delta-VAF and on-treatment VAF. Significant differences were observed among groups (P < 0.0001). §§, P < 0.00001.
In contrast to pretreatment VAF, on-treatment VAF was associated with ORR (P < 0.0001; Fig. 2B), but not with most known clinical prognostic variables (Supplementary Fig. S4A–S4E). We observed that mean change in VAF (delta-VAF) and on-treatment VAF were complementary, nonredundant correlates of improved survival; patients with an increase in ctDNA from pretreatment (delta-VAF > 0) had the worst outcomes, those with decreased but not completely cleared ctDNA (delta-VAF < 0 and on-treatment VAF > 0) had intermediate outcomes, and those with completely cleared on-treatment ctDNA (on-treatment VAF = 0) had the best PFS and OS (P < 0.0001; Fig. 2C and D). Similar PFS and OS associations were observed by stratifying by the median for both delta-VAF and on-treatment VAF (P < 0.0001; Supplementary Fig. S5A and S5B). These results suggest that on-treatment ctDNA dynamics are predictive of benefit with immune checkpoint blockade. Although delta-VAF was able to stratify patients similarly to on-treatment VAF (Supplementary Fig. S6A–S6H), it only assessed relative VAF changes and equated patients with low VAF who had a significant decrease in ctDNA level and patients with a higher VAF and smaller decrease in ctDNA level. Therefore, we sought to implement integrated metrics for the quantification of ctDNA dynamics.
Developing a Framework of “Molecular Response” Using Longitudinal ctDNA Metrics
As both the change in ctDNA and on-treatment ctDNA level were notably associated with efficacy of immune check-point inhibitor therapy, we proposed a framework for determining “molecular response” by integrating information from pretreatment VAF and on-treatment VAF. To test the validity of this framework, a composite score was developed as a linear combination of delta-VAF and on-treatment VAF (Supplementary Materials and Methods), which was ultimately simplified to a ratio of on-treatment VAF to pretreatment VAF when used for stratification.
This ratio-based molecular response metric had a stronger association with RECIST response compared with on-treatment VAF alone (Fig. 3A; AUC = 0.82; 95% CI, 0.71–0.93 for the ratio and AUC = 0.73; 95% CI, 0.61–0.85 for on-treatment VAF). Using the ratio, patients with pretreatment and on-treatment samples in the three cohorts (Study 1108, n = 69; ATLANTIC, n = 66; and Study 10, n = 36) were stratified into ctDNA-defined molecular responders and nonresponders by a cutoff point of 50%, consistent with previous work (34). A similar proportion of patients were defined as molecular responders (41% in Study 1108, 33% in ATLANTIC, and 44% in Study 10, respectively) compared with RECIST-defined responders (36%, 24%, and 44%, respectively). In all three cohorts, molecular response was associated with higher ORRs (Fig. 3B) and improved HRs for PFS and OS (Table 1). Furthermore, molecular response was associated with similar, and numerically more substantial in some cohorts, PFS and OS HR stratification compared with RECIST-defined response at the first CT scan (Fig. 3C), and it generally outperformed stratifications that relied on on-treatment VAF = 0 alone (Table 1; refs. 31, 35). The ratio-based approach yielded similar conclusions to the originally developed composite score approach (Supplementary Materials and Methods; Supplementary Fig. S7A–S7E). Together, these results show that a framework of molecular response produced through the integration of both pretreatment and on-treatment ctDNA levels improved the association with immunotherapy outcomes.
