

Abstract
Purpose:
This study aimed to uncover novel cell types in heterogenous basal limbus of human cornea for identifying LSC at single cell resolution.
Methods:
Single cells of human limbal basal epithelium were isolated from young donor corneas. Single-cell RNA-Sequencing was performed using 10x Genomics platform, followed by clustering cell types through the graph-based visualization method UMAP and unbiased computational informatic analysis. Tissue RNA in situ hybridization with RNAscope, immunofluorescent staining and multiple functional assays were performed using human corneas and limbal epithelial culture models.
Results:
Single-cell transcriptomics of 16,360 limbal basal cells revealed 12 cell clusters belonging to three lineages. A smallest cluster (0.4% of total cells) was identified as LSCs based on their quiescent and undifferentiated states with enriched marker genes for putative epithelial stem cells. TSPAN7 and SOX17 are discovered and validated as new LSC markers based on their exclusive expression pattern and spatial localization in limbal basal epithelium by RNAscope and immunostaining, and functional role in cell growth and tissue regeneration models with RNA interference in cultures. Interestingly, five cell types/states mapping a developmental trajectory of LSC from quiescence to proliferation and differentiation are uncovered by Monocle3 and CytoTRACE pseudotime analysis. The transcription factor networks linking novel signaling pathways are revealed to maintain LSC stemness.
Conclusions:
This human corneal scRNA-Seq identifies the LSC population and uncovers novel cell types mapping the differentiation trajectory in heterogenous limbal basal epithelium. The findings provide insight into LSC concept and lay the foundation for understanding the corneal homeostasis and diseases.
Keywords: Adult stem cells, Cornea, Epithelium, Limbal stem cells, Single-cell transcriptomics
Introduction
The limbal stem cell (LSC) concept that human corneal epithelium is maintained by stem cells residing in the corneal limbus has been recognized for three decades [1–3]. Ocular surface diseases with LSC deficiency, such as Stevens-Johnson syndrome, chemical and thermal burns, radiation injury, extensive microbial infection, and inherited disorders such as aniridia, are sight-threatening and often cause blindness [4–7]. Only transplantation of the corneal limbus that contains the LSCs is capable of regenerating a normal corneal epithelium and restoring vision.
Adult stem cells have been broadly recognized to be present in a variety of human organs and tissues [8]. The unique localization of LSCs in the limbal basal epithelial layer [9,10], a spatially distinct compartment, makes LSCs a great model to study adult stem cell biology. Comprehensive investigations have demonstrated that human LSCs exhibit the key characteristics of epithelial stem cells, which are rare, undifferentiated and quiescent cells with self-renewal ability, high proliferation potential and tissue regeneration capacity [2,10–13]. LSCs respond to corneal epithelial cell turnover by differentiating to produce progenitors and amplifying cells, which divide and migrate toward the central corneal basal layers to replenish the corneal epithelium [14,15]. However, the limbal basal epithelium is heterogeneous, and it remains a mystery what kind of various cell types or subtypes resides in addition to LSCs. it is still challenging and elusive to actually identify and isolate LSCs due to lack of the definitive markers that exclusively identify the stem cells [11,16,17].
The cutting-edge high throughput single-cell RNA-sequencing (scRNA-Seq) technology holds the promise to revolutionize our understanding of diseases and associated biological processes at an unprecedented resolution. It has the power to discover new cell types, to identify unique cell states, and to dissect underlying heterogeneity in a high-throughput and unbiased manner [18–20]. Current knowledge on human limbal basal layer and LSC is mostly based on broad investigations at the tissue and bulk cell population levels. Single-cell transcriptomics opens a new window to uncover the novel cell types of LSCs and their progenies from the heterogeneous limbal basal epithelium of human cornea.
Recently Lavker group reported an insightful study using single-cell RNA transcriptome to define the limbal/corneal epithelial stem/early transit amplifying cells in mice [21]. In the present study, we performed scRNA-Seq using the isolated single-cells from human basal limbus to uncover the various type/subtype cells and to define LSCs at single-cell resolution. We were able to identify the LSC population and uncover multiple cell types mapping the developmental trajectory of LSCs in the heterogeneous limbal basal epithelium. The new marker genes for different cell types were discovered and further validated for their expression pattern in human corneas and tissue regeneration function. Two exclusively expressed genes TSPAN7 and SOX17 were identified as potential new markers for LSCs.
Methods
Donor corneal tissues and single cell isolation from corneal limbal epithelial basal layer
Fresh human corneoscleral tissues were obtained from the Lions Eye Bank of Texas (LEBT, Houston, TX) for this study. The corneal tissues used for single-cell RNA-Seq were from two healthy young donors, one male and one female, without ocular diseases, refractive surgery, chronic disease, cancer and chronic infections such as Hepatitis B, C, and HIV. After central cornea removed and superficial layers scraped, the remaining limbal tissue with basal cells were incubated in dispase II (10 mg/ml) in SHEM at 4 °C overnight [22]. The loose limbal epithelial sheets were peeled gently and digested with 1 ml of 0.05% trypsin/1 mM EDTA at 37 °C for 10 min. The trypan blue was used to test the viability of single cells.
Single-cell RNA-sequencing
Single-cell RNA-Seq was performed at the Single Cell Genomics Core at Baylor College of Medicine. In brief, single cell cDNA library preparation and sequencing were performed following manufacturer’s protocols (https://www.10xgenomics.com). Single cell suspension at 1000 cells/μl in PBS was loaded on a Chromium controller to generate single cell GEMS (Gel Beads-In-Emulsions). The scRNA-Seq library was prepared with chromium single cell 3' reagent kit v2 (10x Genomics). The product was amplified by PCR and sequenced on Illumina Novaseq 6000 (https://www.illumina.com).
Bioinformatic analysis of scRNA-seq data
Cell Ranger software v2.1 (https://www.10xgenomics.com) with default settings was used for alignment, barcode assignment, and UMI counting of the raw sequencing data with genome reference hg19. In initial processing we detected 7,096 cells with 115,983 mean reads per cell and 3,257 median genes per cell from donor1, and 10,491 cells with 82,128 mean reads per cell and 2,731 median genes per cell from donor2.
