

Abstract
Aim
This study aims to assess the potential effects of zanubrutinib on the activity of cytochrome P450 (CYP) enzymes and drug transporter proteins using a cocktail probe approach.
Methods
Patients received single oral doses of probe drugs alone and after at least 8 days of treatment with zanubrutinib 160 mg twice daily in a single‐sequence study in 18 healthy male volunteers. Simultaneous doses of 10 mg warfarin (CYP2C9) and 2 mg midazolam (CYP3A) were administered on Day 1 and Day 14, 0.25 mg digoxin (P‐glycoprotein [P‐gp]) and 10 mg rosuvastatin (breast cancer resistance protein [BCRP]) on Day 3 and Day 16, and 20 mg omeprazole (CYP2C19) on Day 5 and Day 18. Pharmacokinetic (PK) parameters were estimated from samples obtained up to 12 h post dose for zanubrutinib; 24 h for digoxin, omeprazole and midazolam; 48 h for rosuvastatin; and 144 h for warfarin.
Results
The ratios (%) of geometric least squares means (90% confidence intervals) for the area under the concentration–time curve from time zero to the last quantifiable concentration in the presence/absence of zanubrutinib were 99.80% (97.41–102.2%) for S‐warfarin; 52.52% (48.49–56.88%) for midazolam; 111.3% (103.8–119.3%) for digoxin; 89.45% (78.73–101.6%) for rosuvastatin; and 63.52% (57.40–70.30%) for omeprazole. Similar effects were observed for maximum plasma concentrations.
Conclusions
Zanubrutinib 320 mg total daily dose had minimal or no effect on the activity of CYP2C9, BCRP and P‐gp, but decreased the systemic exposure of CYP3A and CYP2C19 substrates (mean reduction <50%).
Keywords: anticancer drugs, cytochrome P450, drug interactions, drug transporter protein, pharmacokinetics
What is already known about this subject
Zanubrutinib, an irreversible BTK inhibitor, has demonstrated promising antitumour activity in patients with B‐cell malignancies.
In vitro studies show that zanubrutinib has an induction and/or inhibition potential on multiple CYPs and transporters.
The drug interaction potential of zanubrutinib with substrates of CYP enzymes and transporters in humans has not been assessed.
What this study adds
The cocktail approach to assess drug–drug interaction (DDI) has been commonly used, and this work was one of few studies utilizing this combination of CYP and transporter substrates.
At clinically relevant concentrations, zanubrutinib did not impact the activity of CYP2C9 and BCRP, but it had a weak induction effect on CYP3A and CYP2C19.
1. INTRODUCTION
Bruton's tyrosine kinase (BTK) is a signalling molecule predominantly expressed in B lymphocytes. Activation of BTK in B cells initiates a series of signalling events that promote B‐cell proliferation and survival.1, 2, 3 Inhibition of BTK has emerged as a promising strategy for targeting B‐cell malignancies. The first‐in‐class BTK inhibitor, ibrutinib, has shown promising antitumour activity in patients with B‐cell malignancies and has become a standard of care in chronic lymphocytic leukaemia/small lymphocytic lymphoma (CLL/SLL), mantle cell lymphoma (MCL), and Waldenström macroglobulinemia (WM).4, 5, 6, 7, 8, 9, 10, 11 Zanubrutinib is a potent second‐generation irreversible BTK inhibitor. Zanubrutinib has been granted a breakthrough therapy designation and recently received accelerated approval by the US Food and Drug Administration (FDA) for the treatment of adult patients with MCL who have received at least one prior therapy.12 Zanubrutinib was also approved by the China National Medical Products Administration (NMPA) for CLL or SLL and MCL in adult patients who have received at least one prior therapy.13 Zanubrutinib has been shown to be more selective than ibrutinib for the inhibition of BTK versus a panel of receptor tyrosine kinases.13 The improved selectivity of zanubrutinib for BTK may minimize off‐target inhibition of other kinases and, therefore, may result in a lower incidence and severity of off‐target toxicities compared with ibrutinib. Emerging clinical data has shown that zanubrutinib is associated with high rates of objective response in patients with B‐cell malignancies, including WM, MCL and CLL.14, 15, 16
Clinical pharmacokinetics (PK) data has shown that zanubrutinib is rapidly absorbed after oral administration; median time to peak zanubrutinib plasma concentration was 2 h and the mean apparent terminal elimination half‐life (t 1/2) was approximately 2–4 h in patients with B‐cell malignancies. Following multiple‐dose administrations of zanubrutinib, limited systemic accumulation is observed, which is consistent with the observed t 1/2. At doses ranging from 40 mg to 320 mg, there is a dose‐proportional increase in both maximum concentration (C max) and area under the plasma concentration versus time curve (AUC).16 Based on in vitro and clinical drug–drug interaction (DDI) studies, CYP3A is considered the primary P450 isoform responsible for zanubrutinib metabolism.17 Results from a human absorption, metabolism, and excretion study (AME) showed that hepatic metabolism and excretion account for a substantial portion of the elimination of zanubrutinib and that renal clearance does not play an important role.
In in vitro studies, zanubrutinib showed potential for reversible inhibition of CYP2C8, CYP2C9 and CYP2C19 in human liver microsomes, with half maximal inhibition concentration (IC50) values of 4.03, 5.69 and 7.80 μM, respectively.18 In vitro, zanubrutinib also induced a ≥2‐fold increase in mRNA (at 3 and 30 μM) of CYP2B6, CYP2C8, CYP2C9 and CYP3A in human hepatocytes. The average R3 values for induction of CYP3A4, CYP2B6, CYP2C8 and CYP2C9 were lower than or near the cutoff value of 0.8 according to FDA DDI guidance. Thus, zanubrutinib could theoretically have the potential to cause induction of these enzymes. In vitro studies showed that zanubrutinib was neither a significant inhibitor of P‐glycoprotein (P‐gp) at concentrations up to 10.0 μM nor a significant inhibitor of breast cancer resistance protein (BCRP), organic anion‐transporting polypeptide (OATP) 1B1, OATP1B3, organic anion transporter (OAT) 1, OAT3, and organic cation transporter (OCT) 2 at concentrations up to 5.0 μM. Based on estimates of ratios following FDA DDI guidance,19 zanubrutinib is not predicted to cause clinically relevant inhibition of the hepatic uptake transporters, OATP1B1 and OATP1B3, or the renal uptake transporters, OAT1, OAT3 and OCT2. However, due to the estimated high concentrations of zanubrutinib in the gastrointestinal (GI) tract associated with the twice‐daily dose (b.i.d.) of 160 mg, there is the potential for zanubrutinib to cause P‐gp‐ or BCRP‐mediated DDI.
