

Abstract
Global warming poses major challenges for plant survival and agricultural productivity. Thus, efforts to enhance stress resilience in plants are key strategies for protecting food security. Gene regulatory networks (GRNs) are a critical mechanism conferring stress resilience. Until recently, predicting GRNs of the individual cells that make up plants and other multicellular organisms was impeded by aggregate population scale measurements of transcriptome and other genome‐scale features. With the advancement of high‐throughput single cell RNA‐seq and other single cell assays, learning GRNs for individual cells is now possible, in principle. In this article, we report on recent advances in experimental and analytical methodologies for single cell sequencing assays especially as they have been applied to the study of plants. We highlight recent advances and ongoing challenges for scGRN prediction, and finally, we highlight the opportunity to use scGRN discovery for studying and ultimately enhancing abiotic stress resilience in plants.
Keywords: abiotic stress, climate change, gene regulatory network, heat stress, high throughput sequencing, resilience, single cell, transcription
Short abstract
Single cell gene regulatory networks can guide research to improve crop resilience to climate change stress.
1. INTRODUCTION
Complex traits are coordinated across diverse cell types and tissues by hormones, metabolites, and mechanical forces in order to generate a coherent plant‐scale response to the environment (Duran‐Nebreda & Bassel, 2019). Underpinning these plant‐scale traits is the regulation of gene expression which occurs principally independently in each cell in the plant body. Gene regulatory networks (GRNs) are used to represent condition specific interactions of regulators of gene expression with the expression of target genes (Sullivan et al., 2014; Wilkins et al., 2016).
There is ample evidence, through direct measurement of transcription factor binding and target gene regulation, that GRNs function as a mechanism of plant resilience. For example, the regulation of submergence tolerance in rice (Xu et al., 2006) and nutrient signalling in Arabidopsis (Para et al., 2014; Taylor‐Teeples et al., 2014) are regulated by GRNs. In the last decades there have been major advances in global GRN prediction methods that aim to map all transcription factor‐target gene interactions from genome‐scale data sets. For example, Weighted Gene Correlation Network Analysis (WGCNA) (Langfelder & Horvath, 2008) predicts GRNs from expression data; ConnecTF (Brooks et al., 2020) and TF2Network (Kulkarni, Vaneechoutte, Van de Velde, & Vandepoele, 2018) predict GRNs using transcription factor binding sequence information; and Arboretum (Roy et al., 2013) integrates genomic and transcriptome data from evolutionarily diverse taxa to predict GRNs.
Because gene regulation occurs principally within single cells, many advances in GRN prediction algorithms have been developed in prokaryotic (Arrieta‐Ortiz et al., 2015; Greenfield, Hafemeister, & Bonneau, 2013), single‐celled eukaryotic organisms (Jackson, Castro, Saldi, Bonneau, & Gresham, 2020; Thompson et al., 2013) or isolated eukaryotic cell types (Ciofani et al., 2012; Miraldi et al., 2019) where populations of synchronized cells could be studied in bulk. Translation of GRN prediction methods for use in multicellular organisms like plants has been more difficult because measurements of bulk tissues on which GRN prediction are based, report aggregate genome‐scale measurements taken across cells with diverse regulatory states.
Recent technological developments such as high‐throughput partitioning of individual cells in aqueous reaction droplets coupled with synthesis of massive, unique barcode libraries have facilitated unbiased sampling of the transcriptomes and chromatin at the resolution of the single cell (Hashimshony, Wagner, Sher, & Yanai, 2012; Macosko et al., 2015; Zheng et al., 2017). The majority of single cell RNA‐seq (scRNA‐seq) studies in plants have examined developmental processes including Arabidopsis roots (Denyer et al., 2019; Jean‐Baptiste et al., 2019; Ryu, Huang, Kang, & Schiefelbein, 2019; Shulse et al., 2019; Wendrich et al., 2020; Zhang, Xu, Shang, & Wang, 2019) and maize shoot apices (Satterlee, Strable, & Scanlon, 2020) and anthers (Nelms & Walbot, 2019). The principal aim of these studies has been to identify different cell types and cell states within otherwise well‐characterized developmental trajectories. The value of single cell scale understanding of molecular mechanisms for plant research has been recognized and is an area of community interest (Rhee, Birnbaum, & Ehrhardt, 2019). The increasing resolution, capture rates, and available assays for single cell sequencing technologies have opened the possibility of studying single cell gene regulatory networks (scGRNs) in plants (Aibar et al., 2017; Jackson et al., 2020; Matsumoto et al., 2017; Van de Sande et al., 2020).
One major goal of GRN discovery in plants is to enhance stress resilience, because resilience is a key strategy for protecting food security during global warming. High temperatures, for example, impact human and environmental health through myriad avenues including increased demands for agricultural inputs (e.g., water, pesticides, fungicides) and through yield loss caused by environmental stressors (e.g., heat, drought, flooding) (Zampieri, Ceglar, Dentener, & Toreti, 2017). The Earth's surface temperature continues to increase, with the decade between 2010 and 2019 being the hottest on record (NOAA National Centers for Environmental Information, 2020). Essentially every biological process can be directly affected by heat because fundamental molecular processes and structures are sensitive to temperature change, including DNA and chromatin organisation, membrane fluidity, formation and stability of protein complexes, and transcription and translation (Vu, Gevaert, & De Smet, 2019). That said, not all tissues or developmental processes are equally sensitive to high temperature stress. Developing floral organs and fruits appear to be especially sensitive to high temperature in many plants, including rice (Shi, Ishimaru, Gannaban, Oane, & Jagadish, 2015), wheat (Narayanan, Prasad, Fritz, Boyle, & Gill, 2015), quinoa (Lesjak & Calderini, 2017; Tovar et al., 2020), and sorghum (Sunoj et al., 2017). High temperatures can affect flower and fruit development through chromatin remodelling leading to delayed flowering (del Olmo, Poza‐Viejo, Piñeiro, Jarillo, & Crevillén, 2019), by disrupting meiotic events in male gamete production (De Storme & Geelen, 2020), and by decreasing pollen production and reception (Prasad, Boote, Allen, Sheehy, & Thomas, 2006), all of which can lead to a decrease in overall yield (Zhao et al., 2017). Understanding the cell‐scale regulatory mechanisms that contribute to plant resilience to climate stressors, including high temperatures and drought, are critical for guiding genetic innovations that will contribute to food security in the future.
In this article, we report on recent advances in experimental and analytical methodologies for scRNA‐seq and other single cell genomic assays especially as they have been applied to the study of plants. We highlight recent advances and ongoing limitation for scGRN prediction, and finally, we highlight the opportunity to use scGRN discovery for studying and ultimately enhancing high temperature and other abiotic stress resilience in plants.
2. SINGLE CELL SEQUENCING IN PLANTS
The scientific value of single cell genomic resolution is recognized across biological systems, such as: diversity of gene expression patterns between cells and cell‐types; identification of rare cell types or cell states; functional characterization of cells (Rhee et al., 2019; Stuart et al., 2019). Although, low throughput single cell sequencing technologies have existed for some time, the focus of this article is on high‐throughput systems that have developed over the last 5 years (Macosko et al., 2015; Zheng et al., 2017).
The most popular single cell library construction tools follow a similar workflow (Figure 1). Briefly, cells are dissociated from one another, then a microfluidics system is used to encapsulate each individual cell within a droplet that contains a system for labelling transcripts with distinct barcodes which identify the cell from which the transcript originated and frequently also with a unique molecular identifier (UMI) sequence which can be used to identify sequencing reads corresponding to unique transcripts within the cell. The sequencing libraries are then prepared in bulk and the libraries are sequenced in bulk on a high throughput sequencing platform. The combined sequencing read data are then partitioned into single cell transcriptomes based on the occurrence of the barcode sequences, and then UMI are used to quantify individual transcripts within each cell.
FIGURE 1.
