

Abstract
Aims
The objectives of this study were to investigate the safety, tolerability, pharmacokinetics and pharmacodynamics of single and multiple oral doses of ONO‐7684, a novel activated factor XI (FXIa) inhibitor, in healthy subjects.
Methods
This was a first‐in‐human (FIH), randomised, placebo‐controlled, double‐blind, single and multiple dose study in healthy subjects under fed and fasted conditions. This study consisted of two parts: single ascending dose (Part A; 1, 5, 20, 80, 150 or 300 mg ONO‐7684 or placebo) and multiple ascending doses (Part B; 80, 150 or 250 mg ONO‐7684 or placebo daily for 14 days). In both parts, subjects were randomised in a 3:1 ratio to receive ONO‐7684 or placebo.
Results
ONO‐7684 was well tolerated at all dose levels tested following both single and repeated doses, with a low overall incidence of treatment‐emergent adverse events. There was no evidence to suggest a bleeding risk. Dose proportionality in exposure was observed for the range of 1–300 mg ONO‐7684 in Part A. In Part A, the half‐life of ONO‐7684 administered in the fasted state ranged from 16.0 to 19.8 hours. In Part B, the half‐life of ONO‐7684 administered in the fed state ranged from 22.1 to 27.9 hours, supporting once daily oral dosing. ONO‐7684 strongly inhibited factor XI coagulation activity (FXI:C) and increased activated partial thromboplastin time (aPTT), with a mean maximum on treatment percentage inhibition versus baseline of 92% and a mean maximum on treatment ratio‐to‐baseline of 2.78, respectively, at 250 mg ONO‐7684 daily.
Conclusions
The data generated in this FIH study demonstrate the promising potential of oral FXIa inhibition and ONO‐7684 for indications requiring anticoagulation.
Keywords: anticoagulants, pharmacodynamics, pharmacokinetics
What is already known about this subject
Thromboembolic conditions are a leading cause of death and disability with direct oral anticoagulants (DOACs) currently being used for the prevention and treatment of these conditions.
Although effective, DOACs still carry a risk of bleeding.
Based on the lack of major bleeding in humans with severe FXI deficiency and preclinical data, FXIa inhibition using an oral agent (ONO‐7684) was investigated as a novel mechanism of anticoagulation therapy.
What this study adds
This study investigated the safety, tolerability, pharmacokinetics and pharmacodynamics of single and multiple oral doses of a novel agent (ONO‐7684) in healthy subjects.
The first clinical data generated for ONO 7684 show that the drug was well tolerated, with an encouraging pharmacodynamic profile and no evidence of a bleeding risk.
The data demonstrate the promising potential of FXIa inhibition and ONO‐7684 as an oral agent for anticoagulation therapy and this study will inform future clinical trials with ONO‐7684 and other agents in this class.
1. INTRODUCTION
Thromboembolic conditions, including deep‐vein thrombosis, pulmonary embolism, acute coronary syndrome, and ischaemic stroke are a leading cause of death and disability.1 Oral anticoagulants are widely used for long‐term prevention and treatment of venous and arterial thromboembolism. Traditionally, vitamin K antagonists such as warfarin have been the only available oral anticoagulants. More recently, direct oral anticoagulants (DOACs), such as dabigatran, rivaroxaban, apixaban and edoxaban, have been approved for treatment and prevention of venous thromboembolism.2 The currently approved DOACs target factor Xa or thrombin within the coagulation cascade (Supplementary Figure S1).
DOACs were designed to overcome the limitations of warfarin, mainly bleeding risk. Haemorrhagic bleeding into the brain is a serious complication of anticoagulation therapy. Although the DOACs come closer to the goal of bleeding reduction than vitamin K antagonists, bleeding is still a major risk with DOACs.3 Therefore, there is an unmet medical need for new oral anticoagulants that offer effective and predictable anticoagulation without increased risk of adverse events, such as bleeding.
Activated factor XI (FXIa) is a serine protease involved in the amplification of thrombin production.4 It is a component of the intrinsic pathway of the coagulation cascade and has been postulated to play an important role in thrombosis but only a minor role in haemostasis, unlike thrombin and FXa. Whereas elevated FXI levels have been associated with an increased risk of thrombosis, FXI deficiency may be associated with a lower risk of cardiovascular events and venous thromboembolism.5 The attractiveness of FXIa as a therapeutic target for anticoagulation is further supported by the lack of significant bleeding associated with FXIa modulation. Spontaneous bleeding in humans with a deficiency in FXI (haemophilia C) is generally known to be rare.6 In addition, cases of haemorrhage following trauma or surgical procedures have been reported at a lower degree and frequency in FXI‐deficient patients compared to patients with other types of coagulation factor deficiency.7 This suggests that inhibition of FXIa poses a reduced risk of bleeding. Several different experimental animal models have also demonstrated robust antithrombotic efficacy with a direct FXIa inhibitor without significant bleeding liability.8 Furthermore, a number of candidate compounds targeting FXIa have been evaluated to date including novel FXI antibodies (Abelacimab [MAA868],9 Osocimab [BAY1213790],10, 11 Xisomab12 [AB203], a FXI‐antisense oligonucleotide,13 parentally administered small molecule FXIa inhibitors [BMS‐262084 and BMS‐654457]14 and orally administered small molecule FXIa inhibitors [JNJ‐70033093 (BMS‐986177)15 and BAY 243333416]). Data with these compounds has shown the promising potential of FXIa as a therapeutic target; however, none have progressed beyond development stage.
ONO‐7684 is a novel orally administered small molecule FXIa inhibitor that inhibits human FXIa in a competitive and reversible manner (Ki = 0.002 μmol/L).17 During preclinical evaluation, ONO‐7684 did not prolong bleeding time in a monkey nail‐cut bleeding model and also significantly inhibited thrombus formation in a monkey arteriovenous shunt model.17 This preclinical pharmacology data combined with the lack of major bleeding in humans with severe FXI deficiency provide a strong rationale for investigating the inhibition of FXIa as a novel mechanism of anticoagulation therapy. Thus, the primary objective of the study reported here was to investigate the safety and tolerability of single and multiple oral doses of ONO‐7684 in healthy subjects.
2. METHODS
This study (EudraCT number: 2018–003160‐32; NCT03919890) was performed in compliance with the ethical principles founded in the Declaration of Helsinki, Good Clinical Practice guidelines, the European Union Directives on Clinical Trials (2001/20/EC and 2005/28/EC), and local laws and regulations. The original protocol, protocol amendments, informed consent forms and the Investigator's Brochure were reviewed and approved by an appropriate ethics committee. All subjects provided written informed consent.
2.1. Study design
This was a first‐in‐human (FIH), randomised, placebo‐controlled, double‐blind, single and multiple dose study conducted at a single site (Hammersmith Medicines Research [HMR], London, UK) in healthy subjects under fed and fasted conditions. The study comprised two parts: single ascending dose (Part A) and multiple ascending doses (Part B). In both parts, for each cohort, subjects were randomised according to a schedule prepared by an independent statistician using an SAS® program (SAS Institute Inc., Cary, NC, USA), in a 3:1 ratio to receive ONO‐7684 or placebo.
For Part A, six cohorts of eight subjects were enrolled. Each subject received a single dose of ONO‐7684 (1, 5, 20, 80, 150, or 300 mg) or placebo by mouth, following an overnight fast of at least 8 hours. One cohort (80 mg ONO‐7684) completed a second study session, during which they took another single dose of ONO‐7684 or placebo after an FDA high‐fat breakfast. There was an interval of at least 7 days between dosing cohorts, or between fasted and fed doses of the same cohort, to allow for detailed review of available safety, pharmacokinetic (PK) and pharmacodynamic (PD) data. As this was an FIH study, in Part A only, two sentinel subjects were dosed first, at least 23 hours before dosing the remaining subjects in the cohort. To maintain the blind, the sentinel subjects were randomised such that one subject received ONO‐7684 and one subject received placebo.
For Part B, three cohorts of eight subjects were enrolled. Each subject received once daily (QD) doses of ONO‐7684 (80, 150 or 250 mg), or placebo, in the fed state (standard meal) for 14 days. Subjects were dosed in the fed state only in Part B following review of food effect data from Part A suggesting a preferential PK profile in the fed state (i.e. blunted peak concentration but with similar overall exposure as in the fasted state). The dose was not escalated unless the safety and tolerability of the previous dose level were considered acceptable, and plasma exposures at the next dose level were not expected to exceed the exposure limits outlined in the protocol.
