

Abstract
Hyperargininemia in patients with arginase 1 deficiency (ARG1‐D) is considered a key driver of disease manifestations, including spasticity, developmental delay, and seizures. Pegzilarginase (AEB1102) is an investigational enzyme therapy which is being developed as a novel arginine lowering approach. We report the safety and efficacy of intravenously (IV) administered pegzilarginase in pediatric and adult ARG1‐D patients (n = 16) from a Phase 1/2 study (101A) and the first 12 weeks of an open‐label extension study (102A). Substantial disease burden at baseline included lower‐limb spasticity, developmental delay, and previous hyperammonemic episodes in 75%, 56%, and 44% of patients, respectively. Baseline plasma arginine (pArg) was elevated (median 389 μM, range 238‐566) on standard disease management. Once weekly repeat dosing resulted in a median decrease of pArg of 277 μM after 20 cumulative doses (n = 14) with pArg in the normal range (40 to 115 μM) in 50% of patients at 168 hours post dose (mean pegzilarginase dose 0.10 mg/kg). Lowering pArg was accompanied by improvements in one or more key mobility assessments (6MWT, GMFM‐D & E) in 79% of patients. In 101A, seven hypersensitivity reactions occurred in four patients (out of 162 infusions administered). Other common treatment‐related adverse events (AEs) included vomiting, hyperammonemia, pruritus, and abdominal pain. Treatment‐related serious AEs that occurred in five patients were all observed in 101A. Pegzilarginase was effective in lowering pArg levels with an accompanying clinical response in patients with ARG1‐D. The improvements with pegzilarginase occurred in patients receiving standard treatment approaches, which suggests that pegzilarginase could offer benefit over existing disease management.
Keywords: ARG1‐D, arginase 1 deficiency, human enzyme, hyperammonemia, hyperargininemia, pegzilarginase, spasticity
1. INTRODUCTION
Arginase 1 deficiency (ARG1‐D) (OMIM number 207800) is a rare, progressive, autosomal‐recessive disease characterized by a marked reduction in the activity of arginase 1, a cytosolic enzyme expressed predominantly in the liver.1, 2, 3 The clinical presentation of ARG1‐D is distinct from the other urea cycle disorders, and includes highly elevated plasma arginine (pArg), spasticity, gait disorders, difficulty walking, developmental delay, failure to thrive, and seizures. The neurological symptom of spasticity is specific to ARG1‐D; this and the other neurological deficits are hypothesized to be caused by the markedly elevated levels of arginine and arginine‐derived metabolites in plasma, and in many cases accompanying elevated levels of arginine in the CSF.4, 5
Affected patients demonstrate markedly elevated levels of arginine and arginine‐derived metabolites with episodic hyperammonemia due to impairment of the urea cycle. Hyperammonemic episodes are typically less frequent and severe relative to other UCDs, but can be both severe and a presenting symptom in ARG1‐D. Spastic diplegia, which typically manifests in early childhood with difficulties in walking and climbing stairs, can sometimes be initially misdiagnosed as cerebral palsy (CP) or hereditary spastic paraplegia (HSP).6 Full‐scale IQ (FSIQ) in the ARG1‐D population is below the population mean, impairing school performance and educational achievement.7 Current treatment is focused on pArg level reduction (as high pArg is the hallmark of ARG1‐D) and prevention of hyperammonemia. Severe dietary protein restriction (below World Health Organization guideline ranges of 1.5 g/kg/day at 2 years of age to 0.83 g/kg/day at 18 years of age) with essential amino acid (EAA) supplementation aims to reduce pArg levels and ammonia scavengers for management of hyperammonemia.8, 9 The rationale for lowering pArg is based on case reports of improvements in disease manifestations following the introduction of a modified diet.10, 11 Although these data provide support for the potential value of reduction of pArg, in practice, dietary protein restriction has a limited impact on pArg levels with persistence of the distinct neurological manifestations, continued disease progression, and early death.2
Given the high unmet medical need in ARG1‐D and the demonstrated medical value and safety of recombinant human enzyme therapies for other metabolic diseases,12 we have developed a novel human enzyme‐based approach for disease management. Pegzilarginase (AEB1102; Aeglea BioTherapeutics, Inc., Austin, Texas) is a cobalt‐substituted, pegylated human recombinant arginase 1 enzyme with a marked increase in catalytic activity and stability relative to the native enzyme. In ARG1 knockout mice, pegzilarginase has been shown to reduce pArg levels after single and repeat doses in a dose‐dependent manner, which supports its therapeutic potential as an arginine‐reducing approach in patients with ARG1‐D.13
This is the first report of the clinical use of pegzilarginase in patients with ARG1‐D. The studies reported herein were designed to evaluate safety, pharmacokinetics (PK), and pharmacodynamic (PD) activity of repeat dosing with pegzilarginase. The PK profile will be reported separately.