After generating UMI count profile, we applied Seurat 3.0 for quality control and downstream analysis [23]. A global-scaling normalization method ‘LogNormalize’ is employed to normalize the gene expression measurements for each cell by the total expression, then the result is multiplied by a scale factor (10,000 by default), and log-transformed in each data set. For data alignment, we selected 1,000 highly variable genes in each data matrix and performed ‘FindIntegrationAnchors’ and ‘IntegrateData’ functions in Seurat 3.0. Next, we performed the clustering using ‘FindClusters’ in Seurat to identify sub-cell type clusters. UMAP of each data set were visualized if each cluster was derived from both donors (Supplementary Fig. 1).
To identify differentially expressed marker genes in each cluster, we used the ‘FindAllMarkers’ function based on the Wilcoxon rank-sum test in Seurat. Top 10 DEGs in each cluster were visualized with a heatmap using Seurat. Cell cycle scoring was performed with cell cycle phase marker genes using ‘CellCycleScoring’ function in Seurat [24,25].
Trajectory and gene regulatory network analysis
Monocle 3 and CytoTRACE (Cellular Trajectory Reconstruction Analysis using gene Counts and Expression) were used to infer the cell differentiation trajectories [26–29]. These algorithms place the cells along a trajectory corresponding to a biological process (in our case, cell differentiation) by taking advantage of an individual cell’s asynchronous progression under an unsupervised framework.
In addition, functional principal component analysis (FPCA) using our own developed R scripts was performed using the dominant modes of variation of functional data [30]. Gene regulatory network (GRN) analysis was conducted using Single-Cell Regulatory Network Inference and Clustering (SCENIC) [31]. Then we identified a set of regulons with transcript factors and their target genes, and examined their expression levels and AUCell scores using R.
RNA in situ hybridization using RNAscope assays
The RNAscope HiPlex Assay (ACD Biosystems) was performed according to the ACD protocol for fresh-frozen tissue. Corneal sections from postmortem human were hybridized with three mRNA probes per experiment. To confirm the mRNA integrity of tissue blocks, positive control probes targeting against human house-keeping genes Polr2a, PPIB, UBC, HPRT were visualized. Additionally, marker probes were used on one section per slide to confirm signal specificity. The probes were amplified according to the manufacturer’s instructions and labeled with the following fluorophores for each experiment: Alexa 488 nm, Atto 550 nm, Atto 647 nm, and AF750. The Vectra® Polaris™ Automated Quantitative Pathology Imaging System was used to visualize the FISH signals.
Primary human limbal epithelial cultures and in vitro regeneration model
HLECs were cultured using explants from donor corneal limbal rims for multiple in vitro models by our previous method [22,32–35]. In brief, the wound incisions were made by scraping cells in 2 mm wide area in confluent primary HLEC cultures or siRNA treated cultures. Cultures at different time periods of epithelial regeneration after wound were used for RNA extraction, or immunofluorescent staining.
WST cell proliferation assay
WST Cell Proliferation assay was performed according to the manufacturer’s protocol and our previous report [32]. In brief, 10 μl of WST-1 cell proliferation agent was added to each well containing 100 μl medium in 96-well plates. The cells were then incubated for 2 h at 37 °C in a 5% CO2 incubator. The plate was measured at a wavelength of 450 nm with a reference wavelength 690 nm in an Tecan Infinite M200 microplate reader (Männedorf, Switzerland).
RNA interference by siRNA
RNA interference was performed using small interfering RNA (siRNA) according to our previous Fast-Forward Transfection method using HiperFect transfection reagent [32]. In brief, primary HCECs (6 × 104 cells/cm2) in 12-well were transfected by Hiperfect reagent (Qiagen) with annealed double-stranded siRNA specific for TSPAN7 (siR-NA-TSPAN7) or SOX17 (siRNA-SOX17) at different concentrations, and a non-coding sequence siRNA-fluorescein (siRNA-F) was used as a negative control (also served as a visible monitor for transfection efficiency). The transfected cells were created a 2 mm wide wound for regeneration models and incubated for 72 h for RNA extraction and immunofluorescent staining. For WST-1 cell proliferation assay, the mixture of siRNA and Hiperfect reagent was added in 96-well plate for 5–10 min incubation at room temperature, and HCECs were then seeded at 6000/well, which made total volume 100 μl/well, and incubated for additional 72 h before WST assay.
Total RNA extraction, reverse transcription, and quantitative real-time PCR (RT-qPCR)
As previously described [32,36], total RNA was isolated from cells using RNeasy Micro Plus kit (Qiagen), quantified by NanoDrop ND-1000, and stored at −80 °C, the first-strand cDNA was synthesized by using Ready-To-Go You-Prime First-Strand Beads, and the real-time PCR was performed in the Applied Biosystems® QuantStudio® 3 Real-Time PCR System using TaqMan® qPCR reagents with TaqMan gene expression assays including human GAPDH (Hs99999905-ml), TSPAN7 (Hs00190284_m1), SOX17 (Hs00751752_s1), and DCN (Hs00370385_m1) (Applied Biosystems). The results were analyzed by the comparative threshold cycle method and normalized by GAPDH.
Immunofluorescent Staining and laser scanning confocal microscopy
Immunofluorescent staining was performed using protocols as previously described [32,34,35]. In brief, human corneal frozen sections or cultured limbal epithelial cells were fixed with cold acetone at −30 °C for 3 min or 4% paraformaldehyde at 4 °C for 10 min. Cultured cells were permeabilized with 0.2% Triton X-100 in phosphate-buffered saline (PBS) at room temperature for 10 min. After blocking with 20% normal goat serum in PBS for 30 min, the samples were incubated with primary antibodies overnight at 4 °C. The primary antibodies against markers for different cell types were TSPAN7, SOX17, DCN, PLIN2, TK1, KI67 and Melan-A (see details in Supplementary Table 1). Alexa-Fluor 488 (1:200) was used as secondary antibody with DAPI for nuclear counterstaining. The digital images were captured with a laser scanning confocal microscope (Nikon A1 RMP, Nikon, Melville, NY) wavelength 400–750 nm and one μm z-step. The images were processed using software NIS Elements 4.20 version (Nikon, Garden City, NY).
