

Abstract
Background
Phosphorylcholine (PC) is an important pro‐inflammatory damage‐associated molecular pattern. Previous data have shown that natural IgM anti‐PC protects against cardiovascular disease. We aimed to develop a monoclonal PC IgG antibody with anti‐inflammatory and anti‐atherosclerotic properties.
Methods
Using various techniques PC antibodies were validated and optimized. In vivo testing was performed in a femoral artery cuff model in ApoE3*Leiden mice. Safety studies are performed in rats and cynomolgus monkeys.
Results
A chimeric anti‐PC (PC‐mAb(T15), consisting of a human IgG1 Fc and a mouse T15/E06 Fab) was produced, and this was shown to bind specifically to epitopes in human atherosclerotic tissues. The cuff model results in rapid induction of inflammatory genes and altered expression of genes associated with ER stress and choline metabolism in the lesions. Treatment with PC‐mAb(T15) reduced accelerated atherosclerosis via reduced expression of endoplasmic reticulum stress markers and CCL2 production. Recombinant anti‐PC Fab fragments were identified by phage display and cloned into fully human IgG1 backbones creating a human monoclonal IgG1 anti‐PC (PC‐mAbs) that specifically bind PC, apoptotic cells and oxLDL. Based on preventing macrophage oxLDL uptake and CCL2 production, four monoclonal PC‐mAbs were selected, which to various extent reduced vascular inflammation and lesion development. Additional optimization and validation of two PC‐mAb antibodies resulted in selection of PC‐mAb X19‐A05, which inhibited accelerated atherosclerosis. Clinical grade production of this antibody (ATH3G10) significantly attenuated vascular inflammation and accelerated atherosclerosis and was tolerated in safety studies in rats and cynomolgus monkeys.
Conclusions
Chimeric anti‐PCs can prevent accelerated atherosclerosis by inhibiting vascular inflammation directly and through reduced macrophage oxLDL uptake resulting in decreased lesions. PC‐mAb represents a novel strategy for cardiovascular disease prevention.
Keywords: atherosclerosis, inflammation, restenosis, therapeutics, vascular disease

Introduction
In atherosclerotic diseases, cellular stress and inflammation generate damage‐associated molecular pattern molecules (DAMPs). One of these DAMPs is phosphorylcholine (PC), the polar headgroup of the dominating membrane phospholipid (PL) phosphatidylcholine [1]. Enzymatic hydrolysis and oxidative modification of phosphatidylcholine in cell and LDL membranes, especially fatty acids in the sn‐2 position, lead to the formation of bioactive PC‐containing lipids (often referred to as oxPL) [2]. In human plasma, the main carrier of oxPLs is lipoprotein a (Lp(a) [2], and these Lp(a)‐associated oxPLs are able to induce arterial inflammation [3].
Many of these PC‐containing lipids are recognized by the innate immune system and stimulate powerful biological processes such as endothelial dysfunction, apoptosis and endoplasmic reticulum (ER) stress [4, 5]. They are considered important mediators of vascular inflammation rendering oxPL as a promising therapeutic target [1]. There are several innate immune system receptors recognizing PC including proteins, scavenger receptors of phagocytic cells and natural antibodies (IgM anti‐PC) [6]; in general, these receptors are part of the system for clearance of damaged particles and cells (efferocytosis) [7]. The murine T15/E06 IgM antibody is well known to inhibit oxLDL uptake into macrophages [8] and enhance efferocytosis [9, 10]. A pathophysiological role of PC‐containing oxPLs in atherosclerosis and ischaemia–reperfusion injury was shown by the protective effect of anti‐PC lacking effector function [11, 12].
Immunization leading to high anti‐PC levels can prevent native and accelerated atherosclerosis in mice by inhibition of oxLDL uptake and inflammatory foam cell formation [13, 14]. Epidemiological data suggest that IgM anti‐PC protects against cardiovascular disease development and that low levels of IgM anti‐PC are associated with increased risks of cardiovascular events [15, 16, 17, 18, 19]. Acute coronary syndrome (ACS) patients with low levels of IgM anti‐PC have a worse prognosis than patients with higher levels [20], although this was not confirmed in another study [21]. Experimental data show that T15/E06 natural IgM PC antibodies have profound anti‐inflammatory properties (unrelated to foam cell formation) and induce enhanced clearance of apoptotic cells [7, 22, 23]. This suggests that anti‐PC could be therapeutically effective.
Human serum contains anti‐PCs of several subclasses [24] and not all have associations with cardiovascular disease [25]. PC is also a pathogen‐associated molecular pattern (PAMP), present, for example, in the polysaccharide capsule of Streptococcus pneumoniae. Whilst T15/E06 was also effective against Streptococcus infections in mice, there was major variability in IgG anti‐PC effects [26, 27, 28]. In man, the relation between carotid atherosclerosis and anti‐PC may depend on the Ig isotype [25]. These results indicate that although PC is a chemically defined entity, the presentation of the PC epitope depends on the local environment such as the exact modification of the fatty acid component leading to the generation of PC epitopes and that different IgG anti‐PCs may be more specific for the variation in presentation of PC than the natural IgM anti‐PC.
IgM antibodies are rapidly eliminated from plasma, and there are currently no therapeutic IgMs available. The aim was to generate a fully human monoclonal IgG1 anti‐PC (PC‐mAb) that has the potential to block vascular inflammation, can inhibit uptake of oxLDL by macrophages and prevent vascular disease. As a reproducible model of such disease, we used cuff‐induced femoral artery lesions in mice [29], an established model that was further validated by transcriptomic analysis at several time‐points after cuff placement, to confirm that lesion development is inflammation‐driven and results in changes in choline metabolism. Drug effects were evaluated at two time‐points, 3 and 14 days after cuff placement. For validation purposes, we first generated a chimeric mouse/human IgG1 (PC‐mAb(T15)) with a human IgG1 Fc and the murine T15/E06 Fab fragment. This antibody prevented vascular inflammation both in vitro and in vivo, providing support that IgG PC‐mAbs can be effective against vascular disease. We then identified multiple fully human monoclonal PC‐mAbs using phage display library screening and cloned the Fab fragments into a human IgG1 backbone. These human monoclonal PC‐mAb IgG1 constructs were tested for their in vitro and in vivo anti‐inflammatory and anti‐atherosclerotic effects. The clinical grade antibody (ATH3G10) showed significant inhibition of vascular inflammation and prevented accelerated atherosclerosis in the murine cuff model and was well‐tolerated in rats and cynomolgus monkeys. ATH3G10 is now being tested in clinical studies (EudraCT Number: 2018‐003676‐12; ClinicalTrials.gov Identifier: NCT03991143).
Materials and methods
An extended version of the material and methods can be found in Appendix S2.
Mice
All mouse experiments were performed in compliance with Dutch government guidelines and conform to the guidelines from Directive 2010/63/EU of the European Parliament on protection of animals used for scientific purposes and were approved by the Institutional Committee for Animal Welfare of the Leiden University Medical Center (LUMC Approval Numbers 09224, 10207, 11208 and 13137). Transgenic male C57BL/6J ApoE*3‐Leiden mice were used due to their specific response to the hypercholesterolaemic diet [30] and their stronger response to vascular interventions [31]. The mice (bred in our own laboratory) were aged 10–12 weeks at the start of the dietary run‐in period and were fed a high‐cholesterol high‐fat diet (AB Diets, Woerden, The Netherlands) to induce hypercholesterolaemia 3 weeks prior to surgery and continued throughout the entire experiment. Mice were randomized based on plasma cholesterol levels (Roche Diagnostics, 1489437, Almere, The Netherlands ) and body weight. All animals received food and water ad libitum.
For the 3‐day antibody testing experiments, mice received one intraperitoneal injection at the time of surgery. For the 14‐day experiments, mice received intraperitoneal injections at the time of surgery and twice weekly (four injections in total). As negative controls, sterile 0.9% NaCl or human IgG1 streptavidin antibodies (control anti‐streptavidin IgG1; Dyax Corp, a Takeda company, Camebridge, MA, USA) were used.
