

Summary
Organic matter degradation in marine environments is essential for the recycling of nutrients, especially under conditions of anoxia where organic matter tends to accumulate. However, little is known about the diversity of the microbial communities responsible for the mineralization of organic matter in the absence of oxygen, as well as the factors controlling their activities. Here, we determined the active heterotrophic prokaryotic community in the sulphidic water column of the Black Sea, an ideal model system, where a tight coupling between carbon, nitrogen and sulphur cycles is expected. Active microorganisms degrading both dissolved organic matter (DOM) and protein extracts were determined using quantitative DNA stable isotope probing incubation experiments. These results were compared with the metabolic potential of metagenome‐assembled genomes obtained from the water column. Organic matter incubations showed that groups like Cloacimonetes and Marinimicrobia are generalists degrading DOM. Based on metagenomic profiles the degradation proceeds in a potential interaction with members of the Deltaproteobacteria and Chloroflexi Dehalococcoidia. On the other hand, microbes with small genomes like the bacterial phyla Parcubacteria, Omnitrophica and of the archaeal phylum Woesearchaeota, were the most active, especially in protein‐amended incubations, revealing the potential advantage of streamlined microorganisms in highly reduced conditions.
Introduction
Organic matter (OM) turnover in the oceans is driven by microbial activity but very little is known of the organisms that are performing this process in anoxic environments. Niches in oxygen‐depleted environments have traditionally been defined by available electron sinks (reviewed in Ulloa et al., 2012; Wright et al., 2012; Bertagnolli and Stewart, 2012), while the role of carbon sources in shaping these communities has not often been addressed. Since sinking OM sustains the microbial communities in the water column (Pomeroy, 1974; Azam et al., 1983, Azam, 1998), the adaptation to degrade different organic molecules is possibly one of the major factors defining the niches of water column organisms (Cottrell and Kirchman, 2000; Kujawinski, 2011; Gomez‐Consarnau et al., 2012).
The advent of sequencing methods and genomic approaches are expanding our understanding of the bacterial and archaeal phyla that are predicted, based on their genome, to be involved in OM processing in the absence of oxygen (Wrighton et al., 2012; Brown et al., 2015; Castelle et al., 2015, 2018; Hug et al., 2016; Solden et al., 2016). The taxonomic groups to which these organisms belong to form a major part of all detected sequences in natural environments (Lloyd et al., 2018). However, these organisms often resist traditional cultivation methods, which could be attributed to their lifestyle in highly connected and co‐dependent microbial communities, and, therefore, novel cultivation‐independent approaches are necessary to describe the functioning of these microbial communities. While (meta)genomics and transcriptomics data can give us an estimation of the potential capabilities of microbial communities and their uncultivated members, the direct observation of activity in OM degradation processes is necessary to understand the relationships and interactions of microbial cells with each other. Methods like DNA‐stable isotope probing (DNA‐SIP) allows us to track the metabolic flow of a substrate through a microbial community and increases our ability to understand the activity of microorganisms in situ. In addition, the combination of DNA‐SIP with the analytical resolution provided by next‐generation sequencing methodologies provides an opportunity to define active microbial members from complex communities (Bell et al., 2011; Hungate et al., 2015; Orsi et al., 2016; Pepe‐Ranney et al., 2016). For example, previous DNA‐SIP studies have given insights on the anaerobic heterotrophic carbon utilization in intertidal sediments (Webster et al., 2010; Graue et al., 2011, 2012), freshwater sediments (Coskun et al., 2018), soils (Pepe‐Ranney et al., 2016), salt marsh sediments (Seyler et al., 2014), and sands (Gihring et al., 2009). For water columns, DNA‐SIP has so far been used to evaluate the degradation of protein and high molecular weight dissolved organic nitrogen only in oxic and hypoxic conditions (Orsi et al., 2016; Liu et al., 2017).
Here, we focused on the microbial community of the anoxic and sulphide‐containing (euxinic) water column of the Black Sea, which is the largest stable anoxic basin in the world. It contains a shallow oxic layer of about 100 m, which overlays sulphidic waters spanning 2000 m in depth (Oguz et al., 2004). OM is preferentially preserved in euxinic conditions. This preservation is hypothesized to be due to the inability of organisms to degrade recalcitrant organic molecules in the absence of oxygen (Canfield, 1989, 1994; Bastviken et al., 2004). However, net dissolved organic carbon (DOC) removal (Margolin et al., 2016), as well as detected high carboxylic acid and amino acid concentrations (Mopper and Kieber, 1991; Albert et al., 1995), point to active anaerobic carbon degradation processes. This makes it probable that the unknown microbial community in the Black Sea water column relies on labile OM for their metabolic needs. In addition, there is evidence that the degradation of nitrogenous compounds is preferred when oxygen is depleted (Van Mooy et al., 2002; Pantoja et al., 2004, 2009; Engel et al., 2017), making organic nitrogen compounds (e.g. proteins) a potentially important substrate type.
In this study, we identify OM‐metabolizing microbial communities in the euxinic Black Sea. Specifically, we examined the changes in the microbial diversity and metabolic preference of the active anaerobic heterotrophic microbial community in anoxic sulphidic waters (at 1000 m depth) by performing incubations with two complex carbon substrates: 13C‐ and 15N‐labelled dissolved organic matter (DOM) and protein extracts. Additionally, we compared the activity of specific microbial groups with the metabolic potential of phylogenetically related metagenome‐assembled genomes (MAGs) obtained from the water column.
Results
Physicochemical conditions and microbial community composition at 1000 m depth
At the time of sampling, the Black Sea water column was fully anoxic starting from 100 m depth (Fig. 1). The sulphide concentration was measured during the cruise in 2013; it reached a maximum of 400 μM at 2000 m depth (Sollai et al., 2018). There was also an accumulation of ammonium reaching approximately 80 μM in the deep waters (Fig. 1). DOC and dissolved organic DON were 140 and 6.91 μM respectively, at 1000 m.
Fig. 1.
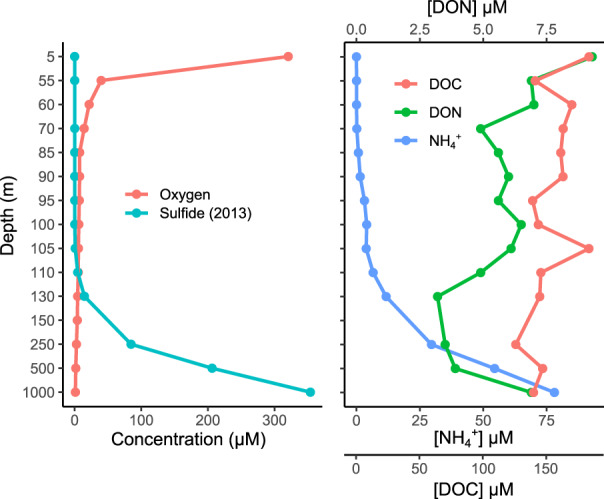
Water column physicochemical measurements at the sampling site. DON: dissolved organic nitrogen, DOC: dissolved organic carbon. Note that the y‐axis is not to scale. [Color figure can be viewed at wileyonlinelibrary.com]
The prokaryotic community composition of the anoxic water at 1000 m was analysed in both 2013 and 2017 with 16S rRNA gene amplicon sequencing (Fig. 2A). In addition, the taxonomic composition of the 16S rRNA genes extracted from the metagenomes in 2013 (see below) was also analysed to estimate possible biases arising from PCR amplification (Fig. 2A). In the amplicon data sets, the only difference between the years was the filter size used for sample collection (0.7 μm‐pore size glass fibre filter and 0.22 μm‐pore size polycarbonate filter respectively). Despite the different pore size filters used, the prokaryotic community composition was similar in both years (Fig. 2A). The most abundant bacterial taxonomic groups present belonged to the phylum Marinimicrobia (28% in 2013% and 27% in 2017 respectively). Chloroflexi were the next most abundant phylum (28% and 20%), mainly belonging to the genus Thermoflexus (12% and 15%). Members of the phylum Cloacimonetes comprised approximately 14% and 13% of the total community and were mostly comprised of the class MSBL2. Other abundant groups were Deltaproteobacteria (9% and 8%), belonging predominantly to the genus Desulfatigans (4.2% and 4.3%) and the uncultured SEEP‐SRB1 group (3.7% and 3.2%), and the phylum Planctomycetes (4.9% and 6.5%), which were dominated by the candidate order MSBL9 in both years (1.7% and 2.2%), and Phycisphaerales in 2017 (2.3%; not detected in 2013). Similarly, the phylum Omnitrophica was more abundant in 2017 (1.1% and 6.6%). Other phyla comprising >1% of the total community were Bacteroidetes (1.8% and 2%), Aminicenantes (1.2% and 0.2%) and Chlorobi (1.1% and 0.6%).
Fig. 2.
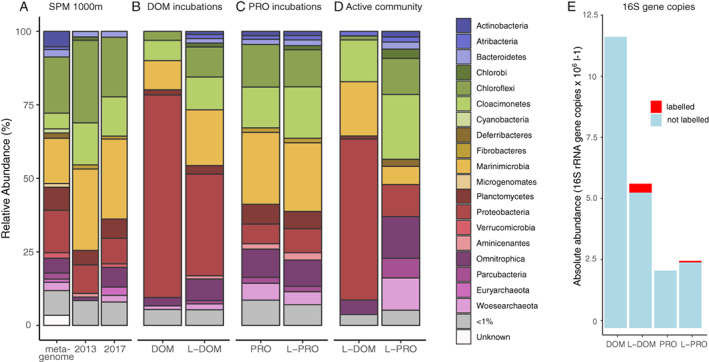
Prokaryotic community composition shown as the relative abundance of 16S rRNA genes in the Black Sea water column at 1000 m with and without the addition of OM.A. Water column community from collected suspended particulate matter (SPM) using the metagenomic data, 2013 and 2017 amplicon data sets.B. Incubations with unlabelled dissolved organic matter (DOM) and 13C/15N labelled dissolved organic matter (L‐DOM).C. Incubations with unlabelled protein (PRO) and 13C/15N labelled protein (L‐PRO).D. The community composition of the active operational taxonomic units (OTUs). E. Absolute abundance of prokaryotes (estimated as 16S rRNA gene copies L−1) for the different treatments, with red areas depicting the amount of copies that were assigned to labelled OTUs calculated by using the relative abundance of OTUs assigned as active. [Color figure can be viewed at wileyonlinelibrary.com]
The most abundant archaeal community members belonged to the anaerobic methane‐oxidizing archaea ANME‐1b within the Euryarchaeota (<1% in 2013, 2.4% in 2017), as well as to the phylum Woesearchaeota (<1% and 2.2%) belonging to the DPANN (Diapherotrites, Parvarchaeota, Aenigmarchaeota, Nanoarchaeota and Nanohaloarchaeota) superphylum.
Microbial community composition after incubations with DOM and protein extracts
DNA‐SIP experiments were conducted with 13C and 15N‐labelled and unlabelled DOM and protein extracts, in a total of four incubations lasting 72 h (Fig. S2). We used a labile carbon source as a proxy for the degradation of compounds from different biochemical categories, which does not reflect the natural OM available at 1000 m in the Black Sea. The analyses resulted in the identification of 15 279 individual operational taxonomic units (OTUs). Compared to the natural microbial community composition, incubations with labelled and unlabelled DOM resulted in an increase from 0.04% (average in 2013 and 2017) to 28%–66% of the relative abundance of 16S rRNA gene sequences closely related to the Gammaproteobacteria genus Psychromonas (Fig. 2B). Other abundant community members belonged to the phyla Marinimicrobia (10% and 19%), Cloacimonetes (6.8% and 11%), Thermoflexales from the phylum Chloroflexi (3.1% and 3.3%) and members of the phylum Omnitrophica (2.9% and 7.3%) (Fig. 2B). However, their relative abundance was slightly lower than in the natural situation because of the huge increase of Psychromonas spp.
In the incubations with unlabelled and labelled protein, the microbial community composition differed less from the natural situation (Fig. 2C); the most common phylum was Marinimicrobia (SAR406) (24% and 23% respectively), followed by Cloacimonetes (13% and 17%) and members of Chloroflexi (15% and 13%), mainly from the order Thermoflexales (5.5% and 3.8%) and from the candidate order GIF9 (1.2% and 2.0%) (Fig. 2C).
The total community composition clearly differed between the two substrates added. In the incubations with labelled substrates, 45% of the OTUs were shared between the two different substrate incubations. The taxonomic composition of the labelled and unlabelled incubations for each substrate was more similar than when those for the different substrates were compared (cf. Fig. 2B and C).