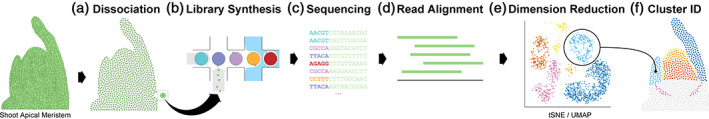
General workflow for single cell sequencing assays. (a) Tissues or organs are dissociated into individual cells through the isolation of protoplasts (small green circles); (b) the protoplasts are loaded into a microfluidics system that encapsulates individual protoplasts (small green circles) with reagents for labelling transcripts with distinct barcodes (larger multi‐coloured circles) which identify the cell from which the transcript originated, other barcodes such as UMIs may be added through this process as well; (c) the barcoded transcripts are then pooled and sequenced using a short read technology; (d) sequencing reads are then processed to assign each transcript to a cell of origin based on the barcode sequence added during library preparation; (e) the transcriptomes of all cells undergo dimension reduction (e.g., tSNE or UMAP) whereby cells with similar transcriptome profiles will be plotted closer together in two‐dimensional space while those with less similar transcriptomes will be plotted farther apart, and clusters of cells with similar transcriptomes can be identified algorithmically. In this example, each point on the plot represents a single cell and the colour of the point represents the cluster to which that cell has been assigned. (f) Clusters of cells may be characterized as a known cell types based on the abundance of known marker genes or on overall similarity to the transcriptomes of established cell types; cell clusters may also be described as unknown or novel if no known markers match the observed transcriptome profiles. In this example, cells in the reconstructed tissue are coloured to reflect the hypothetical transcriptome clusters identified in panel (e) [Colour figure can be viewed at wileyonlinelibrary.com]
In the last 2 years, single cell sequencing technologies, which were initially developed for use with mammalian cells, have been translated for use in the study of plant biology. Unlike animal cells, plant cells have rigid cell walls which must be disrupted to release protoplasts or nuclei for single cell sequencing; they have chloroplasts which can impact chromatin assays; and, they have abundance of secondary metabolites which can affect the efficiency and output of molecular assays. For these reasons, the majority of the first wave of high‐throughput, single cell assays have focussed on the Arabidopsis root for which well‐established protoplasting protocols, an absence of chloroplasts, and decades of experience with cell‐type transcriptome assays exist (Denyer et al., 2019; Jean‐Baptiste et al., 2019; Ryu et al., 2019; Shulse et al., 2019; Wendrich et al., 2020; Zhang et al., 2019).
2.1. Cell type inventories
The drive to functionally classify and characterize cells is fundamental to biology. Ideally scRNA assays would capture every transcript for all cell types in a tissue, to provide a complete and accurate census of gene expression and cell demographics at the moment of sampling. Current scRNA assays do not offer this level of resolution. For scRNA‐seq studies in Arabidopsis, the proportion of inputted cells for which high‐quality single cell sequence data is generated is between 20 and 50% in papers that reported these data (Denyer et al., 2019; Zhang et al., 2019). Though these figures varied between projects (Table 1), the number of transcripts captured was typically less than 10,000 per cell and represented fewer than 5,000 genes. It is unclear if the relative proportion of each cell type was accurately represented by the scRNA‐seq data or if some cell types were more likely to be lost during sample preparation. Studies which included biological replicates showed that the proportion of cells in each cluster was generally conserved across replicates (Ryu et al., 2019; Shulse et al., 2019). Creating accurate cell type inventories from scRNA‐seq data requires recognition of these limiting features of the data.
TABLE 1.
Summary of high throughput scRNA‐seq assays of Arabidopsis roots
| Number of scRNA Librariesa | Median Number of Transcripts/Cell | Median Number of Genes/Cell | Total Number of Genes | Clusters | |
|---|---|---|---|---|---|
| Ryu et al. (2019) | 7,522 | ~24,000 | ~5,000 | >22,000 | 9 |
| Jean‐Baptiste et al. (2019) | 3,121 | 6,152 | 2,445 | 22,419 | 11 |
| Denyer et al. (2019) | 4,727 | 14,758 | 4,276 | 16,975 | 15 |
| Shulse et al. (2019) | 12,198 | 2,291 | 1,216 | 25,324 | 17 |
| Zhang et al. (2019) | 7,695 | 4,556 | 1,875 | 23,161 | 24 |
| Wendrich et al. (2020) | 5,145 | – | 6,781 | 21,492 | 14 |
| Farmer, Thibivilliers, Ryu, Schiefelbein, and Libault (2020)b | 10,608 nuclei | 1,384 | 1,126 | 24,740 | 21 |
The number of scRNA‐libraries is variously reported as the number of transcriptomes, the number of single cells, and the number of STAMPs. In all cases this is taken to mean the number of single cells for which high quality sequencing data were obtained.
Farmer et al. used snucRNA‐seq in this project. All data in this row relate to single nuclei.
The first step in creating a cell‐type or cell‐state inventory is to distribute cells based on the similarities of their transcriptomes using dimension reduction techniques like t‐distributed Stochastic Neighbor Embedding (t‐SNE) (Maaten & Hinton, 2008) or Uniform Manifold Approximation and Projection (UMAP) (McInnes, Healy, & Melville, 2018). Next, an algorithm, such as the Louvain method for community detection (Blondel, Guillaume, Lambiotte, & Lefebvre, 2008), is used to identify discrete clusters of like cells within the overall cell populations. Distinct features and structures of scRNA‐seq data however demand caution when interpreting scRNA‐seq results for data driven cell‐type classifications. For example, the proportion of genes with no detectable expression in scRNA‐seq data is much higher than in bulk tissue RNA‐seq data (Grabski & Irizarry, 2020; Hicks, Townes, Teng, & Irizarry, 2018). The higher number of genes with detected transcripts in the bulk libraries or pseudo‐bulk libraries compared to the median number of genes in the studies of the Arabidopsis root cells reflect this feature of scRNA‐seq data (Table 1). Though biological variation in number of transcribed genes per cell and between cell‐types is expected, the number of transcribed genes is lower and variance in the number of transcribed genes is higher in scRNA‐seq experiments than would be expected from biological variation alone (Buen Abad Najar, Yosef, & Lareau, 2020; Hicks et al., 2018). Even for genes with transcripts which are definitively present in the biological sample, the probability of a non‐zero read count is less than one because only a subset of transcripts present in a cell are ultimately represented in the corresponding scRNA‐seq library (Grabski & Irizarry, 2020). Such elevated proportions of zero‐read genes can lead to overestimation of the distances between cells with low transcript detection rates during the dimension reduction phase of analysis. These effects can lead to an inflated number of predicted cell clusters (Grabski & Irizarry, 2020; Hicks et al., 2018).
Another attribute of scRNA‐seq data related to sparsity that may affect cell‐type classification is the now‐refuted finding that many cells in a homogenous population appeared to make one or another splice variant of a given gene but not both (Shalek et al., 2013; Song et al., 2017). Subsequent analysis of scRNA‐seq studies determined that this result was principally a technical artefact that could be explained by low levels of sequencing for single cells (Buen Abad Najar et al., 2020). Ongoing evaluation of technical sources of variation in data will be required as more data and new types of single cell assays become available.
2.2. Functional classification of cells
The next step in creating a cell inventory is assigning functional roles to clustered cells. This can be accomplished through comparison of the whole transcriptomes of the scRNA‐seq clusters with curated expression data sets for specific cell types. Though effective, the application of this method is generally limited because of the paucity of transcriptome data sets for most cell types, tissues, and species (Grabski & Irizarry, 2020). This approach has been used for assigning cell type identities to scRNA‐seq data from Arabidopsis roots (Denyer et al., 2019; Jean‐Baptiste et al., 2019; Shulse et al., 2019; Zhang et al., 2019) for which curated, cell‐type resolution gene expression data are available (Brady et al., 2006; Li, Yamada, Han, Ohler, & Benfey, 2016). Even in the case of well‐surveyed tissues like the Arabidopsis root, only 8 of the 15 clusters identified by Denyer et al. (2019) could confidently be assigned cell types using this method. A related approach creates an Index of Cell Identity for each cell to assign a label to cells based on the expression of a set informative transcripts from curated cell‐type specific transcriptome data (Efroni, Ip, Nawy, Mello, & Birnbaum, 2015).
