

Abstract
Aims
Clofarabine has recently been evaluated as part of the conditioning regimen for allogeneic hematopoietic stem cell transplantation (HCT) in children. Pharmacokinetic (PK) exposure of different agents commonly used in conditioning regimens is strongly related to HCT outcome. Consequently, the PK of clofarabine may be important for outcome. This report describes the population PK of clofarabine in paediatric patients and one adult.
Methods
From 80 paediatric (0.5–18 years) and 1 adult patient (37 years), 805 plasma concentrations were included in pharmacokinetic analyses using nonlinear mixed effects modelling.
Results
A two‐compartment model adequately described the PK of clofarabine. Body weight and estimated glomerular filtration rate (eGFR) were included as covariates. Clearance was differentiated into nonrenal and renal clearance (approximately 55% of total clearance), resulting in population estimates of 24.0 L/h (95% confidence interval [CI] 13.7–34.4) and 29.8 L/h (95% CI 23.9–36.1) for a patient of 70 kg with normal renal function, respectively. Unexplained interindividual variability in clearance was 17.8% (95% CI 14.6–22.4). A high variability in exposure was observed (range area under the curveT0‐inf 1.8–6.0 mg/L*h) after body surface area (BSA) based dosing. Interestingly, children with low body weight had a lower exposure than children with a higher body weight, which indicates that the currently practised BSA‐based dosing is not adequate for clofarabine.
Conclusion
A clofarabine dosing algorithm based on this PK model, using body weight and eGFR, results in a more predictable exposure than BSA‐based dosing. However, the exact target exposure needs to be further investigated.
Keywords: allogeneic hematopoietic cell transplantation, clinical pharmacology, clofarabine, paediatrics, pharmacokinetics
What is already known about this subject
Clofarabine has recently been added to the conditioning regimen for allogeneic hematopoietic stem cell transplantation (HCT) in paediatric haematological malignancies.
Clofarabine is primarily renally cleared.
Body weight has been shown to be the best predictor of clofarabine clearance in children, but at present clofarabine is still dosed based on body surface area (BSA).
What this study adds
Body weight and estimated glomerular filtration rate (eGFR) appeared as important covariates influencing clofarabine clearance.
A high variability in exposure was observed after BSA‐based dosing, and children with low body weight appeared to have a lower exposure than children with a higher body weight.
A clofarabine dosing algorithm, using body weight and eGFR, was developed, which results in a more predictable exposure than BSA‐based dosing.
1. INTRODUCTION
Clofarabine, a purine nucleoside analogue with anti‐tumour activity, is approved for the treatment of children (<21 years) with relapsed or refractory acute lymphoblastic leukaemia (ALL).1 In addition, clofarabine was recently added to the conditioning regimen for allogeneic hematopoietic stem cell transplantation (HCT) in paediatric haematological malignancies for its capacity to enhance the antileukaemic effect in combination with busulfan and fludarabine (BuFlu).2 These studies concluded that this strategy is safe and promising in high risk (myeloid) leukaemia.
Clofarabine is a prodrug, metabolized intracellularly by phosphorylation to the active metabolite clofarabine‐5′‐triphosphate. Clofarabine‐5′‐triphosphate decreases cell replication and DNA repair leading to cell death. Unchanged clofarabine is mainly renally cleared; approximately 60% is excreted with urine.1 The half‐life of clofarabine is approximately 5 hours, while the half‐life of clofarabine‐5′‐triphosphate is around 24 hours.1
The pharmacokinetics (PK) of clofarabine in children and adults has been studied previously.3, 4, 5 In patients with haematologic malignancies and solid tumours, the clofarabine exposure increased with decreasing estimated glomerular filtration rate (eGFR), and a dose adjustment in case of moderate (eGFR 30–60 mL/min/1.73 m2) and severe (eGFR <30 mL/min/1.73 m2) renal impairment is suggested.4 Recently, a population PK model of clofarabine used in conditioning regimens for HCT in children has been developed.5 No effect of renal function on clofarabine clearance was seen. However, only patients with normal renal function (range eGFR 96–150 mL/min/1.73 m2) were included, so the effect of impaired renal function could not be studied. Additionally, the previous population PK models of clofarabine all found that body weight was the best predictor of clofarabine clearance.3, 4, 5 Taken together, this would indicate that dosing based on weight and renal function would lead to the best predictable exposure. However, at present clofarabine is still dosed based on body surface area (BSA).
Previous work on the PK of busulfan and fludarabine used in conditioning regimens for HCT in paediatric and adult patients showed that optimal individual exposure of both agents is needed to prevent graft failure and relapse and that overexposure leads to an increase in toxicity and delayed immune reconstitution.6, 7 The same is to be expected for clofarabine. More knowledge on the PK of clofarabine used in conditioning regimens of HCT in children is needed to investigate whether the exposure relates to clinical outcome, which parameters predict the clofarabine exposure, and how to adjust the dose to achieve adequate exposure.