Active and placebo treatments could not be distinguished based on labelling, were identical in appearance, and were similar in taste and smell. To maintain the blind, the same number of capsules was given to each subject in a cohort. The Investigators (and clinical team), Sponsor, Sponsor's Medical Officer and clinical monitor remained blinded throughout the relevant part of the study, and the blind remained unbroken throughout.
The sample size was based on practical considerations, with sufficient subjects included to evaluate safety, tolerability and PK whilst exposing as few subjects as possible to the study medication and procedures.
2.2. Subjects
Subjects were healthy volunteers, aged 18 to 55 years who were registered with a general practitioner in the UK. Part A enrolled men only, while Part B enrolled men or women of non‐childbearing potential. Subjects were required to have a body mass index (BMI) of 18.0–30.0 kg/m2, be non‐smokers or ex‐smokers who had not smoked for the previous 6 months and must have agreed to use effective contraception. Subjects had to have sufficient intelligence to understand the nature of the study and any hazards of participating in it, and must have been willing to give written consent to participate and to have data entered into The Over‐Volunteering Prevention System.18
Subjects should not have been taking any strong inhibitors or inducers of cytochrome P450 3A4/5 or P‐glycoprotein or use of anticoagulants (e.g., warfarin or heparin), antiplatelet agents (e.g., clopidogrel), non‐steroidal anti‐inflammatory drugs, and/or acetylsalicylic acid during the 30 days before screening. Subjects should also not have been using any prescription medicine, over‐the‐counter medicine, vitamins, herbal treatments or dietary supplements with the exception of acetaminophen (paracetamol), during the 7 days or 5 half‐lives (whichever was longer) before the first dose of study medication.
2.3. Objectives
The primary objective of the study was to investigate the safety and tolerability of single and multiple oral doses of ONO‐7684 in healthy subjects. The secondary objectives were to assess the PK of ONO‐7684 after single and multiple oral doses, to evaluate the effect of food on the PK of a single dose, and to assess the PD effects of single and multiple doses of ONO‐7684.
2.4. Assessments
Safety assessments in both parts of the study included adverse event (AE) reporting using the Medical Dictionary for Regulatory Activities (version 22.0), clinical laboratory tests (biochemistry, haematology and urinalysis), vital signs, electrocardiograms (ECGs), physical examination and cardiac telemetry (Part A only).
Plasma samples were collected pre‐ and post‐dose in both parts of the study for PK and PD analyses. Blood samples for PK analysis were taken into heparin sodium tubes, placed on ice and centrifuged at 1500g for 10 minutes at 4°C. Plasma samples were frozen at −70°C or below within 2 hours of collection, and stored until dispatch to LGC Ltd, UK for analysis of ONO‐7684 concentration. After solid extraction, the resulting plasma samples were analysed by a fully validated liquid chromatography–mass spectrometry method. Concentrations greater than 1 ng/mL (the lower limit of quantification) were determined with a precision of ≤6.7% and an accuracy of −5.0% to 1.6% relative error.
Coagulation assays (prothrombin time [PT], international normalised ratio [INR], activated partial thromboplastin time [aPTT]) were performed on plasma samples. Factor XI coagulation activity (FXI:C) was performed on whole blood samples. Processing of samples and assaying for FXI:C, aPTT, PT and INR was done by the HMR Analytical Laboratory.
For FXI:C, a HemosIL factor XI deficient plasma kit (Instrumentation Laboratory, Bedford, MA, USA) was used according to the manufacturer's instructions. Samples were analysed using the ACL TOP 300 Coagulation analyser (Instrumentation Laboratory, Bedford, MA, USA).
Blood samples for the aPTT assay were collected in a tube containing 109 mmol/L buffered sodium citrate with a 9:1 ratio (blood to citrate). Samples were then centrifuged at 2000g for 15 minutes at room temperature. The sample was incubated with aPTT Synth ASil® reagent at 37°C, calcium chloride Synth ASil reagent was then added, and the time for clot formation measured automatically using the ACL TOP 300 Coagulation analyser at 37°C.
The percentage coefficient of variation values of the coagulation assays were ≤3.9% (PT) and ≤2.1% (aPTT). The corresponding value for FXI:C was ≤12.4%.
2.5. Statistical reporting
All statistical analyses were performed using SAS 9.4. No formal statistical testing was performed as this was an exploratory study and no null hypotheses were tested. Continuous data were summarised using descriptive statistics and categorical data were summarised in frequency tables with n and percentage. Safety analyses were performed on all subjects who received at least one dose of the study drug, and reported PK and PD data were from all subjects who received at least one dose of the study drug and from whom a PK sample or PD measure was available as appropriate. Summary PD parameters were presented as absolute values and change from baseline, and for aPTT and FXI:C the ratio‐to‐baseline (i.e., fold change from baseline) was also presented.
The following PK parameters were calculated using WinNonlin, version 8.1, using the geometric mean as the primary measure of central tendency for log normally distributed parameters19 and the arithmetic mean or median for other parameters. The standard deviation and between‐subject coefficient of variation were used as measures of variability:
Part A: maximum (peak) plasma concentration (C max), time to reach C max (T max), area under the plasma concentration–time curve (AUC) from time zero to infinity (AUCinf), AUC from time zero to time t (AUCt) and terminal elimination half‐life (t 1/2), of ONO‐7684 in plasma.
Part B: C max, T max and AUCt of ONO‐7684 on Day 1, and C max, T max, AUC during a dosing interval (AUCtau), t 1/2 of ONO‐7684 on Day 14.
In Parts A (fasted subjects only) and B, dose proportionality was assessed using a power model, i.e. a mixed effect model with the logarithm of the PK parameters (AUC and C max) as dependent variables and with subject intercept and subject slope as random effects and dose as a fixed effect. In Part A, the effect of food was assessed using an analysis of variance (ANOVA) model with the logarithm of the PK parameters (AUC and C max) as dependent variables, fed/fasted status as a fixed effect and subject as a random effect. In Part B, the accumulation ratio of ONO‐7684 was assessed using an ANOVA model with the logarithm of the PK parameters (AUC and C max) as dependent variables, dose, day and dose‐by‐day interaction as fixed effects and subject as a random effect.
2.6. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.20
3. RESULTS
3.1. Subject disposition and demographics
The study was conducted between 14 January 2019 and 23 August 2019. In Part A, all 48 subjects completed the study and, in Part B, all 24 subjects completed the study (Figure 1). In both parts of the study, subjects were male, not of Hispanic or Latino ethnicity and predominantly white. Demographics of the subjects are presented in Table 1.
FIGURE 1.
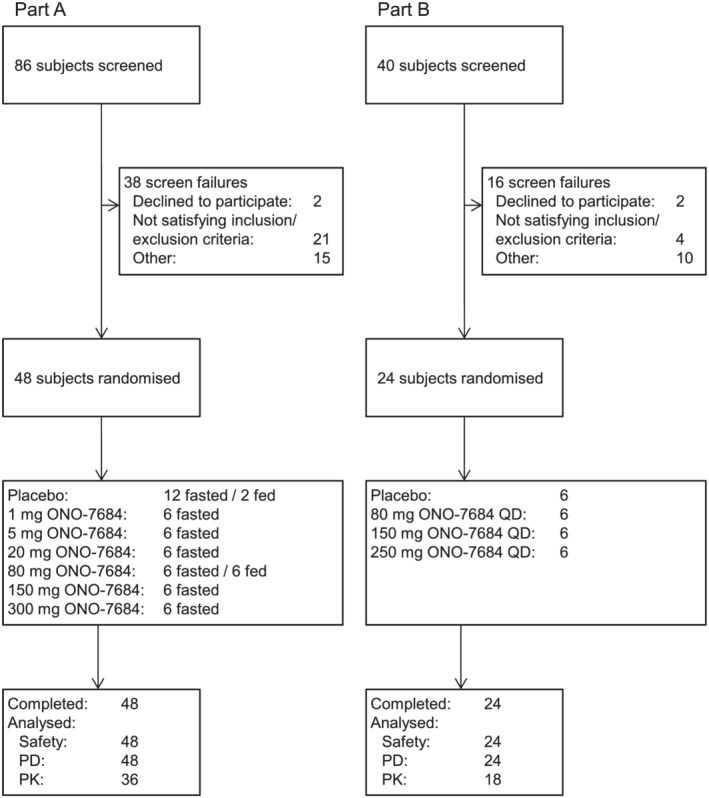
(A) Subject disposition of participants in Part A of the study. (B) Subject disposition of participants in Part B of the study PD, pharmacodynamics; PK, pharmacokinetics; QD, once daily
TABLE 1.
