

Abstract
Catabolite control protein A (CcpA) is a master regulator of carbon source utilization and contributes to the virulence of numerous medically important Gram‐positive bacteria. Most functional assessments of CcpA, including interaction with its key co‐factor HPr, have been performed in nonpathogenic bacteria. In this study we aimed to identify the in vivo DNA binding profile of CcpA and assess the extent to which HPr is required for CcpA‐mediated regulation and DNA binding in the major human pathogen group A Streptococcus (GAS). Using a combination RNAseq/ChIP‐seq approach, we found that CcpA affects transcript levels of 514 of 1667 GAS genes (31%) whereas direct DNA binding was identified for 105 GAS genes. Three of the directly regulated genes encode the key GAS virulence factors Streptolysin S, PrtS (IL‐8 degrading proteinase), and SpeB (cysteine protease). Mutating CcpA Val301 to Ala (strain 2221‐CcpA‐V301A) abolished interaction between CcpA and HPr and impacted the transcript levels of 205 genes (40%) in the total CcpA regulon. By ChIP‐seq analysis, CcpAV301A bound to DNA from 74% of genes bound by wild‐type CcpA, but generally with lower affinity. These data delineate the direct CcpA regulon and clarify the HPr‐dependent and independent activities of CcpA in a key pathogenic bacterium.
Keywords: ChIP‐seq, HPr‐independent CcpA regulation, Streptococcus pyogenes
The carbon catabolite protein, CcpA, regulates carbohydrate utilization and virulence in concert with its phosphorylated co‐factor histine containing protein (HPr~P). In this study we engineered a CcpA isoform that does not interact with HPr~P. Using a combination of ChIPseq and RNAseq we report the in vivo binding sites of CcpA in group A Streptococcus. We also demonstrate that CcpA retains the ability to bind and regulate genes even when it cannot interact with HPr~P.
![]()
1. INTRODUCTION
Carbon catabolite repression (CCR) is a global process by which bacteria prioritize the use of favorable energy sources (Deutscher et al., 2006; Fujita, 2009). The core mechanism of CCR is an alteration in levels of proteins involved in metabolite transport and utilization which in turn is primarily achieved at the transcriptional level (Gorke & Stulke, 2008). The LacI‐GalR family transcriptional regulator catabolite control protein (CcpA) is a key mediator of CCR in many Gram‐positive bacteria (Henkin et al., 1991; Titgemeyer & Hillen, 2002; Warner & Lolkema, 2003). CcpA inactivation impacts ~15%–20% of the transcriptome of a broad array of Gram‐positive bacteria with the majority of impacted genes encoding proteins involved in carbohydrate and nitrogen utilization (Antunes et al., 2012; DebRoy et al., 2016; Seidl et al., 2009; Zeng et al., 2013). Importantly, CcpA also affects the transcript levels of genes encoding known and putative virulence factors in human Gram‐positive pathogens ranging from streptococci to clostridia (Iyer et al., 2005; Mertins et al., 2007; Seidl, Bischoff, et al., 2008; Varga et al., 2004). Consequently, CcpA inactivation in diverse organisms result in altered virulence‐related phenotypes such as extracellular capsule production, biofilm formation, lysis of red blood cells, and inter‐species signaling (Giammarinaro & Paton, 2002; Johnson et al., 2009; Kinkel & McIver, 2008; Seidl, Goerke, et al., 2008; Watson et al., 2013).
Although CcpA is clearly necessary to the virulence of numerous key Gram‐positive pathogens, understanding of CcpA physiologic function is mainly derived from studies in nonpathogenic bacteria (Warner & Lolkema, 2003). Based primarily on investigations in Bacillus species, CcpA is currently thought to impact gene expression by binding cis‐acting DNA known as catabolite response elements (cre) which are composed of the pseudo‐palindromic motif WTGNAANCGNWNNCWW (where W = A or T and N = any base) (Miwa et al., 2000; Schumacher et al., 2011; Stulke & Hillen, 2000). CcpA affinity for cre sites is significantly increased by the co‐effector molecule, histidine‐containing protein (HPr) phosphorylated at Ser46 (HPrSer46~P) (Aung‐Hilbrich et al., 2002; Deutscher et al., 2005; Shelburne et al., 2008). HPr is phosphorylated and dephosphorylated at Ser46 by HPr kinase/phosphorylase (HPrK/P), a bifunctional ATP‐dependent enzyme whose activity is responsive to intracellular energy status (Poncet et al., 2004). Thus, the interaction of CcpA with HPrSer46~P facilitates alteration in gene expression in response to metabolic changes (Deutscher et al., 2006). All major, invasive Gram‐positive pathogens contain highly conserved orthologs of CcpA, HPr, and HPrK/P with amino acid similarities ranging from 70% for CcpA to 85% for HPr. However, Bacillus species also contain Crh (catabolite repression HPr), which is an HPr‐like protein important for CcpA‐mediated gene regulation that is not present in typical Gram‐positive pathogens such as staphylococci and streptococci (Deutscher et al., 2006; Galinier et al., 1997; Schumacher et al., 2006).
Although the pseudo‐palindromic cre sequence seems to be the predominate site of CcpA‐DNA interaction in Bacillus species, there have been suggestions that CcpA may bind other DNA sites. Using in silico approaches, cre sites have been predicted in only a small percentage of genes whose transcript levels are significantly altered by CcpA inactivation (Carvalho et al., 2011; DebRoy et al., 2016). Additionally, a recent study in Clostridium acetobutylicum identified a CcpA binding motif (TGTAA/TTTACA) which is quite distinct from the previous cre motif (Yang et al., 2017). Moreover, in addition to the “classic” cre motif, a genome‐wide CcpA binding study identified a cre2 motif in Streptococcus suis (TTTTYHWDHHWWTTTY, where Y is C or T, H is A or C or T, and D is A or G or T) that was primarily important for CcpA function in the stationary phase (Willenborg et al., 2014). Although CcpA transcriptome analyses have been performed in a wide variety of bacteria, genome‐wide characterization of CcpA‐DNA binding is not currently available for a major, invasive Gram‐positive pathogen other than Streptococcus suis (Antunes et al., 2012; Buescher et al., 2012; Willenborg et al., 2014).
In addition to a sub‐optimal understanding of how CcpA interacts with DNA in invasive Gram‐positive bacteria, analysis of the role of HPr and HPrK/P in pathogenic bacteria has been quite limited (Mertins et al., 2007; Shelburne et al., 2008). In part, this is because in major Gram‐positive pathogens such as group A Streptococcus (GAS), Streptococcus pneumoniae, and Staphylococcus aureus, HPr or even HPrSer46~P appears to be essential, in contrast to what is observed in Bacillus species (Fleming et al., 2015). In Staphylococcus xylosus elimination of HPrSer46~P completely abolished CCR suggesting that CcpA absolutely requires HPrSer46~P to impact gene transcription (Jankovic & Bruckner, 2002). In contrast, CcpA regulation at cre2 sites in S. suis was postulated to be independent of HPr (Willenborg et al., 2014). As pathogens establish and propagate human infections, they are likely to encounter vastly different metabolic conditions, which in turn would be expected to alter HPrSer46~P levels. Thus, establishing whether CcpA can function independently of HPrSer46~P and which genes are regulated by CcpA in the absence of HPrSer46~P is necessary to fully understand the role that nutritional acquisition plays in modulation of pathogenesis and infection outcomes. Herein, we sought to determine the global DNA binding characteristics of CcpA in GAS and analyze the effect of blocking the interaction between CcpA and HPrSer46~P in order to broaden insight into the physiology underlying the critical contribution of CcpA to Gram‐positive pathogenesis.
2. RESULTS
2.1. A CcpA mutant that does not bind HPr
We chose to conduct our study in the emm1 GAS strain MGAS2221 because emm1 strains are leading causes of GAS infections, MGAS2221 is representative of the current emm1 strains causing human disease worldwide, and MGAS2221 has a fully sequenced genome and lacks mutations in known regulators that have been shown to impact the CcpA transcriptome (Shelburne et al., 2010; Sumby et al., 2005). Both HPr and HPrK/P are essential in GAS (Le Breton et al., 2015). Thus, to probe into the possible existence of a CcpA regulon independent of HPrSer46~P, we first attempted to generate an HPr‐S46A mutant. However, we were unable to generate a viable GAS HPr‐S46A mutant despite repeated efforts suggesting that, as in the case of S. pneumoniae (Fleming et al., 2015), a S46A mutation in GAS HPr is lethal, possibly due to HPrSer46~P dependent cellular functions that are essential. We also made several attempts to generate a HPrK‐D197A mutant which would prevent Ser46 phosphorylation of HPr, but this mutation was also not viable. As an alternate approach we sought to generate a GAS mutant which retains the ability for HPr to be phosphorylated, but does not permit interactions between HPrSer46~P and CcpA. In silico alanine scanning mutagenesis of the CcpA‐HPrSer46~P interface, modeled on the crystal structure of the Bacillus megaterium CcpA‐HPrSer46~P‐DNA complex, identified four amino acid residues of CcpA that are critical to CcpA‐HPrSer46~P interaction (Homeyer et al., 2007; Schumacher et al., 2004). We chose to alter one of these residues, valine 301, to alanine given that the V301 residue is conserved in all CcpA family proteins (Schumacher et al., 2004) and gene regulation studies have demonstrated that a CcpAV301 mutant is compromised in glucose‐mediated gene repression (Sprehe et al., 2007).
We first sought to test the hypothesis that the CcpAV301A mutation would impair the interaction of recombinant CcpA and HPr using surface plasmon resonance (SPR). To this end, we expressed and purified recombinant CcpA and CcpAV301A along with HPr. We used recombinant HPrK/P to phosphorylate HPr at Ser46 as previously described (Shelburne et al., 2010). As expected, wild type CcpA had about 30‐fold higher affinity (i.e., bound more tightly) for HPrSer46~P relative to HPr with K D value of 5.8 ± 0.5 versus 188 ± 18 µM (Figure 1). Consistent with our hypothesis that V301 is critical for CcpA‐HPrSer46~P interaction in GAS, recombinant CcpAV301A bound to HPrSer46~P with a K D value of 153 ± 17 µM which closely approximated that observed for wild type CcpA/HPr interaction (K D = 188 ± 18 µM)(Figure 1). The interaction between CcpAV301A and HPr is much weaker and the affinity could not be determined under the conditions used. The equilibrium dissociation constant values (K D) provided in the text are mean ± standard deviation obtained from two experiments while those in Figure 1 are from a representative experiment.
FIGURE 1.
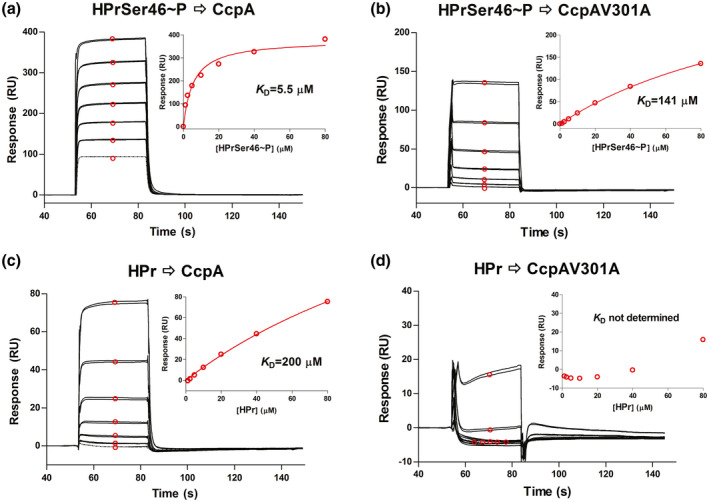
In vitro analysis of CcpA‐HPr interaction. Representative SPR analysis (n = 2) of the binding between CcpA and HPr recombinant proteins. HPrSer46~P and HPr (2‐fold serial dilutions from 1.25 to 80 μM) was injected in duplicate to (a & c) CcpA surface (3,700 RU) and (b & d) CcpAV301A surface (4,200 RU). The SPR response curves of bound protein are shown in black with lower curve corresponding to lower concentration of protein injected. The average responses at steady state (shown in red circles) were plotted as a function of the HPr concentration and the isotherm was fit to a one‐site binding (hyperbola) model (fitted curve shown in red) to determine equilibrium dissociation constant K D (inset)
Next, we used site directed mutagenesis to create the isoallellic mutant strain 2221‐CcpA‐V301A in the same parental background, the serotype emm1 strain MGAS2221, as our previously created 2221∆ccpA isolate (Table 1). Given that deleting ccpA has previously been shown to affect HPr/HPr~P ratios in other bacteria (Leboeuf et al., 2000; Ludwig et al., 2002), we used Phos‐tag gels to analyze HPr and HPr~P levels at the mid‐exponential phase of growth in nutrient rich Todd‐Hewitt media (0.2% glucose present in THY activates the HPr kinase and subsequent phosphorylation of HPrSer46~P). Under these conditions, the majority of HPr in strains MGAS2221 and 2221‐CcpA‐V301A was unphosphorylated with no significant difference of HPr/HPr~P ratios identified between the two strains (Figure 2a,b). Conversely, HPr~P levels were increased in strain 2221∆ccpA relative to the other two strains. A similar increase in HPr~P levels has been observed in ccpA mutants of B. subtilis and E. faecalis, and has been attributed to increased HPr kinase activity (Leboeuf et al., 2000; Ludwig et al., 2002). These data show that the V301A alteration in CcpA did not have significant effects on cellular HPr~P levels (Figure 2a,b). Additionally, the cellular levels of CcpA were similar between MGAS2221 and 2221‐CcpA‐V301A indicating that the amino acid variation did not impact CcpA autoregulation (note similar CcpA band densities for MGAS2221 and 2221‐CcpA‐V301A in Figure 2c, third panel).
TABLE 1.
Strains and plasmids used in this study
| Strain or plasmid | Description | Reference |
|---|---|---|
| Strains | ||
| MGAS2221 | Invasive clinical isolate, reference serotype M1 | Sumby et al. (2006) |
| 2221ΔccpA | MGAS2221 ΔccpA::spc | Shelburne et al. (2010) |
| 2221‐CcpA‐V301A | MGAS2221 with V301A change in CcpA | This study |
| BL21‐pET‐His2‐CcpA | E. coli producing recombinant wild type GAS CcpA | Shelburne et al. (2008) |
| BL21‐pET‐His2‐V301A | E. coli producing recombinant GAS CcpA with V301A change | This study |
| BL21‐pET‐His2‐HPr | E. coli producing recombinant GAS HPr | Shelburne et al. (2010) |
| BL21‐pET21a‐HPrKP | E. coli producing recombinant GAS HPrK/P | Shelburne et al. (2010) |
| Plasmids | ||
| pET‐His2‐CcpA | pET‐His2 plasmid with GAS ccpA gene | Shelburne et al. (2008) |
| pET‐His2‐V301A | pET‐His2 plasmid with GAS ccpA gene encoding V301A change | This study |
| pET‐His2‐HPr | pET‐His2 plasmid with GAS HPr gene | Shelburne et al. (2010) |
| pET21a‐HPrK/P | pET21a plasmid with GAS HPrK/P gene | Shelburne et al. (2010) |
| pBBL740 ccpAV301A | pBBL740 plasmid with GAS ccpA gene encoding V301A change | This study |
FIGURE 2.