2. METHODS
2.1. Study design
A Phase 1/2 open‐label study to investigate the safety, PK, and PD of IV pegzilarginase (CAEB1102‐101A [101A]) was conducted across nine sites in four countries. Patients completing 101A were eligible to enroll in the CAEB1102‐102A open‐label extension study (102A) to further evaluate the effects of pegzilarginase (Figure 1). The research was conducted in accordance with the Declaration of Helsinki and GCP guidelines. The protocols were approved by the appropriate ethics committees or institutional review boards at participating institutions and conducted in compliance with country‐specific regulatory requirements. The trials were registered as NCT02488044: 101A (http://www.clinicaltrials.gov/ct2/show/NCT02488044) and NCT03378531: 102A (http://www.clinicaltrials.gov/ct2/show/NCT03378531). All patients or their substitutes provided written informed consent/assent prior to undergoing study‐specific procedures.
FIGURE 1.
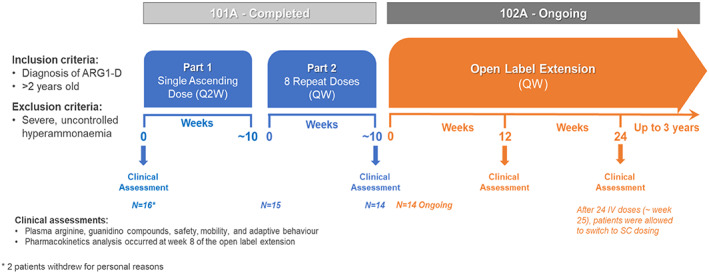
Flowchart diagram of the 101A Phase 1/2 study and the 102A long‐term open label extension. *Two patients withdrew for personal reasons
101A was conducted in two parts. In Part 1, patients received single ascending doses of pegzilarginase IV at 2‐week intervals. The pre‐defined doses were escalated until stopping rules were met for each patient in Part 1 (Appendix 1). In Part 2, patients received 8 weekly repeat doses of pegzilarginase IV and started with a dose chosen based on dose response from Part 1 of the study.
After completion of 101A, patients were eligible to enroll in 102A to evaluate the long‐term effects of pegzilarginase. In this study, patients resumed treatment with weekly administration of pegzilarginase at a dose selected based on 101A data; dose adjustments based on observed pArg levels were permitted.
2.2. Study participants
Eligible patients were ≥2 years old with baseline pArg levels >200 μM. Diagnosis was confirmed by the presence of pathogenic variants in the ARG1 gene or deficiency in red blood cell (RBC) enzyme activity. Exclusion criteria included recent hyperammonemic episode requiring hospitalization or active infection requiring treatment; history of hypersensitivity to polyethylene glycol (PEG); or any comorbid condition or laboratory abnormality that could interfere with study participation or interpretation (Appendix 2). All patients who completed 101A were eligible to participate in 102A.
2.3. Assessments
From the time of enrollment, patients were monitored at regular intervals for safety and tolerability. In 101A, these assessments continued until study participation ended. In 102A, efficacy assessments were analyzed through dose 12, which represents a total of 20 doses through both studies. The safety data cut was on 10 August 2019, providing a greater duration of safety follow‐up relative to the efficacy assessments.
Safety evaluations included monitoring of adverse events (AEs), concomitant medications, physical examinations, vital signs, and clinical laboratory tests. PD assessments included measurement of pArg and guanidino compounds (GCs) (ArgA = R, S‐argininic acid; GAA = guanidinoacetic acid; GVA = α‐keto‐δ‐guanidinovaleric acid; NAArg = Nα‐acetyl‐L‐arginine).
Baseline disease burden and clinical responses were assessed using multiple neuromotor, neurocognitive, and quality‐of‐life measures, including the Six‐Minute Walk Test (6MWT), Gross Motor Function Measures (GMFM), and Adaptive Behavior Systems (General Adaptive Composite, Conceptual, Practical and Social standard scores) and Vineland Adaptive Behavior Scales‐II (Adaptive Behavior Composite, Communication Domain, Daily Living Skills Domain, Motor Skills Domain, Socialization Domain standard scores). The presence of anti‐pegzilarginase and/or anti‐PEG antibodies (ADAs) was assessed using validated assays based on FDA immunogenicity testing guidance for therapeutic protein products (US Department of Health and Human Services FDA, January 2019).
2.4. Statistical analysis
This is an open‐label Phase 1 study; it is descriptive and thus no formal power calculations were applied. All patients who received at least one dose of pegzilarginase were included in the safety analysis. AEs, vital signs, and laboratory tests including PD assessments were summarized. Baseline abnormalities and key mobility assessments were displayed using a heat map‐like display.14 Given the absence of any previous reports of clinical outcome assessment use in ARG1‐D, baseline deficits and clinical response definitions were based on literature insights from other relevant mobility impacting conditions and expert opinions. Baseline deficits were defined as: 6MWT: below the lower fifth percentile; GMFM‐D: <35 of 39; GMFM‐E: <68 of 72; ABAS‐3: ≤85.15, 16, 17 Clinical response was defined using Minimal Clinically Important Difference (MCID) criteria (see Appendix 4). A Responder was defined based on a ≥1 MCID improvement in at least one of the three key mobility assessments (6MWT, GMFM‐D, and GMFM‐E).16, 17
6MWT MCID was considered as a 9% change from baseline.16 Patients were categorized into Gross Motor Function Classification System (GMFCS) classes to determine score changes necessary to attain MCID of medium effect size (Appendix 4). GMFM‐D MCID was taken as medium effect size in relative GMFCS classes I, II, and III of 2.4, 3.3, and 1.5, respectively.17 GMFM‐E MCID was taken as medium effect size in relative GMFCS classes I, II, and III of 4.0, 2.8, and 1.8, respectively.17 In both GMFM‐D and E, GMFCS class IV patients do not have a documented MCID, since they have such severe disability.
