

Abstract
Species with a circannual life cycle need to match the timing of their life history events to the environment to maximize fitness. However, our understanding of how circannual traits such as timing of reproduction are regulated on a molecular level remains limited. Recent studies have implicated that epigenetic mechanisms can be an important part in the processes that regulate circannual traits. Here, we explore the role of DNA methylation in mediating reproductive timing in a seasonally breeding bird species, the great tit (Parus major), using genome‐wide DNA methylation data from individual females that were blood sampled repeatedly throughout the breeding season. We demonstrate rapid and directional changes in DNA methylation within the promoter region of several genes, including a key transcription factor (NR5A1) known from earlier studies to be involved in the initiation of timing of reproduction. Interestingly, the observed changes in DNA methylation at NR5A1 identified here are in line with earlier gene expression studies of reproduction in chicken, indicating that the observed shifts in DNA methylation at this gene can have a regulatory role. Our findings provide an important step towards elucidating the genomic mechanism that mediates seasonal timing of a key life history traits and provide support for the idea that epigenetic mechanisms may play an important role in circannual traits.
Keywords: avian reproductive timing, DNA methylation, ecological epigenetics, life history traits, Parus major
Short abstract
see also the Perspective by Melanie J. Heckwolf and Britta S. Meyer
1. INTRODUCTION
Increasing global temperatures have led to shifts in phenology traits of many species over the last few decades with major ecological impacts (Parmesan & Yohe, 2003; Petchey et al., 1999). Such shifts include leaf unfolding of trees (Fu et al., 2015), spring‐flowering of plants (Fitter & Fitter, 2002), emergence of butterflies (Roy & Sparks, 2000), timing of egg laying in seasonally reproducing birds (Both & Visser, 2001) and hibernation phenology in squirrels (Lane et al., 2012). While temporal shifts in circannual traits are well documented (Parmesan, 2007; Parmesan & Yohe, 2003; Thackeray et al., 2016), we know surprisingly little about the genomic basis of circannual traits (Franks & Hoffmann, 2012).
Epigenetic modifications (i.e., chemical modifications of the DNA sequence or chromatin proteins that affect gene expression and consequently traits without changes in the DNA sequence; Suzuki & Bird, 2008), constitute promising genomic mechanisms for the regulation of circannual traits. Indeed, recent studies in plants (Bastow et al., 2004; Wilschut et al., 2016; You et al., 2017), insects (Pegoraro et al., 2016) and mammals (Stevenson, 2017; Stevenson & Prendergast, 2013) have emphasized the potential for short‐term temporal variation in epigenetic modifications to be involved in mediating the temporal expression of phenology traits across taxa. For example, flowering time in Arabidopsis is characterized by variation in histone methylation of flowering locus C (FLC) (Bastow et al., 2004), photoperiodic diapause in a parasitic wasp (Nasonia vitripennis) is associated with variation in DNA methylation induced by different photoperiods (Pegoraro et al., 2016), and gonadal regression in Siberian hamsters (Phodopus sungorus) is accompanied by photoperiod‐induced and reversible variation in DNA methylation of type III deiodinase (dio3), a gene involved in the regulation of reproduction (Stevenson & Prendergast, 2013).
These studies demonstrate a role for DNA methylation in the regulation of circannual traits and suggest that epigenetic modifications can be an important part of the molecular control of circannual traits (Stevenson & Lincoln, 2017), similar to what is observed for circadian rhythms (Stevenson, 2018). However, the generality of epigenetic modifications being involved in circannual traits is limited to a handful of species and thus further studies examining this would be very useful.
To investigate the potential for DNA methylation to mediate seasonal timing of reproduction, we have conducted experiments using great tits (Parus major). The great tit is well suited for examining whether DNA methylation is involved in controlling circannual rhythms as it is a seasonal breeder which times its onset of breeding with environmental cues such as photoperiod (Dawson et al., 2001; Sharp, 2005), temperature (McClerry & Perrins, 1998; Visser et al., 2009) and possibly the emergence of caterpillars (Jones, 1972; Noordwijk et al., 1995; Schaper et al., 2011) and, like many other species, great tits have advanced their phenology over the last few decades (McClerry & Perrins, 1998; Visser et al., 2003; Winkel & Hudde, 1997). In recent years, the great tit has increasingly been the subject of studies with a focus on molecular ecology and evolution (e.g., Bosse et al., 2017; Laine et al., 2016; Perrier et al., 2018) providing us with species‐specific knowledge on DNA methylation (Derks et al., 2016; Lindner et al., 2021; Sepers et al., 2019; van Oers et al., 2020; Viitaniemi et al., 2019). In vertebrates, methylation predominately occurs on cytosines. In great tit blood cells, 97% of the methylation occurs in a CG context, often referred to as “CpG sites.” In brain cells, however, DNA methylation occurs both in CG and in non‐CpG context (CHH) (Derks et al., 2016). How CpG methylation affects the expression of genes depends strongly on the genomic location; in close proximity to the transcription start site, low levels of DNA methylation associate with lower expression of the respective gene while CpG methylation at other genomic locations such as transposable elements might not necessarily reduce the expression of genes (Derks et al., 2016; Laine et al., 2016).
For DNA methylation to be involved in mediating seasonal timing of reproduction in great tits, DNA methylation must change over a short time (i.e., throughout the breeding season). Although many recent studies have examined DNA methylation in wild species (Heckwolf et al., 2020; Rubenstein et al., 2016; Saino et al., 2017, 2019; Sepers et al., 2019), we still know very little about the temporal stability of DNA methylation. We have previously demonstrated that just over 40,000 CpG sites do indeed display temporal changes in DNA methylation throughout the breeding season in great tits (Viitaniemi et al., 2019). However, individual females vary in their timing of reproduction and hence it is unclear whether these temporal changes in DNA methylation relate to the reproductive timing of females. Based on this previous work, we here focused on whether temporal patterns in DNA methylation vary with the reproductive timing of females (i.e., time relative to when females initiate egg laying) rather than time per se to investigate whether temporal patterns in DNA methylation have the potential to mediate seasonal timing of reproduction. We tested for differential methylation between reproductive timing groups and used an unsupervised approach (comethylation analysis) to examine the association between changes in CpG methylation and reproductive timing. Our results suggest that DNA methylation might act as a molecular switching mechanism of the reproductive cascade by mediating, among other genes, the expression of a key transcription factor. Our findings highlight the potential role for DNA methylation in the genomic mechanism that mediates reproductive timing in great tits.
2. MATERIALS AND METHODS
2.1. Study system
We used the great tit, a well‐known model species in ecology and evolution with a reference genome (Laine et al., 2016) and whole transcriptome and methylome for various tissues (Derks et al., 2016; Laine et al., 2016; Santure et al., 2011). The individuals included are part of a bidirectional selection experiment for early and late reproduction using genomic selection (Gienapp et al., 2019; Verhagen, Gienapp, et al., 2019 details in Methods S1). For the experiment, 18 breeding pairs of the early selection line as well as 18 pairs of the late selection line of the F2 generation were housed in climate‐controlled aviaries mimicking natural temperature and photoperiod patterns of a cold and warm year in the Netherlands (Laine et al., 2019; Verhagen, Laine, et al., 2019), but here we focus on females from the early selection line only. Within the early selection line, we found no significant difference in laying dates between the temperature environments (Kruskal–Wallis test; χ2 = 0.4213, df =1, p = .5163, Table S1). Since the temperature environment was part of the experimental set up, we nevertheless included temperature treatment as a fixed factor in our analyses, but we did not focus on this aspect further in this study.
2.2. Blood sampling and sample selection
Pairs were blood sampled biweekly in two batches from January to July between 8:30 a.m. and 2:30 p.m. from the jugular vein (up to 150 µl) and for DNA extraction red blood cells were separated from the plasma (Mäkinen et al., 2019) (details in Methods S2). We chose samples from 16 females of the early selection line collected at four sampling times based on the females’ realized laying dates (Figure 1; Table S2): the day when (i) day length >12 hr, (ii) 25% of the females exposed to the warm temperature environment had initiated egg laying, (iii) 25% and 50% of the females exposed to the cold and warm temperature environment respectively had initiated egg laying, and (iv) 50% of the females exposed to the cold temperature environment had initiated egg laying. One blood sample is missing for one female (due to the female incubating) at the fourth sampling time, resulting in a total of 63 samples. Although samples of 16 females were sequenced, two females did not initiate egg laying during the experiment and the respective samples were removed from statistical analyses, reducing the data set to 55 samples (see Table S2 for samples used in differential methylation and comethylation analysis).
FIGURE 1.
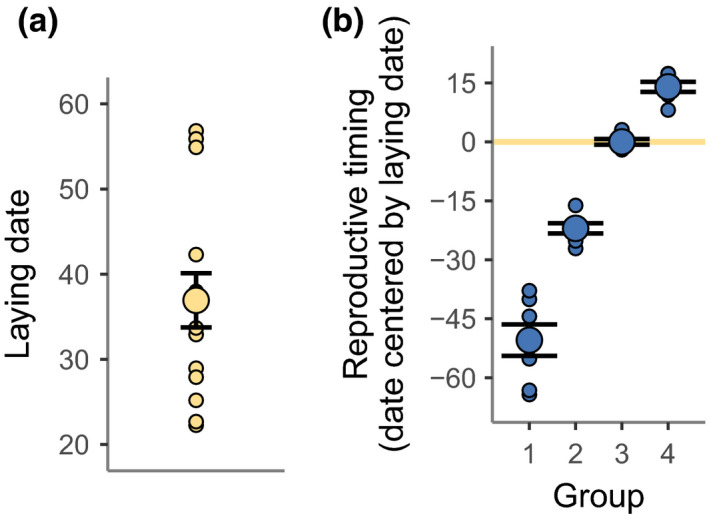
Reproductive timing. (a) Mean laying date with standard errors (in April dates, i.e., 1 = April 1) with the laying date of individual females (n = 14) in the background. (b) The reproductive timing (y‐axis) of the four reproductive timing groups (x‐axis): (1) early pre‐laying group, (2) late pre‐laying group, (3) laying group, (4) post‐laying group. Reproductive timing is calculated as the sampling date centred by females’ laying date (i.e., reproductive timingij = sampling dateij – laying datej for sampling time i and female j). Mean reproductive timing of each group with standard errors is shown and reproductive timing of the individual samples within each group are displayed in the background. Yellow line highlights the reproductive timing that corresponds to females’ laying date
2.3. Sample processing and reduced representation bisulfite sequencing
We used a reduced representation bisulfite sequencing (RRBS) approach, a method that enriches sequenced reads for CpG sites by using the restriction enzyme mspI that nonrandomly cuts DNA in coding regions and in CG‐rich areas such as CpG islands and in this way reduces the number of reads required to obtain high coverage of a reproducible fraction of genome‐wide CpG sites (Gu et al., 2011; Meissner et al., 2008). Library preparation and sequencing was done by the Roy J. Carver Biotechnology Centre (University of Illinois at Urbana‐Champaign, USA). For details on sample processing and sequencing see Mäkinen et al. (2019). In short, DNA was extracted using the FavorPrepT M 96‐well Genomic DNA Kit (Favorgen) and libraries for RRBS were prepared following the manufacturer's protocol (Illumina). Sixteen libraries were randomly pooled into four sets and run on eight lanes such that every set was run on two lanes with 100 bp from single end reads. All lanes were run on the same flow cell on a HiSeq 2500 sequencer using the HiSeq RRBS sequencing kit version 4 (Illumina). RRBS data have been submitted to the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under BioProject PRJNA208335 and accession nos. SRX3209916–SRX3209919.