The aim of this study was to describe the population PK of clofarabine, using a large heterogeneous dataset of paediatric patients, in order to optimize the dosing regimen for clofarabine during conditioning prior to allogeneic hematopoietic cell transplantation in children.
2. METHODS
2.1. Patients and sampling
A retrospective PK analysis was performed with data from patients who received myeloablative conditioning before HCT, between October 2011 and January 2019, at the University Medical Centre Utrecht (UMCU) and the Princess Máxima Center for Pediatric Oncology in the Netherlands, and from whom PK samples were available. No restrictions were applied for comorbidities, age and indication for HCT. Patients were included after written informed consent was acquired. Ethical approval by the institutional Medical Ethics Committee of the UMCU was obtained under protocol number 11/063.
The conditioning regimen consisted of 4 days of chemotherapy (administered from Day −5 to Day −2 relative to HCT). Patients were treated with a 1 hour infusion of clofarabine directly followed by a 1 hour infusion of fludarabine and a 3 hour infusion of busulfan. In the unrelated donor HCT setting, rabbit ATG was added: 4 hour infusions on four consecutive days from Day −9 to Day −6 relative to HCT (10 mg/kg < 30 kg; 7.5 mg/kg > 30 kg) until 2015. After that patients received lymphocyte count‐ and weight‐based dosing of ATG from Day −9 with a maximum of 10 mg/kg in 4 days.8 Patients received a cumulative dose of 120 mg/m2 clofarabine. Fludarabine was given intravenously in a cumulative dose of 40 mg/m2 and busulfan was targeted to a myeloablative cumulative 4‐day exposure of 90 mg*h/L (expressed as area under the curve for all doses [AUCT0−inf]). For patients receiving ATG, clemastine, paracetamol and 2 mg/kg prednisolone (to a maximum of 100 mg) were administered intravenously prior to ATG infusion.
Plasma concentrations of clofarabine were determined in PK samples taken for routine busulfan therapeutic drug monitoring (TDM). According to the local TDM protocol, plasma samples were drawn on the first or second day of the conditioning regimen. If considered necessary for busulfan TDM purposes, samples were also drawn on the following days. Additional samples were taken on the final day of conditioning (Day 4). In general, plasma samples were taken at 5, 6, 7 and 8 hours, after the end of the clofarabine infusion. For a subset of patients, additional samples were collected from 8 to 24 hours post‐infusion. From January 2016 onwards, additional samples were collected between the end of the fludarabine infusion and the start of the busulfan infusion, which equals approximately 1.5 hours after the end of the clofarabine infusion. Clofarabine concentrations were measured using a validated liquid chromatography mass spectrometry method, with a lower limit of quantification (LLOQ) of 1 ng/mL, as described by Punt et al.9
2.2. Model development
Starting point for model development was a two‐compartment model with first order elimination consisting of a renal and non‐renal fraction.
Interindividual variability (IIV) was evaluated for all parameters, according to Equation 1:
| (1) |
where P i represents the individual parameter estimate for individual i, P pop represents the typical population parameters estimate, and η i is assumed to be normally distributed with a mean of zero and a variance of ω 2.
Since data for multiple days of therapy was available, interoccasion variability (IOV) was implemented similarly as IIV, with each dose and subsequent sampling defined as a separate occasion. This variability was evaluated for all parameters to diagnose potential time‐dependent trends and to allow for random unaccounted variability between dosing moments.
Residual unexplained variability was evaluated as a proportional error model or as a combination of a proportional and additive error model.
2.3. Covariate analysis
Following structural model development, the influence of patient‐specific factors for variability in PK parameters were evaluated. Assessed covariates included body weight (BW), body surface area (BSA), fat free mass (FFM), age and renal function. These continuous covariates were evaluated using both a linear function and a power function. To implement body size descriptors on PK parameters, standard allometric scaling was applied, with p fixed at 0.75 (BW, FFM) or 1 (BSA) for clearances, and 1 for distribution volumes (BW, BSA, FFM).10
Renal function was evaluated as a covariate, since clofarabine is partly eliminated renally.1 As creatinine levels were not measured daily, the most recent values of creatinine prior to infusion (maximum 10 days) were used. Subsequently, eGFR was calculated using the Cockcroft–Gault equation, which takes age into account.11 eGFR for patients below the age of 17 years for women and 14 years for men was calculated using the Schwartz equation.12 eGFR was capped to a maximum of 8.4 L/h/1.73 m2 (140 mL/min/1.73 m2) and was assumed to increase to this level from birth until the age of 1.5 years, starting at 2.1 L/h/1.73 m2 (35 mL/min/1.73 m2) (25% of maximum value). The absolute eGFR (in L/h) was standardized to 70 kg as shown in the equation. Relative renal function (RF) was normalized to a standard eGFR (eGFR STD) of 6 L/h (100 mL/min):
| (2) |
where eGFR is the absolute estimated glomerular filtration rate in L/h, BW is body weight in kg and eGFR STD is a standard eGFR (6 L/h was used in this model).