3. RESULTS
3.1. Patients
Baseline characteristics are described in Table 1. Sixteen patients were enrolled into 101A (of 17 screened); two withdrew for personal reasons. All percentages in this section are based on the 16 patients who enrolled in 101A. Patients' median age was 15.0 years (range: 5‐31 years) and 69% of patients were < 18 years of age. Median age at diagnosis was 1.0 year (range: 0‐27 years). One patient initially diagnosed with Cerebral Palsy and 1 with Hereditary Spastic Paraplegia were diagnosed with ARG1‐D at 11 and 25 years of age, respectively. Medical history and clinical biochemical findings were as expected in patients with ARG1‐D. Lower‐limb spasticity was reported in 75% of patients and considered moderate or severe in 56% of patients. Developmental delay was evident in 56% of patients and the median FSIQ was 61.7.
TABLE 1.
Baseline demographics and disease characteristics (n = 16)
| Characteristic | Value |
|---|---|
| Age at screening (years) | |
| Mean (SD), median (range) | 15 (8.5), 15 (5‐31) |
| Sex | |
| Female | 11 (69%) |
| Race | |
| White | 11 (69%) |
| Other | 5 (31%) |
| Height | |
| <Fifth centile | 8 (50%) |
| <10th centile | 12 (75%) |
|
Weight <fifth centile |
1 (6%) |
| <10th centile | 3 (19%) |
| Alanine aminotransferase (U/L) | |
| Mean (SD), median (range) | 47 (40.0), 34 (15‐171) |
| Aspartate aminotransferase (U/L) | |
| Mean (SD), median (range) | 42 (13.0), 38 (25‐63) |
| Ammonia (μmol/L) | |
| Mean (SD), median (range) | 41.1 (21.50), 38.0 (9.0‐77.0) |
|
History of Hyperammonemia (median number in past year, range) |
44% (0, 0‐6) |
| Ammonia scavenger medication use | 14 (88%) |
| Arginine (μmol/L) | |
| Mean (SD), median (range) | 373.4 (91.31), 389.3 (237.8‐565.8) |
| Dietary protein restriction + EAA supplementation | 16 (100%) |
| Seizure history | 7 (44%) |
A history of hyperammonemic episodes was present in 44% of patients; median number of episodes in the year prior to study entry was 0 (range: 0‐6). Seven patients had a history of seizures. All patients were managed with dietary protein restriction and EAA supplementation, and 88% of patients were taking ammonia scavengers. Height was <10th centile in 75% of patients. There were two patients taking anti‐epileptic therapy and four patients on baclofen. pArg levels were markedly elevated in all patients (median 389 μM; range 238‐566) (Figure 2A). Ammonia levels were abnormal in 44% of patients. ALT and AST were abnormal at baseline in 44% and 50% of patients, respectively.
FIGURE 2.
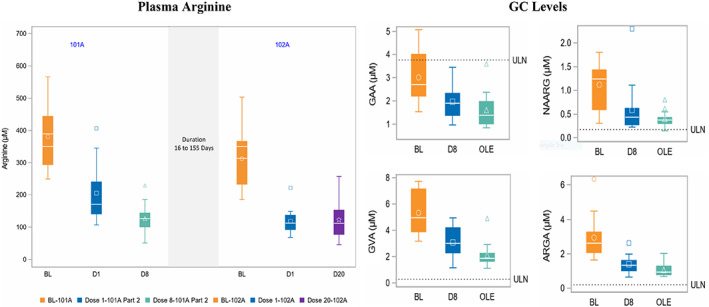
Box plot demonstrating the minimum, first quartile, median, third quartile, and maximum values for, A, plasma arginine at baseline, 7 days after doses 1 and 8 in 101A part 2 and 7 days after doses 1 and 12 in 102A. B, Plasma GC levels for GAA (guanidinoacetic acid), NAA(N‐α‐acetyl‐L‐arginine), GVA (α‐keto‐δ‐guanidinovaleric acid), and ArgA (argininic acid) at baseline and dose 8 in 102A, and as median in open‐label extension study. Note: For arginine, Dose 20 (if available) or Dose 19 (if D20 was not available) was used for 102A. Ten patients had D20 and 4 patients had D19. For GC, the mean of all OLE visits from 102, that is, Doses 9 to 20 was used. ArgA, R,S‐argininic acid; BL, baseline; D, dose; GAA, guanidinoacetic acid; GVA, α‐keto‐δ‐guanidinovaleric acid; NAArg, Nα‐acetyl‐L‐arginine; OLE, mean of doses in 102A (open‐label extension); ULN, upper limit of normal, determined from study of GCs in healthy adults (n = 12), equal to 0.2, 3.7, 0.3, and 0.17 μM for ArgA, GAA, GVA, and NAArg, respectively
At baseline, 88% of patients had at least one mobility deficit (Figure 3) and 88%, 50%, and 56% of patients had a baseline deficit in 6MWT, GMFM‐D, and GMFM‐E, respectively. ABAS‐3 assessments were available for 10 patients and 80% of these patients had baseline deficiencies in ≥1 ABAS‐3 domain. All 14 patients who completed 101A entered 102A.
