

Abstract
African swine fever virus variants with different numbers of a 10‐bp tandem repeat were isolated in South Korea soon after being identified in wild boar. The short emergence periods and sympatric distributions within a narrow geographical region suggest that the variants were sporadically generated in the pre‐existing viral population.
Keywords: African swine fever virus, intergenic region variant, replication slippage, South Korea, tandem repeat, wild boar
1. INTRODUCTION
African swine fever (ASF) is a highly contagious disease occurring in domestic pigs and wild suids, and leads to serious haemorrhage and nearly 100% mortality. ASF is caused by infection with African swine fever virus (ASFV; family: Asfarviridae, genus: Asfivirus) (Dixon et al., 2020). Since its original description in the 1920s in Kenya, this viral disease has been highly prevalent in African and European countries. In Asia, ASF was first reported in a farm near Shenyang City in Liaoning Province in China, in August 2018 (Zhou at al., 2018). Thereafter, it quickly spread to other countries in South‐East Asia, including Myanmar, Laos, Vietnam and the Philippines, as well as nearly all provinces in mainland China, in 2019 (Lu et al., 2020). ASF outbreaks have also been described in North Korea and South Korea in the same year (Kim, et al., 2020; Kim, et al., 2020).
Despite the high mortality rates and socio‐economic impacts of ASF, no vaccines or therapeutic agents are available for controlling its outbreak or its effective treatment (Dixon et al., 2020). Therefore, studies on the routes and patterns of ASF transmission and its early detection are urgently needed. Molecular epidemiology approaches using polymorphic DNA sequences can provide insight into the spatiotemporal patterns of disease transmission throughout the areas in which ASF is prevalent. The genomic DNA of ASFV shows a low evolution rate. Nevertheless, multiple sites show inter‐genomic polymorphisms, particularly those containing short tandem repeats (STRs), which can be selected as informative markers in epidemiological investigations (Goller et al., 2015; Nix et al., 2006).
After ASFV was first isolated from a wild boar in 2 October 2019 in South Korea (Kim, et al., 2020), we conducted a surveillance programme for wild boars in the relevant areas by the National Institute of Environmental Research (NIER). Genotypes of ASFV DNAs obtained during these surveys were investigated via polymerase chain reaction (PCR) and nucleotide sequencing to trace viral transmission in the wild boar population and monitor the probable emergence of viral variants.
2. MATERIAL AND METHODS
During the nationwide comprehensive monitoring of wild boars from 2 October to 30 December 2019, 56 viral isolates were collected from whole blood or tissue of wild boar carcasses in the border area covering Paju and Yeoncheon Counties of Gyeonggi Province and Cheorwon County of Gangwon Province by NIER (Figure 1). The positive rate was highest in Paju (Table 1). Partial segments of B646L (p72) and EP402R (CD2v), as well as an intergenic region (IGR) between I73R and I329L, were amplified from each of the viral DNAs by PCR using specific primer sets, as described previously (Kim, et al., 2020). The PCR products were subject to automated paired‐end sequencing reactions and the resulting contig sequences were used in phylogenetic analyses together with their homologues retrieved from the GenBank database (neighbour‐joining algorithm with the MEGA 6.0 software).
FIGURE 1.
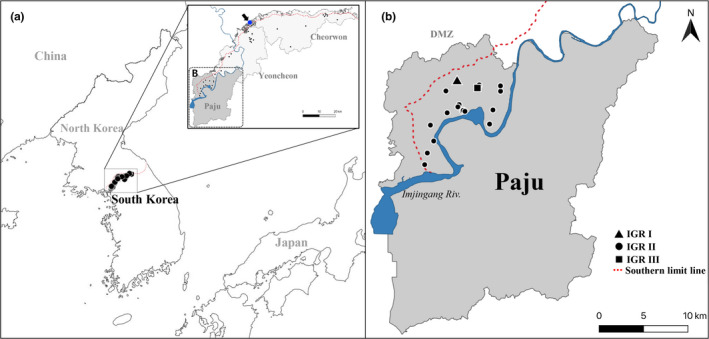
Global (a) and locality (b) maps showing the collection sites of African swine fever virus from wild boars in South Korea from 2 October to 30 December 2019. Local area in Paju County where the IGR variants and pre‐existing strain were isolated (dotted box in panel A) are magnified in panel B. Dot marked with an arrow in the inset map of panel A indicates the place where the first African swine fever case was detected in a wild boar in South Korea. The collection sites of IGR variants I, II and III in Paju County are indicated by a triangle, dot and square, respectively, in panel B [Colour figure can be viewed at wileyonlinelibrary.com]
TABLE 1.