Cuff model
Mice were anaesthetized with an intraperitoneal injection of midazolam (5 mg kg−1, Roche Diagnostics), dexmedetomidine (0.5 mg kg−1, Orion, Espoo, Finland) and fentanyl (0.05 mg kg−1, Janssen Pharmaceutica, Beerse, Belgium). Toe pinching was used to check the depth of anaesthesia. Femoral arteries were isolated and sheathed with a rigid nonconstrictive polyethylene cuff (Portex, Kent, United Kingdom). After the surgery, the anaesthesia was antagonized with a subcutaneous injection of atipamezole (2.5 mg kg−1, Orion) and flumazenil (0.5 mg kg−1, Fresenius Kabi, Bad Homburg vor der Höhe, Germany). Buprenorphine (0.1 mg kg−1, MSD Animal Health, Boxmeer, The Netherlands) was provided (subcutaneous injection) directly after surgery to relieve pain. Mice in the antibody studies were sacrificed at 3 days (acute inflammation) or 14 days (accelerated atherosclerosis) after cuff placement. At sacrifice, mice were anaesthetized with the aforementioned midazolam/dexmedetomidine/fentanyl mix and were euthanized via exsanguination followed by PBS perfusion (transcriptome analysis) and perfusion fixation (histology studies). Cuffed femoral arteries were harvested and snap‐frozen (transcriptome analysis) or fixed overnight in 3.7% formaldehyde and processed to paraffin (histology studies).
Human atherosclerotic material
Endarterectomy tissues were obtained in accordance with guidelines set out by the ‘Code for Proper Secondary Use of Human Tissue’ of the Dutch Federation of Biomedical Scientific Societies (Federa, Rotterdam, The Netherlands) and conform to the principles outlined in the Declaration of Helsinki.
Toxicity studies
Toxicity studies of 4‐ and 26‐week duration were performed (table 1). These studies were conducted at Charles River Laboratories, Edinburgh, and all animal housing and procedures were fully compliant with EU Directive 2010/63, UK legislation and AAALAC (Charles River Labs, Edinburgh, UK). Weekly intravenous doses of 4, 10 and 40 mg kg−1 were used in all studies. The following parameters and end‐points were evaluated in this study: clinical signs, body weights, body weight changes, food consumption, ophthalmology, electrocardiology, blood pressure, respiratory rate, clinical pathology parameters (haematology, coagulation, clinical chemistry and urinalysis), immunogenicity, toxicokinetic parameters, gross necropsy findings, organ weights and histopathologic examinations.
Table 1.
Overview of toxicity studies performed
| Species | Duration | Doses (weekly, iv) | n/group (m + f) | Recovery groups |
|---|---|---|---|---|
| SD rats | 4 weeks | 0; 3; 10 and 40 mg kg−1 bw | 10 + 10 | 5 + 5 |
| Cynomolgus | 4 weeks | 0; 3; 10 and 40 mg kg−1 bw | 3 + 3 | 2 + 2 |
| Cynomolgus | 26 weeks | 0; 3; 10 and 40 mg kg−1 bw | 3 + 3 | 2 + 2 |
For rats, additional groups of rats were treated equally and used for toxicokinetics and immunogenicity studies.
Statistical analysis
All data are presented as mean ± standard deviation, unless otherwise indicated. Overall comparisons between data from groups were performed with one‐way ANOVA, and for nonparametric testing, the Kruskal–Wallis test was used. If significant differences were found, groups were compared using a Mann–Whitney rank‐sum test. All statistical analyses were performed with GraphPad Prism 8. P‐values less than 0.05 were regarded as statistically significant. For RNA‐seq data, to determine differential expression compared with baseline empirical Bayes‐moderated t‐statistics were computed using the limma package (3.28.14). The Benjamini–Hochberg‐adjusted P‐values less than 0.05 were regarded as statistically significant.
Results
Cuff placement results in an early induction of inflammatory and ER stress genes and sustained changes in choline metabolism gene expression
To select a proper model to test the effect of PC antibodies in vivo, we focused on the femoral artery cuff model and profiled the transcriptome of arterial segments in response to injury. Dimension reduction through multidimensional scaling revealed distinct and tight clustering of early time‐points (baseline, 6 h, 1 day and 3 days) and more common clustering of later time‐points (7,14 days) (Fig. 1a). KEGG pathway over‐representation analysis of upregulated genes across time‐points showed an early inflammatory signature at time‐points 6 h, 1 day and 3 days with pathways such as TNF signalling, IL‐17 signalling, NF‐kB signalling and B‐cell receptor signalling all significantly over‐represented (Fig. 1b). Pathways over‐represented in downregulated genes tended to be consistent across all time‐points and included multiple extracellular signalling and second messenger pathways including ECM–receptor interaction, focal adhesion, cGMP PKG signalling and calcium signalling pathways (Fig. 1c). Along with surgical stress, key mediators of endoplasmic reticulum (ER) stress including Atf4, Atf6 and spliced Xbp1 rapidly and transiently increased, peaking at 6 h postsurgery and returning to baseline levels by 3 days (Fig. 1d). Consistent with this increase, numerous transcriptional targets of these key mediators were also significantly increased at early time‐points including Hspa5, Atf4 and Gadd34 (Fig. 1e). Interestingly, genes related to choline metabolism (KEGG mmu05231) showed distinct patterns of expression across the time‐points and this gene set was significantly differentially expressed at both day 3 (Fig. 1f) and day 14 (Fig. 1g) compared with baseline. Genes specifically related to interconversion of choline and phosphorylcholine were also significantly regulated over time. At 6 h, Chka, which converts choline to phosphorylcholine, was significantly upregulated and tended to be upregulated across time‐points (Fig. 1h,i). Across all time‐points, Chpt1, which generates phosphatidylcholine, was significantly downregulated (Fig. 1h,i). Other genes that shunt choline away from phosphorylcholine (Chdh, Ache) tended to either be significantly downregulated or unchanged across time‐points (Fig. 1h,i). Other genes that generate choline from phosphatidylcholine (Gpcpd1, Lypla1, Pla2) tended to either be significantly upregulated or unchanged (Fig. 1h,i). Based on the different gene regulation patterns, further studies were conducted at day 3 after cuff placement to investigate inflammation‐driven processes and at day 14 to investigate vascular remodelling and accelerated atherosclerosis.
Fig. 1.
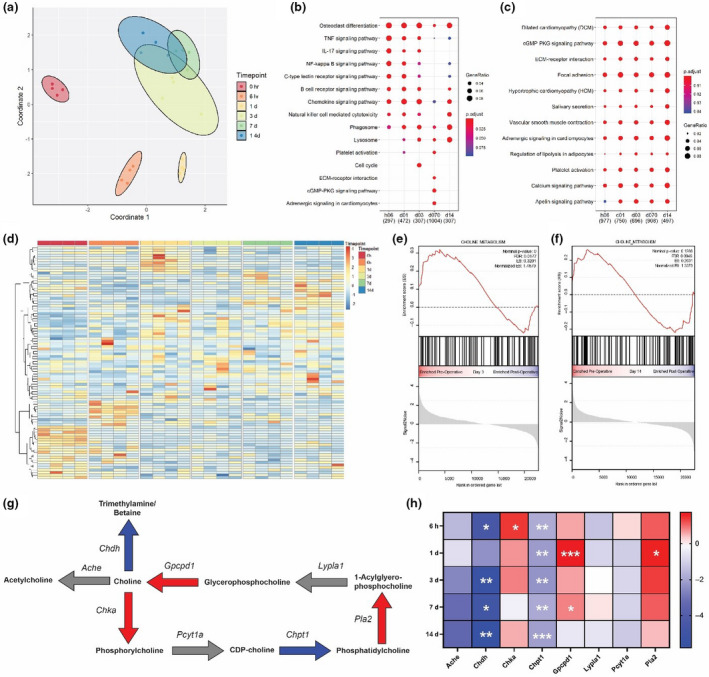
Femoral arterial cuff placement causes rapid and sustained alterations to global transcriptome including ER stress and choline metabolism. (a) Multidimensional scaling plot of transcriptome of cuffed arteries at baseline (0 h) or multiple time‐points after cuffing (n = 4–8 mice/time‐point. (b) KEGG pathway over‐representation test of genes significantly upregulated or downregulated (c) at multiple time‐points after cuffing. (d) Log2 fold changes in key upstream mediators of the ER stress response and downstream targets (e). (f) Gene set enrichment analysis of genes in the choline metabolism pathway at 3 and 14 (g) days after cuffing. (h) Diagram of major choline metabolic reactions, bold indicates metabolites and italic indicates genes carrying out the reactions. Red arrows indicate enzymes significantly upregulated over the time course, blue significantly downregulated and grey unchanged. (i) Log2 fold changes in genes shown in (g). Empirical Bayes‐moderated t‐statistics were computed to determine differential expression compared with baseline; *P < 0.1, **P < 0.05 and ***P < 0.001.