RF was included in the model, using a linear independent combination of renal and non‐renal CL parameters:
| (3) |
where CL overall is the overall population value of parameter for clearance (CL). CL non‐renal is non‐renal CL and CL renal is renal CL.13, 14
Because the dataset contained several infants, the effect of maturation on CL was implemented using the method described by Rhodin et al.15 They showed that maturation of renal clearance across the entire paediatric population was well described using postmenstrual age (PMA) with a sigmoidal Hill equation. The TM50, the PMA at which clearance is 50% of the mature value, was estimated at 55.4 weeks and the Hill coefficient describing the slope of the sigmoidal curve at 3.33.15 For our population, exact PMA was not known, so the PMA was estimated using age in weeks plus mean gestational age (40 weeks):
| (4) |
The Hill coefficient and TM50 were fixed to 3.92 and 54.2 weeks, respectively, according to published models.16
2.4. Model evaluation
Discrimination between models was guided by physiological plausibility, goodness‐of‐fit (GOF) plots, precision of parameter estimates and change in objective function value (dOFV). A drop of ≥3.84 points, corresponding to a P < 0.05 (χ2‐distribution with 1 degree of freedom [df]), was considered a significant improvement of the fit for hierarchical nested models. The adequacy of the models was assessed by GOF plots and visual predictive checks (VPC).17 Parameter precision was assessed by the sampling importance resampling (SIR) procedure.18
2.5. Software
Nonlinear mixed‐effects modelling was performed using NONMEM (version 7.3.0, ICON development Solutions, Ellicott City, MD, USA) and Pearl‐speaks‐NONMEM (PsN, version 4.7.0) with First‐Order Conditional Estimation with interaction (FOCE‐I) as estimation method.19, 20 Pirana (version 2.9.9) was used as graphical user interface for NONMEM.21 R (version 3.4.3) was used for data handling and visualization.22
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.23
3. RESULTS
3.1. Patients and sampling
A total of 81 patients with a median age of 11.1 years (range 0.5–37.8) were included in this study. Five patients were younger than 12 months and one adult (37 years) was included. Of these patients, 805 PK samples were available for analysis. None of these samples was below the lower limit of quantification. Figure 1 displays the observed plasma concentrations over time. Detailed patient characteristics are shown in Table 1.
FIGURE 1.
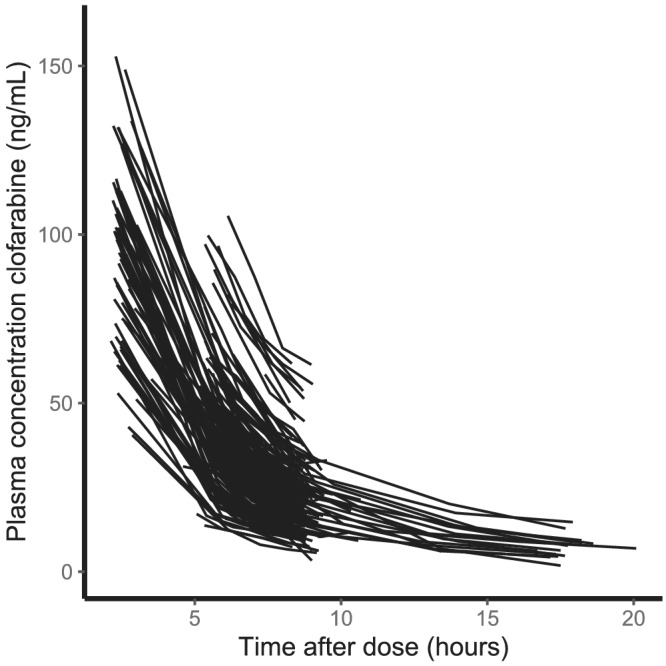
Clofarabine plasma concentrations vs time after dose. Each line corresponds to a single dose
TABLE 1.
Patient characteristics (n = 81)
| Median [range] | |
|---|---|
| Available data | |
| Total no. of PK samples [n] | 805 |
| No. of samples per patient | 10 [3–20] |
| Patient characteristics | |
| Female sex [n (%)] | 30 (37%) |
| Age at transplantation, years | 11.1 [0.5–37.8a, IQR 5.5–14.8] |
| Actual bodyweight, kg | 36.6 [6.6–102.9, IQR 20.1–53.5] |
| Renal function, mL/min/1.73 m2 | 140 [69.3–140, IQR 123.1–140] |
| Indication for transplantation [n (%)] | |
| ALL | 40 (49%) |
| AML | 28 (35%) |
| CML | 2 (2%) |
| Myelodysplastic syndrome | 8 (10%) |
| Other | 3 (4%) |
| Transplant cell source [n (%)] | |
| Cord blood | 46 (57%) |
| Bone marrow | 35 (43%) |
IQR, interquartile range; ALL, acute lymphoblastic leukaemia; AML, acute myeloid leukaemia; CML, chronic myeloid leukaemia
The population consisted of 80 paediatric patients, aged 0.5–18 years, and one adult patient of 37.8 years.