Figure 3.
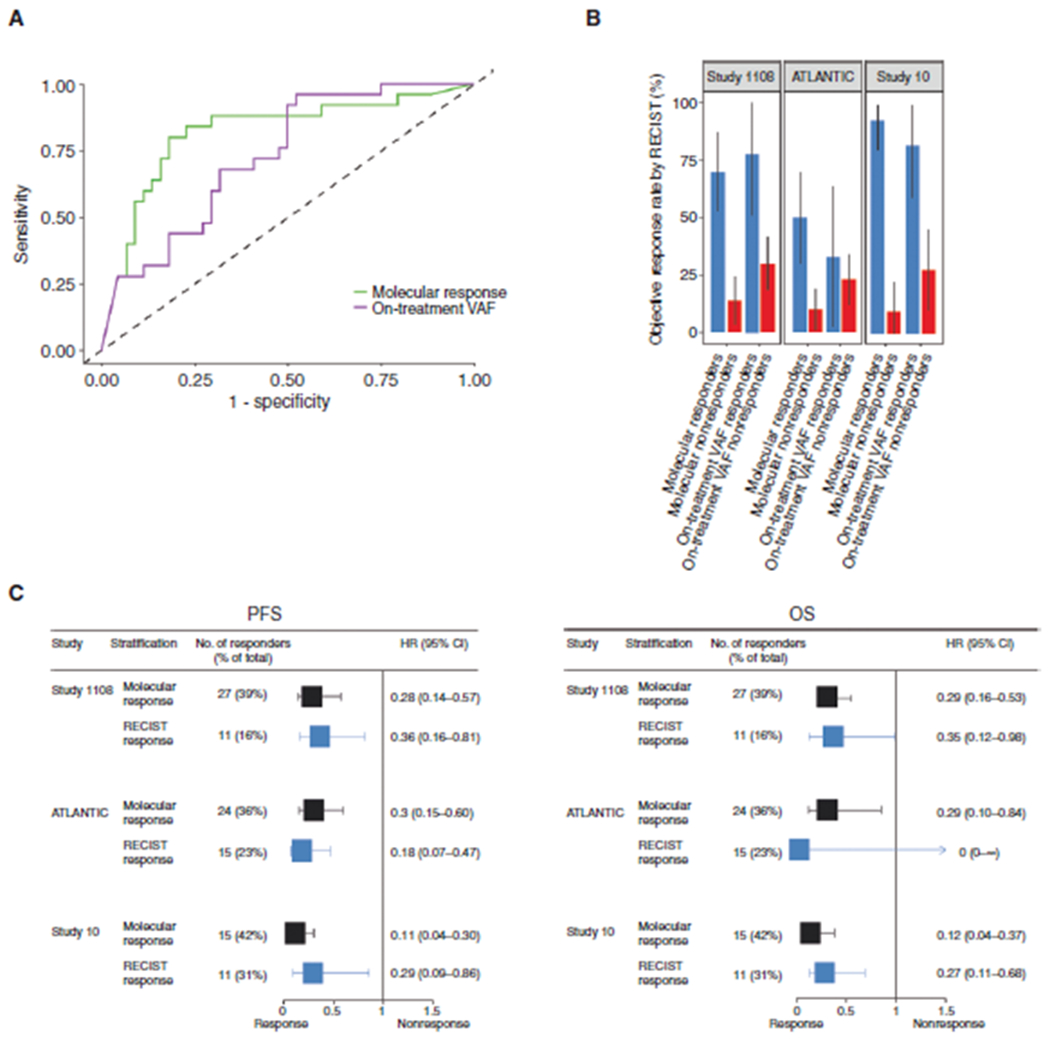
Development of a definition of molecular response with contributions from pretreatment VAF and on-treatment VAF. A, ROC curves of molecular response and on-treatment VAF to predict the best response. B, ORR among patients with and without response as defined by either the ratio (molecular response) or on-treatment VAF. C, Forest plot for PFS and OS based on molecular response or the first RECIST response in each study.
Table 1.
Association between two different ctDNA dynamics measurements and survival in three cohorts of patients with solid tumors receiving immune checkpoint blockade
| Study | Study 1108 (n = 69) | ATLANTIC (n = 66) | Study 10 (n = 36) | |||
|---|---|---|---|---|---|---|
| Treatment | Durvalumab | Durvalumab | Durvalumab + tremelimumab | |||
|
| ||||||
| PFS | ||||||
| n (%) | HR (95% CI) | n (%) | HR (95% CI) | n (%) | HR (95% CI) | |
| On-treatment VAF = 0 | 9 (13) | 0.55 (0.25-1.24) | 9 (14) | 0.72 (0.31-1.69) | 11 (31) | 0.12 (0.04-0.37) |
| Ratio < 50% | 27 (39) | 0.28 (0.14-0.57) | 24 (36) | 0.3 (0.15-0.60) | 15 (42) | 0.11 (0.04-0.30) |
|
| ||||||
| OS | ||||||
| n (%) | HR (95% CI) | n (%) | HR (95% CI) | n (%) | HR (95% CI) | |
| On-treatment VAF = 0 | 9 (13) | 0.31 (0.1-1.02) | 9 (14) | 0 (0-∞)a | 11 (31) | 0.05 (0.01-0.39) |
| Ratio < 50% | 27 (39) | 0.29 (0.16-0.53) | 24 (36) | 0.29 (0.10-0.84) | 15 (42) | 0.12 (0.04-0.37) |
All patients with on-treatment VAF = 0 were censored.
Molecular Response Identifies Long-Term Benefit among Patients with Initially Radiologic SD
A major challenge in drug development and clinical practice is determining whether a patient with radiologic SD is truly benefiting from treatment. We hypothesized that molecular response might discriminate patients with early SD who would ultimately benefit from treatment from those who would not.
Across all three cohorts with on-treatment ctDNA assessments, 74 (43%) patients had radiologic SD at their first assessment (week 5–9; Supplementary Table S3). Of those who eventually went on to have a radiologic response, the majority were molecular responders (19/25, 76%). This proportion was significantly greater than the proportion of molecular responders in patients who did not go on to have a radiologic response (12/49, 24%; P < 0.0001, Fisher test; Fig. 4A). Among patients with initial SD who were molecular responders and eventually achieved radiologic response, the ctDNA assessment predated achieving radiologic response by a median of 8 weeks (Fig. 4B). Among patients with initial SD, maximum tumor shrinkage during treatment was significantly greater in molecular responders than molecular nonresponders (P < 0.0001; Fig. 4C). In addition, molecular response among patients with initially radiologic SD was associated with longer PFS (median 17.4 vs. 5.4 months; HR, 0.31; 95% CI, 0.17–0.56; log-rank P < 0.0001; Fig. 4D) and OS (median 31.2 vs. 18.4 months; HR, 0.36; 95% CI, 0.17–0.79; P = 0.008; Fig. 4E). These data demonstrate the potential of molecular response to adjudicate benefit among patients with initial radiologically defined SD on durvalumab alone and in combination with tremelimumab.