Subject demographics
| Part A | Treatment (placebo or ONO‐7684) | Total n = 48 | ||||||
|---|---|---|---|---|---|---|---|---|
| Placebo (fasted/fed) n = 12 | 1 mg (fasted) n = 6 | 5 mg (fasted) n = 6 | 20 mg (fasted) n = 6 | 80 mg (fasted/fed) n = 6 | 150 mg (fasted) n = 6 | 300 mg (fasted) n = 6 | ||
| Age, years | 31.8 (7.07) | 34.5 (9.97) | 33.5 (10.48) | 39.3 (10.11) | 45.7 (7.45) | 39.8 (8.13) | 32.8 (7.70) | 36.1 (9.29) |
| Race, n (%) black/African American | 1 (8.3) | 1 (16.7) | 0 | 1 (16.7) | 1 (16.7) | 0 | 1 (16.7) | 5 (10.4) |
| White | 10 (83.3) | 5 (83.3) | 6 (100.0) | 5 (83.3) | 5 (83.3) | 6 (100.0) | 4 (66.7) | 41 (85.4) |
| Other | 1 (8.3) | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 2 (4.2) |
| Male, n (%) | 12 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 48 (100.0) |
| BMI, kg/m2 | 23.79 (2.468) | 24.88 (3.434) | 26.97 (2.157) | 23.42 (2.050) | 26.05 (1.946) | 23.43 (2.537) | 24.27 (1.515) | 24.58 (2.546) |
| Part B | Treatment (placebo or ONO‐7684) | Total n = 24 | |||
|---|---|---|---|---|---|
| Placebo n = 6 | 80 mg QD n = 6 | 150 mg QD n = 6 | 250 mg QD n = 6 | ||
| Age, years | 34.5 (9.50) | 38.0 (8.49) | 48.5 (6.25) | 45.2 (10.98) | 41.5 (10.11) |
| Race, n (%) Asian | 2 (33.3) | 1 (16.7) | 0 | 0 | 3 (12.5) |
| Black/African American | 0 | 2 (33.3) | 2 (33.3) | 0 | 4 (16.7) |
| White | 4 (66.7) | 3 (50.0) | 3 (50.0) | 6 (100.0) | 16 (66.7) |
| Other | 0 | 0 | 1 (16.7) | 0 | 1 (4.2) |
| Male, n (%) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 6 (100.0) | 24 (100.0) |
| BMI, kg/m2 | 25.27 (2.192) | 26.45 (3.388) | 25.48 (2.530) | 25.73 (1.925) | 25.73 (2.438) |
Age and BMI are presented as mean (standard deviation).
BMI, body mass index; QD, once daily
3.2. Safety
A total of eight (16.7%) out of 48 fasted subjects and one (12.5%) out of eight fed subjects reported treatment‐emergent adverse events (TEAEs) in Part A, and three (12.5%) of 24 subjects reported TEAEs in Part B (Table 2). There were no severe or serious TEAEs and no TEAEs that led to treatment discontinuation or interruption. No TEAEs were reported in the 150 and 250 mg ONO‐7684 QD groups in Part B.
TABLE 2.
Summary of treatment‐emergent adverse events
| Part A Preferred term, n (%) | Treatment (placebo or ONO‐7684) | Total (fasted) n = 48 | Total (fed) n = 8 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Placebo (fasted) n = 12 | Placebo (fed) n = 2 | 1 mg (fasted) n = 6 | 5 mg (fasted) n = 6 | 20 mg (fasted) n = 6 | 80 mg (fasted) n = 6 | 80 mg (fed) n = 6 | 150 mg (fasted) n = 6 | 300 mg (fasted) n = 6 | |||
| All TEAEs | 2 (16.7) | 0 | 0 | 1 (16.7) | 1 (16.7) | 2 (33.3) | 1 (16.7) | 0 | 2 (33.3) | 8 (16.7) | 1 (12.5) |
| Headache | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (16.7) | 0 | 2 (33.3)a | 3 (6.3) | 1 (12.5) |
| Dizziness | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (2.1) | 0 |
| Lethargy | 0 | 0 | 0 | 1 (16.7)b | 0 | 0 | 0 | 0 | 0 | 1 (2.1) | 0 |
| Gonorrhoea | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 1 (2.1) | 0 |
| Nasopharyngitis | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 0 | 1 (2.1) | 0 |
| Abdominal pain upper | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (2.1) | 0 |
| Nausea | 0 | 0 | 0 | 0 | 1 (16.7) | 0 | 0 | 0 | 0 | 1 (2.1) | 0 |
| Thrombocytopenia | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (16.7) | 1 (2.1) | 0 |
| Pain | 1 (8.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2.1) | 0 |
| Skin laceration | 1 (8.3) | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 0 | 1 (2.1) | 0 |
| Part B Preferred term, n (%),] | Treatment (placebo or ONO 7684) | Totaln = 24 | |||
|---|---|---|---|---|---|
| Placebon = 6 | 80 mg QDn = 6 | 150 mg QDn = 6 | 250 mg QDn = 6 | ||
| All TEAEs | 1 (16.7) | 2 (33.3) | 0 | 0 | 3 (12.5) |
| Dizziness | 0 | 1 (16.7) | 0 | 0 | 1 (4.2) |
| Headache | 0 | 1 (16.7) | 0 | 0 | 1 (4.2) |
| Bleeding time prolonged | 1 (16.7)b | 0 | 0 | 0 | 1 (4.2) |
| Muscle twitching | 0 | 1 (16.7) | 0 | 0 | 1 (4.2) |
QD, once daily; TEAE, treatment‐emergent adverse event.
One subject experienced a treatment‐related TEAE of headache.
Treatment‐related TEAE.
Data are presented as number (%) of subjects who experienced a TEAE.
There were no serious or severe TEAEs during either part of the study and no TEAEs that led to treatment discontinuation or interruption.
Not included in this summary are events that were not considered to be treatment‐emergent. In Part A, these events comprised an event of rhinorrhoea in one subject in the placebo group, an event of headache in one subject in the 80 mg ONO‐7684 (fasted) group and an event of headache in one subject in the 150 mg ONO‐7684 (fasted) group. In Part B, an event of headache in one subject in the placebo group was not considered treatment‐emergent.
The only TEAE reported in more than one subject during Part A of the study was headache, which was reported by three fasted subjects and one fed subject. In Part B, no TEAE was reported by more than one subject (Table 2).
Treatment‐related TEAEs were reported by two subjects (4.2%) in Part A and one subject (4.2%) in Part B. In Part A, one subject (16.7%) experienced a mild TEAE of lethargy in the 5 mg ONO‐7684 group and one subject (16.7%) experienced a moderate TEAE of headache in the 300 mg ONO‐7684 group. The event of lethargy resolved without treatment and the event of headache resolved following use of paracetamol. In Part B, there was one TEAE of prolonged bleeding time in one subject (16.7%) in the placebo group, which occurred following a cut to the skin whilst shaving. The event was mild in severity and resolved without treatment.
Importantly, there was no evidence of a bleeding risk. There were no important changes in vital signs, ECG, cardiac telemetry (Part A only) or physical examinations, and no clinically relevant laboratory values were observed. No individual dose escalation or trial stopping criterion was met.
3.3. Pharmacokinetics
3.3.1. Part A
Mean plasma concentrations peaked at approximately 4 hours post‐dose in each cohort except for the 80 mg ONO‐7684 fed cohort, which had a peak plasma concentration at approximately 8 hours. Plasma concentrations of ONO‐7684 then gradually decreased to baseline levels (Figure 2A and Supplementary Figure S4). T max was comparable across the fasted cohorts (median range: 2.50–4.00 hours) but was delayed in the 80 mg ONO‐7684 fed cohort (median 7.05 hours), as intestinal absorption was delayed by food intake (Table 3). Mean t 1/2 for ONO‐7684 ranged from 16.0 to 19.8 hours.