2.4. Sequence alignment and CpG site calling
Sequence alignment (Mäkinen et al., 2019), CpG site calling (Mäkinen et al., 2019), quantification of DNA methylation (Mäkinen et al., 2019) and general methylation statistics of the data set (Viitaniemi et al., 2019) are described in the respective publications. CpG sites with a minimum of 10× coverage in all samples were included, resulting in methylation information of 522,645 CpG sites covered in each of the 55 samples.
2.5. Promoter region and gene annotation
The RRBS approach enriches for CpG site‐rich regions, which are present through the genome but are particularly common in promoter regions of the great tit genome (Derks et al., 2016). We here focused on CpG sites close to the transcription start site of genes as this is the region for which CpG site methylation has a strong negative effect on gene expression (Laine et al., 2016). We defined a region spanning 2000 bp upstream to 200 bp downstream of a gene's transcription starting site as its promoter region, the region we used to annotate CpG sites using the Parus major reference genome build 1.1 (https://www.ncbi.nlm.nih.gov/assembly/GCF_001522545.2) and R packages genomicfeatures (Lawrence et al., 2013) version 1.30.0 and rtracklayer version 1.42.2 (Lawrence et al., 2009). We found 223,282 CpG sites located within the promoter region of 12,325 genes (out of 18,611 annotated genes) using bedtools version 2.26.0 (Quinlan & Hall, 2010). Average CpG site methylation levels of these 223,282 sites are provided in Table S3 (average over all samples) and Table S4 (average for each sample). Note that one CpG site can be associated to the promoter region of two genes if the two genes are located on opposite strands and have overlapping promoter regions.
2.6. Differential methylation analysis
We calculated the reproductive timing as the sampling date centred by the respective females’ laying date (Table S2) and used this to group samples of all 14 females into four different reproductive timing groups: early pre‐laying group (64–38 days prior to laying), late pre‐laying group (27–16 days prior to laying), laying group (2 days before to 3 days after laying), and post‐laying group (8–17 days after laying) (Figure 1; Table S2). Each reproductive timing group constitutes seven samples (making a total of 28 samples). Although the grouping of samples reduced our sample size, the grouping allows us to test for differences in methylation levels between any of the reproductive timing groups without limiting our analysis to an a priori defined trend for the change in DNA methylation over reproductive timing. To balance the temperature environments across groups, we included four samples from females exposed to one temperature environment and three samples from females exposed to the other temperature environment in each group (Table S5).
We calculated the average methylation level for each CpG site over the samples within each reproductive timing group (with equal weights for all samples) and used these average methylation levels to filter CpG sites such that only sites with at least 10% change in average methylation level between any of the four reproductive groups were included in the differential methylation analysis (Leenen et al., 2016), reducing the data set to 5,097 CpG sites.
We applied a generalized linear mixed model (GLMM) approach to identify sites that were significantly differentially methylated between any of the four reproductive timing groups using R package lme4qtl version 0.1.10 (Ziyatdinov et al., 2018). For each of the 5,097 CpG sites we fitted a GLMM with binomially distributed errors as specified in Equation 1:
| (1) |
with y for the modelled response, a two‐column matrix of methylated and unmethylated counts as the dependent variable to account for variation in CpG site coverage (corresponding to methylation levels weighted by the total number of counts, Lea et al., 2017; Zhang et al., 2016), µ for the intercept term, xRS , xTE and xB for the vector relating samples to their reproductive timing group, temperature environment, and batch, respectively, βRS , βTE and βB for the reproductive timing group, temperature environment, and batch effect, respectively, Z for an incidence matrix relating samples to their observed values, f and r for the random effects for repeated measurements and relatedness, and e for the residuals. Genomic relatedness was calculated using the R package genabel version 1.8.0 (Aulchenko et al., 2007). We used a boundary distance of 0.01, a maximum number of iterations of 2 × 108, and “bobyqa” as an optimizer to speed up computation and optimize convergence (Powell, 2009). For each GLMM (i.e., each CpG site) we calculated the dispersion statistic following Zuur et al. (2013) and calculated the 95% highest density interval (HDI) for the distribution of dispersion statistics using the R package hdinterval version 0.2.2 (Meredith & Kruschke, 2020) as in contrast to symmetric density intervals, the HDI is defined such that all estimates within the interval have a higher probability density than estimates outside the interval also for nonsymmetric distributions (Kruschke, 2011). For each CpG site we used the fitted model to infer the estimated marginal means (EMMs) for each reproductive timing group and tested for an effect of any pairwise contrast between the EMMs of the four reproductive timing groups using the R package emmeans version 1.3.3 (Lenth, 2019). We used a Bonferroni‐corrected α‐threshold based on the number of CpG sites in the promoter region in our data set (αBF =0.05/223,282 = 2.24e‐07) and accepted pairwise contrasts with a p‐value below this threshold as significant. We excluded one CpG site (chr22_2621974) as the respective model failed to converge. Dispersion statistics were normally distributed with a median of 1.09 (95% confidence interval [1.08, 1.10]) and HDI of [0.45, 1.83] (Figure S1 and Table S2) and we excluded the 256 CpG sites outside the 95% HDI from evaluation of the differential methylation analysis. Although quantile–quantile plots indicate a slight “epi‐genomic” inflation (Figure S2), vulcano plots (Figure S3), significance vs. mean coverage plots (Figure S4), and p‐value distributions (Figure S5) do not indicate any inflation of p‐values; the vulcano plots show the expected V‐shaped pattern in which highly significant CpG sites are the sites with the highest difference in methylation levels, significance vs. mean coverage plots did not show a p‐value bias of high coverage towards low p‐values (i.e., high ‐log10[p‐values]), and p‐value distributions show the expected uniform‐like distribution that is enriched towards low p‐values.
2.7. Comethylation analysis
For the weighted comethylation network analysis (comethylation analysis) we used the R package wgcna version 1.66 (Langfelder & Horvath, 2008) to cluster CpG sites into modules based on similarity in methylation pattern throughout the reproductive timing of females. The package implements functions to cluster CpG sites into modules based on similarity in methylation pattern over samples. The modules, in turn, can be correlated to sample traits of interest. The package is mainly applied to expression data (Laine et al., 2019; Langfelder & Horvath, 2008), but is increasingly used for methylation data (Horvath et al., 2012; Lim et al., 2018; Nardone et al., 2017; van Eijk et al., 2012; Wang et al., 2016). In contrast to the differential methylation analysis, all 55 samples of females that have initiated egg laying were used (as no outliers were detected using hierarchical clustering, Figure S6).
We used the same set of 5,097 CpG sites as for the differential methylation analysis as we found a skewed scale‐free topology when using all 223,282 CpG sites located within the regulatory region of genes (Figure S7, but see below). A network is specified by its adjacency matrix aij (symmetric n × n matrix with entries in [0,1]) whose component aij encodes the network connection strength between site i and j. The adjacency, see Equation 2, is constructed from correlations
| (2) |
with power being the soft thresholding power which is used to emphasize strong correlation on the expense of weak correlations. We chose the power based on the scale‐free topology criterion (Zhang & Horvath, 2005). We picked a soft threshold of 14 as this is the lowest power at which scale free topology index R 2 > .9 (truncated R 2 = .99) and mean connectivity decreases to 2.50 (Figure S8). Thus, the weighted networks used here are highly robust with regard to the power. Furthermore, weighted networks allow the adjacency to take on continuous values between 0 and 1 (in unweighted networks adjacency is either 1 or 0, which does not reflect the continuous nature of the underlying DNA methylation levels). We specified a merge cut height of 0.65 to prevent high adjacency between module eigensites (Figure S9, but see Figure S10 for adjacency between module eigensites with a merge cut height of 0.15).
Each detected module was randomly assigned a colour and the methylation profile of each module was summarized as a module's eigensite equivalent to the first principal component of a module based on the methylation profile of all CpG sites within that module. For each detected module, we tested whether the methylation profile of CpG sites within a module covaried with the reproductive timing of females by correlating the module eigensite of a module with reproductive timing. We also tested for correlations of module eigensites with other sampling traits (i.e., laying date per se, sampling date, female identity and temperature environment). We used a Bonferroni‐corrected α‐threshold based on the number of correlations tested (αBF =0.05/50 = 0.001 for correlations of 10 modules with five sampling traits) and accepted correlations with a p‐value below this threshold as significant. Modules with a significant correlation to reproductive timing and CpG sites within such modules that show a significant module membership and a significant trait‐based site significance are potential candidates for further validation (Langfelder & Horvath, 2008). The module membership of a CpG site is based on the correlation of the CpG site‐specific methylation profile with the module eigensite of the respective module. The trait‐based site significance of a CpG site is based on the correlation of the CpG site‐specific methylation profile with the reproductive state. We used a Bonferroni‐corrected α‐threshold based on the number of CpG sites within a module (αBF =0.05/826 = 6.05e‐05 for the turquoise module and αBF =0.05/234 = 2.14e‐04 for the green module) and accepted CpG sites with a p‐value for site significance and module membership below this threshold as significant.
