

Abstract
Aims
Amyloid‐β (Aβ) oligomers trigger synaptic degeneration that precedes plaque and tangle pathology. However, the signalling molecules that link Aβ oligomers to synaptic pathology remain unclear. Here, we addressed the potential role of RAPGEF2 as a novel signalling molecule in Aβ oligomer‐induced synaptic and cognitive impairments in human‐mutant amyloid precursor protein (APP) mouse models of Alzheimer's disease (AD).
Methods
To investigate the role of RAPGEF2 in Aβ oligomer‐induced synaptic and cognitive impairments, we utilised a combination of approaches including biochemistry, molecular cell biology, light and electron microscopy, behavioural tests with primary neuron cultures, multiple AD mouse models and post‐mortem human AD brain tissue.
Results
We found significantly elevated RAPGEF2 levels in the post‐mortem human AD hippocampus. RAPGEF2 levels also increased in the transgenic AD mouse models, generating high levels of Aβ oligomers before exhibiting synaptic and cognitive impairment. RAPGEF2 upregulation activated the downstream effectors Rap2 and JNK. In cultured hippocampal neurons, oligomeric Aβ treatment increased the fluorescence intensity of RAPGEF2 and reduced the number of dendritic spines and the intensities of synaptic marker proteins, while silencing RAPGEF2 expression blocked Aβ oligomer‐induced synapse loss. Additionally, the in vivo knockdown of RAPGEF2 expression in the AD hippocampus prevented cognitive deficits and the loss of excitatory synapses.
Conclusions
These findings demonstrate that the upregulation of RAPGEF2 levels mediates Aβ oligomer‐induced synaptic and cognitive disturbances in the AD hippocampus. We propose that an early intervention regarding RAPGEF2 expression may have beneficial effects on early synaptic pathology and memory loss in AD.
Keywords: Alzheimer's disease, amyloid‐beta, GTPase, memory, Rap, synapse
The authors have identified that the upregulation of RAPGEF2 levels mediates early synaptic and cognitive dysfunction in the 3xTg‐AD mouse model of Alzheimer's disease (AD). They showed that oligomeric Aβ treatment increases the fluorescence intensity of RAPGEF2 and reduces the number of dendritic spines and the intensities of synaptic markers, while silencing RAPGEF2 in the AD mouse hippocampus blocked loss of excitatory synapses and cognitive impairments. This study highlights that early intervention regarding RAPGEF2 expression may have beneficial effects on early synaptic pathology and memory loss in AD.
Abbreviations
- AD
Alzheimer's disease
- APP
amyloid precursor protein
- Aβ
amyloid‐β
- CaMKIIα
calcium/calmodulin‐dependent protein kinase type II subunit alpha
- ERK
extracellular signal‐regulated kinases
- GFP
green fluorescent protein
- JNK
c‐Jun N‐terminal kinases
- MAPK
mitogen‐activated protein kinase
- MAPT
microtubule‐associated protein tau
- PSD
postsynaptic density
- PSEN1
presenilin‐1
- RBD
Ras‐binding domain
- TG
transgenic
- WT
wild‐type
INTRODUCTION
Alzheimer's disease (AD) is the most common and devastating type of dementia, with no treatment currently available to halt its progression. A number of studies support the assumption that AD is the result of synaptic failure1, 2 and that soluble amyloid‐β (Aβ) oligomers cause AD synaptic and behavioural pathogenesis.3, 4 Indeed, synaptic dysfunction precedes Aβ plaque and neurofibrillary tangle pathology as well as cognitive impairments in AD.5, 6 While synapse dysfunction and memory impairments correlate poorly with plaque burden, soluble Aβ oligomer levels and synaptic aberrations are thought to be the best indicators of the early stages of memory loss in AD.7, 8 Several lines of evidence indicate that Aβ oligomer levels are markedly elevated in the brains of AD patients and in AD mouse models expressing human‐mutant amyloid precursor protein (APP).9, 10 These elevated levels trigger synaptic degeneration at the level of the dendritic spines,11, 12 which are tiny membrane protrusions that receive most of the excitatory inputs in the brain.13 It has also been suggested that excessive neuronal activity related to stress or seizures promotes Aβ production in the brain.14, 15, 16 Together, these studies highlight the crucial role of Aβ oligomers in initiating synaptopathy and early memory loss in AD.17 However, the signalling molecules that link Aβ oligomers to synaptic pathology are not well understood.
Rap proteins, members of the Ras family of small GTPases, play pivotal roles in neural development and synaptic plasticity in the brain.18, 19, 20, 21 RAPGEF2 (also named CNrasGEF, nRapGEP, PDZ‐GEF1 and RA‐GEF‐1) is a neural‐specific activator of Rap1 and Rap222, 23 and associates with the synaptic scaffolding protein S‐SCAM.24 Early studies have shown that RAPGEF2‐Rap signalling is involved in the activity‐dependent remodelling of dendritic spines, AMPA receptor trafficking and synaptic plasticity.21, 25, 26, 27, 28 Notably, a recent genetic study identified RAPGEF2 involvement in the pathogenesis of epilepsy.29 Because a high incidence of seizures is commonly observed in AD patients and AD mouse models,30, 31, 32 and neuronal hyperactivity increases Aβ production in vitro and in vivo,14, 15, 16 we postulated that the dysregulation of RAPGEF2 may contribute to the process of synaptic degeneration observed in AD.
Here, we report that RAPGEF2 acts as a molecular link between Aβ oligomers and synaptic aberrations. We observed that RAPGEF2 levels were highly upregulated in the post‐mortem human AD hippocampus and in multiple AD mouse models, generating high levels of Aβ oligomers. This Aβ oligomer‐mediated stimulation of RAPGEF2 activated its downstream targets Rap2 and JNK (c‐Jun N‐terminal kinases) in vivo. In cultured hippocampal neurons, the treatment of Aβ oligomers increased the fluorescence intensity of RAPGEF2, with a concomitant reduction in spine numbers and intensities of synaptic marker proteins compared to the controls. Importantly, silencing RAPGEF2 expression prevented Aβ oligomer‐induced synaptic and cognitive impairments in 3xTg‐AD mouse model. Taken together, our data suggest that early intervention regarding RAPGEF2 expression may be a potential strategy that helps mitigate synaptic and cognitive dysfunctions in AD.
MATERIALS AND METHODS
Antibodies and reagents
The following antibodies were used in this study: mouse monoclonal GFP (#ab1218, Abcam), mouse monoclonal β‐amyloid, 1–16 antibody 6E10 (#803002, BioLegend) and mouse monoclonal Rap2 (#610216, BD Biosciences). The antibodies and their recognition domains against RAPGEFs are listed in Table 1. The specificity of these RAPGEF2 antibodies were tested in hippocampal neurons and heterologous cells overexpressing RAPGEF2 and the highly homologous protein RAPGEF6 (Figure S1). Antibodies against phospho‐p38 (#4511), p38 (#9212), phospho‐JNK (#9255), JNK (#9252), Rap1 (#2399) and β‐tubulin (#2146) were purchased from Cell Signaling Technology. The rabbit polyclonal GFP antibody (#A11122) and Alexa 488, 594 and 555 conjugated goat/donkey anti‐mouse/rabbit secondary antibodies were obtained from Invitrogen. The JNK inhibitor SP600125 was purchased from Calbiochem.
TABLE 1.
RAPGEFs antibodies used in this study
| No. | RAPGEFs antibody | # Catalog | Company | Immunogen (epitope) |
|---|---|---|---|---|
| 1 | Mouse monoclonal RAPGEF1 | TA506838 | OriGene | Full length human recombinant protein of human RAPGEF1 (NP_941372) |
| 2 | Rabbit polyclonal RAPGEF2 | A301‐966A | Bethyl Laboratories | 975–1025 amino acid of human RAPGEF2 (NP_055062.1, GeneID 9693) |
| 3 | Mouse polyclonal RAPGEF2 | H00009693‐A01 | Abnova | 1398–1487 amino acid of human RAPGEF2 (XP_376350) |
| 4 | Mouse monoclonal RAPGEF2 | H00009693‐M01 | ||
| 5 | Rabbit polyclonal RAPGEF3 (Epac1) | ab21236 | Abcam | Amino acids 526‐541 of Epac1 |
| 6 | Rabbit monoclonal RAPGEF4 (Epac2) | ab193665 | Abcam | Recombinant fragment within Human Epac2 aa 900 to the C‐terminus. |
| 7 | Rabbit monoclonal RAPGEF5 | ab129008 | Abcam | Synthetic peptide within Human RAPGEF5 (N terminal) |
| 8 | Goat polyclonal RAPGEF6 | SC‐69596 | Santa Cruz | Internal region of human RAPGEF6 |
Oligomeric Aβ preparation
To prepare Aβ oligomers, lyophilised β‐amyloid (1–42) peptide powder was purchased from AnaSpec. According to the manufacturer's protocol, the β‐amyloid peptide was dissolved in 20 mM HEPES/150 mM NaCl (pH 7.2) at a concentration of 50 μM and sonicated until completely solubilised. Oligomers were acquired by diluting the Aβ peptide solution to 25 μM with phenol red‐free DMEM‐F12 (Caisson Labs) and incubating this diluted Aβ solution at 4°C for 24 h.
Primary neuron culture and transfection
Primary rat hippocampal cultures were prepared from embryonic day (E) 18–19 Sprague–Dawley fetal rats as previously described.25 For mouse cortical and hippocampal neuron cultures, brains from E 16–18 mouse embryos were isolated. Briefly, after dissecting out the hippocampal tissue, trypsinised cells were dissociated by gently triturating with flame‐polished Pasteur pipettes. Cells were seeded on a precoated plate with poly‐D‐lysine (100 μg/ml, Sigma‐Aldrich) and laminin (2 μg/ml, Roche) at a density of 4–5 × 104 cells/well. The neurons were grown in Neurobasal medium (Gibco) supplemented with SM1 (Stem Cell Technologies), 0.5 mM glutamine, and 25 μM glutamate for 18–21 days in vitro (DIV). For transfections, DNA constructs were transfected into hippocampal neurons (18–21 DIV) using Lipofectamine 2000 reagent (Thermo Fisher Scientific) for 2–3 days.