Figure 4.
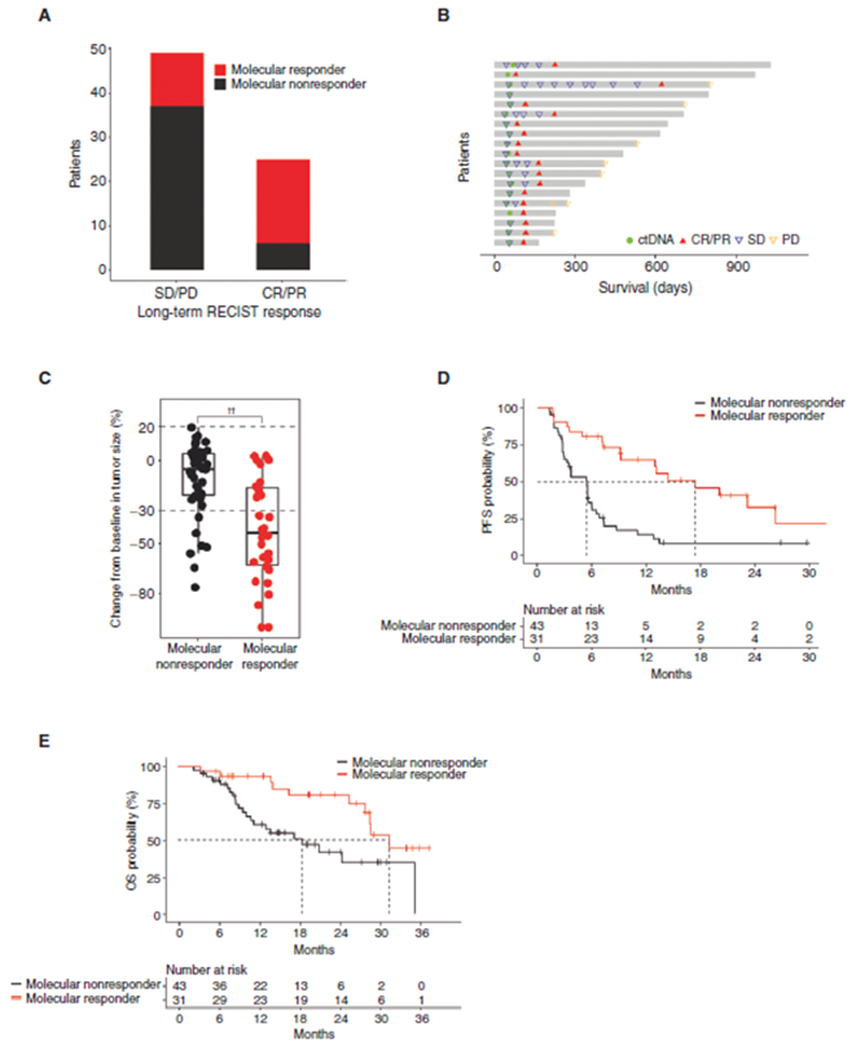
A, Number of molecular responders and molecular nonresponders (per ratio approach) with eventual RECIST response. A higher percentage of eventual radiologic responders was initial molecular responders (19/25, 76%) than the percentage of eventual radiologic nonresponders who were initial molecular responders (12/49, 24%; P < 0.0001, Fisher test). B, Swimmer plot of 14 patients with initial SD who were molecular responders and eventually had a radiologic response. The median time to radiologic response was 114 days from starting of the therapy, a median of 59 days later than the ctDNA assessment. C, Tumor shrinkage from baseline was significantly greater in molecular responders than molecular nonresponders (per ratio approach; P < 0.0001). The horizontal bar represents the mean, box represents the 25th and 75th percentiles, and whiskers ± 1.5 × interquartile range. D and E, Kaplan–Meier analysis of PFS (D) and OS (E) of patients who had RECIST and molecular response assessments stratified by molecular response (per ratio approach). PFS and OS were significantly longer in molecular responders than in molecular nonresponders (HR, 0.31; 95% CI, 0.17–0.56; P = 0.0001 and HR, 0.36; 95% CI, 0.17–0.79; P = 0.008, respectively). ††, P < 0.0001.