FIGURE 3.
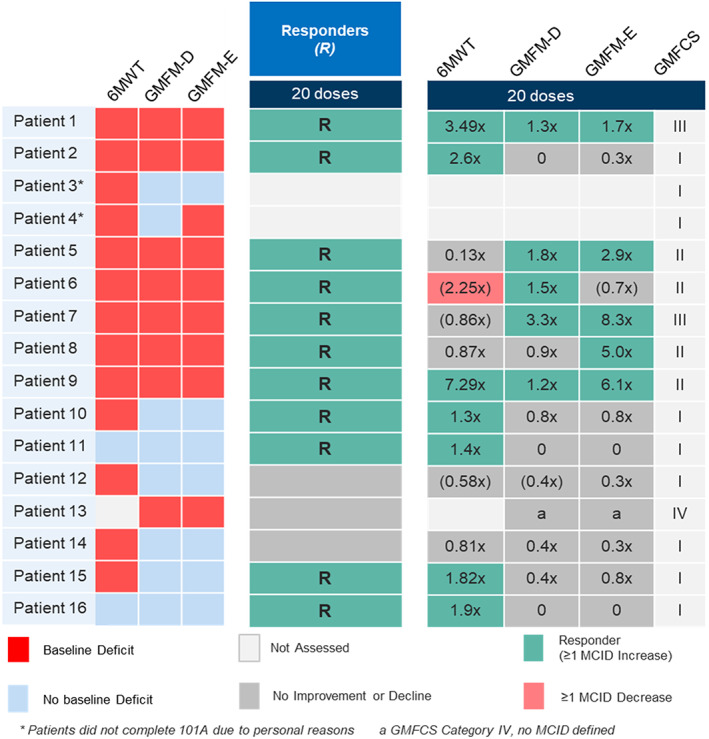
Heat map of baseline deficits in clinical outcomes assessments in all patients & overall clinical responses and individual assessment responses using three key complementary mobility assessments
3.2. Safety
A summary of the treatment‐emergent adverse events (TEAEs) is summarized in Appendix 6.
Overall, there were 162 TEAE, 95 of which were treatment related. Thirty were serious TEAEs, of which 13 were treatment related. None of the TEAEs led to discontinuation. The incidence of treatment‐related AEs is summarized in Table 2.
TABLE 2.
Cumulative summary of treatment‐related adverse events occurred in at least 15% of patients
| All patients (N = 16) Maximum grade n (%) | |||||
|---|---|---|---|---|---|
| System organ class | |||||
| Preferred term | Any severity | Mild | Moderate | Severe | Missing |
| Any event | 13 (81.3) | 12 (75.0) | 10 (62.5) | 1 (6.3) | 0 (0.0) |
| Gastrointestinal disorders | 7 (43.8) | 4 (25.0) | 3 (18.8) | 0 (0.0) | 0 (0.0) |
| Vomiting | 5 (31.3) | 3 (18.8) | 2 (12.5) | 0 (0.0) | 0 (0.0) |
| Abdominal pain | 3 (18.8) | 2 (12.5) | 1 (6.3) | 0 (0.0) | 0 (0.0) |
| Immune system disorders | 4 (25.0) | 1 (6.3) | 3 (18.8) | 0 (0.0) | 0 (0.0) |
| Hypersensitivity | 4 (25.0) | 1 (6.3) | 3 (18.8) | 0 (0.0) | 0 (0.0) |
| Metabolism and nutrition disorders | 3 (18.8) | 2 (12.5) | 0 (0.0) | 1 (6.3) | 0 (0.0) |
| Hyperammonaemia | 3 (18.8) | 2 (12.5) | 0 (0.0) | 1 (6.3) | 0 (0.0) |
| Skin and subcutaneous tissue disorders | 4 (25.0) | 4 (25.0) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
| Pruritus | 3 (18.8) | 3 (18.8) | 0 (0.0) | 0 (0.0) | 0 (0.0) |
All patients experienced at least one AE from 101A through to the safety data cut on 10 August 2019. The majority of AEs and all treatment‐related serious AEs (SAEs) were observed in 101A. The most frequent treatment‐related AEs included vomiting (31%), hypersensitivity (25%), and hyperammonemia, pruritus, and abdominal pain (19% each). In 101A, out of 162 infusions administered, there were seven hypersensitivity reactions experienced by four patients. In all four patients the first hypersensitivity reaction occurred with the second infusion. In 102A through Dose 12, a total of 151 infusions were administered and no hypersensitivity reactions were experienced. ADAs were detected in two of 16 patients at baseline prior to receiving pegzilarginase and in eight of 16 (50%) patients after dosing in Part 1 of 101A. Initial ADA titers ranged from 10 to 400 with no increase in titer after the initial ADA detection for any patient. All ADAs were undetectable by the end of 101A Part 2 with no new ADAs detected in 102A after 12 doses.