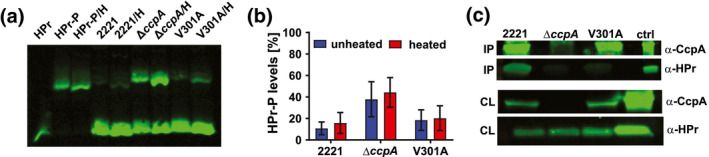
Characterization of the CcpAV301A mutant protein. (a) Representative Phostag Western blot (n = 2) and (b) graphical representation of the levels of phosphorylated HPr in lysates of wild type (2221), ccpA deletion mutant (∆ccpA) and the CcpAV301A mutant (V301A) grown to mid‐exponential phase. Lanes marked with “H” indicate heated samples. Purified recombinant HPr and HPr~P was used as controls. Error bars indicate standard deviation. (c) Co‐immunoprecipitation of HPr from lysates of indicated strains using anti‐CcpA antibody. “IP” and “CL” indicates immunoprecipitated material and cell lysate, respectively. The antibody for each sub‐panel is indicated on the right. Purified recombinant CcpA and HPr proteins were used as controls (ctrl)
To further test our hypothesis that CcpAV301A was not interacting with HPr~P in vivo, we performed immunoprecipitation reactions using anti‐CcpA antibody which detects both the wild type and CcpAV301A mutant protein (Figure 2c, first and third panels). Strains MGAS2221, 2221ΔccpA, and 2221‐CcpA‐V301A were cross‐linked and harvested at mid‐exponential phase. CcpA‐containing complexes were immunoprecipitated using a polyclonal anti‐CcpA antibody, and then, analyzed for the presence of HPr by western blotting using anti‐HPr antibody (Figure 2c). By quantitative analysis, 10 times more HPr was immunoprecipitated by CcpA antibody from strain MGAS2221 compared to strain 2221‐CcpA‐V301A. No HPr was immunoprecipitated by CcpA antibody in strain 2221ΔccpA. Taken together, we conclude that the V301A amino acid change essentially abolishes the ability of CcpA to interact with HPr~P both in vitro and in vivo.
2.2. Inability of CcpA to interact with HPr~P impacts the CcpA transcriptome
Given that CcpA‐HPrSer46~P interaction is thought to be critical for CcpA activity (Deutscher, 2008; Deutscher et al., 2005, 2006; Gorke & Stulke, 2008; Stulke & Hillen, 2000), we next sought to test the hypothesis that the CcpAV301A change would result in a transcriptome similar to that observed for a complete CcpA knockout. To this end, we performed RNAseq analyses, in quadruplicate, for strains MGAS2221, 2221ΔccpA, and 2221‐CcpA‐V301A grown to mid‐exponential phase in THY. The RNAseq data described herein is new and is not that reported in our previously published study (DebRoy et al., 2016). We did not observe any significant changes in ccpA transcript levels between MGAS2221 and 2221‐CcpA‐V301A, confirming that the V301A change does not affect transcription of ccpA itself (Figure S1). Principal component analysis showed that the data were reproducible and, contrary to our hypothesis, that the transcriptomes of the three strains were quite distinct (Figure 3a). We assigned transcript levels as being significantly different when there was a mean difference of at least 1.5‐fold and a p < .05 after accounting for multiple comparisons. We first sought to assess how the transcriptome of MGAS2221differed from 2221ΔccpA. Deletion of ccpA resulted in significantly different transcript levels of 514 genes representing ~31% of the 1667 genes in the MGAS2221 genome with sufficient transcript levels for analysis (Figure 3b; Table S1). More genes (361) had increased transcript levels in strain 2221∆ccpA (i.e., were CcpA repressed) compared to genes (153) whose transcript levels were lower in 2221∆ccpA (i.e., were CcpA activated), which is in accordance with previous observations (Carvalho et al., 2011; DebRoy et al., 2016; Kietzman & Caparon, 2011; Shelburne et al., 2008). The transcript levels of 12 virulence factor encoding genes were impacted by CcpA inactivation including speB, speA2, sic, spd, ska, grab, the streptolysin S (sag) operon, the nga‐slo operon, and prtS (Table 2). Consistent with the central role of CcpA in carbon source acquisition and utilization, the transcript levels of genes encoding 13/14 of the phosphotransferase systems (PTS) present in MGAS2221 were significantly altered by CcpA inactivation as were genes encoding four ATP binding cassette (ABC) carbohydrate transporters (Table 3). Cluster of orthologous group (COG) analysis revealed that genes whose transcript levels were significantly impacted by CcpA inactivation were more likely to be in groups C (energy production and conversion) and G (carbohydrate transport and metabolism) relative to unaffected genes (Figure S2a,b).
FIGURE 3.
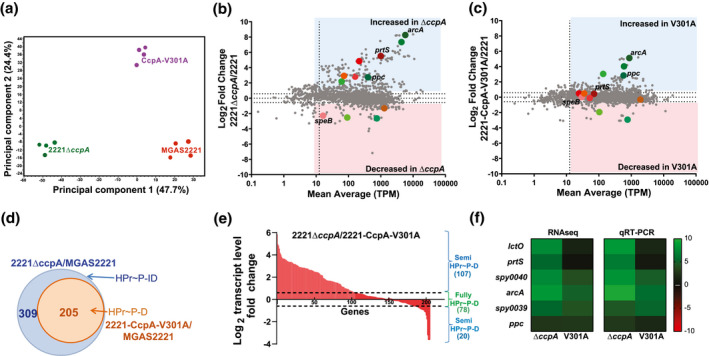
Impact of CcpAV301A mutation on the GAS transcriptome. (a) Principal component analysis showing that the transcriptomes of MGAS2221, 2221∆ccpA and 2221‐CcpA‐V301A are distinct. Each strain has four biological replicates. MA plots displaying transcriptome comparisons of (b) ccpA deletion (∆ccpA/MGAS2221) and (c) isoallelic CcpA V301A mutant (2221‐CcpA‐V301A/MGAS2221) to wild type MGAS2221. Colored quadrants include transcripts that are differentially regulated in each comparison as per the criteria outlined in the text. Select genes are color coded to indicate if they are HPr~P‐dependent (shades of green) or HPr~P‐independent (shades of red). (d) Venn diagram showing the subsets of ccpA‐affected genes that are HPr~P‐independent (HPr‐ID) and HPr~P‐dependent (HPr‐D). The strain comparisons used to generate these subsets are indicated in their respective colors. (e) Waterfall plot showing the gradation in the magnitude of the transcriptional impact of the CcpAV301A mutation on the HPr~P‐dependent genes. The fold change for HPr~P‐dependent genes between strains 2221∆ccpA and 2221‐CcpA‐V301A, as observed in the RNAseq data, is plotted. The genes within the dotted lines are fully HPr~P‐dependent, while those outside are HPr~P semi‐dependent. The number of genes in each category are specified in parentheses. (f) Gene transcript levels of selected CcpA‐impacted genes that exhibit varying transcriptional effects of the CcpAV301A mutation by RNAseq analysis were validated by targeted Taqman qRT‐PCR analysis. Transcript levels in strains 2221∆ccpA and 2221‐CcpA‐V301A are shown relative to wild type MGAS2221. For Taqman qRT‐PCR, data are mean ± standard deviation of two biological replicates, with two technical replicates, done on two separate days (n = 8)
TABLE 2.
Influence of CcpA‐HPr~P interaction on virulence gene regulation and DNA binding
| M5005 spy# | Gene | CcpA repressed vs. activated | HPr~P dependent vs. independent | If HPr~P dependent : semi or fully | Bound by CcpA | Bound by CcpAV301A |
|---|---|---|---|---|---|---|
| 0139‐0141 | nga‐slo | Repressed | Dependent (nga) | Semi | No | No |
| 0341 | prtS | Repressed | Independent | Yes | Yes | |
| 0351 | spyA | Repressed | Independent | No | No | |
| 0562‐70 | Streptolysin S operon | Repressed | Dependent | Semi | Yes | Yes |
| 0996 | speA2 | Repressed | Independent | No | No | |
| 1106 | grab | Activated | Dependent | Semi | No | No |
| 1540 | endoS | Activated | Dependent | Semi | No | No |
| 1684 | ska | Repressed | Independent | No | No | |
| 1711 | lmb | Repressed | Independent | No | No | |
| 1718 | sic | Repressed | Independent | No | No | |
| 1735 | speB | Activated | Independent | Yes | Yes | |
| 1738 | spd | Repressed | Independent | No | No |
TABLE 3.
Influence of CcpA‐HPr~P interaction on regulation and DNA binding of genes encoding carbohydrate transport systems of MGAS2221
| M5005 spy# | Putative transported carbohydrate | Transporter type | CcpA repressed vs. activated | HPr~P dependent vs. independent | Bound by CcpA | Bound by CcpAV301A |
|---|---|---|---|---|---|---|
| 0212‐0219 | Sialic acid | ABC | Repressed | Independent | Yes | No |
| 0475 | Β‐glucoside | PTS | Repressed | Semi‐dependent | No | No |
| 0521 | N‐acetylglucosamine | PTS | Repressed | Independent | No | No |
| 0662 | Fructose | PTS | Repressed | Fully ‐dependent | No | No |
| 0780‐0783 | Mannose/fructose | PTS | Repressed | Semi‐dependent | No | No |
| 1058‐1060 | Maltose | ABC | Repressed | Semi‐dependent | Yes | Yes |
| 1067‐1062 | Maltodextrin | ABC | Repressed | Semi‐dependent | Yes | Yes |
| 1083‐1079 | Cellobiose | PTS | Repressed | Semi‐dependent | Yes | Yes |
| 1310‐1308 | Unknown sugar | ABC | Repressed | Semi‐dependent | No | No |
| 1399‐1401 | Galactose | PTS | Repressed | Semi‐dependent | No | No |
| 1479‐1481 | Mannose | PTS | Repressed | Semi‐dependent | Yes | Yes |
| 1542 | Sucrose | PTS | Activated | Semi‐dependent | No | No |
| 1634‐1633 | Lactose | PTS | Activated | Fully ‐dependent | No | No |
| 1664‐1662 | Mannitol | PTS | Repressed | Semi‐dependent | No | No |
| 1692 | Maltose | PTS | Repressed | Independent | Yes | Yes |
| 1746‐1744 | Cellobiose | PTS | Repressed | Semi‐dependent | Yes | No |
| 1784 | Trehalose | PTS | Repressed | Semi‐Dependent | No | No |
Abbreviations: ABC, ATP binding cassette; PTS, phosphotransferase system;
Next, we compared the transcriptomes of 2221‐CcpA‐V301A and MGAS 2221 (Figure 3c) and analyzed transcript levels in strain 2221‐CcpA‐V301 for genes whose transcript levels were significantly different between strains MGAS2221 and 2221ΔccpA. If the CcpA‐HPr~P interaction is critical to regulation of a particular CcpA‐impacted gene then we would expect that transcript levels for said gene to be significantly different between strains MGAS221 and 2221‐CcpA‐V301A (e.g., compare arcA and ppc between Figure 3b,c). However, of the 514 genes whose transcript levels were significantly affected by CcpA inactivation, only 205 (40%) also had significantly different transcript levels in strain 2221‐CcpA‐V301A relative to strain MGAS2221 (Figure 3d). We considered these to be HPr~P‐dependent genes because eliminating the interaction between CcpA and HPr~P impacted their transcript levels (Table S1). The remaining 309 (60%) genes whose transcript levels were impacted by CcpA inactivation did not have significantly different transcript levels between strains 2221‐CcpA‐V301A and MGAS2221 (e.g., compare prtS and speB between Figure 3b,c), and were, therefore, denoted as HPr~P‐independent (Figure 3d) (Table S1). A significantly higher percentage of the HPr~P‐dependent genes were CcpA repressed (157/205, 77%) compared to HPr~P‐independent genes (204/309, 66%, p = .01 by Fisher's exact test). For the 12 virulence factor encoding genes/operons impacted by CcpA inactivation, only four were HPr~P‐dependent (nga‐slo, sag operon, grab, and endoS). Conversely, of the 17 CcpA‐regulated genes/operons encoding carbohydrate transport systems, 14 were HPr~P‐dependent (Tables 2 and 3). Similarly, COG analysis showed that, the HPr~P‐dependent genes were more likely to be in category G (carbohydrate transport and metabolism) and less likely to be in category L (replication and repair), M (cell wall/membrane/envelop biogenesis), and S (function unknown) relative to HPr~P‐independent genes (Figure S2c,d).
We next sought to determine whether the loss of CcpA‐HPr~P interaction was equivalent to complete CcpA inactivation in terms of the absolute effect on gene expression by using fold‐change (FC) comparison of the HPr~P‐dependent genes. If the CcpA‐HPr~P complex is essential for CcpA‐regulation of a particular gene, then, there should be no significant difference in transcript levels between strains 2221ΔccpA and 2221‐CcpA‐V301A. In fact, similar transcript levels in these two strains were observed for only 78 (38%) of the 205 HPr~P‐dependent genes, which we subsequently considered as fully HPr~P‐dependent (i.e., the effect of eliminating CcpA‐HPr~P interaction was equivalent to the effect of total CcpA inactivation) (Table S1). For the remaining 127/205 (62%) genes, transcript levels were significantly different between strains 2221∆ccpA and 2221‐CcpA‐V301A. Given that CcpA is primarily a repressor, for these 127 genes the loss of interaction with HPr~P resulted in transcript levels that were generally lower than complete CcpA inactivation, but the magnitude of effect varied for individual genes (Figure 3e). We designated these genes as semi‐HPr~P‐dependent (Table S1). All four HPr~P‐dependent virulence factor encoding genes/operons (nga‐slo operon, sag operon, grab, and endoS) were semi‐HPr~P‐dependent (Table 2). Of the 14 HPr~P‐dependent carbohydrate utilization encoding operons, only two (lactose and fructose transporters) were fully HPr~P‐dependent (Table 3). We chose CcpA‐regulated genes that exhibit varying degrees of transcriptional impact upon disrupting the CcpA‐HPr~P interaction and verified the changes in transcript levels using targeted qRT‐PCR (Figure 3f). Taken together, our data show that CcpA influences a significant proportion of the GAS genome even without HPr~P interaction and that the quantitative impact of HPr~P on CcpA‐mediated regulation is distinct for different genes.