Discussion:
ctDNA analysis is emerging as a new noninvasive technique to assess disease status and therapeutic response, although the specific opportunities for application in the context of immunotherapy remain to be clarified. Here, we analyzed ctDNA and clinical data from almost 1,000 patients with locally advanced/metastatic tumors treated with immune checkpoint blockade. We demonstrated that ctDNA is detectable in most patients, although there are important disease-specific differences which may reveal variability in the relative pattern of tumor DNA shedding. Pretreatment ctDNA level appears to be an independent, inversely prognostic variable across tumor types, characterized by an association with OS and other known prognostic variables, but not with ORR. On-treatment ctDNA dynamics appear to be predictive of long-term benefit from immunotherapy across tumor types. Analysis of ctDNA permits early identification of patients with molecular response, which is associated with eventual RECIST response and improved survival among patients with initial radiologic SD. Overall, these results demonstrate that ctDNA analyses can be an important baseline prognostic feature for stratification in clinical trials, can serve as an early biomarker of response, and can complement radiologic assessments to improve the identification of patients benefiting from immunotherapy across different cancers.
Our findings regarding ctDNA as a biomarker for immune checkpoint blockade response are similar to other proof-of-concept studies in the literature on ctDNA and systemic therapies. Pretreatment ctDNA appears to be associated with survival in other treatment settings (25, 36–41). For example, in a group of 88 patients with advanced NSCLC and detectable somatic mutations, Schwaederle and colleagues found that those with a VAF ≥ 5% had significantly shorter median survival than those with a VAF of < 5% (4.2 months vs. not reached; P = 0.012; 39). In addition, on-treatment reductions in ctDNA levels seem to be predictive of response to systemic therapies in a wide range of cancers (28, 31, 32, 35, 42–44).
The use of ctDNA fills an important unmet need as a complement to radiologic assessments of benefit. Radiologic SD is a common and particularly challenging clinical category, composed of patients with slowly progressive disease, indolent nonresponding disease, and radiologically subtle responses to immunotherapy (34, 45). We demonstrate that molecular response, defined by ctDNA dynamics, can help differentiate patients who will ultimately derive benefit from immunotherapy from those with indolent or progressive disease who are unlikely to derive further benefit from treatment. This tool has substantial application in drug development to provide a more nuanced assessment of benefit in early-phase clinical trials, in translational analyses to permit stronger responder versus nonresponder comparisons, and potentially in clinical practice to inform decisions about continuation or early transition of therapy in those with radiologically equivocal benefit.
Limitations of this study include the moderate size of the gene panel used, which hinders reliable estimates of tumor mutation burden as well as other potentially relevant molecular features such as clonality and tumor heterogeneity. In addition, we did not have paired tumor tissue or leukocyte cellular DNA to maximize removal of potential germline and clonal hematopoietic variants (46, 47), although we did filter against several large databases of germline and clonal hematopoietic variants. Although we demonstrated the potential utility of molecular response using a composite ctDNA metric that is similar to the ratio approach used in other studies, the cutoff point applied, the assay resolution around “undetectability,” and adjustment for divergent changes in individual variants will need to be further optimized; these results should therefore be regarded as a proof of concept. Relatedly, ctDNA assays with improved precision to detect low levels of residual disease are being developed, although it should be noted that, in contrast to this study, these assays have (to date) primarily been focused on application to early-stage cancers. Emerging markers of the immune microenvironment are being actively investigated and may add to the predictive power of ctDNA assessment. Further investigations should also evaluate how the assessment of ctDNA dynamics should be utilized in combination with currently available tools, including radiologic and biochemical methods, to refine the assessment of patient response in real time. With continuous improvement in ctDNA assays tailored to important clinical contexts, the understanding of the optimal specific approaches to using ctDNA as a biomarker and outcome in immunotherapy also needs to evolve. Finally, the timing of ctDNA sample collection should be considered, contextualized by the clinical question under investigation. For example, future prospective studies seeking to intervene upon early determination of disease progression may explore earlier time points of ctDNA assessment, whereas efforts to predict long-term responders may benefit from more intermediate or longterm timing of ctDNA assessment. In conclusion, ctDNA measurements are taking on an increasingly important role in the clinic. The data presented here build upon the results of previous studies, representing the largest pan-tumor assessment of the prognostic and predictive utility of ctDNA, with 978 pretreatment and 171 on treatment samples across 16 solid tumor types. In particular, the use of ctDNA dynamics to assess clinical benefit in patients with radiologic SD represents a novel tool addressing an important unmet need, with potential utility across a range of settings. Our analysis contributes to the understanding of the role of ctDNA as a prognostic and predictive biomarker and its potential to complement radiologic endpoints and adjudicate radiologically equivocal benefit.
Methods
Study Design and Patients
We included patients with advanced solid tumors enrolled in three phase I/II studies receiving durvalumab with or without tremelimumab. The three studies were Study 1108 (NCT01693562, durvalumab), ATLANTIC (NCT02087423, durvalumab), and Study 10 (NCT02261220, durvalumab plus tremelimumab; refs. 5, 6, 48, 49). Pretreatment samples were collected before initiation of treatment. On-treatment sample collection at 6 to 8 weeks after the first dose was optional; therefore, only a subset of patients consented to provide paired samples. The studies were conducted in accordance with Good Clinical Practices guidelines and the Declaration of Helsinki and were approved by each institution’s ethical review board. Patients provided written informed consent. The study design, patient demographics, and clinical outcomes of these studies have been reported previously (5, 6, 48, 49).