3.2. Model development
A linear two‐compartment model with first order kinetics was appropriate to describe the PK of clofarabine. Final estimates and 95% confidence intervals (CI) are shown in Table 2. The model was parameterized in terms of volume of distribution of the central (V1) and peripheral (V2) compartment, clearance from the central compartment (CL) and intercompartment clearance between V1 and V2 (Q). BW was a priori included as covariate using allometric scaling on all PK parameters. The exponents for BW on clearance and volume of distribution were fixed to 0.75 and 1, respectively, prior to covariate analyses.
TABLE 2.
Final population PK parameter estimates
| PK parameter | Estimate | 95% CI | |
|---|---|---|---|
|
| |||
|
| |||
| CLnon − renal,70kg (L/h) | 24.0 | 13.7–34.4 | |
| CLrenal,70kg (L/h) | 29.8 | 23.9–36.1 | |
|
| |||
| V170kg (L) | 268 | 234.8–296.6 | |
|
| |||
| V270kg (L) | 186 | 165.4–210.7 | |
|
| |||
| Q70kg (L/h) | 33.2 | 27.5–40.9 | |
| IIV CL (%) | 17.8 | 14.6–22.4 | |
| IIV V1 (%) | 12.6 | 6.8–18.1 | |
| IIV Q (%) | 64.5 | 49.5–83.7 | |
| IOV CL (%) | 9.7 | 7.8–11.5 | |
| IOV V2 (%) | 39.1 | 29.2–53.7 | |
| Proportional residual error (%) | 8.3 | 7.7–8.8 | |
PK, pharmacokinetics; CI, confidence interval obtained by sampling importance resampling; CL, clearance; RF, relative renal function; BW, bodyweight; V1, volume of distribution of the central compartment; V2, volume of distribution of the peripheral compartment; Q, intercompartment clearance between V1 and V2; IIV, interindividual variability; IOV, interoccasion variability.
Population estimates CLrenal,70kg, CLnon‐renal,70kg, V170kg, V270kg, Q70kg correspond to a subject weighing 70 kg and are adjusted to an individual value, according to the corresponding parameter formula in the table.
IIV was added on CL, V1 and Q. Inclusion of IOV on CL and V2 led to a significant improvement in model fit.
3.3. Covariate selection
BSA and FFM were evaluated as metrics for body size, but did not improve the model fit over BW
Renal function was evaluated as covariate on CL. Renal clearance was differentiated from non‐renal clearance by adding an extra parameter for renal clearance, which was normalized to a standard eGFR. Adding renal function resulted in a significant improvement in model fit, with a drop of 34 points in OFV (P < 0.05). The effect of maturation on CL was tested using the method described by Rhodin et al.15 This did not result in a better fit of the model, so maturation was not included in the final model. In the final model CL renal was estimated at 29.8 L/h for a typical patient, which corresponds to 55% of the total clearance in patients with normal renal function. The calculated alpha and beta half‐life (t ½α and t ½β) were 1.7 h and 8.1 h, respectively.
Including BW and eGFR in the model caused a decline in IIV CL from 46.2% to 17.8%
Figure 2A depicts the variability in total exposure (observed AUCT0–inf). As shown in Figure 1, plasma concentrations over time after dose were highly variable, leading to a wide range of observed AUCT0–inf (1.8–6.0 mg/L*h). Figure 2B and C show the exposure at different weight and renal function categories. Low BW seems to be correlated to low exposures, indicating that BSA‐based dosing does not sufficiently account for variability. As expected, patients with a creatinine clearance below 80 mL/min/1.73 m2 have a higher exposure than patients with a better renal function. The renal function of the patients weighing less than 20 kg varied from 70 to 140 mL/min/1.73 m2, 14 patients (70%) had a creatinine clearance >120 mL/min/1.73 m2. This shows that the lower exposure is partly, but not totally, explained by a good renal function throughout this subgroup.
FIGURE 2.