Chimeric PC‐mAb(T15) binds to human atherosclerotic lesions, inhibits in vivo vascular inflammation and prevents accelerated atherosclerotic lesion formation
Immunohistochemical staining with chimeric PC‐mAb(T15) showed strong and specific binding to cells including macrophages within human atherosclerotic lesions (Fig. 2a). Specific binding of PC‐mAb(T15) to PC was confirmed by ELISA (data not shown). Therapeutic effectiveness of PC‐mAb(T15) was tested in the mouse cuff model for postinterventional accelerated atherosclerosis. Plasma cholesterol levels or body weight were not affected by PC‐mAb(T15) (Table S1). PC‐mAb(T15) reduced endothelial adherence and vessel infiltration of monocytes/macrophages to the injured arterial segments 3d after injury (Fig. 2b). Together, this led to a 60% significant reduction in macrophages in the vessel wall compared with both control groups (Fig. 2c & Figure S1A, B). Comparable results were found for CD45+ leucocytes (Figure S1C, D). ER stress‐related proteins are signs of pro‐inflammatory responses as shown in human atherosclerotic lesions in which HSPA5 (Figure S1E) and DDIT3 (Figure S1F) are highly expressed. Both HSPA5 and DDIT3 are also found in murine lesions (Figure S1G, H). PC‐mAb(T15) treatment showed trends towards reduction in HSPA5+ cells (Figure S1I‐J), whereas DDIT3+ cells attached to the endothelium and in the media were significantly reduced with respect to 80 and 67% (Figure S1K‐L). These results indicate that PC‐mAb(T15) prevents early inflammatory responses, well before generation of local foam cells in this model (26).
Fig. 2.
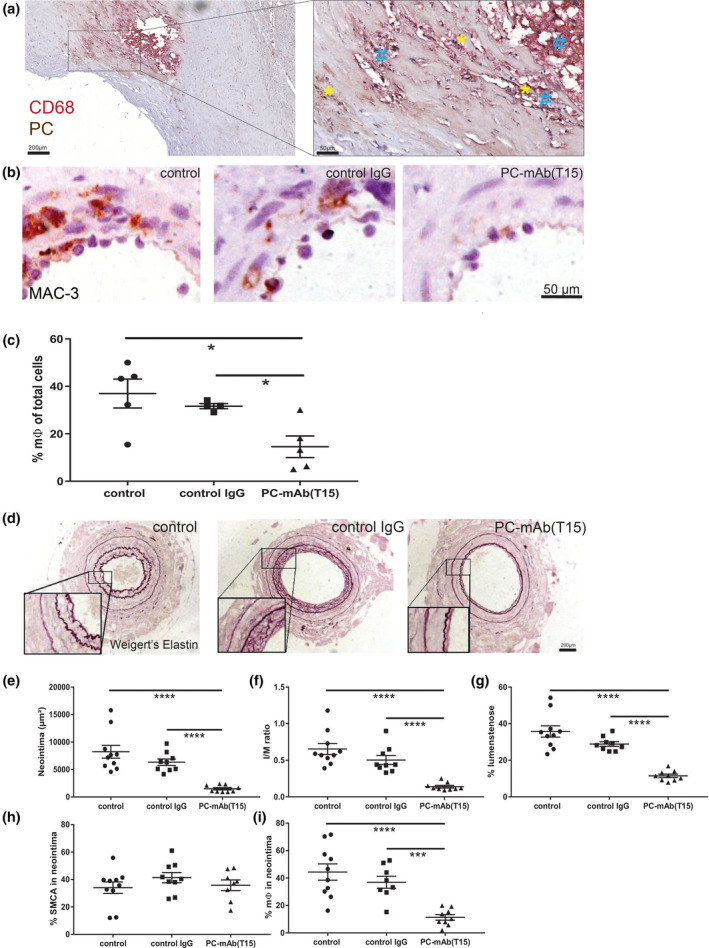
Chimeric PC‐mAb(T15) prevents accelerated atherosclerotic lesion formation. (a) Representative human endarterectomy specimen (n = 12) stained for phosphorylcholine (PC, yellow star) and macrophages (CD68, blue hash). (b) Representative cross sections of macrophages (MAC‐3) in cuffed arteries of ApoE3*Leiden mice receiving vehicle (control), human anti‐streptavidin (control IgG) or anti‐PC(T15) IgG after 3 days (n = 5/group). (c) Quantification of macrophages (t3). (d) Representative cross sections of cuffed arteries at day 14 (n = 9–10/group). Quantification of neointima (e), intima/media ratio (f), % lumen stenosis (g), % smooth muscle cells (h) and % macrophages. (i) Mean ± SD, Mann–Whitney; *P < 0.05, ***P < 0.001 and ****P < 0.0001.
Effects of PC‐mAb(T15) on accelerated atherosclerosis and remodelling were evaluated 14d after cuff placement (Fig. 2d–i, Figure S2). PC‐mAb(T15) in comparison with both the control and IgG control groups (Fig. 2d) significantly reduced neointima formation (82% and 76%, Fig. 2e), intima/media ratio (79% and 72%, Fig. 1f) and luminal stenosis (68% and 60%, Fig. 1g). PC‐mAb(T15) treatment resulted in a significant increase in SMCs in the media (87% compared with control and 37% compared with control IgG) (Figure S2D,E), whereas no differences could be detected in the neointima (Fig. 2h). PC‐mAb(T15) significantly reduced macrophage infiltration in the neointima (resp., 74% and 69%, Fig. 2i, Figure S2F,G). Furthermore, PC‐mAb(T15) reduced the number of CD45+ leucocytes in both media and neointima (Figure S2H–J), as well as cells displaying ER stress marker HSPA5 (Figure S2K–M) and the percentage of cells displaying DDIT3 (Figure S2N–P). Thus, passive immunization with PC‐mAb(T15) prevents vascular inflammation and lesion formation in part by reducing ER stress.