FIGURE 2.

Mean PK and PD data following administration of a single dose of ONO‐7684 (Part A). Time course of mean ONO‐7684 concentrations (A), mean ratio‐to‐baseline values for FXI:C (B), and mean ratio‐to‐baseline values for aPTT (C). ONO‐7684 concentrations were not measured for placebo (fasted) and placebo (fed) groups and are, therefore, not presented in (A)aPTT, activated partial thromboplastin time; FXI:C, factor XI coagulation activity; LLOQ, lower limit of quantification; PD, pharmacodynamics; PK, pharmacokinetics
TABLE 3.
Pharmacokinetic parameters
| Part A Parameter | Treatment (ONO‐7684) | ||||||
|---|---|---|---|---|---|---|---|
| 1 mg fasted) n = 6 | 5 mg (fasted) n = 6 | 20 mg (fasted) n = 6 | 80 mg (fasted) n = 6 | 80 mg (fed) n = 6 | 150 mg (fasted) n = 6 | 300 mg (fasted) n = 6 | |
| Cmax, ng/mL | 6.77 (4.80, 9.54) | 42.3 (33.6, 53.2) | 159 (134, 188) | 942 (760, 1167) | 537 (434, 663) | 1390 (1239, 1559) | 2084 (1343, 3234) |
| Tmax, h | 2.50 (1.00, 4.00) | 3.50 (2.00, 4.00) | 2.50 (2.00, 4.00) | 2.50 (2.00, 4.02) | 7.05 (3.00, 8.03) | 4.00 (3.00, 4.00) | 4.00 (3.00, 4.02) |
| AUC24, h*ng/mL | 89.0 (60.2, 131) | 541 (448, 653) | 2107 (1732, 2563) | 11 072 (9974, 12 541) | 8627 (7708, 9656) | 17 949 (15 046, 21 411) | 34 091 (22 518, 51 612) |
| AUCinf, h*ng/mL | 148 (90.6, 242) | 948 (800, 1124) | 3414 (2716, 4290) | 17 504 (15 986, 19 167) | 15 908 (13 938, 18 157) | 26 953 (21 743, 33 410) | 61 620 (34 340, 110 571) |
| t1/2, h | 16.5 (3.96) | 17.8 (4.58) | 18.7 (2.47) | 17.1 (2.93) | 18.5 (3.10) | 16.0 (1.19) | 19.8 (6.84) |
| Part B Parameter | Treatment (ONO‐7684) | |||||
|---|---|---|---|---|---|---|
| 80 mg QD | 150 mg QD | 250 mg QD | ||||
| Day 1n = 6 | Day 14n = 6 | Day 1n = 6 | Day 14n = 6 | Day 1n = 6 | Day 14n = 6 | |
| Cmax, ng/mL | 553 (471, 649) | 1041 (764, 1418) | 1055 (832, 1338) | 2004 (1382, 2906) | 1771 (1296, 2422) | 3704 (2674, 5131) |
| Tmax, h | 2.50 (1.00, 4.00) | 4.01 (2.00, 12.0) | 4.00 (2.00, 6.00) | 4.01 (3.00, 8.00) | 4.00 (2.00, 4.03) | 4.00 (2.00, 12.0) |
| AUC24, h*ng/mL | 8107 (7179, 9155) | 18 853 (13 987, 25 413) | 16 345 (12 271, 21 772) | 36 290 (23 891, 55 125) | 27 863 (21 305, 36 440) | 67 048 (45 857, 98 030) |
| AUCinf, h*ng/mL | – | 44 712 (29 559, 67 631) | – | 55 773 (37 147, 83 738) | – | 130 935 (85 057, 201 561) |
| t1/2, h | – | 27.9 (5.33) | – | 24.8(17.1) | – | 22.1 (2.56) |
Cmax, AUCinf and AUC24 are presented as geometric mean (95% CI), T max as median (range) and t 1/2 as mean (standard deviation).
AUC24, area under concentration–time curve from time zero to 24 hours; AUCinf, area under concentration–time curve from time zero to infinity; CI, confidence interval; C max, maximum plasma concentration; QD, once daily; t 1/2, terminal elimination half‐life; T max, time to maximum plasma concentration
In Part A, dose proportionality and food effect were assessed using AUCinf and C max. In Part B, dose proportionality and accumulation were assessed using AUC24 and C max.
Dose proportionality in exposure was observed for the range of 1–300 mg ONO‐7684 as follows: C max and AUCinf values increased about 308‐ and 416‐fold with a 300‐fold increase in dose, respectively (geometric means of 6.77–2084 ng/mL for C max and 148–61 620 h*ng/mL for AUCinf; Table 3). Statistical analyses supported dose proportionality with slopes of 1.04 (95% CI: 0.99–1.10) and 1.03 (95% CI: 0.97–1.10) for C max and AUCinf, respectively (Supplementary Table 1).
Evidence of a food effect was provided by statistical analysis which showed a reduced C max (geometric means ratio of 0.57; 90% CI: 0.47–0.70) and a slightly decreased AUCinf (geometric means ratio of 0.91; 90% CI: 0.84–0.98) in the fed cohort (Supplementary Table S1).
3.3.2. Part B
Mean plasma concentrations peaked at approximately 4 to 6 hours after the first dose in each cohort on Day 1 and then gradually decreased to baseline levels. Plasma concentrations of ONO‐7684 increased with daily dosing and reached steady state by Day 6. On Day 14, plasma concentrations increased following the final dose of ONO‐7684, peaking at approximately 4–6 hours post dose. Plasma concentrations of ONO‐7684 then gradually decreased to baseline levels by 72 hours (Figure 3A and Supplementary Figure S5). T max was comparable across the cohorts at both Day 1 (median range: 2.50–4.00 hours) and Day 14 (median range: 4.00–4.01 hours). The mean t 1/2 for ONO‐7684 after Day 14 ranged from 22.1 to 27.9 hours, slightly longer than that observed in Part A.
FIGURE 3.
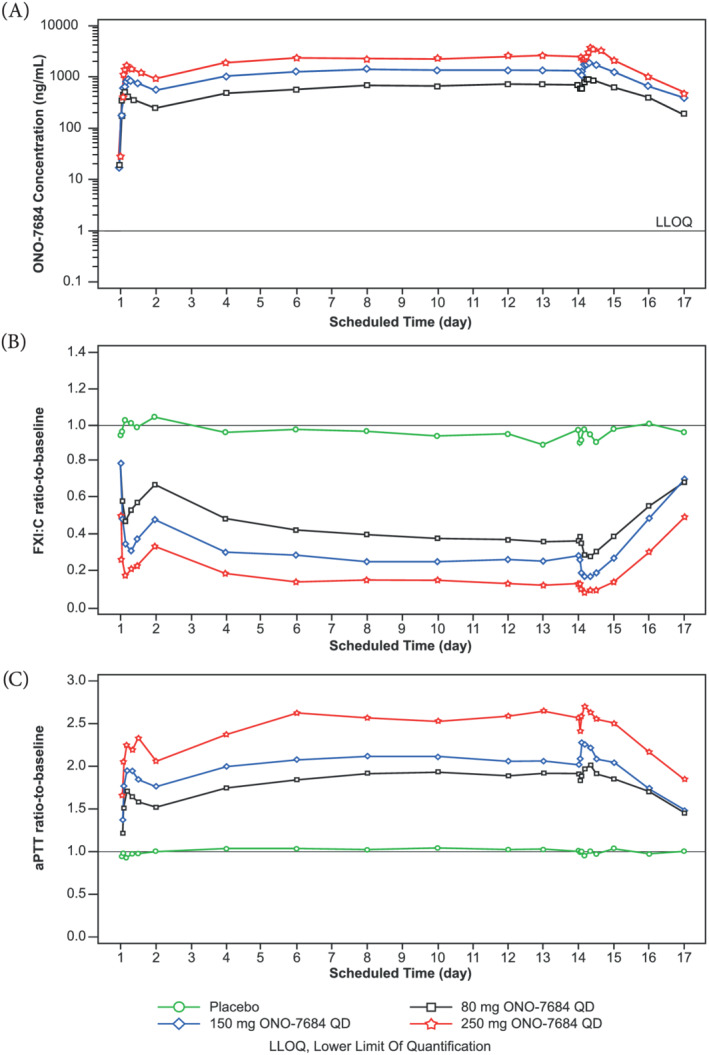
Mean PK and PD data following administration of multiple doses of ONO‐7684 for 14 days (Part B). Time course of mean ONO‐7684 concentrations (A), mean ratio‐to‐baseline values for FXI:C (B), and mean ratio‐to‐baseline values for aPTT (C). ONO‐7684 concentrations were not measured for the placebo group and are, therefore, not presented in (A)aPTT, activated partial thromboplastin time; FXI:C, factor XI coagulation activity; LLOQ, lower limit of quantification; PD, pharmacodynamics; PK, pharmacokinetics; QD, once daily
Dose proportionality in exposure was observed in Part B for the range of 80–250 mg ONO‐7684 QD as follows: C max and AUC24 values both increased approximately 3.6‐fold with a 3.1‐fold increase in dose (geometric mean values of 1041–3704 ng/mL for C max and 18 853–67 048 h*ng/mL for AUC24 on Day 14; Table 3). Statistical analyses supported dose proportionality with slopes of 1.02 (95% CI: 0.77–1.28) and 1.11 (95% CI: 0.75–1.47) for C max at Day 1 and Day 14, respectively, and 1.08 (95% CI: 0.85–1.32) and 1.11 (95% CI: 0.75–1.48) for AUC24 at Day 1 and Day 14, respectively (Supplementary Table 1).