2.8. Functional analysis
We performed gene ontology (GO) analyses for genes identified with the differential methylation analysis and the comethylation analysis (details in Methods S6) using the cluego version 2.5.3 (Bindea et al., 2009) plug‐in for cytoscape version 3.7.1 (Shannon et al., 2003). We used the human annotations, GO categories “biological process,” “cellular components,” “molecular functions” and KEGG pathways (versions from July 3, 2020), and a custom background lists of all genes with a CpG site within their promoter region. We specified the selection criteria for GO terms such that >5% of the genes associated with a GO term and >3 genes associated with the GO term had to be present in the input genes. For the enrichment and functional analysis we used a two‐sided enrichment/depletion test, p‐value correction for multiple testing via Bonferroni step down, and set the network specificity to “medium” ranging from third to tenth GO level. In addition to the GO analyses, we used the string version 1.4.2 (Doncheva et al., 2019) plug‐in for cytoscape version 3.7.1 (Shannon et al., 2003) to construct protein–protein interaction networks for the same genes as in the GO analyses and used a confidence cutoff of 0.7.
2.9. Methylation profiles of most significant findings
For the differential methylation analysis, we ranked genes based on the p‐value of the most significant CpG site within their promoter region in any of the pairwise contrasts between the reproductive timing groups. For the comethylation analysis, we ranked genes for the turquoise and green module separately such that the rank is based on the p‐value for the site significance of the most significant CpG site within the promoter region. We calculated the average rank for genes (i.e., sum of the ranks for the differential methylation analysis and the comethylation analysis divided by two) that had at least one CpG site in their promoter region that was significant in the differential methylation analysis and in the comethylation analysis. We visualized the methylation profiles of significant CpG sites within the promoter region of such genes by plotting the methylation level against the reproductive timing.
3. RESULTS
3.1. Differential methylation analysis
We tested for variation in CpG site methylation between any of the six pairwise contrasts of the four reproductive timing groups (Figure 1) using a differential methylation analysis on 5,097 CpG sites within the promoter region of genes, a region known to affect gene expression in great tits (Laine et al., 2016; and Tables S6 and S7). For the contrast between the early and late pre‐laying group (a), we did not find any CpG sites with significant variation in methylation (Figure 2). However, we identified 35 CpG sites within the promoter region of 11 genes (Figure 2; Table S8) that showed genome‐wide significant variation in methylation in at least one of the other five pairwise contrasts between the reproductive timing groups, 11 CpG sites for the contrast between the early pre‐laying and laying group (b), 29 CpG sites for the contrast between the early pre‐laying and post‐laying group (c), two CpG sites for the contrast between the late pre‐laying and laying group (d), 24 CpG sites for the contrast between the late pre‐laying and post‐laying group (e), and six CpG sites for the contrast between the laying and post‐laying group (f). The p‐values and mean change in DNA methylation level between the respective reproductive timing groups for the 35 CpG sites with genome‐wide significant variation in DNA methylation are presented in Table S8. Genomic locations enriched for CpG sites with significant variation in methylation were shared between the pairwise contrasts and most pronounced for the contrasts of the early (c) and late (d) pre‐laying groups with the post‐laying group (Figure 2).
FIGURE 2.
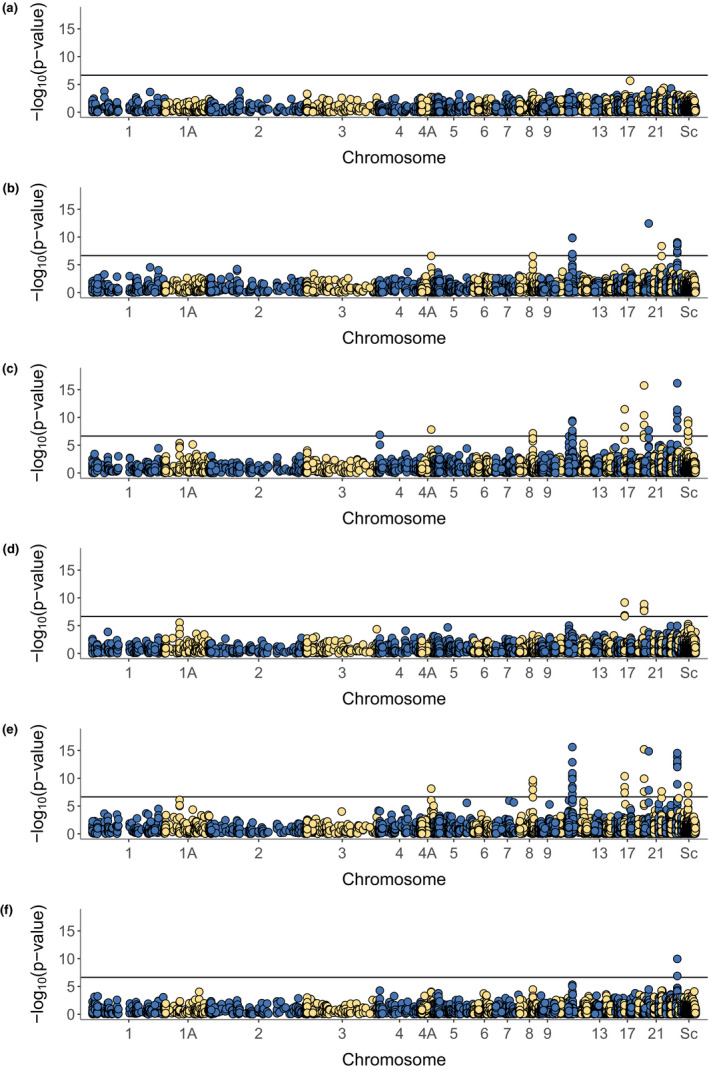
Manhattan plots. p‐values (in −log10 scale) correspond to the significance of difference in DNA methylation level between the respective reproduction timing groups. Black lines mark the genome‐wide significance threshold (Bonferroni‐corrected, −log10(αBF) = −log10(0.05/223,282) =6.65). “Sc” refers to unplaced scaffolds. All pairwise comparison of the four reproductive timing groups are displayed: (a) early vs. late pre‐laying, (b) early pre‐laying vs. laying, (c) early pre‐laying vs. post‐laying, (d) late pre‐laying vs. laying, (e) late pre‐laying vs. post‐laying, and (f) laying vs. post‐laying
3.2. Comethylation analysis
We used all samples independently of the grouping in a comethylation analysis to test for covariation in CpG site methylation with the reproductive timing of females. Thus, while the differential methylation analysis presented above required a priori defined comparisons between groups, in this analysis we used all samples in an unsupervised approach using a weighted comethylation network analysis (comethylation analysis), to cluster CpG sites based on the similarity of their methylation profiles. Out of 5,097 CpG sites we found that 2,347 clustered into nine different modules (Table S9) and assigned all remaining CpG sites to the grey module. We defined the module eigensite of each module as the first principal component of the respective module, which explained between 5% (grey module) and 46% (salmon module) of the variance in CpG site methylation (Figure S11 and Table S11). For two modules, we found a significant correlation between the modules’ eigensite and the reproductive timing of females: a positive correlation for the turquoise module (.66, p = 1.57e‐06, Figure 3b) and a negative correlation for the green module (−.71, p = 4.82e‐08, Figure 3c; Table S11 and Table S12). The module eigensites of the turquoise and green modules did not correlate significantly with the temperature environment or female identity (see Figure S12 and Table S11–Table S13 for the correlation between all module eigensites and additional sampling traits of interest).
FIGURE 3.
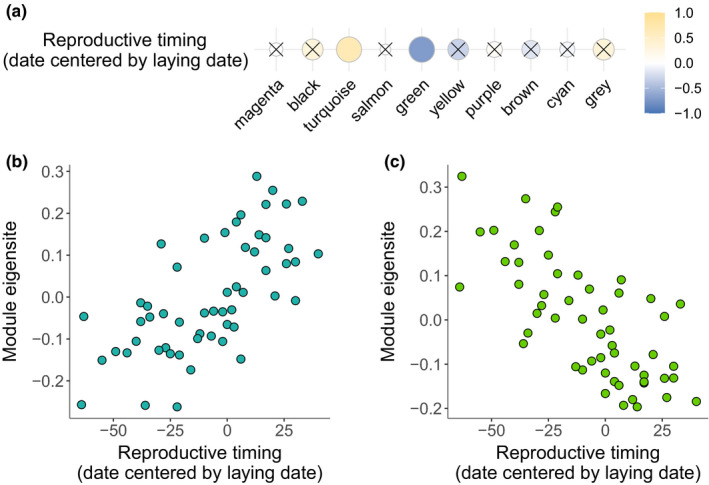
(a) Correlation of the module eigensite of each detected module with reproductive timing. Yellow corresponds to a positive correlation and blue to a negative correlation. The size of spheres indicates the significance of a correlation (i.e., the bigger a sphere, the lower the p‐value). Nonsignificant correlations (p > .001, Bonferroni‐corrected α‐threshold) are crossed. Significant correlation of the module eigensite with reproductive timing for (b) the turquoise module (0.66, p = 1.57e‐06) and (c) the green module (−0.71, p = 4.82e‐08)
We defined the site significance and module membership of each CpG site as the correlation of the CpG site's methylation pattern with the reproductive timing of females and with the module eigensite, respectively. For all 5,097 CpG sites, the site significance and the module membership with all 10 modules are presented in Table S14. For the turquoise and green modules, we found 38 CpG sites within the promoter region of 20 genes and 44 CpG sites within the promoter region of 13 genes with significant site significance and module membership, respectively (Figure S13 and Tables S15–S18).
3.3. Functional analysis
We performed GO and string analyses on the genes found in the differential methylation analysis and comethylation analyses, but we did not detect any significantly enriched functional GO groups or protein–protein interaction networks. Hence, the genes we identified have not been previously described to share biological functions or interact with each other.
3.4. Methylation profiles of most significant findings
To identify the CpG site methylation patterns that most strongly associated with reproductive timing of females, we combined the findings from the differential methylation analysis and comethylation analysis. This resulted in a list of 10 genes with at least one CpG site that was genome‐wide significant in the differential methylation analysis and had significant site significance and module membership in the comethylation analysis (Table S19 and S20). CpG sites within the promoter region of six of these genes showed a decrease in DNA methylation with the reproductive timing of females: six sites in LOC107215054 (MYLK‐like, up to 74% decrease in methylation level within site), eight sites in LOC107209693 (GP2‐like) and DRC7 (up to 86% decrease in methylation level within site), two sites in LOC107213450 (uncharacterized gene, up to 83% decrease in methylation level within site), one site in NUDC (up to 57% decrease in methylation level within site), and one site in DLG3 (up to 67% decrease in methylation level within site, Table S21 and S22, right panel of Figure 4). At most of these CpG sites the decrease in methylation level was initiated 3–4 weeks prior to laying and methylation stabilized at low levels about a week after laying, while methylation levels of the CpG site in NUDC decreased until 3 weeks prior to laying and were consistently low throughout laying (right panel of Figure 4). CpG sites within the promoter region of four genes showed an increase in DNA methylation with the reproductive timing of females: three sites in NR5A1 (up to 87% increase in methylation level within site), five sites in CFAP45 (up to 94% increase in methylation level within site), four sites in SLC6A9 (up to 80% increase in methylation level within site), and four sites in CLUH (up to 87% increase in methylation level within site, Table S21 and Table S22; left panel of Figure 4). At most of these CpG sites the increase in methylation level was initiated approximately 3 weeks prior to laying and methylation stabilized at medium to high levels about 3 weeks after the laying date (left panel of Figure 4).