This clinical drug interaction study was designed to determine the effect of multiple doses of zanubrutinib on the PK of probe substrates of CYP3A (midazolam), CYP2C9 (warfarin), CYP2C19 (omeprazole), P‐gp (digoxin) and BCRP (rosuvastatin) in healthy volunteers using a cocktail approach. Simultaneous single oral doses of probe drugs, warfarin and midazolam were administered followed by a cocktail of digoxin and rosuvastatin 2 days later. Midazolam, warfarin and omeprazole have been extensively used together as probe substrates in studies using the “Modified Cooperstown Cocktail + 1” approach.20 Omeprazole was dosed separately in this study to avoid potential pH‐dependent absorption of zanubrutinib, and an exploratory objective was also included to assess whether a single dose of omeprazole had any impact on the PK of zanubrutinib. Digoxin and rosuvastatin have been safely administered together as part of a recent cocktail transporter study.21 Probe substrates for CYP2C8 and CYP2B6 were not included in this study mainly because of lack of validated probes for these enzymes in cocktail DDI approaches. Results from this study will be used to assess clinical DDI risk for concomitant administration of zanubrutinib with substrates of these CYP enzymes or transporters.
2. METHODS
This cocktail DDI study was registered on ClinicalTrials.gov (NCT03561298). The study protocol was reviewed and approved by the study centres' Independent Ethics Committee and was conducted in compliance with the protocol, and in accordance with the International Conference on Harmonization Good Clinical Practice principles and the Declaration of Helsinki. All participating subjects provided written informed consent prior to screening.
2.1. Subjects
Healthy male subjects of any race aged between 18 and 60 years and with a body mass index (BMI) between 18.0 and 32.0 kg/m2 were selected according to the inclusion criteria listed in the protocol. Subjects were excluded if they had consumed or intended to use any medications/products known to alter the absorption, metabolism or elimination processes of the study drugs, within 30 days prior to the first dose administration, or if they had used any metabolic enzyme inhibitors or inducers that would affect the study drugs, within 30 days of the first dose administration. Eighteen subjects were enrolled to ensure that 12 subjects completed the study. The sample size chosen for this study was based on precedent set by other cocktail DDI studies of similar nature (assuming similar within‐subject variation of substrate drugs). The fixed‐sequence crossover design used in this study is typical for cocktail DDI studies20 because it reduces inter‐subject variability from the comparison between treatments; thus, a relatively small number of subjects are required.
2.2. Study design
This was a phase 1, open‐label, single‐centre, single‐sequence study to investigate the effect of multiple doses of zanubrutinib on the PK of probe drugs in healthy male subjects. All subjects received study drugs in a fixed sequence, all of which were administered orally, first without zanubrutinib and then with zanubrutinib. A summary of the study design is shown in Figure 1. In brief, simultaneous single oral doses of probe drugs, warfarin and midazolam were administered followed by dosing of digoxin and rosuvastatin together 2 days later. Omeprazole was dosed separately on Day 5. Subjects were in a fasted state on the day of probe drug administration and PK collection. On Days 7–19, subjects received twice‐daily doses of 160 mg zanubrutinib; after a sufficient enzyme induction period, the probe drugs (using the same sequence) were administered again starting on Day 13. On Days 1–3 and Days 14–16, subjects received 10 mg of vitamin K to counteract the anticoagulant effect of warfarin. The dose levels of the probe drugs (Figure 1) were selected based on standard doses used in other cocktail DDI studies.21, 22, 23, 24 The selected dose of 320 mg daily (160 mg b.i.d.) for zanubrutinib had been shown to be well tolerated and was the dose used in phase 2 and phase 3 clinical studies. An enzyme induction period of at least 8 days was used in this study (Days 7–14 for CYP3A4 and CYP2C9 and Days 7–18 for CYP2C19). This is based on reports indicating maximal induction of intestinal and hepatic CYP3A4 achieved in >90% of participants within 5 and 10 days, respectively. Furthermore, when considering the design of studies involving induction of CYP3A4, 7 days of oral dosing is routinely accepted, since mean AUC ratios of midazolam with and without rifampin were similar (0.12 vs 0.11) for either 7 or 14 days of rifampin dosing, respectively.25 The half‐life of zanubrutinib (~2–4 h) is similar to that of rifampin, and zanubrutinib steady‐state concentrations are expected to be reached in 1 day. Pharmacogenetic blood samples were collected, and in the event of aberrant PK outliers, genotype testing was conducted.
FIGURE 1.
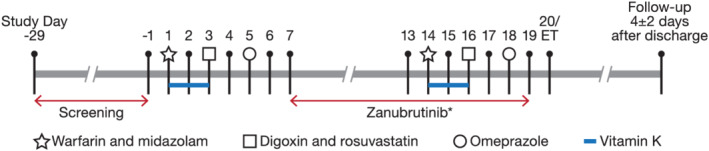
Study design. Abbreviation: ET = end of study (subject discharge). *PK samples for zanubrutinib were collected on Day 13 and Day 18
2.3. Pharmacokinetic sample analysis
To determine the PK of the probe drugs, blood samples were collected for the analysis of plasma concentrations of S‐warfarin, midazolam, digoxin, rosuvastatin, omeprazole and zanubrutinib (Figure 1 and Table 1). The analyses were performed by XenoBiotic Laboratories, Inc. (Plainsboro, NJ) using validated liquid chromatography tandem mass spectrometry (LC–MS/MS) methods.
TABLE 1.