Values for C max and AUC24 were increased at Day 14 compared to Day 1 across all three dose levels, suggesting accumulation of ONO‐7684 in plasma following repeated dosing. Indeed, the accumulation ratios were 1.88 (90% CI: 1.56–2.26), 1.90 (90% CI: 1.58–2.28) and 2.09 (90% CI: 1.74–2.52) for C max and 2.33 (90% CI: 1.99–2.72), 2.22 (90% CI: 1.90–2.60) and 2.41 (90% CI: 2.06–2.82) for AUC24 for the 80, 150 and 250 mg ONO‐7684 QD groups, respectively (Supplementary Table S1).
Values for C max and AUC24 in the 80 mg ONO‐7684 QD group in Part B at Day 1 were similar to those observed following a single dose of 80 mg after an FDA high‐fat breakfast in Part A; however, T max (median: 2.5 h) was similar to that observed following a single dose of 80 mg in the fasted state, suggesting that the high‐fat breakfast had a more significant effect on drug absorption (Table 3).
3.4. Pharmacodynamics
Overall and consistent with the expected mechanism of action and preclinical data, ONO‐7684 decreased FXI:C in a dose‐dependent manner in Parts A and B (Table 4). ONO‐7684 also demonstrated dose‐dependent increases in aPTT in Parts A and B (Table 4), but did not significantly affect PT or INR at any of the dose levels tested in Part A or Part B (Supplementary Figure S6).
TABLE 4.
Effect of ONO‐7684 on FXI:C and aPTT
| Part A Treatment | Treatment (placebo or ONO‐7684) | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Placebo (fasted) n = 12 | Placebo (fed) n = 2 | 1 mg (fasted) n = 6 | 5 mg (fasted) n = 6 | 20 mg (fasted) n = 6 | 80 mg (fasted) n = 6 | 80 mg (fed) n = 6 | 150 mg (fasted) n = 6 | 300 mg (fasted) n = 6 | |
| FXI:C | |||||||||
| Day 1 24 hours RTB | 0.99 ± 0.123 | 1.08 ± 0.250 | 0.95 ± 0.141 | 0.95 ± 0.049 | 0.95 ± 0.185 | 0.61 ± 0.051 | 0.61 ± 0.066 | 0.48 ± 0.057 | 0.26 ± 0.089 |
| Day 1 24 hours inhibition (%) | 1 ± 12.3 | −8 ± 25.0 | 5 ± 14.1 | 5 ± 4.9 | 5 ± 18.5 | 39 ± 5.1 | 39 ± 6.6 | 52 ± 5.7 | 74 ± 8.9 |
| Minimum on treatment RTB | 0.88 ± 0.097 | 0.84 ± 0.036 | 0.87 ± 0.077 | 0.90 ± 0.026 | 0.71 ± 0.048 | 0.31 ± 0.070 | 0.46 ± 0.089 | 0.22 ± 0.034 | 0.15 ± 0.087 |
| Maximum on treatment inhibition (%) | 12 ± 9.7 | 16 ± 3.6 | 13 ± 7.7 | 10 ± 2.6 | 29 ± 4.8 | 69 ± 7.0 | 54 ± 8.9 | 78 ± 3.4 | 85 ± 8.7 |
| aPTT | |||||||||
| Day 1 24 hours RTB | 1.02 ± 0.053 | 1.02 ± 0.249 | 1.00 ± 0.034 | 1.10 ± 0.061 | 1.15 ± 0.119 | 1.54 ± 0.111 | 1.61 ± 0.128 | 1.63 ± 0.031 | 1.99 ± 0.139 |
| Maximum on treatment RTB | 1.04 ± 0.057 | 1.14 ± 0.082 | 1.04 ± 0.047 | 1.18 ± 0.054 | 1.40 ± 0.016 | 1.83 ± 0.172 | 1.74 ± 0.163 | 1.97 ± 0.078 | 2.22 ± 0.223 |
| Part B Treatment | Treatment (placebo or ONO‐7684) | |||
|---|---|---|---|---|
| Placebon = 6 | 80 mg QDn = 6 | 150 mg QDn = 6 | 250 mg QDn = 6 | |
| FXI:C | ||||
| Day 1 24 hours RTB | 1.05 ± 0.068 | 0.67 ± 0.035 | 0.48 ± 0.112 | 0.33 ± 0.065 |
| Day 1 24 hours inhibition (%) | −5 ± 6.8 | 33 ± 3.5 | 52 ± 11.2 | 67 ± 6.5 |
| Day 14 24 hours RTB | 0.98 ± 0.054 | 0.39 ± 0.111 | 0.27 ± 0.096 | 0.14 ± 0.053 |
| Day 14 24 hours inhibition (%) | 2 ± 5.4 | 61 ± 11.1 | 73 ± 9.6 | 86 ± 5.3 |
| Minimum on treatment RTB | 0.83 ± 0.034 | 0.25 ± 0.077 | 0.15 ± 0.052 | 0.08 ± 0.025 |
| Maximum on treatment inhibition (%) | 17 ± 3.4 | 75 ± 7.7 | 85 ± 5.2 | 92 ± 2.5 |
| aPTT | ||||
| Day 1 24 hours RTB | 1.02 ± 0.039 | 1.53 ± 0.119 | 1.78 ± 0.174 | 2.06 ± 0.091 |
| Day 14 24 hours RTB | 1.05 ± 0.042 | 1.87 ± 0.163 | 2.06 ± 0.261 | 2.53 ± 0.146 |
| Maximum on treatment RTB | 1.09 ± 0.026 | 2.07 ± 0.152 | 2.35 ± 0.275 | 2.78 ± 0.147 |
FXI:C and aPTT values are presented as mean ± SD.
Percentage inhibition values are calculated using the appropriate ratio‐to‐baseline value.
aPTT, activated partial thromboplastin time; FXI:C, factor XI coagulation activity; QD, once daily; RTB, ratio‐to‐baseline; SD, standard deviation
3.4.1. Part A
Decreases in FXI:C ratio‐to‐baseline were exposure‐dependent, with a mean (±SD) minimum on treatment ratio‐to‐baseline of 0.87 (±0.077) in the 1 mg ONO‐7684 group and 0.15 (±0.087) in the 300 mg ONO‐7684 group (Table 4). The corresponding ratio in the placebo fasted group was 0.88 (±0.097). The mean minimum on treatment ratio‐to‐baseline value in the 300 mg ONO‐7684 group corresponded to a mean maximum on treatment percentage inhibition vs baseline of 85%. Increases in aPTT ratio‐to‐baseline were also exposure‐dependent, with a mean (±SD) maximum on treatment ratio‐to‐baseline of 1.04 (±0.047) in the 1 mg ONO‐7684 group and 2.22 (±0.223) in the 300 mg ONO‐7684 group (Table 4). The corresponding ratio in the placebo fasted group was 1.04 (±0.057). ONO‐7684 did not significantly affect PT or INR.