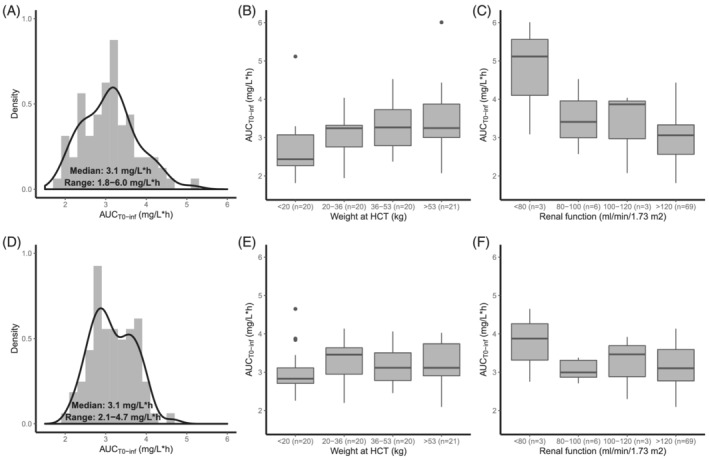
Exposure variability after dosing in the trial (A, B, C) and after dosing with suggested dosing algorithm (D, E, F). (A) Histogram (grey area) and density plot (black solid line) of the observed AUCT0–inf. (B) Boxplots of the observed AUCT0–inf per body weight quartile. (C) Boxplots of the observed AUCT0–inf per renal function category. (D) Histogram (grey area) and density plot (black solid line) of the calculated AUCT0–inf. (E) Boxplots of the calculated AUCT0–inf per body weight quartile. (F) Boxplots of the calculated AUCT0–inf per renal function category
3.4. Model evaluation
The GOF plots (Supplementary Figure S1A and B) showed accurate population and individual predictions, without any signs for over‐ or underprediction. CWRES are evenly distributed over the whole plasma concentration range (Supplementary Figure S1C) and time interval (Supplementary Figure S1D). No trends were observed for CWRES vs. renal function (Supplementary Figure S1E) or actual body weight (Supplementary Figure S1F).
The VPC demonstrated that the median and the 95% CI of the observed data were in line with those from the simulation‐based predictions from the model for all age and BW strata (Figure 3), except for the early time points, where the median and the 95% CI of the observations were slightly lower than the predictions, indicating underprediction. However, only 61 samples (7.6%) with a time after dose <4 h were included in the model.
FIGURE 3.
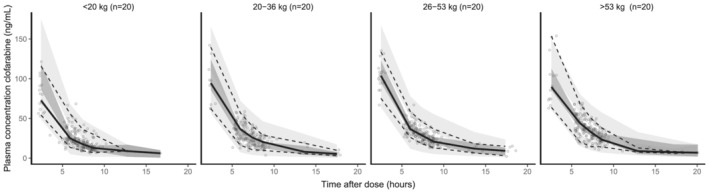
Body weight stratified prediction‐corrected visual predictive check. Black lines depict the observed median (solid) and 2.5% and 97.5% percentile (dashed) concentrations. Dark‐ and light‐grey areas represent 95% prediction intervals of the simulated mean and the 2.5 and 97.5% percentiles, respectively. Round dots represent observations
3.5. Dosing regimen
As mentioned before, previous publications showed that exposure of busulfan and fludarabine used in conditioning regimens for HCT relates to clinical outcome.6, 7 Target AUCs for these agents have been established. Even though a target AUC for clofarabine has not yet been described, a dose algorithm could be extracted from this PK model:
| (5) |
where Dose is the cumulative clofarabine dose for four days in mg, AUC Target is the cumulative target AUC, eGFR is the absolute estimated glomerular filtration rate in L/h and BW is body weight in kg.
Using this algorithm and the median AUC of 3.1 mg/L*h, a new dose was calculated for each patient. Following this, the exposure using this new dose was calculated based on the individual estimated CL values of our patients. Figure 2D depicts the variability in total exposure under the new dosing regimen (calculated AUCT0–inf). This figure shows that the range of the calculated AUCT0–inf was smaller than with BSA‐based dosing (2.1–4.7 mg/L*h resp. 1.8–6.0 mg/L*h). Figure 2E and F show the calculated exposure at different weight and renal function categories. In Figure 4, a line plot of the 4 day cumulative clofarabine dose as a function of the body weight for several relative renal function values and an AUC target of 3.1 mg/L*h is presented.
FIGURE 4.