Another widely used strategy for assigning cell identities is to survey the expression of previously characterized marker genes across the cell clusters. This approach is limited by the sparsity of scRNA‐seq data as described above; even if a marker gene is uniformly expressed in a given cell type it is unlikely to be measured in the majority of scRNA‐seq libraries. This is because only a subset of transcripts present in each cell are captured for sequencing, though higher abundance transcripts are more likely to be consistently detected than are less abundant ones (Zheng et al., 2017). A systematic investigation of a synthetic scRNA‐seq data found that even very well established marker genes were limited in their use for assigning cell type identities (Grabski & Irizarry, 2020). Cell type classification based on the expression of marker genes has been used in plant studies and has been used to identify cells to the level of cell types if not the level of cell states. One outcome of scRNA‐seq studies has been the emergence of data‐driven, unbiased marker gene selection methods, whereby genes that are both specific and sensitive to a cluster of cells are defined for each cluster. These methods have the advantage of being applicable to cell clusters for which no a priori marker genes are known and so they can be used to characterize novel cell types or cell states as well as developing more robust marker sets for cells of known identity. The application of these methods in plants (Denyer et al., 2019; Shulse et al., 2019) has led to the identification of new cell type specific markers for known cell types, many of which are common between studies.
2.3. Expansion and diversification of single cell genome scale assays
In addition to the widely used scRNA‐seq assay, a number of other single cell, genome scale assays have recently become available, only some of which have been applied to questions in plant biology. These assays can provide orthogonal functional data which will contribute to the more accurate assignment of functional cell identities (Stuart et al., 2019), and ultimately to more accurate predictions of scGRNs (Jackson et al., 2020).
Single cell Assay for Transposase Accessible Chromatin (scATAC‐seq) has been used to show that regions of accessible regulatory chromatin vary between classes of Arabidopsis root cells, thereby indicating a distinct cell type specific regulatory logic (Dorrity et al., 2020). This finding was consistent with the cell‐type specific ATAC‐seq studies of bulked root hair and non‐root hair protoplasts derived from Fluorescence Activated Cell Sorting (FACS) (Maher et al., 2018) and it greatly increased the number of cell types for which these data are available.
Single nucleus RNA‐seq (snucRNA‐seq) and snucATAC‐seq analysis of Arabidopsis roots have identified many of the same cell types and states as identified using scRNA‐seq, and identified several additional cell states that had not previously been described (Farmer et al., 2020). Nuclear transcriptome studies have the distinct advantages of rapid sample preparation time relative to protoplast‐based protocols, and may be particularly useful for the study of tissues for which protoplast isolation may not be possible or convenient. Moreover, nuclear assays provide transcriptome data that are distinct from whole cell assays. Previous studies of the nuclear transcriptome in rice, showed that the nuclear transcriptome, relative to the cytosolic transcriptome, was enriched for regulatory and nascent RNAs (Reynoso et al., 2018). Similar enrichments were detected in the snucRNA‐seq study of Arabidopsis root (Farmer et al., 2020), but these gains are made at the expense of the capture of fewer transcripts per cell (Table 2).
TABLE 2.
Summary of experimental considerations for sgGRN prediction
| Tissue Selection | Stress Selection |
|---|---|
|
Does the tissue include diverse cell types or cell states? Can enough cells or nuclei be harvested sufficiently quickly to perform the assay? Is the tissue sensitive to the proposed treatment? |
How severe and long lasting will the stress be? When will the stress be applied in development? In the circadian period? Will the stress be applied in isolation, in combination or in series with other relevant stressors? |
| Assay Selection | Network Algorithm Selection |
|
Can protoplasts or nuclei be quickly and efficiently isolated from the tissue? Do isolated protoplasts or nuclei represent the full diversity of cells in the tissue? Is knowledge of the spatial arrangement of cells important for the analysis? |
Is multimodal single cell sequencing data available either in data repositories or in the proposed experiment? Does the proposed experiment incorporate time series sampling? What complementary data exists for the tissue and/or treatment? |
Patterns of open chromatin as detected by ATAC‐seq are not direct predictors of transcript abundance in single cell, cell‐type enriched, or bulk tissue experiments (Farmer et al., 2020; Maher et al., 2018; Wilkins et al., 2016). New techniques that permit simultaneous measurement of transcriptome and chromatin accessibility in the same individual cell (Reyes, Billman, Hacohen, & Blainey, 2019) may provide clearer insight into the relationship between these genomic features and may assist in refining classifications of cell states and cell types (Hao et al., 2020). The power of multimodal single cell 'omics technologies, wherein multiple genome‐scale measurements are made on single cells are widely appreciated (Teichmann & Efremova, 2020) and the expanding diversity of single cell multimodal assays are reviewed elsewhere (Zhu, Preissl, & Ren, 2020).
Finally, spatially resolved transcriptome analysis, wherein information about the physical origins of genomic information is preserved in the sequence data, has been used to study transcription in plant tissues for which less cell‐resolved gene expression data is available, including the Arabidopsis inflorescence meristem, Populus tremula leaf buds, and Picea abies female cones (Giacomello et al., 2017). Spatially resolved scRNA‐seq are lower throughput than scRNA‐seq assays of dissociated cells, but they provide information about tissue organisation which cannot be inferred from dissociated cell data (Rodriques et al., 2019; Ståhl et al., 2016). With the wealth of single cell assays available as well as their optimization for use in plants, uncovering regulatory mechanisms in multicellular organisms is increasingly tractable.
3. USING scOMICS TO STUDY COMPLEX TRAITS: METHODS FOR scGRN PREDICTION
Transcription is regulated by multiple factors that determine when, where, and how much of each transcript is synthesized. These factors include proteins (transcription factors and RNA binding species) and small RNAs that interact with accessible conserved regulatory DNA elements (e.g., cis‐regulatory elements, and enhancers). A major goal of transcriptome research is to identify the regulatory mechanisms that have created the transcriptional or genomic snapshots provided by sequencing. The diversity of transcriptional states discovered in scRNA‐seq assays provides support for the simultaneous existence of a diversity of gene regulatory networks (GRNs) between cells. A complete inventory of gene regulatory events in a cell would be represented by stacks of spatially and temporally resolved GRNs showing all regulatory interactions and their functional outputs. However, this level of measurement is not yet achievable thus necessitating the use of computational methods to predict global GRNs from incomplete data. In this section, we describe examples of two promising and widely used algorithms for the discovery of scGRNs and discuss some of the particular challenges and opportunities related to predicting scGRNs compared to predicting GRNs in bulk tissue.
3.1. scGRN prediction
Network prediction methods explore statistical relationships between genes, and then test which of these statistical relationships have the highest likelihood of being regulatory (Chan, Stumpf, & Babtie, 2017). There are many methods that have been developed to accomplish this task using high‐throughput sequencing data (Bonneau et al., 2006; Castro, de Veaux, Miraldi, & Bonneau, 2019; Kulkarni et al., 2018; Roy et al., 2013; Van de Velde, Heyndrickx, & Vandepoele, 2014). Targeted molecular analyses have revealed how enormous the scale of the task is given the large number of molecular interactors and the diverse temporal and spatial scales on which the regulation of gene expression occurs. Many genes are regulated through the transient interactions between transcription complexes and their targets which in turn may be controlled through reversible post‐translational modifications of the transcription factors that form them (Para et al., 2014).
Moreover, there is the added complexity that transcriptional regulators may act synergistically, additively, or antagonistically to modulate gene expression. Considering the diverse mechanism of regulation, it is not surprising that many of the most promising computational approaches for predicting global gene regulatory networks rely on measurements of multiple complementary genome‐scale events (e.g., transcriptome, chromatin accessibility, etc.). The application of GRN prediction experience developed for population‐based analysis has contributed to the development of a variety of new and improved tools to overcome the challenges of single cell analysis (Aibar et al., 2017; Chan et al., 2017; Jackson et al., 2020; Matsumoto et al., 2017). These include sparsity of the expression matrices, the presence of technical noise and transcriptional stochasticity, and the predicted heterogeneity of GRNs in different cells within an experiment. Different methods have been developed for learning scGRN that vary in both the input data they require, and on the algorithms used to link regulators to target genes (Figure 2).
FIGURE 2.