FIGURE 4.
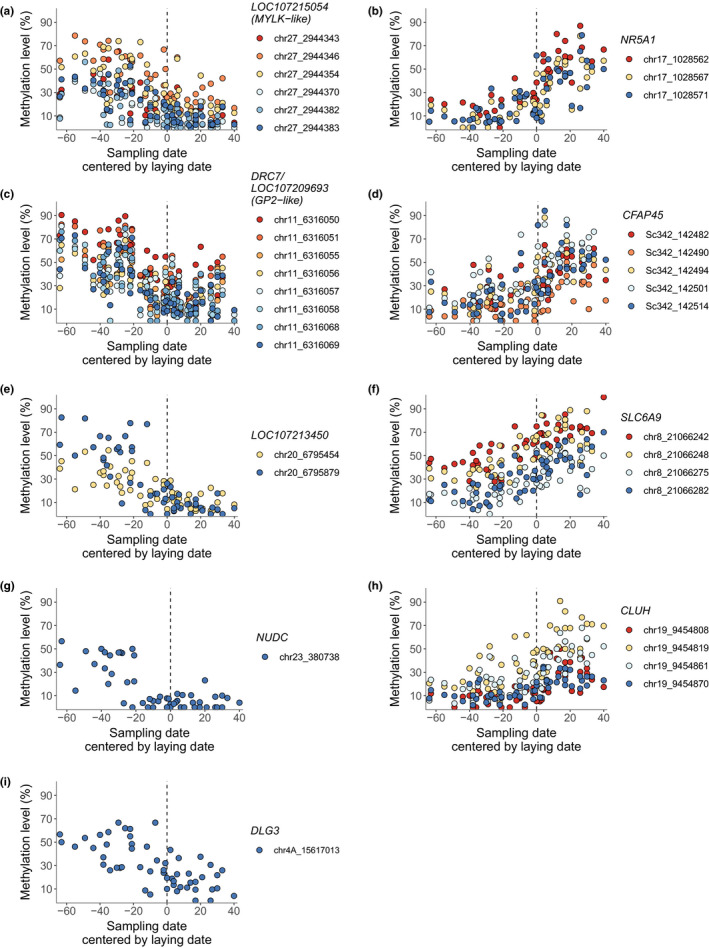
Variation of DNA methylation with reproductive timing across all samples (n = 55). Only genes with CpG sites significant in both analyses, i.e., differential methylation and comethylation analysis, are displayed: (a) LOC107215054 (MYLK‐like), (b) NR5A1, (c) DRC7 and LOC107209693 (GP2‐like), (d) CFAP45, (e) LOC107213450, (f) SLC6A9, (g) NUDC, (h) CLUH and (i) DLG3
4. DISCUSSION
The genomic mechanism that mediates the timing of phenology traits remains largely unknown (Caro et al., 2013), but recent studies on plants (Bastow et al., 2004; Wilschut et al., 2016; You et al., 2017), insects (Pegoraro et al., 2016) and mammals (Stevenson, 2017; Stevenson & Prendergast, 2013) emphasize the potential for epigenetic modifications, such as DNA methylation, to be involved (Stevenson & Lincoln, 2017). Here, we add support to this idea by demonstrating that rapid and directional changes in DNA methylation associate with reproductive timing in a wild songbird species, the great tit. A differential methylation analysis and comethylation analysis identified rapid changes at CpG sites within the promoter region of a handful of genes (Table S19). Some of these genes have well‐known function in mediating reproduction, which suggests that DNA methylation might act as a molecular switching mechanism of the reproductive cascade. Our study therefore illustrates the potential role for DNA methylation as a genomic mechanism that mediates reproductive timing.
4.1. Methylation profiles of significant genes from both differential methylation and comethylation analysis
When we combined findings from the differential methylation analysis and comethylation analysis, we identified 10 genes that displayed a consistent and replicable change in DNA methylation in relation to the reproductive status of females. Some of these genes are well known from earlier studies to be involved in reproduction such as LOC107215054 (MYLK‐like) and NR5A1.
LOC107215054 (MYLK‐like) is predicted as a myosin light chain kinase, smooth muscle‐like and shares sequence regions with the actual myosin light chain kinase (MYLK), a kinase that facilitates phosphorylation of the myosin light chain, a process essential for shell gland contractile activity during oviposition, that is egg laying (Johnson, 2015; Kupittayanant et al., 2009). In chicken ovaries, MYLK is up‐regulated during egg laying (Liu et al., 2018), a pattern consistent with the observed decrease in methylation at CpG sites within the promoter region of LOC107215054 (MYLK‐like) in the weeks prior to laying. A recent study on the methylation landscape of chicken found that DNA methylation of MYLK putatively controls MYLK gene expression (Höglund et al., 2020), further supporting the idea that identified changes in DNA methylation at the promoter region of this gene can have a regulatory function by mediating gene expression. Whether both genes function as myosin light chain kinases, however, remains to be established.
LOC107213450 is a protein‐coding but uncharacterized gene, which means that, in contrast to LOC107215054 (MYLK‐like), no reliable gene prediction is available. As the gene network and its individual genes that mediate the timing of laying date in great tit females are currently unknown (Gienapp et al., 2017; Laine et al., 2019), a gene of yet uncharacterized biological function with CpG site hypomethylation in the weeks prior to the initiation of laying date constitutes an interesting candidate for being involved in this gene network.
LOC107209693 (GP2‐like) is predicted as pancreatic secretory granule membrane major glycoprotein (GP2), which is a component of the inner perivitelline layer surrounding the avian ovum during ovulation that helps to maintain the structural integrity of the vitelline membrane (Kido et al., 1977; Wishart & Horrocks, 2000). The inner perivitelline layer mechanically supports the ovum and mediates the initial interaction between the ovum and spermatozoa in which glycopeptides are suggested to play an essential role (Wishart & Horrocks, 2000) and consequently GP2 might have a function in fertilization. Furthermore, GP2 was reported as a modulator for immune response (Werner et al., 2012), such as by binding pathogenic enterobacteria (Hase et al., 2009). However, whether LOC107209693 (GP2‐like) and GP2 share their biological function remains to be established. Furthermore, the promoter region of LOC107209693 (GP2‐like) and DRC7 overlap such that the CpG sites identified here are located within the promoter region of both genes and hence the observed decrease in DNA methylation can be functional for both, one or none of the genes.
NR5A1 codes for a transcription factor that regulates the expression of many important genes within all levels of the reproductive axis (Ingraham et al., 1994; Meinsohn et al., 2019) and most genes involved in gonadal steroidogenesis (Jameson, 2004). For example, NR5A1 modulates the expression of the steroidogenic acute response protein (STAR) that transfers cholesterol to initiate an enzymatic cascade that comprises steroid synthesis, essential for folliculogenesis (Murayama et al., 2012). NR5A1 is expressed in the major steroidogenic tissues, such as theca and granulosa cells in the ovary (Ikeda et al., 1994, 1996; Ingraham et al., 1994) or hypothalamus (e.g., regulating hypothalamic pituitary gonadotroph organization and function; Shinoda et al., 1995). Consequently, NR5A1 plays a key role in female reproduction such as ovarian functioning (Lourenço et al., 2009; Meinsohn et al., 2019) and steroidogenesis (Parker, 2002). Expression studies of NR5A1 in chickens are concordant with the hypermethylation observed here as NR5A1 is up‐regulated during egg laying relative to brooding (Shen et al., 2016). The observed DNA methylation pattern, in combination with the essential role of NR5A1 in ovarian functioning and steroidogenesis, suggests that DNA methylation might act as a molecular switching mechanism of the reproductive cascade by mediating the expression of a key transcription factor, NR5A1.
The remaining genes are linked to general biological processes and functions: cilia and flagella motor activity (DRC7 and CFAP45; Li et al., 1999; Heuser et al., 2009; Fu et al., 2018), mitosis, cytokinesis, ciliogenesis, neuronal migration, platelet production and inflammatory response (NUDC; Aumais et al., 2003; Fu et al., 2016), neurotransmission and cellular and whole body homeostasis (SLC6A9; Bröer & Gether, 2012), synaptic transmission, development and plasticity (DLG3; Tarpey et al., 2004; Qu et al., 2009), and control of cell energetic and metabolic status (CLUH; Wakim et al., 2017). A more detailed description of these functions and, if applicable, their potential involvement in mediating reproduction is given in Text S1.
We have previously demonstrated temporal changes in DNA methylation over the breeding season in great tits at around 40,000 CpG sites (Viitaniemi et al., 2019) and several of the genes that we identify here were on the list of genes that displayed temporal change over the breeding season. However, in the study by Viitaniemi et al. (2019), we were not able to link the changes in methylation directly to the reproductive status of females, as we only examined within‐female change in DNA methylation over time per se. As females vary in their reproductive timing, such temporal changes in methylation can be due to numerous factors (change in age, environmental conditions, etc.) and not necessarily relate to reproductive timing. By instead focusing on the association between the reproductive timing of females and changes in DNA methylation, we can now demonstrate their potential involvement in the initiation of egg laying in this species.
4.2. DNA methylation as a general mechanism for regulation of circannual phenotypes
In contrast to the well‐described circadian clock gene pathways, the gene and regulatory pathways underlying the circannual expression of traits are less well described and experimental data confirming a functional role of DNA methylation in producing and maintaining circannual rhythms are limited (Stevenson, 2018). In Siberian hamsters, the expression of enzymes that mediate DNA methylation, DNA methyltransferases (dnmts), decreased in response to short photoperiod and coincided with the decrease in DNA methylation within the promoter region of dio3, a gene known to inhibit the reproductive cascade (Stevenson & Prendergast, 2013). This decrease in promoter DNA methylation of dio3, in turn, was accompanied by an increase and dio3 expression and gonadal regression. This study provided the first evidence for DNA methylation and enzymes that mediate DNA methylation to underly a photoperiod‐induced response on the phenotypic level. Also, nonmammalian systems provide evidence for a role of epigenetics in mediating circannual phenotypes; in Arabidopsis flowering time was characterized by variation in histone methylation of flowering locus C (FLC) (Bastow et al., 2004) and in a parasitic wasp photoperiodic diapause was associated with photoperiod‐induced variation in DNA methylation (Pegoraro et al., 2016). In avian systems few studies have examined the association between DNA methylation patterns and circannual phenotypes, so the potential generality of our finding is difficult to assess. The only other study we are aware of used a candidate gene approach to demonstrate that DNA methylation at the CLOCK gene is correlated with laying date and other circannual phenotypes in barn swallows (Hirundo rustica) (Saino et al., 2017).