Investigational product administration and blood sampling times
| Investigational product | Dose | CYPs/transporters of interest | Blood sampling time |
|---|---|---|---|
| Zanubrutinib | 320 mg daily (160 mg b.i.d.) | Not applicable | Predose and 0.5, 1, 2, 3, 4, 6, 8, and 12 h postdose (Day 13 and Day 18) |
| Midazolam | 2 mg | CYP3A | Predose and 0.5, 1, 1.5, 2, 3, 4, 8, 12, and 24 h postdose (Days 1–2, Days 14–15) |
| Warfarin | 10 mg | CYP2C9 (for S‐warfarin) | Predose and 0.5, 1, 1.5, 2, 3, 4, 8, 12, 24, 48, 72, 96, 120, and 144 h postdose (Days 1–7, Days 14–20) |
| Vitamin K | 10 mg | Not applicable | NA |
| Omeprazole | 20 mg | CYP2C19 | Predose and 0.5, 1, 1.5, 2, 3, 4, 8, 12, and 24 h postdose (Days 5–6, Days 18–19) |
| Digoxin | 0.25 mg | P‐gp | Predose and 0.5, 1, 1.5, 2, 3, 4, 8, 12, and 24 h postdose (Days 3–4, Days 16–17) |
| Rosuvastatin | 10 mg | OATP1B1, OATP1B3, BCRP | Predose and 0.5, 1, 1.5, 2, 3, 4, 8, 12, 24, and 48 h postdose (Days 3–4, Days 16–17) |
Abbreviations: BCRP = breast cancer resistance protein; b.i.d. = twice daily; CYP = cytochrome P450; NA = not applicable; OATP = organic anion‐transporting polypeptide; P‐gp = P‐glycoprotein; PK = pharmacokinetic.
The solid phase extraction procedure was utilized to extract digoxin with internal standards (IS), and the protein precipitation extraction procedures were utilized to extract other analytes with IS from human plasma containing dipotassium ethylenediaminetetraacetic acid (K2EDTA) as an anticoagulant. These methods utilize a reversed‐phase high‐performance liquid chromatography column to elute analytes with IS and an LC–MS/MS instrument with positive ESI‐MRM mode or negative ESI‐MRM mode to quantify each analyte. The calibration range of analytes in plasma was 0.100–20.0 ng/mL for midazolam, 2.00–1000 ng/mL for omeprazole, 10.0–5000 pg/mL for digoxin, 2.00–1000 ng/mL for S‐warfarin, 0.05–25.0 ng/mL for rosuvastatin, and 1.00–1000 ng/mL for zanubrutinib, respectively. The Inter‐Run Precision over the concentration ranges of the analytes were all lower than 11.59% (coefficient of variation, % CV) and the Inter‐Run Accuracy (% Bias) of the analytes were all between 95.33% and 107.00%. Additional details on the bioanalytical method are included in the Supporting Information.
2.4. Pharmacokinetic data analyses
The PK parameters included C max, time to C max (T max), area under the plasma concentration–time curve from time zero to last measurable timepoint (AUC0‐t ), AUC from time zero extrapolated to infinity (AUC0‐∞), and t 1/2, using noncompartmental methods with Phoenix WinNonlin (Certara USA, Inc.) Version 6.4. Pharmacokinetic analysis was carried out using actual postdose times. C max and T max values were obtained directly from the plasma concentration–time profiles. Area under the concentration–time curve over a dosing interval τ (AUC0–12, where τ = 12 h) for zanubrutinib was also calculated when it was administered alone or co‐administered with omeprazole. Regression‐based parameters (AUC0‐∞, t 1/2) were calculated only if the R2‐adjusted value of the regression line was ≥0.7. Values for AUC0‐∞ where the percentage extrapolation was greater than 30% were reported but excluded from descriptive statistics.
2.5. Pharmacokinetic statistical analysis
The effect of zanubrutinib on the PK of each probe drug was evaluated using a confidence interval (CI) approach. In each statistical analysis, zanubrutinib given in combination with each probe drug was the test treatment, and each probe drug given alone was the reference treatment. The PK parameters AUC0‐t , AUC0‐∞ (if allowed by the available data), and C max of each probe drug were log‐transformed prior to analysis and analysed using a mixed model26 for each part of the study. The model included treatments as fixed effects and subject as a random effect. For AUC0‐t , AUC0‐∞, and C max, the geometric least squares (LS) means were calculated for the test and reference treatments; the test‐to‐reference geometric LS means ratios (GMRs) and the corresponding 90% CI were also calculated. The T max was analysed nonparametrically using the Wilcoxon signed‐rank test.
2.6. Safety analyses
Clinical chemistry, haematology, urinalysis, vital signs and electrocardiogram (ECG) assessments were collected for safety evaluation. Adverse events (AEs) that occurred following the administration of any probe drug on Day 1 until the administration of zanubrutinib on Day 7 were assigned to be associated with “probe drugs only”. Adverse events that occurred following the administration of zanubrutinib on Day 7 until the administration of warfarin, vitamin K or midazolam on Day 14 were assigned to be associated with “zanubrutinib only”. Any AEs that occurred at the administration of warfarin, vitamin K or midazolam on Day 14 through the follow‐up visit were assigned to be associated with “probe drugs plus zanubrutinib”. Treatment‐emergent AEs (TEAEs) were graded according to the National Cancer Institute Common Terminology Criteria for Adverse Events v4.03.
2.7. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, the common portal for data from the IUPHAR/BPS Guide to PHARMACOLOGY, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.27
3. RESULTS
3.1. Study population
A total of 18 subjects were enrolled and 17 completed the study. All 18 enrolled subjects were included in the safety and PK populations. One subject withdrew consent prior to dosing on Day 3 of the study. Demographic data are summarized in Table 2. Subjects enrolled in the study were males between the ages of 21 and 58 years with BMI values ranging from 22.6 to 32.0 kg/m2.
TABLE 2.
Summary of demographics
| Overall (n = 18) | |
|---|---|
| Mean age, years (SD) | 37 (12.2) |
| Sex, n (%) | |
| Male | 18 (100%) |
| Race, n (%) | |
| Black or African American | 5 (27.8%) |
| White | 13 (72.2%) |
| Ethnicity, n (%) | |
| Hispanic or Latino |
9 (50.0%) |
| Not Hispanic or Latino | 9 (50.0%) |
| Mean weight, kg (SD) | 80.9 (9.31) |
| Mean height, cm (SD) | 172 (5.5) |
| Mean BMI, kg/m2 (SD) | 27.2 (2.42) |
Abbreviations: BMI = body mass index; SD = standard deviation.