2.3. In vivo interaction of CcpA with GAS chromatin
There are limited genome wide assessments of CcpA binding to DNA in pathogenic bacteria and none for a strain in which CcpA‐HPr~P interaction has been abrogated (Willenborg et al., 2014). Therefore, we performed ChIP‐seq analysis for strains MGAS2221 and 2221‐CcpA‐V301A grown to mid‐exponential phase in THY media to analyze CcpA chromatin occupancy profiles and assess how the reduced ability of CcpA to interact with HPr~P affects CcpA‐DNA binding. The 2221∆ccpA strain was used as a control to ensure the specificity of our findings.
We identified 76 CcpA binding sites in strain MGAS2221, including in the promoter region of ccpA itself which is consistent with a previous in vitro analysis of CcpA binding (Almengor et al., 2007) (Table 4). The genome‐wide distribution of these sites is shown in Figure 4a. The lack of significant enrichment at any of these sites in strain 2221∆ccpA indicates that these are true in vivo binding sites for the CcpA protein. The CcpA binding sites were mostly (67%) in predicted promoter regions (i.e., within 500 bps of a translational start site) (Fujita, 2009), which is in accord with other CcpA ChIP‐seq analyses (Antunes et al., 2012; Buescher et al., 2012; Willenborg et al., 2014). Of the 76 enriched regions, 46 sites (61%) were associated with genes whose transcript levels were significantly altered in strain 2221∆ccpA relative to strain MGAS221. These 46 sites were associated with 45 operons containing 107 genes. The discrepancy between numbers of binding sites and operons affected is due to the occasional identification of multiple distinct binding sites for the same gene. Thus, only 107 (21%) of the 514 genes whose transcript levels were significantly impacted by CcpA inactivation in strain MGA2221were identified as being directly regulated by CcpA (Figure 4b). The vast majority of sites enriched in the ChIP‐seq analysis (88%) correlated with genes which had increased transcript levels in strain 2221∆ccpA relative to MGAS2221 (i.e., were CcpA‐repressed). By COG analysis, genes directly regulated by CcpA were significantly more likely to be in category C (energy production and conversion) and G (carbohydrate transport and metabolism) (Figure 4c).
TABLE 4.
CcpA binding sites identified by ChIP‐seq analysis and associated genes
| CBSa | Bound by WT | Bound by V301A | cre | cre locationb | cre sequence | Locus tagc | Gene | Annotation | COG | Regulated by CcpA | HPr‐dependency |
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Yes | Yes | Yes | Prom | AAAGAAAGCGGTTTCA | M5005_Spy_0039 | adh2 | Bifunctional acetaldehyde‐CoA/alcohol dehydrogenase | C | Yes | Semi dependent |
| 2 | Yes | Yes | Yes | Prom | ACAGAAAACGATTTCA | M5005_Spy_0094 | ackA | Acetate kinase | F | Yes | Independent |
| 3 | Yes | No | Yes | ORF | ATGGTGGCGTTTTCTT | M5005_Spy_0128 | ntpE | V‐type ATP synthase subunit E | C | Yes | Independent |
| 4 | Yes | Yes | Yes | Prom | AACGAAAACCTTTTCA | M5005_Spy_0180 | – | Hypothetical protein | D | No | NA |
| 4 | Yes | Yes | Yes | Prom | AACGAAAACCTTTTCA | M5005_Spy_0185 | pgi | pgi | G | No | NA |
| 5 | Yes | No | Yes | Prom | TTGAAAGCGCTTTATT | M5005_Spy_0212 | – | N‐acetylmannosamine‐6‐phosphate 2‐epimerase | G | Yes | Independent |
| 6 | Yes | Yes | Yes | Prom | AAAGAAAGCCCTTTCC | M5005_Spy_0233 | plr | Aldehyde dehydrogenase | G | No | NA |
| 7 | Yes | Yes | Yes | Prom | AATGTAAGCGCTAACAAAAT | M5005_Spy_0339 | exoA | Exodeoxyribonuclease | L | Yes | Independent |
| 7 | Yes | Yes | Yes | Prom | AATGTAAGCGCTAACAAAAT | M5005_Spy_0340 | lctO | L‐lactate oxidase | C | Yes | Semi dependent |
| 8 | Yes | Yes | Yes | ORF | TAGGAAGCGTTTTCTT | M5005_Spy_0341 | prtS | Peptidase S8 | O | Yes | Independent |
| 9 | Yes | No | Yes | ORF | GTGCAAGCGCTTTGAT | M5005_Spy_0362 | gcaD | Glucosamine‐1‐phosphate acetyltransferase | M | No | NA |
| 10 | Yes | No | No | – | – | M5005_Spy_0416 | Glutamine cyclotransferase | O | No | NA | |
| 10 | Yes | No | No | – | – | M5005_Spy_0417 | pcp | Pyrrolidone‐carboxylate peptidase | O | No | NA |
| 11 | Yes | No | No | – | – | M5005_Spy_0417 | pcp | Pyrrolidone‐carboxylate peptidase | O | No | NA |
| 12 | Yes | No | Yes | Prom | TTGAAAACTTTTTCAA | M5005_Spy_0424 | ccpA | Catabolite control protein A | K | Yes | Independent |
| 13 | Yes | No | No | – | – | M5005_Spy_0495 | lysS | Lysine‐‐tRNA ligase | J | No | NA |
| 13 | Yes | No | No | – | – | M5005_Spy_0496 | Haloacid dehalogenase | S | No | NA | |
| 14 | Yes | No | No | – | – | M5005_Spy_0504 | pepF | Oligoendopeptidase F | E | No | NA |
| 15 | Yes | Yes | Yes | ORF | TTTGGGAACGATTTCTCAAG | M5005_Spy_0771 | – | CRISPR‐associated endonuclease Cas2 | L | No | NA |
| 16 | Yes | Yes | Yes | Prom | AAATAAAGCGCTTACT | M5005_Spy_0505 | ppc | Phosphoenolpyruvate carboxylase | H | Yes | Fully dependent |
| 17 | Yes | Yes | Yes | Prom | GAGAAAACGTTTTAGT | M5005_Spy_0512 | – | Sugar phosphate phosphatase | S | Yes | Fully dependent |
| 18 | Yes | No | Yes | Prom | TTGACACCGTTTTCAT | M5005_Spy_0533 | – | Hypothetical protein | S | Yes | Independent |
| 18 | Yes | No | Yes | Prom | TTGACACCGTTTTCAT | M5005_Spy_0534 | – | Acetoin reductase | IQ | Yes | Semi dependent |
| 19 | Yes | No | Yes | ORF | GAAGATATCGCTTCTA | M5005_Spy_0556 | eno | Phosphopyruvate hydratase | F | No | NA |
| 20 | Yes | No | Yes | Prom | TATTATATCGATTTCT | M5005_Spy_0555 | – | Hypothetical protein | S | No | NA |
| 20 | Yes | No | Yes | Prom | TATTATATCGATTTCT | M5005_Spy_0556 | eno | Phosphopyruvate hydratase | F | No | NA |
| 21 | Yes | Yes | Yes | Prom | AAGAAAGGGTTTACAT | M5005_Spy_0562 | sagA | Streptolysin S family bacteriocin | Yes | Semi dependent | |
| 22 | Yes | No | Yes | ORF | ATGGAAGCTTTTTCAG | M5005_Spy_0622 | – | Alkaline phosphatase | M | No | NA |
| 22 | Yes | No | Yes | ORF | ATGGAAGCTTTTTCAG | M5005_Spy_0625 | aroF | Chorismate synthase | E | No | NA |
| 23 | Yes | No | Yes | ORF | GTGAAGGGTTTATCAT | M5005_Spy_0772 | – | Type II‐A CRISPR‐associated protein Csn2 | S | No | NA |
| 24 | Yes | No | No | – | – | M5005_Spy_0778 | msrB | Peptide‐methionine (R)‐S‐oxide reductase | O | No | NA |
| 25 | Yes | Yes | Yes | ORF | GAAGATAACGATTTCA | M5005_Spy_0817 | dacA1 | D‐alanyl‐D‐alanine carboxypeptidase | M | No | NA |
| 26 | Yes | No | Yes | ORF | TTGTAAGCGCTACCGA | M5005_Spy_0823 | folQ | Dihydroneopterin aldolase | H | No | NA |
| 27 | Yes | Yes | Yes | Prom | AAGAAAGGGTTTTCAA | M5005_Spy_0834 | – | Alcohol dehydrogenase | E | Yes | Semi dependent |
| 27 | Yes | Yes | Yes | Prom | AAGAAAGGGTTTTCAA | M5005_Spy_0835 | – | Acid phosphatase/phosphotransferase | S | Yes | Semi dependent |
| 28 | Yes | No | Yes | Prom | ACTGATAACGCTTCCAA | M5005_Spy_0873 | ldh | L‐lactate dehydrogenase | C | No | NA |
| 28 | Yes | No | Yes | Prom | ACTGATAACGCTTCCAA | M5005_Spy_0874 | gyrA | DNA gyrase subunit A | L | No | NA |
| 29 | Yes | No | No | – | – | M5005_Spy_0925 | rnhB | Hypothetical protein | F | No | NA |
| 30 | Yes | Yes | Yes | Prom | CTTGAAACCGCTTGCT | M5005_Spy_0934 | ‐ | Lipoate‐‐protein ligase | H | Yes | Fully dependent |
| 31 | Yes | No | Yes | Prom | AATGAAAGCGTTTATA | M5005_Spy_0938 | pgmA | Phosphoglucomutase | G | Yes | Semi dependent |
| 32 | Yes | No | No | – | – | M5005_Spy_0938 | pgmA | Phosphoglucomutase | G | Yes | Semi dependent |
| 33 | Yes | No | No | – | – | M5005_Spy_0938 | pgmA | Phosphoglucomutase | G | Yes | Semi dependent |
| 34 | Yes | Yes | Yes | ORF | TAAGATACCGCTTGCA | M5005_Spy_1055 | glgP | Maltodextrin phosphorylase | G | Yes | Independent |
| 34 | Yes | Yes | Yes | ORF | TAAGATACCGCTTGCA | M5005_Spy_1058 | malE | Maltose/maltodextrin‐binding protein | G | Yes | Independent |
| 35 | Yes | Yes | Yes | Prom | CTGCAAGCGGTTGCAT | M5005_Spy_1057 | malR | LacI family transcriptional regulator | K | Yes | Independent |
| 35 | Yes | Yes | Yes | Prom | CTGCAAGCGGTTGCAT | M5005_Spy_1058 | malE | Maltose/maltodextrin‐binding protein | G | Yes | Independent |
| 36 | Yes | Yes | Yes | Prom | ATCGTAATCGCTTTCA | M5005_Spy_1067 | malX | Sugar ABC transporter substrate‐binding protein | G | Yes | Semi dependent |
| 37 | Yes | Yes | Yes | Prom | TTAGAAAACGCTTTCT | M5005_Spy_1083 | bglG | Transcription antiterminator BglG | G | Yes | Semi dependent |
| 38 | Yes | Yes | Yes | ORF | CTAAAAGCGTTTTCTC | M5005_Spy_1096 | – | Thioesterase | Q | No | NA |
| 39 | Yes | No | Yes | Prom | CATGATAACCCTTACA | M5005_Spy_1122 | nrdH | NrdH‐redoxin | O | Yes | Independent |
| 39 | Yes | No | Yes | Prom | CATGATAACCCTTACA | M5005_Spy_1121 | ptsH | Phosphocarrier protein HPr | G | Yes | NA |
| 40 | Yes | Yes | Yes | Prom | CAAGAAATCGCTTTCT | M5005_Spy_1235 | – | Phosphomannomutase | G | Yes | Semi dependent |
| 41 | Yes | No | Yes | ORF | CAGAAAACTCTTTCTT | M5005_Spy_1250 | ftsA | Cell division protein FtsA | D | No | NA |
| 42 | Yes | No | Yes | Prom | ATGGAATCGCTTTCTA | M5005_Spy_1258 | – | Hypothetical protein | S | Yes | Independent |
| 43 | Yes | No | Yes | ORF | ATCGTAAGCGCCTCCA | M5005_Spy_1265 | – | Ribose operon repressor | No | NA | |
| 44 | Yes | No | Yes | ORF | GTAAAATCTTTTTCTG | M5005_Spy_1272 | – | Arginine:ornithine antiporter | S | Yes | Semi dependent |
| 45 | Yes | No | No | – | – | M5005_Spy_1274 | N‐acetyltransferase | K | Yes | Semi dependent | |
| 46 | Yes | No | No | – | – | M5005_Spy_1274 | N‐acetyltransferase | K | Yes | Semi dependent | |
| 47 | Yes | No | No | – | – | M5005_Spy_1274 | N‐acetyltransferase | K | Yes | Semi dependent | |
| 48 | Yes | Yes | Yes | Prom | TGAGTAATCGCTTACA | M5005_Spy_1275 | arcA | Arginine deiminase | E | Yes | Semi dependent |
| 48 | Yes | Yes | Yes | Prom | TGAGTAATCGCTTACA | M5005_Spy_1277 | ahrC.2 | Arginine regulator | K | Yes | Semi dependent |
| 49 | Yes | Yes | Yes | ORF | CTGCAATCGTTTACTT | M5005_Spy_1319 | – | RNA methyltransferase | J | No | NA |
| 49 | Yes | Yes | Yes | ORF | CTGCAATCGTTTACTT | M5005_Spy_1323 | – | Hypothetical protein | L | No | NA |
| 50 | Yes | Yes | Yes | Prom | CTTGAAGCGCTTACTT | M5005_Spy_1328 | – | YigZ family protein | S | Yes | Independent |
| 50 | Yes | Yes | Yes | Prom | CTTGAAGCGCTTACTT | M5005_Spy_1325 | – | Ribosome‐associated factor Y | J | Yes | Fully dependent |
| 51 | Yes | No | No | – | – | M5005_Spy_1366 | Penicillin‐binding protein 2X | M | No | NA | |
| 52 | Yes | Yes | Yes | ORF | GAGAAAAGGATTTCAT | M5005_Spy_1367 | ftsL | Cell division protein FtsL | D | No | NA |
| 53 | Yes | Yes | Yes | Prom | AAGTAAGCGTTTTCCT | M5005_Spy_1381 | glpK | Glycerol kinase | F | Yes | Independent |
| 54 | Yes | No | No | – | – | M5005_Spy_1382 | Hypothetical protein | T | Yes | Independent | |
| 55 | Yes | Yes | Yes | Prom | CTGTAAGCGATTACTT | M5005_Spy_1387 | – | 2,5‐diketo‐D‐gluconic acid reductase | C | Yes | Independent |
| 56 | Yes | No | No | – | – | M5005_Spy_1448 | Nuclease | S | No | NA | |
| 57 | Yes | Yes | Yes | Prom | GTGAAAACGTTTTAAA | M5005_Spy_1477 | – | NCS2 family permease | S | Yes | Semi dependent |
| 57 | Yes | Yes | Yes | Prom | GTGAAAACGTTTTAAA | M5005_Spy_1479 | manL | PTS mannose transporter subunit EIIAB | G | Yes | Semi dependent |
| 58 | Yes | Yes | Yes | Prom | AAAGAAAACGTTTTCT | M5005_Spy_1496 | phaB | Enoyl‐CoA hydratase | I | Yes | Independent |
| 59 | Yes | No | No | – | – | M5005_Spy_1497 | dnaJ | Chaperone DnaJ | O | Yes | Fully dependent |
| 60 | Yes | No | Yes | Prom | AAAGAAAACACTTGCA | M5005_Spy_1503 | – | Histidine phosphatase family protein | G | Yes | Independent |
| 61 | Yes | No | No | – | – | M5005_Spy_1513 | Aminotransferase | E | No | NA | |
| 61 | Yes | No | No | – | – | M5005_Spy_1514 | Universal stress protein UspA | T | No | NA | |
| 62 | Yes | Yes | Yes | Prom | TGGGAAAACGTTTCCT | M5005_Spy_1569 | pfl | Formate acetyltransferase | C | Yes | Independent |
| 63 | Yes | No | No | – | – | M5005_Spy_1575 | norA | MFS transporter | EGP | Yes | Fully dependent |
| 64 | Yes | No | Yes | Prom | TTTAAAGCTTTTTAA | M5005_Spy_1599 | pgk | Phosphoglycerate kinase | F | No | NA |
| 65 | Yes | Yes | Yes | Prom | ATAAAAGCGTTATCTC | M5005_Spy_1624 | – | Hypothetical protein | No | NA | |
| 66 | Yes | Yes | Yes | ORF | AGAGAAACCGGTACCA | M5005_Spy_1627 | salY | ABC transporter permease | V | No | NA |
| 67 | Yes | Yes | Yes | Prom | TGCGCAAGCGCTTGCA | M5005_Spy_1680 | pulA | Pullulanase | G | No | NA |
| 68 | Yes | Yes | Yes | Prom | GATGCAATCGCTTGCA | M5005_Spy_1692 | – | PTS maltose | G | Yes | Independent |
| 69 | Yes | Yes | Yes | ORF | GTGATAGCGCTATCTT | M5005_Spy_1734 | speB | Streptopain | M | Yes | Independent |
| 69 | Yes | Yes | Yes | ORF | GTGATAGCGCTATCTT | M5005_Spy_1736 | – | Hypothetical protein | Yes | Independent | |
| 70 | Yes | No | Yes | Prom | TTGTAATCGTTTACAT | M5005_Spy_1746 | – | PTS cellobiose transporter subunit IIA | G | Yes | Semi dependent |
| 71 | Yes | No | No | – | – | M5005_Spy_1758 | Dipeptidase | M | Yes | Semi dependent | |
| 72 | Yes | Yes | Yes | Prom | GTGAAAGCGTTATCGT | M5005_Spy_1758 | – | Dipeptidase | M | Yes | Semi dependent |
| 73 | Yes | Yes | Yes | Prom | ATGTAAGCGTTATCTAA | M5005_Spy_1772 | – | Glutamate formimidoyltransferase | E | Yes | Independent |
| 73 | Yes | Yes | Yes | Prom | ATGTAAGCGTTATCTAA | M5005_Spy_1770 | hutI | Imidazolonepropionase | Q | Yes | Semi dependent |
| 74 | Yes | Yes | Yes | Prom | CATGAAAACGCCTCCA | M5005_Spy_1779 | rpsB | ATP‐binding protein | T | Yes | Semi dependent |
| 75 | Yes | No | No | – | – | M5005_Spy_1807 | argR2 | Arginine repressor | K | Yes | Independent |
| 76 | Yes | Yes | Yes | ORF | ACAGATAACGCTTACT | M5005_Spy_1865 | htrA | Serine protease | O | No | NA |
Abbreviation: NA, not applicable.