Comparison of SCENIC and the Inferelator, two scGRN prediction algorithms. The SCENIC and Inferelator algorithms both use scRNA‐seq data as their primary input. SCENIC uses a random forest clustering algorithm to identify target genes that are co‐expressed with transcription factors. It then filters the putative regulatory clusters to retain only those whose targets are enriched for the occurrence of a priori known cisregulatory elements for the relevant transcription factor. Inferelator uses a multi‐task learning algorithm to learn scGRNs from transcriptome and complementary data types that are used to estimate the activity of transcription factors. The Inferelator can accept a wide variety of complementary inputs including Chromatin Immunoprecipitation ‐ sequencing (ChIP‐seq), protein–protein interaction and can explicitly use time series data. Both algorithms produce a matrix or graph of transcription factor target interactions [Colour figure can be viewed at wileyonlinelibrary.com]
Single‐Cell Regulatory Network Inference and Clustering (SCENIC) uses a three‐step workflow to predict GRNs from single cell data (Aibar et al., 2017; Van de Sande et al., 2020). First it identifies genes that are co‐expressed with transcription factors using GENIE3 (Huynh‐Thu, Irrthum, Wehenkel, & Geurts, 2010) or GRNBoost (Aibar et al., 2017). These algorithms use random‐forest regression to determine which transcription factor's expression profile best explains the expression profile of each target gene (Figure 2). Because they are based only on co‐expression, these methods are likely to include false positives and indirect targets in the co‐expression clusters (Aibar et al., 2017). The next step of SCENIC effectively filters these clusters by determining which of them are enriched for genes with relevant transcription factor binding sites. Through this process it defines “regulons” which include transcription factors and target genes that are both co‐expressed and enriched for the cis‐regulatory element to which the predicted regulatory transcription factor may bind. The third step of this workflow identifies cells in which the “regulons” defined in step two are active and in so doing identifies GRNs for each individual cell. This method uses the strengths of scRNA‐seq data, namely the high number of samples to overcome some of weaknesses of scRNA‐seq data, namely the sparsity of expression data for each cell.
A second method, called the Inferelator, was first developed for bulk cell GRN prediction (Bonneau et al., 2006) and has now also been adapted for use with scRNA‐seq data in yeast (Jackson et al., 2020). This method explicitly incorporates complementary a priori knowledge into the prediction of the GRNs from scRNA‐seq data rather than as a post hoc filter of co‐expression clusters (Figure 2). A key step in the Inferelator workflow is the estimation of a latent biophysical parameter termed Transcription Factor Activity (TFA). TFA is an estimated value that represents the effect of a transcription factor binding to DNA on modulating transcription of its target genes. Estimation of TFAs requires the construction of known prior transcription factor‐target network(s) (Arrieta‐Ortiz et al., 2015; Bonneau et al., 2006; Ciofani et al., 2012; Wilkins et al., 2016). Prior transcription‐factor‐target networks may be constructed using complementary genome scale features such as open chromatin, protein–protein interaction networks, cis‐regulatory element maps, or validated transcription factor‐DNA interactions. For example, a prior network of regulatory interactions of transcription factors and target genes could be constructed from ChIP‐seq or yeast 1‐hybrid data for a variety of transcription factors or by known cis‐regulatory motifs in regions of open chromatin. In the case of scGRN, scATAC‐seq data could be directly incorporated into a prior network. The prior network(s) are then used to estimate the TFA for each transcription factor based on the expression of the target genes in the prior networks. The Inferelator algorithm then uses multitask learning to infer regulatory interactions between transcription factors and their target genes based on the premise that the profile of a target gene can be expressed as the weighted sum of the TFAs of the transcription factors that regulate it. For the scGRN application of the Inferelator, the authors have implemented a multitask learning method through which separate GRNs can be learned for each cluster of cells identified by the scRNA‐seq analysis.
The use of TFA rather than transcript abundance to infer regulatory targets overcomes challenges resulting from the low transcriptional rates of many transcription factor genes. It also incorporates a measure of the potential consequences of post‐translational regulation of transcription factors that may temporally uncouple the production of transcription factor message from the generation of active protein transcription factors. The multitask learning approach also partially overcomes the limitations of sparsity typical of RNA‐seq data, by transferring information from one cell cluster to another.
3.2. scGRN discovery in plants
Neither the SCENIC nor the Inferelator method have been implemented with plant data, and they will likely require several adjustments to overcome obstacles when they are first used in these systems. First, the DNA binding sequence for most transcription factors in plants are not known and so the filtering step of SCENIC could presumably filter out the vast majority of co‐expression clusters because unknown cis‐regulatory sequences will not be enriched. Similarly, construction of a robust network prior based on occurrence of cis‐regulatory sequences for the Inferelator may not yet be possible for many plants as a consequence of the limited knowledge of true regulatory interactions. Methods that either directly measure transcription factor binding, like DNA Affinity Purification sequencing (DAP‐seq) (Bartlett et al., 2017; O'Malley et al., 2016), or predict them algorithmically, like cisBP (Weirauch et al., 2014), have greatly increased the number of transcription factors and plant species for which DNA binding sequences are available. However, experimentally validated binding sequence data remains sparse for most plant species.
A second obstacle is that plant transcription factors exist in large families wherein many members have identical or as yet undistinguishable binding sequences (Weirauch et al., 2014; Wilkins et al., 2016). For this reason, it may not be possible to assign a single transcription factor regulator to clusters where several members of the same transcription factor family are co‐expressed or to suitably divide targets between related regulators in a network prior. Third, transcription and translation of many transcription factors, including those involved in stress response, are often uncoupled from their regulatory activity. In these cases, post‐translational modifications regulate the entry of transcription factors into the nucleus and thereby their interactions with target genes. For these genes, the utility of co‐expression‐based GRN prediction methods may be limited.
Finally, SCENIC uses only transcription factor binding sequences that are proximal to transcriptional start sites. There is growing evidence of the importance of long‐distance regulatory elements, such as enhancers, playing important roles in plant gene expression (Joly‐Lopez et al., 2020; Ricci et al., 2019). In theory, long‐distance regulatory sequences can be incorporated into a prior network, but this will be dependent on greater knowledge of these regulators and their targets than are presently known in plants. Nonetheless, these methods will be highly valuable for scGRN prediction in projects for which scRNA‐seq data, thorough genome annotation, and knowledge of cis‐regulatory element sequences are available.
4. ENHANCING HIGH TEMPERATURE RESILIENCE BY TARGETING scGRNs
There are a number of outstanding questions for how to optimally use single cell sequencing technologies for studying stress induced changes in GRNs in plants: Which tissues will be amenable to different single cell platforms and assays; how many cells will be required to make meaningful inference; and, how many replicates are required for a robust analysis? These questions are in addition to the universal problems related to the analysis of single cell data outlined above.
Only a few published scRNA‐seq studies have incorporated perturbations into their analyses. These include environmental stressors (heat stress) (Jean‐Baptiste et al., 2019), nutrient treatments (Shulse et al., 2019) and genetic lesions (Denyer et al., 2019; Ryu et al., 2019). Much remains to be determined with regards to the most effective ways to use single cell sequencing technologies for learning environmental scGRNs. The goal of this section is to present a pathway for researchers to begin using single cell sequencing technologies to enhance crop resilience using the power of scGRN prediction and targeted genome editing. Below, we identify some of the decisions that researchers might contend with as they plan and implement these studies and some of the challenges that lay ahead. Throughout, we use the example of high temperature stress on the development of floral meristems. We have selected this example because high temperature stress is pervasive in agricultural and in less managed ecological settings, it is a tractable experimental question, and it is a trait predicted to involve the activity of many genes.
4.1. Designing experiments for scGRN prediction
Prerequisite to scGRN prediction is appropriate tissue, stress, and assay selection. What follows are guiding considerations that may be useful to researchers planning scGRN discovery projects (Table 2), and examples of how these decisions could be addressed in the context of high temperature stress on rice flowers.
4.1.1. Tissue selection
To perform single cell sequencing assays, a sufficient number of high‐quality protoplasts or nuclei must be isolated. For the 10X Genomics platform, for example, between 500 and 10,000 viable cells or nuclei are required per replicate. To obtain the desired number of cells, a large number of individual plants, or organs may be required. Shulse et al. (2019) used thousands of Arabidopsis roots to generate just over 12,000 scRNA‐seq libraries, and Satterlee et al. (2020) used hundreds of maize meristems to generate just over 250 scRNA‐seq libraries. Based on this, it is anticipated that hundreds of rice floral meristems would be required to obtain sufficient materials for a scGRN study.