Summary of results for wild boars examined for ASFV infection in 2019 in South Korea
| County | No. of wild boars tested | No. of infected wild boars (%) | No. of positive whole blood/tissue samples |
|---|---|---|---|
| Yeoncheon | 302 | 19 (6.3) | 4/15 |
| Cheorwon | 473 | 17 (3.4) | 6/14 |
| Paju | 117 | 20 (17.1) | 8/9 |
| Other areas | 3,782 | 0 (0) | ‐ |
| Total | 4,674 | 56 (1.0) | 18/38 |
3. RESULTS AND DISCUSSION
All partial B646L and EP402R sequences of the ASFV isolates were identical to those of the original Korean isolate, Korea/19S804/wb/2019 (GenBank Accession Nos. MN817977 and MN817978). These sequences were categorized into the genotype II (Figure 2a) and serogroup 8 (Figure 2b) groups, respectively. Most IGR fragments also showed 100% sequence identity to the corresponding region of the Korea/19S804/wb/2019 isolate (GenBank Accession No. MN817979), except for those of two isolates collected in Paju (Korea/19S3965/wb/2019 on 3 December and Korea/19S5464/wb/2019 on 30 December). The IGR fragment contained an STR (5′‐GGAATATATA‐3′), with a repeat time varying in or among ASFV populations (Goller et al., 2015; Nix et al., 2006). The 10‐bp STR was inserted three times in the corresponding regions of the major Korean ASFV isolates from the wild boar (IGR variant II), similar to those in the Russia/Volgograd/wb/2014 (GenBank Accession No. KP137637), Belgium/Etalle/wb/2018 (GenBank Accession No. MH998359.1) and China/2018/Domestic pig (GenBank Accession No. MH735144) strains. The genome of another Korean isolate from a domestic pig (Korea/2019/Domestic pig, GenBank Accession No. MN603969 [Kim, et al., 2020]) also belonged to the variant II group. On the other hand, the nucleotide stretch was repeated two and four times in the genomes of the Korea/19S3965/wb/2019 (IGR variant I) and Korea/19S5464/wb/2019 (IGR variant III) isolates, respectively (Table 2).
FIGURE 2.
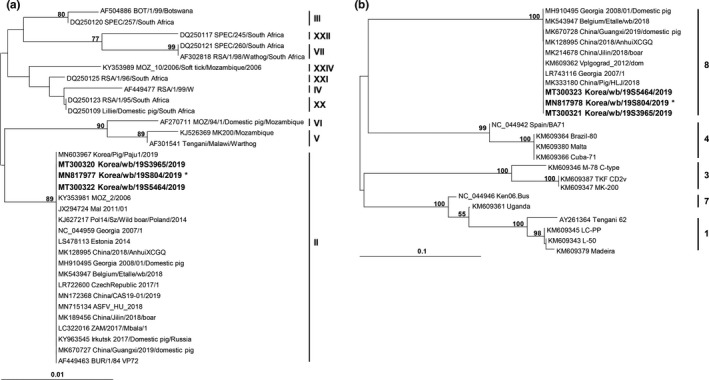
Phylogenetic analyses of partial B646L (a) and EP402R (b) sequences of African swine fever virus isolates obtained in this study. The neighbour‐joining trees were constructed with MEGA 6 based on the Kimura 2‐parameter model. Numerals on branching nodes indicate the bootstrap values obtained with 1,000 replicates (>50%). The sequences obtained in this study are distinguished by bold letters, while the phylogenetic positions of 54 sequences other than those of 19S3965 and 19S5464 isolates are represented by that of the pre‐existing Korea/19S804/wb/2019 isolate (marked with asterisk)
TABLE 2.
Comparison of intergenic region (IGR) sequences between I73R and I329L encompassing a short tandem repeat (STR)
| GenBank No. | Strain | Partial nucleotide sequence encompassing the STR in IGR between I73R and I329L * | Type |
|---|---|---|---|
| MT300324 | Korea/19S3965/wb/2019 | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | IGR I |
| FR682485 | Georgia 2007/1 | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | |
| KY982843 | Russia/Irkutsk/2017/Domestic pig | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | |
| MK189457 | China/Jilin/2018/boar | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | |
| MG939584 | Poland/Pol16/2016/wb | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | |
| KP137637 | Russia/Volgograd/wb/2014 | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | IGR II |
| MH998359 | Belgium/Etalle/wb/2018 | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | |
| MH735144 | China/2018/Domestic pig | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | |
| MN603969 | Korea/2019/Domestic pig | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGGAATATATA GAAATATATAGAAATAGCTAA | |
| MN817979 * | Korea/19S804/wb/2019 | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | |
| MK670729 | China/Guangxi/2019/Domestic pig | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA | IGR III |
| MT300325 | Korea/19S5464/wb/2019 | GCAATAAATAACAAGTATATAGGAATATATAGGAATATATAGGAATATATAGGAATATATAGAAATATATAGAAATAGCTAA |
The nucleotide sequences obtained in this study (bold sequences) were compared to those of other African swine fever virus (ASFV) strains representing each IGR genotype. Of the 56 ASFV sequences, 54 sequences, excluding the 19S3965 and 19S5464 isolates, were represented by the pre‐existing Korea/19S804/wb/2019 isolate (marked with asterisk), showing 100% identity. The 10‐bp STR unit (5′‐GGAATATATA‐3′) is underlined.