Human brain tissue preparation
The human hippocampal tissue samples used in this study were obtained from the Netherlands Brain Bank (Project #1009). Experiments using human tissue were approved by the Institutional Review Board of the Korea Brain Research Institute (201610‐BR‐004‐01). The hippocampal samples were stored at −80°C until use. Information about the human tissue used in this study is provided in Table 2.
TABLE 2.
Post‐mortem human hippocampal samples
| Group | No. | Clinical Diagnosis | Gender | Age | Amyloid | Braak | ApoE (Presence of ApoE alleles) |
|---|---|---|---|---|---|---|---|
| Controls | 1 | Nondemented control | M | 89 | 0 | 2 | Unknown |
| 2 | Nondemented control | F | 92 | 0 | 3 | 43 | |
| 3 | Nondemented control | F | 78 | A | 1 | 33 | |
| 4 | Nondemented control | F | 60 | 0 | 0 | 32 | |
| 5 | Nondemented control | F | 93 | 0 | 2 | 33 | |
| 6 | Nondemented control | F | 82 | A | 1 | 33 | |
| 7 | Nondemented control | M | 73 | A | 2 | 33 | |
| 8 | Nondemented control | F | 72 | A | 1 | 33 | |
| AD patients | 1 | Alzheimer's disease | F | 85 | C | 6 | 44 |
| 2 | Alzheimer's disease | M | 60 | C | 6 | 43 | |
| 3 | Alzheimer's disease | M | 84 | C | 6 | 43 | |
| 4 | Alzheimer's disease | M | 74 | C | 6 | 43 | |
| 5 | Alzheimer's disease | F | 61 | C | 6 | 44 | |
| 6 | Alzheimer's disease | F | 71 | C | 6 | 44 | |
| 7 | Alzheimer's disease | F | 91 | C | 6 | 42 | |
| 8 | Alzheimer's disease | M | 81 | C | 6 | 43 | |
| 9 | Alzheimer's disease | F | 88 | C | 6 | 43 | |
| 10 | Alzheimer's disease | F | 78 | C | 6 | 43 | |
| 11 | Alzheimer's disease | F | 90 | C | 6 | 44 | |
| 12 | Alzheimer's disease | M | 78 | C | 6 | 43 |
This table shows the characteristics of the human (control and AD) individuals whose brain tissues were analysed in this study. [Amyloid]: 0, absent; A, mild (basal frontal and temporal lobes); B, moderate (association neocortex and hippocampus); C, severe (primary cortices, subcortical nuclei and cerebellum).
Western blot analysis
Cultured hippocampal neurons and brain tissue from mice and humans were lysed in ice‐cold RIPA buffer (150 mM NaCl, 10 mM Na2HPO4, pH 7.2, 0.5% sodium deoxycholate and 1% Nonidet P‐40) containing a protease inhibitor cocktail (GenDepot) and a phosphatase inhibitor cocktail (Sigma‐Aldrich). The lysates were centrifuged at 15,000×g at 4°C for 20 min. Protein concentrations of the supernatants were determined using BCA reagent (Thermo Fisher Scientific). The samples were separated on 8%–12% polyacrylamide gels, transferred to a nitrocellulose membrane (GE Healthcare Life Sciences) and blocked with 5% non‐fat dry milk or 3% bovine serum albumin (Sigma‐Aldrich) in 1× TBS buffer solution containing 0.1% Tween‐20. Blots were incubated with primary antibodies at 4°C overnight, followed by horseradish peroxidase‐conjugated secondary antibodies (Cell Signaling Technology) or infrared dye‐conjugated secondary antibodies (Li‐Cor Bioscience) at room temperature for 1 h. Blots were visualised using a Fusion Fx7 ECL system (Vilber) or an Odyssey CLx Infrared Imaging System (Li‐Cor Bioscience). Protein band intensities in the western blot were quantitatively measured using ImageJ software (National Institutes of Health).
Active Rap pull‐down assay
Mouse cortical tissue was homogenised in ice‐cold RIPA buffer containing a protease inhibitor cocktail (GenDepot) and a phosphatase inhibitor cocktail (Sigma‐Aldrich). After collecting supernatants from the brain homogenates by centrifugation, the lysates (1.5 mg) were incubated with 40 μl RalGDS RBD agarose beads (Abcam) at 4°C for 3 h. Pellets were washed three times and resuspended in 40 μl 2× reducing SDS sample buffer (62.5 mM Tris‐HCl, pH 6.8, 2% SDS, 10% glycerol, 0.1% bromophenol blue and 0.5% β‐mercaptoethanol). GTP‐bound active Rap1 and Rap2 levels were detected by western blotting.
Immunocytochemistry
Primary cultured hippocampal neurons were fixed with 4% paraformaldehyde (PFA)/4% sucrose for analysis of the spine or with 4% PFA/4% sucrose followed by methanol (−20°C) for immunofluorescent labelling of RAPGEF2. Neurons were incubated with primary antibodies in a GDB solution (30 mM phosphate buffer, pH 7.4, containing 0.1% gelatine, 0.3% Triton X‐100, 450 mM NaCl) at 4°C overnight and then with Alexa 488‐ and Alexa 594‐conjugated secondary antibodies (Thermo Fisher Scientific) at room temperature for 1 h; neurons were then mounted on a glass slide with VECTASHIELD mounting solution (Vector Labs).
To quantify the immunofluorescence intensity of RAPGEF2, VGLUT1 and PSD‐95, we selected at least two dendritic segments (30 μm in length each) in the individual GFP‐positive neurons. The integrated intensity was measured at a constant threshold value using the region measurement tool of MetaMorph Software (Molecular Devices). For the analysis of dendritic spine structures, we focused on linear secondary dendritic segments of pyramidal neurons (at least 30 μm in length). Images were acquired with a TI‐RCP confocal microscope (Nikon). Z‐stack images were collected at 0.4 μm intervals and then compressed into a 2D image with maximal intensity projection.
Animals
All mutants and their corresponding control mice were purchased from the Jackson Laboratory. The 3xTg‐AD transgenic mice (MMRRC Stock No: 34830‐JAX) were maintained as homozygotes. The 5xFAD mice (MMRRC Stock No: 34840‐JAX) were maintained as hemizygotes by crossing with wild‐type (WT) mice on a C57BL/6 J Χ SJL background strain. To generate rTg (TauP301L) 4510 mice, tetO‐MAPT*P301L mice (FVB‐Fgf14Tg(tetO‐MAPT*P301L)4510 Kha/JlwsJ, Stock No. 015815) were crossed with mice from the CaMKII‐tTA mouse line (B6.Cg‐Tg(Camk2a‐tTA)1Mmay/DboJ, Stock No. 007004). All mice were housed 4 to 5 per cage in a room maintained at 21 ± 1°C, with an alternating 12/12 h light/dark cycle and free access to food and water.
shRNA constructs and virus production
The following oligonucleotides (5′‐3′) were inserted into the pLL 3.7 vector, which simultaneously expresses RNAi‐inducing shRNAs and GFP under the U6 and CMV promoters, respectively (Rubinson et al., 2003): scrambled shRNA, GCAAACGCTCGACATTAA; rat RAPGEF2‐shRNA, GGACCCAACATTCATAGA (NM_001107684.1; 709–726 bp) and mouse RAPGEF2‐shRNA, GCTGGAACCATTGTGTTA (NM_001099624.3; 520–537 bp). Lentiviral particles were produced according to the manufacturer's instructions (ViraPower Lentiviral Expression System, Thermo Fisher Scientific). Briefly, a mixture of plasmids was transfected into HEK293FT packaging cells. The virus‐containing medium was harvested 48 or 72 h after transfection and subsequently precleaned with 3000×g centrifugation and 0.45 μm filtration (Merck Millipore). The virus‐containing medium was overlaid on sucrose‐containing buffer and centrifuged at 120,000×g at 4 °C for 2 h. After ultracentrifugation, the supernatants were carefully removed and resuspended in phosphate buffered saline.
Stereotaxic virus injection
Two‐month‐old non‐transgenic B6129SF2/J control and 3xTg‐AD transgenic mice were anaesthetised with isoflurane, and 1 µl (pll3.7, 8.9 × 10^10 IU/ml; shRAPGEF2, 8.3 × 10^10 IU/ml) of the viral solution was injected bilaterally into the hippocampal CA1 region with a glass pipette at a flow rate of 0.2 µl/min according to stereotaxic coordinates (Bregma: antero‐posterior −2.0 mm, medial‐lateral ±1.5 mm, dorsoventral −1.4 mm from the dura). Glass pipettes used for microinjection were kept in place for 5 min after the injection had been completed to ensure complete absorption of the viral particles. After recovery in a heated chamber, the mice were returned to their home cages. Behavioural tests were performed 8 weeks after virus injections. Mice that had incorrect injection sites were excluded from the behavioural data analysis.
Behavioural testing
Mouse behaviours were recorded using the video tracking software SMART 3.0 (Panlab). For contextual fear conditioning, the sound‐attenuating apparatus was equipped with a stainless steel foot‐shock grid and interchangeable walls to allow context changes. All mice were habituated in the conditioning chamber for 3 min to reduce basal freezing levels. On the conditioning day, 24 h after habituation, all subjects were placed back into the same chamber. Mice were allowed to explore the chamber for 3 min, and then, they received three electric foot shocks (0.5 mA, 2 s) at intervals of 1 min as an unconditioned stimulus (US). After the last shock, the mice were removed from the chamber. On the contextual test day, all subjects were placed in the same chamber and were exposed to the same context without exposure to the adverse stimulus. The duration of freezing behaviours was automatically calculated for 5 min (freezing criterion: 1 s). Mice with freezing ratio values of 5 or less were excluded.