CcpA binding site number
Prom, binding site located in noncoding promoter region upstream of ATG. ORF, binding site identified within coding region of gene.
Locus tag numbers are based on the MGAS5005 genome.
FIGURE 4.
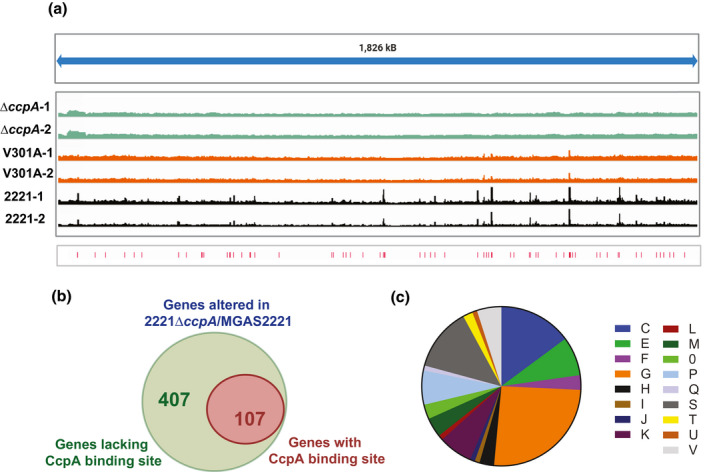
Characterization of in vivo DNA binding of CcpA. (a) Linear representation of indicated strains showing the CcpA binding sites as determined by ChIP‐seq analysis. Replicates of each strain analyzed are shown and peak positions are indicated by tally marks in the bottom sub‐panel. (b) Venn diagram showing proportion of genes identified as CcpA‐regulated by RNAseq that have CcpA binding sites as identified in our ChIP‐seq analysis. (c) COG distribution of genes that have CcpA binding sites and are transcriptionally altered in 2221∆ccpA. [C] Energy production and conversion; [D] Cell cycle control, cell division, chromosome partitioning; [E] Amino acid transport and metabolism; [F] Nucleotide transport and metabolism; [G] Carbohydrate transport and metabolism; [H] Coenzyme transport and metabolism; [I] Lipid transport and metabolism; [J] Translation, ribosomal structure and biogenesis; [K] Transcription; [L] Replication, recombination and repair; [M] Cell wall/membrane/envelope biogenesis; [N] Cell motility; [O] Posttranslational modification, protein turnover, chaperones; [P] Inorganic ion transport and metabolism; [Q] Secondary metabolites biosynthesis, transport, and catabolism; [S] Function unknown; [T] Signal transduction mechanisms; [U] Intracellular trafficking, secretion, and vesicular transport and [V]Defense mechanisms
Specific CcpA binding was identified for genes encoding four PTS systems known and putatively involved in the transport and utilization of maltose, cellobiose (two operons), and mannose, which stands in contrast to the 13 PTS gene/operons whose transcript levels were affected by CcpA inactivation. We also observed CcpA binding sites in genes/operons encoding three ABC carbohydrate transport systems for maltose, maltodextrin, and sialic acid. The central role of maltose/maltodextrin utilization in GAS pathophysiology is reflected by the five distinct CcpA binding sites for genes involved in the acquisition of these prevalent carbohydrates including that in the promoter region of the pulA gene encoding a cell‐surface pullulanase important for both nutrient acquisition and adherence (Figure 5a). In addition to transporters, genes encoding glycerol kinase, phosphoglucomutase, and HPr, which are also involved in carbohydrate metabolism, had CcpA binding sites and were directly regulated by CcpA.
FIGURE 5.
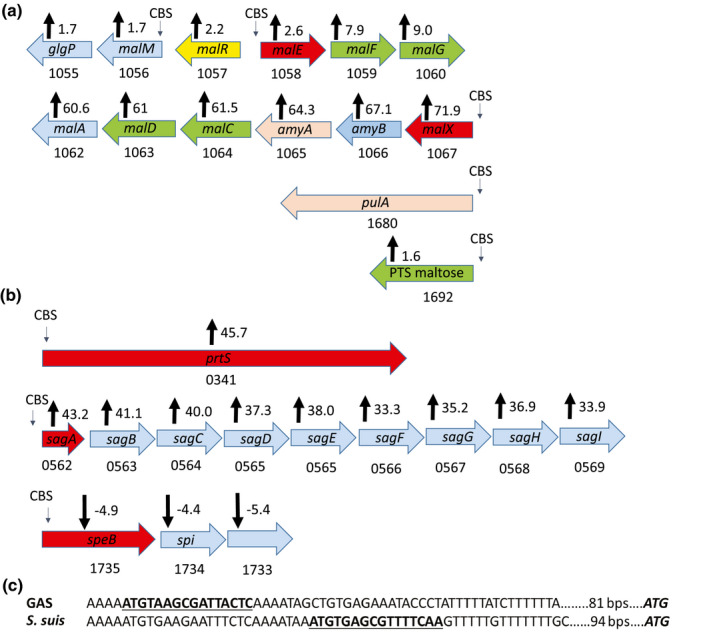
CcpA‐bound sites in MGAS2221. Schematic representation of the binding sites for CcpA (CBS) in key (a) carbohydrate transport and (b) virulence factor encoding genes. Fold change in transcript levels upon CcpA inactivation is displayed with black arrows. Positive numbers indicate higher transcript levels in 2221∆ccpA compared to MGAS2221. Genes in the schematic diagram are color coded to indicate function: blue—intracellular carbohydrate processing protein; yellow—transcriptional regulator; red—substrate binding lipoprotein; green—transport protein and beige—cell surface/secreted protein. (c) Sequence variation in the promoter region of the arginine deiminase (arcA) gene in GAS and S. suis. Enriched site for GAS and putative CcpA binding site for S. suis are highlighted
CcpA enriched sites were also identified in the promoters of genes encoding various amino acid uptake and utilization pathways such as those involved in arginine, histidine, and glutamate metabolism. Although a recent CcpA study in Streptococcus suis did not identify in vivo CcpA binding for arcA (Willenborg et al., 2014), the initial gene in the arginine catabolism operon, arcA showed the strongest CcpA‐mediated DNA enrichment for the entire MGAS2221 data set (Figure S3). A comparison of the arcA promoters showed that whereas a CcpA binding site is present in GAS and predicted for S. suis, these sites are quite variable both in terms of composition and location, which may explain the divergent results (Figure 5c).
Importantly, DNA enrichment was observed for the promoter and/or 5’ coding regions of three key GAS virulence factor encoding genes (Figure 5b). Namely we observed in vivo binding of CcpA for sagA, the first gene in the operon encoding the key cytotoxin Streptolysin S, speB, which encodes an actively secreted broad‐spectrum cysteine proteinase, and prtS which encodes an IL‐8 degrading enzyme. No CcpA binding or effect on gene transcript level was observed for cfa (M5005_spy0981) which was previously reported to be directly regulated by CcpA (Kietzman & Caparon, 2010). Similarly, we did not observe enrichment for the other nine virulence factor encoding genes whose transcript levels were varied by CcpA inactivation (Table 2) suggesting an indirect mechanism for CcpA impact on these genes.
Given the large numbers of genes impacted by CcpA inactivation relative to the number of identified CcpA binding sites, it was reasonable to suspect that many of the genes might be secondarily affected by regulators under CcpA control. Indeed, RNAseq analysis showed that the transcript levels of 21 genes/operons encoding known and putative transcriptional regulators, including four two‐component gene regulatory systems (TCS), were significantly affected by CcpA inactivation (Table S1). Of these, however, we identified direct CcpA binding for only three genes encoding the transcriptional regulators BglG, ArgR2, and Spy1495 (Table 4). While the bglG and argR2 genes had a CcpA‐binding site in their promoters, spy1495 was in an operon where the first gene (spy1496) was CcpA‐bound. As previously noted, there was also CcpA enrichment in the intergenic region between malE and malR (Figure 5a), which encodes a maltose regulatory protein. Hence, malR could also be directly regulated by CcpA although the location of the binding site suggests that CcpA is more likely to influence malE transcription. There was no DNA enrichment for any TCS or the master regulator Mga (multigene activator), which was identified to be directly regulated by CcpA in a previous study (Almengor et al., 2007). Given that none of the regulators directly controlled by CcpA are known or expected to have large transcriptomes (Shelburne et al., 2011), these data suggest that the broad impact of CcpA inactivation on GAS gene transcript levels is unlikely to be primarily mediated by regulators directly under CcpA control.
2.4. Analysis of CcpAV301A in vivo DNA Binding
Of the 76 DNA loci bound by CcpA, 37 enriched regions which encompass 75 genes (12% of the CcpA transcriptome) were also enriched in strain 2221‐CcpA‐V301A (Figure 4a). All of the enriched sites in 2221‐CcpA‐V301 were associated with genes that also evidenced enriched sites in MGAS2221. All three of the virulence factor encoding genes bound by CcpA in strain MG22221, prtS, sagA, and speB, also evidenced binding in strain 2221‐CcpA‐V301A (Tables 2 and 4). Similarly, five of the seven PTS/ABC carbohydrate transport encoding genes/operons (Tables 3 and 4) bound by CcpA were also bound by CcpAV301A as was arcA (Table 4). In contrast, spy1746, which encodes the first gene in a cellobiose PTS operon, and spy0212, which encodes the first gene in the sialic acid uptake ABC operon were not bound by CcpAV301A. We also did not observe binding of the CcpAV301A protein to the promoter of either ccpA, ptsH (HPr) or the operon encoding the ATP synthase genes (Table 4). When considering all DNA binding sites for the CcpA protein in strain MGAS2221, those sites that were also bound in strain 2221‐CcpA‐V301A were more likely to have significantly different transcript levels following CcpA inactivation (p < .001 by Fisher's exact test). To validate the findings from our ChIP‐seq, six sites that were enriched in both the wild type MGAS2221 and the 2221‐CcpA‐V301A strain were confirmed by SYBR qRT PCR (Figure 7a).
FIGURE 7.