Statistical analysis
In addition to bioinformatics analysis for scRNA-Seq data, Student t-test or Mann-Whitney U test was used for all biological experiments to make comparisons between 2 groups. ANOVA or the Kruskal-Wallis tests were used to evaluate 3 or more groups with the appropriate post-test to compare pairs of group means.
Data and code availability
All raw data are available throughout Gene Expression Omnibus (GEO) with accession number GSE153515 (https://urldefense.proofpoint.com/v2/url?u=https-3A__www.ncbi.nlm.nih.gov_geo_query_acc.cgi-3Facc-3DGSE153515&d=DwIBAg&c=ZQs-KZ8oxEw0p81sqgiaRA&r=0ttAf5n03KatecVFHf58mw&m=FGyZl_yfIX4uoDe0fAXWlLVMKCgHTkLuKKcbI1dzx4U&s=7x8PSA8eY8Wg4vi2IoVrwtJZW5xNkg_qk92jJJI6wzk&e=). R scripts used to analyze are freely available at the github (https://github.com/sangbaekim/10xRNAseq_LSC).
Results
Single cell transcriptomic profiles reveal heterogeneous cell types in limbal basal epithelium of human cornea
It is well accepted that human LSCs are a small subpopulation residing in the limbal basal epithelial layer. The number of LSCs is unclear but it had been estimated to be about 100 cells per cornea based on holoclone number using limbal clonal cell cultures [14,37]. It would be difficult to obtain LSCs if the single cells are isolated from entire corneal or limbal epithelium. To enrich for LSCs, we isolated single cells from basal limbus (Fig. 1A). These basal cells are small in size at 10–20 μm in diameter and accounted for ~15% of all limbal epithelial cells (Fig. 1B and C), with high viability (90–95%) from donor tissues.
Fig. 1.
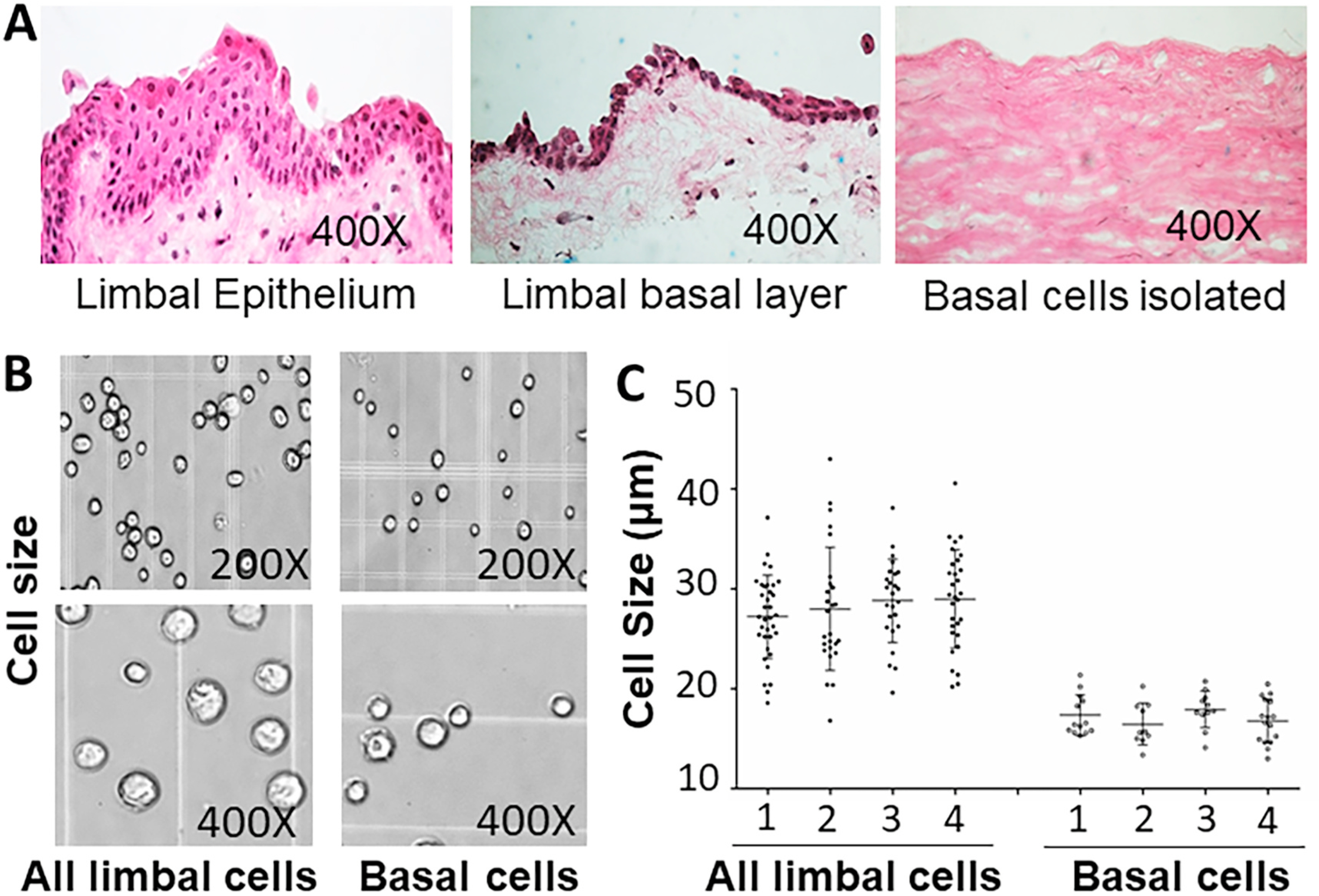
Isolation of viable limbal basal epithelial cells from donor corneas. A. The palisades of Vogt are distinctive normal features of the human corneal limbus with 5–7 layers of limbal epithelial cells. Only 1–2 layers of limbal basal cell remain after removing the superficial layers. The limbal basal cells are isolated to be single cells through treatment of Dispase II and trypsin/EDRA. B–C. The isolated limbal basal single cells are 10–20 μm in diameter, significantly smaller than all limbal epithelial cells based on four separate experiments (P < 0.01, n = 4).