The SAEs in five patients were assessed as treatment related. Three of these five patients experienced a single hypersensitivity reaction that was managed with interruption of pegzilarginase administration, medications including antihistamines, and successful re‐initiation of the infusion at a slower rate. Two patients experienced hyperammonemia that was assessed as related to pegzilarginase by the investigator as the patients did not have a documented history of HA events. In both cases alterations were made to their individualized disease management (IDM), specifically changes to ammonia‐scavenging therapies and dietary protein intake, prior to and during these episodes.
3.3. Efficacy endpoints
pArg levels were rapidly and sustainably reduced by pegzilarginase (Figure 2A). In 101A, dose‐dependent decreases in pArg levels were seen with single and repeat doses. Repeat dosing resulted in marked and sustained reductions with a median reduction of 274 and 277 μM after 8 and 20 doses, respectively. The mean dose of pegzilarginase at Dose 8 was 0.09 mg/kg (range 0.04‐0.20) and at Dose 20 was 0.10 mg/kg (range 0.04‐0.20). Reductions in pArg from baseline to Dose 1, Dose 8, (101A Part 2), and 102A were statistically significant (P < .001). By Dose 20, 13 of 14 patients had pArg levels <200 μM prior to the next dose and 7 patients had pArg in the normal range (40‐115 μM). Reductions in pArg were accompanied by reductions in measured plasma GCs, which included GVA, ArgA, NAA, and GAA (Figure 2B).
Overall clinical response and the relative contribution of the three key mobility assessments in individual patients are shown in Figure 3. Eleven of 14 (79%) patients were defined as overall clinical responders at Dose 20 based on a ≥1 MCID improvement in at least one of the 6MWT, GMFM‐D, or GMFM‐E assessments. Four patients (29%) showed a ≥1 MCID improvement in ≥2 mobility assessments. For the 6MWT, 7 of 13 (54%) patients were responders with a mean change of 32 m across all patients and 66 m in the 7 responders. For the GMFM‐D, 5 of 8 (63%) patients with a baseline deficit were responders on this component alone (mean MCID 1.84, range 1.21‐3.33). For the GMFM‐E, five of eight (63%) patients with a baseline deficit were responders on this component alone (mean MCID 4.79, range 1.67‐8.33).
4. DISCUSSION
ARG1‐D is a rare enzyme deficiency disorder characterized by marked hyperargininemia with spastic diplegia and other distinct neurological manifestations.2 Given the likely relationship between high pArg levels and disease progression, current disease management is based on severe protein restriction to limit dietary contribution to the body arginine pool and the use of ammonia scavenger medications to prevent hyperammonemia. The impact of this restriction on pArg levels is limited as normal tissue turnover and not diet is the major contributor to pArg flux.18 Although the cytosolic localization of arginase 1 precludes the application of enzyme replacement approaches that have been successfully utilized for the treatment of lysosomal storage diseases,12 we hypothesized that an enzyme approach could have a favorable impact on arginine levels in patients with ARG1‐D. The results of this first‐in‐human clinical study demonstrate the safety and effectiveness of a human enzyme‐based approach in adults and children with ARG1‐D using a pegylated human arginase with enhanced catalytic activity and plasma stability.
Patients with ARG1‐D enrolled into 101A showed a range of clinical abnormalities typical for this patient population. Baseline biochemical findings were in alignment with literature reports with markedly elevated pArg levels and infrequent hyperammonemic episodes.1, 8 Disease manifestations and concomitant medication use were also as expected with frequent findings of spastic diplegia (75% patients), developmental delay (56.3% patients), and seizure history (44% patients). Standardized assessments at baseline provided insights for the first time on outcome assessments with potential utility as tools for both quantifying disease burden and assessing the clinical benefits of more effective pArg control. The 6MWT, GMFM‐D, and GMFM‐E mobility assessments were found to be useful in capturing disease burden with measurable baseline deficits in 88%, 50%, and 56% of 16 patients, respectively.