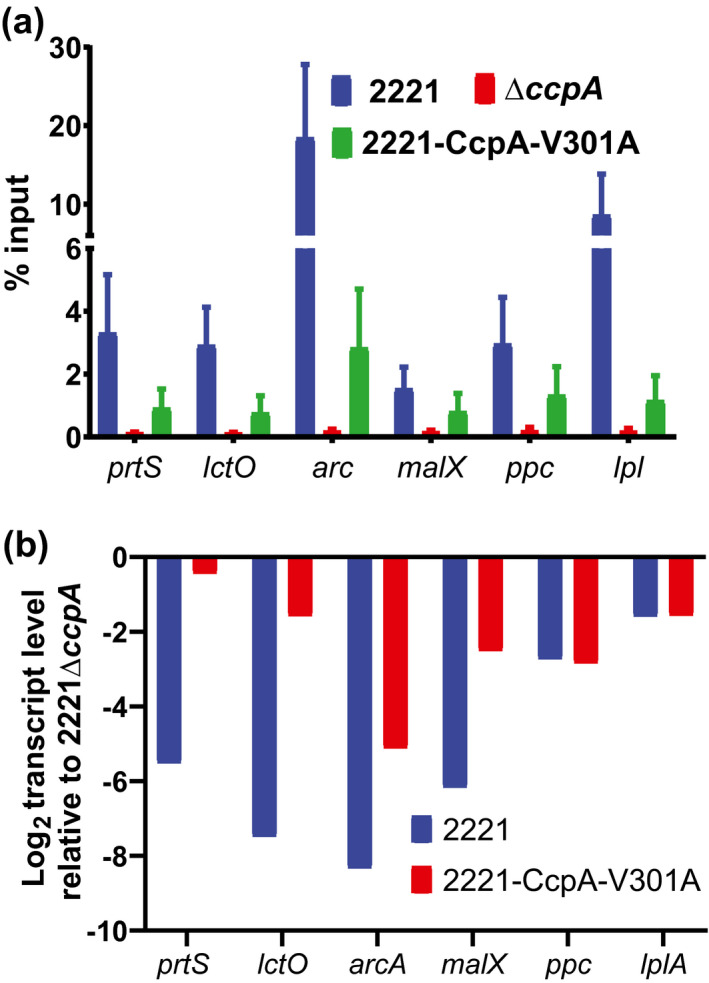
Quantitative impact of CcpAV301A alteration on gene transcript level and DNA binding. (a) SYBR qRT PCR analysis of DNA precipitated using anti‐CcpA antibody from indicated strains (legend inset) for specific promoters (indicated on x‐axis). (b) Transcript level changes as reported in our RNAseq data for the six genes in panel a
2.5. Characterization of CcpA binding sites not associated with differentially expressed genes
For 29 CcpA binding sites, we did not observe a significant difference in transcript levels for a nearby gene following CcpA inactivation. The majority of these binding sites were located in the middle of genes suggesting that the observed CcpA binding is likely nonfunctional. For nine genes, the DNA enrichment site overlapped with promoter regions including four genes whose function would suggest regulation by CcpA (Table 4). These four genes were pgi, which encodes a glucose utilization protein, plr, which encodes an aldehyde dehydrogenase, ldh, which encodes an enzyme that converts lactate to pyruvate, and the previously mentioned pulA. Of these 29 CcpA enriched sites that were not associated with transcript level variation after CcpA inactivation, 11 were also enriched in strain 2221‐CcpA‐V301 including three genes encoding carbohydrate utilization proteins (pgi, plr, and pulA) as well as one site in a CRISPR operon and the salY gene, which encodes an ABC transport protein involved in lantibiotic secretion (Table 4). The combination of CcpA binding site location and putative function suggests that these genes may be directly regulated by CcpA under conditions distinct from those studied herein.
2.6. Identification of CcpA binding motif
Using the ChIP‐seq data, we identified a single consensus motif in the enriched sites for MGAS2221 (Figure 6b) which was highly consistent with previously identified cre motifs from Bacillus species (Marciniak et al., 2012; Schumacher et al., 2004). This motif was present in 57 of the 76 CcpA enriched sites (75%). For the remaining 19 sites, no particular consensus motif was identified. We did not identify the cre2 sequence of S. suis or the flexible binding site of Clostridium acetobutylicum that were recently described in any of the enriched sites in MGAS2221 (Willenborg et al., 2014; Yang et al., 2017). Compared to enriched areas with cre sites, enriched areas lacking cre sites were less likely to be in genes/gene promoters whose transcript levels were significantly affected by CcpA inactivation (p = .01 by Fisher's exact test), suggesting that the cre‐containing enriched DNA sites were more biologically impactful under the studied conditions. The cre sites that were associated with genes whose transcript levels were affected by CcpA inactivation, were significantly more likely to be located within 500 nt upstream and 50 nt downstream of the start codon compared to cre sites that were near genes not impacted by CcpA inactivation (p < .001 by Fisher's exact test) (Figure 6a). Relative to our previously described in silico derived cre (Figure 6e, (DebRoy et al., 2016)), the major difference for our ChIP‐seq derived cre was a lack of predominance of T at positions 1 and 2 (Figure 6b). We compared the cre sites that we found using the ChIP method in this study to those that we had previously predicted in MGAS2221 using in silico analysis of RNAseq data (DebRoy et al., 2016). Both studies were conducted on mid‐exponential cells grown in THY. We had predicted 72 cre sites in silico compared to the 57 we found by ChIP‐seq in this study. Of these sites, only 29 were in common with 20 being transcriptionally altered upon CcpA inactivation in both studies. Importantly, the ChIP‐seq analysis enabled identification of 14 additional binding sites that are near genes that have altered transcript levels in a ∆ccpA mutant but were not predicted by the in silico approach. These include the cre sites identified in the promoter of prtS, sagA and the three cre sites in the PTS and ABC transport systems for maltose/maltodextrin. Conversely, the in silico method predicted 23 DNA loci that were transcriptionally impacted by CcpA inactivation in our study but were not bound by CcpA in our ChIP‐seq analysis. These genes included those encoding the transcriptional regulator FruR, an alcohol dehydrogenase, and the ABC transporter Spy1310.
FIGURE 6.
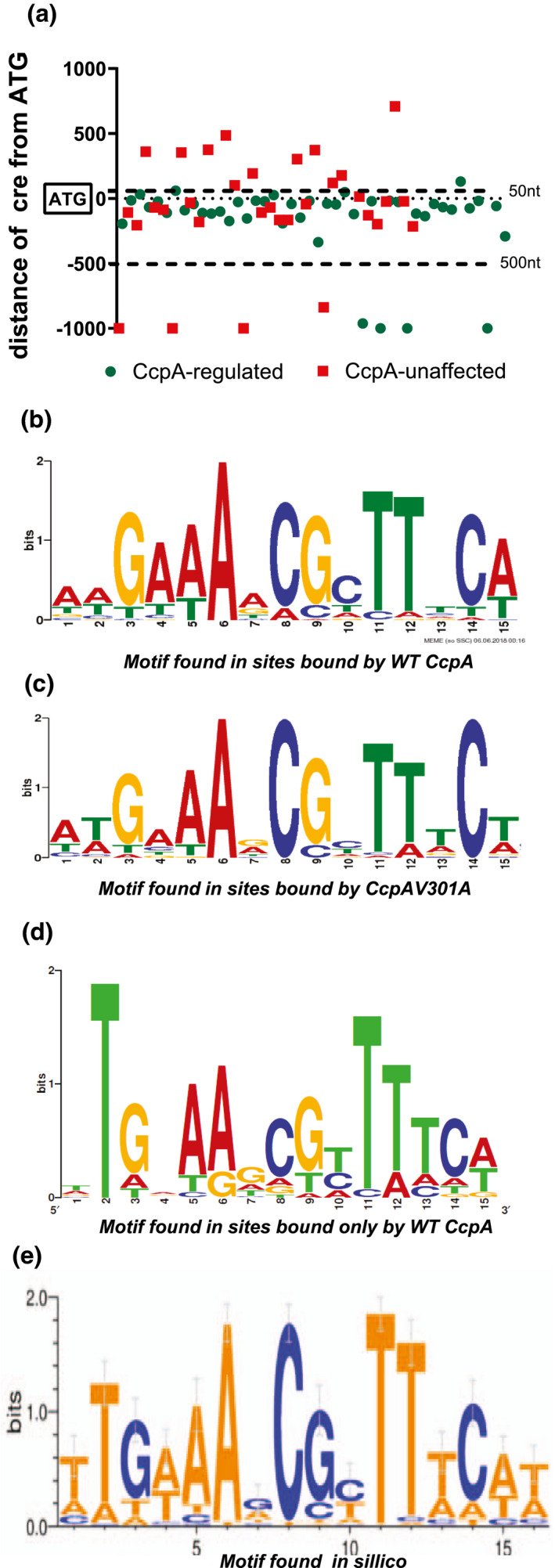
Consensus motifs identified in CcpA binding sites. (a) Scatter plot showing the distance from the translational start site (TSS) of enriched sites bound by the wild type CcpA protein that contain a consensus motif. Genes associated with the enriched sites are color coded to indicate whether they are (CcpA‐regulated) or not (CcpA‐unaffected) transcriptionally impacted upon CcpA inactivation. Enriched sites that are located farther than 1,000 nt from the TSS are not plotted. WebLogo representation of the consensus cre motif identified from CcpA binding sites in strains (b) MGAS2221 and (c) 2221‐CcpA‐V301A. (d) The cre motif found in sites that were bound by the wild type CcpA but not the CcpAV301A mutant. (e) The consensus motif identified in our previous study by in silico analysis of CcpA‐regulated genes in three different GAS serotypes (DebRoy et al., 2016)
When considering sites enriched in 2221‐CcpA‐V301A only (Figure 6c), we identified a motif that was highly similar to that for strain MGAS2221. The occurrence of an A at position 15 was reduced in the cre consensus derived from DNA loci in strain 2221‐CcpA‐V301A compared to that derived from MGAS2221. Of 57 cre sites in MGAS2221, 37 were bound in strain 2221‐CcpA‐V301, and we did not identify any enriched cre containing sites that were present in strain 2221‐CcpA‐V301 but not in MGAS2221. For cre sites that were bound only in strain MGAS2221 but not in the strain 2221‐CcpA‐V301A (Figure 6d), the occurrence of a T at position 2 was absolute and markedly increased relative to all cre sites in MGAS2221. Thus, the ChIP‐seq data show that although GAS cre sites have a similar architecture to other Gram‐positive bacteria, in silico prediction of GAS CcpA binding using motifs is problematic.
2.7. Impact of CcpAV301A mutation on magnitude of transcript level variation and degree of DNA binding
Given our finding of the differential transcript level impact of the CcpAV301A change (Figure 3e), we sought to further study how interruption of CcpA‐HPr~P interaction impacted CcpA function by assessing whether the CcpAV301A amino acid variation quantitatively impacted CcpA gene regulation and DNA binding. For clarity of analysis, we only examined genes whose transcript levels were significantly increased by CcpA inactivation in MGAS2221 (e.g., were CcpA repressed), that evidenced in vivo DNA binding by CcpA, and were monocistronic or the first gene in an operon. We identified 33 genes that met these criteria, all but one of which (argR2) was associated with a cre (Table S2). Using these genes, we tested the hypothesis that the CcpAV301A polymorphism would result in less release of catabolite repression compared with total CcpA inactivation. Indeed, for 27/33 genes, transcript levels were significantly higher in strain 2221∆ccpA relative to 2221‐CcpA‐V301A, up to 57‐fold but with a broad range (Table S2).
Next, we sought to determine whether this differential impact on transcript levels correlated with a change in DNA binding affinity as determined by amount of DNA immunoprecipitated using anti‐CcpA antibody. We used SYBR qPCR to quantify precipitated DNA from the promoter regions of six genes that evidenced CcpA binding for both MGAS2221 and 2221‐V301A‐CcpA strains by ChIP‐seq. Location of predicted cre sites in the enriched CcpA binding sites identified in the promoters of these six genes are shown in Figure 5a,b and in Figure S4. For all six genes, we observed significantly more DNA precipitation in MGAS2221 compared to 2221‐CcpA‐V301A and compared to strains 2221∆ccpA (Figure 7a). However, we did not identify a consistent relationship between quantitative differences in the amount of DNA precipitated and the gene transcript level variation observed in our RNAseq results (Figure 7b). For example, there was markedly more DNA from the ppc and lplA promoters precipitated in strain MGAS2221 compared to 2221‐CcpA‐V301A (Figure 7a), yet, there was no significant difference in ppc and lplA gene transcript levels between strains 2221 and 2221‐CcpA‐V301A relative to 2221∆ccpA (Figure 7b). Conversely, there was only a modest difference in the amount of malX promoter DNA precipitated from MGAS2221 compared to 2221‐CcpA‐V301A (Figure 7a), yet, we observed marked variation in transcript levels between MGAS2221 and 2221‐CcpA‐V301A relative to 2221ΔccpA (Figure 7b). Taken together, we conclude that abrogation of CcpA‐HPr~P interaction quantitatively impacts CcpA function both in terms of the effect of CcpA on gene transcript levels and DNA binding affinity, but decreases in CcpA affinity for promoter DNA due to the V301A alteration does not strictly correlate with a subsequent impact on gene transcript level under the studied conditions.
3. DISCUSSION
The complex interplay between basic metabolic processes and bacterial pathophysiology is being increasingly appreciated (Eisenreich et al., 2010; Pacheco et al., 2012; Rohmer et al., 2011). For Gram‐positive bacteria, CcpA stands squarely at this interface given its central role in controlling preferred carbon utilization pathways and regulating virulence factors in a diverse array of human pathogens (Iyer et al., 2005; Mendez et al., 2012; Shelburne et al., 2008; Vega et al., 2016). The primary link of CcpA to the energy status of the bacterial cell is via its interaction with HPr, but study of HPr in many important Gram‐positive bacteria has been limited due to its essential nature (Fleming et al., 2015; Willenborg et al., 2014). Herein, we sought to extend our knowledge about how CcpA links metabolic and virulence processes through a comprehensive identification of in vivo CcpA binding sites in the major human pathogen group A Streptococcus. Additionally, we created a strain in which CcpA cannot interact with HPr~P, to delineate HPr~P‐dependent and HPr~P‐independent aspects of CcpA function. Our findings show that CcpA directly regulates the genes encoding several critical GAS virulence factors and that >50% of genes in the GAS CcpA transcriptome are impacted by CcpA independent of HPr~P.
A key contribution of our study is the first genome wide analysis of in vivo CcpA DNA binding for a β‐hemolytic streptococci. We identified 76 CcpA binding sites in GAS, the specificity of which were demonstrated by the lack of enrichment in a CcpA knockout strain. The only other available CcpA ChIP‐seq analysis in streptococci was performed in Streptococcus suis (Willenborg et al., 2014) and identified 58 DNA loci bound by CcpA at the mid‐exponential phase of growth as was studied herein. Some loci, such as those in the promoters of glpK, malX, and pgmA were identified in both studies, while others like arcA, manLMN, pgi, and eno were not. In MGAS2221 we found the arcA site to be the most strongly enriched peak and the most derepressed gene upon ccpA deletion. In contrast, the S. suis study did not identify enrichment of the arcA or the manLMN promoters even though these genes were impacted by CcpA inactivation. Additionally, we did not identify the cre2 motif present in the S. suis data set, perhaps because that motif was identified at the stationary phase of growth, which we did not investigate.