Microbial community composition with 13C and 15N labelled substrates
A quantitative SIP (qSIP) approach (Hungate et al., 2015) was applied to determine the labelled—and hence active—organisms in the incubations. Briefly, with this method, the identity of labelled organisms was determined by comparing the weighted average density of the 16S rRNA gene from unlabelled to labelled incubations (Fig. 3B and D). A shift to a heavier density of a specific OTU is considered to be due to uptake of the label and, hence, activity. In addition, the GC content of the gene is evaluated based on the density in unlabelled incubations and this can be used to estimate the percentage of labelling compared to the theoretical maximum density if all atoms of the DNA molecule were labelled. The quantitative level of labelling was therefore evaluated by calculating the theoretical molecular weight excess (MWE; see experimental procedures section for full explanation of all calculations). During the incubations, a relatively small fraction of the OTUs became labelled. In incubations with labelled DOM and protein, 6.1% (210 OTUs) and 2.2% (357 OTUs) respectively became labelled (Table 1; Fig. 3). Additionally, absolute copy numbers were calculated by multiplying the relative abundance of each OTU with the total 16S rRNA copies as determined by quantitative PCR (qPCR) analysis (Fig. 2E). The weighted average density of the active community was 1.716 g ml−1 for the labelled DOM incubations and 1.721 g ml−1 for labelled protein incubations, while the corresponding mean values considering all OTUs were 1.710 and 1.708 g ml−1, showing a clear shift in the 16S rRNA gene density (Fig. 3; see Table S1 for all calculated qSIP values). Most of the labelled OTUs were only detected in incubations with one of the substrates; only 2% of the labelled OTUs were shared between labelled communities of the DOM and PRO incubations. However, the taxonomic positioning of the labelled OTUs was predominantly across the same phylogenetic groups in both incubation types and predominantly belonged to the Marinimicrobia, Cloacimonetes, Chloroflexi, Deltaproteobacteria and Omnitrophica (Fig. 4).
Fig. 3.

Comparison of the distributions of two individual and all (stacked) active OTUs across the density gradient in the qSIP experiments for the unlabelled and labelled incubations. Panels A–C and D–F show two examples (i.e. Psychromonas and Woesearcheaota OTUs respectively) of the results obtained during qSIP calculations aimed to determine labelled OTUs. Panels A and D show the relative abundance (in fractional abundance) of the example OTUs relative to total read counts obtained from sequencing of each fraction for the unlabelled (blue) and labelled (red) incubations for the different density fractions obtained. Panels B and E show the distribution of the absolute copy number of the example OTUs across the density gradient obtained from the data in (A) and (D) and the measured total copy number in each density fraction. Dashed lines depict calculated weighted average densities of the OTU used for qSIP calculations. Panels C and F show the distribution of copy numbers across the density gradient as the relative abundance of the maximum copy number (values between 0 and 1). Panels G and H reveal the stacked distributions (as shown in C and F) of all active OTUs in incubations with DOM (G) and protein (H). [Color figure can be viewed at wileyonlinelibrary.com]
Table 1.
The taxonomic affiliations and abundance values of the most common OTUs determined active in incubations with 13C/15N labelled DOM and 13C/15N labelled protein substrate.
| Phylum | Class | Labelled DOM incubations | Labelled protein incubations | |||||
|---|---|---|---|---|---|---|---|---|
| Number of active OTUs | Total sequences | Active OTUs (%) | Number of active OTUs | Total sequences | Active OTUs (%) | |||
| Bacteria | Actinobacteria | 0 | 0 | 0 | 11 | 2613 | 3.1 | |
| Chlorobi | 5 | 8386 | 2.4 | 17 | 3375 | 4.8 | ||
| Chloroflexi | Anaerolineae | 1 | 5575 | 0.5 | 3 | 138 | 0.8 | |
| Dehalococcoidia | 15 | 9213 | 7.1 | 21 | 10 650 | 5.9 | ||
| Thermoflexia | 0 | 0 | 0 | 6 | 2088 | 1.7 | ||
| Cloacimonetes | MSBL2 | 19 | 137 700 | 9.0 | 18 | 25 713 | 5.1 | |
| MSBL8 | 5 | 18 312 | 2.4 | 3 | 3913 | 0.8 | ||
| Marinimicrobia | 18 | 202 225 | 8.6 | 16 | 17 788 | 4.5 | ||
| Omnitrophica | 24 | 55 513 | 11.4 | 43 | 14 925 | 12.1 | ||
| Parcubacteria | 3 | 1950 | 1.4 | 28 | 6025 | 7.9 | ||
| Proteobacteria | Alphaproteobacteria | 1 | 138 | 0.5 | 1 | 125 | 0.3 | |
| Deltaproteobacteria | 16 | 47,400 | 7.6 | 30 | 10 975 | 8.4 | ||
| Epsilonproteobacteria | 1 | 2338 | 0.5 | 0 | 0 | 0 | ||
| Gammaproteobacteria | 59 | 549 075 | 28.1 | 0 | 0 | 0 | ||
| Archaea | Woesearchaeota (DHVEG‐6) | 5 | 6088 | 2.4 | 73 | 11 175 | 20.5 | |
| Total | 210 | 1 × 106 | 100 | 357 | 1.3 × 105 | 100 | ||
Fig. 4.
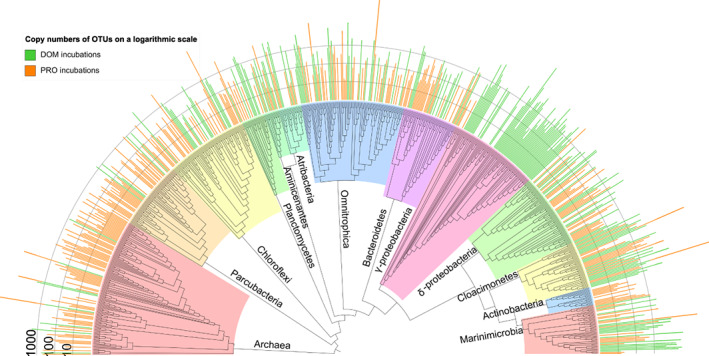
Taxonomic positioning of selected OTUs determined as active in the incubations with 13C/15N labelled dissolved organic matter (DOM, green bars) and 13C/15N labelled protein (PRO, orange bars). Colours on phylogenetic tree correspond to groupings at the Phylum level. Abundance bars are shown on a logarithmic scale. [Color figure can be viewed at wileyonlinelibrary.com]
For the DOM‐utilizing microorganisms, 16S rRNA gene sequences affiliated to the Gammaproteobacteria of the Psychromonas genus were dominant (28%) among the labelled OTUs, and a total of 5.4 × 105 copies, with a single OTU with 3.8 × 105 copies (Table 1). The next most abundant labelled OTUs were from Omnitrophica (11.4%), followed by the group SEEP‐SRB1 and Desulfatigans from the Deltaproteobacteria (both 3.8%), Marinimicrobia (8.6%) and the class MSBL2 of Cloacimonetes (11.4%). In contrast, the most abundant OTUs in terms of relative copy numbers after Gammaproteobacteria were those affiliated to Marinimicrobia (2 × 105 copies) and to Cloacimonetes (1.4 × 105 copies). There were only five OTUs with an MWE of >35%; three Proteobacteria, one Omnitrophica and one Marinimicrobia.
Regarding the protein‐utilizing microorganisms, Woesearchaeota was the most active phylum with a 20.5% relative abundance of the active community. However, its total abundance in copy numbers was relatively low (1 × 104 copies, Table 1). The next most active taxa in terms of labelled OTUs were Omnitrophica (12.1%), followed by Parcubacteria (7.9%), the Chloroflexi class Dehalococcoidia (5.9%), Cloacimonetes MSBL2 (5.1%), the Deltaproteobacterial genera SEEP‐SRB1 (4.5%) and Desulfatiglans (3.6%), Chlorobi (4.8%) and Marinimicrobia (4.5%). The majority of relative copy numbers were from Cloacimonetes (3 × 104 copies), followed by the Marinimicrobia (2 × 104 copies) (Table 1). Organisms with an MWE of >35% (33 OTUs) belonged predominantly to the candidate phyla Parcubacteria and the DPANN archaeal phyla Woesearchaeota.
Analysis of MAGs taxonomically related to the active community
We compared the active microbial community detected both in DOM and in protein‐amended incubations with the genomic potential for OM degradation of MAGs obtained from the Black Sea water column in 2013. We chose the analysed MAGs based on their quality, completeness and affiliation to the phyla that were determined as active in the DNA‐SIP incubations performed during the 2017 campaign. Phylogenetic trees showing the relationship of closely related genomes with 16S rRNA genes to the active OTUs can be found in Figs S3‐S8. We analysed metabolic genes of the selected MAGs using a selection of protein databases (i.e. KAAS and TIGRFAM; see section Experimental procedure for detailed methods). Additionally, we searched for genes encoding for carbohydrate‐active enzymes (CAZymes) and extracellular peptidases (MEROPS and SignalP or PRED‐SIGNAL) to estimate the substrate degradation potential of the MAGs. We analysed three MAGs affiliated to the bacterial phyla Chloroflexi (bin31, bin47 and bin52), two from Proteobacteria (bin40 and bin81 both belonging to the Deltaproteobacteria), and one each from Marinimicrobia (bin1), Omnitrophica (bin146) and Cloacimonetes (bin17). Two MAGs belonging to the archaeal phylum Woesearchaeota (bin111 and bin61) were also analysed (Table 2). The MAGs ranged from 56% to 96% in completeness with a contamination between 0% and 10% (Table 2). The Woesearcheaota MAGs as well the Omnitrophica MAG had a small estimated genome size of about 1.5 Mb, while the largest estimated genome sizes belonged to Chloroflexi bin30 (5.7 Mb) and the Deltaproteobacteria bin40 (4.6 Mb). The abundance of the co‐assembled reads belonging to each MAG at the 1000 m depth metagenome ranged from 8% (Marinimicrobia bin1) to 0.001% (Woesearchaeota bin111) (Table 2). Of the annotated proteins (n = 821–4286) a larger percentage of genes were assigned hypothetical for archaea (mean 58%) than for bacteria (mean 43%).
Table 2.
Assembly statistics of MAGs acquired from the metagenomes obtained in 2013.
| Bin No. | Phylum | Class | Family/Genus | Compla (%) | Contb (%) | Estimated genome size (Mbp)c | % reads mapped | No. of contigs | No. of annotated proteins | IMG accession |
|---|---|---|---|---|---|---|---|---|---|---|
| bin1 | Marinimicrobia | 89.0 | 1.0 | 2.24 | 8.005 | 232 | 1708 | Pending | ||
| bin146 | Omnitrophica | 56.5 | 9.6 | 1.43 | 0.045 | 230 | 821 | 2814122937 | ||
| bin17 | Cloacimonetes | 95.6 | 0.0 | 2.03 | 2.141 | 230 | 1612 | 2737471814 | ||
| bin31 | Chloroflexi | 80.9 | 4.6 | 5.71 | 2.346 | 612 | 4132 | 2806311051 | ||
| bin47 | Chloroflexi | Dehalococcoidia | 71.0 | 2.0 | 1.95 | 0.890 | 234 | 1355 | 2806311052 | |
| bin52 | Chloroflexi | Anaerolineales | 60.9 | 7.0 | 4.01 | 1.621 | 615 | 2500 | 2806311053 | |
| bin40 | Proteobacteria | Deltaproteobacteria | Desulfatiglans | 94.7 | 4.7 | 4.58 | 1.289 | 487 | 4286 | 2814122938 |
| bin81 | Proteobacteria | Deltaproteobacteria | Desulfobulboceae | 96.4 | 0.0 | 3.13 | 0.007 | 227 | 2828 | 2814122939 |
| bin111 | Woesearchaeota | 84.6 | 5.8 | 1.46 | 0.001 | 113 | 1440 | 2806311056 | ||
| bin61 | Woesearchaeota | 86.6 | 0.5 | 1.46 | 0.054 | 39 | 1369 | 2806311055 |
The most detailed known taxonomic information is shown.
Compl: completeness.
Cont: contamination.
Genome size was estimated from the bin size corrected for the degree of contamination and degree of completeness; Mbp: Millions of base pairs.
Central metabolic pathways of the MAGs
A range of 416–1645 genes were categorized into cell functions from the KEGG database (Fig. 5, Table S2). A near‐complete glycolysis pathway was found in all MAGs, as well as a diverse range of genes involved in fermentative metabolism, such as 2‐oxoglutarate/2‐oxoacid and pyruvate ferredoxin oxidoreductases (korAB, porABDG), formate dehydrogenases (fdoGH), methylmalonyl‐CoA mutase (mcmA) and alcohol dehydrogenases (adh).
Fig. 5.