This study shows dose‐dependent reductions in pArg levels after single and repeat doses of pegzilarginase. Repeat dosing was highly effective in controlling arginine levels with 50% of patients achieving levels into the normal range after just 20 weekly IV doses, with a mean pegzilarginase dose of only 0.10 mg/kg. Most of the published literature on pArg lowering has focused on severe protein restriction. While these approaches have reduced pArg levels, they have not come close to the level of control achieved with pegzilarginase.8, 10, 11 Proof of concept that enhancement of blood arginase activity could decrease pArg levels with a potentially favorable impact on disease manifestations was demonstrated in 1984 in a 5‐year‐old affected with severe spastic quadriplegia.19 This patient underwent an exchange transfusion with normal RBCs containing arginase with lowering of pArg levels from 445 to 154 μM and an accompanying improvement in clinical status. The results with pegzilarginase show that a human enzyme‐based approach with a weekly dosing schedule can achieve pArg control to levels that have not been possible using currently available treatment approaches. The pegzilarginase‐mediated reductions in pArg levels were also accompanied by reductions in the levels of four known arginine‐derived GCs that accumulate in ARG1‐D patients.4 Although the tissue of origin and enzymology contributing to the increases in GC levels in patients with ARG1‐D is not known, the reductions in these metabolites with pegzilarginase suggests that pArg reductions may be reducing intracellular availability of arginine for subsequent GC formation. Although not well studied in humans, data from other species demonstrate tissue and plasma reductions in GCs as a consequence of reductions in plasma and intracellular arginine availability.20
In addition to documenting sustained reductions in pArg levels, this study provides evidence that the improved pArg control when pegzilarginase is added to current standard of care, is accompanied by improvements in important disease manifestations. Although the limited clinical improvements in response to the modest reductions in pArg achieved by protein restriction support the importance of pArg reduction, none of these studies have utilized standardized assessments to quantify any of the effects observed with diet.4, 6 Given the absence of any previous reports of clinical outcome assessment use in ARG1‐D, this study provides evidence for the first time of the potential utility of 6MWT, GMFM‐D, and GMFM‐E as tools for assessing clinical response in patients with ARG1‐D. The data supports the value of a combinatorial approach for capturing clinical benefit with a high overall clinical responder rate of 79% after 20 doses. The overall mean change in distance walked after 20 doses of 32 m exceeds what is considered an MCID based on insights from a range of other diseases.21 Clinically meaningful improvements were also seen with GMFM‐D and GMFM‐E, based on responder definitions which were originally developed in cerebral palsy patients who like ARG1‐D patients are similarly impacted by spastic diplegia.
The safety profile of pegzilarginase is supportive of further development as a therapy for ARG1‐D. There were no safety‐related discontinuations and hypersensitivity reactions were manageable with standard measures (Figure 4). Although low‐titer ADAs were detected after initial doses of pegzilarginase in Part 1 of 101A, the ADAs were transient and undetectable in all patients by Part 2 follow‐up and at Dose 12 in 102A. Hyperammonemia is a recognized manifestation of ARG1‐D and was observed during the study. Most cases of hyperammonemia were considered unrelated to pegzilarginase. In two patients hyperammonemia was assessed as related to pegzilarginase by the investigator. In both cases, alterations were made to the patient's IDM, specifically changes to ammonia scavenging therapies and dietary protein intake, prior to and during these episodes.
FIGURE 4.
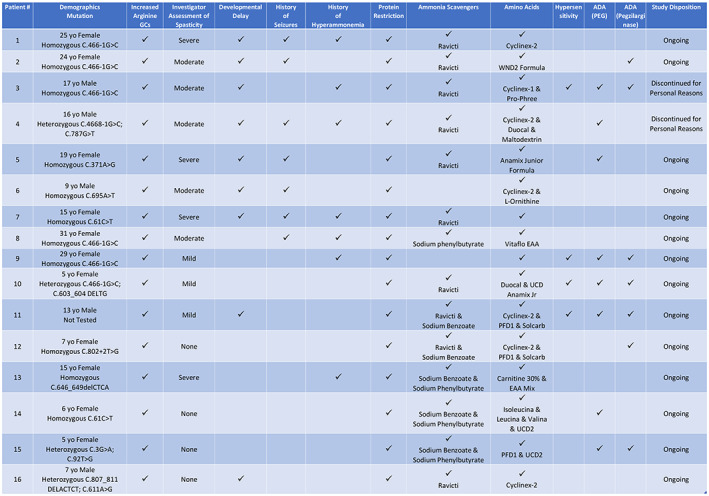
Patient profiles outlining baseline deficits, therapeutic & disease history, and occurrence of hypersensitivity and ADA
The 101A and 102A studies have some limitations. 101A was designed to assess the safety and tolerability of escalating doses of pegzilarginase, which means that none of the patients initiated dosing with the 0.1 mg/kg dose, which is anticipated, based on the insights from these studies, to be a dose that provides good pArg control in the majority of patients with ARG1‐D. In addition, both studies were open label and did not include a control group. Although insights from other mobility impacting diseases strongly support the clinical relevance of the observed improvements in 6MWT, GMFM‐D, and GMFM‐E that accompany the marked reduction in pArg, the reliance on orthogonal data is a limitation of this study and confirmation of their relevance in a controlled study in patients with ARG1‐D will be important.