We constructed two scRNA-seq libraries for the isolated cells from two healthy young donors, and obtained the transcriptomic profiles of 7,096 and 10,491 cells respectively using the 10x Genomics platform (Fig. 2A). Mean reads per cell were 115,983 and 82,128, and median genes per cell were 3,257 and 2,731 respectively (Supplementary Table 2). Cells from both samples contribute to these clusters with a minimal batch effect observed (Supplementary Fig. 2). After quality assessment and filtering [23,38], a total number of 16,360 cells with good quality data were retained for downstream analysis.
Fig. 2.
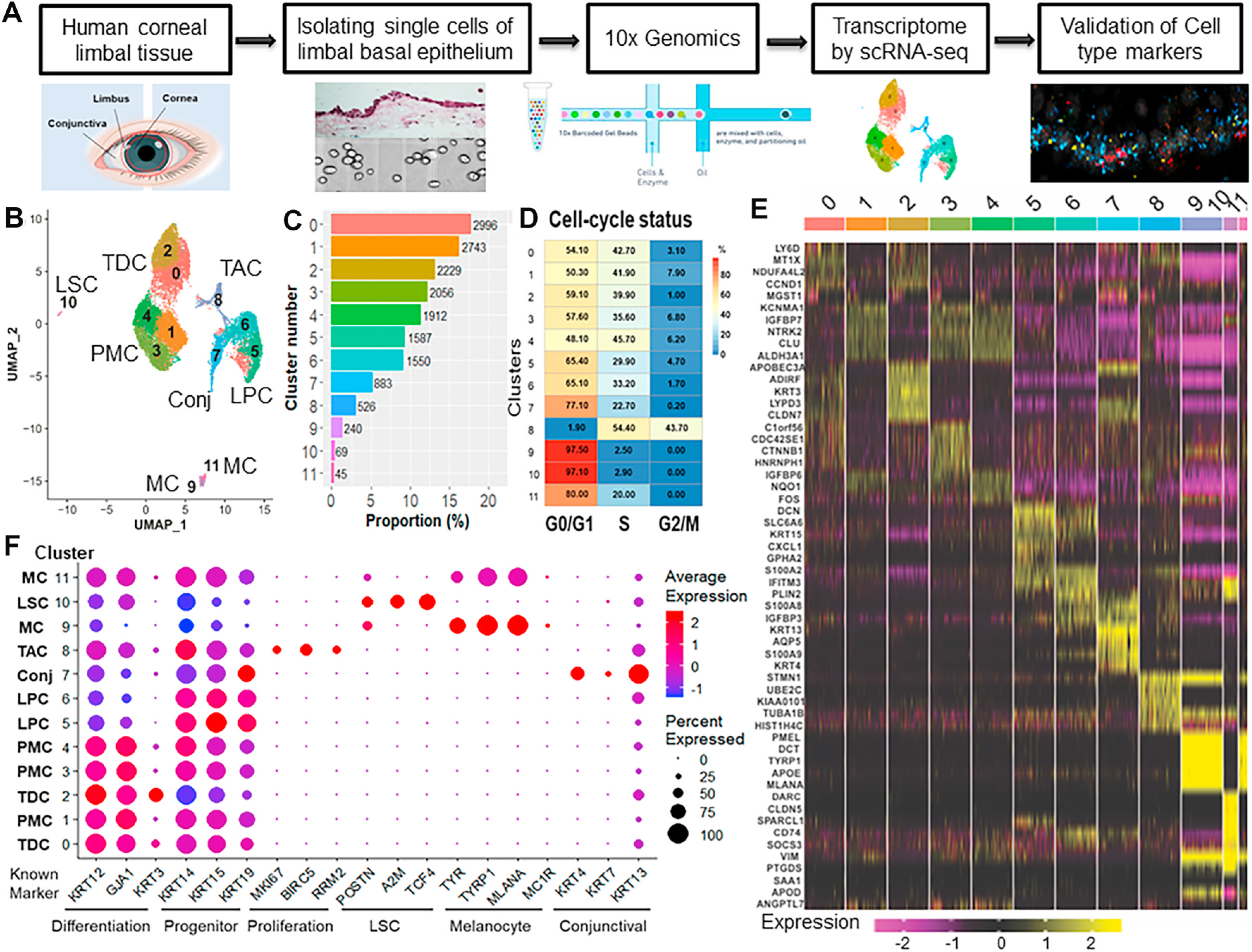
scRNA-seq revealed the heterogeneous cell types in limbal basal epithelium of human cornea. A. Experimental design for scRNA-seq analysis. B. UMAP of limbal basal cells from two donor tissues generated single-cell transcriptomic profiles of total16,360 cells with 12 differential cell clusters using Seurat package (v3.1.0). C. Proportion of cell populations in each cluster. Numbers on bar indicate the number of cells. D. The cell cycle stages for single cells per cluster were predicted by subpopulation using the Cell Cycle Scoring function of the Seurat package. E. Heatmap of the top 5 differentially expressed genes from each cluster showed distinguishable transcriptomic profiles among 12 cell populations. F. Dot plot for known marker genes indicated specific cell types for terminally differentiated cell (TDC), post-mitotic cell (PMC), transient amplifying cell (TAC), limbal progenitor cell (LPC), limbal stem cell (LSC), melanocyte (MC), conjunctival cell (Conj).