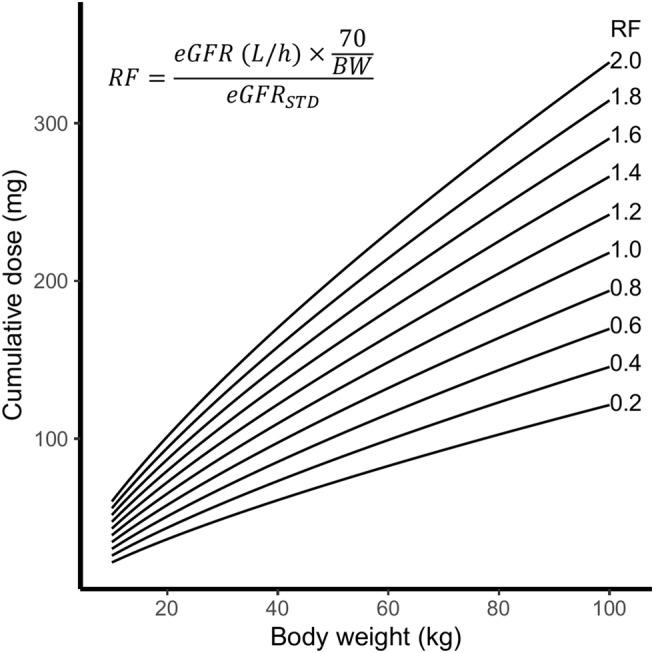
Line plot of the 4‐day cumulative clofarabine dose as a function of the body weight (kg) for several relative renal function values (RF)
4. DISCUSSION
In this analysis, a population PK model was developed, using a large and diverse dataset including paediatric patients from the age of 0.5 years with a variety of haematological diagnoses requiring HCT. A two‐compartment model was appropriate to describe the clofarabine PK in these patients, which is in line with previous published clofarabine PK models.3, 4, 5
Two covariates were identified as predictor for clofarabine CL. Body weight was included using allometric scaling, which is in line with the previous described PK models. In addition, renal function was included as covariate, which significantly improved the model fit and reduced the IIV CL significantly (from 46.2% to 17.8%). Bonate et al. also found a significant improvement in the model after including renal function as covariate, while Wang et al. did not find a significant effect.4, 5
By including renal function into our model, we estimated that clofarabine is renally cleared to approximately 55% in a typical patient. In addition, we found that the exposure to clofarabine is higher in patients with a creatinine clearance below 80 mL/min/1.73 m2. This is in accordance with information that can be found in the literature,1 and is in line with the advice of Bonate et al. to reduce the dose in case of renal impairment.3 However, we did not include any patients with moderate (eGFR 30–60 mL/min/1.73 m2) or severe (eGFR <30 mL/min/1.73 m2) renal impairment. Therefore, one must be careful translating the results of this population PK model to patients with moderate or severe renal impairment. On the other hand, moderate and/or severe renal impairment is exceptional in paediatric patients undergoing HCT, so the results of this PK model might be sufficient for this specific patient population.
The median clofarabine exposure was 3.1 mg/L*h with a range of 1.8–6.0 mg/L*h. This is in accordance with the median cumulative AUC of 3.3 mg/L*h (range 1.5–5.5 mg/L*h) after a cumulative dose of 120 mg/m2 as described by Wang et al.5 We observed a decreased exposure in children with low BW (<20 kg). Accordingly, younger children also seemed to have a lower exposure than older children. Previous population PK models found that CL increases with increasing BW or increasing age (for patients <20 years).4, 5 This is in line with the results of our study, but it does not correlate with a low exposure in children with low BW or age. Bonate et al. simulated the effect of age, BW and eGFR on clofarabine exposure after BSA‐based dosing and showed that the exposure is lower in younger children than in older children with comparable eGFR.4 In contrast, Wang et al. suggested a lower dose for younger children, based on their simulation of CL values.5 Our results show, however, that reducing the dose in younger children could lead to underexposure of those patients. In contrast to the advice of Wang et al., younger children may require a higher clofarabine dose. Nevertheless, these results need to be carefully interpreted in the case of very young infants. Especially up to the age of 3 months, the metabolic capacity and renal elimination undergo substantial developmental changes.24 This population model did not include any patients below the age of 6 months, so lower doses might be needed for these children because of this maturation phase.
The results of this PK study show that renal function and body weight are two important covariates for clearance, and should, therefore, be considered as components on which to base the clofarabine dose. Clofarabine is still dosed based on BSA, while renal function is not taken into account, apart from exceptional cases of renal impairment (according to the label of clofarabine for non‐conditioning for HCT indications, a dose reduction of 50% needs to be made in patients with moderate renal impairment, while clofarabine is contraindicated in patients with severe renal impairment). The decrease in clofarabine clearance relating to renal function is a gradual process and even an eGFR below 120 mL/min/1.73 m2 is associated with a decrease in clofarabine clearance and concomitant higher exposures,4 which makes a dose algorithm, taking renal function into account, more suitable to decide which dose should be administered. This approach has been described previously for other drugs, for example carboplatin and fludarabine.25, 26, 27 Such a dosing algorithm for clofarabine was derived from this PK model (Equation 5); however, a target AUC is needed to calculate the conventional clofarabine dose. A target AUC for clofarabine during conditioning prior to HCT has not yet been determined. The relation between clofarabine exposure and clinical outcome after HCT in children needs to be studied in order to determine the target AUC, but this could be challenging since these children were treated with multiple agents (busulfan and fludarabine), all contributing to clinical outcome. Despite this, when the target AUC for clofarabine is set, the dose algorithm can be used to calculate the conventional dose for an individual patient. The median of the observed AUC was used for calculations of the new dose and exposure in this paper. These calculations illustrate that the range of the clofarabine exposure is smaller when this dosing algorithm is used, while the median exposure is similar. The number of very young patients in our population was limited and, therefore, the benefits of weight and renal function‐based dosing in this population need further investigation.