The challenges of managing pArg levels in patients with ARG1‐D represent a significant unmet medical need given the disease burden due to persistent hyperargininemia. Pegzilarginase allowed the majority of patients in this study to achieve predose/trough pArg levels <200 μM, with control into the normal range in half of the patients. The accompanying clinical improvements after a relatively short duration of dosing demonstrate that pegzilarginase has the potential to improve patient care and the long‐term outcomes for this devastating disease. The improvements in arginine control and evidence of clinical benefit following pegzilarginase treatment support the key endpoints and other design elements of the pivotal Phase 3 PEACE trial (NCT03921541).
5. CONCLUSION
The data from these two studies clearly demonstrate that pegzilarginase is highly effective in lowering pArg levels with demonstrated evidence of improvements in important disease‐related manifestations. pArg reductions were seen in all patients and 79% of patients with ARG1‐D were defined as responders based on clinically meaningful improvements in one or more of the three key mobility assessments. Notably, the observed clinical improvements with pegzilarginase occurred in patients who had experienced persistent and/or progressive disease manifestations while receiving standard current treatment approaches. This suggests that pegzilarginase has the potential to improve disease outcomes over existing disease management.
CONFLICT OF INTEREST
G. A. D. has performed as a consultant and received travel and clinical study support from Aeglea. A. Schulze has received funds from Aeglea for the clinical study and for investigator‐initiated research. M. C. McNutt has received funds from Aeglea for clinical studies, honoraria, and travel reimbursement. E. Leão‐Teles has received financial support for educational activities and consulting and has received funds from Aeglea for the clinical study. J. L. Merritt II has received funds from Aeglea for the clinical study. G. M. Enns has received funds from Aeglea for the clinical study. S. Batzios has received funds from Aeglea for the clinical study, acted as a speaker on behalf of Aeglea. A. Bannick has received funds from Aeglea for the clinical study. R. T. Zori has performed as a consultant and received travel support and clinical study funding from Aeglea. L. S. Sloan, S. L. Potts, G. Bubb, and A. G. Quinn are employees of Aeglea BioTherapeutics Inc.
AUTHOR CONTRIBUTIONS
The first author and representatives of the sponsor made the decision to submit the manuscript for publication. All the authors had access to all the data and vouch for the accuracy and completeness of the data and analyses and for the fidelity of the study to the protocol.
G. A. Diaz: Reviewed and edited manuscript as first/lead author; aided in the revision; written first draft of the manuscript. A. Schulze: Reviewed and edited manuscript; aided in the revision. M. C. McNutt: Conducted the study; reviewed and edited the manuscript; aided in the revision. E. Leão‐Teles: Reviewed and edited manuscript; aided in the revision. J. L. Merritt: Reviewed and edited manuscript; aided in the revision. G. M. Enns: Reviewed and edited manuscript; aided in the revision. S. Batzios: Reviewed and edited manuscript; aided in the revision. A. Bannick: Reviewed and edited manuscript; aided in the revision. R. T. Zori: Reviewed and edited manuscript; aided in the revision. L. S. Sloan: reviewed and edited the manuscript; aided in the revision. S. L. Potts: reviewed and edited the manuscript; aided in the revision. G. Bubb: reviewed and edited the manuscript; aided in the revision. A. G. Quinn: reviewed and edited the manuscript; aided in the revision; written first draft of the manuscript.
Supporting information
Appendix S1: Supporting Information
ACKNOWLEDGMENTS
The study reported in this publication was funded by Aeglea BioTherapeutics Inc., Austin, Texas.
The authors would like to acknowledge: Dr Julien Baruteau, Consultant in Pediatric Metabolic Medicine from GOSH, Sub‐Investigator for the study. Clinical Research Facility staff members, Great Ormond Street Hospital, London, UK. Research co‐ordinators Liora Caspi and Ashley Wilson for running operational aspects of the study at Toronto Sick Kids. Dr. Michal Inbar‐Feigenberg—Clinical Investigator Toronto Sick Kids. Esmeralda Rodrigues, MD, Clinical Investigator and cared for study patients at Centro Hospitalar de São João, Porto. Dr Robert Conway—Clinical Investigator and cared for study patients at Children's Hospital of Michigan, Wayne State University, Detroit. Thu Quan (Study coordinator) and Chung Lee, MD (Clinical Investigator), who cared for study patients at Stanford University, Stanford. Susan Alters—developed Arginine assay and performed analysis of PK/PD/ADAs and reviewed manuscript. Mark Bechter facilitated manuscript development and communication with external authors. Susan Potts—statistical analysis and figures and tables. Adrienne Motion—reviewed and managed development of the manuscript. Kim Norris—editorial support.
Diaz GA, Schulze A, McNutt MC, et al. Clinical effect and safety profile of pegzilarginase in patients with arginase 1 deficiency. J Inherit Metab Dis. 2021;44:847–856. 10.1002/jimd.12343
Trial registration number: NCT02488044 and NCT03378531.