PC‐mAb(T15) and human polyclonal IgG anti‐PC do not block oxLDL uptake by macrophages, unlike polyclonal anti‐PC IgM
The murine IgM T15/E06 natural antibody and polyclonal human IgM anti‐PC [32] are known to block scavenger receptor‐mediated uptake of oxLDL [13]. As expected, polyclonal anti‐PC IgM showed an inhibition of Dil‐labelled Cu‐oxLDL uptake by human monocyte‐derived macrophages (Fig. 3a), which was dose‐dependent (Figure S3A). In contrast to anti‐PC IgM, serum‐derived polyclonal anti‐PC IgGs did not show an inhibition of Cu‐oxLDL uptake (Fig. 3b, Figure S3B). PC‐mAb(T15) also did not prevent Cu‐oxLDL uptake in human macrophages. Unlike IgM, the Fc region of IgG bears a highly conserved N‐glycosylation site that is essential for Fc receptor‐mediated activity by macrophages and could lead to increased oxLDL uptake of oxLDL–immune complexes by macrophages via Fc receptors. As such a mechanism could mask inhibition of scavenger receptor‐mediated uptake, we excluded this possibility by using both IgG1 isotype chimeric PC‐mAb(T15) and IgG4 isotype chimeric PC‐mAb(T15) (Fig. 3c). As IgG4 antibodies are less prone to bind Fc receptors, the experiment was also performed including Fc receptor blockade by anti‐CD32 and anti‐CD64 antibodies. However, this had no effect on Cu‐ox‐LDL uptake (Fig. 3d). Even though PC‐mAb(T15) has anti‐inflammatory effects in vivo, transfer of the T15/E06 variable region from IgM to the IgG format abolished the scavenger receptor blocking effects.
Fig. 3.
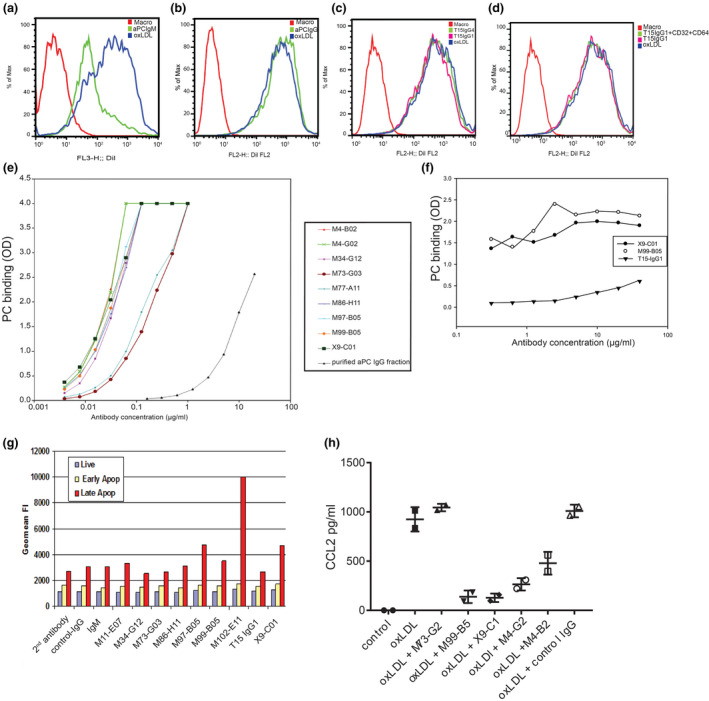
Development and screening of monoclonal human PC‐mAbs Flow cytometry demonstrates that unlike murine IgM anti‐PC(A), polyclonal anti‐PC IgG does not block oxLDL uptake by macrophages (b). Both IgG1 and IgG4 antibodies fail to display an inhibiting effect (c), excluding the potential enhanced Fc receptor‐regulated uptake of oxLDL (d) including Fc‐receptor blockade by anti‐CD32 and anti‐CD64 antibodies. Relative ranking of antibody affinity to PC (e) and oxLDL (f), measured through ELISA (expressed as OD). Flow cytometry of antibody affinity (g) for live cells (annexin V−PI−), early apoptotic cells (annexin V+PI−) and late apoptotic cells (annexin V+PI+), expressed as mean fluorescent intensity of each cell population. (h) CCL2 release assay of human monocytes stimulated with oxLDL alone or in combination with antibody clones, results of 2 individual experiments. One‐way ANOVA.
Development of monoclonal human PC‐mAbs
Because the ability to block oxLDL uptake is important, we decided to optimize the PC‐mAb using phage display and binding to PC‐BSA as an initial screen, with secondary screens including inhibition of oxLDL uptake and binding to apoptotic cells in vitro. Screening of 10 660 different phages yielded 1511 positive hits, defined as a >3‐fold stronger signal by the individual phage for binding to BSA‐ or ferritin‐conjugated PC compared with IgG controls. 54 antibodies were recovered after recombinant reformatting to full‐length IgG. Binding specificity was assessed using a Biacore SPR assay and ELISA for binding to either control, linker, PC‐BSA and PC‐KLH, allowing selection of antibodies with potential therapeutic effectiveness. 27 antibodies with the best binding affinities to PC‐BSA and PC‐ferritin were investigated for their effects on macrophage oxLDL uptake (data not shown). Nine recombinant anti‐PC IgG antibodies inhibited oxLDL uptake similarly or better than polyclonal IgM anti‐PC on a weight basis (Figure S3C,D).
These nine monoclonal antibodies displayed an approximately 1000‐fold higher affinity for PC than polyclonal IgG anti‐PC (Fig. 3e). M99‐B05 and X9‐C01 were analysed for binding to Cu‐oxidized LDL, and both displayed profoundly increased binding affinity compared with chimeric PC‐mAb(T15) (Fig. 3f). The high avidity of murine IgM T15/E06 to PC compared with the IgG isotype explains the effectiveness of the IgM T15/E06 [22] on oxLDL uptake.
Antibodies with high affinity for PC and oxLDL were tested for binding to apoptotic Jurkat cells using FACS analysis. Only marginally increased binding to cells considered as early apoptotic (annexin A5+ PI‐) was found. Some antibodies, including M99‐B05 and X9‐C01, were found to bind strongly to late apoptotic (annexin A5+ PI+) Jurkat cells (Fig. 3g, Figure S3E–G). Five selected antibodies were finally tested for their ability to block oxLDL‐induced release of CCL2 from monocytes of which M99‐B05 and X9‐C01 effectively blocked this release in a dose‐dependent fashion with an IC50 in the 1–3 nM range (data not shown), whilst other tested antibodies were less effective (Fig. 3h).
PC‐mAbs are effective against vascular inflammation and remodelling in vivo
The anti‐inflammatory properties of four antibodies (M4B2 was not continued due to low yields during production) were tested in vivo in the cuff model, with PC‐mAb(T15) as positive control. Treatment with M99‐B05 and PC‐mAb(T15) reduced endothelial adherence and extravasation of CD45+ leucocytes (Fig. 4a) and macrophages (Fig. 4b) significantly, whilst the other PC‐mAbs did not (images; Figure S4A). Interestingly, the number of CCL2+ cells in the vessel wall was significantly reduced by all three antibodies: PC‐mAb(T15) 82%, M99‐B05 82% and X9‐C01 (74%) (Fig. 4c,d). Next, the effects of M99‐B05 and X9‐C01 on accelerated atherosclerosis in the 14‐day femoral artery cuff model were analysed. Both antibodies significantly prevented neointima formation (42% and 44%, resp.) and reduced intima/media ratio (both 43%) and lumen stenosis (56% and 64%, resp.) in comparison with control IgG (Fig. 4e–g, Figure S4B–D). Based on all these previous data, M99‐B05 was selected for further optimization.
Fig. 4.