5. CONCLUSION
In summary, the results of this clofarabine population PK model, using data from the largest paediatric dataset reported to date, showed that BW and eGFR appeared as important covariates influencing the clofarabine CL. Unexplained interindividual variability in exposure clearance was observed (17.8%, 95% CI 14.6–22.4). In the included population a high variability in exposure was observed (range AUCT0–inf 1.8–6.0 mg/L*h). Interestingly, children with low BW have a lower exposure than children with a higher BW, which indicates that BSA‐based dosing is not adequate for clofarabine. Younger children may require higher doses than older children. A dosing algorithm based on this PK model, using BW and eGFR, was developed, which would result in a more predictable exposure than BSA‐ based dosing. However, the exact target exposure needs to be further investigated.
COMPETING INTERESTS
There are no conflicts of interest to declare.
CONTRIBUTORS
S.N., C.L., J.B., M.B. and B.V. were responsible for protocol development, implementation of the study and enrolling patients. Acquisition of the data was performed by all authors. Population PK modelling was performed by L.N. A.H. supervised this work. The first draft of the manuscript was written by L.N., and all the authors commented on previous versions of the manuscript. All the authors read and approved the final manuscript.
Supporting information
Figure S1. Goodness of fit plots.
ACKNOWLEDGEMENTS
This paper was written in memory of Dr. E.M. van Maarseveen. The authors would like to thank him for his expertise and work on this project. This work was partly supported by Children Cancer‐free Foundation (KiKa) project number 190.
Nijstad AL, Nierkens S, Lindemans CA, et al. Population pharmacokinetics of clofarabine for allogeneic hematopoietic cell transplantation in paediatric patients. Br J Clin Pharmacol. 2021;87:3218–3226. 10.1111/bcp.14738
Principal investigator statement: The authors confirm that the PI for this paper is Dr. C.A. Lindemans and that she had direct clinical responsibility for patients.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Genzyme Europe BV . Summary of Product Characteristics: Evoltra. 2016. https://www.ema.europa.eu/en/documents/product-information/evoltra-epar-product-information_en.pdf. Accessed January 18, 2021.
- 2.Andersson BS, Valdez BC, de Lima M, et al. Clofarabine ± fludarabine with once daily i.v. busulfan as pretransplant conditioning therapy for advanced myeloid leukemia and MDS. Biol Blood Marrow Transplant. 2011;17(6):893‐900. 10.1016/j.bbmt.2010.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bonate PL, Craig A, Gaynon P, et al. Population pharmacokinetics of clofarabine, a second‐generation nucleoside analog, in pediatric patients with acute leukemia. J Clin Pharmacol. 2004;44(11):1309‐1322. 10.1177/0091270004269236 [DOI] [PubMed] [Google Scholar]
- 4.Bonate PL, Cunningham CC, Gaynon P, et al. Population pharmacokinetics of clofarabine and its metabolite 6‐ketoclofarabine in adult and pediatric patients with cancer. Cancer Chemother Pharmacol. 2011;67(4):875‐890. 10.1007/s00280-010-1376-z [DOI] [PubMed] [Google Scholar]
- 5.Wang H, Jones AK, Dvorak CC, et al. Population pharmacokinetics of clofarabine as part of pretransplantation conditioning in pediatric subjects before hematopoietic cell transplantation. Biol Blood Marrow Transplant. 2019;25(8):1603‐1610. 10.1016/j.bbmt.2019.04.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bartelink IH, Lalmohamed A, van Reij EML, et al. Association of busulfan exposure with survival and toxicity after haemopoietic cell transplantation in children and young adults: a multicentre, retrospective cohort analysis. Lancet Haematol. 2016;3(11):e526‐e536. 10.1016/S2352-3026(16)30114-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Langenhorst JB, Van Kesteren C, Van Maarseveen EM, et al. Fludarabine exposure in the conditioning prior to allogeneic hematopoietic cell transplantation predicts outcomes. Blood Adv. 2019;3(14):2179‐2187. 10.1182/bloodadvances.2018029421 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Admiraal R, van Kesteren C, Jol‐van der Zijde CM, et al. Population pharmacokinetic modeling of thymoglobulin® in children receiving allogeneic‐hematopoietic cell transplantation: towards improved survival through individualized dosing. Clin Pharmacokinet. 2015;54(4):435‐446. 10.1007/s40262-014-0214-6 [DOI] [PubMed] [Google Scholar]