Transmission electron microscopy
Four‐month‐old male WT and 3xTg‐AD mice transduced with lentivirus expressing either control or RAPGEF2‐shRNA (n = 3 per group) were perfused with 2% PFA and 2.5% glutaraldehyde in 0.15 M cacodylate buffer (pH 7.4). Using a vibratome, 150‐µm coronal sections were obtained, and the hippocampal CA1 stratum radiatum was dissected out. Tissue blocks were prepared using standard procedures for TEM as described previously.33 Briefly, tissue was post‐fixed in 2% OsO4 for 1 h, en bloc stained with 1% uranyl acetate, dehydrated and then embedded in Epon 812 resin (EMS). Seventy‐nanometre‐thick sections were collected on 200‐mesh grids and stained with uranyl acetate followed by lead citrate. For each animal, 30 images were recorded on a Tecnai F20 TEM (FEI) (2500× magnification, 120 kV). The counting frame (6 × 6 μm2) was placed on each image, and synapses with a clear PSD and presynaptic vesicles were identified. Synapses were classified into either asymmetric (excitatory) or symmetric (inhibitory) types based on their distinct PSD shapes. All analyses were performed blinded to the experimental conditions.
Statistical analysis
All data represent the mean ± SEM of at least three independent experiments, unless otherwise indicated. For two‐sample comparisons, two‐tailed unpaired Student's t‐tests were used. For four‐sample comparisons, one‐way ANOVA, two‐way ANOVA and Tukey's multiple‐comparisons test were used as explained in the figure legends. Statistically significant differences were determined as p < 0.05.
RESULTS
RAPGEF2 is upregulated in human and mouse AD brains
RAPGEF2 expression is highly enriched in the hippocampus,22, 34 and is critical for learning and memory making it especially vulnerable to damage in the early stages of AD. To test whether RAPGEF2 is involved in the pathophysiological mechanisms of AD, we examined the expression levels of RAPGEF2 in the lysates of post‐mortem human AD hippocampal tissue using western blot analysis (Figure 1A,B). Compared to non‐AD controls, RAPGEF2 levels were upregulated in hippocampal lysates from AD patients (Figure 1A). The quantitation of RAPGEF2 band intensity normalised to housekeeping GAPDH signals displayed a significant increase in the levels of RAPGEF2 in human AD brains by approximately twofold (Figure 1B).
FIGURE 1.
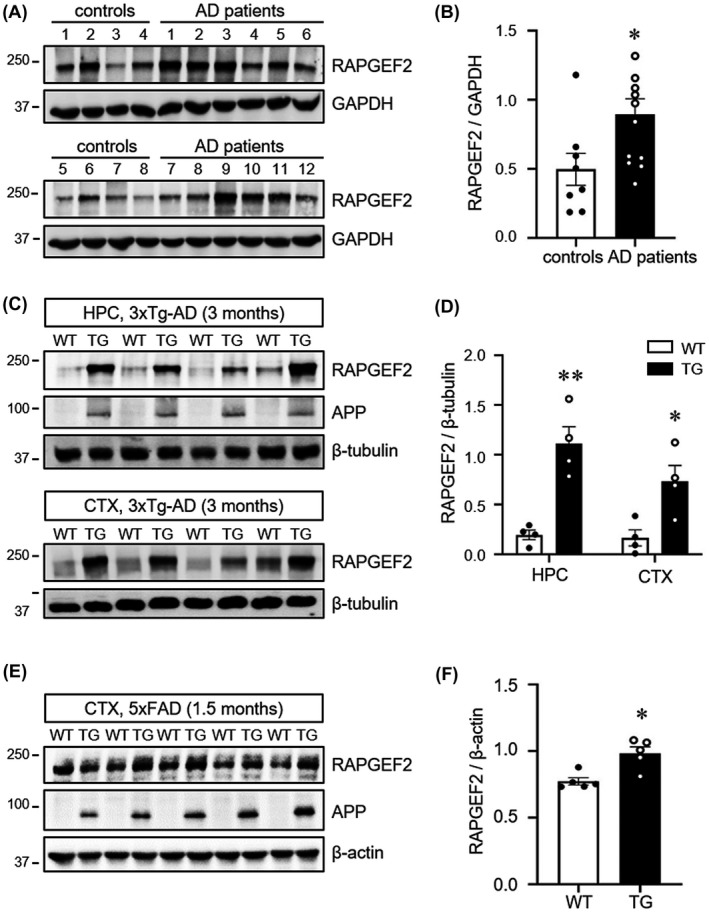
Elevated levels of RAPGEF2 in the post‐mortem human AD hippocampus and AD mouse brains. A, Post‐mortem human hippocampal tissue from non‐patients (controls) and AD patients was subjected to western blot analysis for the RAPGEF2 levels. B, Quantification of RAPGEF2 levels normalised to GAPDH (control, n = 8; AD patients, n = 12). C, RAPGEF2 expression levels were analysed at 3 months of age in the hippocampus (HPC) and cortex (CTX) of wild‐type (WT) and 3xTg‐AD mice (TG). D, Quantification of RAPGEF2 levels in the HPC and CTX of 3‐month‐old WT (n = 4; males), and TG mice (n = 4; males). E, RAPGEF2 expression levels were analysed in the cortical lysates of 1.5‐month‐old WT and 5xFAD (TG) mice. Note that the levels of APP are markedly higher in the TG mice. F, Quantification of RAPGEF2 levels normalised to β‐actin (n = 5; 4 males, 1 female). All data are shown as the mean ± SEM. **p < 0.01, *p < 0.05; two‐tailed unpaired Student's t‐test
To identify whether RAPGEF2 levels were also altered in the transgenic mouse models of AD, we performed western blot analysis of the cortical and hippocampal lysates obtained from 3‐ and 12‐month‐old 3xTg‐AD mice (Figure 1C,D and Figure S2), one of the most well‐characterised and widely used AD mouse models that overexpresses three mutations associated with familial AD [APP Swedish, microtubule‐associated protein tau (MAPT) P301L, and presenilin‐1 (PSEN1) M146 V].6 The neuropathological phenotypes of this model include extracellular Aβ deposits in the frontal cortex by 6 months and neurofibrillary tangles in the hippocampus by 12 months and then later in the cortex.6, 35 Hippocampus‐dependent synaptic and cognitive deficits are observed at 4–6 months of age.35, 36 Intriguingly, we found a significant increase in RAPGEF2 levels in 3‐month‐old 3xTg‐AD mouse brains (Figure 1C,D). RAPGEF2 levels in 12‐month‐old mutants also showed an increasing trend without statistical significance, possibly due to the high variability of RAPGEF2 levels in older 3xTg‐AD mice (Figure S2). These observations suggest that the upregulation of RAPGEF2 before the onset of synaptic and cognitive impairments may contribute to the early pathogenic mechanisms of AD.
To corroborate these results and to narrow down the genetic mutations responsible for the upregulation of RAPGEF2, we next employed two additional AD mouse models expressing mutations associated with familial AD (5xFAD for APP and PSEN1 mutations; rTg4510 for MAPT P301L).37, 38 In 5xFAD mice, intraneuronal Aβ was observed in cortical pyramidal neurons at 1.5 months of age, and extracellular amyloid plaques were detected in the hippocampus and cortex of 2‐month‐old animals.39 Evidence of synaptic deficits in 5xFAD mice was obtained at 2–3 months using electrophysiology.40 Furthermore, the loss of synapses and some cognitive impairments were observed at 3–6 months in the 5xFAD mice.41 However, rTg4510 mice expressing human P301L mutant tau in the forebrain by the CaMKIIα (calcium/calmodulin‐dependent protein kinase type II subunit alpha) promoter developed progressive neurofibrillary tangles by 4–5.5 months, impaired CA1 long‐term potentiation at 4.5 months, loss of hippocampal and cortical neurons by >5.5 months, and behavioural impairments between 2 and 6 months of age.37, 42, 43, 44
Western blot analysis revealed significantly increased RAPGEF2 levels in the cortical lysates of the 1.5‐month 5xFAD mice (Figure 1E‐F). In contrast, there were no differences in the RAPGEF2 levels in the cortex and hippocampus between 2.5 and 5.5 months in the rTg4510 and age‐matched control animals (Figure S3). These data indicate that amyloid, but not tau, pathology contributes to enhanced RAPGEF2 expression in 3xTg‐AD brains. Taken together, these results demonstrate that RAPGEF2 levels are commonly upregulated in sporadic human AD and APP transgenic mouse brains and suggest that RAPGEF2 may participate in the early pathological mechanisms of AD.
Oligomeric Aβ induces the upregulation of RAPGEF2
Because amyloid plaques and neurofibrillary tangles have not yet been identified in 3‐month‐old 3xTg‐AD mice or in 1.5‐month‐old 5xFAD mutants,6, 38 we speculated that either full‐length APP or Aβ oligomers may trigger the upregulation of RAPGEF2 expression. Indeed, western blot analysis using the 6E10 antibody (anti‐Aβ, also recognises APP) showed formation of oligomeric Aβ in the cortical lysates of the 1.5‐month‐old 5xFAD mice (Figure S4A).
To further provide direct evidence, we next generated Aβ oligomers by incubating the Aβ peptide for 24 h (Figure 2A) and then examined the RAPGEF2 levels in cultured neurons treated with Aβ oligomers (1 μM) for 6 h. Both western blotting and immunofluorescence staining confirmed heightened levels of endogenous RAPGEF2 in the cortical and hippocampal neurons following Aβ oligomer treatment (Figure 2B‐E). In contrast, we observed an inverse relationship between the amount of full‐length APP (APP770) and RAPGEF2 levels in heterologous cells (Figure S4B), supporting the role of oligomeric Aβ rather than full‐length APP in the upregulation of RAPGEF2. Thus, our results indicate that Aβ oligomers are sufficient to elevate RAPGEF2 expression in neurons.