Many scGRN studies will want to examine the variation in regulatory interactions across diverse cell types. In these cases it will be necessary to select a tissue type from which a diversity of cells can be isolated. The Arabidopsis root was a tractable first tissue for scRNA‐seq in part because of the extensive knowledge of the number, diversity, developmental trajectories, and transcriptomes of the diverse cell types they comprise. For most plant varieties and tissues, cell type transcriptomes are not available. In these cases, anatomical or histological evidence of cell types may be used to guide tissue selection for single cell assays. Rice floral meristems are a suitable tissue in these regards because there is extensive knowledge of the anatomy and development of the constituent cell types (Wang & Li, 2005). Moreover, transcriptionally distinct cell populations in micro‐dissected maize floral meristems have been identified (Knauer et al., 2019); transcriptional signatures of these cell types could be useful in characterizing single cells from rice meristems.
4.1.2. Stress selection
The duration, timing, and intensity of a stress treatment will elicit different physiological responses from the plant and tissue and will expose different aspects of stress responsive GRNs. Similarly, stresses experienced alone, in combination (e.g., simultaneous drought and heat) or in series (e.g., drought followed by heat) will query different aspects of plant stress responses. Likewise, transcriptional responses to heat stress in the field differs from responses in controlled environmental growth chambers (Plessis et al., 2015; Wilkins et al., 2016). For developing rice flowers, extensive physiological, agronomic, and anatomical examinations of heat stress responses have identified sensitive developmental stages and tissues (Cheabu, Moung‐Ngam, Arikit, Vanavichit, & Malumpong, 2018; Jagadish, Craufurd, & Wheeler, 2007). Moreover, rice flowers have a noted sensitivity to high nighttime temperatures (Desai et al., 2019). Selecting appropriate sampling times will have to consider these various aspects of stress response.
4.1.3. Assay selection
Selecting the appropriate assays to use for scGRN project will be influenced by technical constraints related to the selected tissue and treatment. In most cases, transcriptome will be the principal data used for scGRN prediction, and so the first decision will be whether to sequence single cells or single nuclei. This decision will be guided in part by the ability to quickly and efficiently dissociate the tissue for library preparation. For Arabidopsis roots, which have efficient and well‐established protoplast isolation protocols scRNA‐seq has been a valuable assay. However, for many other tissues, for which protoplast isolation protocols as less established or which are less amenable to protoplast isolation, snucRNA‐seq may be more appropriate. For example, protoplasts can be isolated from shoot and floral meristems in maize, but the protoplast are extremely delicate and are easily ruptured in handling (Satterlee et al., 2020). Moreover, it is unclear if protoplasts of all cell types within the meristem are equally susceptible to rupture, and so bias in the assayed cell types may be introduced at this stage. As such, in many cases it may be more pragmatic to isolate nuclei rather than protoplasts for sequencing. However, the number of reads per cell may be lower and the libraries will be biased for nascent transcripts over older transcripts when nuclear transcriptomes are assayed (Farmer et al., 2020; Reynoso et al., 2018).
In addition to transcriptome data, scGRN prediction will benefit from inclusion of additional genomic measurements, especially chromatin accessibility data. To date, multimodal assays have been applied to scGRN discovery in mammalian cells only (Hao et al., 2020; Mimitou et al., 2019); however, the increasing availability of off‐the‐shelf multimodal assays will expand their application into plant research. If little is known about constituent cells in a tissue, there may be particular value in using a spatial genomics assay. These assays provide complementary data about arrangement of transcriptome profiles which could contribute to the accurate description of cell function.
4.2. Target selection for genome editing
After predicting the network, the basic steps for using it to engineer improved crops are prioritizing regulatory interactions, using genome editing to alter regulatory interactions, and testing plants for improved resistance to stressor (Figure 3). While most GRN prediction methods rank interactions based on metrics such as variance explained, there are presently no reliable formulas for ranking interactions according to their likely impact on a physiological process. Prioritization of regulatory interactions for experimental characterization may be done through post hoc assessment of transcription factors and of co‐regulated target genes (Figure 3b). For example, transcription factors that regulate a large number of target genes or that regulate genes that are strongly differentially expressed in response to the treatment may be prioritized. Similarly, regulators of target genes that share a conserved cis‐regulatory element in their promoter sequences, or that are enriched for a biological process or Gene Ontology (GO) related to the stress response may be prioritized. In many cases, prioritizing interactions will require knowledge about the function of some genes or complementary genome analyses like GWAS or QTL, which can support educated guesses about which regulatory interactions may have the greatest impact on stress response.
FIGURE 3.
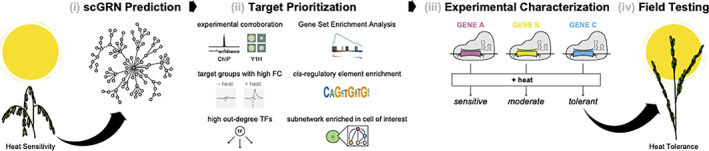
General workflow for using scGRNs for enhancing crop resilience. (a) The predicted scGRN will be a network of directed edges connecting transcription factors to the target genes they regulate. (b) post hoc assessment of the scGRN is used to prioritize regulatory targets for experimental characterization. This may include the identification of subnetworks that enriched in a cell type of interest; identification of co‐regulated genes that are enriched for biological processes of interest using Gene Set Enrichment Analysis or which are enriched for known cis‐regulatory elements; identification of regulatory interactions with corroborating experimental data, for example ChIP or yeast 1‐hybrid data; identification of co‐regulated genes that are strongly differentially expressed in response to the stress treatment; and characterization of transcription factors (TFs) that regulate many target genes. (c) Experimental characterization of prioritized components of the scGRN can be undertaken using genome editing approaches such as CRISPR. The coloured bars indicate different genomic regions that are targeted for editing. We anticipate that editing different interactions in the scGRN will influence plant resilience to varying degrees. (d) The most promising genome edited lines can then be tested in the field to determine the full effects of the modified scGRNs on stress resilience [Colour figure can be viewed at wileyonlinelibrary.com]
In the context of single cells, prioritization may be aided by the a priori knowledge of which cells types are most sensitive to the stressor and then focusing on regulatory interactions found in these cell types. Prioritized interactions can then be tested using inducible genome editing that enable cell type specific genomic changes (Wang et al., 2020) to determine what contributions they make to stress resilience (Figure 3c). As additional experiments are performed and network functions are experimentally characterized, more effective and established pipelines for target prioritization will be developed.
5. CONCLUSIONS
Although there are many uncertainties about how to most effectively predict and engineer gene regulatory networks in single plant cells, the value of regulatory knowledge at this resolution is unmistakeable. There will undoubtedly be trial and error as new ways of predicting and prioritizing regulatory interactions are developed and as higher throughput genome editing assays come on line. That said, the rapid uptake of the single cell sequencing technology by the plant science community as outlined above and the existence of a number of projects that aim connect different scales of mechanistic knowledge in plants (Marshall‐Colon et al., 2017; Rhee et al., 2019; Zhu et al., 2016) suggest that progress in this area is forthcoming. In this article, we have described recent application of single cell sequencing technologies to plant biology, new developments in scGRN prediction algorithms, and we have proposed a framework for using scGRN prediction for directing stress resilience studies in crops. While this workflow was developed around the question of rice response to heat stress, the framework could equally be used for the study of other abiotic stress responses and developmental programs in a variety of plants and tissues.
ACKNOWLEDGEMENTS
We wish to thank Drs. Z. Joly Lopez and J.A. Wilkins for their thoughtful comments and productive discussion on earlier drafts of this article. We also wish to thank R. Rahni for translating our rough sketches into the graphics included herein. Finally, we wish to thank the authors of many of the works cited herein for their open and frank discussions with us about their ground breaking work. This work was supported by a Discovery Grant awarded to OW from Natural Science and Engineering Research Council of Canada.
Tripathi RK, Wilkins O. Single cell gene regulatory networks in plants: Opportunities for enhancing climate change stress resilience. Plant Cell Environ. 2021;44:2006–2017. 10.1111/pce.14012
Funding information Natural Sciences and Engineering Research Council of Canada, Grant/Award Number: RGPIN‐2016‐04966; Science and Engineering Research Council
DATA AVAILABILITY STATEMENT
Data sharing not applicable ‐ no new data generated, or the article describes entirely theoretical research.