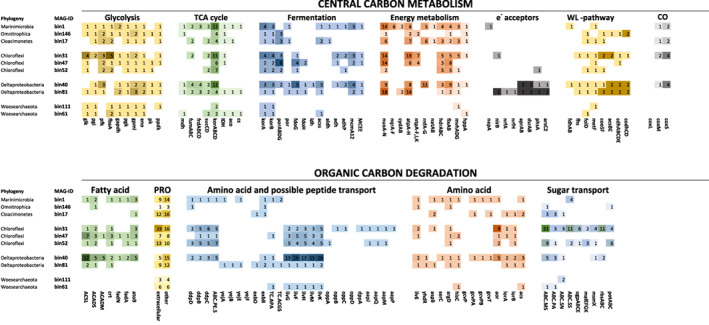
Comparison of selected genes involved in central carbon metabolism, energy metabolism and protein degradation between the MAGs discussed in this article. See full explanation of gene names and counts in Table S2. Darker colours correspond to higher counts in each category, while the lightest colour is always 1 copy of each gene. WL‐pathway: Wood‐Ljungdahl pathway of acetogenesis, CO: carbon monoxide processing genes, PRO: Proteases. [Color figure can be viewed at wileyonlinelibrary.com]
Energy conservation genes involved in oxidative phosphorylation, such as NADH oxidoreductases (nuoA‐N) and ATP synthases (atpA‐H/ntpA‐K) were found in all MAGs, except in the Woesearchaeota MAGs (bin111, bin61). In addition, a Na+ transporting NADH‐quinone oxidoreductase (nqrA‐F) was found in bin1 (Marinimicrobia). Only a few genes suggesting the use of terminal electron acceptors were identified. Genes for nitrogen cycling (nitrite reductase nirB, nrfAH; nitrogenase nifDHK) were mainly found in bin81 (Deltaproteobacteria) and a periplasmic nitrate reductase (napA) was found in bin1 (Marinimicrobia). Dissimilatory sulphur cycling genes (sulfite reductase, dsrAB, polysulfide reductase phsA) were present in Deltaproteobacteria (bin40 and bin81). Potential arsenate reductase genes (arsC2) were found in the Deltaproteobacteria MAGs (bin40, bin81) and possible selenate reductase genes (ygfKM) in bin31 (Chloroflexi). Oxygen‐reducing ubiquinol oxidases (cydAB) were found in bin1 (Marinimicrobia) and bin81 (Deltaproteobacteria). Potential for using hydrogen with NiFe ‐hydrogenase (hyaABD) was found in bin47 (Chloroflexi) and bin40 (Deltasulfatiglans). Bin40 also contained a second NiFe ‐hydrogenase (hydAB).
Several complexes were found that drive alternative intracellular redox cycling through the electron carriers NADH and ferredoxin (Fd). An Rnf–complex was found in bin1, bin17 and bin40. Heterodisulfide reductase (hdrABC) was encoded in all bacterial MAGs except bin146 (Omnitrophica). In addition, the mvhADG gene complex was encoded in MAGs bin1, bin17 and bin31. The fixAB electron transfer flavoprotein was encoded in all MAGs except in the Woesearchaeota (bin111 and bin61). We also found the Metf methylenetetrahydrofolate reductase in most MAGs as well as the glycine cleavage system in bin1 and bin40. The full Wood‐Ljungdahl pathway was encoded in the MAGs of Chloroflexi bin31 and the Deltaproteobacterial MAGs bin40 and bin81.
OM degradation potential of the MAGs
The CAZyme and MEROPS databases were used to evaluate the genomic potential for the degradation of different groups of organic compounds in the MAGs (Fig. 6A, Table S2). The most common carbohydrate esterases (CEs) detected belonged to the families 4 and 14. The first gene potentially encoding the esterase was detected three times in the Woesearchaeota bin111 and twice in the Omnitrophica bin146 and includes acetyl xylan esterases and chitin as well as peptidoglycan deacetylases. On the other hand, family 14 CEs acting on C─N bonds in linear amides were detected in the Chloroflexi bin31 and bin52. The most common carbohydrate‐binding module was from family 50, commonly found in enzymes cleaving chitin or peptidoglycan and mainly detected in the Chloroflexi bin31. The most abundant auxiliary activity was from family 4, which catalyses the conversion of a wide range of phenolic compounds and was mainly detected in the Desulfatiglans bin40. The quantity of identified CAZy was highest in one of the Chloroflexi MAGs (bin31, total assigned CAZy; n = 71) and the MAG classified as Desulfatiglans (bin40, n = 33, Fig. 6A).
Fig. 6.
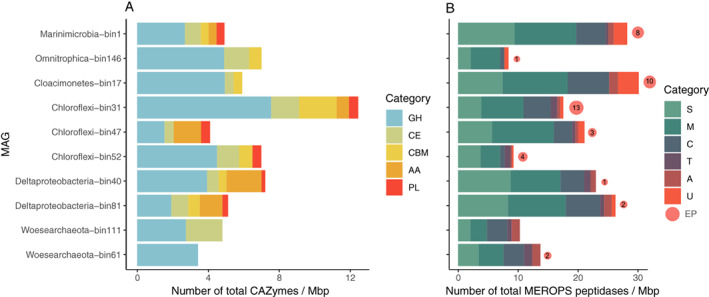
Number of genes encoding for substrate processing categories by MAG normalized to estimated genome size (Mbp: Millions of base pairs). A. Carbohydrate active enzymes, AA; auxiliary activities, CBM; carbohydrate‐binding modules, CE; carbohydrate esterases, GH; glucoside hydrolases, PL; polysaccharide lyases, and B. Peptidases identified by the MEROPS database and extracellular peptidases with Signal‐P or PRED‐SIGNAL, S; serine peptidases, M; metallopeptidases, C; cysteine peptidases, T; threonine peptidases, A; aspartic peptidases, U; unknown peptidases, and EP; extracellular proteases, amount shown with the size and total number inside the red circles (not normalized). [Color figure can be viewed at wileyonlinelibrary.com]
The identified peptidases were diverse, with few overlaps between the analysed MAGs. Most peptidases (also extracellular) were found in the Chloroflexi, Cloacimonetes and Marinimicrobia MAGs (n = 13, 10 and 8 extracellular peptidases respectively, Fig. 6B). Commonly found extracellular peptidases were serine endopeptidases like subtilisin (S8A), chymotrypsin (S1C), and cysteine endopeptidases like gingipain (C25). Three different types of bacterial cell wall hydrolysing peptidases (C82A, M23A and M23B) were found in Chloroflexi bin 31.
In addition, we searched for substrate degradation pathways and related transporters (Fig. 5). Genes for fatty acid degradation through the beta‐oxidation pathway were more complete in the MAGs Chloroflexi bin47 (19 copies of seven different genes; Fig. 5) and Desulfatiglans bin40 (32 copies of seven different genes; Fig. 5). General capabilities for amino acid degradation through the use of aminotransferases to produce 2‐keto acids and ferredoxin oxidoreductases (aor, iorAB) to metabolize these to aldehydes or acetyl‐coA were found in most bins, with the exception of the Woesearchaeota MAGs (bin111 and bin61). Branched‐chain amino acid transporters were the most common ABC transporters (livGHMK) with up to 16 copies found in MAGs affiliated to the phyla Chloroflexi (bin31, bin47 and bin52) and Deltaproteobacteria, (bin40 and bin81). The ABC‐type peptide/nickel transporters (ddpBCD) were also identified in multiple copies in Chloroflexi (bin31), Deltaproteobacteria (bin31, bin52, bin40) and in single copies in Marinimictobia (bin1) and in Dehalococcoidia (bin47). In addition, the most complete Chloroflexi MAG (bin31) had multiple sugar transporters matching its diverse CAZyme repertoire.
Discussion
The aim of this study was to identify the microbial community degrading labile OM in a marine euxinic water column and to determine if this community is defined by the available substrate type. With the combination of exploring the genetic potential using metagenomics and an incubation study to define active taxonomic groups, we are able to better understand the metabolic roles of the uncultivated majority of the diverse microbes in this environment. These complementary, cultivation‐independent approaches were applied in tandem to estimate how well metabolic predictions from genomic information match actual activity detected in the specific process of OM degradation.
The microbial community at 1000 m in the Black Sea was comprised of uncultivated microbial taxa ubiquitously occurring in anoxic environments like Chloroflexi (Hug et al., 2013a) and Deltaproteobacteria (Jochum et al., 2018; Skennerton et al., 2017), or specific to oceanic oxygen minimum zones worldwide, like Marinimicrobia (Hawley et al., 2017), as well as of microbial groups that remain poorly characterized like Cloacimonetes (Pelletier et al., 2008) and Omnitrophica (Rotaru et al., 2012). The composition of this community was similar in 2013 and 2017 (Fig. 2A), showing that the biogeochemical factors defining the microbial community do not vary much with time in the deep waters of the Black Sea, as would be expected from the strong stratification of the large basin. We compared the active OTUs in an incubation to the MAGs obtained from the same site that were affiliated to the same phyla. Though they are not directly linked, we consider the obtained MAGs to be the closest available genetic information to the studied OTUs, and to give valuable evidence on the genetic potential of the microbial community in the Black Sea.
DOM‐utilizing microbes
Few phyla were found to be labelled in only one of the two provided substrate incubations. Moreover, these substrate‐specific groups made up a major part (10%–30%) of the active OTUs in each incubation type, indicating that their activity was directly linked to the compound type of the provided substrate. In the active organisms in DOM incubations Gammaproteobacteria genus Psychromonas was the most abundant taxa (Fig. 2B). These organisms have previously been connected to rapid sugar fermentation in RNA‐SIP incubations with tidal‐flat sediments (Graue et al., 2011). Additionally, members of the closely related genus in Gammaproteobacteria are well‐known copiotrophs and have been detected in bottle incubations with or without substrate additions (Nelson and Carlson, 2012, Stewart et al. 2012). This phenomenon is deemed the bottle effect and possibly has to do with physical changes induced by sampling strategies in marine research. However, the OM provided in our incubations is labile and represents fresh phytoplankton biomass, which is not expected to be abundant at 1000 m depth in the sulphidic water column. The total absence of the Gammaproteobacteria from the active protein‐degrading community (and minor contribution to the total community) gives an indication that in our incubations the bioavailability of the substrate and the dominance of labile molecules enabled the growth of a copiotrophic organism, not significantly present in the deeper reduced water column of the Black Sea. The labelling of this genus after only half a day in previous anoxic experiments supports this conclusion (Graue et al., 2011). In addition to this, the DOM incubations had eightfold the amount of 16S rRNA gene sequences as determined by qPCR (Fig. 2E) compared to the protein incubations, also indicating a more rapidly growing microbial community and therefore possibly more readily usable carbon sources in the DOM carbon pool.
Protein‐utilizing organisms
The active groups that were specific to the protein‐amended incubations at the phylum level are from organisms with conspicuously small genomes; the DPANN archaea Woesearchaeota, and the bacterial phyla Parcubacteria (Nelson and Stegen, 2015; Liu et al., 2017; Castelle et al., 2018). In fact, the most active phylum, the Woesearchaeota, had about double the number of active OTUs than the next most abundant phylum. In addition, the most labelled organisms in terms of MWE were also predominantly affiliated to the Woesearchaeota. This high level of labelling would suggest that this organism could be utilizing the protein substrate for cellular growth, and therefore incorporating its carbon and nitrogen into its DNA. However, the MAGs classified as Woesearchaeota retrieved from the study site have relatively few genes assigned to CAZymes and few extracellular peptidases. This group belongs to the recently described DPANN superphylum of archaea, which is found worldwide in diverse habitats, with a preference for anoxic environments (Liu et al., 2018). Similar to previous studies on DPANN genomes, the two Woesearchaeota MAGs had considerably small genomes, seemingly lacking many essential metabolic genes, and in the case of bin111 also lacking atricarboxylic acid cycle and amino acid transporters, pointing to nutritional dependencies on other organisms (Castelle et al., 2015, 2018; Liu et al., 2018; Dombrowski et al., 2019). It is also possible that these genes were missed as the MAGs are not 100% complete. Conclusive genomic information to explain the success of this group is not easy to acquire as the taxonomic diversity and also metabolic versatility of the genomes acquired is large and a majority of the detected genes remain uncharacterized. Furthermore, more targeted studies will be required to understand the mechanism by which these organisms can incorporate the substrate into their cells.