4.3. Caveats and future outlook
The interpretation of our findings is based on the assumption that CpG site methylation within the promoter region of a gene affects the transcription of the gene, which in turn has the potential to change the expression of phenotypes (Bossdorf et al., 2008; Rubenstein et al., 2016; Verhoeven et al., 2016). This is generally true and also the case in the great tit where DNA methylation in the promoter region is negatively correlated with gene expression, and especially close to the transcription start site low levels of DNA methylation are sufficient to shut down the expression of the respective gene (Laine et al., 2016). We therefore restricted our analyses to CpG sites within the promoter region of genes to specifically look at CpG sites that we expect to have an effect on gene expression. However, this also means that we miss any impact of CpG sites in genomic locations outside this region which also can have effects on gene expression (Anastasiadi et al., 2018). Furthermore, recent studies suggest that variation in DNA methylation may not exclusively acts as a cause of gene expression, but can be a downstream consequence of gene expression (Pacis et al., 2019). We acknowledge that the data used here does not allow to test whether DNA methylation acts as a cause or consequence of gene expression. Equally, it is important to keep in mind that, as with any association study, we are, of course, unable to demonstrate a causal link between the change in DNA methylation and timing of egg laying. For this experimental validation, using functional tools is needed and at this point this is not feasible on nonmodel organisms such as the great tit.
On question is also to what degree the observed changes in DNA methylation are dependent on the DNA sequence. Recent studies have demonstrated that genetic variants can underly local and distant variation in DNA methylation in a variety of species: Arabidopsis thaliana (Dubin et al., 2015), maize (Xu et al., 2019), reef‐building corals (Liew et al., 2020), inter‐crosses between wild derived junglefowl and domestic chickens (Höglund et al., 2020), and humans (Heyn et al., 2013). While we controlled for relatedness among individuals in our differential methylation analysis using single nucleotide polymorphism (SNP) genotype data, the number of females sampled is too small to detect any genome‐wide significant effects of SNPs on local or distant variation in DNA methylation. Hence, we do not know whether identified patterns in DNA methylation are dependent on local genetic variation or genetic variation elsewhere in the genome.
RRBS has become a popular approach for methylation profiling as it enriches for CpG‐rich regions of the genome such as the transcription start site and the promoter region and in this way reduces the number of reads required to obtain a high coverage of a reproducible fraction of CpG sites per sample (Gu et al., 2011; Husby, 2020; Meissner et al., 2008). The downside of RRBS, like all reduced approaches, is that it covers only a small fraction of the genome and thus we might miss changes in methylation at regions not sequenced.
A difficulty with all ecological epigenetic studies is to determine which tissue type to sample when associating DNA methylation with a phenotype of interest (Derks et al., 2016; Husby, 2020; Verhulst et al., 2016). We repeatedly blood sampled great tit females and used isolated red blood cells to examine within‐female patterns in CpG site methylation. The strength of this approach is that we are able to examine how DNA methylation covaries with the reproductive timing within females. More informative tissues such as gonads, hypothalamus or liver cannot be sampled repeatedly and do not allow a direct correlation with the reproductive timing of females as some females would be sampled before the initiation of egg laying and hence do not yet express the phenotype of interest.
Many ecological epigenetic studies measure DNA methylation from blood samples (see table 1 in Husby, 2020), but the relevance of methylation in blood compared to methylation patterns in other tissues is only starting to be understood. It is well known that methylation is tissue‐ and even cell type‐specific (Schilling & Rehli, 2007), but some recent studies have also demonstrated correlations in methylation levels across tissues (Tylee et al., 2013). For example, in humans nearly 10,000 genomic regions with a correlated methylation pattern between different tissues were identified that also varied between individuals (Gunasekara et al., 2019). In great tits red blood cell and liver DNA methylation change predictably in both a tissue‐specific and a tissue‐general manner (Derks et al., 2016; Lindner et al., 2021), suggesting that tissue‐general changes in DNA methylation can be expressed in a tissue‐specific manner (Lindner et al., 2021). Future studies examining tissue‐specific correlations in DNA methylation and RNA expression in ecological model organisms will be highly valuable for the field.
5. CONCLUSION
We demonstrate that CpG site methylation within the promoter region of genes covaried with the reproductive timing of females and that the observed DNA methylation patterns are in line with expression pattern of the respective genes in chicken. This highlights the potential for an epigenetic modification to play an important role in the genomic mechanism that mediates phenology in this species, although, as for any association study, experimental work is ultimately needed to verify this.
AUTHOR CONTRIBUTIONS
M.E.V., K.v.O. and A.H. designed the experiment, I.V. conducted the experiments, H.M.V. conducted the alignments, M.L. conducted the statistical analysis with the help of V.N.L., and M.L. and A.H. wrote the manuscript with input from all authors.
Supporting information
Supplementary Material
Table S1‐S22
Supplementary Material
ACKNOWLEDGEMENTS
We thank Koen Verhoeven for commenting on an earlier draft of the manuscript, Christa Mateman and colleagues at NIOO‐KNAW for laboratory assistance, Bart van Lith and Ruben de Wit for assistance during the experiments, Jeroen Laurens and Gilles Wijlhuizen for technical assistance prior and during the experiments, and the animal carers at the NIOO‐KNAW for taking care of the birds. We thank Tyler Stevenson, two anonymous reviewers and Victoria Sork for constructive comments that improved the manuscript.
Funding information
This work was supported by the Research Council of Norway through its Centre of Excellence funding (223257), a personal grant to A.H. (239974) from the Norwegian Research Council and a European Research Council Advanced grant (ERC‐2013‐AdG 339092) to M.E.V.
Contributor Information
Melanie Lindner, Email: M.Lindner@nioo.knaw.nl.
Arild Husby, Email: arild.husby@ebc.uu.se.
DATA AVAILABILITY STATEMENT
The data underlying this article are available in the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under BioProject PRJNA208335 and accession nos. SRX3209916–SRX3209919, and the code for statistical analyses is provided in the Supporting Informationn.
REFERENCES
- Anastasiadi, D., Codina, A. E., & Piferrer, F. (2018). Consistent inverse correlation between DNA methylation of the first intron and gene expression across tissues and species. Epigenetics & Chromatin, 11, 37. 10.1186/s13072-018-0205-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aulchenko, Y. S., Ripke, S., Isaacs, A., & Van Duijn, C. M. (2007). GenABEL: An R library for genome‐wide association analysis. Bioinformatics, 23(10), 1294–1296. 10.1093/bioinformatics/btm108 [DOI] [PubMed] [Google Scholar]
- Aumais, J. P., Williams, S. N., Luo, W., Nishino, M., Caldwell, K. A., Caldwell, G. A., Lin, S.‐H. & Yu‐Lee, L. Y. (2003). Role of NudC, a dynein‐associated nuclear movement protein, in mitosis and cytokinesis. Journal of Cell Science, 116(10), 1991–2003. 10.1242/jcs.00412 [DOI] [PubMed] [Google Scholar]
- Bastow, R., Mylne, J. S., Lister, C., Lippman, Z., Martienssen, R. A., & Dean, C., & 1Department (2004). Vernalization requires epigenetic silencing of FLC by histone methylation. Nature, 427(8), 164–167. 10.1038/nature02269 [DOI] [PubMed] [Google Scholar]
- Bindea, G., Mlecnik, B., Hackl, H., Charoentong, P., Tosolini, M., Kirilovsky, A., Fridman, W.‐H., Pagès, F., Trajanoski, Z., & Galon, J. (2009). ClueGO: A Cytoscape plug‐in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics, 25(8), 1091–1093. 10.1093/bioinformatics/btp101 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bossdorf, O., Richards, C. L., & Pigliucci, M. (2008). Epigenetics for ecologists. Ecology Letters, 11(2), 106–115. 10.1111/j.1461-0248.2007.01130.x [DOI] [PubMed] [Google Scholar]
- Bosse, M., Spurgin, L. G., Laine, V. N., Cole, E. F., Firth, J. A., Gienapp, P., Gosler, A. G., McMahon, K., Poissant, J., Verhagen, I., Groenen, M. A. M., van Oers, K., Sheldon, B. C., Visser, M. E., & Slate, J. (2017). Recent natural selection causes adaptive evolution of an avian polygenic trait. Science, 358(6361), 365–368. 10.1126/science.aal3298 [DOI] [PubMed] [Google Scholar]
- Both, C., & Visser, M. E. (2001). Adjustment to climate change is constrained by arrival date in a long‐distance migrant bird. Nature, 411, 296–298. [DOI] [PubMed] [Google Scholar]
- Bröer, S., & Gether, U. (2012). The solute carrier 6 family of transporters. British Journal of Pharmacology, 167(2), 256–278. 10.1111/j.1476-5381.2012.01975.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro, S. P., Schaper, S. V., Hut, R. A., Ball, G. F., & Visser, M. E. (2013). The case of the missing mechanism: How does temperature influence seasonal timing in endotherms? PLoS Biology, 11(4), e1001517. 10.1371/journal.pbio.1001517 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawson, A., King, V. M., Bentley, G. E., & Ball, G. F. (2001). PHOTOPERIODIC control of seasonality in birds alistair. Journal of Biological Rhythms, 16(4), 365–380. [DOI] [PubMed] [Google Scholar]