Characterization of ctDNA
ctDNA was extracted centrally as described previously (50). A next-generation sequencing–based targeted panel (Guardant360) was used to characterize somatic genomic alterations [single-nucleotide variants (SNV), insertion/deletion mutations (indels), gene amplifications, and gene fusions] in 73 genes with known somatic cancer gene variants (34). All steps, including ctDNA isolation, targeted sequencing, and variant calls, were performed at Guardant Health, a CLIA/CAP-accredited laboratory. Potential germline and clonal hematopoietic variants were also filtered against previously described variants (46, 47).
Genomic Analysis
For each sample, somatic SNVs and indels from the Guardant360 report were used to calculate the mean and maximum VAF and total mutation count. A VAF of 0.3% was the limit of detection of the Guardant360 test (31), with the exception of variants detected in both pretreatment and on-treatment samples such that variants with VAF < 0.3% in one sample were retained if the exact variant was also detected in the paired sample with VAF ≥ 0.3%.
The delta-VAF was calculated per patient as the sum of the on-treatment VAF minus the pretreatment VAF for each detected SNV or indel divided by the number of detected SNVs or indels, as shown in the following equation:
here, onVAFi,j represents the on-treatment VAF for variant i in patient j, prVAFi,j is the pretreatment VAF for variant i in patient j, and n is the number of variants with VAF ≥ 0.3% at either the pretreatment or on-treatment time point in patient j. Delta-VAF < 0 reflects a decline in mean ctDNA levels. The mean on-treatment VAF for each patient was defined as the average VAF of variants detected at the on-treatment timepoint, per the following equation:
here, m represents the number of variants with onVAF ≥ 0.3% in patient j. If m = 0 or no on-treatment variant was reported, the mean on-treatment VAF was set to 0. If a variant was only detected pretreatment, the on-treatment VAF was set to 0, and vice versa.
Development of a Definition of Molecular Response
We initially approached the question of defining a ctDNA-based response metric (molecular response) using a linear “composite score” model of delta-VAF and on-treatment VAF (Supplementary Materials and Methods). During initial investigations, we found that the “composite score” model could generally be represented in simpler terms through a ratio of on-treatment VAF to pretreatment VAF with similar performance:
We defined molecular response as a >50% decrease in VAF. This cutoff point was similar to that used in previous studies (34, 42). These findings were then independently examined using ATLANTIC and Study 10 as validation cohorts.
Statistical Analysis
The Kaplan–Meier method was used to calculate survival estimates for PFS and OS. A multivariable Cox proportional hazards model was used to define the association of the ctDNA-derived metrics with PFS and OS, after adjusting for tumor type and known prognostic factors, including baseline liver metastases, baseline lymph node disease, tumor burden, smoking status (patients with NSCLC only), and baseline ECOG performance status. P values were assessed using the log-rank test. We used the Wilcoxon rank-sum test and Kruskal–Wallis test when comparing continuous variables. All P values were two-sided. Analyses were performed using R (version 3.4.3, R Foundation). The clinical dataset analyzed here is available and may be obtained in accordance with AstraZeneca’s data sharing policy, which is described at https://astrazenecagrouptrials.pharmacm.com/ST/Submission/Disclosure.
Supplementary Material
SIGNIFICANCE:
In a pan-cancer analysis of immune checkpoint blockade, pretreatment ctDNA levels appeared prognostic and on-treatment dynamics predictive. A “molecular response” metric identified long-term responders and adjudicated benefit among patients with initially radiologically stable disease. Changes in ctDNA may be more dynamic than radiographic changes and could complement existing trial endpoints.
Acknowledgments
These studies (NCT01693562, NCT02087423, and NCT02261220) were sponsored by AstraZeneca. The study was supported by Memorial Sloan Kettering Cancer Center Support grant/core grant no. P30 CA008748 and the Druckenmiller Center for Lung Cancer Research at Memorial Sloan Kettering Cancer Center; M.D. Hellmann is a Damon Runyon Clinical Investigator supported in part by the Damon Runyon Cancer Research Foundation grant no. CI-98-18 and is a member of the Parker Institute for Cancer Immunotherapy. J. Luo was supported in part by T32-CA009207 and K30-UL1TR00457 grants from the NIH. Medical writing support was provided by James Holland and Susanne Gilbert of Cirrus Communications, an Ashfield Company, and was funded by AstraZeneca.