3.2. Effect of zanubrutinib on PK of probe drugs
Statistical comparisons for each probe drug administered alone or co‐administered with zanubrutinib are presented in Table 3, and concentration–time profiles are shown in Figure 2. In all statistical comparisons of T max for each probe drug analyte, no statistically significant differences were observed when the probe drugs were co‐administered with zanubrutinib compared to when administered alone. In general, within‐subject variation in C max and AUC was low for S‐warfarin (≤14.2%), midazolam (≤17.6%), digoxin (≤24.4%) and rosuvastatin (≤27.5%), and low to moderate for omeprazole (15% and 35% for AUC and C max, respectively).
TABLE 3.
Summary of the pharmacokinetic parameters of probe drugs administered alone and with zanubrutinib 160 mg b.i.d.
| Probe drug | Parameter (unit) | Probe drugs only | Probe drugs + zanubrutinib 160 mg b.i.d. | Ratio of GLSM (%) | 90% CI (%) | ||
|---|---|---|---|---|---|---|---|
| n | Mean (cv%) | n | Mean (cv%) | ||||
| S‐warfarin | AUC0‐t (h*ng/mL) | 18 | 18 150 (35.17) | 16 | 18 040 (18.76) | 99.8 | 97.41–102.2 |
| AUC0‐∞ (h*ng/mL) | 16 | 19 150 (19.73) | 16 | 19 220 (19.58) | 100.4 | 97.92–102.9 | |
| Cmax (ng/mL) | 18 | 698 (14.9) | 16 | 666 (22.2) | 95.25 | 87.6–104.0 | |
| Tmax (h)a | 18 | 1.00 (0.500–4.00) | 16 | 1.00 (0.500–2.00) | — | — | |
| t1/2 (h)b | 18 | 39.3 (18.5) | 16 | 34.3 (3.71) | — | — | |
| Midazolam | AUC0‐t (h*ng/mL) | 18 | 26.93 (42.27) | 17 | 14.27 (35.44) | 52.52 | 48.49–56.88 |
| AUC0‐∞ (h*ng/mL) | 18 | 28.22 (43.64) | 17 | 14.97 (36.23) | 52.58 | 48.50–57.02 | |
| Cmax (ng/mL) | 18 | 10.2 (28.7) | 17 | 7.12 (35.6) | 70.17 | 63.20–77.91 | |
| Tmax (h)a | 18 | 0.500 (0.500–1.03) | 17 | 0.500 (0.500–1.00) | — | — | |
| t1/2 (h)b | 18 | 4.87 (2.29) | 17 | 2.51 (0.653) | — | — | |
| Digoxin | AUC0‐t (h*pg/mL) | 17 | 6544 (19.21) | 17 | 7281 (20.11) | 111.3 | 103.8–119.3 |
| Cmax (pg/mL) | 17 | 1160 (33.1) | 17 | 1560 (43.2) | 134.1 | 116.1–154.8 | |
| Tmax (h)a | 17 | 1.00 (0.500–2.00) | 17 | 1.00 (0.500–2.00) | — | — | |
| Rosuvastatin | AUC0‐t (h*ng/mL) | 17 | 36.79 (44.65) | 17 | 32.90 (37.38) | 89.45 | 78.73–101.6 |
| AUC0‐∞ (h*ng/mL) | 15 | 40.98 (42.72) | 15 | 34.39 (36.84) | 89.29 | 79.15–100.7 | |
| Cmax (ng/mL) | 17 | 3.47 (62.5) | 17 | 3.75 (47.6) | 108 | 91.92–127.0 | |
| Tmax (h)a | 17 | 3.00 (2.00–8.00) | 17 | 3.00 (1.50–4.05) | — | — | |
| t1/2 (h)b | 15 | 12.9 (4.28) | 15 | 11.2 (4.42) | — | — | |
| Omeprazole | AUC0‐t (h*ng/mL) | 16 | 451.8 (74.32) | 15 | 282.2 (90.92) | 63.52 | 57.40–70.30 |
| Cmax (ng/mL) | 17 | 229 (63.4) | 17 | 182 (78.4) | 79.5 | 64.97–97.28 | |
| Tmax (h)a | 17 | 2.00 (1.00–8.00) | 17 | 2.00 (1.50–4.00) | — | — | |
Abbreviations: AUC0‐∞ = area under the concentration‐time curve from time zero to infinity; AUC0‐t = area under the concentration–time curve from time zero to the time of the last quantifiable concentration; b.i.d. = twice daily; CI = confidence interval; C max = maximum observed plasma concentration; CV = coefficient of variation; GLSM = geometric least squares mean; t 1/2 = apparent terminal elimination half‐life; T max = time of the maximum observed plasma concentration.
Note: Geometric mean (CV%) data are presented;
Median (min‐max) data are provided for T max;
Arithmetic mean (SD) data are provided for t 1/2.
FIGURE 2.

Plasma concentration–time profile of (A) S‐warfarin, (B) midazolam, (C) digoxin, (D) rosuvastatin, (E) omeprazole after administration of single doses of probe drugs alone or in combination with zanubrutinib 160 mg b.i.d. Arithmetic mean with standard deviation is shown
The PK data showed that one subject had a much longer S‐warfarin half‐life (109 h compared to the mean of 39.3 h) and had predose S‐warfarin values >5% of C max on Day 14. Thus, genotyping for CYP2C9 was conducted and the data suggested that this subject was a CYP2C9‐poor metabolizer (*3/*3 [rs1057910]). For these reasons, this subject's concentration and parameter data from Day 14 onwards were excluded from descriptive and inferential statistics. Thus, descriptive and inferential statistics for S‐warfarin included four CYP2C9‐intermediate metabolizers (*1/*2 [rs1799853] or *1/*3 [rs1057910]) and 12 normal metabolizers. The mean concentration profiles indicated S‐warfarin was rapidly absorbed (with or without co‐administration of 160 mg zanubrutinib b.i.d.), with a median T max of 1 h postdose. After reaching C max, the disposition of S‐warfarin appeared to be biphasic.