While the Woesearchaeota were an abundant phylum in the environmental samples based on 16S rRNA gene amplicon sequencing, the Parcubacteria groups were not detected in significant amounts in natural Black Sea waters, and, hence, we did not obtain a Parcubacteria‐related MAG from metagenomes sequenced from the study site. This may highlight the disconnection between abundance and activity and shows the importance of activity measurements in characterizing microbial processes. In this case, the activity of this group could also be caused by a lability of the substrate that is not relevant in the deeper Black Sea similar to the most active phylum in the DOM incubations. The similarities between these two substrate‐specific phyla in the protein incubations can, however, indicate a metabolic strategy that was induced in our experiment. Like Woesearchaeota, the members of the Parcubacteria phylum, which is part of the candidate phyla radiation (CPR), are hypothesized to have a lifestyle that is dependent on other organisms for many essential metabolites (Kantor et al., 2013; Brown et al., 2015; Nelson and Stegen, 2015; Hug et al., 2016; Castelle et al., 2018). These groups had also relatively low copy numbers that can be the result of their slow growth, though it is also possible that it is an artefact of PCR bias, arising from the fact that these taxonomic groups have usually only one copy of the 16S rRNA gene in their genomes (Castelle et al., 2018). Our results indicate that the DPANN archaea and CPR bacteria, which seem to share a metabolic strategy (Castelle et al., 2015, 2018; Nelson and Stegen, 2015; Paul et al., 2017; Liu et al., 2018; Dombrowski et al., 2019), form an important part of the protein‐degrading microbial communities in the anoxic waters of the Black Sea. The role of archaea in degrading proteins especially in anoxic marine sediments has been hypothesized (Lloyd et al., 2013; Castelle et al., 2015). Orsi et al. (2016) reported that MGII Euryarchaeota were active in protein degradation in surface waters. On the other hand, protein was the only substrate out of several tested that was not assimilated by Crenarchaeota in salt marsh sediments (Seyler et al., 2014). The activity of DPANN Woesearchaeota in our incubations would, therefore, be confirming the role of an archaeal phylum in protein degradation in an anoxic environment for the first time.
Previous studies testing the microbial communities degrading proteins in marine environments found completely different microorganisms from those detected in our incubations (cf. Orsi et al., 2016 ; Liu et al., 2017). A diverse community was detected with abundant members of Gamma‐ and Alphaproteobacteria as well as members of the Flavobacteria and Verrucomicrobia in oxic waters (Orsi et al., 2016; Liu et al., 2017). The only similarity we find to the oxic incubations is a considerable activity of Actinobacteria in the assimilation of protein substrate (Table 1), as also detected by the studies of Orsi et al. (2016) and Liu et al. (2017). The Actinobacteria sequences in our incubations belonged almost completely (10 out of 11 OTUs) to the uncultured group OPB41, which surprisingly previously has been linked to the degradation of sugars in subsurface sediments (Bird et al., 2019). Our results could suggest a more widespread role for members of the Actinobacteria in protein degradation in the anoxic marine water columns. Liu et al. (2017) found a strong difference in the active microbial community degrading peptides when incubations with oxic waters were compared to that under hypoxic conditions but it was still very different from the one in our experiment. This difference may well be explained as the sulphidic waters incubated in our experiments have a much lower redox potential than the hypoxic waters incubated in their experiments. This would mean that, at least in the case of protein degradation, environmental conditions would be a more defining factor for the community than the substrate type provided. More comparison across the full water column and with more specific substrate sources would help answer these questions.
Taxonomic groups active in both substrate incubations
In addition to the main degraders forming a major part of the active community discussed in the previous section, several uncultivated microbial groups that belong to the most abundant taxonomic groups in the original water column were found to be active at both substrate incubations. Members of the phyla Omnitrophica, Marinimicrobia, Cloacimonetes, Chloroflexi and the order Deltaproteobacteria, each comprised 5%–12% of the total active OTUs in each incubation experiment.
Omnitrophica spp. were the second most active in both incubations with 11%–12% of active OTUs. We detected only a few genes for polymer degradation in the Omnitrophica MAG (bin146; Fig. 6), making it unlikely that these organisms are capable of the initial degradation of the provided substrate (i.e. algal proteins or DOM). Omnitrophica spp. have been linked to a host‐attached lifestyle in methanogenic reactors (Rotaru et al., 2012), and therefore its success in both substrate incubations indicates a metabolic strategy not linked to a specific carbon source but possibly to an actively growing host population (Rotaru et al., 2012; Kizina et al., 2017). The Omnitrophica MAG acquired from the Black Sea was the least complete of our analysed genomes (56%), making it difficult to infer the presence of full degradation pathways. However, we find partial pathways for OM degradation through glycolysis to acetate as well as evidence for hydrogen production with formate hydrogenlyase, and electron transfer with fixAB. Genes related to hydrogen production in this group has previously been detected from hydrothermal sediments (Dombrowski et al., 2017). Similar to Kizina et al. (2017), we find genes for the Type II secretion/Type IV pilus assembly system possibly linked to attachment to a host cell. Our Omnitrophica MAG could possibly perform hydrogen and acetate producing fermentation by scavenging low‐molecular‐weight OM from its environment.
The Marinimicrobia and Cloacimonetes phyla were the next most abundant shared organism groups in our DNA‐SIP incubations. The MAGs affiliated to these groups contained the genes encoding complete central metabolic pathways like glycolysis and enzymes for the degradation of amino acids, with several extracellular peptidases and the largest collection of energy metabolism genes of the assessed MAGs (Figs 5 and 6). They encode the electron transferring complexes and hydrogenases (Rnf, MvhHdr and FixAB) known from highly reduced conditions in methanogenic reactors (Nobu et al., 2015). The Rnf complex is capable of either coupling the reduction of NAD+ to the oxidation of reduced Fd and translocation of a proton across the membrane or the reverse, and is sometimes found in connection with the electron‐transfer‐flavoprotein‐oxidizing hydrogenase FixAB (Nobu et al., 2015). On the other hand, MvhHdr is an electron bifurcating hydrogenase, which couples the reduction of ferredoxin and a heterodisulfide from CoM‐CoB to the oxidation of hydrogen. This enables an organism to drive a thermodynamically unfavourable endergonic reaction with the reduction of two electron acceptors (Poudel et al., 2018). While Marinimicrobia spp. have been described with diverse capabilities to use alternative electron acceptors (Wright et al., 2014; Hawley et al., 2017; Plominsky et al., 2018), recently obtained MAGs from oxygen minimum zones and the hypoxic northern Gulf of Mexico suggested also a niche for degradation of macromolecular OM and peptides (Bertagnolli et al., 2017; Thrash et al., 2017). On the other hand, the elusive Cloacimonetes have been mainly linked to amino acid (Pelletier et al., 2008), cellulose (Limam et al., 2014) and propionate degradation (Nobu et al., 2015; Dyksma and Gallert, 2019). The Cloacimonetes MAG encodes pathways for the degradation of several amino acids but only one gene of those necessary for propionate degradation (pccB) was identified on a short contig (Fig. S11). Despite the considerable amount of extracellular peptidases encoded by the MAGs belonging to both phyla (Fig. 6), our DNA‐SIP study did not indicate a specific preference for the proteinaceous substrate. Instead, the Marinimicrobia and Cloacimonetes spp. could act as initial degraders of high‐molecular‐weight OM (including protein), and therefore have a role as generalist fermentative heterotrophs, ensuring their survival and persistence with many carbon sources. Their success could be based on being able to use complex carbon substrates while using diverse strategies to discard electrons in a highly reduced environment.
The Deltaproteobacteria and (Chloroflexi) Dehalococcoidia spp. also occurred in similar abundances in the DNA‐SIP incubations with the different substrates. The obtained MAGs affiliated to these phyla suggest capabilities for the degradation of intermediate products in an interaction with primary degraders. The Deltaproteobacterial phylum was comprised of two main known sulphate‐reducing genera, Desulfatiglans from the Desulfurellales class (Jochum et al., 2018), and the SEEP‐SRB1 group from the Desulfobacterales (Skennerton et al., 2017). The MAG affiliated to Desulfatiglans (bin40) contained, in addition to dsrAB, 10 copies of branched‐chain amino acid ABC transporter genes, and genes encoding formate dehydrogenases, a full pathway for fatty acid degradation and the enzymes required for the utilization of propanoate and butanoate, all important indicators for the degradation of intermediate carbon compounds resulting from primary fermentation (Schink and Stams, 2013; Narihiro et al., 2016). The other detected Deltaproteobacteria MAG (bin81) also contained a dsrAB, genes of the propanoate conversion pathway, as well as the branched‐chain amino acid transporters and genes for the degradation pathways similar to those of the Desulfatiglans MAG. Dehalococcoidia, on the other hand, have been defined by their capabilities for dehalogenation as a terminal reduction pathway, though recently genomes retrieved from the deep biosphere have classified these bacteria as anaerobic acetogens capable of fermentation of plant polymers as well as amino acids and organosulphur compounds (Hug et al., 2013a; Wasmund et al., 2014). In the Dehaloccoidia MAG (bin47), we did not find indications of complex substrate degradation, the Wood‐Ljungdahl pathway, or a reductive dehalogenase gene, but several copies of genes for the beta‐oxidation of fatty acids (fadD, fabG), for formate (fdoGH) and hydrogen utilization (hdrABC, fixAB), and propionate degradation (mcmA1/2, MCEE), connecting it to a similar metabolic role in the community as the Deltaproteobacteria. Based on this metabolic analysis, the MAGs belonging to these phyla suggest capabilities for the degradation of intermediate fermentation products in close interaction with other organisms. The Deltaproteobacteria most likely use sulfuric compounds as electron sinks, while the possible electron acceptor used by Dehalococcoidia is not evident from the MAG analysis. Based on the ubiquity of dehalogenation in this taxonomic group, could be utilizing organohalide compounds (Hug et al., 2013b; Biderre‐Petit et al., 2016). In contrast with the Dehalococcoidia, other taxonomic groups belonging to the phylum Chloroflexi were low in abundance in the active community as evident from the qSIP experiments. The other Chloroflexi MAGs (bin31 and bin52) contained by far the most CAZy and extracellular peptidases of all evaluated MAGs. Therefore, the lack of labelling with the provided substrates is surprising and attests to a different metabolic strategy that possibly does not benefit from the addition of the labile carbon sources supplied. In addition, in a previous SIP study, members of the phyla Chloroflexi were active in both heterotrophic and autotrophic incubations with freshwater sediments (Coskun et al., 2018), so it is possible that these organisms would have an autotrophic or mixotrophic strategy for carbon assimilation. This confirms the importance of activity measurements in addition to the analysis of the metabolic potential of a microbial community.
Many of these phyla that were active in both substrate incubations belonged to the most common taxonomic groups from the original water column microbial community. These organisms seem to be able to persist though not proliferate in incubations with both protein and DOM. This could be an indication of a metabolic strategy based on the potential to utilise diverse complex carbon sources or intermediates of the degradation process to sustain cellular activity. The labelling of organisms in both substrate types that are possibly responsible for the degradation of intermediate fermentation products supports the findings from the genetic analysis, as these secondary fermenters would presumably be less reliant on the composition of the original substrate.
Conclusions
Our study has revealed that the euxinic water column of the Black Sea harbours a microbial community highly adapted to the utilization of diverse electron cycling metabolisms while oxidizing the organic carbon compounds available in their environment. Many genomic features common to those reported for methanogenic reactors (Nobu et al., 2015) and deep subsurface environments (Poudel et al., 2018) are found in MAGs from our study, showing that utilisation of low redox potentials is one of the main defining features of life in this euxinic water column. In combination with the genomic information, our DNA‐SIP experiment indicates that members of the Marinimicrobia and Cloacimonetes phyla are generalists capable of utilizing macromolecular OM of different forms, while possibly relying on close interactions with other organisms to process fermentation products. According to our results, the degraders of these fermentation products could be organisms affiliated to Deltaproteobacteria as well as Dehalococcoidia from the phylum Chloroflexi. However, none of these organisms became dominant with the added labile carbon pools, suggesting that, as could be expected, the OM available in the Black Sea sulphidic zone at 1000 m is more diverse and/or recalcitrant than the OM provided in our incubations. In addition, the activity of cells affiliated to Woesearcheota, Parcubacteria and Omnitrophica, especially in the protein‐amended incubations, is an interesting phenomenon, which shows for the first time the potential competitive advantage these organisms with small genomes and lacking many essential metabolic pathways might have in a natural microbial community. The exact reason why these organisms are able to become the most active members in our incubations remains to be answered, but it likely requires that their exploitation of the metabolism of other organisms or leaking metabolites is efficient. Nonetheless, a streamlined metabolism appears to be an important part of the degradation of OM in low‐energy euxinic conditions. It is possible that the role of competition is less important as a shaping force of the community than environmental pressures; something that has been suggested for oxygen‐depleted water columns previously (Bryant et al., 2012). When competition for limiting nutrients or substrate is not the defining factor of the community, but rather the limitations exposed by energetic constraints and possibly the sulphide concentration, it is more likely that redundant metabolic strategies can coexist within organisms that can cope with the environmental constraints. This could explain why organisms that are phylogenetically distant express the same metabolic strategy. Future incubations with pure amino acids or isotopic confirmation with microscopic techniques would give more insight into their ecological strategy and potential interactions with other organisms.