FIGURE 2.
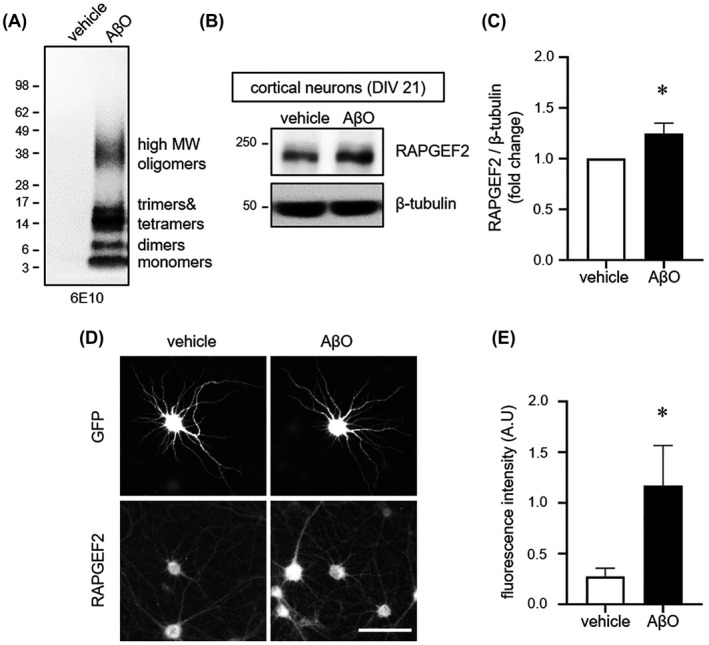
Oligomeric Aβ increases RAPGEF2 levels. A, SDS‐PAGE analysis of oligomeric Aβ. Oligomers were separated by western blotting on a 4%–12% gradient Bis‐Tris gel and immunoblotted with the 6E10 antibody. Oligomeric Aβ (AβO) consisted of monomers (~4 kDa), dimers, trimers, tetramers and high molecular weight (MW) oligomers. No fibrils (>75 kDa) were detected. B, Cultured cortical neurons (DIV 21) treated with vehicle or AβO (1 μM) for 6 h and immunoblotted for RAPGEF2. C, Relative fold change in RAPGEF2 levels (n = 7). D, Representative RAPGEF2 fluorescence images of cultured hippocampal neurons. Neurons (DIV 21) treated with vehicle or oligomeric AβO (1 μM) for 6 h and immunolabelled for GFP and RAPGEF2. Scale, 100 μm. E, Quantification of the integrated intensity of RAPGEF2 in proximal dendrites (n = 12). All data are shown as the mean ± SEM. *p < 0.05; two‐tailed unpaired Student's t‐test
Oligomeric Aβ‐mediated upregulation of RAPGEF2 activates the Rap2‐JNK pathway
The small GTPases Rap1 and Rap2 are well‐established downstream effectors of RAPGEF2.22, 23 At neuronal synapses, Rap1 and Rap2 are involved in different forms of synaptic plasticity through the regulation of specific signalling pathways.28 To test whether Aβ oligomer‐induced RAPGEF2 may selectively activate its downstream signalling targets, active Rap pull‐down assays were performed on cortical lysates of 2.5‐month‐old 3xTg‐AD mice using RalGDS‐RBD agarose beads that pull down active GTP‐bound Rap.
We found a significant increase in the levels of active Rap2 in the 3xTg‐AD mice compared to the WT animals. In contrast, active Rap1 levels were not significantly different between genotypes, due to the high variability of active Rap1 levels (Figure 3A‐D). Because JNK is a known downstream effector of Rap2,27, 28 we next investigated the phosphorylation status of JNK in the 3xTg‐AD and 5xFAD mice. As expected, JNK phosphorylation levels were significantly higher in the cortex of the 2.5‐month‐old 3xTg‐AD and 1.5‐month‐old 5xFAD mice than in the WT controls (Figure 3E,G; Figure S5A‐B). In contrast, the phosphorylation and total levels of p38 MAPK (mitogen‐activated protein kinase), a downstream target of Rap1, did not change in the 3xTg‐AD mice compared to the controls (Figure 3F,H). Additionally, decreased phosphorylation levels of ERK were observed in the 1.5‐month‐old 5xFAD mice, in agreement with a previous finding showing that active Rap2 also reduced extracellular signal‐regulated kinase (ERK) signalling27 (Figure S5C–D).
FIGURE 3.
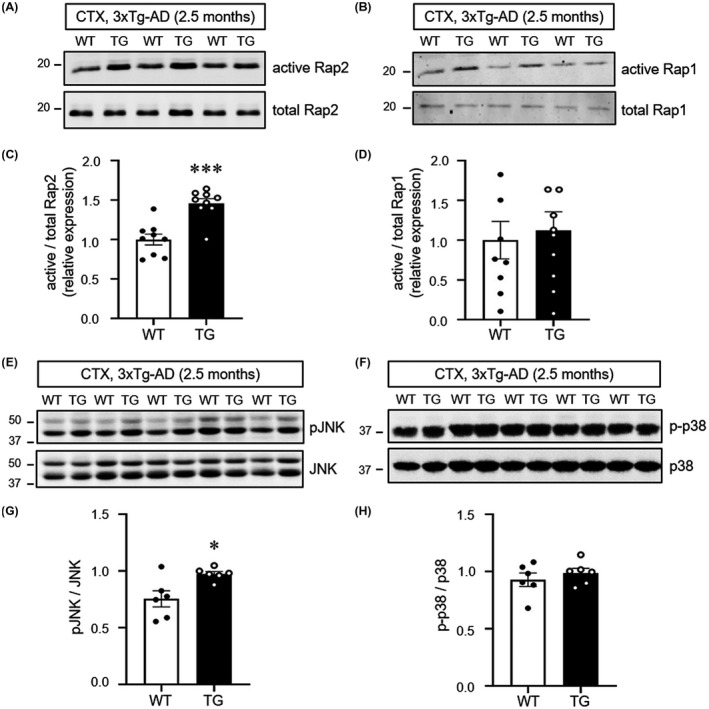
Activation of the small GTPase Rap2‐JNK pathway in 3xTg‐AD mice. A and B, Cortical lysates from 2.5‐month‐old wild‐type (WT) and 3xTg‐AD (TG) mice were incubated with RalGDS‐RBD agarose beads, followed by immunoblotting for Rap2 (A) or Rap1 (B), respectively. C and D, Quantification of active Rap2 (C) and active Rap1 (D) levels normalised to total Rap2 and Rap1 respectively (n = 9; 6 males, 3 females). E and F, Cortical lysates from 2.5‐month‐old wild‐type (WT) and 3xTg‐AD (TG) mice were collected and subjected to western blotting for activation (phosphorylation) of JNK (E) and p38 (F). G and H, Quantification of pJNK (G, n = 6; 5 males, 1 female) and p‐p38 (H, n = 6; 5 males, 1 female) levels normalised to total expression. All data are shown as the mean ± SEM. *p < 0.05, ***p < 0.001; two‐tailed unpaired Student's t‐test
To examine if RAPGEF2 knockdown could decrease JNK activation in neuronal cultures from 3xTG‐AD mice, we transduced lentiviral particles expressing either GFP (pll3.7) or GFP plus shRAPGEF2 (validated in Figure 4A,B and Figure S6) into cultured cortical neurons at DIV 7. After 8 days, cell lysates from WT and 3xTg‐AD mice were used for western blot analysis of RAPGEF2 and MAPK levels. Again, we found that neurons cultured from the 3xTg‐AD mice showed increased levels of RAPGEF2 and phosphorylated JNK compared to cultures from the WT animals (Figure 4A–C). Knockdown of RAPGEF2 in the 3xTg‐AD neurons significantly reduced the elevated levels of phosphorylated JNK (Figure 4A,C). Interestingly, silencing of RAPGEF2 in cultures from the WT animals did not change JNK activation, possibly reflecting very low levels of active JNK in basal conditions to prevent synapse loss. In addition, the phosphorylation of p38 and ERK were largely unaffected by silencing RAPGEF2 in neurons from both the WT and 3xTG‐AD mice (Figure 4D–E). These results further support our finding that RAPGEF2‐mediated activation of JNK may contribute to the early pathogenesis of AD.
FIGURE 4.
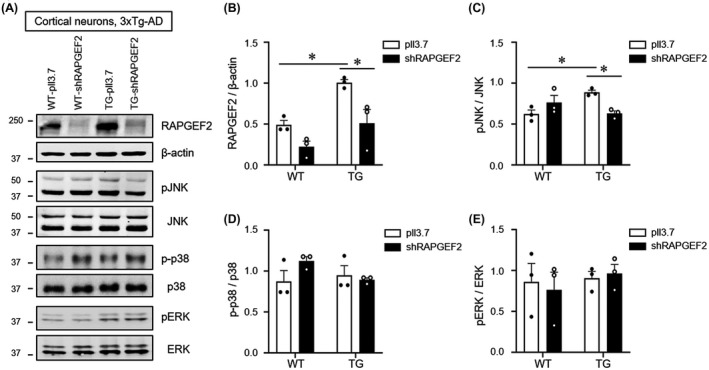
Knockdown of RAPGEF2 decreases JNK activation in 3xTg‐AD mice. A, Lentiviral particles expressing either shRAPGEF2 or a control vector (pll3.7) were transduced into cultured cortical neurons at DIV 7. After 8 days, cell culture lysates from wild‐type (WT) and 3xTg‐AD (TG) mice were used for western blot analysis of RAPGEF2 and MAPK levels (n = 3). B–E, Quantification of RAPGEF2 (B), pJNK (C), pp38 (D), and pERK (E) levels. All data are shown as the mean ± SEM. *p < 0.05; Two‐way ANOVA, Tukey's multiple‐comparison test
Silencing of RAPGEF2 prevents Aβ oligomer‐induced spine loss
Aβ oligomer activates JNK before synaptopathy in hippocampal neurons, and the inhibition of JNK blocks spine loss induced by Aβ oligomer.45, 46 Moreover, transgenic mice expressing constitutively active Rap2 had shorter and fewer dendritic spines in the hippocampus.27 Thus, we reasoned that the Aβ oligomer‐mediated upregulation of RAPGEF2 may participate in the early synaptic pathology of AD.