REFERENCES
- Aibar, S., González‐Blas, C. B., Moerman, T., Huynh‐Thu, V. A., Imrichova, H., Hulselmans, G., … Aerts, S. (2017). SCENIC: Single‐cell regulatory network inference and clustering. Nature Methods, 14(11), 1083–1086. 10.1038/nmeth.4463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrieta‐Ortiz, M. L., Hafemeister, C., Bate, A. R., Chu, T., Greenfield, A., Shuster, B., … Eichenberger, P. (2015). An experimentally supported model of the Bacillus subtilis global transcriptional regulatory network. Molecular Systems Biology, 11(11), 839. 10.15252/msb.20156236 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett, A., O'Malley, R. C., Huang, S. C., Galli, M., Nery, J. R., Gallavotti, A., & Ecker, J. R. (2017). Mapping genome‐wide transcription factor binding sites using DAP‐seq. Nature Protocols, 12(8), 1659–1672. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blondel, V. D., Guillaume, J.‐L., Lambiotte, R., & Lefebvre, E. (2008). Fast unfolding of communities in large networks. Journal of Statistical Mechanics: Theory and Experiment, 2008(10), P10008. 10.1088/1742-5468/2008/10/P10008 [DOI] [Google Scholar]
- Bonneau, R., Reiss, D. J., Shannon, P., Facciotti, M., Hood, L., Baliga, N. S., & Thorsson, V. (2006). The Inferelator: An algorithm for learning parsimonious regulatory networks from systems‐biology data sets de novo. Genome Biology, 7(5), R36. 10.1186/gb-2006-7-5-r36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brady, S. M., Orlando, D. A., Lee, J.‐Y., Wang, J. Y., Koch, J., Dinneny, J., … Benfey, P. N. (2006). A high‐resolution root spatiotemporal map reveals dominant expression patterns. Science (New York, N.Y.), 318(5851), 801–806. [DOI] [PubMed] [Google Scholar]
- Brooks, M. D., Juang, C.‐L., Katari, M. S., Alvarez, J. M., Pasquino, A., Shih, H.‐J., … Coruzzi, G. M. (2020). ConnecTF: A platform to integrate transcription factor‐gene interactions and validate regulatory networks. Plant Physiology, kiaa012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buen Abad Najar, C. F., Yosef, N., & Lareau, L. F. (2020). Coverage‐dependent bias creates the appearance of binary splicing in single cells. eLife, 9, 1–24. 10.7554/eLife.54603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castro, D. M., de Veaux, N. R., Miraldi, E. R., & Bonneau, R. (2019). Multi‐study inference of regulatory networks for more accurate models of gene regulation. PLoS Computational Biology, 15(1), e1006591. 10.1371/journal.pcbi.1006591 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan, T. E., Stumpf, M. P. H., & Babtie, A. C. (2017). Gene regulatory network inference from single‐cell data using multivariate information measures. Cell Systems, 5(3), 251–267.e3. 10.1016/j.cels.2017.08.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheabu, S., Moung‐Ngam, P., Arikit, S., Vanavichit, A., & Malumpong, C. (2018). Effects of heat stress at vegetative and reproductive stages on spikelet fertility. Rice Science, 25(4), 218–226. 10.1016/J.RSCI.2018.06.005 [DOI] [Google Scholar]
- Ciofani, M., Madar, A., Galan, C., Sellars, M., Mace, K., Pauli, F., … Littman, D. R. (2012). A validated regulatory network for Th17 cell specification. Cell, 151(2), 289–303. 10.1016/j.cell.2012.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Storme, N., & Geelen, D. (2020). High temperatures alter cross‐over distribution and induce male meiotic restitution in Arabidopsis thaliana . Communications Biology, 3(1), 187. 10.1038/s42003-020-0897-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- del Olmo, I., Poza‐Viejo, L., Piñeiro, M., Jarillo, J. A., & Crevillén, P. (2019). High ambient temperature leads to reduced FT expression and delayed flowering in Brassica rapa via a mechanism associated with H2A.Z dynamics. The Plant Journal, 100(2), 343–356. 10.1111/tpj.14446 [DOI] [PubMed] [Google Scholar]
- Denyer, T., Ma, X., Klesen, S., Scacchi, E., Nieselt, K., & Timmermans, M. C. P. (2019). Spatiotemporal developmental trajectories in the Arabidopsis root revealed using high‐throughput single‐cell RNA sequencing. Developmental Cell, 48(6), 840–852.e5. 10.1016/J.DEVCEL.2019.02.022 [DOI] [PubMed] [Google Scholar]
- Desai, J. S., Lawas, L. M. F., Valente, A. M., Leman, A. R., Grinevich, D. O., Jagadish, S. V. K., & Doherty, C. J. (2019). Warm nights disrupt global transcriptional rhythms in field‐grown rice panicles. BioRxiv, 702183, 1–44. 10.1101/702183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dorrity, M. W., Alexandre, C., Hamm, M., Vigil, A.‐L., Fields, S., Queitsch, C., & Cuperus, J. (2020). The regulatory landscape of Arabidopsis thaliana roots at single‐cell resolution. BioRxiv, 204792, 1–41. 10.1101/2020.07.17.204792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duran‐Nebreda, S., & Bassel, G. W. (2019). Plant behaviour in response to the environment: Information processing in the solid state. Philosophical Transactions of the Royal Society B: Biological Sciences, 374(1774), 20180370. 10.1098/rstb.2018.0370 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Efroni, I., Ip, P.‐L., Nawy, T., Mello, A., & Birnbaum, K. D. (2015). Quantification of cell identity from single‐cell gene expression profiles. Genome Biology, 16(1), 9. 10.1186/s13059-015-0580-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Farmer, A., Thibivilliers, S., Ryu, K. H., Schiefelbein, J., & Libault, M. (2020). The impact of chromatin remodeling on gene expression at the single cell level in Arabidopsis thaliana . Molecular Plant, 1. 10.1101/2020.07.27.223156 [DOI] [PubMed] [Google Scholar]
- Giacomello, S., Salmén, F., Terebieniec, B. K., Vickovic, S., Navarro, J. F., Alexeyenko, A., … Lundeberg, J. (2017). Spatially resolved transcriptome profiling in model plant species. Nature Plants, 3, 17061. 10.1038/nplants.2017.61 [DOI] [PubMed] [Google Scholar]
- Grabski, I. N., & Irizarry, R. A. (2020). Probabilistic gene expression signatures identify cell‐types from single cell RNA‐seq data. BioRxiv, 895441, 1–12. 10.1101/2020.01.05.895441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenfield, A., Hafemeister, C., & Bonneau, R. (2013). Robust data‐driven incorporation of prior knowledge into the inference of dynamic regulatory networks. Bioinformatics, 29(8), 1060–1067. 10.1093/bioinformatics/btt099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hao, Y., Hao, S., Andersen‐Nissen, E., Mauk, W. M., III, Zheng, S., Butler, A., … Satija, R. (2020). Integrated analysis of multimodal single‐cell data. BioRxiv, 335331, 1–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimshony, T., Wagner, F., Sher, N., & Yanai, I. (2012). CEL‐Seq: Single‐cell RNA‐Seq by multiplexed linear amplification. Cell Reports, 2(3), 666–673. 10.1016/j.celrep.2012.08.003 [DOI] [PubMed] [Google Scholar]
- Hicks, S. C., Townes, F. W., Teng, M., & Irizarry, R. A. (2018). Missing data and technical variability in single‐cell RNA‐sequencing experiments. Biostatistics, 19(4), 562–578. 10.1093/biostatistics/kxx053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huynh‐Thu, V. A., Irrthum, A., Wehenkel, L., & Geurts, P. (2010). Inferring regulatory networks from expression data using tree‐based methods. PLoS One, 5(9), e12776. 10.1371/journal.pone.0012776 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jackson, C. A., Castro, D. M., Saldi, G.‐A., Bonneau, R., & Gresham, D. (2020). Gene regulatory network reconstruction using single‐cell RNA sequencing of barcoded genotypes in diverse environments. eLife, 9, 1–34. 10.7554/eLife.51254 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jagadish, S., Craufurd, P., & Wheeler, T. (2007). High temperature stress and spikelet fertility in rice (Oryza sativa L.). Journal of Experimental Botany, 58(7), 1627–1635. 10.1093/jxb/erm003 [DOI] [PubMed] [Google Scholar]