Finally, our study shows the importance of estimating the activity of microbial communities to enhance our understanding of the meaning of genetic adaptations to specific environmental conditions. Solely genomic data can give a good overview of metabolisms available in the community but to understand the defining dynamics of natural microbial communities more studies are required that also consider the consequences of these genomic adaptations. Therefore, the use of novel methods to follow activity in microbial communities is paramount to broaden our understanding of natural phenomena and metabolic versatility, as well as the evolutionary pressures that affect life on earth.
Experimental procedures
Sample collection
Water samples were collected from the western basin of the Black Sea in Spring 2017 during R/V Pelagia cruise number 64PE418 from station 2 (42° 53.8′ N 30° 40.7′ E). Two hundred litres of water from 1000 m depth was collected with an ultra‐clean conductivity‐temperature‐density (CTD) system, which was equipped with among others a SBE3plus thermometer, SBE4 conductivity sensor and SBE43 dissolved oxygen sensor (Sea‐Bird Electronics, Bellevue, WA). Water column samples for nutrient analyses were collected from the same CTD bottles and preserved on board for chemical analyses. About 5 ml of the sample was filtered through a 0.2 μm Acrodisc syringe filter with a Supor membrane (Pall Corporation, New York, NY). Samples for NO3 −, NO2 − and NH4 were frozen at −20°C in a pre‐rinsed pony vial. Samples for DIC were preserved at +4°C, and samples for HS were fixed with 40 μl 1 N NaOH, and frozen at −20°C. About 20 ml of water was collected and filtered for DOC and TN measurements into precombusted glass vials. Water column samples for DNA analysis were collected by filtration of approximately 6 L of suspended particulate matter directly from sample bottles through Sterivex 0.22 μm membrane filters (Merck, Burlington, MA) and immediately frozen in −80°C after collection. Water for incubations was collected from the bottom of the 27 L sample bottles, while purging with nitrogen, therefore minimizing contact with ambient atmosphere. Incubations were carried through in 50 L round PP carboys (216‐1738, VWR, Radnor, PA). Carboys used for the incubation experiments were acid‐washed, rinsed once with site‐water and purged for about 10 min with nitrogen with 10 cycles of overpressurising before collecting the incubation water. Over time oxygen will leach through the plastic material used, but considering the short time periods of incubation, the large volume of water and high sulphide concentrations, the oxygen available for organisms should be minimal.
Additionally, samples for metagenomics analyses were collected on the R/V Pelagia 64PE371 in June–July 2013. Sampling methods, chemical and microbiological analyses of the water column during this cruise have been previously reported by Sollai et al. (2018). The main difference to the sampling campaign in 2017 was that samples were collected with a 0.7 μm pore size glass fibre filter (GF/F).
Substrate preparation
Two separate substrates were prepared from cultures of the diatom Thalassiosira pseudonana CYY9928. The diatom culture was grown in normal MDV media (Javaheri et al., 2015, Table S5) to generate unlabelled biomass, as well as grown in the same media supplemented with 30% 13C‐NaHCO3 (Cambridge Isotope Laboratories, Andover, MA), and 15% 15N‐NaNO3 (Sigma Aldrich, St. Louis, MO) to generate 13C/15N‐labelled biomass, for 4 weeks before biomass was collected by centrifugation. Pellets were washed three times with artificial seawater to remove residual labelled substrates, and freeze‐dried. An extract of DOM was prepared by adding 10 ml MilliQ water, 50 mg Devarda's alloy and 25 mg MgO for each 0.2 g freeze‐dried biomass to burst the freeze‐dried cells and eliminate any remaining dissolved inorganic nitrogen. The mixture was shaken for 48 h at room temperature (RT) and the supernatant was collected by centrifugation. The supernatant was subsequently filtered through a pre‐combusted 47 mm glass fibre filter (Whatman, Little Chalfont, UK), and a 0.2 μm syringe filter (Minisart®, Sartorius, Gottingen, Germany), before being freeze‐dried again and used as DOM substrate. Protein was extracted from the same collected biomass with a Trizol‐based method (Kirkland et al., 2006), and subsequently freeze‐dried to be used as the protein substrate. Protein amounts in each substrate were measured by the Bradford dye‐binding method with the Bio‐Rad protein assay kit (Bio‐Rad Laboratories, Hercules, CA). DNA contamination in the protein extracts was checked fluorometrically with Qubit™ dsDNA HS assay kit (Thermo Fisher Scientific, Waltham, MA). About 1 ng/μl DNA was remaining amounting to 0.02% w/w of the extract; this minor amount was considered to have minor effects on the substrate‐utilizing community. The remainder of protein extract weight is assumed to be mainly salts brought through from the extraction protocol. The atom‐% of label in the substrate was measured by EA‐IRMS (Flash 2000 EA with a DeltaV Advantage; Thermo Fisher Scientific, Bremen, Germany) equipped with a NC/NCS 3 m column, indicating a labelling of 11% 13C of the DOM, and 16% 13C of the measured protein in the protein extract (12% w/w).
Incubation conditions
The substrate was added up to 2 h after the collection of the water. 67 mg of either freeze‐dried unlabelled DOM or 13C/15N DOM were dissolved in 10 ml of sterile‐filtered site seawater. Sixty‐seven milligrams of the freeze‐dried unlabelled protein extract and 13C/15N protein extract were dissolved into 15 ml sterile‐filtered site water, with 1% sodium dodecyl sulphate (SDS) and repeated heating in near‐boiling water. SDS denatures and dissolves proteins in water, changing the charge and exposing amino acid chains that would otherwise be hidden. The final concentration of SDS was diluted with an addition of about 3000× and, therefore, its effect on the incubations is considered minimal. Each of the substrates was dissolved in an ambient atmosphere and added through syringes to the 50 L incubation bottles while purging with nitrogen. The final concentration of the added substrate was 1.34 mg L−1, which is equivalent to the amount of DOC measured at these depths in the Black Sea (Ducklow et al., 2007). The bottles were thoroughly mixed and the headspaces were purged with nitrogen. The carboys were incubated on their side to minimize any gas leakage and shaken carefully after 1 day. The container was set to the in situ temperature of 8°C and kept in the dark. After 72 h of incubation, water was filtered on to 142 mm 0.2 μm polycarbonate filters (Isopore™, Merck, Burlington, MA) with a 0.45 μm nitrocellulose support filter (MF membrane, Sigma‐Aldrich, St. Louis, MO). The connecting tubing was acid‐washed in the beginning and rinsed thoroughly with milliq water between each treatment. The filters were collected and immediately frozen at −80°C until further processing back in the lab.
DNA extractions
Water column samples collected in Sterivex filters were extracted with the Powersoil DNA Isolation kit (MoBio, QIAGEN, Carlsbad, CA, USA), according to the manufacturer's instructions but with an elution step with 50 μl TE. In addition, DNA was extracted from the 142 mm 0.2 μm polycarbonate incubation filters with a phenol/chloroform method (Sambrook and Russell, 2006). Briefly, filters were cut into small pieces with a sterile scalpel, and bead‐beaten with a vortex (VWR VV3, Radnor, PA) equipped with a custom adapter for 50 ml falcon tubes for 10 min at maximum speed with 1 g 0.1 mm zirconia beads (Biospec, Bartlesville, OK) and 15 ml of TE/sucrose buffer (6.7% sucrose, pH 8.0, 4°C). Supernatant was recovered, incubated at 37°C for 30 min with 0.8 mg ml−1 lysozyme (Thermo Scientific™, Waltham, MA), and further incubated at 65°C for 15 min 0.2 mg ml−1 Proteinase K (Invitrogen™, Carlsbad, CA) and 0.5% SDS. The mixture was cooled to 37°C and incubated again at 37°C for 30 min with 0.08 mg ml−1 RNase A. DNA was further extracted with one volume of phenol:chloroform:isoamyl alcohol (25:24:1), washed with chloroform:isoamyl alcohol (24:1) and chloroform. DNA was precipitated with 50 μg ml−1 glycogen, 0.3 M sodium acetate (pH 5.5), and 2.5 volumes of cold 70% ethanol. The pellet was dried and redissolved in PCR‐grade water. DNA concentrations were measured fluorometrically with Qubit™ dsDNA HS assay kit (Thermo Fisher Scientific), and extracts were stored at −80°C.
Density gradient centrifugation and gradient fractionation
A density gradient centrifugation was done with CsCl gradients following the protocol of Dunford and Neufeld (2010). Briefly, 4 μg of DNA from each sample was added to a CsCl solution to the final density of 1.725 g ml−1 in 5.1 ml QuickSeal Polyallomer tubes (Beckman Coulter, Brea, CA). The samples were centrifuged for 60 h at 44 000 rpm (177 000 g av) at 20°C using a Vti 65.2 rotor (Beckman Coulter). The samples were then fractionated to 13 equal fractions, and the gradient formation was checked by measuring the density of each fraction from 10 μl of sample with a digital refractometer (AR2000 Reichert Technologies, Buffalo, NY). DNA was precipitated by adding 2 volumes of PEG solution (30% PEG6000, 1.6 M NaCl) and 20 μg of polyacrylamide as a carrier, incubating at RT for 2 h and pelleting the DNA by centrifugation at 13 000 ×rcf for 30 min at 4°C. Pellets were washed with 70% ethanol, let dry and resuspended in PCR‐grade water.
16S rRNA gene amplicon sequencing and analysis
The V4 region of the 16S SSU rRNA gene was amplified from DNA extracted both from the water column and from all fractions collected in the density gradient fractionation of the DNA‐SIP experiment (see above) with the forward primer 515F‐Y and the reverse primer 806RB (Caporaso et al., 2011; Apprill et al., 2015; Parada et al., 2016). PCR reactions were made in Phusion HF buffer with 0.2 mM dNTPs, 800 μg ml−1 BSA, 0.6 μM primers, 1 unit Phusion High‐Fidelity Taq polymerase (Thermo Fisher Scientific, Waltham, MA) and 5 μl of DNA template in a total of 50 μl. The conditions were 98°C for 30 s, 25 rounds of 98°C for 10 s, 50°C for 20 s, 72°C for 30 s, and finally, 7 min of extension at 72°C. All reactions were performed in triplicates and negative controls were added to each round. PCR products were run on 1% agarose gels for 50 min at 80 V with SmartLadder small fragment (100–1000 bp, Kaneka Eurogentec S.A., Seraing, Belgium) as a size marker. Target bands were cut out of the gel, and DNA was purified with the QIAquick Gel Extraction Kit (QIAGEN, Hilden, Germany). Samples were combined at equal concentrations, concentrated with the MinElute PCR Purification Kit (QIAGEN, Hilden, Germany) and sequenced at the University of Utrecht (Netherlands) on an Illumina Miseq platform as paired‐end reads of 250 bp.