To test whether silencing of RAPGEF2 could prevent Aβ oligomer‐induced synaptopathy, hippocampal neurons (DIV 18) were transfected with either scrambled shRNA (scrambled) or shRAPGEF2 for 3 days and treated with oligomeric Aβ (AβO, 1 μM) for 10 h before immunostaining. As expected, the results showed that Aβ oligomers significantly reduced spine density, length and size in the control neurons. In contrast, knockdown of RAPGEF2 prevented Aβ oligomer‐mediated reduction in spine number and morphology. In addition, silencing of RAPGEF2 in the absence of Aβ oligomer had no effect on dendritic spine morphology (Figure 5), possibly reflecting very weak RAPGEF2 expression under basal conditions (Figure 5).
FIGURE 5.
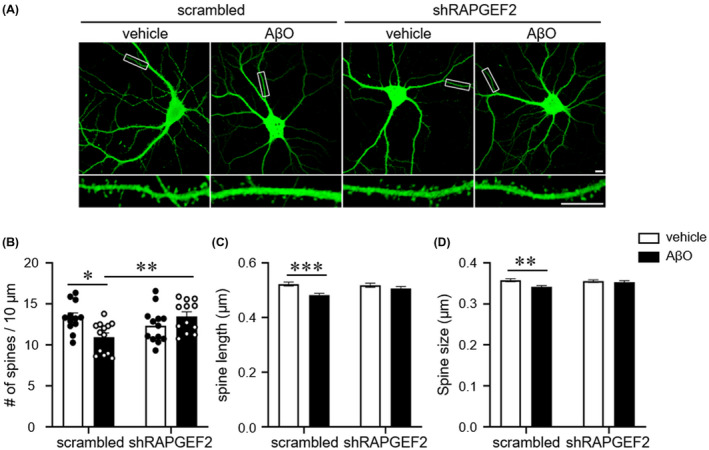
Knockdown of RAPGEF2 halts the AβO‐induced spine loss. A, Representative GFP fluorescence images of cultured hippocampal neurons. Neurons (DIV 18) were transfected with either scrambled shRNA or shRAPGEF2 for 3 days and treated with oligomeric Aβ (AβO, 1 μM) for 10 h before immunostaining. Bottom, Enlarged images of the data enclosed in rectangles at the top. Scale, 10 μm. B–D, Quantification of spine density (B), length (C), and head size (D) (n = 12 neurons in scrambled‐vehicle and shRAPGEF2‐AβO; n = 13 neurons in shRAPGEF2‐vehicle and scrambled‐AβO). All data are shown as the mean ± SEM. *p < 0.05; ** p < 0.01, *** p < 0.001; one‐way ANOVA, Tukey's multiple‐comparison test
To further examine whether Aβ oligomer with or without RAPGEF2 knockdown affects the pre‐ and post‐synaptic structure, we performed immunocytochemistry using the presynaptic marker VGLUT1 as well as the postsynaptic marker PSD‐95. Our results showed that Aβ oligomer treatment significantly decreased the fluorescence intensity of both VGLUT1 and PSD‐95 in the control neurons, while silencing of RAPGEF2 halted the Aβ oligomer‐mediated reduction of these pre‐ and post‐synaptic markers (Figure S7). Together, these results demonstrate that RAPGEF2 knockdown blocks Aβ oligomer‐mediated synaptic impairments in hippocampal neurons.
We next examined whether the JNK inhibitor SP600125 could block the loss of dendritic spines in hippocampal cultures from 3xTg‐AD mice. We found that either knockdown of RAPGEF2 or inhibition of JNK was sufficient to prevent a reduction in synaptic density in cultures from the 3xTg‐AD mice (Figure S8). Furthermore, silencing of RAPGEF2 in the presence of SP600125 failed to further increase the number of spines, indicating that RAPGEF2 and JNK operate by overlapping mechanisms.
RAPGEF2 knockdown alleviates synaptic and cognitive deficits in the AD hippocampus
We then examined whether the in vivo silencing of RAPGEF2 in the hippocampus could prevent synaptic and cognitive impairments in 3xTg‐AD mice. Lentiviral particles expressing RAPGEF2‐shRNA were bilaterally delivered to the hippocampal CA1 regions of 2‐month‐old 3xTg‐AD mice using stereotaxic surgery (Figure 6A,B). At 8 weeks after the viral injections, the mice were subjected to a hippocampus‐dependent contextual fear‐conditioning test.48, 49 Following the behavioural test, the extent of RAPGEF2 downregulation was evaluated using western blot analysis with lysates from the hippocampal CA1 area. We found that viral expression of RAPGEF2 shRNAs for 2 months significantly reduced the levels of RAPGEF2 in both the WT and 3xTg mice (Figure 6C,D). Notably, in vivo silencing of RAPGEF2 in the 3xTg‐AD mice induced no compensatory changes in the levels of other RAPGEFs (Figure S9). In separate groups of animals, we observed a significant deficit in freezing behaviour in the relatively young 4‐month‐old 3xTg‐AD mice compared to their matched WT animals (Figure 6E). More importantly, the knockdown of endogenous RAPGEF2 expression successfully blocked the impairment of context‐dependent freezing behaviour in the 3xTg‐AD mice (Figure 6F), implying that modulation of RAPGEF2 expression prior to synaptic dysfunction is sufficient to retain long‐term fear memory in 3xTg‐AD mice. The WT animals with RAPGEF2 knockdown displayed comparable freezing ratio with the WT controls (Figure 6F).
FIGURE 6.
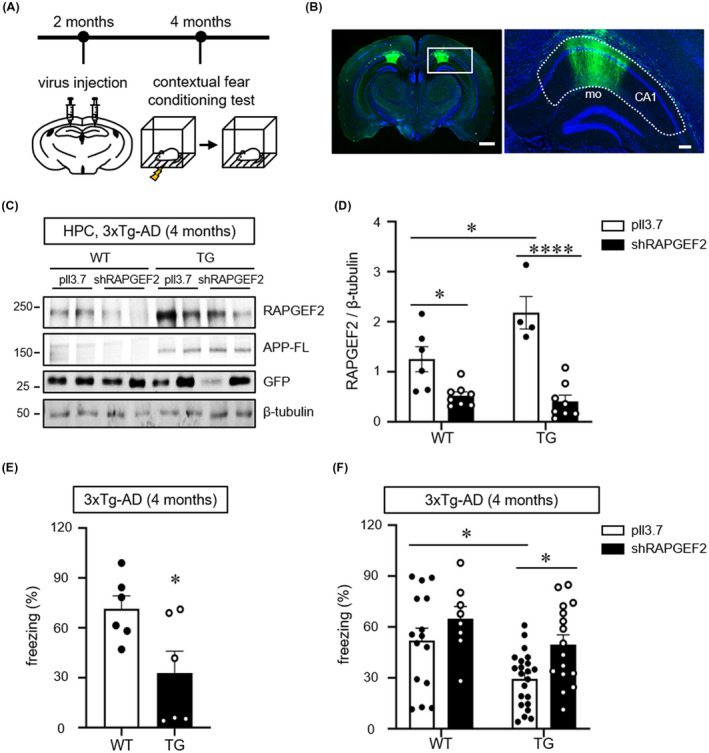
Knockdown of RAPGEF2 rescues fear memory deficits in 4‐month‐old 3xTg‐AD mice. A, Time course of stereotaxic delivery of lentiviral particles and behavioural tests. B, Representative image of GFP expression in the hippocampal CA1 area. Lentiviral particles simultaneously expressing shRAPGEF2 and GFP were bilaterally injected into the hippocampal CA1 area. Hoechst dye was used to stain neuronal nuclei (blue). Scale, 10 μm. Right, higher magnification image enclosed in rectangles at the left. A dotted line indicates the CA1 (mo: molecular layer in dentate gyrus). 53% ± 0.9% area of CA1 region was GFP‐positive (virus transduced) (n = 3). Scale, 200 μm. C, Virus‐mediated knockdown extent of RAPGEF2 in vivo. Lentiviral particles expressing either shRAPGEF2 or a control empty vector (pll3.7) were transduced into hippocampal CA1 area in 2 months of wild‐type (WT) and 3xTg‐AD (TG) mice. After 2 months, hippocampal lysates were used for western blot analysis for RAPGEF2. APP‐FL (full length APP) were used as the marker of TG mice. D, Quantification of RAPGEF2 levels normalised to β‐Tubulin. pll3.7 in WT, n = 6 (5 males, 1 female); shRAPGEF2 in WT, n = 8 (7 males, 1 female); pll3.7 in TG, n = 4 (3 males, 1 female); shRAPGEF2 in TG, n = 8 (7 males, 1 female). E, The contextual fear memory test in 4‐month‐old wild‐type (WT) and 3xTg‐AD (TG) mice (n = 6, males). F, Control (pll3.7) or shRAPGEF2‐expressing viral particles were injected into the hippocampal CA1 area in 2‐month‐old WT and 3xTg‐AD mice. After 2 months, the contextual fear memory test was performed (pll3.7 in WT, n = 15 males; shRAPGEF2 in WT, N = 8 males; pll3.7 in 3xTg‐AD mice, n = 22 males; shRAPGEF2 in 3xTg‐AD, n = 17 males). All data are shown as the mean ± SEM. *p < 0.05, **p < 0.01; ****p < 0.0001; two‐tailed unpaired Student's t‐test (E) and Two‐way ANOVA, Tukey's multiple‐comparison test (D and F)
Finally, we examined whether RAPGEF2 knockdown‐mediated fear memory retention in 3xTg‐AD mice could be due to synaptic preservation in the hippocampal CA1 area. Transmission electron microscopy revealed that the density of excitatory (asymmetric) synapses, but not inhibitory (symmetric) synapses, was significantly decreased in the CA1 stratum radiatum of the 3xTg‐AD mice compared to the WT animals (Figure 7A–D). In contrast, the knockdown of RAPAGEF2 in 3xTg‐AD mice blocked the reduction in excitatory synaptic density (Figure 7B,C). Again, the number of inhibitory synapses was not affected by the modulation of RAPGEF2 expression (Figure 7B,D). These results demonstrate that the loss of the RAPGEF2 protein halts Aβ oligomer‐mediated reduction in excitatory synapses in vivo without altering inhibitory synapses. Collectively, our data suggest that early intervention regarding RAPGEF2 expression could prevent Aβ oligomer‐mediated synaptic and behavioural dysfunction in the AD hippocampus.