- Derks, M. F. L., Schachtschneider, K. M., Madsen, O., Schijlen, E., Verhoeven, K. J. F., & van Oers, K. (2016). Gene and transposable element methylation in great tit (Parus major) brain and blood. BMC Genomics, 17(1), 332. 10.1186/s12864-016-2653-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doncheva, N. T., Morris, J. H., Gorodkin, J., & Jensen, L. J. (2019). Cytoscape StringApp: Network analysis and visualization of proteomics data. [Research‐article]. Journal of Proteome Research, 18(2), 623–632. 10.1021/acs.jproteome.8b00702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubin, M. J., Zhang, P., Meng, D., Remigereau, M.‐S., Osborne, E. J., Paolo Casale, F., Drewe, P., Kahles, A., Jean, G., Vilhjálmsson, B., Jagoda, J., Irez, S., Voronin, V., Song, Q., Long, Q., Rätsch, G., Stegle, O., Clark, R. M., & Nordborg, M. (2015). DNA methylation in Arabidopsis has a genetic basis and shows evidence of local adaptation. Elife, 4, e05255. 10.7554/eLife.05255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitter, A. H., & Fitter, R. S. R. (2002). Rapid changes in flowering time in British plants. Science, 296(5573), 1689–1691. 10.1126/science.1071617 [DOI] [PubMed] [Google Scholar]
- Franks, S. J., & Hoffmann, A. A. (2012). Genetics of climate change adaptation. Annual Review of Genetics, 46, 185–208. [DOI] [PubMed] [Google Scholar]
- Fu, C., Luo, J., Ye, S., Yuan, Z., & Li, S. (2018). Integrated lung and tracheal mRNA‐Seq and miRNA‐Seq analysis of dogs with an avian‐like H5N1 canine influenza virus infection. Frontiers in Microbiology, 9(MAR), 1–14. 10.3389/fmicb.2018.00303 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fu, Q., Wang, W., Zhou, T., & Yang, Y. (2016). Emerging roles of NudC family: From molecular regulation to clinical implications. Science China Life Sciences, 59(5), 455–462. 10.1007/s11427-016-5029-2 [DOI] [PubMed] [Google Scholar]
- Fu, Y. H., Zhao, H., Piao, S., Peaucelle, M., Peng, S., Zhou, G., Ciais, P., Huang, M., Menzel, A., Peñuelas, J., Song, Y., Vitasse, Y., Zeng, Z., & Janssens, I. A. (2015). Declining global warming effects on the phenology of spring leaf unfolding. Nature, 526, 104–107. 10.1038/nature15402 [DOI] [PubMed] [Google Scholar]
- Gienapp, P., Calus, M. P. L., Laine, V. N., & Visser, M. E. (2019). Genomic selection on breeding time in a wild bird population. Evolution Letters, 1–10, 10.1002/evl3.103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gienapp, P., Laine, V. N., Mateman, A. C., van Oers, K., & Visser, M. E. (2017). Environment‐dependent genotype‐phenotype associations in avian breeding time. Frontiers in Genetics, 8, 102. 10.3389/fgene.2017.00102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu, H., Smith, Z. D., Bock, C., Boyle, P., Gnirke, A., & Meissner, A. (2011). Preparation of reduced representation bisulfite sequencing libraries for genome‐scale DNA methylation profiling. Nature Protocols, 6(4), 468–481. 10.1038/nprot.2010.190 [DOI] [PubMed] [Google Scholar]
- Gunasekara, C. J., Scott, C. A., Laritsky, E., Baker, M. S., MacKay, H., Duryea, J. D., Kessler, N. J., Hellenthal, G., Wood, A. C., Hodges, K. R., Gandhi, M., Hair, A. B., Silver, M. J., Moore, S. E., Prentice, A. M., Li, Y., Chen, R., Coarfa, C., & Waterland, R. A. (2019). A genomic atlas of systemic interindividual epigenetic variation in humans. Genome Biology, 20(1), 1–12. 10.1186/s13059-019-1708-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hase, K., Kawano, K., Nochi, T., Pontes, G. S., Fukuda, S., Ebisawa, M., Kadokura, K., Tobe, T., Fujimura, Y., Kawano, S., Yabashi, A., Waguri, S., Nakato, G., Kimura, S., Murakami, T., Iimura, M., Hamura, K., Fukuoka, S.‐I., Lowe, A. W., … Ohno, H. (2009). Uptake through glycoprotein 2 of FimH + bacteria by M cells initiates mucosal immune response. Nature, 462(7270), 226–230. 10.1038/nature08529 [DOI] [PubMed] [Google Scholar]
- Heckwolf, M. J., Meyer, B. S., Häsler, R., Höppner, M. P., Eizaguirre, C., & Reusch, T. B. H. (2020). Two different epigenetic information channels in wild three‐spined sticklebacks are involved in salinity adaptation. Science Advances, 6(12), eaaz1138. 10.1126/sciadv.aaz1138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heuser, T., Raytchev, M., Krell, J., Porter, M. E., & Nicastro, D. (2009). The dynein regulatory complex is the nexin link and a major regulatory node in cilia and flagella. Journal of Cell Biology, 187(6), 921–933. 10.1083/jcb.200908067 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heyn, H., Moran, S., Hernando‐herraez, I., Sayols, S., Gomez, A., Sandoval, J., & Esteller, M. (2013). DNA methylation contributes to natural human variation. Genome Research, 23, 1363–1372. 10.1101/gr.154187.112.Freely [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höglund, A., Henriksen, R., Fogelholm, J., Churcher, A. M., Guerrero‐Bosagna, C. M., Martinez‐Barrio, A., Johnsson, M., Jensen, P., & Wright, D. (2020). The methylation landscape and its role in domestication and gene regulation in the chicken. Nature Ecology and Evolution, 10.1038/s41559-020-01310-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horvath, S., Zhang, Y., Langfelder, P., Kahn, R. S., Boks, M. P., van Eijk, K., van den Berg, L. & Ophoff, R. A. (2012). Aging effects on DNA methylation modules in human brain and blood tissue. Genome Biology, 12, R97. 10.1016/j.jbspin.2011.02.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Husby, A. (2020). On the use of blood samples for measuring DNA methylation in ecological epigenetic studie. Integrative and Comparative Biology, 60, 1558–1566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda, Y., Shen, W. H., Ingraham, H. A., & Parker, K. L. (1994). Developmental expression of mouse steroidogenic factor‐1, an essential regulator of the steroid hydroxylases. Molecular Endocrinology, 8, 654–662. 10.1210/mend.8.5.8058073 [DOI] [PubMed] [Google Scholar]
- Ikeda, Y., Swain, A., Weber, T. J., Hentges, K. E., Zanaria, E., Lalli, E., Tamai, K. T., Sassone‐Corsi, P., Lovell‐Badge, R., Camerino, G., & Parker, K. L. (1996). Steroidogenic factor 1 and Dax‐1 colocalize in multiple cell lineages: potential links in endocrine development. Molecular Endocrinology, 10, 1261–1272. 10.1210/mend.10.10.9121493 [DOI] [PubMed] [Google Scholar]
- Ingraham, H. A., Lala, D. S., Ikeda, Y., Luo, X., Shen, W. H., Nachtigal, M. W., Abbud, R., Nilson, J. H., & Parker, K. L. (1994). The nuclear receptor steroidogenic factor 1 acts at multiple levels of the reproductive axis. Genes and Development, 8(19), 2302–2312. 10.1101/gad.8.19.2302 [DOI] [PubMed] [Google Scholar]
- Jameson, J. L. (2004). Editorial: Of mice and men. The tale of steroidogenic factor‐1. Journal of Clinical Endocrinology and Metabolism, 89(12), 5927–5929. 10.1210/jc.2004-2047 [DOI] [PubMed] [Google Scholar]
- Johnson, A. L. (2015). Reproduction in the female. In Scanes C. G. (Ed.), Sturkie’s avian physiology (6th ed., pp. 635–665). Elsevier. 10.1016/B978-0-12-407160-5.00028-2 [DOI] [Google Scholar]
- Jones, P. J. (1972). Food as a proximate factor regulating the breeding season of the great tit (Parus major). Proceedings of the International Ornithological Congress, 15, 657–658. [Google Scholar]
- Kido, S., Janado, M., & Nunoura, H. (1977). Macromolecular of components of the vitelline membrane Hen’ s ‐ III. physiochemical properties of glycoprotein II. Journal of Biochemistry, 81, 1543–1548. [PubMed] [Google Scholar]
- Kruschke, J. K. (2011). Doing Bayesian data analysis: A tutorial with R and BUGS. Elsevier. [Google Scholar]
- Kupittayanant, S., Kupittayanant, P., & Suwannachat, C. (2009). Mechanisms of uterine contractility in laying hens. Animal Reproduction Science, 115(1–4), 215–224. 10.1016/j.anireprosci.2008.10.020 [DOI] [PubMed] [Google Scholar]
- Laine, V. N., Gossmann, T. I., Schachtschneider, K. M., Garroway, C. J., Madsen, O., Verhoeven, K. J. F., de Jager, V., Megens, H.‐J., Warren, W. C., Minx, P., Crooijmans, R. P. M. A., Corcoran, P., Sheldon, B. C., Slate, J., Zeng, K., van Oers, K., Visser, M. E., & Groenen, M. A. M. (2016). Evolutionary signals of selection on cognition from the great tit genome and methylome. Nature Communications, 7, 1–9. 10.1038/ncomms10474 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laine, V. N., Verhagen, I., Mateman, A. C., Pijl, A., Williams, T. D., Gienapp, P., van Oers, K., & Visser, M. E. (2019). Exploration of tissue‐specific gene expression patterns underlying timing of breeding in contrasting temperature environments in a song bird. BMC Genomics, 20(1), 693. 10.1186/s12864-019-6043-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lane, J. E., Kruuk, L. E. B., Charmantier, A., Murie, J. O., & Dobson, F. S. (2012). Delayed phenology and reduced fitness associated with climate change in a wild hibernator. Nature, 489(7417), 554–557. 10.1038/nature11335 [DOI] [PubMed] [Google Scholar]
- Langfelder, P., & Horvath, S. (2008). WGCNA: An R package for weighted correlation network analysis. BMC Bioinformatics, 9, 559. 10.1186/1471-2105-9-559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, M., Gentleman, R., & Carey, V. (2009). rtracklayer: An R package for interfacing with genome browsers. Bioinformatics, 25(14), 1841–1842. 10.1093/bioinformatics/btp328 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence, M., Huber, W., Pagès, H., Aboyoun, P., Carlson, M., Gentleman, R., Morgan, M. T., & Carey, V. J. (2013). Software for computing and annotating genomic ranges. PLoS Computational Biology, 9(8), 1–10. 10.1371/journal.pcbi.1003118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lea, A. J., Vilgalys, T. P., Durst, P. A. P., & Tung, J. (2017). Maximizing ecological and evolutionary insight in bisulfite sequencing data sets. Nature Ecology & Evolution, 1(8), 1074–1083. 10.1038/s41559-017-0229-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenen, F. A. D., Muller, C. P., & Turner, J. D. (2016). DNA methylation: Conducting the orchestra from exposure to phenotype? Clinical Epigenetics, 8(1), 1–15. 10.1186/s13148-016-0256-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenth, R. (2019). emmeans: Estimated Marginal Means, aka Least‐Squares Means. [Google Scholar]