- 9.Punt AM, Langenhorst JB, Egas AC, Boelens JJ, van Kesteren C, van Maarseveen EM. Simultaneous quantification of busulfan, clofarabine and F‐ARA‐A using isotope labelled standards and standard addition in plasma by LC–MS/MS for exposure monitoring in hematopoietic cell transplantation conditioning. J Chromatogr B Analyt Technol Biomed Life Sci. 2017;1055–1056(D):81‐85. 10.1016/j.jchromb.2017.04.025 [DOI] [PubMed] [Google Scholar]
- 10.West GB, Brown JH, Enquist BJ. A general model for the origin of allometric scaling laws in biology. Science. 1997;276(5309):122‐126. 10.1126/science.276.5309.122 [DOI] [PubMed] [Google Scholar]
- 11.Cockcroft DW, Gault MH. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16(1):31‐41. 10.1159/000180580 [DOI] [PubMed] [Google Scholar]
- 12.Schwartz GJ, Haycock GB, Edelmann CM, Spitzer A. A simple estimate of glomerular filtration rate in children derived from body length and plasma creatinine. Pediatrics. 1976;58(2):259‐263. 10.1055/s-2004-830943 [DOI] [PubMed] [Google Scholar]
- 13.Mould D. Population pharmacokinetic and adverse event analysis of topotecan in patients with solid tumors. Clin Pharmacol Ther. 2002;71(5):334‐348. 10.1067/mcp.2002.123553 [DOI] [PubMed] [Google Scholar]
- 14.Matthews I, Kirkpatrick C, Holford N. Quantitative justification for target concentration intervention—parameter variability and predictive performance using population pharmacokinetic models for aminoglycosides. Br J Clin Pharmacol. 2004;58(1):8‐19. 10.1111/j.1365-2125.2004.02114.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Rhodin MM, Anderson BJ, Peters AM, et al. Human renal function maturation: a quantitative description using weight and postmenstrual age. Pediatr Nephrol. 2009;24(1):67‐76. 10.1007/s00467-008-0997-5 [DOI] [PubMed] [Google Scholar]
- 16.Knøsgaard KR, Foster DJR, Kreilgaard M, Sverrisdóttir E, Upton RN, van den Anker JN. Pharmacokinetic models of morphine and its metabolites in neonates: systematic comparisons of models from the literature, and development of a new meta‐model. Eur J Pharm Sci. 2016;92:117‐130. 10.1016/j.ejps.2016.06.026 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bergstrand M, Hooker AC, Wallin JE, Karlsson MO. Prediction‐corrected visual predictive checks for diagnosing nonlinear mixed‐effects models. AAPS J. 2011;13(2):143‐151. 10.1208/s12248-011-9255-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Dosne AG, Bergstrand M, Karlsson MO. An automated sampling importance resampling procedure for estimating parameter uncertainty. J Pharmacokinet Pharmacodyn. 2017;44(6):509‐520. 10.1007/s10928-017-9542-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Lindbom L, Ribbing J, Jonsson EN. Perl‐speaks‐NONMEM (PsN)—a Perl module for NONMEM related programming. Comput Methods Programs Biomed. 2004;75(2):85‐94. 10.1016/j.cmpb.2003.11.003 [DOI] [PubMed] [Google Scholar]
- 20.Boeckmann AJ, Sheiner LB, Beal SL. NONMEM User Guide. NONMEM Part V. San Francisco, CA: University of California; 2011. [Google Scholar]
- 21.Keizer RJ, van Benten M, Beijnen JH, Schellens JHM, Huitema ADR. Piraña and PCluster: a modeling environment and cluster infrastructure for NONMEM. Comput Methods Programs Biomed. 2011;101(1):72‐79. 10.1016/j.cmpb.2010.04.018 [DOI] [PubMed] [Google Scholar]
- 22.R Core Team . R: a language and environment for statistical computing. Vienna: R Foundation for Statistical Computing; 2009. [Google Scholar]
- 23.Alexander SPH, Kelly E, Mathie A, et al. The Concise Guide to PHARMACOLOGY 2019/20. Br J Pharmacol. 2019;176(S1):S1‐S493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kearns GL, Abdel‐Rahman SM, Alander SW, Blowey DL, Leeder JS, Kauffman RE. Developmental pharmacology—drug disposition, action, and therapy in infants and children. N Engl J Med. 2003;349(12):1157‐1167. [DOI] [PubMed] [Google Scholar]
- 25.Calvert AH, Newell DR, Gumbrell LA, et al. Carboplatin dosage: prospective evaluation of a simple formula based on renal function. J Clin Oncol. 1989;7(11):1748‐1756. 10.1200/JCO.1989.7.11.1748 [DOI] [PubMed] [Google Scholar]
- 26.Langenhorst JB, Dorlo TPC, van Maarseveen EM, et al. Population pharmacokinetics of fludarabine in children and adults during conditioning prior to allogeneic hematopoietic cell transplantation. Clin Pharmacokinet. 2019;58(5):627‐637. 10.1007/s40262-018-0715-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Langenhorst JB, Dorlo TPC, van Kesteren C, et al. Clinical trial simulation to optimize trial design for fludarabine dosing strategies in allogeneic hematopoietic cell transplantation. CPT Pharmacometrics Syst Pharmacol. 2020;9(5):272‐281. 10.1002/psp4.12486 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Goodness of fit plots.
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.