- Jean‐Baptiste, K., McFaline‐Figueroa, J. L., Alexandre, C. M., Dorrity, M. W., Saunders, L., Bubb, K. L., … Cuperus, J. T. (2019). Dynamics of gene expression in single root cells of Arabidopsis thaliana . The Plant Cell, 31(5), 993–1011. 10.1105/TPC.18.00785 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joly‐Lopez, Z., Platts, A. E., Gulko, B., Choi, J. Y., Groen, S. C., Zhong, X., … Purugganan, M. D. (2020). An inferred fitness consequence map of the rice genome. Nature Plants, 6, 1–12. 10.1038/s41477-019-0589-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knauer, S., Javelle, M., Li, L., Li, X., Ma, X., Wimalanathan, K., … Timmermans, M. C. P. (2019). A high‐resolution gene expression atlas links dedicated meristem genes to key architectural traits. Genome Research, 29, 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kulkarni, S. R., Vaneechoutte, D., Van de Velde, J., & Vandepoele, K. (2018). TF2Network: Predicting transcription factor regulators and gene regulatory networks in Arabidopsis using publicly available binding site information. Nucleic Acids Research, 46(6), e31–e31. 10.1093/nar/gkx1279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langfelder, P., & Horvath, S. (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics, 9, 1–13. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesjak, J., & Calderini, D. F. (2017). Increased night temperature negatively affects grain yield, biomass and grain number in Chilean quinoa. Frontiers in Plant Science, 8, 352. 10.3389/fpls.2017.00352 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li, S., Yamada, M., Han, X., Ohler, U., & Benfey, P. N. (2016). High‐resolution expression map of the Arabidopsis root reveals alternative splicing and lincRNA regulation. Developmental Cell, 39(4), 508–522. 10.1016/j.devcel.2016.10.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Maaten, L., & Hinton, G. (2008). Visualizing data using t‐SNE. Journal of Machine Learning Research, 9(Nov), 2579–2605.Retrieved from. https://www.jmlr.org/papers/v9/vandermaaten08a.html [Google Scholar]
- Macosko, E. Z., Basu, A., Satija, R., Nemesh, J., Shekhar, K., Goldman, M., … McCarroll, S. A. (2015). Highly parallel genome‐wide expression profiling of individual cells using nanoliter droplets. Cell, 161(5), 1202–1214. 10.1016/j.cell.2015.05.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maher, K. A., Bajic, M., Kajala, K., Reynoso, M., Pauluzzi, G., West, D. A., … Deal, R. B. (2018). Profiling of accessible chromatin regions across multiple plant species and cell types reveals common gene regulatory principles and new control modules. The Plant Cell, 30(1), 15–36. 10.1105/tpc.17.00581 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marshall‐Colon, A., Long, S. P., Allen, D. K., Allen, G., Beard, D. A., Benes, B., … Zhu, X.‐G. (2017). Crops in silico: Generating virtual crops using an integrative and multi‐scale modeling platform. Frontiers in Plant Science, 8, 786. 10.3389/fpls.2017.00786 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsumoto, H., Kiryu, H., Furusawa, C., Ko, M. S. H., Ko, S. B. H., Gouda, N., … Nikaido, I. (2017). SCODE: An efficient regulatory network inference algorithm from single‐cell RNA‐Seq during differentiation. Bioinformatics, 33(15), 2314–2321. 10.1093/bioinformatics/btx194 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McInnes, L., Healy, J., & Melville, J. (2018). UMAP: Uniform manifold approximation and projection for dimension reduction. Retrieved from http://arxiv.org/abs/1802.03426
- Mimitou, E. P., Cheng, A., Montalbano, A., Hao, S., Stoeckius, M., Legut, M., … Smibert, P. (2019). Multiplexed detection of proteins, transcriptomes, clonotypes and CRISPR perturbations in single cells. Nature Methods, 16(5), 409–412. 10.1038/s41592-019-0392-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miraldi, E. R., Pokrovskii, M., Watters, A., Castro, D. M., De Veaux, N., Hall, J. A., … Bonneau, R. (2019). Leveraging chromatin accessibility for transcriptional regulatory network inference in T helper 17 cells. Genome Research, 29(3), 449–463. 10.1101/gr.238253.118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Narayanan, S., Prasad, P. V. V., Fritz, A. K., Boyle, D. L., & Gill, B. S. (2015). Impact of high night‐time and high daytime temperature stress on winter wheat. Journal of Agronomy and Crop Science, 201(3), 206–218. 10.1111/jac.12101 [DOI] [Google Scholar]
- Nelms, B., & Walbot, V. (2019). Defining the developmental program leading to meiosis in maize. Science, 364(6435), 52–56. 10.1126/SCIENCE.AAV6428 [DOI] [PubMed] [Google Scholar]
- NOAA National Centers for Environmental Information . (2020). State of the climate: Global climate report for annual 2019. Retrieved from https://www.ncdc.noaa.gov/sotc/global/201913
- O'Malley, R. C., Huang, S. C., Song, L., Lewsey, M. G., Bartlett, A., Nery, J. R., … Ecker, J. R. (2016). Cistrome and epicistrome features shape the regulatory DNA landscape. Cell, 166(6), 1598. 10.1016/j.cell.2016.08.063 [DOI] [PubMed] [Google Scholar]
- Para, A., Li, Y., Marshall‐Colón, A., Varala, K., Francoeur, N. J., Moran, T. M., … Coruzzi, G. M. (2014). Hit‐and‐run transcriptional control by bZIP1 mediates rapid nutrient signaling in Arabidopsis. Proceedings of the National Academy of Sciences of the United States of America, 111(28), 10371–10376. 10.1073/pnas.1404657111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Plessis, A., Hafemeister, C., Wilkins, O., Gonzaga, Z. J. Z., Meyer, R. S., Pires, I., … Purugganan, M. D. (2015). Multiple abiotic stimuli are integrated in the regulation of rice gene expression under field conditions. eLife, 4, 1–27. 10.7554/eLife.08411 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prasad, P. V. V., Boote, K. J., Allen, L. H., Sheehy, J. E., & Thomas, J. M. G. (2006). Species, ecotype and cultivar differences in spikelet fertility and harvest index of rice in response to high temperature stress. Field Crops Research, 95(2–3), 398–411. 10.1016/j.fcr.2005.04.008 [DOI] [Google Scholar]
- Reyes, M., Billman, K., Hacohen, N., & Blainey, P. C. (2019). Simultaneous profiling of gene expression and chromatin accessibility in single cells. Advanced Biosystems, 3(11), 1900065. 10.1002/adbi.201900065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynoso, M. A., Pauluzzi, G. C., Kajala, K., Cabanlit, S., Velasco, J., Bazin, J., … Bailey‐Serres, J. (2018). Nuclear transcriptomes at high resolution using retooled INTACT. Plant Physiology, 176(1), 270–281. 10.1104/pp.17.00688 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rhee, S. Y., Birnbaum, K. D., & Ehrhardt, D. W. (2019). Towards building a plant cell atlas. Trends in Plant Science, 24(4), 303–310. 10.1016/J.TPLANTS.2019.01.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ricci, W. A., Lu, Z., Ji, L., Marand, A. P., Ethridge, C. L., Murphy, N. G., … Zhang, X. (2019). Widespread long‐range cis‐regulatory elements in the maize genome. Nature Plants, 5(12), 1237–1249. 10.1038/s41477-019-0547-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriques, S. G., Stickels, R. R., Goeva, A., Martin, C. A., Murray, E., Vanderburg, C. R., … Macosko, E. Z. (2019). Slide‐seq: A scalable technology for measuring genome‐wide expression at high spatial resolution. Science (New York, N.Y.), 363(6434), 1463–1467. 10.1126/science.aaw1219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, S., Wapinski, I., Pfiffner, J., French, C., Socha, A., Konieczka, J., … Regev, A. (2013). Arboretum: Reconstruction and analysis of the evolutionary history of condition‐specific transcriptional modules. Genome Research, 23(6), 1039–1050. 10.1101/gr.146233.112 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ryu, K. H., Huang, L., Kang, H. M., & Schiefelbein, J. (2019). Single‐cell RNA sequencing resolves molecular relationships among individual plant cells. Plant Physiology, 179(4), 1444–1456. 10.1104/pp.18.01482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Satterlee, J. W., Strable, J., & Scanlon, M. J. (2020). Plant stem‐cell organization and differentiation at single‐cell resolution. Proceedings of the National Academy of Sciences, 117(52), 33689–33699. 10.1101/2020.08.25.267427 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalek, A. K., Satija, R., Adiconis, X., Gertner, R. S., Gaublomme, J. T., Raychowdhury, R., … Regev, A. (2013). Single‐cell transcriptomics reveals bimodality in expression and splicing in immune cells. Nature, 498(7453), 236–240. 10.1038/nature12172 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi, W., Ishimaru, T., Gannaban, R. B., Oane, W., & Jagadish, S. V. K. (2015). Popular rice ( Oryza sativa L.) cultivars show contrasting responses to heat stress at gametogenesis and anthesis. Crop Science, 55(2), 589–596. 10.2135/cropsci2014.01.0054 [DOI] [Google Scholar]