There was a decrease in the FXI:C ratio‐to‐baseline (Figure 2B) and an increase in aPTT ratio‐to‐baseline (Figure 2C) following a single dose of 20–300 mg ONO‐7684, with maximal inhibition of FXI:C and maximal prolongation of aPTT generally at approximately 4 hours post‐dose. However, in the 80 mg ONO‐7684 fed group, the corresponding maximal values were at approximately 8 hours post‐dose, consistent with the delayed T max following a high‐fat meal. Subsequently, FXI:C and aPTT ratio‐to‐baseline values gradually returned towards baseline levels. At the higher dose levels (80, 150 and 300 mg ONO‐7684), the effects on FXI:C and aPTT were maintained up to 24 hours, with both parameters showing a gradual return to near baseline levels by 72 hours post‐dose. There was little change in FXI:C and aPTT ratio‐to‐baseline values over time in the 1 and 5 mg ONO‐7684 groups, and in the placebo group (Figure 2B and C). These data suggest an exposure‐dependent relationship for changes in both FXI:C and aPTT following administration of ONO‐7684 (Supplementary Figures S7 and S8).
3.4.2. Part B
Following repeated administration in Part B, ONO‐7684 demonstrated inhibition of FXI:C (Table 4). In the 250 mg ONO‐7684 QD group, there was a mean (±SD) minimum on treatment ratio‐to‐baseline of 0.08 (±0.025), which corresponded to a mean maximum on treatment percentage inhibition vs baseline of 92%. Daily dosing of ONO‐7684 also increased aPTT, with a mean (±SD) maximum on treatment ratio‐to‐baseline of 2.78 (±0.147) in the 250 mg ONO‐7684 QD group (Table 4). ONO‐7684 did not significantly affect PT or INR.
Similar to Part A, there was a decrease in the FXI:C ratio‐to‐baseline values and an increase in aPTT ratio‐to‐baseline values following the first dose of 80, 150 and 250 mg ONO‐7684, with maximal inhibition of FXI:C and maximal aPTT prolongation at approximately 4 hours post‐dose (Figure 3B and C, Supplementary Figure S2B and C). Subsequently, FXI:C and aPTT ratio‐to‐baseline gradually returned towards baseline levels up to the 24 hour timepoint. Daily administration of ONO‐7684 for 14 days caused a further decrease of FXI:C and further increase in aPTT compared to Day 1, with these effects maintained from Day 6 to Day 14 (Figure 3B and C).
Following the final dose of ONO‐7684 on Day 14, there was a further decrease of FXI:C and further increase in aPTT (Figure 3B and C, and Supplementary Figure S3B and C). Prolongation of aPTT and suppression of FXI:C was maintained at 24 hours post‐last dose. After this time, there was a gradual trend towards baseline levels with values for both parameters returning close to baseline levels by the time of the follow‐up assessment. In the placebo group, FXI:C and aPTT ratio‐to‐baseline values remained unchanged over time (Figure 3B and C). Similar to Part A, these data suggest an exposure‐dependent relationship for changes in FXI:C and aPTT following administration of ONO‐7684 (Supplementary Figures S9 and S10).
4. DISCUSSION
There is an unmet medical need for new DOAC that offers effective and predictable anticoagulation without increased risk of adverse events, such as bleeding. The lack of major bleeding in humans with severe FXI deficiency and the preclinical pharmacology data provided a strong rationale to investigate the inhibition of FXIa as a novel mechanism of anticoagulation therapy.
Other studies have reported the use of various FXI inhibitors. An antisense oligonucleotide demonstrated efficacy superior to enoxaparin in total knee arthroplasty13; however, due to the long half‐life (60–80 h), it is likely to take a couple of weeks to achieve a targeted level of plasma FXI reduction and thus a clinically relevant antithrombotic effect. In addition, the time course for the loss of antithrombotic effect after stopping treatment is likely to be protracted (i.e. weeks) and may require the use of specific coagulation products to manage bleeding risk. A parenteral agent envisioned for use in the hospital setting (BMS‐962212; intravenous infusion) demonstrated a fast onset of action (1–2 h).14 However, 17.6% of patients in the active treated groups reported AEs, with 5.9% reporting infusion site pain or irritation. Osocimab (BAY 1213790), a fully human IgG1 monoclonal antibody, reduced apparent factor XIa activity and prolonged aPTT in a dose‐dependent manner.10 However, based on the results of this study, it was not possible to conclude that the risk of bleeding using osocimab was less than that of currently available anticoagulants or to conclude that osocimab was any more effective in preventing venous thromboembolism than apixaban. MAA868, a fully human antibody which binds both FXI and FXIa, induced a robust and sustained prolongation of aPTT and FXI suppression following subcutaneous administration.15 Similar to the antisense oligonucleotide, antibodies will intrinsically have a slow offset duration and thus may present challenges if the antithrombotic effect needed to be reversed rapidly. The oral FXIa inhibitor, BAY 2433334, showed promising preclinical results21 and is currently undergoing evaluation in patients with atrial fibrillation.22 Therefore, a highly selective FXIa inhibitor with a fast onset and offset of action that can be used in a wide range of thromboembolic conditions and clinical settings remains an attractive option.
ONO‐7684 has shown a promising profile during preclinical evaluation. ONO‐7684 was shown to be highly selective for FXIa relative to other coagulation/fibrinolysis factors including FXa, FIXa, FXIIa, FVIIa, thrombin, tPA, urokinase and plasmin.17 During in vivo experiments, including a rabbit FeCl3‐induced arterial thrombosis model and rabbit abdominal incision bleeding model, ONO‐7684 was shown to have a lower bleeding risk than rivaroxaban when used in combination with clopidogrel.17
In the current study, the first clinical evaluation, ONO‐7684 was well tolerated in healthy subjects under fed and fasted conditions, with all randomised subjects completing the study as planned without any significant tolerability issues. Importantly, there was no evidence of a bleeding risk, which remains a concern with existing anticoagulant treatments. The pharmacokinetics of ONO‐7684 were well characterized; a high‐fat meal reduced C max and delayed T max, but had a limited impact on AUCinf. The t 1/2 of ONO‐7684 (mean 22.1–27.9 h) observed in Part B supports once daily oral dosing offering the possibility of dosing at home in addition to the hospital setting.
Although ONO‐7684 strongly inhibited FXI:C at the highest daily dose tested (250 mg) during repeated dosing, some residual FXI:C remained. This observation suggests that the threshold of complete factor XI inhibition is likely to lie only marginally beyond clinically effective levels, thus the possibility of a maximum anticoagulant effect not far outside these desired levels, even in the case of overdose. Concomitant with the extensive FXI:C inhibition, ONO‐7684 resulted in a clinically significant prolongation of aPTT. Notably, whilst ONO‐7684 demonstrated dose‐dependent increases in aPTT in Parts A and B, ONO‐7684 administration did not significantly affect INR. This reduction of FXI:C and prolongation of aPTT without significantly affecting PT or INR would appear consistent with the observation that spontaneous bleeding in humans with a deficiency in FXI is rare6 and may indicate no increased bleeding risk on ONO‐7684 administration.
For both FXI:C and aPTT, the effects of ONO‐7684 dosing were exposure‐dependent and, moreover, the time course of the prolongation of aPTT closely mirrored the reduction in FXI:C. Consistent with current NOACs,23 ONO‐7684 demonstrated predictable pharmacokinetics. The close relationship of PK parameters with FXI:C activity and aPTT provides the potential for ONO‐7684 concentration to be estimated by measuring the FXI:C or aPTT prolongation.
The clinical profile of ONO‐7684 appears to be comparable in terms of pharmacokinetics and pharmacodynamics with another oral FXIa inhibitor being developed (BAY 2433334), that recently reported results from a first clinical study.15 It is hoped that these oral compounds in development will offer a more convenient and safer approach to anticoagulation for patients in a wide array of thromboembolic conditions. In addition, recent in vitro data showed that the anticoagulant activity of ONO‐7684 was effectively neutralised using approved blood coagulation factor products (recombinant factor VIIa, NovoSeven®; and anti‐inhibitor coagulant complex; FEIBA),24 demonstrating that bleeding risk could be clinically managed in the case of an accidental overdose.