Illumina amplicon data were analysed with an in‐house pipeline. Briefly, sequence quality was checked at several process points with FastQC (v. 0.11.3, Andrews, 2010). Paired‐end reads were extended with PEAR (v. 0.9.8, Zhang et al., 2013) with a minimum length of reads of 20, minimum overlap set at 7 and a p‐value threshold of 0.05. Barcodes were removed (rules: extract_barcodes –bc1_len 12 and correct_barcodes –bc_missmatch 2) and libraries were split (rule: split_libraries, max unacceptable phred –q 10, and maximum number of consecutive low base calls –r 5) and reads outside of 250–350 bp length were discarded (rule: remove_short_long_reads) with QIIME (v. 1.9.1, Caporaso et al., 2010). OTUs were picked with uclust with a 97% threshold and the most common sequence of each OTU cluster was picked as representative. The taxonomy of the representative sequences were assigned using uclust (75% cut‐off) against the SILVA database release 128 (Quast et al., 2012). As many sequences do not have more detailed taxonomic classification in the database, a cut‐off of 75% for phylum level taxonomy was used (Yarza et al., 2014). The taxonomic positioning of the sequences was confirmed with a phylogenetic analysis that included the alignment and trimming of sequences in ARB (v. 6.0.2, Westram et al., 2011) against the SILVA database release 128 (Quast et al., 2012). Sequences and selected neighbours of each taxonomic group were further aligned with MAFFT (v. 7.310, Katoh and Standley, 2013) and trimmed with TRIMAL (v. 1.2rev59, Capella‐Gutierrez et al., 2009) with the automatic method selection. A phylogenetic tree was then built from trimmed sequences with IQ‐TREE using automatic model selection and ultrafast bootstrap approximation (v. 1.6.7, Nguyen et al., 2015, Kalyaanamoorthy et al., 2017, Hoang et al., 2017) and visualized with iTOL (Letunic and Bork, 2016) (Supplementary Figs 3–8). 16S rRNA genes longer than 400 bp were collected from the metagenomes with barrnap (v. 0.9, https://github.com/tseemann/barrnap) with an e‐value cut‐off of 1e −20. The taxonomy of these genes was analysed the same way as the amplicon data. The number of reads mapping to a gene was normalized by the total length of the gene and further to a relative abundance of all normalized reads Fig. 2A. All further analyses were carried out in the R package Phyloseq (v. 1.22.3, McMurdie and Holmes, 2013). For water column amplicon data sets, singletons were removed. For the substrate incubations, OTUs that had less than 13 sequences in each incubation (13 fractions per substrate) were removed prior to analysis as well as OTUs belonging to Cyanobacteria, as these could not be separated from remaining DNA contaminants from substrate addition. Finally, the remaining OTUs were normalized to the total read count per sample to obtain the relative abundance. The high numbers of OTUs we identified in our data sets may have to do with the clustering method (UCLUST) which has been known to result in overinflating OTU numbers when compared with mock communities (Edgar, 2017). Therefore, we will discuss the differences between treatments mainly in terms of relative abundances and do not consider an OTU as directly representing a microbial species. The 16S rRNA gene amplicon reads (raw data) have been deposited in the NCBI Sequence Read Archive under the BioProject number PRJNA596220.
qPCR and qSIP analysis
For all fractions collected in the density gradient fractionation of the DNA‐SIP experiment where the sample was remaining, 16S rRNA gene copy numbers were estimated by performing a qPCR. qPCR was performed on a Biorad CFX96™ Real‐Time System/C1000™ Thermal cycler equipped with CFX Manager™ Software. qPCR reactions of a final volume of 25 μl were made in Phusion HF buffer, 0.2 mM, 0.6 μM of each of the primers used for the 16S rRNA gene amplicon sequencing analysis, 0.8 mg ml−1 of BSA, 0.2× SYBRGreen, 0.5 units of HF Phusion Taq polymerase and 4 μl of 10× diluted DNA template. All reactions were performed in triplicate and threshold values were compared to a calibration curve with concentrations ranging from 4.06 × 107 to 4.06 × 102 gene copies. Non‐template controls were included in every run. The PCR reactions were carried out with an initial denaturation step of 98°C for 30 s followed by 45 cycles of denaturation at 98°C for 10 s, annealing at 52°C for 20 s with plate read and elongation at 72°C for 30 s and a final elongation step at 80°C for 25 s. The specificity of the reactions was checked by a melting curve analysis. Copy numbers used for qSIP calculations are 16S rRNA gene copies found in total in each fraction of the density gradient. The abundance of prokaryotes (16S rRNA gene copy numbers l−1) were estimated by using the total copy numbers of all fractions in one sample normalized with the amount of DNA extracted from each incubation to estimate the seawater that was used to collect 4 μg of DNA used for each centrifugation. These are estimates, and should not be considered absolute, as it is probable that most error in this case comes from the amount of DNA originally extracted from the incubation filters.
We chose the qSIP analysis method (Hungate et al., 2015) to evaluate the incorporation of labelled substrate into the DNA, and consequently to identify ‘active’ OTUs with labelled 13C/15N DNA. Calculations were done using the QSIP function of the HTSSIP R package (Youngblut et al. 2018) with some modifications to also take into account 15N‐nitrogen assimilation. When calculating maximum heavy DNA molecular weight, we added the effect of nitrogen labelling to the functions in the R package. Since the nucleotides A and G have five nitrogen atoms, a maximum of 4.985 g mol−1 was added if fully labelled. For T with two nitrogen atoms the addition would be 1.994 g mol−1 and for C with three nitrogen atoms 2.991 g mol−1. Therefore, we modified equation (9) from Hungate et al. (2015) to account for nitrogen:
The final calculation is thus not atom fraction excess (AFE) but MWE. In addition, we did not take into account the effect of natural isotope fractions in the calculation of MWE, as these have a minimal effect and in principal are already included in the calculations for unlabelled controls (W light and GC content). Bootstrapping was not performed as we only had one replicate 13C/15N‐labelled and unlabelled control of each substrate. Because of the lack of statistical tests, we chose a stringent cut‐off value of the estimated MWE of 0.14, to assign OTUs as active. This matches approximately the proportion of label in the added substrate and, moreover, is considerably higher than the values Hungate et al. (2015) and Coskun et al. (2018) reported as average AFE of OTUs assigned as active in their experimental data. For more discussion on considerations for the use of this method, see Supplementary File 1, section 2. Phylogenetic analysis of the active OTUs was done with selected closest neighbours retrieved with the SILVA ACT tool. Alignment, trimming and tree‐building were done as previously described. Supplementary Fig. S2 shows the total workflow from incubations to determine active community members.
Metagenomic and phylogenetic analysis of the MAGs
Sample collection, DNA extraction and metagenome sequencing and sequence analysis of water column samples collected from the Black Sea water column in a previous campaign in 2013 (64PE371, see above) is described in Villanueva et al. (2018). For some of the MAGs discussed here, 16S rRNA genes were identified with the CheckM ‘ssu_finder’ utility (v. 1.0.12; Parks et al., 2014). For the rest of the MAGs, Phylosift (v. 1.0.1; Darling et al., 2014) was used to extract marker genes for their phylogenetic placement as described in Dombrowski et al. (2018). The MAGs were then compared to all publicly available genomes in the NCBI database from the same taxonomic group, and the closest available 16S rRNA gene from a related MAG was added to the phylogenetic tree with the 16S rRNA genes of active OTUs to compare to the phylogeny of the active community (Figs S3‐S8). To determine the relative abundance, we mapped the scaffolds from each MAG against the metagenomics reads from the 1000 m water column sample using BBmap (v. 34.x, BBtools, Bushnell, B., sourceforge.net/projects/bbmap/). The percentage of reads mapped to each MAG was considered as an indicator of the abundance of the specific MAG at 1000 m depth. Genes were called using prodigal (v. 1.14‐dev, Seemann, 2014), and gene annotations performed against several databases. These include KAAS (KEGG automatic annotation server, v. 2.1; Moriya et al., 2007), TIGRFAM (v. 15.0; Haft et al., 2001), Pfam (v. 31.0; El‐Gebali et al., 2018 and Engel et al., 2017), CAZymes (v. 2; Lombard et al., 2014), Merops (version of November 2018; Rawlings et al., 2018), COG (version of November 2018; Tatusova et al., 2000) or arCOG (Makarova et al., 2015) and ncbi_nr (version of October 2018; O'Leary et al., 2016) databases with an e‐score threshold of 1e−20 for all except ncbi_nr (1e−5), MEROPS (1e−80), Pfam (1e−10) and COG (1e−10) databases. Annotations were performed using Hmmsearch (v. 3.1b2; Potter et al., 2018) and Blastp (v. 2.7.1; Altschul et al., 1997) or Diamond (v. 0.9.22; Buchfink et al., 2015). MAG comparisons were done with the identified KO hits for major carbon degradation and central metabolic genes (Dombrowski et al., 2018; Table S2), and an estimation of full pathways in each MAG separately was done by comparing the hits of all databases as well as localization in contigs. Extracellular peptidases were determined for MEROPS peptidase hits of MAGs using SignalP (Petersen et al., 2011) for bacterial MAGs and PRED‐SIGNAL (Bagos et al., 2009) for archaeal MAGs and manually curated by comparing to other database hits. All annotations can be found in Table S3. The MAGs described and discussed here are deposited in the Joint Genome Institute's integrated microbial genomes database (Table 2).
Supporting information
Supplementary file 1 Section 1, A description of the main metabolic potential found in each evaluated MAG
Section 2, Considerations for DNA‐SIP
Supplementary Fig. 1. Above: Measured chemical parameters from the water column in μM; DOC: dissolved organic carbon, DIC: dissolved inorganic carbon, TN: total nitrogen. Below: CTD profiles across the water column.
Supplementary Fig. 2. Diagram of workflow from incubations to DNA‐SIP experiments
Supplementary Figs 3–8. Phylogenetic tree showing the 16S rRNA of active OTUs by phyla and a closest neighbouring 16S rRNA gene obtained from a publicly available genome closely related to the MAG of that phyla (based on 34 single‐copy marker genes) is shown.
Supplementary Table 5. List of 34 single‐copy marker genes used for phylogenetic analysis of MAGs
Supplementary Fig. 9. Chemical measurements from incubations at 72 h
Supplementary Table 6. Recipe for MDV media used for culturing the diatom biomass
Supplementary Figs 10–13. Reconstructed metabolic pathways of selected MAGs
Supplementary file 2 Supplementary Table 1. DNA‐SIP calculated parameters for all active OTUs in DOM incubations (Sheet 1), and PRO incubations (Sheet 2)
Supplementary Table 2. DNA‐SIP calculated parameters for all active OTUs in PRO incubations
Supplementary Table 3. List of KO annotations used for comparisons in Fig. 5.
Supplementary Table 4. Final list of extracellular peptidases bacteria MAGs
Supplementary Table 5. Final list of extracellular peptidases archaea MAGs
Supplementary Table 6. List of annotated CAZymes
Supplementary Table 7. List of annotated MEROPS peptidases
Supplementary Table 8. List of genes used for Supplementary Fig. 10
Supplementary file 3 Supplementary Table 9. All annotated genes in order by contigs for bacterial MAGs
Supplementary Table 10. All annotated genes in order by contigs for archaeal MAGs
Acknowledgements
The authors would like to thank the crew and scientific parties of the R/V Pelagia cruises 64PE371 and 64PE418 for support in the sampling. Sanne Vreugdenhil, Maartje Brouwer, and Iris Schahner are acknowledged for practical help in the molecular laboratory. We also acknowledge Alejandro Abdala and Julia Engelmann for help in establishing the amplicon sequencing pipelines as well as for constructive input. This research is supported by the Soehngen Institute of Anaerobic Microbiology (SIAM) Gravitation Grant (024.002.002) of the Netherlands Ministry of Education, Culture and Science (OCW) and the Netherlands Organization for Scientific Research (NWO) to J.S.S.D. and L.V.
References
- Albert, D.B., Taylor, C., and Martens, C.S. (1995) Sulfate reduction rates and low molecular weight fatty acid concentrations in the water column and surficial sediments of the Black Sea. Deep‐Sea Res I 42: 1239–1260. [Google Scholar]
- Altschul, S.F., Madden, T.L., Schäffer, A.A., Zhang, J., Zhang, Z., Miller, W., and Lipman, D.J. (1997) Gapped BLAST and PSI‐BLAST: a new generation of protein database search programs. Nucleic Acids Res 25: 3389–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrews S. (2010) FastQC: a quality control tool for high throughput sequence data. URL http://www.bioinformatics.babraham.ac.uk/projects/fastqc.