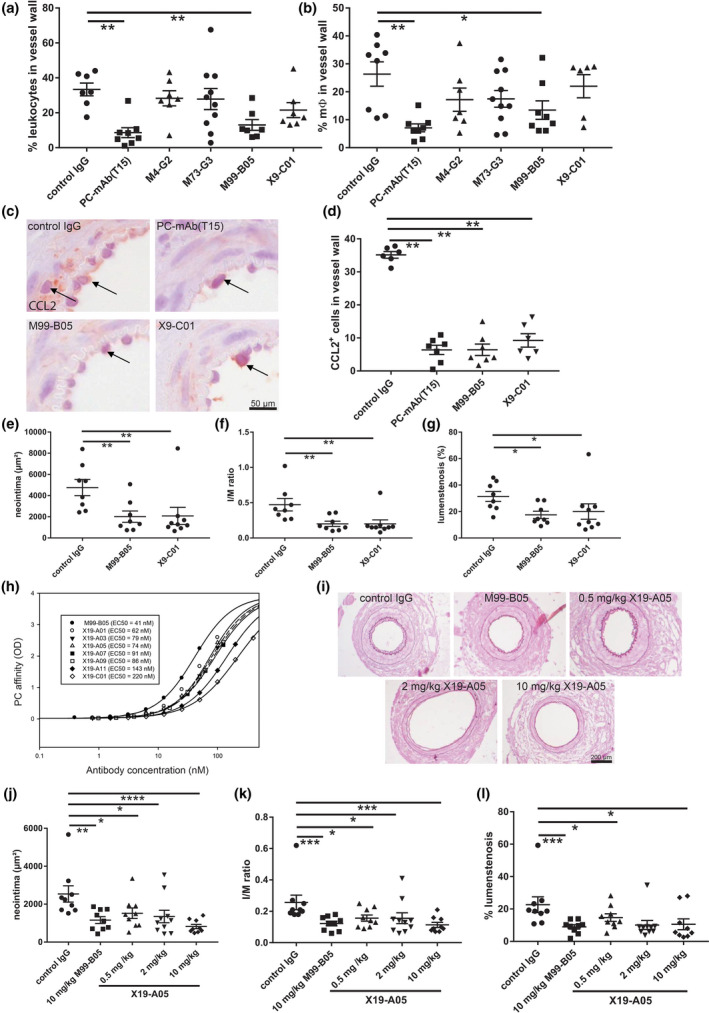
In vivo validation of PC‐mAb antibodies The anti‐inflammatory properties of PC‐mAbs were tested in vivo in the cuff model (3 days), with PC‐mAb(T15) as positive control (n = 7–10). Quantification of (a) % leucocytes and (b) % macrophages. M99‐B05 and X9‐C01 were selected for further analysis. (c) Representative cross sections of CCL2 staining and (d) quantification of CCL2+ cells. M99‐B05 and X9‐C01 were tested in a 14‐day cuff experiment (n = 8–9). Quantification of neointima (e), intima/media ratio (f) and % lumen stenosis (g). After codon optimization, the PC affinity of seven M99‐B05 clones was tested (h). X19‐A05 was selected for further testing in a 14‐day cuff experiment. Representative cross sections (i) of cuffed arteries of ApoE3*Leiden mice receiving human anti‐streptavidin (control IgG), M99‐B05 or X19‐A05 (0.5, 2, 10 mg kg−1) (n = 9–10). Quantification of neointima (j), intima/media ratio (k) and % lumen stenosis (l). Mean ± SD, Mann–Whitney; *P < 0.05, **P < 0.01, ***P < 0.001 and ****P < 0.0001.
Optimization of monoclonal PC‐mAb(M99‐B05) into PC‐mAb(X19‐A05)
To optimize the properties of PC‐mAb(M99‐B05), we employed germlining, codon optimization and amino acid replacements for potential de‐amidation sites, which resulted in a series of mutants. These were tested for binding to PC, and it was observed that some modifications of M99‐B05 negatively affected binding affinity to PC (Fig. 4h). The mutant X19‐A05 was selected as the optimal antibody, as it combines several beneficial modifications of M99‐B05 whilst retaining good binding affinity to PC.
Two‐week treatment with X19‐A05 twice weekly with 0.5, 2 and 10 mg kg−1 dosages significantly prevented neointima formation, and reduced I/M ratio and lumen stenosis, comparable to M99‐B05 (Fig. 4i–l, Figure S4E–G).
X19‐A05 is potent, as a low dosage of 0.5 mg kg−1 twice weekly is already sufficient to produce a 35% inhibition of neointima formation (Fig. 4j).
ATH3G10 is the selected drug product for clinical development
PC‐mAb(X19‐A05) was selected for further clinical development resulting in PC‐mAb (ATH3G10). ATH3G10 is the IgG1za/kappa antibody clinical grade product, produced using a stable cell line with the same Fab fragments as X19‐A05. Biological activity of ATH3G10 was investigated in the 14‐day femoral artery cuff model using twice weekly i.p. injections in the doses 0.1 and 1 mg kg−1 (Fig. 5a, Figure S5A–D). Treatment resulted in significantly reduced neointima formation by both concentrations of, resp., 22% and 27% compared with the control group (Fig. 5b). A 26% reduction in I/M ratio was observed for both groups (Fig. 5c). Furthermore, ATH3G10 reduced the macrophage content in both media and neointima (Fig. 5d,e, Figure S5E,F). ATH3G10 thus has anti‐inflammatory and vascular protective properties.
Fig. 5.
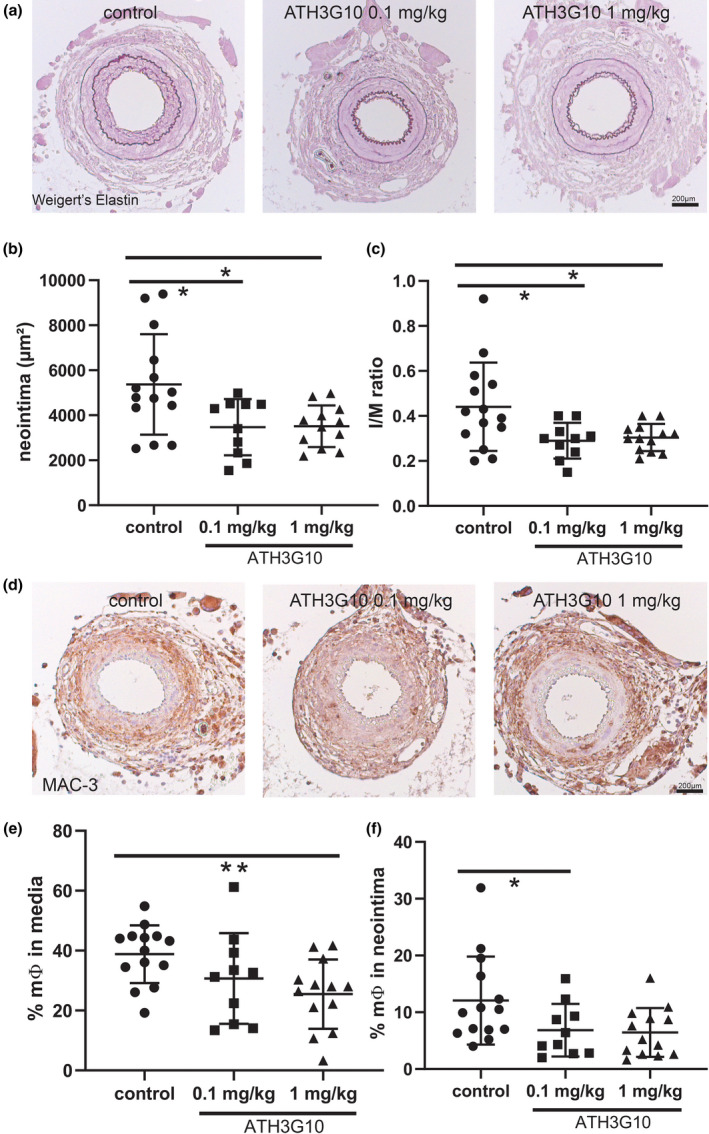
ATH3G10 prevents accelerated atherosclerosis ATH3G10 was tested in a 14‐day cuff experiment (n = 10–12) in ApoE3*Leiden mice receiving vehicle (control), or ATH3G10 (0.1 mg kg−1 or 1 mg kg−1). (a) Representative cross sections of elastin stained cuffed arteries after 14 days. (b) Quantification of neointima (c), intima/media ratio and (d) representative cross sections of macrophage (MAC‐3) staining. Quantification of (e) % macrophages in media and (f) % macrophages in neointima. Mean ± SD, Mann–Whitney; *P < 0.05 and **P < 0.01.