Disclosure of Potential Conflicts of Interest
J. Luo reports personal fees from Targeted Oncology (honoraria), and grants from NIH (T32-CA009207 and K30-UL1TR00457) and Conquer Cancer, the ASCO Foundation (Young Investigator Award) during the conduct of the study. S. Wu reports employment with AstraZeneca and owns AstraZeneca stocks. H. Si reports other from AstraZeneca (salary) during the conduct of the study. S.E. Abdullah reports other from AstraZeneca (employment with AstraZeneca when the article was drafted) during the conduct of the study. B.W. Higgs reports other from AstraZeneca (stock and former employee) during the conduct of the study and outside the submitted work, as well as has a patent for AstraZeneca pending. P.A. Dennis reports other from AstraZeneca (full-time employee) outside the submitted work. M.S. van der Heijden reports other from AstraZeneca (clinical trial fees) during the conduct of the study, grants and personal fees from AstraZeneca (research grant and consultancy, to institute), BMS (research grant and consultancy, to institute), and Roche (research grant and consultancy, to institute), and personal fees from MSD (consultancy, to institute), Seattle Genetics (consultancy, to institute), and Janssen (consultancy, to institute) outside the submitted work. N.H. Segal reports personal fees from Revitope Oncology, PsiOxus, Immunocore PureTech Ventures, Amgen, GlaxoSmithKline, CStone Pharmaceuticals, Synlogic, Pieris, AstraZeneca, Gritstone Oncology, TRM Oncology, Roche/Genentech, Kyn Therapeutics, Aduro, Boehringer Ingelheim, and Pfizer (consulting) outside the submitted work and research funding from Roche/Genentech, Pfizer, Merck, BMS, AstraZeneca, Incyte, and Immunocore. J.E. Chaft reports personal fees from AstraZeneca (consultant), BMS (consultant), Genentech (consultant), and Merck (consultant) outside the submitted work. T. Hembrough reports other from AstraZeneca (employment during this work) during the conduct of the study and NantOmics (employment during past 36 months) outside the submitted work. J.C. Barrett reports personal fees from AstraZeneca (employment and stockholder) during the conduct of the study and outside the submitted work.
M.D. Hellmann reports personal fees and nonfinancial support from AstraZeneca, grants, personal fees, and nonfinancial support from Bristol Myers Squibb, personal fees from Merck, Genentech/Roche, Nektar, Syndax, and Mirati, Blueprint Medicines, and Achilles, personal fees and other from Immunai (including options), Arcus (including options), and Shattuck Labs (including options), and nonfinancial support from Eli Lilly and during the conduct of the study, as well as has a patent related to use of tumor mutation burden to predict response to immunotherapy pending and licensed to PGDx (PCT/US2015/062208). No potential conflicts of interest were disclosed by the other authors.
REFERENCES
- 1.Wei SC, Duffy CR, Allison JP. Fundamental mechanisms of immune checkpoint blockade therapy. Cancer Discov 2018;8:1069–86. [DOI] [PubMed] [Google Scholar]
- 2.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Durvalumab after chemoradiotherapy in stage III non–small-cell lung cancer. N Engl J Med 2017;377:1919–29. [DOI] [PubMed] [Google Scholar]
- 3.Reck M Pembrolizumab as first-line therapy for metastatic non-small-cell lung cancer. Immunother 2018;10:93–105. [DOI] [PubMed] [Google Scholar]
- 4.Brahmer J, Reckamp KL, Baas P, Crinò L, Eberhardt WEE, Poddubskaya E, et al. Nivolumab versus docetaxel in advanced squamous-cell non–small-cell lung cancer. N Engl J Med 2015;373:123–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Garassino MC, Cho BC, Kim JH, Mazieres J, Vansteenkiste J, Lena H, et al. Durvalumab as third-line or later treatment for advanced non-small-cell lung cancer (ATLANTIC): an open-label, single-arm, phase 2 study. Lancet Oncol 2018;19:521–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Powles T, O’Donnell PH, Massard C, Arkenau HT, Friedlander TW, Hoimes CJ, et al. Efficacy and safety of durvalumab in locally advanced or metastatic urothelial carcinoma: updated results from a phase 1/2 open-label study. JAMA Oncol 2017;3:e172411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bellmunt J, Bajorin DF. Pembrolizumab for advanced urothelial carcinoma. N Engl J Med 2017;376:2304. [DOI] [PubMed] [Google Scholar]
- 8.Powles T, Duran I, van der Heijden MS, Loriot Y, Vogelzang NJ, De Giorgi U, et al. Atezolizumab versus chemotherapy in patients with platinum-treated locally advanced or metastatic urothelial carcinoma (IMvigor211): a multicentre, open-label, phase 3 randomised controlled trial. Lancet 2018;391:748–57. [DOI] [PubMed] [Google Scholar]
- 9.Hellmann MD, Antonia SJ, Balmanoukian AS, Brahmer J, Ou S-H, Kim S-W, et al. Updated overall survival and safety profile of durvalumab monotherapy in advanced NSCLC. J Clin Oncol 2018; 36:169. [Google Scholar]