A common observation of both this and the S. suis study (Willenborg et al., 2014), and of CcpA investigations in other pathogens (Antunes et al., 2012), is that direct CcpA binding accounts for only a small percentage of genes affected by CcpA inactivation. For example, of the genes encoding the 14 known carbohydrate PTS systems in MGAS2221, the transcript levels of 13 were affected by CcpA inactivation, but only four are directly regulated. This narrow direct impact of CcpA on the PTS systems was also observed in S. suis, where CcpA directly impacted only two of the 14 PTS systems present. The manLMN operon in GAS and S. pneumoniae has been shown to impact a diverse array of other carbohydrate transporters (Abranches et al., 2003, 2006; Fleming & Camilli, 2016; Vadeboncoeur & Pelletier, 1997). We herein established that in GAS, CcpA binds the promoter of the manLMN operon in vivo (Table 3), and thus, could indirectly account for some of the broad impact of CcpA inactivation on carbohydrate transport systems despite only directly binding a small number of genes encoding these systems. While it has been postulated that the broad CcpA transcriptome may be due to the impact of CcpA on other regulators (Antunes et al., 2012; Carvalho et al., 2011; DebRoy et al., 2016; Seidl et al., 2009), we found that CcpA only bound the promoters of a small number of other regulator encoding genes (Table 4) which is not consistent with a broad effect of CcpA on the GAS regulatory network. The marked increase in HPr~P observed following CcpA inactivation may account for a substantial proportion of the indirect effects of CcpA given the known role of HPr~P in a broad array of regulatory processes (Deutscher et al., 2005). There has been increasing evidence of the role of sRNAs in gene regulation (Dutta & Srivastava, 2018). While sRNAs have been identified to impact GAS virulence, there is no direct evidence of the impact of sRNAs on metabolic pathways or on any of the highly regulated CcpA targets (Danger et al., 2015; Pappesch et al., 2017; Perez et al., 2009). However, CcpA‐regulated sRNAs have been identified in S. aureus and it remains formally possible that sRNAs might mediate some of the regulation observed in GAS upon CcpA inactivation (Bronesky et al., 2019).
Another key finding of this work was our identification that CcpA directly binds to genes encoding three critical GAS virulence factors, sagA, prtS, and speB. How CcpA mechanistically impacts the expression of the sag operon, and subsequent production of the key cytotoxin streptolysin S, has been an object of study for the past 12 years since the original observation that CcpA inactivation strikingly increases streptolysin S production (Kinkel & McIver, 2008; Shelburne et al., 2008). Our group as well as others have identified in vitro binding of CcpA to the sagA promoter (Kinkel & McIver, 2008; Shelburne et al., 2008) whereas an in vivo study showed no precipitation of the sagA promoter using a CcpA antibody (Kietzman & Caparon, 2010). Although both MGAS2221 and HSC5, the strain used in the aforementioned study (Kietzman & Caparon, 2010), both contain the sagA cre site we identified, the area upstream of the cre site is quite divergent for the two strains which could explain the differential in vivo binding results. It has been shown that the manLMN operon and the β‐glucoside PTS (Table 3), which we identified herein as being directly and indirectly regulated by CcpA, respectively, impacts streptolysin production (Braza et al., 2020; Sundar et al., 2017) suggesting that CcpA may regulate streptolysin S production both directly and indirectly. In concert with our data, CcpA was identified as binding in vivo to the gene encoding suilysin, the pore‐forming toxin of S. suis (Willenborg et al., 2014) and to bind in vitro to the sagA promoter of S. anginosus (Bauer et al., 2018). We identified direct binding of CcpA to speB, in accordance with previous studies that have used DNA pulldown and fluorescence polarization methods to demonstrate this interaction (Kietzman & Caparon, 2010; Shelburne et al., 2010). Interestingly, speB was one of the very few genes directly activated by CcpA. Additionally, our identification that CcpA directly regulates prtS, which encodes a critical IL‐8 degrading enzyme, marks the first identification of in vivo binding of a regulator to this important GAS virulence factor. The evolutionary rationale for having such critical and diverse virulence factor encoding genes under direct control of a global metabolic regulator is not clear, but CcpA has also been shown to directly and indirectly impact the expression of key virulence factors in a range of Gram‐positive bacteria from clostridia to staphylococci (Abranches et al., 2008; Chiang et al., 2011; Seidl et al., 2006; Varga et al., 2004).
Although HPr has long been recognized as critical to CcpA function, study of HPr in major human pathogens such as staphylococci and streptococci has been limited due to its essential nature, including the critical Ser46 residue (Fleming et al., 2015; Willenborg et al., 2014). We also were unable to affect HPrSer46~P through a mutagenesis approach, and thus, sought to interrupt CcpA‐HPr~P interaction by modifying the critical CcpAV301 residue. Surprisingly, although the V301A mutation abrogated CcpA‐HPr~P interaction both in vitro and in vivo, more than half of the genes in the CcpA transcriptome still demonstrated CcpA‐based repression in strain 2221‐CcpA‐V301A, indicating the capacity of CcpA to function independently of HPrSer46~P in GAS. We were also able to correlate the preserved function of the CcpAV301A protein through ChIP‐seq analysis which revealed continued enrichment for numerous DNA promoters in strain 2221‐CcpA‐V301A. Our identification of CcpA functioning independent of HPr~P echoes findings from a recent ChIP‐seq study in S. suis which found that CcpA can bind to promoters of and regulate several genes in both exponential and stationary phase of growth (Willenborg et al., 2014). Given the absence of HPrSer46~P in the stationary phase, the authors postulated that the CcpA binding and regulation observed in the stationary phase probably occurred independent of HPr~P (Willenborg et al., 2014). These findings stand in contrast to those from Staphylococcus xylosus in which HPrSer46~P is absolutely essential for CcpA‐mediated catabolite repression (Jankovic & Bruckner, 2002). Our findings are more in concert with studies in B. subtilis, the nonpathogenic gram positive model bacterium, in which the impact of an HPrS46A mutation is essentially an overall dampening of gene regulation in terms of magnitude (Lorca et al., 2005). Indeed, our ChIP‐seq analysis revealed that enrichment of promoter DNA was typically reduced in strain 2221‐CcpA‐V301A compared to the wild‐type even when significant changes in transcript levels were identified, suggesting that the CcpAV301A bound to cre sites with lower affinity. Similarly, we found that the magnitude of effect on gene transcript levels due to the inability of the CcpAV301A mutant protein to bind HPrSer46~P varied such that genes could be grouped into classes that are strongly, mildly or not impacted by the abrogation of CcpA‐HPr~P interaction.
One possible purpose of the varied magnitude of effects on gene transcript levels observed in the 2221‐CcpA‐V301A strain would be to facilitate a broad array of transcriptional responses to carbohydrate availability and intracellular energy status (Paluscio et al., 2018). GAS infects numerous different infection sites such as skin, oropharynx, blood, and muscle (Cole et al., 2011; Cunningham, 2000; Walker et al., 2014). Each of these sites have unique nutritional and environmental makeup. The dependence/independence of CcpA from HPr~P could assist with fine‐tuning regulation of pertinent genes by CcpA such that site specific resources can be utilized optimally to aid the pathogen. For example, a recent study compared the GAS transcriptome during necrotizing fasciitis to growth in standard laboratory medium (Kachroo et al., 2020). Some of the most highly upregulated genes in Kachroo et al. including sagA, arcA, and the cellobiose PTS operon, were directly bound by CcpA in our study, all of which we classified as HPr~P‐dependent. Conversely, the transcript levels of the directly CcpA bound, HPr~P‐independent genes of the sialic acid operon, prtS, ackA, and hpr showed no significant upregulation during necrotizing fasciitis (Kachroo et al., 2020). These findings are consistent with the concept that CcpA can maintain repression of these genes even under metabolically unfavorable conditions when HPrSer46~P levels would be expected to be low.
In conclusion, herein, we present a genome wide analysis of chromatin occupancy by CcpA which reveals that CcpA directly regulates multiple key GAS virulence factors in addition to a broad array of critical metabolic genes. Through use of a CcpA isoform incapable of interacting with HPr~P, we have delineated not only the role of HPr in CcpA‐mediated gene regulation but also the ability of CcpA to function at certain sites independently of its key co‐factor. These findings extend the mechanistic understanding of how CcpA contributes to the pathophysiology of Gram‐positive bacteria.
4. EXPERIMENTAL PROCEDURES
4.1. Bacterial strains, media, and growth conditions
All strains used in this study are listed in Table 1. Group A Streptococcus (GAS) strains were routinely grown in Todd‐Hewitt media supplemented with 0.2% yeast extract (THY) at 37°C with 5% CO2. A single amino acid change in ccpA was engineered into the wild type MGAS2221 strain by homologous recombination using the pBBL740 plasmid as described previously (Horstmann et al., 2018), to construct the isoallelic strain 2221‐CcpA‐V301A.
4.2. Recombinant proteins and antibodies
Site directed mutagenesis (primers in Table S3) was used to introduce the V301A mutation into an existing clone of GAS ccpA (Shelburne et al., 2008) in the pET‐His2 vector to generate the pET‐His2‐V301A plasmid, which was transformed into E. coli BL21/pLysS. Wild type CcpA and V301A mutant protein were induced overnight at 18°C, and purified to >95% homogeneity as described previously (Shelburne et al., 2010) and extensively buffer exchanged to 20 mM Tris/HCl pH 7.5, 200 mM NaCl. Recombinant GAS HPr was overexpressed, phosphorylated, and purified as described previously (Shelburne et al., 2010). For the generation of polyclonal antibodies purified wild type CcpA and HPr proteins were used to immunize rabbits and affinity‐purified antibody was obtained (Covance, Denver, PA).
4.3. Protein‐protein interaction analysis
Surface Plasmon Resonance (SPR) analysis was performed at 25°C using BIAcore T200 instrument (GE Healthcare). Wild type CcpA and V301A mutant proteins were immobilized on sensor chip CM5 via amine coupling. PBS (phosphate‐buffered saline [8.06 mM Na2HPO4 and 1.94 mM KH2PO4, pH 7.4, 2.7 mM KCl, 137 mM NaCl]) was used as running buffer for immobilization and binding experiment. Sensor chip surface was activated by injecting a 1:1 mixture of 0.4 M of 1‐ethyl‐3‐(3‐dimethylaminopropyl) carbodiimide hydrochloride and 0.1 M of N‐hydroxysuccinimide over the flow cell surface at 5 µl/min for 7 min. After the surface was activated, CcpA (10 µg/ml in 10 mM NaOAc pH 5.0) and CcpAV301A (20 µg/ml in 10 mM NaOAc pH 5.0) were injected over different flow cells. Sensor surface was deactivated with an injection of 1.0 M ethanolamine‐HCl pH 8.5 at 5 µl/min for 7 min. Approximately, 3,700 response units (RU) of CcpA and 4200 RU of CcpAV301A were immobilized. A reference flow cell was prepared with activation and deactivation steps but no protein was immobilized. Two‐fold dilutions of HPr or HPrSer46~P from 1.25 to 80 µM in PBS were flown over the immobilized proteins at 20 µl/min for 30 s. All SPR responses were baseline corrected by subtracting the response generated from the corresponding reference surface. Double‐referenced SPR response curves (with the buffer blank run further subtracted) were used for affinity determination. The equilibrium response of each injection was collected and plotted against the concentration of injected protein. A one‐site binding (hyperbola) model was fitted to the data (GraphPad Prism 4) to obtain the equilibrium dissociation constant KD.
4.4. PhosTag gel
Recombinant HPr protein was phosphorylated and purified as described previously (Shelburne et al., 2010). Cell lysates of GAS strains were prepared, separated on 12.5% SuperSep Phos‐tag gels and detected using a polyclonal anti‐HPr antibody (Horstmann et al., 2014) as described previously (Horstmann et al., 2018). Experiments were repeated at least twice using samples collected on separate days.
4.5. Co‐immunoprecipitation
Strains MGAS2221, 2221ΔccpA, and 2221‐CcpA‐V301A were grown to mid‐exponential phase in THY, cross‐linked with EGS [ethylene glycol bis(succinic acid N‐hydroxysuccinimide ester)] and formaldehyde and harvested. Pellets were sonicated and CcpA‐containing complexes were immunoprecipitated using a polyclonal anti‐CcpA antibody, and then, analyzed for the presence of HPr by western blotting using a polyclonal anti‐HPr antibody and the Odyssey imaging system as described previously (Horstmann et al., 2015).
4.6. Analysis of transcript levels
For RNA seq analysis, RNA was isolated from four replicate cultures for each strain grown to mid‐exponential phase in THY (OD ~ 0.5) using the RNeasy kit (Qiagen) and processed as previously described (Horstmann et al., 2014). RNAseq data analysis was performed as previously described (Sanson & Flores, 2020). As the MGAS2221 genome is not publicly available, we used the MGAS5005 genome as the reference genome, which is identical to MGAS2221 (Sumby et al., 2005) in gene content. MA plots comparing log2 fold‐change and transcripts per kilobase million (TPM) were generated for individual comparisons of the wild‐type (MGAS2221) and the isoallelic strain 2221‐CcpA‐V301A to the ∆ccpA deletion strain. For each strain, TPM values from replicate samples for each gene within the reference MGAS5005 genome were averaged. Generated plots in combination with differential gene expression values produced by CLC Genomics Workbench (version 20) were used to identify targets with significantly altered transcript levels in individual comparisons. For Taqman real‐time qRT‐PCR, strains were grown in duplicate on two separate occasions to mid‐exponential phase in THY and processed as described previously (Horstmann et al., 2014). The gene transcript levels between MGAS2221 and either 2221∆ ccpA or 2221‐CcpA‐V301A were compared using an ordinary one way ANOVA. Primers and probes used are listed in Table S3.