ATH3G10 toxicology studies in rats and cynomolgus monkeys
To proceed towards human studies, we performed the required toxicity testing in rats and a nonrodent species the cynomolgus monkey, which is the most commonly used species for antibody testing. For these types of human antibody studies, the expectation is that mammals develop an immune response in the form of anti‐drug antibodies (ADAs) [33, 34]. The repeated toxicity studies in rats and monkeys treated up to 4 weeks did not show any drug‐related toxicity in any organ, and ATH3G10 was considered well‐tolerated. In the 4‐week toxicity studies, the highest tested dose 40 mg kg−1 was considered as the no observable adverse effect level (NOAEL) in both species. In the 26‐week toxicity study in monkeys, most female but no male monkeys developed ADA after several months of drug exposure. Two female monkeys in the highest dose group had severe reactions due to an immune reaction to the human protein [35] leading to precipitation of immune complexes between ATH3G10 and complement in several tissues along with inflammation and bleeding in some organs. In the lower dose groups, no immune complexes were seen in the animals even if ADAs were detected. In the 4‐week studies, ADA was observed in one female rat at termination and in no cynomolgus monkeys. Thus, in rats and cynomolgus monkeys ATH3G10‐limited immunogenicity to the human protein was observed. These toxicity studies allowed ATH3G10 to enter clinical studies that are currently ongoing (EudraCT Number: 2018‐003676‐12; ClinicalTrials.gov Identifier: NCT03991143).
Discussion
Human PC‐binding antibodies selected by phage library display and converted to fully human monoclonal IgG1 targeting PC were used to demonstrate for the first time the therapeutic efficacy of IgG anti‐PC. It has been shown in several studies that there is an inverse correlation of atherosclerosis and IgM as well as polyclonal IgG1 (but not IgG2) PC antibodies [13, 14, 25]. PC antibodies are known to prevent cardiovascular diseases via targeting of various processes; they are anti‐apoptotic and anti‐thrombotic, target oxLDL, are anti‐inflammatory by targeting the NFƙB pathway and prevent smooth muscle cell proliferation and migration [14, 15, 16, 17, 18, 19, 36, 37, 38–14, 15–14, 15, 16, 17, 18, 19, 36, 37, 38. One or several mechanisms of anti‐PC might contribute to a potential benefit, and the mechanism may actually differ between different disease situations. The current project aimed to develop a fully human monoclonal PC antibody (PC‐mAb) for the treatment of inflammation‐associated vascular disease. Therefore, the strategy was to select a drug for clinical trials that has beneficial effects on arterial inflammation and neointima/lesion formation in vivo, rather than a prespecified molecular mechanism. Passive in vivo immunization with chimeric PC‐mAb(T15) and selected fully human PC‐mAbs prevented cuff‐induced arterial inflammation and accelerated atherosclerosis. The selected human PC‐mAb binds to apoptotic cells, has high affinity for oxLDL and inhibited oxLDL uptake by macrophages in vitro, a beneficial effect of polyclonal IgM anti‐PC, but surprisingly not of human polyclonal IgG anti‐PC or PC‐mAb(T15).
PC plays a key role as DAMP in the inflammatory reactions in both native atherosclerosis and following vascular intervention strategies due to acute events [11, 39]. In the latter setting, where rapid onset of action is desired, passive rather than active vaccination is preferred. Up until now, the experimental work validating anti‐PC as a therapeutic strategy has largely been performed using the mouse T15/E06 IgM, whilst for clinical use, IgG‐based antibody formulations are preferred [40]. To test whether this format is effective, we used the chimeric PC‐mAb(T15). Specific binding was observed in human atherosclerotic lesions, proving antibody binding to the target tissue. To minimize the risk of an immunologic reaction to mouse/human chimeric and human proteins in mice, we used an accelerated in vivo model of atherosclerosis [29].
Transcriptomic analysis demonstrated a rapid induction of inflammatory signalling pathways, as well as a rapid induction of key transcriptional mediators of the ER stress response. Expression of choline metabolic genes was robustly altered in response to cuff placement. PC‐mAb(T15) effectively reduced monocyte/macrophage adherence and extravasation with notably less CCL2 expression. Defective inflammatory resolution including ER stress is a known lead to the progression of atherosclerotic lesions [41]. Here demonstrated by expression of ER stress markers HSPA5 and DDIT3 in human and murine atherosclerotic lesions. PC‐mAb(T15) treatment led to retarded accelerated atherosclerosis and a less inflammatory phenotype, involving reduction in ER stress markers. In line with this, recent findings indicate that a fully human IgG1 clone against PC promotes clearance of dead cells and that IgG1 but not IgG2 anti‐PC is associated with protection in relation to atherosclerosis [42].
Polyclonal human IgM anti‐PC inhibited oxLDL uptake by macrophages in vitro, whereas polyclonal IgG anti‐PC and PC‐mAb(T15) did not, probably due to low affinity to oxLDL (Fig. 4b). Postinterventional accelerated atherosclerosis is mainly triggered by injury‐induced local inflammatory factors including cellular (ER) stress and DAMP expression and is not primarily the result of oxLDL uptake [43, 44]. Although this concept supports why PC‐mAb(T15) was effective in the murine cuff model, scavenger receptor‐mediated uptake of oxLDL does contribute to the progression of atherosclerosis [3], and blocking oxLDL uptake should be beneficial, therefore selected IgGs combining high‐affinity binding to the PC epitope on both oxLDL and apoptotic cells, and with inhibition of macrophage oxLDL uptake. Out of a total of 10 660 phage clones, these selection criteria yielded four promising antibody clones. Further in vivo selection resulted in M99‐B05 and X9‐C1 which both reduced CCL2 expression in the vessel wall and reduced accelerated atherosclerosis, proving its long‐term efficacy.
Codon‐optimized M99‐B05, designated X19‐A05, effectively inhibited neointima formation. A stable CHO cell line capable of producing ATH3G10, identical to X19‐A05 in its Fab regions, was developed for the production of clinical grade material. ATH3G10 had preserved PC affinity and therapeutic efficacy shown by significantly reduced neointima formation and I/M ratios in low dosages (0.1–1 mg kg−1). The observed effect of ATH3G10 seemed not as effective as the X19‐A05; however, the cholesterol levels in the ATH3G10 study were higher (Table S1), making it a more robust study. The present findings show that anti‐PC has prominent effects on accelerated lesion development and also support the concept of PC as an important target for new therapeutics in vascular disease as also shown in recently published studies. PC‐mAb preserves coronary flow reserve and attenuates atherosclerotic inflammation as determined by the uptake of 18F‐fluorodeoxyglucose in atherosclerotic LDLR−/−ApoB100/100 mice [45]. Furthermore, PC‐mAb showed preserved cardiac function and reduced infarct size in hypercholesterolaemic mice [46]. ATH3G10 did not cause adverse reactions in toxicity studies opening up the step towards clinical studies. These results combined with the strong epidemiologic support for the concept suggest that ATH3G10 is a promising therapeutic candidate for the prevention of cardiovascular events in situations of increased inflammatory stress, and it is currently in clinical development.
Funding
This study was funded by the European Union Program Grant CVDIMMUNE (037227) and CARDIMMUN (601728).
Conflict of interest statement
KP, AB and ID are consultants to Athera Biotechnologies. JF, KP and AB are minor shareholder of Athera Biotechnologies, Stockholm, Sweden. JF and KP are named inventors on anti‐PC‐related patents. DS is a former employee of Dyax Pharmaceuticals, Cambridge, MA, USA. None of the other authors have conflicts of interest.