- Li, Z., Yao, K., & Cao, Y. (1999). Molecular cloning of a novel tissue‐specific gene from human nasopharyngeal epithelium. Gene, 237(1), 235–240. 10.1016/S0378-1119(99)00234-6 [DOI] [PubMed] [Google Scholar]
- Liew, Y. J., Howells, E. J., Wang, X., Michell, C. T., Burt, J. A., Idaghdour, Y., & Aranda, M. (2020). Intergenerational epigenetic inheritance in reef‐building corals. Nature Climate Change, 10(3), 254–259. 10.1038/s41558-0190687-2. [DOI] [Google Scholar]
- Lim, E., Xu, H., Wu, P., Posner, D., Wu, J., Peloso, G. M., Pitsillides, A. N., DeStefano, A. L., Adrienne Cupples, L., & Liu, C.‐T. (2018). Network analysis of drug effect on triglyceride‐associated DNA methylation. BMC Proceedings, 12(Suppl. 9), 109–116. 10.1186/s12919-018-0130-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lindner, M., Verhagen, I., Viitaniemi, H. M., Laine, V. N., Visser, M. E., Husby, A., & van Oers, K. (2021). Temporal changes in DNA methylation and RNA expression in a small song bird: Within‐ and between‐tissue comparisons. BMC Genomics, 22, 36. 10.1186/s12864-020-07329-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu, L., Xiao, Q., Gilbert, E. R., Cui, Z., Zhao, X., Wang, Y., Yin, H., Li, D., Zhang, H., & Zhu, Q. (2018). Whole‐transcriptome analysis of atrophic ovaries in broody chickens reveals regulatory pathways associated with proliferation and apoptosis. Scientific Reports, 8(1), 1–14. 10.1038/s41598-018-25103-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lourenço, D., Brauner, R., Lin, L., Perdigo, A. D., Weryha, G., Muresan, M., Guerra‐Junior, G., Maciel‐Guerra A. T., & Bashamboo, A. (2009). Mutations in NR5A1 associated with ovarian insufficiency. The New England Journal of Medicine, 360(12), 1200–1210. 10.1128/JB.185.18.5602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mäkinen, H., Viitaniemi, H. M., Visser, M. E., Verhagen, I., van Oers, K., & Husby, A. (2019). Temporally replicated DNA methylation patterns in great tit using reduced representation bisulfite sequencing. Scientific Data, 6(1), 1–7. 10.1038/s41597-019-0136-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McClerry, R. H., & Perrins, C. M. (1998). …temperature and egg‐laying trends. Nature, 391, 30–31. 10.1111/j.1748-7692.1988.tb00545.x [DOI] [Google Scholar]
- Meinsohn, M.‐C., Smith, O. E., Bertolin, K., & Murphy, B. D. (2019). The orphan nuclear receptors steroidogenic factor‐1 and liver receptor homolog‐1: structure, regulation, and essential roles in mammalian reproduction. Physiological Reviews, 99(2), 1249–1279. 10.1152/physrev.00019.2018 [DOI] [PubMed] [Google Scholar]
- Meissner, A., Mikkelsen, T. S., Gu, H., Wernig, M., Hanna, J., Sivachenko, A., Zhang, X., Bernstein, B. E., Nusbaum, C., Jaffe, D. B., Gnirke, A., Jaenisch, R., & Lander, E. S. (2008). Genome‐scale DNA methylation maps of pluripotent and differentiated cells. Nature, 454(7205), 766–770. 10.1038/nature07107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meredith, M., & Kruschke, J. (2020). HDInterval: Highest (Posterior) Density Intervals. R Package Version, (2), 2. Retrieved from https://cran.r‐project.org/package=HDInterval [Google Scholar]
- Murayama, C., Miyazaki, H., Miyamoto, A., & Shimizu, T. (2012). Luteinizing hormone (LH) regulates production of androstenedione and progesterone via control of histone acetylation of StAR and CYP17 promoters in ovarian theca cells. Molecular and Cellular Endocrinology, 350(1), 1–9. 10.1016/j.mce.2011.11.014 [DOI] [PubMed] [Google Scholar]
- Nardone, S., Sams, D. S., Zito, A., Reuveni, E., & Elliott, E. (2017). Dysregulation of cortical neuron DNA methylation profile in autism spectrum disorder. Cerebral Cortex, 27(12), 5739–5754. 10.1093/cercor/bhx250 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacis, A., Mailhot‐Léonard, F., Tailleux, L., Randolph, H. E., Yotova, V., Dumaine, A., Grenier, J.‐C., & Barreiro, L. B. (2019). Gene activation precedes DNA demethylation in response to infection in human dendritic cells. Proceedings of the National Academy of Sciences of the United States of America, 116(14), 6938–6943. 10.1073/pnas.1814700116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker, K. L. (2002). Steroidogenic factor 1: An essential mediator of endocrine development. Recent Progress in Hormone Research, 57(1), 19–36. 10.1210/rp.57.1.19 [DOI] [PubMed] [Google Scholar]
- Parmesan, C. (2007). Influences of species, latitudes and methodologies on estimates of phenological response to global warming. Global Change Biology, 13(9), 1860–1872. 10.1111/j.1365-2486.2007.01404.x [DOI] [Google Scholar]
- Parmesan, C., & Yohe, G. (2003). A globally coherent fingerprint of climate change impacts across natural systems. Nature, 421(6918), 37–42. 10.1038/nature01286 [DOI] [PubMed] [Google Scholar]
- Pegoraro, M., Bafna, A., Davies, N. J., Shuker, D. M., & Tauber, E. (2016). DNA methylation changes induced by long and short photoperiods in Nasonia. Genome Research, 26, 203–210. 10.1101/gr.196204.115.Freely [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perrier, C., Lozano del Campo, A., Szulkin, M., Demeyrier, V., Gregoire, A., & Charmantier, A. (2018). Great tits and the city: Distribution of genomic diversity and gene–environment associations along an urbanization gradient. Evolutionary Applications, 11(5), 593–613. 10.1111/eva.12580 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petchey, O. L., McPhearson, P. T., Gasey, T. M., & Morin, P. J. (1999). Environmental warming alters food‐web structure and ecosystem function. Nature, 402, 69–72. 10.1038/47023 [DOI] [Google Scholar]
- Powell, M. J. D. (2009). The BOBYQA algorithm for bound constrained optimization without derivatives. In Report No. DAMTP 2009/NA06, Centre for Mathematical Sciences, University of Cambridge, UK. [Google Scholar]
- Qu, M., Aronica, E., Boer, K., Fällmar, D., Kumlien, E., Nistér, M., Wester, K., Pontén, F., & Smits, A. (2009). DLG3/SAP102 protein expression in malformations of cortical development: A study of human epileptic cortex by tissue microarray. Epilepsy Research, 84(1), 33–41. [DOI] [PubMed] [Google Scholar]
- Quinlan, A. R., & Hall, I. M. (2010). BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26(6), 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy, D. B., & Sparks, T. H. (2000). Phenology of British butterflies and climate change. Global Change Biology, 6(4), 407–416. 10.1046/j.1365-2486.2000.00322.x [DOI] [Google Scholar]
- Rubenstein, D. R., Skolnik, H., Berrio, A., Champagne, F. A., Phelps, S., & Solomon, J. (2016). Sex‐specific fitness effects of unpredictable early life conditions are associated with DNA methylation in the avian glucocorticoid receptor. Molecular Ecology, 25, 1714–1728. 10.1111/mec.13483 [DOI] [PubMed] [Google Scholar]
- Saino, N., Albetti, B., Ambrosini, R., Caprioli, M., Costanzo, A., Mariani, J., Parolini, M., Romano, A., Rubolini, D., Formenti, G., Gianfranceschi, L., & Bollati, V. (2019). Inter‐generational resemblance of methylation levels at circadian genes and associations with phenology in the barn swallow. Scientific Reports, 9(1), 1–16. 10.1038/s41598-019-42798-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saino, N., Ambrosini, R., Albetti, B., Caprioli, M., De Giorgio, B., Gatti, E., Liechti, F., Parolini, M., Romano, A., Romano, M., Scandolara, C., Gianfranceschi, L., Bollati, V., & Rubolini, D. (2017). Migration phenology and breeding success are predicted by methylation of a photoperiodic gene in the barn swallow. Scientific Reports, 7(February), 1–10. 10.1038/srep45412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santure, A. W., Gratten, J., Mossman, J. A., Sheldon, B. C., & Slate, J. (2011). Characterisation of the transcriptome of a wild great tit Parus major population by next generation sequencing. BMC Genomics, 12, 283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schaper, S. V., Rueda, C., Sharp, P. J., Dawson, A., & Visser, M. E. (2011). Spring phenology does not affect timing of reproduction in the great tit (Parus major). Journal of Experimental Biology, 214(21), 3664–3671. 10.1242/jeb.059543 [DOI] [PubMed] [Google Scholar]
- Schilling, E., & Rehli, M. (2007). Global, comparative analysis of tissue‐specific promoter CpG methylation. Genomics, 90(3), 314–323. 10.1016/j.ygeno.2007.04.011 [DOI] [PubMed] [Google Scholar]
- Sepers, B., van den Heuvel, K., Lindner, M., Viitaniemi, H., Husby, A., & van Oers, K. (2019). Avian ecological epigenetics: Pitfalls and promises. Journal of Ornithology, 160, 1183–1203. 10.1007/s10336-019-01684-5 [DOI] [Google Scholar]
- Shannon, P., Markiel, A., Ozier, O., Baliga, N. S., Wang, J. T., Ramage, D., & Ideker, T. (2003). Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Research, 13, 2498–2504. 10.1101/gr.1239303.metabolite [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp, P. J. (2005). Photoperiodic regulation of seasonal breeding in birds. Annals of the New York Academy of Sciences, 1040, 189–199. 10.1196/annals.1327.024 [DOI] [PubMed] [Google Scholar]