Because the genomes of DNA viruses, including ASFV, show relatively low evolutionary divergence, few informative molecular markers have been detected within the genic regions of the viral genomes, as observed by analysis of the B646L and EP402R sequences in this study. However, the lengths of genomic STR are readily expanded or contracted during DNA replication largely by slipped strand mispairing (slippage mechanism) (Levinson & Gutman, 1987). If the affected STR is in an intergenic non‐coding region, the allelic variant can be fixed in the population because of the low levels of purifying selection (Gemayel et al., 2010). ASFV strains with different IGR genotypes (I and II) have been allopatrically discovered in Russia (Goller et al., 2015) and China (Ge et al., 2019; Li et al., 2019). In China, the major IGR genotype prevalent in domestic pigs was variant II, whereas an ASFV strain with variant III genotype was isolated in a domestic pig farm in Guangxi Province on 7 March 2019. The variant I genotype was detected in an ASFV isolate from a wild boar in Jilin Province on 16 November 2018 (Ge et al., 2019 and references therein). However, unlike the IGR variants in these countries, those identified in this study were sympatrically distributed with the pre‐existing type (variant II) in a small county (Figure 1b). The time intervals for their emergence were also very short (approximately 2 and 3 months after the first outbreak in wild boars). Civilian access to the concerned region is strictly controlled because the area is near or inside the military operation area. Furthermore, after the first ASF outbreak in domestic pigs on 16 September 2019, quarantine measures have been strictly enforced, and fences have been installed to prevent any possible transmission of the virus. Taken together, these facts may suggest that the Korean IGR variants I and III were sporadically generated rather than being independently transported from other countries through replication error in the pre‐existing IGR variant II population.
The probable transmission routes of ASF can be predicted by analysing the spatiotemporal distributions of ASFV with distinct IGR genotypes (Goller et al., 2015). Therefore, the polymorphic STR was suggested as an informative marker to discriminate closely related ASFV strains (Ge et al., 2019). Currently, we have no evidence supporting clonal expansion of these variants in the relevant region, which may be because of their recent emergence. The short lifespan of infected wild boars and the slow spatial spread of ASFV within wild boar subpopulations (European Food Safety Authority et al., 2017) may also be involved in the protected/delayed expansion of the variant ASFVs. Surveillance of wild boars will be continued until the viral disease is eliminated. If simultaneous propagation of these IGR variants is observed in the near future, our data will provide a highly informative genetic marker for molecular epidemiological approaches to trace both local and global transmission of ASFV.
In conclusion, we identified ASFV variants with different IGR genotypes during the comprehensive survey of wild boars in small counties of South Korea surrounding the original ASF outbreak point. Considering the short emergence periods of <3 months and sympatric distributions within a narrow geographical region, these variant strains are likely to have spontaneously emerged in the local viral population through a molecular mechanism(s) such as replication slippage. Nevertheless, we cannot exclude the hypothesis that the IGR I and IGR III variants were simultaneously transmitted into the local area from the respective foreign countries. Future investigations adapting additional epidemiologically informative markers are needed, to get more conclusive pieces of evidence and to better understand the origin(s) of the ASFV variants.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
This manuscript was written by S‐H Kim and W‐H Jheong, experiment and data analysis were performed by S‐H Kim, S‐I Lee, H‐G Jeong, J‐Yoo, K Son and H‐S‐Jeong, and study was designed by S‐H Kim and W‐H Jheong. Isolate partial sequence data from this study were deposited in GenBank with the Accession Numbers MT300320–MT300325.
ETHICAL APPROVAL
Not applicable.
ACKNOWLEDGEMENTS
This study was funded (Grant Number: NIER‐2019‐01‐01‐006) by the National Institute of Environmental Research (NIER), Ministry of Environment, Republic of Korea.
Kim S‐H, Lee S‐I, Jeong H‐G, et al. Rapid emergence of African swine fever virus variants with different numbers of a tandem repeat sequence in South Korea. Transbound Emerg Dis.2021;68:1726–1730. 10.1111/tbed.13867
Kim and Lee these authors contributed equally to this article.