4.7. Chromatin immunoprecipitation (ChIP) and sequencing (ChIP‐seq)
For ChIP analysis, MGAS2221, 2221∆ccpA, and 2221‐CcpA‐V301A strains were grown to mid‐exponential phase in THY. Crosslinking was performed with 1% formaldehyde and quenched with 0.125 M glycine. DNA was sheared using a Diagenode Bioruptor Plus and immunoprecipitated with anti‐CcpA antibody. We performed ChIP‐seq for two independent replicates of each strain. ChIP sequencing was performed in the Advanced Technology Genomics Core (ATGC) at MD Anderson Cancer Center. Briefly, Illumina compatible Indexed libraries were prepared from 12 ng of Diagenode Bioruptor Pico sheared ChIP DNA using the KAPA Hyper Library Preparation Kit (Roche). Libraries were enriched with 2 cycles of PCR then assessed for size distribution using the 4200 TapeStation High Sensitivity D1000 ScreenTape (Agilent Technologies) and quantified using the Qubit dsDNA HS Assay Kit (Thermo Fisher). Equimolar quantities of the indexed libraries were multiplexed, 9 libraries per pool. The pool was quantified by qPCR using the KAPA Library Quantification Kit (Roche) then sequenced on the Illumina NextSeq500 sequencer using the 75 nt high‐output flow cell. Raw FASTQ files from DNA sequencing were trimmed through Trimmomatic v. 0.36 (Bolger et al., 2014). A sliding window quality trimming was performed, cutting once the average quality of a window of three bases fell <30. Reads shorter than 32 bp after trimming were discarded. Resulting data were aligned to GenBank reference genome of Streptococcus pyogenes MGAS5005 (NC_007297.2) using Bowtie2 v. 2.3.0 (Langmead & Salzberg, 2012), without allowing mismatch. Resulting SAM files were converted to BED format using sam2bed v. 2.4.33 from BEDOPS (Neph et al., 2012). In order to identify peaks from Chip‐seq data, we used MACS2 v. 2.1.1.2 (Zhang et al., 2008). First, we estimated the coverage for each base using coverageBed v. 2.27.1 from BEDTools (Quinlan & Hall, 2010). Next, we compared data from the wild type MGAS2221 and 2221‐CcpA‐V301A mutant to the ∆ccpA deletion strain (which serves as the control) to remove sequencing noise background based on a coverage ≤2 of fold change. Finally, we applied MACS2 on the clean data using a cutoff q‐value of 0.05 with nonmodel option and MFOLD of “2 50.” The effective genome size was set to 1,838,562. The sequences of assembled peaks were retrieved and submitted to Multiple Em for Motif Elicitation v. 5.0.0 (MEME) (Bailey et al., 2015) for motif searching. The expected number of sites was set to one per sequence and the minimum motif width was set to 15 bp (Sharma et al., 2017). A single statistically significant motif (E‐value < 1e‐12) was recovered for MGAS2221 and 2221‐CcpA‐V301A and these matched the known CcpA binding consensus (DebRoy et al., 2016). The E‐value is derived by MEME, from the motif's log likelihood ratio, taking the motif length and background DNA sequence into account. Manual visualization of identified motifs and closest coding genes (CDS) on S. pyogenes MGAS5005 genome was performed using Integrative Genomic Viewer v. 2.3 (IGV) (Thorvaldsdottir et al., 2013).
To verify selected sites of enrichment observed by ChIP‐seq, ChIP DNA was analyzed by SYBR qRT PCR. Primers (Table S3) were designed to flank the respective enriched sites. Each reaction contained 1 µl of ChIP or input DNA, 10 mM primers and 5 µl of PowerUp SYBR Green Master Mix (Applied Biosystems). Reaction were amplified as per manufacturer's instructions followed by a melting curve of the product.
CONFLICT OF INTEREST
The authors have no conflict of interests.
AUTHOR CONTRIBUTIONS
S.D., S.A.S., V.A., G.G., M.L., and M.H. designed the research, S.D., S.A., N.H., and S.A.S. did the experiments, S.D., X.L., S.A.S., A.R.F, V.A., G.G., V.M., M.L., and M.H. performed data analysis and S.D., X.L., M.L., M.H., and S.A.S. wrote the manuscript. All authors reviewed the manuscript.
Supporting information
Fig S1‐S4
Table S1
Table S2
Table S3
ACKNOWLEDGMENTS
This work was supported by RO1 AI089891 (S.A.S) from the National Institutes of Health, CONICYT‐PIA Program AFB170001 of the Center for Mathematical Modeling (M.L), FONDECYT N° 1190742 (M.L), Center for Genome Regulation FONDAP 15090007 (M.L), Mesa Minería—Consorcio de Universidades del Estado de Chile—CUECH (M.L) and Fondo de Innovación para la Competitividad—FIC2018—6ta región, Gobierno Regional Chile (M.L). We acknowledge the CCSG P30 CA016672 funds for the Bioinformatics Shared Resource and the Sequencing and Microarray facility at MD Anderson Cancer Center. We would like to acknowledge Kunal Rai and Christopher Terranova for their knowledge and guidance in developing ChIP‐seq methodology for GAS.
DebRoy S, Aliaga‐Tobar V, Galvez G, et al. Genome‐wide analysis of in vivo CcpA binding with and without its key co‐factor HPr in the major human pathogen group A Streptococcus . Mol Microbiol.2021;115:1207–1228. 10.1111/mmi.14667
DATA AVAILABILITY STATEMENT
The data that support the findings of this study will be shared upon publication. RNA‐Seq and Chip‐seq data will be deposited at the sequence‐read archive.
REFERENCES
- Abranches, J., Candella, M.M., Wen, Z.T., Baker, H.V. & Burne, R.A. (2006) Different roles of EIIABMan and EIIGlc in regulation of energy metabolism, biofilm development, and competence in Streptococcus mutans . Journal of Bacteriology, 188, 3748–3756. 10.1128/JB.00169-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abranches, J., Chen, Y.Y. & Burne, R.A. (2003) Characterization of Streptococcus mutans strains deficient in EIIAB Man of the sugar phosphotransferase system. Applied and Environment Microbiology, 69, 4760–4769. 10.1128/AEM.69.8.4760-4769.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abranches, J., Nascimento, M.M., Zeng, L., Browngardt, C.M., Wen, Z.T., Rivera, M.F. et al. (2008) CcpA regulates central metabolism and virulence gene expression in Streptococcus mutans . Journal of Bacteriology, 190, 2340–2349. 10.1128/JB.01237-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almengor, A.C., Kinkel, T.L., Day, S.J. & McIver, K.S. (2007) The catabolite control protein CcpA binds to Pmga and influences expression of the virulence regulator Mga in the group A Streptococcus . Journal of Bacteriology, 189, 8405–8416. 10.1128/JB.01038-07 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antunes, A., Camiade, E., Monot, M., Courtois, E., Barbut, F., Sernova, N.V. et al. (2012) Global transcriptional control by glucose and carbon regulator CcpA in Clostridium difficile. Nucleic Acids Research, 40, 10701–10718. 10.1093/nar/gks864 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aung‐Hilbrich, L.M., Seidel, G., Wagner, A. & Hillen, W. (2002) Quantification of the influence of HPrSer46P on CcpA‐cre interaction. Journal of Molecular Biology, 319, 77–85. 10.1016/S0022-2836(02)00245-0 [DOI] [PubMed] [Google Scholar]
- Bailey, T.L., Johnson, J., Grant, C.E. & Noble, W.S. (2015) The MEME suite. Nucleic Acids Research, 43, W39–W49. 10.1093/nar/gkv416 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer, R., Mauerer, S. & Spellerberg, B. (2018) Regulation of the beta‐hemolysin gene cluster of Streptococcus anginosus by CcpA. Scientific Reports, 8, 9028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger, A.M., Lohse, M. & Usadel, B. (2014) Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics, 30, 2114–2120. 10.1093/bioinformatics/btu170 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braza, R.E., Silver, A.B., Sundar, G.S., Davis, S.E., Razi, A., Islam, E. et al. (2020) PTS uptake and metabolism of the ss‐glucoside salicin impacts group a streptococcal bloodstream survival and soft tissue infection. Infection and Immunity. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bronesky, D., Desgranges, E., Corvaglia, A., Francois, P., Caballero, C.J., Prado, L. et al. (2019) A multifaceted small RNA modulates gene expression upon glucose limitation in Staphylococcus aureus . EMBO Journal, 38, e99363 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buescher, J.M., Liebermeister, W., Jules, M., Uhr, M., Muntel, J., Botella, E. et al. (2012) Global network reorganization during dynamic adaptations of Bacillus subtilis metabolism. Science, 335, 1099–1103. 10.1126/science.1206871 [DOI] [PubMed] [Google Scholar]
- Carvalho, S.M., Kloosterman, T.G., Kuipers, O.P. & Neves, A.R. (2011) CcpA ensures optimal metabolic fitness of Streptococcus pneumoniae . PLoS One, 6, e26707. 10.1371/journal.pone.0026707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiang, C., Bongiorni, C. & Perego, M. (2011) Glucose‐dependent activation of Bacillus anthracis toxin gene expression and virulence requires the carbon catabolite protein CcpA. Journal of Bacteriology, 193, 52–62. 10.1128/JB.01656-09 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cole, J.N., Barnett, T.C., Nizet, V. & Walker, M.J. (2011) Molecular insight into invasive group A streptococcal disease. Nature Reviews Microbiology, 9, 724–736. 10.1038/nrmicro2648 [DOI] [PubMed] [Google Scholar]
- Cunningham, M.W. (2000) Pathogenesis of group A streptococcal infections. Clinical Microbiology Reviews, 13, 470–511. 10.1128/CMR.13.3.470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danger, J.L., Cao, T.N., Cao, T.H., Sarkar, P., Trevino, J., Pflughoeft, K.J. et al. (2015) The small regulatory RNA FasX enhances group A Streptococcus virulence and inhibits pilus expression via serotype‐specific targets. Molecular Microbiology, 96, 249–262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DebRoy, S., Saldana, M., Travisany, D., Montano, A., Galloway‐Pena, J., Horstmann, N. et al. (2016) A multi‐serotype approach clarifies the catabolite control protein A regulon in the major human pathogen group A streptococcus. Scientific Reports, 6, 32442. 10.1038/srep32442 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher, J. (2008) The mechanisms of carbon catabolite repression in bacteria. Current Opinion in Microbiology, 11, 87–93. 10.1016/j.mib.2008.02.007 [DOI] [PubMed] [Google Scholar]
- Deutscher, J., Francke, C. & Postma, P.W. (2006) How phosphotransferase system‐related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiology and Molecular Biology Reviews, 70, 939–1031. 10.1128/MMBR.00024-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deutscher, J., Herro, R., Bourand, A., Mijakovic, I. & Poncet, S. (2005) P‐Ser‐HPr–a link between carbon metabolism and the virulence of some pathogenic bacteria. Biochimica et Biophysica Acta, 1754, 118–125. 10.1016/j.bbapap.2005.07.029 [DOI] [PubMed] [Google Scholar]
- Dutta, T. & Srivastava, S. (2018) Small RNA‐mediated regulation in bacteria: A growing palette of diverse mechanisms. Gene, 656, 60–72. 10.1016/j.gene.2018.02.068 [DOI] [PubMed] [Google Scholar]
- Eisenreich, W., Dandekar, T., Heesemann, J. & Goebel, W. (2010) Carbon metabolism of intracellular bacterial pathogens and possible links to virulence. Nature Reviews Microbiology, 8, 401–412. 10.1038/nrmicro2351 [DOI] [PubMed] [Google Scholar]
- Fleming, E. & Camilli, A. (2016) ManLMN is a glucose transporter and central metabolic regulator in Streptococcus pneumoniae . Molecular Microbiology, 102, 467–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fleming, E., Lazinski, D.W. & Camilli, A. (2015) Carbon catabolite repression by seryl phosphorylated HPr is essential to Streptococcus pneumoniae in carbohydrate‐rich environments. Molecular Microbiology, 97, 360–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujita, Y. (2009) Carbon catabolite control of the metabolic network in Bacillus subtilis . Bioscience, Biotechnology, and Biochemistry, 73, 245–259. [DOI] [PubMed] [Google Scholar]
- Galinier, A., Haiech, J., Kilhoffer, M.C., Jaquinod, M., Stulke, J., Deutscher, J. et al. (1997) The Bacillus subtilis crh gene encodes a HPr‐like protein involved in carbon catabolite repression. Proceedings of the National Academy of Sciences, 94, 8439–8444. 10.1073/pnas.94.16.8439 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Giammarinaro, P. & Paton, J.C. (2002) Role of RegM, a homologue of the catabolite repressor protein CcpA, in the virulence of Streptococcus pneumoniae . Infection and Immunity, 70, 5454–5461. 10.1128/IAI.70.10.5454-5461.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorke, B. & Stulke, J. (2008) Carbon catabolite repression in bacteria: Many ways to make the most out of nutrients. Nature Reviews Microbiology, 6, 613–624. 10.1038/nrmicro1932 [DOI] [PubMed] [Google Scholar]
- Henkin, T.M., Grundy, F.J., Nicholson, W.L. & Chambliss, G.H. (1991) Catabolite repression of alpha‐amylase gene expression in Bacillus subtilis involves a trans‐acting gene product homologous to the Escherichia coli lacl and galR repressors. Molecular Microbiology, 5, 575–584. [DOI] [PubMed] [Google Scholar]
- Homeyer, N., Essigke, T., Meiselbach, H., Ullmann, G.M. & Sticht, H. (2007) Effect of HPr phosphorylation on structure, dynamics, and interactions in the course of transcriptional control. Journal of Molecular Modeling, 13, 431–444. 10.1007/s00894-006-0162-7 [DOI] [PubMed] [Google Scholar]
- Horstmann, N., Sahasrabhojane, P., Saldana, M., Ajami, N.J., Flores, A.R., Sumby, P. et al. (2015) Characterization of the effect of the histidine kinase CovS on response regulator phosphorylation in group A Streptococcus. Infection and Immunity, 83, 1068–1077. 10.1128/IAI.02659-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstmann, N., Saldana, M., Sahasrabhojane, P., Yao, H., Su, X., Thompson, E. et al. (2014) Dual‐site phosphorylation of the control of virulence regulator impacts group a streptococcal global gene expression and pathogenesis. PLoS Path, 10, e1004088. 10.1371/journal.ppat.1004088 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Horstmann, N., Tran, C.N., Brumlow, C., DebRoy, S., Yao, H., Nogueras Gonzalez, G. et al. (2018) Phosphatase activity of the control of virulence sensor kinase CovS is critical for the pathogenesis of group A streptococcus. PLoS Path, 14, e1007354. 10.1371/journal.ppat.1007354 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iyer, R., Baliga, N.S. & Camilli, A. (2005) Catabolite control protein A (CcpA) contributes to virulence and regulation of sugar metabolism in Streptococcus pneumoniae . Journal of Bacteriology, 187, 8340–8349. 10.1128/JB.187.24.8340-8349.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jankovic, I. & Bruckner, R. (2002) Carbon catabolite repression by the catabolite control protein CcpA in Staphylococcus xylosus. Journal of Molecular Microbiology and Biotechnology, 4, 309–314. [PubMed] [Google Scholar]