In summary, ONO‐7684 was well tolerated when given orally as a tablet for up to 14 days in the fed state in healthy subjects, with no safety or tolerability concerns. There was no significant impact on PT or INR and no evidence of an associated bleeding risk. Furthermore, clinically relevant increases in aPTT were observed in the dose range explored. These results suggest that ONO‐7684 has the potential to be an effective oral anticoagulant warranting further clinical evaluation.
COMPETING INTERESTS
All authors currently work or have previously worked for Ono Pharma UK Ltd or Hammersmith Medicines Research at the time of publication of this article.
CONTRIBUTORS
D.B., M.Bruce, J.D. and M.Boyce designed the research. M.Boyce and J.D. performed the research, and D.B., M.Bruce, N.H., P.S., F.M. and J.D. analysed the data. All authors contributed to the writing of the manuscript.
Supporting information
Table S1. Analysis of pharmacokinetic parameters
Figure S1. Schematic showing the coagulation cascade and the targets of various direct oral anticoagulants
Figure S2. Mean PK and PD data following administration of the first dose of ONO‐7684 on Day 1 in Part B. Time course of mean ONO‐7684 concentrations (A), mean ratio‐to‐baseline values for FXI:C (B) and mean ratio‐to‐baseline values for aPTT (C). ONO‐7684 concentrations were not measured for the placebo group and are, therefore, not presented in (A)
aPTT, activated partial thromboplastin time; FXI:C, factor XI coagulation activity; LLOQ, lower limit of quantification; PD, pharmacodynamics; PK, pharmacokinetics; QD, once daily.
Figure S3. Mean PK and PD data following administration of the final dose of ONO‐7684 on Day 14 in Part B. Time course of mean ONO‐7684 concentrations (A), mean ratio‐to‐baseline values for FXI:C (B) and mean ratio‐to‐baseline values for aPTT (C). ONO‐7684 concentrations were not measured for the placebo group and are, therefore, not presented in (A)
aPTT, activated partial thromboplastin time; FXI:C, factor XI coagulation activity; LLOQ, lower limit of quantification; PD, pharmacodynamics; PK, pharmacokinetics; QD, once daily.
Figure S4. Mean (SD) PK data following administration of a single dose of ONO‐7684 (Part A). Time course of mean (± SD) ONO‐7684 concentrations by dose group (semi‐log). Concentrations below the LLOQ are not shown. LLOQ, lower limit of quantification; PK, pharmacokinetics; SD, standard deviation.
Figure S5. Mean (SD) PK data following administration of the first dose and the final dose of ONO‐7684 on Day 1 and 14 respectively (Part B). Time course of mean (± SD) ONO‐7684 concentrations (semi‐log). LLOQ, lower limit of quantification; PK, pharmacokinetics; QD, once daily; SD, standard deviation.
Figure S6. Mean PD data following administration of a single dose (Part A) or multiple doses (Part B) of ONO‐7684. Time course of mean PT time (upper panel) and mean PT‐INR ratio (bottom panel). PT, prothrombin; PT‐INR, prothrombin time‐international normalised ratio; QD, once daily.
Figure S7. Mean (SD) PK and PD data (aPTT) following administration of a single dose of ONO‐7684 (Part A). Time course of mean (± SD) ONO‐7684 concentrations and aPTT ratio to baseline values by dose group. Notes: Placebo [fasted] was pooled over all cohorts for each fasted plot and placebo [fed] was used in the 80 mg fed plot; the time period for PD sampling was increased to 72 h from the 80 mg fed dose group onwards. aPTT, activated partial thromboplastin time; PD, pharmacodynamics; PK, pharmacokinetics.
Figure S8. Mean (SD) PK and PD data (FXI:C) following administration of a single dose of ONO‐7684 (Part A). Time course of mean (± SD) ONO‐7684 concentrations and FXI:C ratio to baseline values by dose group. Notes: Placebo [fasted] was pooled over all cohorts for each fasted plot and placebo [fed] was used in the 80 mg fed plot; the time period for PD sampling was increased to 72 h from the 80 mg fed dose group onwards. PD, pharmacodynamics; PK, pharmacokinetics; FXI:C, factor XI coagulation activity.
Figure S9. Mean (SD) PK and PD data (aPTT) following administration of the first dose and the final dose of ONO‐7684 on Day 1 and 14 respectively (Part B). Time course of mean (± SD) ONO‐7684 concentrations and aPTT ratio‐to‐baseline values by dose group. aPTT, activated partial thromboplastin time; PD, pharmacodynamics; PK, pharmacokinetics.
Figure S10. Mean (SD) PK and PD data (FXI:C) following administration of the first dose and the final dose of ONO‐7684 on Day 1 and 14 respectively (Part B). Time course of mean (± SD) ONO‐7684 concentrations and FXI:C ratio‐to‐baseline values by dose group. FXI:C, factor XI coagulation activity; PD, pharmacodynamics; PK, pharmacokinetics.
ACKNOWLEDGEMENTS
The authors would like to thank and acknowledge the contribution of the following: The volunteers for their time and commitment; Insight Medical Writing, Kidlington, UK for medical writing support; and Hammersmith Medicines Research, London UK for providing CRO services to support the study, including clinical, medical, data management, statistical and laboratory services. The study was sponsored by Ono Pharmaceutical Co. Ltd.
Beale D, Dennison J, Boyce M, et al. ONO‐7684 a novel oral FXIa inhibitor: Safety, tolerability, pharmacokinetics and pharmacodynamics in a first‐in‐human study. Br J Clin Pharmacol. 2021;87:3177–3189. 10.1111/bcp.14732
Principal investigator statement: The authors confirm that the Principal Investigator for this paper was initially Malcolm Boyce and then subsequently Jeremy Dennison and that they had direct clinical responsibility for patients.
DATA AVAILABILITY STATEMENT
Ono Pharmaceutical Co. Ltd. may share data from our sponsored clinical studies in patients, which are included in the NDA documents for our products that have been approved for marketing or additional indications by the US Food and Drug Administration (FDA) or the European Medicines Agency (EMA) since 1 January 2014 (https://www.ono.co.jp/eng/rd/policy.html).