- Shen, X. U., Bai, X., Xu, J., Zhou, M., Xu, H., Nie, Q., Lu, X., & Zhang, X. (2016). Transcriptome sequencing reveals genetic mechanisms underlying the transition between the laying and brooding phases and gene expression changes associated with divergent reproductive phenotypes in chickens. Molecular Biology Reports, 43(9), 977–989. 10.1007/s11033-016-4033-8 [DOI] [PubMed] [Google Scholar]
- Shinoda, K., Lei, H., Yoshii, H., Nomura, M., Nagano, M., Shiba, H., Sasaki, H., Osawa, Y., Ninomiya, Y., Niwa, O., Morohashi, K.‐I., & Li, E. N. (1995). Developmental defects of the ventromedial hypothalamic nucleus and pituitary gonadotroph in the Ftz‐F1 disrupted mice. Developmental Dynamics, 204(1), 22–29. 10.1002/aja.1002040104 [DOI] [PubMed] [Google Scholar]
- Stevenson, T. J. (2017). Environmental and hormonal regulation of epigenetic enzymes in the hypothalamus. Journal of Neuroendocrinology, 29(5), 1–9. 10.1111/jne.12471 [DOI] [PubMed] [Google Scholar]
- Stevenson, T. J. (2018). Epigenetic regulation of biological rhythms: An evolutionary ancient molecular timer. Trends in Genetics, 34(2), 90–100. 10.1016/j.tig.2017.11.003 [DOI] [PubMed] [Google Scholar]
- Stevenson, T. J., & Lincoln, G. (2017). Epigenetic mechanisms regulating circannual rhythms. In Kumar V. (Ed.), Biological timekeeping: Clocks, rhythms and behaviour (pp. 607–624). Springer. [Google Scholar]
- Stevenson, T. J., & Prendergast, B. J. (2013). Reversible DNA methylation regulates seasonal photoperiodic time measurement. Proceedings of the National Academy of Sciences, 110(41), 16651–16656. 10.1073/pnas.1402460111 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki, M. M., & Bird, A. (2008). DNA methylation landscapes: Provocative insights from epigenomics. Nature Reviews Genetics, 9, 465–476. 10.1038/nrg2341 [DOI] [PubMed] [Google Scholar]
- Tarpey, P., Parnau, J., Blow, M., Woffendin, H., Bignell, G., Cox, C., Cox, J., Davies, H., Edkins, S., Holden, S., Korny, A., Mallya, U., Moon, J., O’Meara, S., Parker, A., Stephens, P., Stevens, C., Teague, J., Donnelly, A., … Raymond, F. L. (2004). Mutations in the DLG3 gene cause nonsyndromic X‐linked mental retardation. American Journal of Human Genetics, 75(2), 318–324. 10.1086/422703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thackeray, S. J., Henrys, P. A., Hemming, D., Bell, J. R., Botham, M. S., Burthe, S., Helaouet, P., Johns, D. G., Jones, I. D., Leech, D. I., Mackay, E. B., Massimino, D., Atkinson, S., Bacon, P. J., Brereton, T. M., Carvalho, L., Clutton‐Brock, T. H., Duck, C., Edwards, M., … Wanless, S. (2016). Phenological sensitivity to climate across taxa and trophic levels. Nature, 535(7611), 241–245. 10.1038/nature18608 [DOI] [PubMed] [Google Scholar]
- Tylee, D. S., Kawaguchi, D. M., & Glatt, S. J. (2013). On the outside, looking in: A review and evaluation of the comparability of blood and brain “‐omes”. American Journal of Medical Genetics Part B: Neuropsychiatric Genetics, 162(7), 595–603. 10.1002/ajmg.b.32150 [DOI] [PubMed] [Google Scholar]
- van Eijk, K. R., De, J. S., Boks, M. P., Langeveld, T., Colas, F., Veldink, J. H., Ophoff, R. A. (2012). Genetic analysis of DNA methylation and gene expression levels in whole blood of healthy human subjects. BMC Genomics, 13(1), 1471–2164. 10.1186/1471-2164-13-636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Noordwijk, A. J., McCleery, R. H., & Perrins, C. M. (1995). Selection for the timing of great tit breeding in relation to caterpillar growth and temperature. The Journal of Animal Ecology, 64(4), 451. 10.2307/5648 [DOI] [Google Scholar]
- van Oers, K., Sepers, B., Sies, W., Gawehns, F., Verhoeven, K. J. F., & Laine, V. N. (2020). Epigenetics of animal personality: DNA methylation cannot explain the heritability of exploratory behavior in a songbird. Integrative and Comparative Biology, 60(6), 1517–1530. 10.1093/icb/icaa138 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhagen, I., Gienapp, P., Laine, V. N., Van Grevenhof, E. M., Mateman, A. C., Oers, K. V., & Visser, M. E. (2019). Genetic and phenotypic responses to genomic selection for timing of breeding in a wild songbird. Functional Ecology, 33, 1708–1721. 10.1111/1365-2435.13360 [DOI] [Google Scholar]
- Verhagen, I., Laine, V. N., Mateman, A. C., Pijl, A., de Wit, R., van Lith, B., Kamphuis, W., Viitaniemi, H. M., Williams, T. D., Caro, S. P., Meddle, S. L., Gienapp, P., van Oers, K., & Visser, M. E. (2019). Fine‐tuning of seasonal timing of breeding is regulated downstream in the underlying neuro‐endocrine system in a small songbird. Journal of Experimental Biology, 222, jeb202481. [DOI] [PubMed] [Google Scholar]
- Verhoeven, K. J. F., VonHoldt, B. M., & Sork, V. L. (2016). Epigenetics in ecology and evolution: What we know and what we need to know. Molecular Ecology, 25(8), 1631–1638. 10.1111/mec.13617 [DOI] [PubMed] [Google Scholar]
- Verhulst, E. C., Mateman, A. C., Zwier, M. V., & Caro, S. P. (2016). Evidence from pyrosequencing indicates that natural variation in animal personality is associated with DRD4 DNA methylation. Molecular Ecology, 25, 1801–1811. 10.1111/mec.13519 [DOI] [PubMed] [Google Scholar]
- Viitaniemi, H. M., Verhagen, I., Visser, M. E., Honkela, A., van Oers, K., & Husby, A. (2019). Seasonal variation in genome‐wide DNA methylation patterns and the onset of seasonal timing of reproduction in great tits. Genome Biology and Evolution, 11(3), 970–983. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser, M. E., Adriaensen, F., Van Balen, J. H., Dhondt, A., Dongen, S. V., Feu, C., Ivankina, E. V., Kerimov, A. B., de Laet, J., Matthysen, E., McCleery, R., & Orell, M. & Thomson, D. L. (2003). Variable responses to large‐scale climate change in European Parus populations. Proceedings of the Royal Society B: Biological Sciences, 270, 367–372. 10.1098/rspb.2002.2244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Visser, M. E., Holleman, L. J. M., & Caro, S. P. (2009). Temperature has a causal effect on avian timing of reproduction. Proceedings of the Royal Society B: Biological Sciences, 276(1665), 2323–2331. 10.1098/rspb.2009.0213 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakim, J., Goudenege, D., Perrot, R., Gueguen, N., Desquiret‐Dumas, V., Chao de la Barca, J. M., Dalla Rosa, I., Manero, F., Le Mao, M., Chupin, S., Chevrollier, A., Procaccio, V., Bonneau, D., Logan, D. C., Reynier, P., Lenaers, G., & Khiati, S. (2017). CLUH couples mitochondrial distribution to the energetic and metabolic status. Journal of Cell Science, 130(11), 1940–1951. 10.1242/jcs.201616 [DOI] [PubMed] [Google Scholar]
- Wang, F., Xu, H., Zhao, H., Gelernter, J., & Zhang, H. (2016). DNA co‐methylation modules in postmortem prefrontal cortex tissues of European Australians with alcohol use disorders. Scientific Reports, 6(January), 1–11. 10.1038/srep19430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Werner, L., Paclik, D., Fritz, C., Reinhold, D., Roggenbuck, D., & Sturm, A. (2012). Identification of pancreatic glycoprotein 2 as an endogenous immunomodulator of innate and adaptive immune responses. The Journal of Immunology, 189(6), 2774–2783. 10.4049/jimmunol.1103190 [DOI] [PubMed] [Google Scholar]
- Wilschut, R. A., Oplaat, C., Snoek, L.‐B., Kirschner, J., & Verhoeven, K. J. F. (2016). Natural epigenetic variation contributes to heritable flowering divergence in a widespread asexual dandelion lineage. Molecular Ecology, 25, 1759–1768. 10.1111/mec.13502 [DOI] [PubMed] [Google Scholar]
- Winkel, W., & Hudde, H. (1997). Long‐term trends in reproductive traits of tits (Parus major, P. caeruleus) and pied flycatchers ficedula hypoleuca. Journal of Avian Biology, 28(2), 187–190. [Google Scholar]
- Wishart, G. J., & Horrocks, A. J. (2000). Fertilization in birds. In Tarín J. J., & Cano A. (Eds.), Fertilization in protozoa and metazoan animals: Cellular and molecular aspects. Springer‐Verlag. [Google Scholar]
- Xu, J., Chen, G., Hermanson, P. J., Xu, Q., Sun, C., Chen, W., Kan, Q., Li, M., Crisp, P. A., Yan, J., Li, L., Springer, N. M., & Li, Q. (2019). Population‐level analysis reveals the widespread occurrence and phenotypic consequence of DNA methylation variation not tagged by genetic variation in maize. Genome Biology, 20(1), 1–16. 10.1186/s13059-019-1859-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- You, Y., Sawikowska, A., Neumann, M., Posé, D., Capovilla, G., Langenecker, T., Neher, R. A., Krajewski, P., & Schmid, M. (2017). Temporal dynamics of gene expression and histone marks at the Arabidopsis shoot meristem during flowering. Nature Communications, 8(May). 10.1038/ncomms15120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, B., & Horvath, S. (2005). A general framework for weighted gene co‐expression network analysis. Statistical Applications in Genetics and Molecular Biology, 4(1), 17. [DOI] [PubMed] [Google Scholar]
- Zhang, Y., Baheti, S., & Sun, Z. (2016). Statistical method evaluation for differentially methylated CpGs in base resolution next‐generation DNA sequencing data. Briefings in Bioinformatics, 19(3), 1–13. 10.1093/bib/bbw133 [DOI] [PubMed] [Google Scholar]
- Ziyatdinov, A., Vázquez‐santiago, M., Brunel, H., Martinez‐perez, A., Aschard, H., & Soria, J. M. (2018). lme4qtl: Linear mixed models with flexible covariance structure for genetic studies of related individuals. BMC Bioinformatics, 19(1), 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuur, A. F., Hilbe, J. M., & Ieno, E. N. (2013). A beginner’s guide to GLM and GLMM with R ‐ a frequentist and Bayesian perspective for ecologists. Highland Statistics Ltd. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Material
Table S1‐S22
Supplementary Material
Data Availability Statement
The data underlying this article are available in the NCBI BioProject database (http://www.ncbi.nlm.nih.gov/bioproject/) under BioProject PRJNA208335 and accession nos. SRX3209916–SRX3209919, and the code for statistical analyses is provided in the Supporting Informationn.