- Apprill, A., McNally, S., Parsons, R., and Weber, L. (2015) Minor revision to V4 region SSU rRNA 806R gene primer greatly increases detection of SAR11 bacterioplankton. Aquat Microb Ecol 75: 129–137. [Google Scholar]
- Azam, F. (1998) Microbial control of oceanic carbon flux: the plot thickens. Science 280: 694–696. [Google Scholar]
- Azam, F., Fenchel, T., Field, J., Gray, J., Meyer‐Reil, L., and Thingstad, F. (1983) The ecological role of water‐column microbes in the sea. Mar Ecol Prog Ser 10: 257–263. [Google Scholar]
- Bagos, P.G., Tsirigos, K.D., Plessas, S.K., Liakopoulos, T.D., and Hamodrakas, S.J. (2009) Prediction of signal peptides in archaea. Protein Eng Des Sel 22: 27–35. [DOI] [PubMed] [Google Scholar]
- Bastviken, D., Persson, L., Odham, G., and Tranvik, L. (2004) Degradation of dissolved organic matter in oxic and anoxic lake water. Limnol Oceanogr 49: 109–116. [Google Scholar]
- Bell, T.H., Yergeau, E., Martineau, C., Juck, D., Whyte, L.G., and Greer, C.W. (2011) Identification of nitrogen‐incorporating bacteria in petroleum‐contaminated Arctic soils by using [15N]DNA‐based stable isotope probing and pyrosequencing. Appl Environ Microbiol 77: 4163–4171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertagnolli, A.D., Padilla, C.C., Glass, J.B., Thamdrup, B., and Stewart, F.J. (2017) Metabolic potential and in situ activity of marine Marinimicrobia bacteria in an anoxic water column. Environ Microbiol 19: 4392–4416. [DOI] [PubMed] [Google Scholar]
- Biderre‐Petit, C., Dugat‐Bony, E., Mege, M., Parisot, N., Adrian, L., Moné, A., et al. (2016) Distribution of Dehalococcoidia in the anaerobic deep water of a remote Meromictic crater Lake and detection of Dehalococcoidia‐derived reductive dehalogenase homologous genes. PLoS One 11: e0145558. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bird, J.T., Tague, E.D., Zinke, L., Schmidt, J.M., Steen, A.D., Reese, B., et al. (2019) Uncultured microbial phyla suggest mechanisms for multi‐thousand‐year subsistence in Baltic Sea sediments. MBio 10: 1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown, C.T., Hug, L.A., Thomas, B.C., Sharon, I., Castelle, C.J., Singh, A., et al. (2015) Unusual biology across a group comprising more than 15% of domain bacteria. Nature 523: 208–211. [DOI] [PubMed] [Google Scholar]
- Bryant, J.A., Stewart, F.J., Eppley, J.M., and Delong, E.F. (2012) Microbial community phylogenetic and trait diversity declines with depth in a marine oxygen minimum zone. Ecology 93: 1659–1673. [DOI] [PubMed] [Google Scholar]
- Buchfink, B., Xie, C., and Huson, D.H. (2015) Fast and sensitive protein alignment using DIAMOND. Nat Methods 12: 59–60. [DOI] [PubMed] [Google Scholar]
- Canfield, D.E. (1989) Sulfate reduction and oxic respiration in in marine sediments: implications for organic carbon preservation in euxinic environments. Deep Sea Res 36: 121–138. [DOI] [PubMed] [Google Scholar]
- Canfield, D.E. (1994) Factors influencing organic carbon preservation in marine sediments. Chem Geol 114: 315–329. [DOI] [PubMed] [Google Scholar]
- Capella‐Gutierrez, S., Silla‐Martinez, J.M., and Gabaldon, T. (2009) trimAl: a tool for automated alignment trimming in large‐scale phylogenetic analyses. Bioinformatics 25: 1972–1973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J.G., Kuczynski, J., Stombaugh, J., Bittinger, K., Bushman, F.D., Costello, E.K., et al. (2010) QIIME allows analysis of high‐throughput community sequencing data. Nat Methods 7: 335–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caporaso, J.G., Lauber, C.L., Walters, W.A., Berg‐Lyons, D., Lozupone, C.A., Turnbaugh, P.J., et al. (2011) Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc Natl Acad Sci U S A 108: 4516–4522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Castelle, C.J., Brown, C.T., Anantharaman, K., Probst, A.J., Huang, R.H., and Banfield, J.F. (2018) Biosynthetic capacity, metabolic variety and unusual biology in the CPR and DPANN radiations. Nat Rev Microbiol 16: 629–645. [DOI] [PubMed] [Google Scholar]
- Castelle, C.J., Wrighton, K.C., Williams, K.H., and Banfield, J.F. (2015) Genomic expansion of domain Archaea highlights roles for organisms from new phyla in anaerobic carbon cycling. Curr Biol 25: 690–701. [DOI] [PubMed] [Google Scholar]
- Coskun, Ö.K., Pichler, M., Vargas, S., Gilder, S., and Orsi, W.D. (2018) Linking uncultivated microbial populations with benthic carbon turnover using quantitative stable isotope probing. Appl Environ Microbiol 84: e01083–e01018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cottrell, M.T., and Kirchman, D.L. (2000) Natural assemblages of marine Proteobacteria and members of the Cytophaga‐Flavobacter cluster consuming low‐ and high‐molecular‐weight dissolved organic matter. Appl Environ Microbiol 66: 1692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Darling, A.E., Jospin, G., Lowe, E., Matsen, F.A.I.V., Bik, H.M., and Eisen, J.A. (2014) PhyloSift: phylogenetic analysis of genomes and metagenomes. PeerJ 2: e243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski, N., Lee, J.‐H., Williams, T.A., Offre, P., and Spang, A. (2019) Genomic diversity, lifestyles and evolutionary origins of DPANN archaea. FEMS Microbiol Lett 366: fnz008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski, N., Seitz, K.W., Teske, A.P., and Baker, B.J. (2017) Genomic insights into potential interdependencies in microbial hydrocarbon and nutrient cycling in hydrothermal sediments. Microbiome 5: 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dombrowski, N., Teske, A.P., and Baker, B.J. (2018) Expansive microbial metabolic versatility and hydrothermal sediments. Nat Commun 9: 4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ducklow, H.W., Hansell, D.A., and Morgan, J.A. (2007) Dissolved organic carbon and nitrogen in the Western Black Sea. Mar Chem 105: 140–150. [Google Scholar]
- Dyksma, S., and Gallert, C. (2019) Candidatus Syntrophosphaera thermopropionivorans: a novel player in syntrophic propionate oxidation during anaerobic digestion. Environ Microbiol Rep 11: 558–570. [DOI] [PubMed] [Google Scholar]
- Edgar, R.C. (2017) Accuracy of microbial community diversity estimated by closed‐ and open‐reference OTUs. PeerJ 5: e3889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gihring, T.M., Humphrys, M., Mills, H.J., Huettel, M., and Kostka, J.E. (2009) Identification of phytodetritus‐degrading microbial communities in sublittoral Gulf of Mexico sands. Limnol Oceanogr 54: 1073–1083. [Google Scholar]
- Gomez‐Consarnau, L., Lindh, M.V., Gasol, J.M., and Pinhassi, J. (2012) Structuring of bacterioplankton communities by specific dissolved organic carbon compounds. Environ Microbiol 14: 2361–2378. [DOI] [PubMed] [Google Scholar]
- Graue, J., Engelen, B., and Cypionka, H. (2012) Degradation of cyanobacterial biomass in anoxic tidal‐flat sediments: a microcosm study of metabolic processes and community changes. ISME J 6: 660–669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Graue, J., Kleindienst, S., Lueders, T., Cypionka, H., and Engelen, B. (2011) Identifying fermenting bacteria in anoxic tidal‐flat sediments by a combination of microcalorimetry and ribosome‐based stable‐isotope probing. FEMS Microbiol Ecol 81: 78–87. [DOI] [PubMed] [Google Scholar]
- Haft, D.H., Loftus, B.J., Richardson, D.L., Yang, F., Eisen, J.A., Paulsen, I.T., and White, O. (2001) TIGRFAMs: a protein family resource for the functional identification of proteins. Nucleic Acids Res 29: 41–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hawley, A.K., Nobu, M.K., Wright, J.J., Durno, W.E., Morgan‐Lang, C., Sage, B., et al. (2017) Diverse Marinimicrobia bacteria may mediate coupled biogeochemical cycles along eco‐thermodynamic gradients. Nat Commun 8: 1507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoang, D.T., Chernomor, O., von Haeseler, A., Minh, B.Q., and Vinh, L.S. (2017) UFBoot2: improving the ultrafast bootstrap approximation. Mol Biol Evol 35: 518–522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug, L.A., Baker, B.J., Anantharaman, K., Brown, C.T., Probst, A.J., Castelle, C.J., et al. (2016) A new view of the tree of life. Nat Microbiol 1: 16048. [DOI] [PubMed] [Google Scholar]
- Hug, L.A., Castelle, C.J., Wrighton, K.C., Thomas, B.C., Sharon, I., Frischkorn, K.R., et al. (2013a) Community genomic analyses constrain the distribution of metabolic traits across the Chloroflexi phylum and indicate roles in sediment carbon cycling. Microbiome 1: 22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hug, L.A., Maphosa, F., Leys, D., Löffler, F.E., Smidt, H., Edwards, E.A., and Adrian, L. (2013b) Overview of organohalide‐respiring bacteria and a proposal for a classification system for reductive dehalogenases. Philos Trans R Soc B 368: 20120322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hungate, B.A., Mau, R.L., Schwartz, E., Caporaso, J.G., Dijkstra, P., van Gestel, N., et al. (2015) Quantitative microbial ecology through stable isotope probing. Appl Environ Microbiol 81: 7570–7581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Javaheri, N., Dries, R., Burson, A., Stal, L.J., Sloot, P.M.A., and Kaandorp, J.A. (2015) Temperature affects the silicate morphology in a diatom. Sci Rep 5: 11652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jochum, L.M., Schreiber, L., Marshall, I.P.G., Jørgensen, B.B., Schramm, A., and Kjeldsen, K.U. (2018) Single‐cell genomics reveals a diverse metabolic potential of uncultivated Desulfatiglans‐related Deltaproteobacteria widely distributed in marine sediment. Front Microbiol 9: 1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalyaanamoorthy, S., Minh, B.Q., Wong, T.K.F., von Haeseler, A., and Jermiin, L.S. (2017) ModelFinder: fast model selection for accurate phylogenetic estimates. Nat Methods 14: 589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kantor, R.S., Wrighton, K.C., Handley, K.M., Sharon, I., Hug, L.A., Castelle, C.J., et al. (2013) Small genomes and sparse metabolisms of sediment‐associated bacteria from four candidate phyla. MBio 4: e00708–e00713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K., and Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland, A., Busby, J., Stevens, S., and Maupin‐Furlow, J.A. (2006) Trizol‐based method for sample preparation and isoelectric focusing of halophilic proteins. Anal Biochem 351: 254–259. [DOI] [PubMed] [Google Scholar]
- Kizina, J., Jordan, S., Resteu, A., Probian, C., Littmann, S., Markert, S.et al. (2017) Candidatus Vampirococcus archaeovorus and a transfer of a bacterial intron ribonucleic acid into archaeal cells. Insights into the biology of Candidate Division OP3 LiM populations. PhD thesis. Bremen, Germany, University of Bremen, pp. 85–160.