DATA AVAILABILITY STATEMENT
The data that support the finding of this study are available from the corresponding author upon reasonable request.
REFERENCES
- Dixon, L. K., Stahl, K., Jori, F., Vial, L., & Pfeiffer, D. U. (2020). African swine fever epidemiology and control. Annual Review of Animal Biosciences, 15, 221–246. 10.1146/annurev-animal-021419-083741 [DOI] [PubMed] [Google Scholar]
- European Food Safety Authority , Abrahantes, J. C., Gogin, A., Richardson, K., & Gervelmeyer, A. (2017). Epidemiological analyses on African swine fever in the Baltic countries and Poland. European Food Safety Authority Journal, 15, 4732. 10.2903/j.efsa.2017.4732 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ge, S., Liu, Y., Li, L., Wang, Q., Li, J., Ren, W., Liu, C., Bao, J., Wu, X., & Wang, Z. (2019). An extra insertion of tandem repeat sequence in African swine fever virus, China, 2019. Virus Genes, 55, 843–847. 10.1007/s11262-019-01704-9 [DOI] [PubMed] [Google Scholar]
- Gemayel, R., Vinces, M. D., Legendre, M., & Verstrepen, K. J. (2010). Variable tandem repeats accelerate evolution of coding and regulatory sequences. Annual Review of Genetics, 44, 445–477. 10.1146/annurev-genet-072610-155046 [DOI] [PubMed] [Google Scholar]
- Goller, K. V., Malogolovkin, A. S., Katorkin, S., Kolbasov, D., Titov, I., Höper, D., Beer, M., Keil, G. M., Portugal, R., & Blome, S. (2015). Tandem repeat insertion in African swine fever virus, Russia, 2012. Emerging Infectious Diseases, 21, 731–732. https://dx.doi.org/10.3201%2Feid2104.141792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim, H. J., Cho, K. H., Lee, S. K., Kim, D. Y., Nah, J. J., Kim, H. J., Kim, H. J., Hwang, J. Y., Sohn, H. J., Choi, J. G., Kang, H. E., & Kim, Y. J. (2020). Outbreak of African swine fever in South Korea, 2019. Transboundary and Emerging Diseases, 67, 473–475. 10.1111/tbed.13483 [DOI] [PubMed] [Google Scholar]
- Kim, S. H., Kim, J., Son, K., Choi, Y., Jeong, H. S., Kim, Y. K., Park, J. E., Hong, Y. J., Lee, S. I., Wang, S. J., Lee, Y. S., Kim, W. M., & Jheong, W. H. (2020). Wild boar harbouring African swine fever virus in the demilitarized zone in South Korea, 2019. Emerging Microbes & Infections, 9, 628–630. 10.1080/22221751.2020.1738904 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levinson, G., & Gutman, G. A. (1987). Slipped‐strand mispairing: A major mechanism for DNA sequence evolution. Molecular Biology and Evolution, 4, 203–221. 10.1093/oxfordjournals.molbev.a040442 [DOI] [PubMed] [Google Scholar]
- Li, L., Ren, Z., Wang, Q., Ge, S., Liu, Y., Liu, C., Liu, F., Hu, Y., Li, J., Bao, J., Ren, W., Zhang, Y., Xu, T., Sun, C., Li, L., Wang, S., Fan, X., Wu, Z., Huang, B., Guo, H., Wu, X., & Wang, Z. (2019). Infection of African swine fever in wild boar, China, 2018. Transboundary and Emerging Diseases, 66, 1395–1398. 10.1111/tbed.13114 [DOI] [PubMed] [Google Scholar]
- Lu, G., Pan, J., & Zhang, G. (2020). African swine fever virus in Asia: Its rapid spread and potential threat to unaffected countries. Journal of Infection, 80, 350–371. 10.1016/j.jinf.2019.11.011 [DOI] [PubMed] [Google Scholar]
- Nix, R. J., Gallardo, C., Hutchings, G., Blanco, E., & Dixon, L. K. (2006). Molecular epidemiology of African swine fever virus studied by analysis of four variable genome regions. Archives of Virology, 151, 2475–2494. 10.1007/s00705-006-0794-z [DOI] [PubMed] [Google Scholar]
- Zhou, X., Li, N., Liu, Y., Miao, F., Chen, T., Zhang, S., Cao, P., Li, X., Tian, K., Qiu, H. J., & Hu, R. (2018). Emergence of African Swine Fever in China. Transboundary and Emerging Diseases, 65, 1482–1484. 10.1111/tbed.12989 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The data that support the finding of this study are available from the corresponding author upon reasonable request.