FIGURE 7.
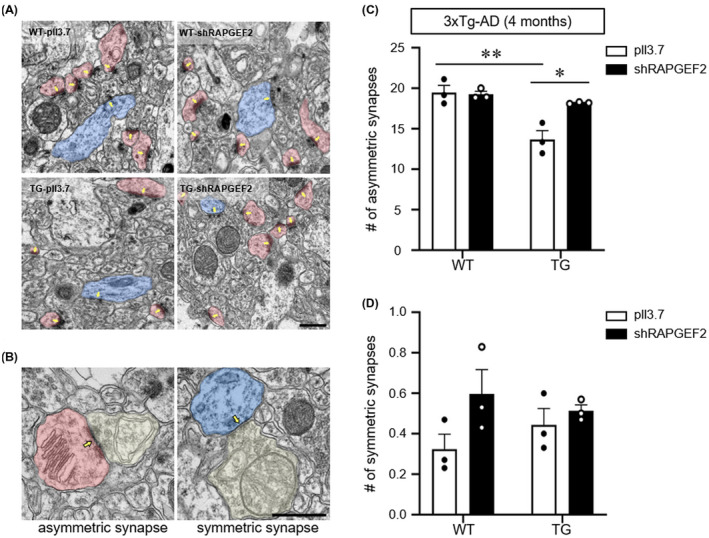
Knockdown of RAPGEF2 preserves excitatory synapses in 3xTg‐AD mice. A, Representative electron microscopic images of the CA1 stratum radiatum of wild‐type (WT) and 3xTg‐AD mice injected with either control or shRAPGEF2‐expressing viral particles. The arrow indicates postsynaptic density. Scale, 1 μm. B, Representative images of asymmetric (left) and symmetric (right) synapses. The arrow indicates postsynaptic density. Scale, 0.5 μm. C and D, Quantification of the number of asymmetric (C) and symmetric (D) synapses for each condition. The data are shown as the mean ± SEM (n = 95–97 images from three male mice per group). *p < 0.05, **p < 0.01; Two‐way ANOVA, Tukey's multiple‐comparison test
DISCUSSION
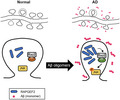
The accumulation of soluble Aβ from the sequential proteolytic processing of APP and the hyperphosphorylation of tau is a primary hallmark of AD pathogenesis.4, 47 In this study, we discovered that RAPGEF2 acts as a novel signalling component involved in Aβ oligomer‐mediated synaptic and cognitive impairments in AD. Using multiple AD mouse models and post‐mortem human AD hippocampal tissue, our results indicate that Aβ oligomers trigger the upregulation of RAPGEF2 levels in AD brains. Increased RAPGEF2 expression induces the loss of synapses via activation of the Rap2‐JNK pathway. Furthermore, the in vivo knockdown of endogenous RAPGEF2 prevented oligomeric Aβ‐induced synaptic and cognitive dysfunction. This scenario showing the involvement of RAPGEF2 in the early synaptopathy of AD is summarised in Figure 8.
FIGURE 8.
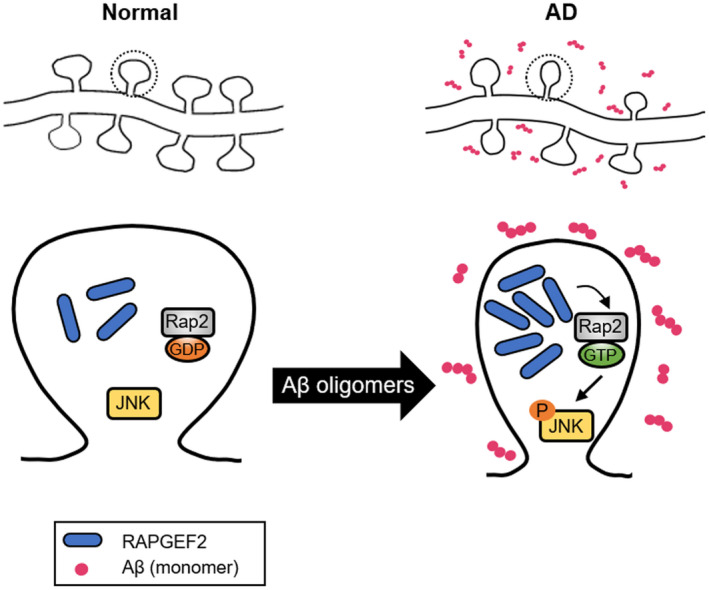
Schematic model for the role of RAPGEF2 in Aβ oligomer‐induced synaptic degeneration. In the AD hippocampus (right), synaptotoxic Aβ oligomers stimulate the upregulation of RAPGEF2 levels and lead to the activation of small GTPase Rap2. Rap2 activation, in turn, phosphorylates its downstream signalling target JNK. The Aβ oligomer‐mediated upregulation of RAPGEF2 induces the loss of excitatory synapses and subsequent memory impairment
One of the best‐characterised familial AD models is the 3xTg‐AD mouse model, which develops Aβ deposits prior to tangle formation, consistent with the amyloid cascade hypothesis.6 Interestingly, we observed heightened levels of RAPGEF2 in 3xTg‐AD mice at 3 months of age before the onset of synaptic and cognitive deficits (Figure 1). A similar result was found in 1.5‐month‐old 5xFAD mice expressing APP and PSEN1 mutations (Figure 1) but not in rTg4510 tau mutants at 2.5 and 5.5 months of age (Figure S3). These results indicate that either full‐length APP or its proteolytic product oligomeric Aβ augments RAPGEF2 expression in early AD brains, suggesting a potentially causative role of RAPGEF2 in subsequent synaptic and cognitive disturbances. Our results revealed that the Aβ oligomer, rather than full‐length APP, induced the upregulation of RAPGEF2 levels (Figure S4). Furthermore, we provided direct evidence that oligomeric Aβ treatment stimulated RAPGEF2 expression in cultured hippocampal neurons (Figure 2).
Importantly, augmented RAPGEF2 levels were also identified in the post‐mortem human AD hippocampus (Figure 1). Because most cases of AD are sporadic (late‐onset) and ~5% of cases are genetic (early onset) in origin,48 we assume that the increased RAPGEF2 levels in the human AD hippocampus may reflect the relatively slow and gradual increase in Aβ oligomers in sporadic AD compared to familial AD. Although it is unclear when RAPGEF2 expression begins to be stimulated in human AD patients, our data show that RAPGEF2 levels are commonly induced in the brains of early‐ and late‐onset AD brains.
It is noteworthy that cross‐reactivity with the highly homologous protein RAPGEF6 may occur with some RAPGEF2 antibodies.49 We also found that some anti‐RAPGEF2 antibodies used in this study exhibited a very weak but discernible cross‐reactivity in cells overexpressing RAPGEF6 (Figure S1A). Nonetheless, this cross‐reactivity issue is unlikely to affect our conclusion that RAPGEF2 mediates Aβ‐induced synapse loss, as Aβ oligomer treatment did not significantly change the levels of other RAPGEFs in the hippocampal neurons (Figure S1B). Similarly, in vivo silencing of RAPGEF2 in 3xTg‐AD mice induced no compensatory changes in the levels of other RAPGEFs (Figure S9).
Rap1 and Rap2, the downstream effectors of RAPGEF2, are members of the Ras family of small GTPases.22 Rap1 and Rap2 share close to 60% sequence homology,50 but they seem to exert distinct functions by activating different signalling pathways. For instance, Rap1 and Rap2 regulate the synaptic removal of AMPA‐type glutamate receptors via the activation of p38 MAPK during long‐term depression and JNK in synaptic depotentiation respectively.20, 26, 28 Furthermore, the activation of Rap2 causes the loss of excitatory synapses, whereas Rap1 has no effect on axonal or dendritic morphology in spiny neurons.21 In line with these findings, our data show that the Aβ oligomer‐mediated stimulation of RAPGEF2 expression selectively activates Rap2‐JNK (Figure 3), suggesting the distinct roles of Rap proteins in the pathogenetic mechanisms of AD.