- Kujawinski, E.B. (2011) The impact of microbial metabolism on marine dissolved organic matter. Ann Rev Mar Sci 3: 567–599. [DOI] [PubMed] [Google Scholar]
- Letunic, I., and Bork, P. (2016) Interactive tree of life (iTOL) v3: an online tool for the display and annotation of phylogenetic and other trees. Nucleic Acid Res 44: W242–W245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limam, R.D., Chouari, R., Mazéas, L., Wu, T.‐D., Li, T., Grossin‐Debattista, J., et al. (2014) Members of the uncultured bacterial candidate division WWE1 are implicated in anaerobic digestion of cellulose. Microbiol Open 3: 157–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, S., Wawrik, B., and Liu, Z. (2017) Different bacterial communities involved in peptide decomposition between normoxic and hypoxic coastal waters. Front Microbiol 8: 353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, X., Li, M., Castelle, C.J., Probst, A.J., Zhou, Z., Pan, J., et al. (2018) Insights into the ecology, evolution, and metabolism of the widespread Woesearchaeotal lineages. Microbiome 6: 102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lloyd, K.G., Schreiber, L., Petersen, D.G., Kjeldsen, K.U., Lever, M.A., Steen, A.D., et al. (2013) Predominant archaea in marine sediments degrade detrital proteins. Nature 496: 215–218. [DOI] [PubMed] [Google Scholar]
- Lloyd, K.G., Steen, A.D., Ladau, J., Yin, J., and Crosby, L. (2018) Phylogenetically novel uncultured microbial cells dominate earth microbiomes. MSystems 3: 1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lombard, V., Ramulu, H.G., Drula, E., Coutinho, P.M., and Henrissat, B. (2014) The carbohydrate‐active enzymes database (CAZy) in 2013. Nucleic Acids Res 42: D490–D495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova, K.S., Wolf, Y.I., and Koonin, E.V. (2015) Archaeal clusters of orthologous genes (arCOGs): an update and application for analysis of shared features between Thermococcales, Methanococcales, and Methanobacteriales . Life 5: 818–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margolin, A.R., Gerringa, L.J.A., Hansell, D.A., and Rijkenberg, M.J.A. (2016) Net removal of dissolved organic carbon in the anoxic waters of the Black Sea. Mar Chem 183: 13–24. [Google Scholar]
- McMurdie, P.J., and Holmes, S. (2013) Phyloseq: an R package for reproducible interactive analysis and graphics of microbiome census data. PLoS One 8: e61217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mopper, K., and Kieber, D.J. (1991) Distribution and biological turnover of dissolved organic compounds in the water column of the Black Sea. Deep‐Sea Res 38: S1021–S1047. [Google Scholar]
- Moriya, Y., Itoh, M., Okuda, S., Yoshizawa, A., and Kanehisa, M. (2007) KAAS: an automatic genome annotation and pathway reconstruction server. Nucleic Acids Res 35: W182–W185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nelson, C.E., and Carlson, C.A. (2012) Tracking differential incorporation of dissolved organic carbon types among diverse lineages of Sargasso Sea Bacterioplankton. Environ Microbiol 14: 1500–1516. [DOI] [PubMed] [Google Scholar]
- Nelson, W.C., and Stegen, J.C. (2015) The reduced genomes of Parcubacteria (OD1) contain signatures of a symbiotic lifestyle. Front Microbiol 6: 1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nguyen, L.‐T., Schmidt, H.A., von Haeseler, A., and Minh, B.Q. (2015) IQ‐TREE: a fast and effective stochastic algorithm for estimating maximum likelihood phylogenies. Mol Biol Evol 32: 268–274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nobu, M.K., Narihiro, T., Rinke, C., Kamagata, Y., Tringe, S.G., Woyke, T., and Liu, W.‐T. (2015) Microbial dark matter ecogenomics reveals complex synergistic networks in a methanogenic bioreactor. ISME J 9: 1710–1722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Leary, N.A., Wright, M.W., Brister, J.R., Ciufo, S., Haddad, D., McVeigh, R., et al. (2016) Reference sequence (RefSeq) database at NCBI: current status, taxonomic expansion, and functional annotation. Nucleic Acids Res 44: D733–D745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oguz, T., Tugrul, S., Kideys, A.E., Ediger, V., and Kubilay, N. (2004) Physical and biochemical characteristics of the Black Sea. In The Sea, Vol. 14, Robinson, A.R., and Brink, K.H. (eds). Cambridge, MA: Harvard University Press, pp. 1331–1369. [Google Scholar]
- Orsi, W.D., Smith, J.M., Liu, S., Liu, Z., Sakamoto, C.M., Wilken, S., et al. (2016) Diverse, uncultivated bacteria and archaea underlying the cycling of dissolved protein in the ocean. ISME J 10: 2158–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantoja, S., Rossel, P., Castro, R., Cuevas, L.A., Daneri, G., and Córdova, C. (2009) Microbial degradation rates of small peptides and amino acids in the oxygen minimum zone of Chilean coastal waters. Deep‐Sea Res II 56: 1019–1026. [Google Scholar]
- Pantoja, S., Sepúlveda, J., and González, H.E. (2004) Decomposition of sinking proteinaceous material during fall in the oxygen minimum zone off northern Chile. Deep‐Sea Res I 51: 55–70. [Google Scholar]
- Parada, A.E., Needham, D.M., and Fuhrman, J.A. (2016) Every base matters: assessing small subunit rRNA primers for marine microbiomes with mock communities, time series and global field samples. Environ Microbiol 18: 1403–1414. [DOI] [PubMed] [Google Scholar]
- Parks, D.H., Imelfort, M., Skennerton, C.T., Hugenholtz, P., and Tyson, G.W. (2014) Assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res 25: 1043–1055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paul, B.G., Burstein, D., Castelle, C.J., Handa, S., Arambula, D., Czornyj, E., et al. (2017) Retroelement‐guided protein diversification abounds in vast lineages of bacteria and Archaea. Nat Microbiol 2: 17045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelletier, E., Kreimeyer, A., Bocs, S., Rouy, Z., Gyapay, G., Chouari, R., et al. (2008) “Candidatus Cloacamonas acidaminovorans”: genome sequence reconstruction provides a first glimpse of a new bacterial division. J Bacteriol 190: 2572–2579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pepe‐Ranney, C., Campbell, A.N., Koechli, C.N., Berthrong, S., and Buckley, D.H. (2016) Unearthing the ecology of soil microorganisms using a high resolution DNA‐SIP approach to explore cellulose and xylose metabolism in soil. Front Microbiol 7: 1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petersen, T.N., Brunak, S., von Heijne, G., and Nielsen, H. (2011) SignalP 4.0: discriminating signal peptides from transmembrane regions. Nat Methods 8: 785–786. [DOI] [PubMed] [Google Scholar]
- Plominsky, A.M., Trefault, N., Podell, S., Blanton, J.M., De la Iglesia, R., Allen, E.E., et al. (2018) Metabolic potential and in situ transcriptomic profiles of previously uncharacterized key microbial groups involved in coupled carbon, nitrogen and sulfur cycling in anoxic marine zones. Environ Microbiol 20: 2727–2742. [DOI] [PubMed] [Google Scholar]
- Pomeroy, L.R. (1974) The ocean's food web, a changing paradigm. Bioscience 24: 499–504. [Google Scholar]
- Potter, S.C., Luciani, A., Eddy, S.R., Park, Y., Lopez, R., and Finn, R.D. (2018) HMMER web server: 2018 update. Nucleic Acids Res 46: W200–W204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poudel, S., Dunham, E.C., Lindsay, M.R., Amenabar, M.J., Fones, E.M., Colman, D.R., and Boyd, E.S. (2018) Origin and evolution of flavin‐based electron bifurcating enzymes. Front Microbiol 9: 1–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quast, C., Pruesse, E., Yilmaz, P., Gerken, J., Schweer, T., Yarza, P., et al. (2012) The SILVA ribosomal RNA gene database project: improved data processing and web‐based tools. Nucleic Acids Res 41: D590–D596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rawlings, N.D., Barrett, A.J., Thomas, P.D., Huang, X., Bateman, A., and Finn, R.D. (2018) The MEROPS database of proteolytic enzymes, their substrates and inhibitors in 2017 and comparison with the peptidases in the PANTHER database. Nucleic Acids Res 46: D624–D632. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rotaru, A.E., Schauer, R., Probian, C., Mussmann, M., and Harder, J. (2012) Visualization of candidate division OP3 cocci in limonene‐degrading methanogenic cultures. J Microbiol Biotechnol 22: 457–461. [DOI] [PubMed] [Google Scholar]
- Schink, B., and Stams, A.J.M. (2013) Syntrophism among prokaryotes. In The Prokaryotes: Prokaryotic Communities and Ecophysiology, Rosenberg, E., DeLong, E.F., Lory, S., Stackebrandt, E., and Thompson, F. (eds). Berlin, Heidelberg: Springer‐Verlag, pp. 471–493 Chap. 21. [Google Scholar]
- Seemann, T. (2014) Prokka: rapid prokaryotic genome annotation. Bioinformatics 30: 2068–2069. [DOI] [PubMed] [Google Scholar]
- Seyler, L.M., McGuinness, L.M., and Kerkhof, L.J. (2014) Crenarchaeal heterotrophy in salt marsh sediments. ISME J 8: 1534–1543. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skennerton, C.T., Chourey, K., Iyer, R., Hettich, R.L., Tyson, G.W., and Orphan, V.J. (2017) Methane‐fueled syntrophy through extracellular electron transfer: uncovering the genomic traits conserved within diverse bacterial partners of anaerobic methanotrophic archaea. mBio 8: e00530–e00517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solden, L., Lloyd, K., and Wrighton, K. (2016) The bright side of microbial dark matter: lessons learned from the uncultivated majority. Curr Opin Microbiol 31: 217–226. [DOI] [PubMed] [Google Scholar]
- Sollai, M., Villanueva, L., Hopmans, E.C., Reichart, G.‐J., and Sinninghe‐Damsté, J.S. (2018) A combined lipidomic and 16S rRNA gene amplicon sequencing approach reveals archaeal sources of intact polar lipids in the stratified Black Sea water column. Geobiology 17: 91–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart, F.J., Dalsgaard, T., Young, C.R., Thamdrup, B., Revsbech, N.P., Ulloa, O., et al. (2012) Experimental incubations elicit profound changes in community transcription in OMZ bacterioplankton. PLoS One 7: e37118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tatusova, R.L., Galperin, M.Y., Natale, D.A., and Koonin, E.V. (2000) The COG database: a tool for genome‐scale analysis of protein functions and evolution. Nucleic Acids Res 28: 33–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash, J.C., Seitz, K.W., Baker, B.J., Temperton, B., Gillies, L.E., Rabalais, N.N., et al. (2017) Metabolic roles of uncultivated bacterioplankton lineages in the northern Gulf of Mexico “dead zone”. mBio 8: e01017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Mooy, B.A.S., Keil, R.G., and Devol, A.H. (2002) Impact of suboxia on sinking particulate organic carbon: enhanced carbon flux and preferential degradation of amino acids via denitrification. Geochim Cosmochim Acta 66: 457–465. [Google Scholar]
- Villanueva, L., von Meijenfeldt, B., Westbye, A.B., Hopmans, E.C., Dutilh, B.E. and Sinninghe Damsté, J.S (2018) Bridging the divide: bacteria synthesizing archaeal membrane lipids. bioRxiv 448035.
- Wasmund, K., Schreiber, L., Lloyd, K.G., Petersen, D.G., Schramm, A., Stepanauskas, R., et al. (2014) Genome sequencing of a single cell of the widely distributed marine subsurface Dehalococcoidia, phylum Chloroflexi . ISME J 8: 383–397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Webster, G., Rinna, J., Roussel, E.G., Fry, J.C., Weightman, A.J., and Parkes, R.J. (2010) Prokaryotic functional diversity in different biogeochemical depth zones in tidal sediments of the Severn estuary, UK, revealed by stable‐isotope probing. FEMS Microbiol Ecol 72: 179–197. [DOI] [PubMed] [Google Scholar]
- Westram, R., Bader, K., Pruesse, E., Kumar, Y., Meier, H., Glöckner, F.O., and Ludwig, W. (2011) ARB: a software environment for sequence data. In Handbook of Molecular Microbial Ecology I: Metagenomics and Complementary Approaches, de Bruijn, F.J. (ed). Hoboken, NJ: John Wiley & Sons, pp. 399–406. [Google Scholar]
- Wright, J.J., Mewis, K., Hanson, N.W., Konwar, K.M., Maas, K.R., and Hallam, S.J. (2014) Genomic properties of marine group a bacteria indicate a role in the marine sulfur cycle. ISME J 8: 455–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wrighton, K.C., Thomas, B.C., Sharon, I., Miller, C.S., Castelle, C.J., VerBerkmoes, N.C., et al. (2012) Fermentation, hydrogen, and sulfur metabolism in multiple uncultivated bacterial phyla. Science 337: 1661–1665. [DOI] [PubMed] [Google Scholar]
- Yarza, P., Yilmaz, P., Pruesse, E., Glöckner, F.O., Ludwig, W., Schleifer, K.H., et al. (2014) Uniting the classification of cultured and uncultured bacteria and archaea using 16S rRNA gene sequences. Nat Rev Microbiol 12: 635–645. [DOI] [PubMed] [Google Scholar]
- Zhang, J., Kobert, K., Flouri, T., and Stamatakis, A. (2013) PEAR: a fast and accurate Illumina paired‐end reAd mergeR. Bioinformatics 30: 614–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary file 1 Section 1, A description of the main metabolic potential found in each evaluated MAG
Section 2, Considerations for DNA‐SIP
Supplementary Fig. 1. Above: Measured chemical parameters from the water column in μM; DOC: dissolved organic carbon, DIC: dissolved inorganic carbon, TN: total nitrogen. Below: CTD profiles across the water column.
Supplementary Fig. 2. Diagram of workflow from incubations to DNA‐SIP experiments
Supplementary Figs 3–8. Phylogenetic tree showing the 16S rRNA of active OTUs by phyla and a closest neighbouring 16S rRNA gene obtained from a publicly available genome closely related to the MAG of that phyla (based on 34 single‐copy marker genes) is shown.
Supplementary Table 5. List of 34 single‐copy marker genes used for phylogenetic analysis of MAGs
Supplementary Fig. 9. Chemical measurements from incubations at 72 h
Supplementary Table 6. Recipe for MDV media used for culturing the diatom biomass
Supplementary Figs 10–13. Reconstructed metabolic pathways of selected MAGs
Supplementary file 2 Supplementary Table 1. DNA‐SIP calculated parameters for all active OTUs in DOM incubations (Sheet 1), and PRO incubations (Sheet 2)
Supplementary Table 2. DNA‐SIP calculated parameters for all active OTUs in PRO incubations
Supplementary Table 3. List of KO annotations used for comparisons in Fig. 5.
Supplementary Table 4. Final list of extracellular peptidases bacteria MAGs
Supplementary Table 5. Final list of extracellular peptidases archaea MAGs
Supplementary Table 6. List of annotated CAZymes
Supplementary Table 7. List of annotated MEROPS peptidases
Supplementary Table 8. List of genes used for Supplementary Fig. 10
Supplementary file 3 Supplementary Table 9. All annotated genes in order by contigs for bacterial MAGs
Supplementary Table 10. All annotated genes in order by contigs for archaeal MAGs