- Shulse, C. N., Cole, B. J., Ciobanu, D., Lin, J., Yoshinaga, Y., Gouran, M., … Dickel, D. E. (2019). High‐throughput single‐cell transcriptome profiling of plant cell types. Cell Reports, 27(7), 2241–2247.e4. 10.1016/J.CELREP.2019.04.054 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song, Y., Botvinnik, O. B., Lovci, M. T., Kakaradov, B., Liu, P., Xu, J. L., & Yeo, G. W. (2017). Single‐cell alternative splicing analysis with expedition reveals splicing dynamics during neuron differentiation. Molecular Cell, 67(1), 148–161.e5. 10.1016/J.MOLCEL.2017.06.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ståhl, P. L., Salmén, F., Vickovic, S., Lundmark, A., Navarro, J. F., Magnusson, J., … Frisén, J. (2016). Visualization and analysis of gene expression in tissue sections by spatial transcriptomics. Science (New York, N.Y.), 353(6294), 78–82. 10.1126/science.aaf2403 [DOI] [PubMed] [Google Scholar]
- Stuart, T., Butler, A., Hoffman, P., Hafemeister, C., Papalexi, E., Mauck, W. M., … Satija, R. (2019). Comprehensive integration of single‐cell data. Cell, 177(7), 1888–1902.e21. 10.1016/j.cell.2019.05.031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan, A. M., Arsovski, A. A., Lempe, J., Bubb, K. L., Weirauch, M. T., Sabo, P. J., … Stamatoyannopoulos, J. A. (2014). Mapping and dynamics of regulatory DNA and transcription factor networks in A . thaliana. Cell Reports, 8(6), 2015–2030. 10.1016/j.celrep.2014.08.019 [DOI] [PubMed] [Google Scholar]
- Sunoj, V. S. J., Somayanda, I. M., Chiluwal, A., Perumal, R., Prasad, P. V. V., & Jagadish, S. V. K. (2017). Resilience of pollen and post‐flowering response in diverse sorghum genotypes exposed to heat stress under field conditions. Crop Science, 57(3), 1658–1669. 10.2135/cropsci2016.08.0706 [DOI] [Google Scholar]
- Taylor‐Teeples, M., Lin, L., de Lucas, M., Turco, G., Toal, T. W., Gaudinier, A., … Brady, S. M. (2014). An Arabidopsis gene regulatory network for secondary cell wall synthesis. Nature, 517, 571–575. 10.1038/nature14099 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teichmann, S., & Efremova, M. (2020). Method of the year 2019: Single‐cell multimodal omics. Nature Methods, 17(1), 1–1. 10.1038/s41592-019-0703-5 [DOI] [PubMed] [Google Scholar]
- Thompson, D. A., Roy, S., Chan, M., Styczynsky, M. P., Pfiffner, J., French, C., … Regev, A. (2013). Evolutionary principles of modular gene regulation in yeasts. eLife, 2, e00603. 10.7554/eLife.00603 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tovar, J. C., Quillatupa, C., Callen, S. T., Castillo, S. E., Pearson, P., Shamin, A., … Gehan, M. A. (2020). Heating quinoa shoots results in yield loss by inhibiting fruit production and delaying maturity. The Plant Journal, 102(5), 1058–1073. 10.1111/tpj.14699 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van de Sande, B., Flerin, C., Davie, K., De Waegeneer, M., Hulselmans, G., Aibar, S., … Aerts, S. (2020). A scalable SCENIC workflow for single‐cell gene regulatory network analysis. Nature Protocols, 15(7), 2247–2276. 10.1038/s41596-020-0336-2 [DOI] [PubMed] [Google Scholar]
- Van de Velde, J., Heyndrickx, K. S., & Vandepoele, K. (2014). Inference of transcriptional networks in Arabidopsis through conserved noncoding sequence analysis. The Plant Cell, 26(7), 2729–2745. 10.1105/tpc.114.127001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vu, L. D., Gevaert, K., & De Smet, I. (2019). Feeling the heat: Searching for plant thermosensors. Trends in Plant Science, 24(3), 210–219. 10.1016/j.tplants.2018.11.004 [DOI] [PubMed] [Google Scholar]
- Wang, X., Ye, L., Lyu, M., Ursache, R., Löytynoja, A., & Mähönen, A. P. (2020). An inducible genome editing system for plants. Nature Plants, 6(7), 766–772. 10.1038/s41477-020-0695-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang, Y., & Li, J. (2005). The plant architecture of rice (Oryza sativa). Plant Molecular Biology, 59, 75–84. [DOI] [PubMed] [Google Scholar]
- Weirauch, M. T., Yang, A., Albu, M., Cote, A. G., Montenegro‐Montero, A., Drewe, P., … Hughes, T. R. (2014). Determination and inference of eukaryotic transcription factor sequence specificity. Cell, 158(6), 1431–1443. 10.1016/j.cell.2014.08.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wendrich, J. R., Yang, B., Vandamme, N., Verstaen, K., Smet, W., Van de Velde, C., … De Rybel, B. (2020). Vascular transcription factors guide plant epidermal responses to limiting phosphate conditions. Science, 370(6518), eaay4970. 10.1126/science.aay4970 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilkins, O., Hafemeister, C., Plessis, A., Holloway‐Phillips, M.‐M., Pham, G. M., Nicotra, A. B., … Purugganan, M. (2016). EGRINs (environmental gene regulatory influence networks) in rice that function in the response to water deficit, high temperature, and agricultural environments. The Plant Cell, 28(10), 2365–2384. 10.1105/TPC.16.00158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu, K., Xu, X., Fukao, T., Canlas, P., Maghirang‐Rodriguez, R., Heuer, S., … Mackill, D. J. (2006). Sub1A is an ethylene‐response‐factor‐like gene that confers submergence tolerance to rice. Nature, 442(7103), 705–708. 10.1038/nature04920 [DOI] [PubMed] [Google Scholar]
- Zampieri, M., Ceglar, A., Dentener, F., & Toreti, A. (2017). Wheat yield loss attributable to heat waves, drought and water excess at the global, national and subnational scales. Environmental Research Letters, 12(6), 064008. 10.1088/1748-9326/aa723b [DOI] [Google Scholar]
- Zhang, T.‐Q., Xu, Z.‐G., Shang, G.‐D., & Wang, J.‐W. (2019). A single‐cell RNA sequencing profiles the developmental landscape of Arabidopsis root. Molecular Plant, 12(5), 648–660. 10.1016/J.MOLP.2019.04.004 [DOI] [PubMed] [Google Scholar]
- Zhao, C., Liu, B., Piao, S., Wang, X., Lobell, D. B., Huang, Y., … Asseng, S. (2017). Temperature increase reduces global yields of major crops in four independent estimates. Proceedings of the National Academy of Sciences, 114(35), 9326–9331. 10.1073/PNAS.1701762114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng, G. X. Y., Terry, J. M., Belgrader, P., Ryvkin, P., Bent, Z. W., Wilson, R., … Bielas, J. H. (2017). Massively parallel digital transcriptional profiling of single cells. Nature Communications, 8(1), 14049. 10.1038/ncomms14049 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu, C., Preissl, S., & Ren, B. (2020). Single‐cell multimodal omics: The power of many. Nature Methods, 17(1), 11–14. 10.1038/s41592-019-0691-5 [DOI] [PubMed] [Google Scholar]
- Zhu, X.‐G., Lynch, J. P., LeBauer, D. S., Millar, A. J., Stitt, M., & Long, S. P. (2016). Plants in silico: Why, why now and what?‐an integrative platform for plant systems biology research. Plant, Cell & Environment, 39(5), 1049–1057. 10.1111/pce.12673 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data sharing not applicable ‐ no new data generated, or the article describes entirely theoretical research.