Co‐administration of zanubrutinib did not significantly affect the PK of S‐warfarin or rosuvastatin; GMRs and 90% confidence intervals (CIs) of AUC0‐t , AUC0‐∞ and C max remained within the predefined equivalence range of 80–125% for S‐warfarin, and the 90% CIs of rosuvastatin (78.73–127%) fell just outside the prespecified range. Co‐administration of zanubrutinib decreased the systemic exposure of midazolam with mean changes to AUC0‐t , AUC0‐∞ and C max less than 50%. For omeprazole, although zanubrutinib co‐administration resulted in mean omeprazole AUC0‐t and C max values being approximately 36% and 21% lower, respectively, it was noted that there was a high between‐subject variability in the concentration time profile of omeprazole with or without zanubrutinib. Omeprazole was absorbed with a wide range of T max, which is consistent with the delayed‐release capsules of the drug used in the study.
Co‐administration with zanubrutinib increased the C max of digoxin, with a GMR of 134.1%. The AUC0‐t values, with a GMR of 111.3%, were considered equivalent with or without co‐administration of zanubrutinib since the 90% CIs (103.8–119.3%) were between 80 and 125%. A summary plot detailing the effects of zanubrutinib on each probe drug analyte is presented in Figure 3.
FIGURE 3.
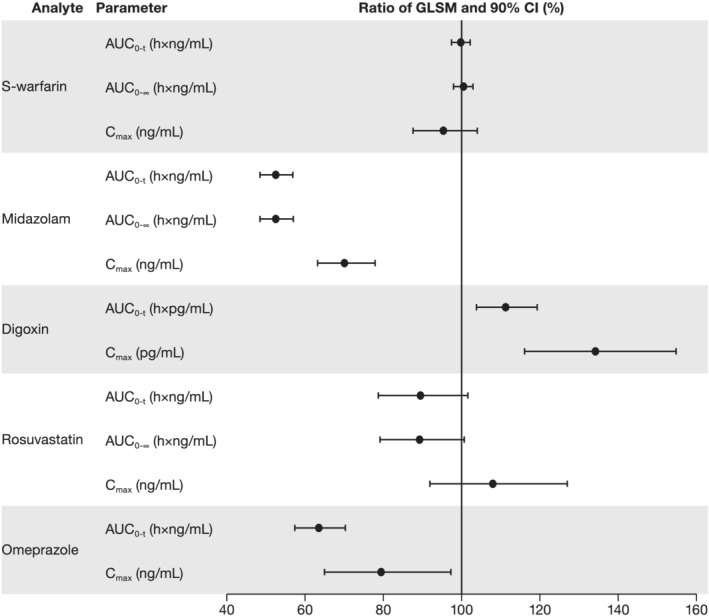
Summary plot of the effect of zanubrutinib on the pharmacokinetic parameters of probe drugs. Abbreviations: AUC0‐∞ = area under the concentration–time curve from time zero to infinity; AUC0‐t = area under the concentration–time curve from time zero to the time of the last quantifiable concentration; CI = confidence interval; C max = maximum observed plasma concentration; GLSM = geometric least squares mean
3.3. Plasma PK profile of zanubrutinib
The PK data of zanubrutinib are available from 17 subjects (Table 4). The geometric mean AUC0‐t and C max values of zanubrutinib administered alone were similar to those of zanubrutinib co‐administered with omeprazole. Zanubrutinib showed a similar median T max and a similar arithmetic mean t 1/2 when administered alone or co‐administered with omeprazole.
TABLE 4.
Summary of the pharmacokinetic parameters of zanubrutinib administered alone and with omeprazole 20 mg
| Parameter | Zanubrutinib 160 mg b.i.d. only (Day 13) (n = 17) | Probe drugs + zanubrutinib 160 mg b.i.d. (Day 18) (n = 17) |
|---|---|---|
| AUC0‐t (h*ng/mL) | 1032 (34.49) | 1073 (33.75) |
| AUC0‐τ (h*ng/mL)a | 1048 (35.18) | 1076 (33.69) |
| Cmax (ng/mL) | 231 (48.6) | 233 (40.4) |
| Tmax (h)b | 2.00 (1.00–4.00) | 2.00 (1.00–3.00) |
| t1/2 (h)c | 2.70 (0.851) | 2.93 (1.17) |
Abbreviations: AUC0‐t = area under the concentration–time curve from time zero to the time of the last quantifiable concentration; AUC0‐τ = area under the concentration–time curve over a dosing interval τ (where τ = 12 h); b.i.d. = twice daily; C max = maximum observed plasma concentration; CV = coefficient of variation; t 1/2 = apparent terminal elimination half‐life; T max = time of the maximum observed plasma concentration.
Note: Geometric mean (CV%) data are presented;
n = 16;
Median (min‐max) are provided for T max;
Arithmetic mean (SD) data are provided for t 1/2
3.4. Safety
During the administration of the probe drugs alone, one subject discontinued the study; therefore, the safety population was 18 subjects for the probe drugs only, 17 subjects for zanubrutinib only, and 17 subjects for probe drugs plus zanubrutinib. Overall, six subjects (33.3%) experienced nine TEAEs during the study, all of which were grade 1 in severity. The most common TEAE was petechiae (two subjects [11.1%]), which occurred following co‐administration of zanubrutinib with the probe drugs. The TEAEs of petechiae were considered to be related to zanubrutinib, warfarin and omeprazole. Three TEAEs (abdominal discomfort, diarrhoea and dizziness) in two subjects (11.8%) were considered to be related to zanubrutinib alone. The incidence of TEAEs [percentage] was similar after administration of the probe drugs only (three subjects [16.7%]), zanubrutinib only (two subjects [11.8%]), or probe drugs plus zanubrutinib (two subjects [11.8%]). All TEAEs were resolved by the end of the study and no subjects discontinued due to a TEAE. No deaths or serious AEs were reported during the study.