A total of 12 cell clusters were identified after dimensional reduction and using principal component analysis (PCA) and graph-based visualization method Uniform Manifold Approximation and Projection (UMAP) in Seurat (v3.1.0) [38] (Fig. 2A–C, Supplementary Table 3). The distinguishable transcriptomic profiles were revealed among 12 cell populations, as shown by a representative heatmap with the top 5 differentially expressed genes (DEGs) from each cluster (Fig. 2E).
The clustering result was annotated by the known markers including KRT12, KRT3 and GJA1 for differentiated cell; KRT14, KRT15 and KRT19 for progenitor; MKI67, BIRC5 and RRM2 for amplifying cell; TCF4, POSTN and A2M for LSC; TYR, TYRP1 and MLANA for melanocyte; and KRT4, KRT7 and KRT13 for conjunctival cells (Fig. 2F). Based on the expression pattern of these known markers, we were able to assign clusters (C) 0–4 as differentiated cells, C5–C6 as potentially progenitor cells, C8 as amplifying cells, C9/C11 as melanocyte, C7 as conjunctival cells, and C10 as the putative LSC. Thus, our scRNA-seq data reveal that the basal limbal epithelium is heterogeneous and contains multiple different cell types.
Define the subtypes of the nine clusters of limbal epithelial cells based on expression patterns of marker genes known to be related to epithelial stem/progenitor cells
LSCs are known to be less differentiated, slow-cycling quiescent cells with the capability of self-renewal and tissue regeneration [2,10–13,37]. Among 12 clusters, nine (C0–C6, C8 and C10) were annotated as LSC and its progenies. To further identify these 9 clusters, the expression pattern of an expanded set of 36 genes, which have been reported to be related to the cell properties in differentiation (10 genes) [1,34,36, 39–41], proliferation (14 genes) [1,36,42–44] and putative epithelial SCs (12 genes) [10,22,32–36,45], were examined as visualized by a heatmap (Fig. 3A) and the distribution maps (Fig. 3B–D).
Fig. 3.
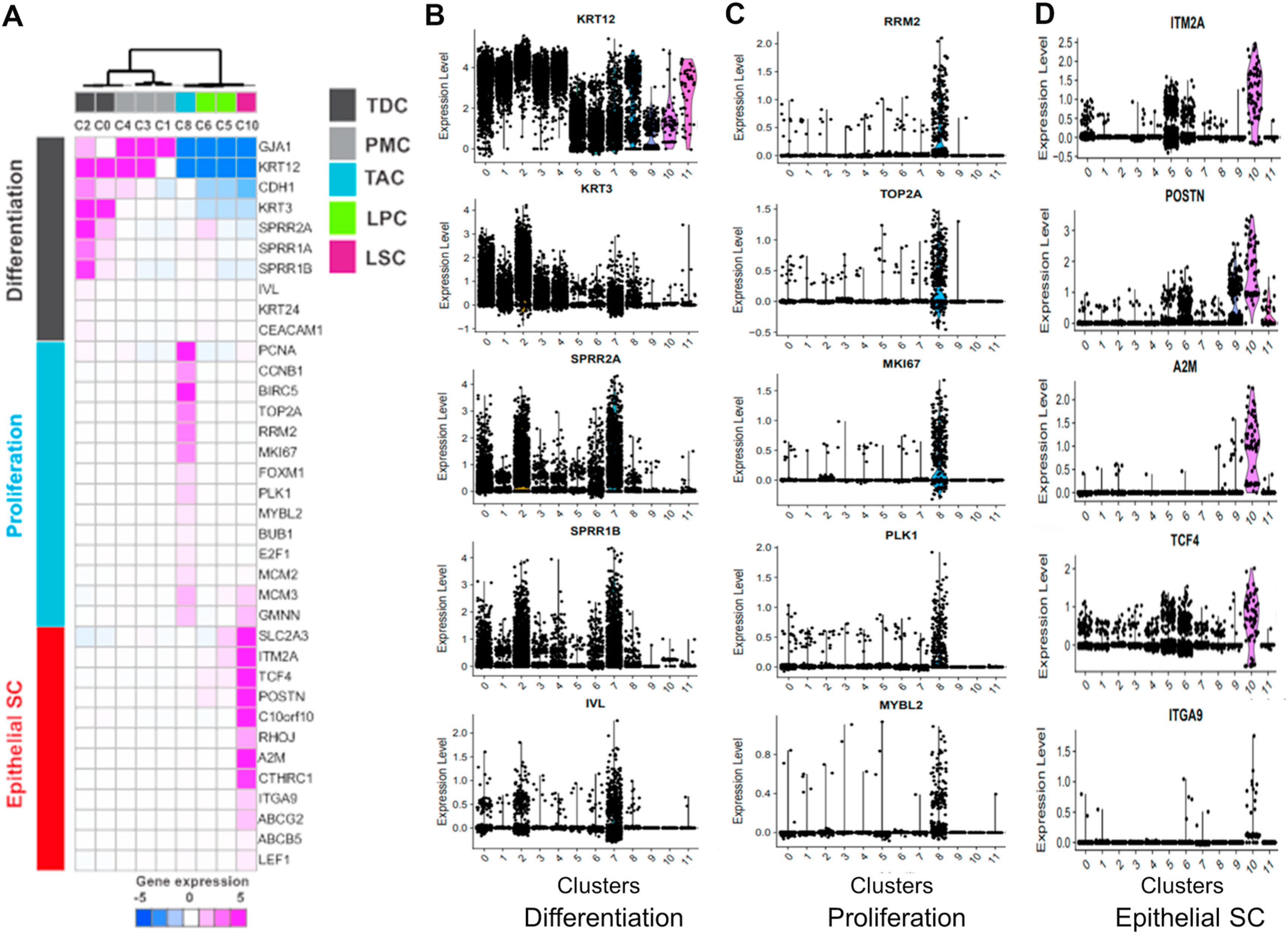
Identification of the subtypes of limbal cells. Known markers (n = 36) classified based on limbal cell differentiation status: Differentiation, proliferation, and epithelial stem cell. A. Unsupervised clustering of the expression profile of 36 known markers. Expression level was median centered of z scaled expression. B-D. Violin plot shows marker genes of differentiation, proliferation, and epithelial stem cells respectively.