- 10.Wolchok JD, Chiarion-Sileni V, Gonzalez R, Rutkowski P, Grob J-J, Cowey CL, et al. Overall survival with combined nivolumab and ipilimumab in advanced melanoma. N Engl J Med 2017;377: 1345–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Rini BI, Plimack ER, Stus V, Gafanov R, Hawkins R, Nosov D, et al. Pembrolizumab plus axitinib versus sunitinib for advanced renal-cell carcinoma. N Engl J Med 2019;380:1116–27. [DOI] [PubMed] [Google Scholar]
- 12.Robert C, Schachter J, Long GV, Arance A, Grob JJ, Mortier L, et al. Pembrolizumab versus ipilimumab in advanced melanoma. N Engl J Med 2015;372:2521–32. [DOI] [PubMed] [Google Scholar]
- 13.Ansell SM, Lesokhin AM, Borrello I, Halwani A, Scott EC, Gutierrez M, et al. PD-1 blockade with nivolumab in relapsed or refractory Hodgkin’s lymphoma. N Engl J Med 2014;372:311–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Fuchs CS, Doi T, Jang RW, Muro K, Satoh T, Machado M, et al. Safety and efficacy of pembrolizumab monotherapy in patients with previously treated advanced gastric and gastroesophageal junction cancer: phase 2 clinical KEYNOTE-059 trial. JAMA Oncol 2018;4:e180013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schmid P, Adams S, Rugo HS, Schneeweiss A, Barrios CH, Iwata H, et al. Atezolizumab and nab-paclitaxel in advanced triple-negative breast cancer. N Engl J Med 2018;379:2108–21. [DOI] [PubMed] [Google Scholar]
- 16.El-Khoueiry AB, Sangro B, Yau T, Crocenzi TS, Kudo M, Hsu C, et al. Nivolumab in patients with advanced hepatocellular carcinoma (CheckMate 040): an open-label, non-comparative, phase 1/2 dose escalation and expansion trial. Lancet 2017;389:2492–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Cohen EEW, Soulières D, Le Tourneau C, Dinis J, Licitra L, Ahn M-J, et al. Pembrolizumab versus methotrexate, docetaxel, or cetuximab for recurrent or metastatic head-and-neck squamous cell carcinoma (KEYNOTE-040): a randomised, open-label, phase 3 study. Lancet 2019;393:156–67. [DOI] [PubMed] [Google Scholar]
- 18.Reck M, Rodríguez-Abreu D, Robinson AG, Hui R, Csőszi T, Fülöp A, et al. Pembrolizumab versus chemotherapy for PD-L1–positive non–small-cell lung cancer. N Engl J Med 2016;375:1823–33. [DOI] [PubMed] [Google Scholar]
- 19.Le DT, Durham JN, Smith KN, Wang H, Bartlett BR, Aulakh LK, et al. Mismatch repair deficiency predicts response of solid tumors to PD-1 blockade. Science 2017;357:409–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Kluger HM, Zito CR, Turcu G, Baine MK, Zhang H, Adeniran A, et al. PD-L1 studies across tumor types, its differential expression and predictive value in patients treated with immune checkpoint inhibitors. Clin Cancer Res 2017;23:4270–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Maron SB, Chase LM, Lomnicki S, Kochanny S, Moore KL, Joshi S, et al. Circulating tumor DNA sequ6encing analysis of gastroesophageal adenocarcinoma. Clin Cancer Res 2019;25: 7098–112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Cohen JD, Li L, Wang Y, Thoburn C, Afsari B, Danilova L, et al. Detection and localization of surgically resectable cancers with a multianalyte blood test. Science 2018;359:926–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early detection of molecular residual disease in localized lung cancer by circulating tumor DNA profiling. Cancer Discov 2017;7:1394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sabari JK, Offin M, Stephens D, Ni A, Lee A, Pavlakis N, et al. A prospective study of circulating tumor DNA to guide matched targeted therapy in lung cancers. J Natl Cancer Inst 2018;111: 575–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Gandara DR, Paul SM, Kowanetz M, Schleifman E, Zou W, Li Y, et al. Blood-based tumor mutational burden as a predictor of clinical benefit in non-small-cell lung cancer patients treated with atezolizumab. Nat Med 2018;24:1441–8. [DOI] [PubMed] [Google Scholar]
- 26.Hyman DM, Smyth LM, Donoghue MTA, Westin SN, Bedard PL, Dean EJ, et al. AKT inhibition in solid tumors with AKT1 mutations. J Clin Oncol 2017;35:2251–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature 2017;545:446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Cabel L, Proudhon C, Romano E, Girard N, Lantz O, Stern MH, et al. Clinical potential of circulating tumour DNA in patients receiving anticancer immunotherapy. Nat Rev Clin Oncol 2018;15: 639–50. [DOI] [PubMed] [Google Scholar]
- 29.Takahashi T, Tateishi A, Bychkov A, Fukuoka J. Remarkable alteration of PD-L1 expression after immune checkpoint therapy in patients with non-small-cell lung cancer: two autopsy case reports. Int J Mol Sci 2019;20:2578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Snyder A, Morrissey MP, Hellmann MD. Use of circulating tumor DNA for cancer immunotherapy. Clin Cancer Res 2019;25:6909–15. [DOI] [PubMed] [Google Scholar]
- 31.Raja R, Kuziora M, Brohawn PZ, Higgs BW, Gupta A, Dennis PA, et al. Early reduction in ctDNA predicts survival in patients with lung and bladder cancer treated with durvalumab. Clin Cancer Res 2018;24:6212–22. [DOI] [PubMed] [Google Scholar]