Doses of zanubrutinib 160 mg b.i.d. were generally well tolerated when administered alone or when co‐administered with warfarin, vitamin K and midazolam; with digoxin and rosuvastatin; or with omeprazole. Overall, there were no clinically relevant changes or findings noted in clinical laboratory evaluations, vital signs measurements, ECG results or physical examinations during the study. No TEAEs higher than grade 1 in severity were reported and no AEs observed in this study were considered as a new safety signal for zanubrutinib compared with clinical data observed in studies of patients with B‐cell malignancies.12
4. DISCUSSION
The results of this cocktail DDI study indicate that at clinically relevant concentrations, zanubrutinib had minimal or no effect on the activity of CYP2C9, BCRP and P‐gp, but decreased the systemic exposure of CYP3A‐ and CYP2C19‐sensitive substrates by less than 50%. While the cocktail approach to assess DDI has been commonly used, to our knowledge, the current study was one of the first studies to use this particular combination of probe substrates to assess DDI liability of these CYP enzymes and transporters together in one study. The cocktail approach used in this study is built upon reports of previous cocktail studies, including the “Modified Cooperstown Cocktail + 1” approach20 (midazolam, warfarin and omeprazole probes) and the Geneva phenotyping cocktail approach (six CYP isoforms and P‐gp probes).28 Digoxin (P‐gp) was safely used in evaluating clinical DDIs with metabolic enzymes.29 In a recent cocktail transporter study, the utility of using digoxin (P‐gp) and rosuvastatin (BCRP) probe drugs together has been validated.21 Building upon these previous studies, the current cocktail study incorporated BCRP and P‐gp transporter protein together with CYP substrate probes for comprehensive DDI assessment for zanubrutinib.
Due to the interplay of intestinal and hepatic metabolism and transporter activity, it is worth noting that changes in midazolam AUC in this study following oral zanubrutinib administration reflects both intestinal and hepatic CYP3A activity. Changes in digoxin C max would reflect activity of intestinal P‐gp. Rosuvastatin is the recommended probe substrate for both intestinal and hepatic BCRP.30 While rosuvastatin is a non‐specific BCRP substrate also transported by OATP1B1 and OATP1B3, in vitro studies showed that zanubrutinib had minimal inhibition potential of the hepatic uptake transporters, OATP1B1 and OATP1B3. Therefore, rosuvastatin was used to assess relatively specific effects of zanubrutinib on BCRP in this study. Since study results indicate minimal interaction during co‐administration of zanubrutinib and rosuvastatin (BCRP), therefore diminishing the probability of the worst‐case clinical BCRP DDI scenario from occurring in subjects harbouring a specific BCRP genotype (c.421C/C).30 Furthermore, the study did not include cholesterol monitoring regarding potential hepatic accumulation of rosuvastatin from inhibition of hepatic BCRP, since the inhibition potential was primarily at intestinal BCRP. This was mainly due to the estimated high concentrations of zanubrutinib in the GI tract ([I]2) associated with 160 mg zanubrutinib b.i.d. The ratios of intestinal concentration and in vitro IC50 ([I]2/IC50) for P‐gp and BCRP were estimated to be higher than the FDA‐recommended cutoff value of 10 for clinical inhibition. This clinical study confirmed that zanubrutinib does not have a clinically relevant impact on intestinal BCRP. Additionally, the ratio ([I]2/IC50) has been much higher than the cutoff value of 10 for compounds that have shown clinical DDI with BCRP, including tyrosine kinase inhibitors such as erlotinib, gefitinib, lapatinib, nilotinib and sunitinib.30
Zanubrutinib had no significant effect on the PK of CYP2C9 probe substrate, warfarin, indicating that zanubrutinib is neither a clinically relevant inhibitor nor an inducer of CYP2C9. Many patients with B‐cell malignancies, the target population of BTK inhibitors, have a history or risk of cardiovascular disease and have been allowed to take antithrombotic agents for primary or secondary prevention. Results from this study showed that zanubrutinib does not have an impact on the PK of CYP2C9 probe substrate, which would further support concomitant use of zanubrutinib with warfarin. From clinical DDI perspectives, the results of the current study also support concomitant use of zanubrutinib with direct oral anticoagulants (DOAC). CYP3A4 is an important metabolizer for apixaban and rivaroxaban but not the other DOACs. It has been shown that the fractions of rivaroxaban and apixaban metabolized by CYP3A4/5 was ~18% and 15%, respectively.31 Co‐administration of rivaroxaban with the strong CYP3A4 inducer rifampicin led to an approximate 50% decrease in mean rivaroxaban AUC, with parallel decreases in its pharmacodynamic effects. Product labelling includes a recommendation that the concomitant use of rivaroxaban (or apixaban) with strong CYP3A4 inducers (e.g., rifampicin, phenytoin, carbamazepine, phenobarbital and St. John's Wort) should be avoided.32 Given that the extent of CYP3A induction by zanubrutinib is far less than that of rifampicin, it can be concluded that zanubrutinib can be safely used with DOACs due to its low DDI potential.
There are limitations in the current study design. Due to the number of PK samples planned for each probe substrate and zanubrutinib (six together) and the limited blood volume that can be drawn per subject, the current PK sampling schedule may not be ideal to capture the C max of substrate drugs. This was reflected in the wide confidence interval in the mean estimate of C max for omeprazole and digoxin. More intensive PK sampling would have been ideal to characterize the effects of zanubrutinib on omeprazole, which was absorbed with a wide T max range with the delayed‐release capsules used in the study. Another limitation of this study is that substrates of CYP2B6 and CYP2C8 were not included in this study due to the lack of sensitive and validated probes for these enzymes to be used in a cocktail DDI study. Since the inhibitory IC50 values and induction potential are comparable for CYP2C8 and CYP2C9, the effect of zanubrutinib on the exposure of CYP2C8 substrates is unlikely to be clinically significant based on the lack of interaction with warfarin, a CYP2C9 substrate in this study. Based on the R3 values according to FDA guidance, the induction potential for CYP2B6 (R3 = 0.55) is relatively weak compared with CYP3A (R3 = 0.23) in vitro. Co‐induction of CYP3A4 has been observed for all CYP2B6 inducers due to crosstalk by the xenobiotic receptors constitutive androstane receptor (CAR) and Pregnane X receptor (PXR). It has been noted that if compounds exhibit CYP2B6 and CYP3A4 induction in vitro, a clinical CYP3A DDI study could serve as a surrogate for identifying the potential risk for CYP2B6 induction in the clinical setting.33 Furthermore, if the CYP3A4 clinical induction study is negative or mild, it can be concluded that the likelihood of CYP2B6 clinical induction is low.33 Thus, based on the clinical results in this study, it can be concluded that the likelihood of CYP2B6 clinical induction is low.