- Johnson, B.P., Jensen, B.J., Ransom, E.M., Heinemann, K.A., Vannatta, K.M., Egland, K.A. et al. (2009) Interspecies signaling between Veillonella atypica and Streptococcus gordonii requires the transcription factor CcpA. Journal of Bacteriology, 191, 5563–5565. 10.1128/JB.01226-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kachroo, P., Eraso, J.M., Olsen, R.J., Zhu, L., Kubiak, S.L., Pruitt, L. et al. (2020) New pathogenesis mechanisms and translational leads identified by multidimensional analysis of necrotizing myositis in primates. mBio, 11(1), e03363‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kietzman, C.C. & Caparon, M.G. (2010) CcpA and LacD.1 affect temporal regulation of S. pyogenes virulence genes. Infection and Immunity, 78, 241–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kietzman, C.C. & Caparon, M.G. (2011) Distinct time‐resolved roles for two catabolite‐sensing pathways during Streptococcus pyogenes infection. Infection and Immunity, 79, 812–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kinkel, T.L. & McIver, K.S. (2008) CcpA‐mediated repression of streptolysin S expression and virulence in the group A Streptococcus . Infection and Immunity, 76, 3451–3463. 10.1128/IAI.00343-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead, B. & Salzberg, S.L. (2012) Fast gapped‐read alignment with Bowtie 2. Nature Methods, 9, 357–359. 10.1038/nmeth.1923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Le Breton, Y., Belew, A.T., Valdes, K.M., Islam, E., Curry, P., Tettelin, H. et al. (2015) Essential genes in the core genome of the human pathogen Streptococcus pyogenes . Scientific Reports, 5, 9838. 10.1038/srep09838 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leboeuf, C., Leblanc, L., Auffray, Y. & Hartke, A. (2000) Characterization of the ccpA gene of Enterococcus faecalis: Identification of starvation‐inducible proteins regulated by ccpA. Journal of Bacteriology, 182, 5799–5806. 10.1128/JB.182.20.5799-5806.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorca, G.L., Chung, Y.J., Barabote, R.D., Weyler, W., Schilling, C.H. & Saier, M.H.Jr (2005) Catabolite repression and activation in Bacillus subtilis: Dependency on CcpA, HPr, and HprK. Journal of Bacteriology, 187, 7826–7839. 10.1128/JB.187.22.7826-7839.2005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ludwig, H., Rebhan, N., Blencke, H.M., Merzbacher, M. & Stulke, J. (2002) Control of the glycolytic gapA operon by the catabolite control protein A in Bacillus subtilis: A novel mechanism of CcpA‐mediated regulation. Molecular Microbiology, 45, 543–553. 10.1046/j.1365-2958.2002.03034.x [DOI] [PubMed] [Google Scholar]
- Marciniak, B.C., Pabijaniak, M., de Jong, A., Duhring, R., Seidel, G., Hillen, W. et al. (2012) High‐ and low‐affinity cre boxes for CcpA binding in Bacillus subtilis revealed by genome‐wide analysis. BMC Genomics, 13, 401. 10.1186/1471-2164-13-401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendez, M.B., Goni, A., Ramirez, W. & Grau, R.R. (2012) Sugar inhibits the production of the toxins that trigger clostridial gas gangrene. Microbial Pathogenesis, 52, 85–91. 10.1016/j.micpath.2011.10.008 [DOI] [PubMed] [Google Scholar]
- Mertins, S., Joseph, B., Goetz, M., Ecke, R., Seidel, G., Sprehe, M. et al. (2007) Interference of components of the phosphoenolpyruvate phosphotransferase system with the central virulence gene regulator PrfA of Listeria monocytogenes . Journal of Bacteriology, 189, 473–490. 10.1128/JB.00972-06 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miwa, Y., Nakata, A., Ogiwara, A., Yamamoto, M. & Fujita, Y. (2000) Evaluation and characterization of catabolite‐responsive elements (cre) of Bacillus subtilis . Nucleic Acids Research, 28, 1206–1210. 10.1093/nar/28.5.1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neph, S., Kuehn, M.S., Reynolds, A.P., Haugen, E., Thurman, R.E., Johnson, A.K. et al. (2012) BEDOPS: High‐performance genomic feature operations. Bioinformatics, 28, 1919–1920. 10.1093/bioinformatics/bts277 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pacheco, A.R., Curtis, M.M., Ritchie, J.M., Munera, D., Waldor, M.K., Moreira, C.G. et al. (2012) Fucose sensing regulates bacterial intestinal colonization. Nature, 492, 113–117. 10.1038/nature11623 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paluscio, E., Watson, M.E.Jr & Caparon, M.G. (2018) CcpA coordinates growth/damage balance for Streptococcus pyogenes pathogenesis. Scientific Reports, 8, 14254. 10.1038/s41598-018-32558-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pappesch, R., Warnke, P., Mikkat, S., Normann, J., Wisniewska‐Kucper, A., Huschka, F. et al. (2017) The regulatory small RNA MarS supports virulence of Streptococcus pyogenes . Scientific Reports, 7, 12241. 10.1038/s41598-017-12507-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez, N., Trevino, J., Liu, Z., Ho, S.C., Babitzke, P. & Sumby, P. (2009) A genome‐wide analysis of small regulatory RNAs in the human pathogen group A Streptococcus. PLoS One, 4, e7668. 10.1371/journal.pone.0007668 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poncet, S., Mijakovic, I., Nessler, S., Gueguen‐Chaignon, V., Chaptal, V., Galinier, A. et al. (2004) HPr kinase/phosphorylase, a Walker motif A‐containing bifunctional sensor enzyme controlling catabolite repression in Gram‐positive bacteria. Biochimica et Biophysica Acta, 1697, 123–135. 10.1016/j.bbapap.2003.11.018 [DOI] [PubMed] [Google Scholar]
- Quinlan, A.R. & Hall, I.M. (2010) BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics, 26, 841–842. 10.1093/bioinformatics/btq033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rohmer, L., Hocquet, D. & Miller, S.I. (2011) Are pathogenic bacteria just looking for food? Metabolism and microbial pathogenesis. Trends in Microbiology, 19, 341–348. 10.1016/j.tim.2011.04.003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanson, M. & Flores, A.R. (2020) Group A Streptococcus transcriptome analysis. Methods in Molecular Biology, 2136, 113–133. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Allen, G.S., Diel, M., Seidel, G., Hillen, W. & Brennan, R.G. (2004) Structural basis for allosteric control of the transcription regulator CcpA by the phosphoprotein HPr‐Ser46‐P. Cell, 118, 731–741. 10.1016/j.cell.2004.08.027 [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Seidel, G., Hillen, W. & Brennan, R.G. (2006) Phosphoprotein Crh‐Ser46‐P displays altered binding to CcpA to effect carbon catabolite regulation. Journal of Biological Chemistry, 281, 6793–6800. [DOI] [PubMed] [Google Scholar]
- Schumacher, M.A., Sprehe, M., Bartholomae, M., Hillen, W. & Brennan, R.G. (2011) Structures of carbon catabolite protein A‐(HPr‐Ser46‐P) bound to diverse catabolite response element sites reveal the basis for high‐affinity binding to degenerate DNA operators. Nucleic Acids Research, 39, 2931–2942. 10.1093/nar/gkq1177 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl, K., Bischoff, M. & Berger‐Bachi, B. (2008) CcpA mediates the catabolite repression of tst in Staphylococcus aureus . Infection and Immunity, 76, 5093–5099. 10.1128/IAI.00724-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl, K., Goerke, C., Wolz, C., Mack, D., Berger‐Bachi, B. & Bischoff, M. (2008) Staphylococcus aureus CcpA affects biofilm formation. Infection and Immunity, 76, 2044–2050. 10.1128/IAI.00035-08 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl, K., Muller, S., Francois, P., Kriebitzsch, C., Schrenzel, J., Engelmann, S. et al. (2009) Effect of a glucose impulse on the CcpA regulon in Staphylococcus aureus . BMC Microbiology, 9, 95. 10.1186/1471-2180-9-95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seidl, K., Stucki, M., Ruegg, M., Goerke, C., Wolz, C., Harris, L. et al. (2006) Staphylococcus aureus CcpA affects virulence determinant production and antibiotic resistance. Antimicrobial Agents and Chemotherapy, 50, 1183–1194. 10.1128/AAC.50.4.1183-1194.2006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma, P., Haycocks, J.R.J., Middlemiss, A.D., Kettles, R.A., Sellars, L.E., Ricci, V. et al. (2017) The multiple antibiotic resistance operon of enteric bacteria controls DNA repair and outer membrane integrity. Nature Communications, 8, 1444. 10.1038/s41467-017-01405-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne, S.A.3rd, Keith, D., Horstmann, N., Sumby, P., Davenport, M.T., Graviss, E.A. et al. (2008) A direct link between carbohydrate utilization and virulence in the major human pathogen group A Streptococcus . Proceedings of the National Academy of Sciences of the United States of America, 105, 1698–1703. 10.1073/pnas.0711767105 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne, S.A., Olsen, R.J., Suber, B., Sahasrabhojane, P., Sumby, P., Brennan, R.G. et al. (2010) A combination of independent transcriptional regulators shapes bacterial virulence gene expression during infection. PLoS Path, 6, e1000817. 10.1371/journal.ppat.1000817 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shelburne, S.A.3rd, Sahasrobhajane, P., Suber, B., Keith, D.B., Davenport, M.T., Horstmann, N. et al. (2011) Niche‐specific contribution to streptococcal virulence of a MalR‐regulated carbohydrate binding protein. Molecular Microbiology, 81, 500–514. 10.1111/j.1365-2958.2011.07708.x [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sprehe, M., Seidel, G., Diel, M. & Hillen, W. (2007) CcpA mutants with differential activities in Bacillus subtilis . Journal of Molecular Microbiology and Biotechnology, 12, 96–105. [DOI] [PubMed] [Google Scholar]
- Stulke, J. & Hillen, W. (2000) Regulation of carbon catabolism in Bacillus species. Annual Review of Microbiology, 54, 849–880. [DOI] [PubMed] [Google Scholar]
- Sumby, P., Porcella, S.F., Madrigal, A.G., Barbian, K.D., Virtaneva, K., Ricklefs, S.M. et al. (2005) Evolutionary origin and emergence of a highly successful clone of serotype M1 group A Streptococcus involved multiple horizontal gene transfer events. Journal of Infectious Diseases, 192, 771–782. [DOI] [PubMed] [Google Scholar]
- Sumby, P., Whitney, A.R., Graviss, E.A., DeLeo, F.R. & Musser, J.M. (2006) Genome‐wide analysis of group A streptococci reveals a mutation that modulates global phenotype and disease specificity. PLoS Path, 2, e5. 10.1371/journal.ppat.0020005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar, G.S., Islam, E., Gera, K., Le Breton, Y. & McIver, K.S. (2017) A PTS EII mutant library in Group A Streptococcus identifies a promiscuous man‐family PTS transporter influencing SLS‐mediated hemolysis. Molecular Microbiology, 103, 518–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thorvaldsdottir, H., Robinson, J.T. & Mesirov, J.P. (2013) Integrative Genomics Viewer (IGV): High‐performance genomics data visualization and exploration. Briefings in Bioinformatics, 14, 178–192. 10.1093/bib/bbs017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titgemeyer, F. & Hillen, W. (2002) Global control of sugar metabolism: A gram‐positive solution. Antonie Van Leeuwenhoek, 82, 59–71. [PubMed] [Google Scholar]
- Vadeboncoeur, C. & Pelletier, M. (1997) The phosphoenolpyruvate:sugar phosphotransferase system of oral streptococci and its role in the control of sugar metabolism. FEMS Microbiology Reviews, 19, 187–207. 10.1111/j.1574-6976.1997.tb00297.x [DOI] [PubMed] [Google Scholar]
- Varga, J., Stirewalt, V.L. & Melville, S.B. (2004) The CcpA protein is necessary for efficient sporulation and enterotoxin gene (cpe) regulation in Clostridium perfringens . Journal of Bacteriology, 186, 5221–5229. 10.1128/JB.186.16.5221-5229.2004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vega, L.A., Malke, H. & McIver, K.S. (2016) Virulence‐related transcriptional regulators of Streptococcus pyogenes . In: Ferretti, J.J., Stevens, D.L. & Fischetti, V.A. (Eds).Streptococcus pyogenes: Basic Biology to Clinical Manifestations. : University of Oklahoma Health Sciences Center. [PubMed] [Google Scholar]
- Walker, M.J., Barnett, T.C., McArthur, J.D., Cole, J.N., Gillen, C.M., Henningham, A. et al. (2014) Disease manifestations and pathogenic mechanisms of Group A Streptococcus. Clinical Microbiology Reviews, 27, 264–301. 10.1128/CMR.00101-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warner, J.B. & Lolkema, J.S. (2003) CcpA‐dependent carbon catabolite repression in bacteria. Microbiology and Molecular Biology Reviews, 67, 475–490. 10.1128/MMBR.67.4.475-490.2003 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watson, M.E.Jr, Nielsen, H.V., Hultgren, S.J. & Caparon, M.G. (2013) Murine vaginal colonization model for investigating asymptomatic mucosal carriage of Streptococcus pyogenes . Infection and Immunity, 81, 1606–1617. 10.1128/IAI.00021-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Willenborg, J., de Greeff, A., Jarek, M., Valentin‐Weigand, P. & Goethe, R. (2014) The CcpA regulon of Streptococcus suis reveals novel insights into the regulation of the streptococcal central carbon metabolism by binding of CcpA to two distinct binding motifs. Molecular Microbiology, 92, 61–83. [DOI] [PubMed] [Google Scholar]
- Yang, Y., Zhang, L., Huang, H., Yang, C., Yang, S., Gu, Y. et al. (2017) A flexible binding site architecture provides new insights into CcpA global regulation in gram‐positive bacteria. mBio, 8(1), e02004‐16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng, L., Choi, S.C., Danko, C.G., Siepel, A., Stanhope, M.J. & Burne, R.A. (2013) Gene regulation by CcpA and catabolite repression explored by RNA‐Seq in Streptococcus mutans . PLoS One, 8, e60465. 10.1371/journal.pone.0060465 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang, Y., Liu, T., Meyer, C.A., Eeckhoute, J., Johnson, D.S., Bernstein, B.E. et al. (2008) Model‐based analysis of ChIP‐Seq (MACS). Genome Biology, 9, R137. 10.1186/gb-2008-9-9-r137 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig S1‐S4
Table S1
Table S2
Table S3
Data Availability Statement
The data that support the findings of this study will be shared upon publication. RNA‐Seq and Chip‐seq data will be deposited at the sequence‐read archive.