- 32.Cristescu R, Mogg R, Ayers M, Albright A, Murphy E, Yearley J, et al. Pan-tumor genomic biomarkers for PD-1 checkpoint blockade-based immunotherapy. Science 2018;362:eaar3593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Taube JM, Anders RA, Young GD, Xu H, Sharma R, McMiller TL, et al. Colocalization of inflammatory response with B7-h1 expression in human melanocytic lesions supports an adaptive resistance mechanism of immune escape. Sci Transl Med 2012;4:127–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Goldberg SB, Narayan A, Kole AJ, Decker RH, Teysir J, Carriero NJ, et al. Early assessment of lung cancer immunotherapy response via circulating tumor DNA. Clin Cancer Res 2018;24:1872–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Anagnostou V, Forde PM, White JR, Niknafs N, Hruban C, Naidoo J, et al. Dynamics of tumor and immune responses during immune checkpoint blockade in non-small cell lung cancer. Cancer Res 2019; 79:1214–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dawson S-J, Tsui DWY, Murtaza M, Biggs H, Rueda OM, Chin S-F, et al. Analysis of circulating tumor DNA to monitor metastatic breast cancer. N Engl J Med 2013;368:1199–209. [DOI] [PubMed] [Google Scholar]
- 37.Khagi Y, Goodman AM, Daniels GA, Patel SP, Sacco AG, Randall JM, et al. Hypermutated circulating tumor DNA: correlation with response to checkpoint inhibitor-based immunotherapy. Clin Cancer Res 2017;23:5729–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Pécuchet N, Zonta E, Didelot A, Combe P, Thibault C, Gibault L, et al. Base-position error rate analysis of next-generation sequencing applied to circulating tumor DNA in non-small cell lung cancer: a prospective study. PLoS Med 2016;13:e1002199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Santiago-Walker A, Gagnon R, Mazumdar J, Casey M, Long GV, Schadendorf D, et al. Correlation of BRAF mutation status in circulating-free DNA and tumor and association with clinical outcome across four BRAFi and MEKi clinical trials. Clin Cancer Res 2016;22:567–74. [DOI] [PubMed] [Google Scholar]
- 40.Schwaederlé MC, Patel SP, Husain H, Ikeda M, Lanman RB, Banks KC, et al. Utility of genomic assessment of blood-derived circulating tumor DNA (ctDNA) in patients with advanced lung adenocarcinoma. Clin Cancer Res 2017;23:5101–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hrebien S, Citi V, Garcia-Murillas I, Cutts R, Fenwick K, Kozarewa I, et al. Early ctDNA dynamics as a surrogate for progression-free survival in advanced breast cancer in the BEECH trial. Ann Oncol 2019;30:945–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Moding EJ, Liu Y, Nabet BY, Chabon JJ, Chaudhuri AA, Hui AB, et al. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small-cell lung cancer. Nat Cancer 2020;1:176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Lipson EJ, Velculescu VE, Pritchard TS, Sausen M, Pardoll DM, Topalian SL, et al. Circulating tumor DNA analysis as a real-time method for monitoring tumor burden in melanoma patients undergoing treatment with immune checkpoint blockade. J Immunother Cancer 2014;2:42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lee JH, Long GV, Menzies AM, Lo S, Guminski A, Whitbourne K, et al. Association between circulating tumor DNA and pseudoprogression in patients with metastatic melanoma treated with anti–programmed cell death 1 antibodies. JAMA Oncol 2018;4: 717–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Rizvi H, Sanchez-Vega F, La K, Chatila W, Jonsson P, Halpenny D, et al. Molecular determinants of response to anti-programmed cell death (PD)-1 and anti-programmed death-ligand 1 (PD-L1) blockade in patients with non-small-cell lung cancer profiled with targeted next-generation sequencing. J Clin Oncol 2018;36:633–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ptashkin RN, Mandelker DL, Coombs CC, Bolton K, Yelskaya Z, Hyman DM, et al. Prevalence of clonal hematopoiesis mutations in tumor-only clinical genomic profiling of solid tumors. JAMA Oncol 2018;4:1589–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Slavin TP, Banks KC, Chudova D, Oxnard GR, Odegaard JI, Nagy RJ, et al. Identification of incidental germline mutations in patients with advanced solid tumors who underwent cell-free circulating tumor DNA sequencing. J Clin Oncol 2018;36:3459–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Balar A, Mahipal A, Grande E, Villalobos VM, Salas S, Kang TW, et al. Abstract CT112: Durvalumab + tremelimumab in patients with metastatic urothelial cancer. Cancer Res 2018;78:CT112. [Google Scholar]
- 49.Antonia SJ, Balmanoukian A, Brahmer J, Ou S-HI, Hellmann MD, Kim S-W, et al. Clinical activity, tolerability, and long-term follow-up of durvalumab in patients with advanced NSCLC. J Thoracic Oncol 2019;14:1794–806. [DOI] [PubMed] [Google Scholar]
- 50.Odegaard JI, Vincent JJ, Mortimer S, Vowles JV, Ulrich BC, Banks KC, et al. Validation of a plasma-based comprehensive cancer genotyping assay utilizing orthogonal tissue- and plasma-based methodologies. Clin Cancer Res 2018;24:3539–49. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.