Author contribution
M. R. de Vries: Conceptualization (equal); data curation (equal); formal analysis (equal); investigation (lead); methodology (lead); project administration (lead); validation (lead); visualization (lead); writing – original draft (lead). M. M. Ewing: Formal analysis (equal); investigation (equal); writing – original draft (supporting). R. C. M. de Jong: Data curation (supporting); formal analysis (supporting); investigation (supporting); writing – review and editing (supporting). M. R. MacArthur: Data curation (supporting); formal analysis (supporting); writing – review and editing (supporting). J. C. Karper: Data curation (supporting); formal analysis (supporting); writing – review and editing (supporting). E. A. B. Peters: Formal analysis (supporting); investigation (equal). M. Nordzell: Investigation (supporting); methodology (supporting); resources (equal); writing – review and editing (equal). S. A. P. Karabina: Data curation (supporting); investigation (supporting); methodology (supporting); writing – review and editing (supporting). D. Sexton: Investigation (supporting); methodology (supporting); resources (supporting); writing – review and editing (supporting). I. Dahlbom: Data curation (supporting); investigation (supporting); methodology (supporting); writing – review and editing (supporting). A. Bergman: Data curation (supporting); formal analysis (supporting); funding acquisition (supporting); investigation (supporting); methodology (supporting); validation (supporting); visualization (supporting); writing – review and editing (supporting). J. R. Mitchell: Resources (supporting); supervision (supporting); writing – review and editing (supporting). J. Frostegård: Conceptualization (supporting); investigation (supporting); methodology (supporting); writing – review and editing (supporting). J. Kuiper: Data curation (supporting); investigation (supporting); methodology (supporting); writing – review and editing (supporting). E. Ninio: Investigation (supporting); methodology (supporting); writing – review and editing (supporting). J. W. Jukema: Conceptualization (equal); validation (supporting); writing – review and editing (supporting). K. Pettersson: Conceptualization (equal); formal analysis (equal); funding acquisition (equal); investigation (equal); methodology (equal); resources (equal); supervision (equal); writing – original draft (supporting). P. H. A. Quax: Conceptualization (equal); funding acquisition (lead); investigation (equal); resources (equal); supervision (equal); writing – original draft (supporting).
Supporting information
Appendix S1. Table S1 and Figures S1–S5.
Table S1. Study characteristics.
Figure S1. Chimeric PC‐mAb(T15) prevents vascular inflammation.
Figure S2. Chimeric PC‐mAb(T15) prevents accelerated atherosclerotic lesion formation.
Figure S3. Development and screening of monoclonal human anti‐PC IgG antibodies.
Figure S4. In vivo validation of anti‐PC antibodies.
Figure S5. ATH3G10 prevents accelerated atherosclerosis.
Appendix S2. Extended version of the material and methods.
Acknowledgements
None.
de Vries MR, Ewing MM, de Jong RCM, MacArthur MR, Karper JC, Peters EAB, Nordzell M, Karabina SAP, Sexton D, Dahlbom I, Bergman A, Mitchell JR, Frostegård J, Kuiper J, Ninio E, Jukema JW, Pettersson K, Quax PHA (LUMC, Leiden, The Netherlands; Harvard T.H. Chan School of Public Health, Boston; Athera Biotechnologies; Sorbonne Université, Paris, France; Takeda Pharmaceutical, Cambridge, MA, USA; Karolinska University Hospital Huddinge and Karolinska Institutet, Stockholm, Sweden; LACDR, Leiden, The Netherlands). Identification of IgG1 isotype phosphorylcholine antibodies for the treatment of inflammatory cardiovascular diseases. J Intern Med. 2021;290:141–156. 10.1111/joim.13234
References
- 1.Lee S, Birukov KG, Romanoski CE, Springstead JR, Lusis AJ, Berliner JA. Role of phospholipid oxidation products in atherosclerosis. Circ Res 2012; 111: 778–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bergmark C, Dewan A, Orsoni Aet al. A novel function of lipoprotein [a] as a preferential carrier of oxidized phospholipids in human plasma. J Lipid Res 2008; 49: 2230–9. [DOI] [PubMed] [Google Scholar]
- 3.van der Valk FM, Bekkering S, Kroon Jet al. Oxidized phospholipids on lipoprotein(a) elicit arterial wall inflammation and an inflammatory monocyte response in humans. Circulation 2016; 134: 611–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Gora S, Maouche S, Atout Ret al. Phospholipolyzed LDL induces an inflammatory response in endothelial cells through endoplasmic reticulum stress signaling. FASEB J 2010; 24: 3284–97. [DOI] [PubMed] [Google Scholar]
- 5.Miller YI, Tsimikas S. Oxidation‐specific epitopes as targets for biotheranostic applications in humans: biomarkers, molecular imaging and therapeutics. Curr Opin Lipidol 2013; 24: 426–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Miller YI, Choi SH, Wiesner Pet al. Oxidation‐specific epitopes are danger‐associated molecular patterns recognized by pattern recognition receptors of innate immunity. Circ Res 2011; 108: 235–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Silverman GJ. Protective natural autoantibodies to apoptotic cells: evidence of convergent selection of recurrent innate‐like clones. Ann N Y Acad Sci 2015; 1362: 164–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Horkko S, Bird DA, Miller Eet al. Monoclonal autoantibodies specific for oxidized phospholipids or oxidized phospholipid‐protein adducts inhibit macrophage uptake of oxidized low‐density lipoproteins. J Clin Investig 1999; 103: 117–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Elkon KB, Silverman GJ. Naturally occurring autoantibodies to apoptotic cells. Adv Exp Med Biol 2012; 750: 14–26. [DOI] [PubMed] [Google Scholar]
- 10.Zou J, Wang G, Li H, Yu X, Tang C. IgM natural antibody T15/E06 in atherosclerosis. Clinica Chimica Acta 2020; 504: 15–22. [DOI] [PubMed] [Google Scholar]
- 11.Que X, Hung MY, Yeang Cet al. Oxidized phospholipids are proinflammatory and proatherogenic in hypercholesterolaemic mice. Nature 2018; 558: 301–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Iseme RA, McEvoy M, Kelly Bet al. A role for autoantibodies in atherogenesis. Cardiovasc Res 2017; 113: 1102–12. [DOI] [PubMed] [Google Scholar]
- 13.Binder CJ, Horkko S, Dewan Aet al. Pneumococcal vaccination decreases atherosclerotic lesion formation: molecular mimicry between Streptococcus pneumoniae and oxidized LDL. Nat Med 2003; 9: 736–43. [DOI] [PubMed] [Google Scholar]
- 14.Caligiuri G, Khallou‐Laschet J, Vandaele Met al. Phosphorylcholine‐targeting immunization reduces atherosclerosis. J Am Coll Cardiol 2007; 50: 540–6. [DOI] [PubMed] [Google Scholar]
- 15.Su J, Georgiades A, Wu R, Thulin T, de Faire U, Frostegard J. Antibodies of IgM subclass to phosphorylcholine and oxidized LDL are protective factors for atherosclerosis in patients with hypertension. Atherosclerosis 2006; 188: 160–6. [DOI] [PubMed] [Google Scholar]
- 16.Gronwall C, Akhter E, Oh C, Burlingame RW, Petri M, Silverman GJ. IgM autoantibodies to distinct apoptosis‐associated antigens correlate with protection from cardiovascular events and renal disease in patients with SLE. Clin Immunol 2012; 142: 390–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Gleissner CA, Erbel C, Haeussler Jet al. Low levels of natural IgM antibodies against phosphorylcholine are independently associated with vascular remodeling in patients with coronary artery disease. Clin Res Cardiol 2015; 104: 13–22. [DOI] [PubMed] [Google Scholar]