The activation of JNK, a downstream target of Rap2, has been reported in human AD brains.51 Our results also revealed that phosphorylation levels of JNK were significantly higher in cortical lysates from the 3xTg‐AD and 5xFAD mice during the same period of RAPGEF2 upregulation (Figure 3E; Figure S5A), which is consistent with the Aβ‐mediated activation of the JNK pathway in the cultured cortical neurons.45, 52 Conversely, JNK phosphorylates APP at the Thr688 residue and facilitates Aβ aggregation,53, 54 and the inhibition of JNK reduces soluble Aβ oligomers.55 Thus, it is likely that Aβ‐RAPGEF2‐JNK could create a reciprocal activation circuit that further exacerbates Aβ pathology. In line with this hypothesis, the knockdown of RAPGEF2 or inhibition of JNK in neuron cultures from the 3xTg‐AD mice were sufficient to restore the elevated JNK activity to control levels and to halt the Aβ‐induced spine loss (Figure 4 and Figure S8).
Which upstream molecules trigger the upregulation of RAPGEF2? Polo‐like kinase 2 (Plk2) could be one of the candidates involved in Aβ‐mediated synaptopathy, as Plk2 has been shown to promote RAPGEF2 activity in the hippocampus.25 In support of this idea, increased levels of Plk2 were also observed in the APP‐SwD1 AD mouse brain and in the human AD brain.56 Furthermore, the pharmacological inhibition of Plk2 function ameliorates synapse loss and memory decline in an AD mouse model.56 Thus, these findings suggest that Plk2 may act as an upstream regulator in RAPGEF2‐mediated synaptic dysfunction and cognitive deficits. Further studies are warranted to identify additional upstream signalling molecules that trigger the upregulation of RAPGEF2.
Previous studies have reported that constitutively active Rap2 reduces the number and length of dendritic spines in CA1 hippocampal neurons,27 and JNK inhibition rescues Aβ oligomer‐mediated spine loss.45 Thus, it has been speculated that the Aβ oligomer‐mediated upregulation of RAPGEF2 levels may induce changes in the number and morphology of dendritic spines by activating the Rap2‐JNK signalling cascade. Indeed, we found that the knockdown of RAPGEF2 prevented the Aβ oligomer‐mediated reduction in spine numbers, length, head size and intensities of synaptic marker proteins such as VGLUT1 and PSD‐95 (Figure 5 and Figure S7). In neuron cultures from 3xTg‐AD mice, silencing of either RAPGEF2 or pharmacological inhibition of JNK also halted the loss of dendritic spines (Figure S8). Aβ oligomers may induce multiple signalling pathways that act in parallel to affect distinct morphological aspects of dendritic spines. Considering that a recent study showed that the Pyk2‐Graf1‐RhoA pathway also plays a role in Aβ‐mediated dendritic spinopathy,57 it would be of interest to investigate how these multiple signalling pathways interact with each other in follow‐up studies.
Surprisingly, in vivo silencing of RAPGEF2 in the hippocampal CA1 area was sufficient to preserve contextual fear memory retention (Figure 6), which requires the intact functioning of the dorsal hippocampus.58 Notably, electron microscopy analysis revealed that RAPGEF2 knockdown was sufficient to block the reduction in excitatory synapses in the hippocampal CA1 stratum radiatum of 3xTG‐AD mice (Figure 7).
In summary, our results demonstrate that RAPGEF2 mediates Aβ oligomer‐induced synaptic and cognitive deficits in the AD hippocampus. Therefore, early intervention regarding RAPGEF2 expression might be a potential therapeutic option to help mitigate Aβ oligomer‐induced synaptic and behavioural impairments in AD.
ETHICS APPROVAL
All animal experiments were carried out in compliance with the Guide for Care and Use of Laboratory Animals of the National Institutes of Health and were approved by the Institutional Animal Care and Use Committee of the Korea Brain Research Institute (IACUC 17‐00017, 17‐00018, 18‐00016, and 18‐00026). Human hippocampal tissue samples used in this study were obtained from the Netherlands Brain Bank (Project #1009, Amsterdam, Netherlands). Experiments using human tissue were approved by the Institutional Review Board at the Korea Brain Research Institute (201610‐BR‐004‐01).
CONFLICT OF INTEREST
The authors declare no competing financial interests.
AUTHORS’ CONTRIBUTIONS
K.J.L. designed the experiments, wrote the manuscript and supervised the project. Y.‐N.J., H.J., J.E.N., and K.‐A.C. performed biochemical, imaging and behavioural experiments and analysed the data. G.H.K. performed transmission electron microscopy and analysed the data. Y.‐N.J. wrote the initial draft of the manuscript. All authors have read and approved the final manuscript.
Peer Review
The peer review history for this article is available at https://publons.com/publon/10.1111/nan.12686.
Supporting information
ACKNOWLEDGEMENTS
The authors thank the members of the Brain Research Core Facilities in KBRI for their assistance with the confocal and electron microscopy experiments. This work was supported by the KBRI basic research program (20‐BR‐01‐08) funded by the Ministry of Science and ICT and grants from the Korean NRF funded by the Ministry of Education (NRF‐2014R1A1A2058740) and Ministry of Science and ICT (NRF‐2017M3C7A1048086).
Jang YN, Jang H, Kim GH, Noh JE, Chang KA, Lee KJ. RAPGEF2 mediates oligomeric Aβ-induced synaptic loss and cognitive dysfunction in the 3xTg-AD mouse model of Alzheimer’s disease. Neuropathol Appl Neurobiol. 2021;47:625–639. 10.1111/nan.12686
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Selkoe DJ. Alzheimer's disease is a synaptic failure. Science. 2002;298:789‐791. [DOI] [PubMed] [Google Scholar]
- 2.Sivanesan S, Tan A, Rajadas J. Pathogenesis of Abeta oligomers in synaptic failure. Curr Alzheimer Res. 2013;10:316‐323. [DOI] [PubMed] [Google Scholar]
- 3.Selkoe DJ. Soluble oligomers of the amyloid beta‐protein impair synaptic plasticity and behavior. Behav Brain Res. 2008;192:106‐113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cavallucci V, D'Amelio M, Cecconi F. Abeta toxicity in Alzheimer's disease. Mol Neurobiol. 2012;45:366‐378. [DOI] [PubMed] [Google Scholar]
- 5.DeKosky ST, Scheff SW. Synapse loss in frontal cortex biopsies in Alzheimer's disease: correlation with cognitive severity. Ann Neurol. 1990;27:457‐464. [DOI] [PubMed] [Google Scholar]
- 6.Oddo S, Caccamo A, Shepherd JD, et al. Triple‐transgenic model of Alzheimer's disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 2003;39:409‐421. [DOI] [PubMed] [Google Scholar]
- 7.Dickson DW, Crystal HA, Bevona C, Honer W, Vincent I, Davies P. Correlations of synaptic and pathological markers with cognition of the elderly. Neurobiol Aging. 1995;16:285‐298; discussion 298–304. [DOI] [PubMed] [Google Scholar]
- 8.Masliah E, Mallory M, Alford M, et al. Altered expression of synaptic proteins occurs early during progression of Alzheimer's disease. Neurology. 2001;56:127‐129. [DOI] [PubMed] [Google Scholar]
- 9.Kuo YM, Emmerling MR, Vigo‐Pelfrey C, et al. Water‐soluble Abeta (N‐40, N‐42) oligomers in normal and Alzheimer disease brains. J Biol Chem. 1996;271:4077‐4081. [DOI] [PubMed] [Google Scholar]
- 10.Gong Y, Chang L, Viola KL, et al. Alzheimer's disease‐affected brain: presence of oligomeric A beta ligands (ADDLs) suggests a molecular basis for reversible memory loss. Proc Natl Acad Sci USA. 2003;100:10417‐10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Klyubin I, Walsh DM, Cullen WK, et al. Soluble Arctic amyloid beta protein inhibits hippocampal long‐term potentiation in vivo. Eur J Neurosci. 2004;19:2839‐2846. [DOI] [PubMed] [Google Scholar]
- 12.Wang Q, Walsh DM, Rowan MJ, Selkoe DJ, Anwyl R. Block of long‐term potentiation by naturally secreted and synthetic amyloid beta‐peptide in hippocampal slices is mediated via activation of the kinases c‐Jun N‐terminal kinase, cyclin‐dependent kinase 5, and p38 mitogen‐activated protein kinase as well as metabotropic glutamate receptor type 5. J Neurosci. 2004;24:3370‐3378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harris KM, Kater SB. Dendritic spines: cellular specializations imparting both stability and flexibility to synaptic function. Annu Rev Neurosci. 1994;17:341‐371. [DOI] [PubMed] [Google Scholar]
- 14.Kamenetz F, Tomita T, Hsieh H, et al. APP processing and synaptic function. Neuron. 2003;37:925‐937. [DOI] [PubMed] [Google Scholar]
- 15.Cirrito JR, Yamada KA, Finn MB, et al. Synaptic activity regulates interstitial fluid amyloid‐beta levels in vivo. Neuron. 2005;48:913‐922. [DOI] [PubMed] [Google Scholar]
- 16.Lee Y, Lee JS, Lee KJ, Turner RS, Hoe HS, Pak DTS. Polo‐like kinase 2 phosphorylation of amyloid precursor protein regulates activity‐dependent amyloidogenic processing. Neuropharmacology. 2017;117:387‐400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Chong YH, Shin YJ, Lee EO, Kayed R, Glabe CG, Tenner AJ. ERK1/2 activation mediates Abeta oligomer‐induced neurotoxicity via caspase‐3 activation and tau cleavage in rat organotypic hippocampal slice cultures. J Biol Chem. 2006;281:20315‐20325. [DOI] [PubMed] [Google Scholar]
- 18.Knox AL, Brown NH. Rap1 GTPase regulation of adherens junction positioning and cell adhesion. Science. 2002;295:1285‐1288. [DOI] [PubMed] [Google Scholar]