REFERENCES
- 1.Wendelboe AM, Raskob GE. Global burden of thrombosis: epidemiologic aspects. Circ Res. 2016;118(9):1340‐1347. [DOI] [PubMed] [Google Scholar]
- 2.Sikorska J, Uprichard J. Direct oral anticoagulants: a quick guide. Eur Cardiol. 2017;12(1):40‐45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cervera A, Amaro S, Chamorro A. Oral anticoagulant‐associated intracerebral hemorrhage. J Neurol. 2012;259(2):212‐224. [DOI] [PubMed] [Google Scholar]
- 4.Schumacher WA, Luettgen JM, Quan ML, Seiffert DA. Inhibition of factor XIa as a new approach to anticoagulation. Arterioscler Thromb Vasc Biol. 2010;30(3):388‐392. [DOI] [PubMed] [Google Scholar]
- 5.Yang DT, Flanders MM, Kim H, Rodgers GM. Elevated factor XI activity levels are associated with an increased odds ratio for cerebrovascular events. Am J Clin Pathol. 2006;126(3):411‐415. [DOI] [PubMed] [Google Scholar]
- 6.James P, Salomon O, Mikovic D, Peyvandi F. Rare bleeding disorders—bleeding assessment tools, laboratory aspects and phenotype and therapy of FXI deficiency. Haemophilia. 2014;20(Suppl 4):71‐75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Wheeler AP, Gailani D. Why factor XI deficiency is a clinical concern. Expert Rev Hematol. 2016;9(7):629‐637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Löwenberg EC, Meijers JC, Monia BP, Levi M. Coagulation factor XI as a novel target for antithrombotic treatment. J Thromb Haemost. 2010;8(11):2349‐2357. [DOI] [PubMed] [Google Scholar]
- 9.Koch AW, Schiering N, Melkko S, Ewertet S. MAA868, a novel FXI antibody with a unique binding mode, shows durable effects on markers of anticoagulation in humans. Blood. 2019;133(13):1507‐1516. [DOI] [PubMed] [Google Scholar]
- 10.Thomas D, Thelen K, Kraff S, Schwers S. BAY 1213790, a fully human IgG1 antibody targeting coagulation factor XIa: first evaluation of safety, pharmacodynamics, and pharmacokinetics. Res Pract Thromb Haemost. 2019;3(2):242‐253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Weitz JI, Bauersachs R, Becker B, Berkowitz SD. Effect of osocimab in preventing venous thromboembolism among patients undergoing knee arthroplasty: the FOXTROT randomized clinical trial. JAMA. 2020;323(2):130‐139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lorentz CU, Verbout NG, Wallisch M, Hagen MW. Contact activation inhibitor and factor XI antibody, AB023, produces safe, dose‐dependent anticoagulation in a Phase 1 first‐in‐human trial. Arterioscler Thromb Vasc Biol. 2019;39(4):799‐809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Buller HR, Bethune C, Bhanot S, Gailani D. Factor XI antisense oligonucleotide for prevention of venous thrombosis. N Engl J Med. 2015;372(3):232‐240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perera V, Luettgen JM, Wang Z, Frost CE. First‐in‐human study to assess the safety, pharmacokinetics and pharmacodynamics of BMS‐962212, a direct, reversible, small molecule factor XIa inhibitor in non‐Japanese and Japanese healthy subjects. Br J Clin Pharmacol. 2018;84(5):876‐887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Thomas D, Kanefendt FS, Schwers S, Unger S. First evaluation of the safety, pharmacokinetics and pharmacodynamics of BAY 2433334 a small molecule targeting coagulation factor XIa in healthy young male participants. Res Pract Thromb Haemost. 2020;4(Suppl 1).83. 10.1002/rth2.12393 [DOI] [Google Scholar]
- 16.Wang X, Li Q, Du F, Shukla N. Antithrombotic effects of a novel small molecular FXIa inhibitor BMS‐986177/JNJ‐70033093 in a rabbit AV‐shunt model of thrombosis. Res Pract Thromb Haemost. 2020;4(Suppl 1):52. 10.1002/rth2.12393 [DOI] [Google Scholar]
- 17.Kouyama S, Ono T, Hagio T, et al. Discovery of ONO‐5450598, a highly orally bioavailable small molecule factor XIa inhibitor: the pharmacokinetic and pharmacological profiles. Res Pract Thromb Haemostasis. 2017;1(Suppl. 1):1038. 10.1002/rth2.12012 [DOI] [Google Scholar]
- 18.The Over‐Volunteering Prevention System. https://www.hra.nhs.uk/about-us/committees-and-services/the-over-volunteering-prevention-system. Updated 18 February 2020. Accessed 12 January 2021.
- 19.Julious SA, Debarnot CA. Why are pharmacokinetic data summarized by arithmetic means? J Biopharm Stat. 2000;10(1):55‐71. [DOI] [PubMed] [Google Scholar]
- 20.Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol. 2019;176(S1):S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heitmeier S, Visser M, Gäfke A, Harwardt M. Preclinical pharmacology of BAY 2433334, a small molecule inhibitor of coagulation factor XIa. Res Pract Thromb Haemost. 2020;4(Suppl 1):83. 10.1002/rth2.12393 [DOI] [Google Scholar]
- 22.Study to gather information about the proper dosing of the oral FXIa inhibitor BAY 2433334 and to compare the safety of the study drug to apixaban, a non‐vitamin K oral anticoagulant (NOAC) in patients with irregular heartbeat (atrial fibrillation) that can lead to heart‐related complications. https://www.clinicaltrials.gov/ct2/show/NCT04218266. Updated 21 December 2020. Accessed 12 January 2021.
- 23.Scaglione F. New oral anticoagulants: comparative pharmacology with vitamin K antagonists. Clin Pharmacokinet. 2013;52(2):69‐82. [DOI] [PubMed] [Google Scholar]
- 24.Kouyama S, Ono T. Investigation of pharmacodynamic biomarkers and neutralizers for ONO‐7684 (ONO‐5450598), an oral FXIa inhibitor. Res Pract Thromb Haemost. 2019;3(Suppl.1):172‐173. 10.1002/rth2.12229 [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1. Analysis of pharmacokinetic parameters
Figure S1. Schematic showing the coagulation cascade and the targets of various direct oral anticoagulants
Figure S2. Mean PK and PD data following administration of the first dose of ONO‐7684 on Day 1 in Part B. Time course of mean ONO‐7684 concentrations (A), mean ratio‐to‐baseline values for FXI:C (B) and mean ratio‐to‐baseline values for aPTT (C). ONO‐7684 concentrations were not measured for the placebo group and are, therefore, not presented in (A)
aPTT, activated partial thromboplastin time; FXI:C, factor XI coagulation activity; LLOQ, lower limit of quantification; PD, pharmacodynamics; PK, pharmacokinetics; QD, once daily.
Figure S3. Mean PK and PD data following administration of the final dose of ONO‐7684 on Day 14 in Part B. Time course of mean ONO‐7684 concentrations (A), mean ratio‐to‐baseline values for FXI:C (B) and mean ratio‐to‐baseline values for aPTT (C). ONO‐7684 concentrations were not measured for the placebo group and are, therefore, not presented in (A)
aPTT, activated partial thromboplastin time; FXI:C, factor XI coagulation activity; LLOQ, lower limit of quantification; PD, pharmacodynamics; PK, pharmacokinetics; QD, once daily.
Figure S4. Mean (SD) PK data following administration of a single dose of ONO‐7684 (Part A). Time course of mean (± SD) ONO‐7684 concentrations by dose group (semi‐log). Concentrations below the LLOQ are not shown. LLOQ, lower limit of quantification; PK, pharmacokinetics; SD, standard deviation.
Figure S5. Mean (SD) PK data following administration of the first dose and the final dose of ONO‐7684 on Day 1 and 14 respectively (Part B). Time course of mean (± SD) ONO‐7684 concentrations (semi‐log). LLOQ, lower limit of quantification; PK, pharmacokinetics; QD, once daily; SD, standard deviation.
Figure S6. Mean PD data following administration of a single dose (Part A) or multiple doses (Part B) of ONO‐7684. Time course of mean PT time (upper panel) and mean PT‐INR ratio (bottom panel). PT, prothrombin; PT‐INR, prothrombin time‐international normalised ratio; QD, once daily.
Figure S7. Mean (SD) PK and PD data (aPTT) following administration of a single dose of ONO‐7684 (Part A). Time course of mean (± SD) ONO‐7684 concentrations and aPTT ratio to baseline values by dose group. Notes: Placebo [fasted] was pooled over all cohorts for each fasted plot and placebo [fed] was used in the 80 mg fed plot; the time period for PD sampling was increased to 72 h from the 80 mg fed dose group onwards. aPTT, activated partial thromboplastin time; PD, pharmacodynamics; PK, pharmacokinetics.
Figure S8. Mean (SD) PK and PD data (FXI:C) following administration of a single dose of ONO‐7684 (Part A). Time course of mean (± SD) ONO‐7684 concentrations and FXI:C ratio to baseline values by dose group. Notes: Placebo [fasted] was pooled over all cohorts for each fasted plot and placebo [fed] was used in the 80 mg fed plot; the time period for PD sampling was increased to 72 h from the 80 mg fed dose group onwards. PD, pharmacodynamics; PK, pharmacokinetics; FXI:C, factor XI coagulation activity.
Figure S9. Mean (SD) PK and PD data (aPTT) following administration of the first dose and the final dose of ONO‐7684 on Day 1 and 14 respectively (Part B). Time course of mean (± SD) ONO‐7684 concentrations and aPTT ratio‐to‐baseline values by dose group. aPTT, activated partial thromboplastin time; PD, pharmacodynamics; PK, pharmacokinetics.
Figure S10. Mean (SD) PK and PD data (FXI:C) following administration of the first dose and the final dose of ONO‐7684 on Day 1 and 14 respectively (Part B). Time course of mean (± SD) ONO‐7684 concentrations and FXI:C ratio‐to‐baseline values by dose group. FXI:C, factor XI coagulation activity; PD, pharmacodynamics; PK, pharmacokinetics.
Data Availability Statement
Ono Pharmaceutical Co. Ltd. may share data from our sponsored clinical studies in patients, which are included in the NDA documents for our products that have been approved for marketing or additional indications by the US Food and Drug Administration (FDA) or the European Medicines Agency (EMA) since 1 January 2014 (https://www.ono.co.jp/eng/rd/policy.html).