Since zanubrutinib exhibits pH‐dependent solubility, the potential impact of omeprazole on the exposure of zanubrutinib was assessed. The AUC and C max values of zanubrutinib administered alone were similar to those when co‐administered with omeprazole 20 mg. Although the interaction was assessed after only a single dose of omeprazole, studies have shown its effect on gastric pH occurs within 1 h, with the maximum effect (increasing the gastric pH to approximately 4.5) occurring within 2 h. The lack of impact for omeprazole on the PK of zanubrutinib is consistent with results from a population PK analysis, showing that co‐administration with proton‐pump inhibitors and acid‐reducing agents does not appear to significantly impact the PK of zanubrutinib.
In conclusion, at clinically relevant concentrations, zanubrutinib does not affect the PK of drugs metabolized by CYP2C9 or transported by BCRP. This result supports the recommendation that no dosage adjustments are necessary for drugs that are substrates of CYP2C9 and BCRP when given concomitantly with zanubrutinib. Zanubrutinib decreased the systemic exposure of CYP3A‐ and CYP2C19‐sensitive substrates with a mean reduction less than 50%. The results from this study provide important information on the potential clinical impact of zanubrutinib on these CYPs and transporter activities.
COMPETING INTERESTS
Y.C.O., Z.T., W.N., M.T., T.L. and S.S. are employees and own stock in BeiGene, Inc. H.A.C. is an employee of Covance.
CONTRIBUTORS
Y.C.O., Z.T., W.N., M.T., T.L. and S.S. designed the study. Y.C.O., Z.T., W.N., M.T., T.L., H.A.C. and S.S. conducted the study. Y.C.O., Z.T. and T.L. analysed the data. Y.C.O. and Z.T. wrote the manuscript. All of the authors reviewed and approved the manuscript.
Supporting information
TABLE S1 Summary of bioanalytical methods for zanubrutinib and probe drugs
ACKNOWLEDGEMENTS
The authors wish to acknowledge the investigative centre study staff, the study patients, and their families. BeiGene, Ltd. provided financial support for this paper, including writing and editorial assistance by Agnieszka Laskowski, PhD, and Elizabeth Hermans, PhD (OPEN Health Medical Communications, Chicago, IL). The study protocol was developed by BeiGene, Ltd. in collaboration with the study investigator. BeiGene, Ltd. was also involved in data collection, analysis and interpretation of results.
All authors were in agreement regarding the submission of this manuscript and vouch for the completeness and accuracy of the data. Professional medical writers, funded by BeiGene, Ltd., assisted with the development and submission of this manuscript under the authors' guidance. The corresponding author had full access to all of the study data and was responsible for the decision to submit the manuscript for publication.
Ou YC, Tang Z, Novotny W, et al. Evaluation of drug interaction potential of zanubrutinib with cocktail probes representative of CYP3A4, CYP2C9, CYP2C19, P‐gp and BCRP. Br J Clin Pharmacol. 2021;87:2926–2936. 10.1111/bcp.14707
The authors confirm that the Principal Investigator for this paper is Hugh A. Coleman, DO, and that he had direct clinical responsibility for the healthy male subjects.
Funding information BeiGene, Ltd., Grant/Award Number: N/A
DATA AVAILABILITY STATEMENT
Upon request, and subject to certain criteria, conditions and exceptions, BeiGene will provide access to individual de‐identified participant data from BeiGene‐sponsored global interventional clinical studies conducted for medicines (1) for indications that have been approved or (2) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary and statistical analysis plan. Data requests may be submitted to medicalinformation@beigene.com.
REFERENCES
- 1.Humphries LA, Dangelmaier C, Sommer K, et al. Tec kinases mediate sustained calcium influx via site‐specific tyrosine phosphorylation of the phospholipase Cgamma Src homology 2‐Src homology 3 linker. J Biol Chem. 2004;279(36):37651‐37661. [DOI] [PubMed] [Google Scholar]
- 2.Advani RH, Buggy JJ, Sharman JP, et al. Bruton tyrosine kinase inhibitor ibrutinib (PCI‐ 32765) has significant activity in patients with relapsed/refractory B‐cell malignancies. J Clin Oncol. 2013;31(1):88‐94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Petro JB, Khan WN. Phospholipase C‐gamma 2 couples Bruton's tyrosine kinase to the NF‐kappaB signaling pathway in B lymphocytes. J Biol Chem. 2001;276(3):1715‐1719. [DOI] [PubMed] [Google Scholar]
- 4.Burger JA, Tedeschi A, Barr PM, et al. Ibrutinib as initial therapy for patients with chronic lymphocytic leukemia. N Engl J Med. 2015;373(25):2425‐2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Byrd JC, Furman RR, Coutre SE, et al. Targeting BTK with ibrutinib in relapsed chronic lymphocytic leukemia. N Engl J Med. 2013;369(1):32‐42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Byrd JC, Brown JR, O'Brien S, et al. Ibrutinib versus ofatumumab in previously treated chronic lymphoid leukemia. N Engl J Med. 2014;371(3):213‐223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Farooqui MZ, Valdez J, Martyr S, et al. Ibrutinib for previously untreated and relapsed or refractory chronic lymphocytic leukaemia with TP53 aberrations: a phase 2, single‐arm trial. Lancet Oncol. 2015;16(2):169‐176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Maddocks K, Jones JA. Bruton tyrosine kinase inhibition in chronic lymphocytic leukemia. Semin Oncol. 2016;43(2):251‐259. [DOI] [PubMed] [Google Scholar]
- 9.Wang ML, Blum KA, Martin P, et al. Long‐term follow‐up of MCL patients treated with single‐agent ibrutinib: updated safety and efficacy results. Blood. 2015;126(6):739‐745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang ML, Rule S, Martin P, et al. Targeting BTK with ibrutinib in relapsed or refractory mantle‐cell lymphoma. N Engl J Med. 2013;369(6):507‐516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Treon SP, Tripsas CK, Meid K, et al. Ibrutinib in previously treated Waldenstrom's macroglobulinemia. N Engl J Med. 2015;372(15):1430‐1440. [DOI] [PubMed] [Google Scholar]
- 12.Brukinsa [package insert]. San Mateo, CA: BeiGene USA, Inc; 2019. [Google Scholar]
- 13.BeiGene announces the approval of BRUKINSA™ (Zanubrutinib) in China for patients with relapsed/refractory chronic lymphocytic leukemia or small lymphocytic lymphoma and relapsed/refractory mantle cell lymphoma. June 3, 2020. http://ir.beigene.com/news‐releases/news‐release‐details/beigene‐announces‐approval‐brukinsatm‐zanubrutinib‐china. Accessed June 26, 2020.