Based on the differentiation status the 5 differentiated cell clusters (C0–C4) can be divided into two subgroups (Fig. 3A and B). C0/2 were terminally differentiated cells (TDCs) as they expressed the highest levels of differentiation markers including corneal epithelial specific differentiation markers, cytokeratin 3 (KRT3) and KRT12, and keratinocyte cornification markers SPRR2A, SPRR1B and IVL, but did not express the proliferation and epithelial SC makers (Fig. 3C and D). In contrast, the expression levels of KRT3, SPRR2A, SPRR1B and IVL by C1/3/4 were significantly lower than that of C0/2, suggesting that C1/3/4 represent post-mitotic cells (PMCs), an early stage of differentiated cells.
Among 4 clusters expressing a low level of differentiation markers, C8 was the only cluster highly expressing major proliferation markers, including BIRC5, RRM2, TOP2A, MKI67 and PLK1 (Fig. 3A,C). This is consistent with the cell cycle analysis (Fig. 2D), which shows 98% of C8 cells in S and G2/M phases. Therefore, C8 is likely to be transiently amplifying cells (TACs) that are about to exit cell cycle and differentiate into corneal epithelial cells. C5 and C6 expressed some putative SC markers, such as SLC2A3, ITM2A, POSTN, and TCF4, at a middle level (Fig. 3A,D), indicating they are likely limbal progenitor cells (LPCs).
C10 is identified as the putative LSC because it has the lowest levels of differentiation markers and the highest expression of putative SC markers, including ITM2A, TCF4, POSTN, A2M, ABCG2 and ITGA9, as shown in Fig. 3A,D. These genes are known to play a critical role in maintaining stem cell properties and multi-lineage potential of LSCs [12, 32–35,46–48]. Furthermore, C10 comprise a small percentage of the limbal basal cells (0.4% in our study) and is in the quiescent state with 97% cells at G0/G1 phases of the cell cycle (Fig. 2B–D).
Thus, single cell transcriptomics reveals that 9 of 12 clusters appear to represent different cell types mapping the trajectory of LSC developing stages from LSC (C10) to LPC (C5/6) and TAC (C8), ultimately differentiating to PMC (C1/3/4) and TDC (C0/2).
Pseudotemporal and functional PCA analysis confirmed the trajectory of LSC differentiation
To investigate the developmental progression from LSC to TDC clusters described above, we ordered these clusters along the differentiation stages by performing the trajectory analysis using Monocle 3 and CytoTRACE packages. The pseudotemporal analysis by Monocle 3, which uses the reversed graph embedding technique to quantitatively estimate the pseudotime and calculate the trajectory path of biological progression [26–28], revealed a putative developmental stages of the LSC differentiation (Fig. 4A–C). Consistent with the Seurat analysis, Monocle 3 reconstructed a similar trajectory, displaying the major cell clusters corresponding to the progenitor, transiently amplifying, and differentiated cell clusters. Interestingly, LSC cluster was well separated from other cell types, indicating that the expression signature of LSC is significantly distinct from other cell types. Moreover, the estimated pseudotime from Monocle 3 by assigning the LSC clusters as the root nodes elucidated the single-cell trajectory path in the order of clusters 10, 5, 6, 8, 4, 1, 3, 0, and 2 (Fig. 4B and C).
Fig. 4.
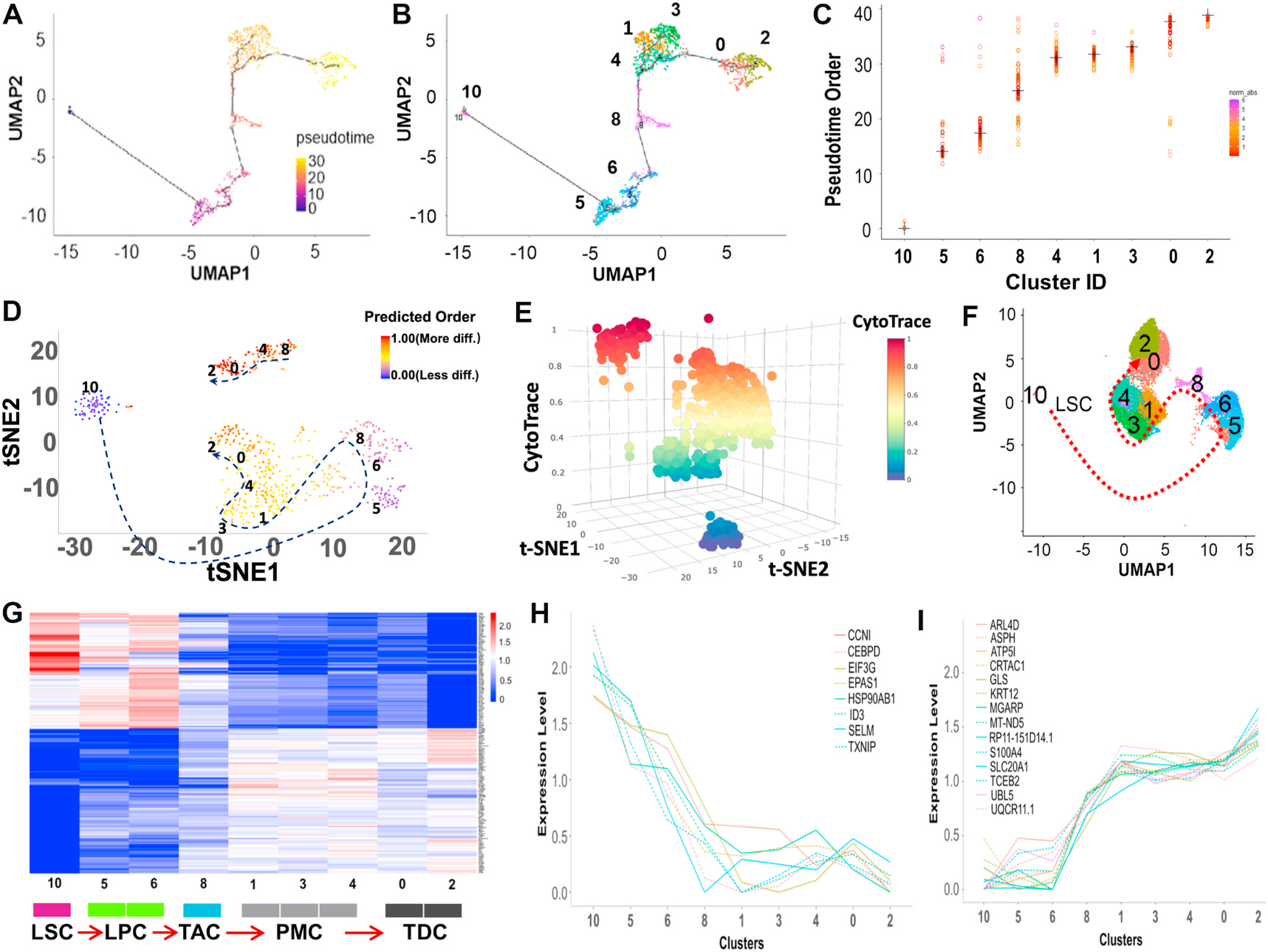