Communicating Editor: Johannes Häberle
Funding information Stanford University; Wayne State University
REFERENCES
- 1.Cederbaum SD, Shaw KN, Spector EB, Verity MA, Snodgrass PJ, Sugarman GI. Hyperargininemia with arginase deficiency. Pediatr Res. 1979;13:827‐833. [DOI] [PubMed] [Google Scholar]
- 2.Schlune A, vom Dahl S, Häussinger D, Ensenauer R, Mayatepek E. Hyperargininemia due to arginase I deficiency: the original patients and their natural history, and a review of the literature. Amino Acids. 2015;47:1751‐1762. [DOI] [PubMed] [Google Scholar]
- 3.Sin YY, Baron G, Schulze A, Funk CD. Arginase‐1 deficiency. J Mol Med. 2015;93(12):1287‐1296. [DOI] [PubMed] [Google Scholar]
- 4.De Deyn P, Marescau B, Qureshi I, Mori A.Hyperargininemia: a treatable inborn error of metabolism? The Fourth International Symposium on Guanidino Compounds in Biology and Medicine: Guanidino Compounds in Biology & Medicine II; 1997: 53‐69.
- 5.Scholl‐Bürgi S, Baumgartner Sigl S, Häberle J, et al. Amino acids in CSF and plasma in hyperammonaemic coma due to arginase1 deficiency. J Inherit Metab Dis. 2008;31(S2):S323‐S328. [DOI] [PubMed] [Google Scholar]
- 6.Carvalho DR, Brand GD, Brum JM, Takata RI, Speck‐Martins CE, Pratesi R. Analysis of novel ARG1 mutations causing hyperargininemia and correlation with arginase I activity in erythrocytes. Gene. 2012;509(1):124‐130. [DOI] [PubMed] [Google Scholar]
- 7.Waisbren SE, Cuthbertson D, Burgard P, Holbert A, McCarter R, Cederbaum S. Members of the urea cycle disorders consortium. Biochemical markers and neuropsychological functioning in distal urea cycle disorders. J Inherit Metab Dis. 2018;41(4):657‐667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Huemer M, Carvalho DR, Brum JM, et al. Clinical phenotype, biochemical profile, and treatment in 19 patients with arginase 1 deficiency. J Inherit Metab Dis. 2016;39(3):331‐340. [DOI] [PubMed] [Google Scholar]
- 9.Häberle J, Burlina A, Chakrapani A, et al. Suggested guidelines for the diagnosis and management of urea cycle disorders: first revision. J Inherit Metab Dis. 2019;42(6):1192‐1230. [DOI] [PubMed] [Google Scholar]
- 10.Cederbaum SD, Moedjono SJ, Shaw KN, Carter M, Naylor E, Walzer M. Treatment of hyperargininaemia due to arginase deficiency with a chemically defined diet. J Inherit Metab Dis. 1982;5:95‐99. [DOI] [PubMed] [Google Scholar]
- 11.Lambert MA, Marescau B, Desjardins M, et al. Hyperargininemia: intellectual and motor improvement related to changes in biochemical data. J Pediatr. 1991;118(3):421‐424. [DOI] [PubMed] [Google Scholar]
- 12.Desnick RJ, Schuchman EH. Enzyme replacement therapy for lysosomal diseases: lessons from 20 years of experience and remaining challenges. Annu Rev Genomics Hum Genet. 2012;13:307‐335. [DOI] [PubMed] [Google Scholar]
- 13.Burrage LC, Sun Q, Elsea SH, et al. Human recombinant arginase enzyme reduces plasma arginine in mouse models of arginase deficiency. Hum Mol Genet. 2015;24(22):6417‐6427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Haller C, Song W, Cimms T, et al. Individual heat map assessments demonstrate vestronidase alfa treatment response in a highly heterogeneous mucopolysaccharidosis VII study population. JIMD Rep. 2019;49(1):53‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.NIH . ToolboxR Scoring and Interpretation Guide for the iPad; 2016.
- 16.Schrover R, Evans K, Giugliani R, Noble I, Bhattacharya K. Minimal clinically important difference for the 6‐min walk test: literature review and application to Morquio a syndrome. Orphanet J Rare Dis. 2017;12(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oeffinger D, Bagley A, Rogers S, et al. Outcome tools used for ambulatory children with cerebral palsy: responsiveness and minimum clinically important differences. Dev Med Child Neurol. 2008;50(12):918‐925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu G, Morris SM. Arginine metabolism: nitric oxide and beyond. Biochem J. 1998;336(Pt 1):1‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Sakiyama T, Nakabayashi H, Shimizu H, Kondo W, Kodama S, Kitagawa T. A successful trial of enzyme replacement therapy in a case of argininemia. Tohoku J Exp Med. 1984;142(3):239‐248. [DOI] [PubMed] [Google Scholar]
- 20.Deshmukh DR, Sarnaik AP, Marescau B, et al. Guanidino compound metabolism in arginine‐free diet induced hyperammonemia. Enzyme. 1991;45(3):128‐136. [DOI] [PubMed] [Google Scholar]
- 21.Geiger R, Strasak A, Treml B, et al. Six‐minute walk test in children and adolescents. J Pediatr. 2007;150(4):395‐399. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1: Supporting Information