- 19.Franco SJ, Martinez‐Garay I, Gil‐Sanz C, Harkins‐Perry SR, Muller U. Reelin regulates cadherin function via Dab1/Rap1 to control neuronal migration and lamination in the neocortex. Neuron. 2011;69:482‐497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Zhu JJ, Qin Y, Zhao M, Van Aelst L, Malinow R. Ras and Rap control AMPA receptor trafficking during synaptic plasticity. Cell. 2002;110:443‐455. [DOI] [PubMed] [Google Scholar]
- 21.Fu Z, Lee SH, Simonetta A, Hansen J, Sheng M, Pak DT. Differential roles of Rap1 and Rap2 small GTPases in neurite retraction and synapse elimination in hippocampal spiny neurons. J Neurochem. 2007;100:118‐131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.de Rooij J, Boenink NM, van Triest M, Cool RH, Wittinghofer A, Bos JL. PDZ‐GEF1, a guanine nucleotide exchange factor specific for Rap1 and Rap2. J Biol Chem. 1999;274:38125‐38130. [DOI] [PubMed] [Google Scholar]
- 23.Liao Y, Kariya K, Hu CD, et al. RA‐GEF, a novel Rap1A guanine nucleotide exchange factor containing a Ras/Rap1A‐associating domain, is conserved between nematode and humans. J Biol Chem. 1999;274:37815‐37820. [DOI] [PubMed] [Google Scholar]
- 24.Ohtsuka T, Hata Y, Ide N, et al. nRap GEP: a novel neural GDP/GTP exchange protein for rap1 small G protein that interacts with synaptic scaffolding molecule (S‐SCAM). Biochem Biophys Res Commun. 1999;265:38‐44. [DOI] [PubMed] [Google Scholar]
- 25.Lee KJ, Lee Y, Rozeboom A, et al. Requirement for Plk2 in orchestrated ras and rap signaling, homeostatic structural plasticity, and memory. Neuron. 2011;69:957‐973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Zhu Y, Pak D, Qin Y, et al. Rap2‐JNK removes synaptic AMPA receptors during depotentiation. Neuron. 2005;46:905‐916. [DOI] [PubMed] [Google Scholar]
- 27.Ryu J, Futai K, Feliu M, Weinberg R, Sheng M. Constitutively active Rap2 transgenic mice display fewer dendritic spines, reduced extracellular signal‐regulated kinase signaling, enhanced long‐term depression, and impaired spatial learning and fear extinction. J Neurosci. 2008;28:8178‐8188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Zhang L, Zhang P, Wang G, et al. Ras and Rap signal bidirectional synaptic plasticity via distinct subcellular microdomains. Neuron. 2018;98(783‐800):e784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ishiura H, Doi K, Mitsui J, et al. Expansions of intronic TTTCA and TTTTA repeats in benign adult familial myoclonic epilepsy. Nat Genet. 2018;50:581‐590. [DOI] [PubMed] [Google Scholar]
- 30.Minkeviciene R, Rheims S, Dobszay MB, et al. Amyloid beta‐induced neuronal hyperexcitability triggers progressive epilepsy. J Neurosci. 2009;29:3453‐3462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Born HA, Kim JY, Savjani RR, et al. Genetic suppression of transgenic APP rescues Hypersynchronous network activity in a mouse model of Alzeimer's disease. J Neurosci. 2014;34:3826‐3840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Amatniek JC, Hauser WA, DelCastillo‐Castaneda C, et al. Incidence and predictors of seizures in patients with Alzheimer's disease. Epilepsia. 2006;47:867‐872. [DOI] [PubMed] [Google Scholar]
- 33.Lee KJ, Park IS, Kim H, Greenough WT, Pak DT, Rhyu IJ. Motor skill training induces coordinated strengthening and weakening between neighboring synapses. J Neurosci. 2013;33:9794‐9799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Hisata S, Sakisaka T, Baba T, et al. Rap1‐PDZ‐GEF1 interacts with a neurotrophin receptor at late endosomes, leading to sustained activation of Rap1 and ERK and neurite outgrowth. J Cell Biol. 2007;178:843‐860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Billings LM, Oddo S, Green KN, McGaugh JL, LaFerla FM. Intraneuronal Abeta causes the onset of early Alzheimer's disease‐related cognitive deficits in transgenic mice. Neuron. 2005;45:675‐688. [DOI] [PubMed] [Google Scholar]
- 36.Stover KR, Campbell MA, Van Winssen CM, Brown RE. Early detection of cognitive deficits in the 3xTg‐AD mouse model of Alzheimer's disease. Behav Brain Res. 2015;289:29‐38. [DOI] [PubMed] [Google Scholar]
- 37.Ramsden M, Kotilinek L, Forster C, et al. Age‐dependent neurofibrillary tangle formation, neuron loss, and memory impairment in a mouse model of human tauopathy (P301L). J Neurosci. 2005;25:10637‐10647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jawhar S, Trawicka A, Jenneckens C, Bayer TA, Wirths O. Motor deficits, neuron loss, and reduced anxiety coinciding with axonal degeneration and intraneuronal Abeta aggregation in the 5XFAD mouse model of Alzheimer's disease. Neurobiol Aging. 2012;33(196):e129‐e140. [DOI] [PubMed] [Google Scholar]
- 39.Richard BC, Kurdakova A, Baches S, Bayer TA, Weggen S, Wirths O. Gene dosage dependent aggravation of the neurological phenotype in the 5XFAD mouse model of Alzheimer's disease. J Alzheimers Dis. 2015;45:1223‐1236. [DOI] [PubMed] [Google Scholar]
- 40.Buskila Y, Crowe SE, Ellis‐Davies GC. Synaptic deficits in layer 5 neurons precede overt structural decay in 5xFAD mice. Neuroscience. 2013;254:152‐159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Crowe SE, Ellis‐Davies GC. In vivo characterization of a bigenic fluorescent mouse model of Alzheimer's disease with neurodegeneration. J Comp Neurol. 2013;521:2181‐2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Helboe L, Egebjerg J, Barkholt P, Volbracht C. Early depletion of CA1 neurons and late neurodegeneration in a mouse tauopathy model. Brain Res. 2017;1665:22‐35. [DOI] [PubMed] [Google Scholar]
- 43.Spires TL, Orne JD, SantaCruz K, et al. Region‐specific dissociation of neuronal loss and neurofibrillary pathology in a mouse model of tauopathy. Am J Pathol. 2006;168:1598‐1607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blackmore T, Meftah S, Murray TK, et al. Tracking progressive pathological and functional decline in the rTg4510 mouse model of tauopathy. Alzheimers Res Ther. 2017;9:77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sclip A, Arnaboldi A, Colombo I, et al. Soluble Abeta oligomer‐induced synaptopathy: c‐Jun N‐terminal kinase's role. J Mol Cell Biol. 2013;5:277‐279. [DOI] [PubMed] [Google Scholar]
- 46.Sclip A, Tozzi A, Abaza A, et al. c‐Jun N‐terminal kinase has a key role in Alzheimer disease synaptic dysfunction in vivo. Cell Death Dis. 2014;5:e1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Zhang H, Ma Q, Zhang YW, Xu H. Proteolytic processing of Alzheimer's beta‐amyloid precursor protein. J Neurochem. 2012;120(Suppl 1):9‐21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bali J, Gheinani AH, Zurbriggen S, Rajendran L. Role of genes linked to sporadic Alzheimer's disease risk in the production of beta‐amyloid peptides. Proc Natl Acad Sci USA. 2012;109:15307‐15311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Jiang SZ, Xu W, Emery AC, Gerfen CR, Eiden MV, Eiden LE. NCS‐Rapgef2, the protein product of the neuronal Rapgef2 Gene, is a specific activator of D1 dopamine receptor‐dependent ERK phosphorylation in mouse brain. eNeuro. 2017;4:ENEURO.0248‐0217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ohba Y, Mochizuki N, Matsuo K, et al. Rap2 as a slowly responding molecular switch in the Rap1 signaling cascade. Mol Cell Biol. 2000;20:6074‐6083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhu X, Raina AK, Rottkamp CA, et al. Activation and redistribution of c‐jun N‐terminal kinase/stress activated protein kinase in degenerating neurons in Alzheimer's disease. J Neurochem. 2001;76:435‐441. [DOI] [PubMed] [Google Scholar]
- 52.Morishima Y, Gotoh Y, Zieg J, et al. Beta‐amyloid induces neuronal apoptosis via a mechanism that involves the c‐Jun N‐terminal kinase pathway and the induction of Fas ligand. J Neurosci. 2001;21:7551‐7560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Ahn JH, So SP, Kim NY, Kim HJ, Yoon SY, Kim DH. c‐Jun N‐terminal Kinase (JNK) induces phosphorylation of amyloid precursor protein (APP) at Thr668, in okadaic acid‐induced neurodegeneration. BMB Rep. 2016;49:376‐381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Muresan Z, Muresan V. c‐Jun NH2‐terminal kinase‐interacting protein‐3 facilitates phosphorylation and controls localization of amyloid‐beta precursor protein. J Neurosci. 2005;25:3741‐3751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Sclip A, Antoniou X, Colombo A, et al. c‐Jun N‐terminal kinase regulates soluble Abeta oligomers and cognitive impairment in AD mouse model. J Biol Chem 2011;286:43871‐43880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Lee JS, Lee Y, Andre EA, et al. Inhibition of polo‐like kinase 2 ameliorates pathogenesis in Alzheimer's disease model mice. PLoS One 2019;14:e0219691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lee S, Salazar SV, Cox TO, Strittmatter SM. Pyk2 signaling through Graf1 and RhoA GTPase is required for amyloid‐beta oligomer‐triggered synapse loss. J Neurosci. 2019;39:1910‐1929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.McNish KA, Gewirtz JC, Davis M. Evidence of contextual fear after lesions of the hippocampus: a disruption of freezing but not fear‐potentiated startle. J Neurosci. 1997;17:9353‐9360. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.