Reconstructing the developmental pseudotime trajectory of scRNA-seq data. A. The single-cell trajectory was predicted by Monocle 3 and visualized by UMAP. Cells are ordered in pseudotime colored in a gradient from purple to yellow. B. Monocle 3 generated pseudotemporal trajectory of 9 clusters identified in scRNA-seq clustering from Seurat analysis. Number and color represent each cluster. C. Plot of the pseudotime order for the 9 clusters from Monocle 3 analysis. Each dot represents a cell ordered in pseudotime grouped by clusters. Cluster median is marked as a cross. Distribution of pseudotime in each cluster is shown by the gradient color plotting each cell. D, E. CytoTRACE was used to predict the trajectory of the differentiation state of cells from nine clusters. Predicted differentiation state was visualized in 2D and 3D t-SNE plots based on the differentiation scores with the dotted arrow. F. Differentiation model on UMAP based on the trajectory analysis using Monocle 3 and CytoTRACE. The development and differentiation of the cells along the trajectory are in the order of clusters 10, 5, 6, 8, 1, 3, 4, 0, and 2, as shown in Fig. 4A–F. G. Heatmap of genes whose expression shows a significant trend along this trajectory using functional PCA in R. H, I. Different expression patterns of significant genes on differentiation status from LSC to TDC predicted by trajectory analysis.
In addition, we applied CytoTRACE, a recently developed method [29] to predict differentiation status from scRNA-seq data by considering the number of expressed genes per cell in combination with the number of RNA copies per gene. CytoTRACE analysis further confirmed the pseudotime order (clusters 10, 5, 6, 8, 1, 3, 4, 0, and 2) for the trajectory of LSC differentiation (Fig. 4D–F). The results from pseudotemporal analysis are consistent with the cell lineage order based on the known markers associated with stem cell properties (Fig. 3).
Functional PCA analysis [30] further showed that the top DEGs in these 9 clusters follow a trend along this trajectory (Fig. 4G). The different trends of selected genes along the trajectory are clearly shown in Fig. 4H and I. For example, the highly expressed genes in C10 (Fig. 4H), such as ID3, CCNI, EIF3G, EPAS1, HSP90AB1, SELM, have been reported to involve in maintaining stem cell property and function [49]. Especially, ID3 (Inhibitor of DNA binding 3) is implicated in regulating a variety of cellular processes including cell growth, differentiation, apoptosis, and angiogenesis. ID3 could promote the stemness via activation of β-catenin and regulate the differentiation of neural stem cells, and it is required for the survival and regeneration of bone marrow to maintain HSC development [50,51]. These DEGs provide new potential to investigate LSC markers and pathways.
Genes distinctively expressed in LSC and LPC may provide new markers to identify LSC
To identify the marker genes for each cell cluster, we performed PCA and unsupervised hierarchical clustering with 200 highly variable genes (HVGs) across clusters (Fig. 5A–C, Supplementary Table 4). We identified DEGs in individual clusters using Wilcoxon rank sum test in Seurat (Fig. 5D–F, the top 50 DEGs listed in Supplementary Table 5). For LSC (C10), exclusively expressed markers, such as TSPAN7, SOX17, SELE, ECSCR, RAMP3, AC011526.1, RNASE1, NPDC1, C10orf10, IGFBP4, SLC2A3, NNMT, KLF2, and PDK4, were revealed (Fig. 5D). While these markers need further investigation for their potential function in LSCs, several of these marker genes are reported to play important roles in other stem cells. These new DEGs for LSC revealed by scRNA-Seq provide novel targets in searching definitive markers for human LSC.
Fig. 5.