- 14.Dimopoulos M, Opat S, Lee HP, et al. Major responses in MYD88 wildtype (MYD88WT) Waldenstrom macroglobulinemia (WM) patients treated with Bruton tyrosine kinase inhibitor zanubrutinib (BGB‐3111). Poster presented at 24th Annual Congress of the European Hematology Association (EHA), June 13–16, 2019, Amsterdam, Netherlands.
- 15.Song Y, Zhou K, Zou D, et al. Zanubrutinib in patients with relapsed or refractory mantle cell lymphoma: a single‐arm, multicenter, pivotal phase 2 study. Presented at 15th International Conference on Malignant Lymphoma, June 18–22, 2019, Lugano, Switzerland.
- 16.Tam CS, Trotman J, Opat S, et al. Phase 1 study of the selective BTK inhibitor zanubrutinib in B‐cell malignancies and safety and efficacy evaluation in CLL. Blood. 2019;134(11):851‐859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Mu S, Tang Z, Novotny W, et al. Effect of rifampin and itraconazole on the pharmacokinetics of zanubrutinib (a Bruton's tyrosine kinase inhibitor) in Asian and non‐Asian healthy subjects. Cancer Chemother Pharmacol. 2020;85(2):391‐399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.US Food and Drug Administration, Center for Drug Evaluation and Research. NDA/BLA multi‐disciplinary review and evaluation NDA 213217 BRUKINSA (zanubrutinib). https://www.accessdata.fda.gov/drugsatfda_docs/nda/2019/213217Orig1s000MultidisciplineR.pdf. Accessed October 28, 2020.
- 19.US Food and Drug Administration: Guidance for Industry. in vitro metabolism and transporter‐mediated drug–drug interaction studies. October 24, 2017. https://www.fda.gov/media/108130/download. Accessed August 12, 2020.
- 20.Chainuvati S, Nafziger AN, Leeder JS, et al. Combined phenotypic assessment of cytochrome p450 1A2, 2C9, 2C19, 2D6, and 3A, N‐acetyltransferase‐2, and xanthine oxidase activities with the “Cooperstown 5+1 cocktail”. Clin Pharmacol Ther. 2003;74(5):437‐447. [DOI] [PubMed] [Google Scholar]
- 21.Stopfer P, Giessmann T, Hohl K, et al. Pharmacokinetic evaluation of a drug transporter cocktail consisting of digoxin, furosemide, metformin, and rosuvastatin. Clin Pharmacol Ther. 2016;100(3):259‐267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gibbons JA, de Vries M, Krauwinkel W, et al. Pharmacokinetic drug interaction studies with enzalutamide. Clin Pharmacokinet. 2015;54(10):1057‐1069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhuang Y, de Vries DE, Xu Z, et al. Evaluation of disease‐mediated therapeutic protein drug interactions between an anti interleukin‐6 monoclonal antibody (Sirukumab) and cytochrome P450 activities in a phase 1 study in patients with rheumatoid arthritis using a cocktail approach. J Clin Pharmacol. 2015;55(12):1386‐1394. [DOI] [PubMed] [Google Scholar]
- 24.Ebner T, Ishiguro N, Taub ME. The use of transporter probe drug cocktails for the assessment of transporter‐based drug–drug interactions in a clinical setting—proposal of a four component transporter cocktail. J Pharm Sci. 2015;104(9):3220‐3228. [DOI] [PubMed] [Google Scholar]
- 25.Kapetas AJ, Sorich MJ, Rodrigues AD, Rowland A. Guidance for rifampin and midazolam dosing protocols to study intestinal and hepatic cytochrome P450 (CYP) 3A4 induction and de‐induction. AAPS J. 2019;21(5):78. [DOI] [PubMed] [Google Scholar]
- 26.Brown H, Prescott R. Cross‐over trials. In: Applied mixed models in medicine. 3rd ed.New York, NY: John Wiley & Sons; 1999:271‐308. [Google Scholar]
- 27.Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to Pharmacology 2019/20: Enzymes. Br J Pharmacol. 2019;176(S1):S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bosilkovska M, Samer CF, Déglon J, et al. Geneva cocktail for cytochrome p450 and P‐glycoprotein activity assessment using dried blood spots. Clinical Pharmacol Ther. 2014;96(3):349‐359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Kim MG, Kim Y, Jeon JY, Kim DS. Effect of fermented red ginseng on cytochrome P450 and P‐glycoprotein activity in healthy subjects, as evaluated using the cocktail approach. Br J Clin Pharmacol. 2016;82(6):1580‐1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lee CA, O'Connor MA, Ritchie TK, et al. Breast cancer resistance protein (ABCG2) in clinical pharmacokinetics and drug interactions: practical recommendations for clinical victim and perpetrator drug–drug interaction study design. Drug Metab Dispos. 2015;43(4):490‐509. [DOI] [PubMed] [Google Scholar]
- 31.Foerster KI, Hermann S, Mikus G, Haefeli WE. Drug–drug interactions with direct oral anticoagulants. Clin Pharm. 2020;59(8):967‐980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Xarelto [package insert]. Titusville, NJ: Janssen Pharmaceuticals, Inc.; 2020. [Google Scholar]
- 33.Fahmi OA, Shebley M, Palamanda J, et al. Evaluation of CYP2B6 induction and prediction of clinical drug–drug interactions: considerations from the IQ consortium induction working group—an industry perspective. Drug Metab Dispos. 2016;44(10):1720‐1730. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
TABLE S1 Summary of bioanalytical methods for zanubrutinib and probe drugs
Data Availability Statement
Upon request, and subject to certain criteria, conditions and exceptions, BeiGene will provide access to individual de‐identified participant data from BeiGene‐sponsored global interventional clinical studies conducted for medicines (1) for indications that have been approved or (2) in programs that have been terminated. BeiGene will also consider requests for the protocol, data dictionary and statistical analysis plan. Data requests may be submitted to medicalinformation@beigene.com.