- 18.Frostegard AG, Su J, Hua X, Vikstrom M, de Faire U, Frostegard J. Antibodies against native and oxidized cardiolipin and phosphatidylserine and phosphorylcholine in atherosclerosis development. PLoS One 2014; 9: e111764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Gronlund H, Hallmans G, Jansson JHet al. Low levels of IgM antibodies against phosphorylcholine predict development of acute myocardial infarction in a population‐based cohort from northern Sweden. Eur J Cardiovas Prev Rehabil 2009; 16: 382–6. [DOI] [PubMed] [Google Scholar]
- 20.Sjoberg BG, Su J, Dahlbom Iet al. Low levels of IgM antibodies against phosphorylcholine‐A potential risk marker for ischemic stroke in men. Atherosclerosis 2009; 203: 528–32. [DOI] [PubMed] [Google Scholar]
- 21.Geller BJ, Mega JL, Morrow DAet al. Autoantibodies to phosphorylcholine and cardiovascular outcomes in patients with acute coronary syndromes in the ATLAS ACS‐TIMI 46 trial. J Thromb Thrombolysis 2014; 37: 310–6. [DOI] [PubMed] [Google Scholar]
- 22.Chang MK, Binder CJ, Miller YIet al. Apoptotic cells with oxidation‐specific epitopes are immunogenic and proinflammatory. J Exp Med 2004; 200: 1359–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Chou MY, Fogelstrand L, Hartvigsen Ket al. Oxidation‐specific epitopes are dominant targets of innate natural antibodies in mice and humans. J Clin Investig 2009; 119: 1335–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sampi M, Veneskoski M, Ukkola O, Kesaniemi YA, Horkko S. High plasma immunoglobulin (Ig) A and low IgG antibody titers to oxidized low‐density lipoprotein are associated with markers of glucose metabolism. J Clin Endocrinol Metab 2010; 95: 2467–75. [DOI] [PubMed] [Google Scholar]
- 25.Fiskesund R, Su J, Bulatovic I, Vikstrom M, de Faire U, Frostegard J. IgM phosphorylcholine antibodies inhibit cell death and constitute a strong protection marker for atherosclerosis development, particularly in combination with other auto‐antibodies against modified LDL. Results Immunol 2012; 2: 13–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Gearhart PJ, Johnson ND, Douglas R, Hood L. IgG antibodies to phosphorylcholine exhibit more diversity than their IgM counterparts. Nature 1981; 291: 29–34. [DOI] [PubMed] [Google Scholar]
- 27.Briles DE, Forman C, Hudak S, Claflin JL. Anti‐phosphorylcholine antibodies of the T15 idiotype are optimally protective against Streptococcus pneumoniae . J Exp Med 1982; 156: 1177–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Szu SC, Clarke S, Robbins JB. Protection against pneumococcal infection in mice conferred by phosphocholine‐binding antibodies: specificity of the phosphocholine binding and relation to several types. Infect Immun 1983; 39: 993–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lardenoye JH, Delsing DJ, de Vries MRet al. Accelerated atherosclerosis by placement of a perivascular cuff and a cholesterol‐rich diet in ApoE*3Leiden transgenic mice. Circ Res 2000; 87: 248–53. [DOI] [PubMed] [Google Scholar]
- 30.van Vlijmen BJ, van't Hof HB, Mol MJ, et al. Modulation of very low density lipoprotein production and clearance contributes to age‐ and gender‐ dependent hyperlipoproteinemia in apolipoprotein E3‐Leiden transgenic mice. J Clin Investig 1996; 97: 1184–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Mendelsohn ME, Karas RH. The protective effects of estrogen on the cardiovascular system. N Engl J Med 1999; 340: 1801–11. [DOI] [PubMed] [Google Scholar]
- 32.de Faire U, Su J, Hua Xet al. Low levels of IgM antibodies to phosphorylcholine predict cardiovascular disease in 60‐year old men: effects on uptake of oxidized LDL in macrophages as a potential mechanism. J Autoimmun 2010; 34: 73–9. [DOI] [PubMed] [Google Scholar]
- 33.Loeffler DA, Smith LM, Klaver ACet al. Development of antihuman IgG antibodies and hematologic deficits but not clinical abnormalities in C57BL/6 mice after repeated administration of human intravenous immunoglobulin. Comp Med 2012; 62: 31–6. [PMC free article] [PubMed] [Google Scholar]
- 34.Fülber I, Bacher M, Dodel R, Röskam S. Evaluating the murine anti‐human antibody response and assessment of general activity and cognition after treatment with human intravenous immunoglobulins in healthy adult C57/B6J mice. Eur J Inflamm 2014; 12: 489–97. [Google Scholar]
- 35.Rojko JL, Evans MG, Price SAet al. Formation, clearance, deposition, pathogenicity, and identification of biopharmaceutical‐related immune complexes: review and case studies. Toxicol Pathol 2014; 42: 725–64. [DOI] [PubMed] [Google Scholar]
- 36.Gigante B, Leander K, Vikstrom Met al. Low levels of IgM antibodies against phosphorylcholine are associated with fast carotid intima media thickness progression and cardiovascular risk in men. Atherosclerosis 2014; 236: 394–9. [DOI] [PubMed] [Google Scholar]
- 37.Rahman M, Sing S, Golabkesh Zet al. IgM antibodies against malondialdehyde and phosphorylcholine are together strong protection markers for atherosclerosis in systemic lupus erythematosus: Regulation and underlying mechanisms. Clin Immunol 2016; 166–167: 27–37. [DOI] [PubMed] [Google Scholar]
- 38.Sun J, Lundström SL, Zhang Bet al. IgM antibodies against phosphorylcholine promote polarization of T regulatory cells from patients with atherosclerotic plaques, systemic lupus erythematosus and healthy donors. Atherosclerosis 2018; 268: 36–48. [DOI] [PubMed] [Google Scholar]
- 39.Yeang C, Hasanally D, Que Xet al. Reduction of myocardial ischaemia‐reperfusion injury by inactivating oxidized phospholipids. Cardiovasc Res 2019; 115: 179–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Beck A, Wurch T, Bailly C, Corvaia N. Strategies and challenges for the next generation of therapeutic antibodies. Nat Rev Immunol 2010; 10: 345–52. [DOI] [PubMed] [Google Scholar]
- 41.Tabas I, Bornfeldt KE. Macrophage phenotype and function in different stages of atherosclerosis. Circ Res 2016; 118: 653–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thiagarajan D, Fiskesund R, Frostegård Aet al. Immunoglobulin G1 antibodies against phosphorylcholine are associated with protection in systemic lupus erythematosus and atherosclerosis: potential underlying mechanisms. ACR Open Rheumatol 2020; 2: 344–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jukema JW, Verschuren JJ, Ahmed TA, Quax PH. Restenosis after PCI. Part 1: pathophysiology and risk factors. Nat Rev Cardiol 2012; 9: 53–62. [DOI] [PubMed] [Google Scholar]
- 44.de Vries MR, Simons KH, Jukema JW, Braun J, Quax PH. Vein graft failure: from pathophysiology to clinical outcomes. Nat Rev Cardiol 2016; 13: 451–70. [DOI] [PubMed] [Google Scholar]
- 45.Ståhle M, Silvola JMU, Hellberg Set al. Therapeutic antibody against phosphorylcholine preserves coronary function and attenuates vascular (18)F‐FDG uptake in atherosclerotic mice. JACC Basic Transl Sci 2020; 5: 360–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Pettersson K, Atsma DE, Jukema JW. Phosphorylcholine Antibodies Preserve Cardiac Function and Reduce Infarct Size by Attenuating the Post‐Ischemic Inflammatory Response. JACC: Basic to Translational Science 2020; 5: 1228–39. 10.1016/j.jacbts.2020.09.012 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Table S1 and Figures S1–S5.
Table S1. Study characteristics.
Figure S1. Chimeric PC‐mAb(T15) prevents vascular inflammation.
Figure S2. Chimeric PC‐mAb(T15) prevents accelerated atherosclerotic lesion formation.
Figure S3. Development and screening of monoclonal human anti‐PC IgG antibodies.
Figure S4. In vivo validation of anti‐PC antibodies.
Figure S5. ATH3G10 prevents accelerated atherosclerosis.
Appendix S2. Extended version of the material and methods.