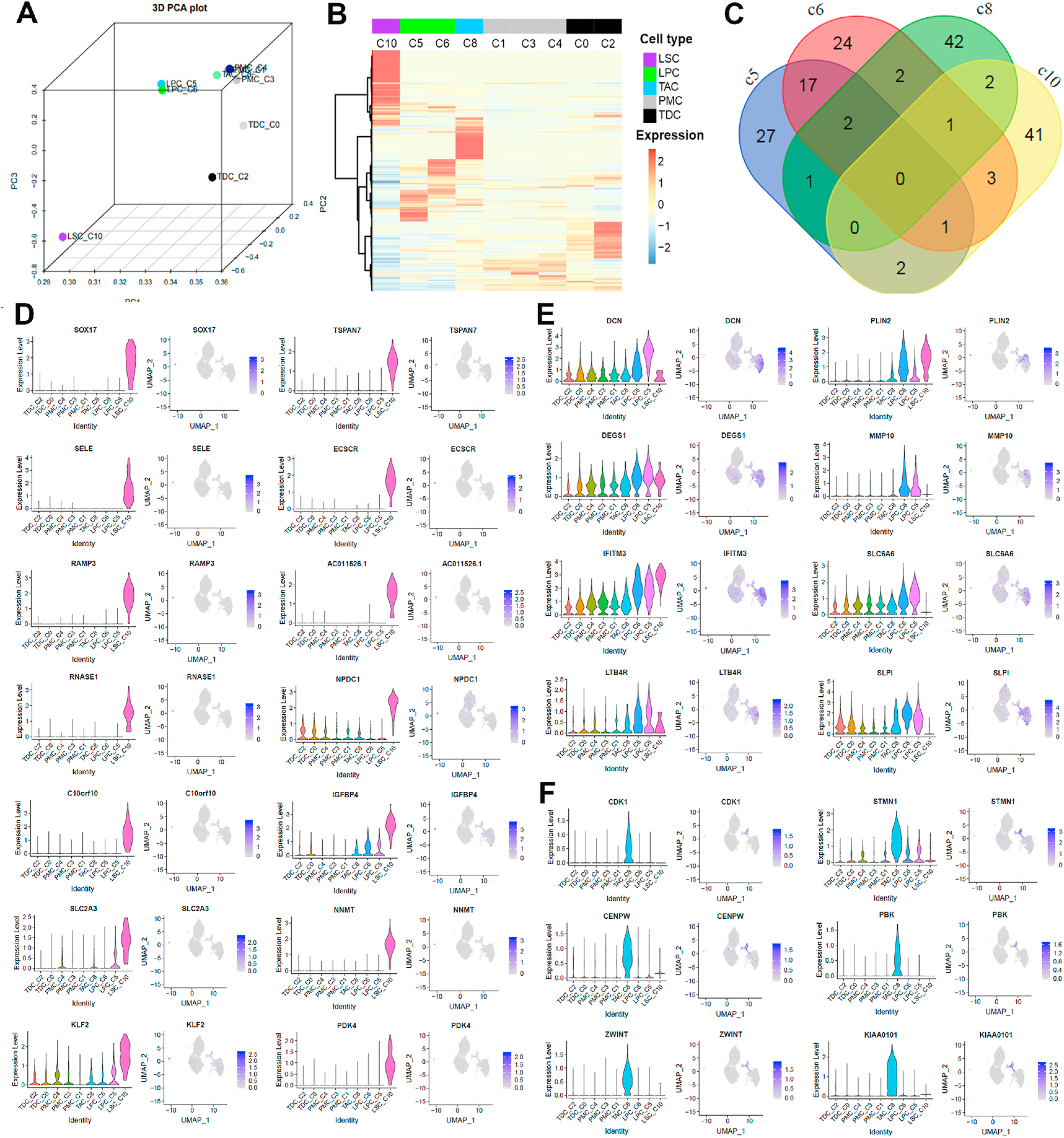
Identifying new markers in LSC (C10), LPC (C5, 6), and TAC (C8). A. 3D PCA of 9 clusters with the averaged expression of the 1240 highly variable genes (HVGs) using scatterplot3d package (https://cran.r-project.org/web/packages/scatterplot3d). Each dot is annotated with cell type and cluster. B. Unsupervised hierarchical clustering of HVGs with averaged expression. Row indicates each of the top 200 HVGs and columns indicate clusters in pseudotime order. Colored bars on columns represent cell type. Expression values were centered and z scaled. C. Venn diagram displaying the numbers of top 50 DEGs intersected among clusters assigned as LSC, LPC and TAC. D-F. Violin and feature plots for new marker genes in LSC (D), LPC (E), and TAC (F) clusters. DEGs were identified using a Wilcoxon rank sum test in Seurat, and ranked by adj-P value.
For example, TSPAN7 encodes a cell surface glycoprotein, a member of the tetraspanin family. TSPAN29 (CD9) was identified as a definitive maker that captures all hematopoietic stem cells in murine bone marrow in the absence of contaminating populations that lack authentic stem cell function [52]. The isolated TSPAN1+ neoblasts was reported to be adult pluripotent stem cells underlying planaria regeneration, and transplantation of single TSPAN1+ cells rescued lethally irradiated animals [53]. TSPAN7-mediated signal transductions also play a role in the regulation of cell morphology, activation, growth, motility, and development [54]. SOX17 encodes a member of the SOX (SRY-related HMG-box) family of transcription factors involved in the regulation of embryonic development and in the determination of the cell fate. It has been known as a critical regulator in proliferation and differentiation of embryonic, hematopoietic cells [55,56], and is related to Wnt signaling pathway during embryogenesis and during adult tissue homeostasis [57].
Unlike the marker genes in LSC, the marker genes in LPC (C5 and C6), such as DCN, PLIN2 DEGS1, MMP10, IFITM3, SLC6A6, LTB4R, and SLPI, do not show exclusive expression pattern but rather have a higher expression level in C5/6 compared with other clusters (Fig. 5E). DCN (decorin) is an extracellular proteoglycan and plays a role in proliferation, spreading, migration, and differentiation of progenitor cells, maintaining human hair follicle SCs, and inhibiting cancer SCs [58]. PLIN2 is a major hepatic lipid droplet protein associated with intracellular neutral lipid storage droplets, WNT signaling pathway [59], and early adipocyte differentiation [60].
Spatial localization of different cell types in human cornea tissue was validated by RNAscope and immunofluorescent staining
To validate different cell types revealed by scRNA-Seq, a combination of tissue RNA in situ hybridization and immunofluorescent staining was performed to determine the spatial location of different cell types in human corneal limbus tissue at mRNA and protein levels. Marker genes specifically expressed in each cell type were selected as such: TSPAN7 and SOX17 for LSC, DCN and PLIN2 for LPC, TK1 for TAC, HTRA1 for PMC, and TYR and MLANA for MC, with known marker KRT3 for TDC as control (Fig. 6A).
Fig. 6.
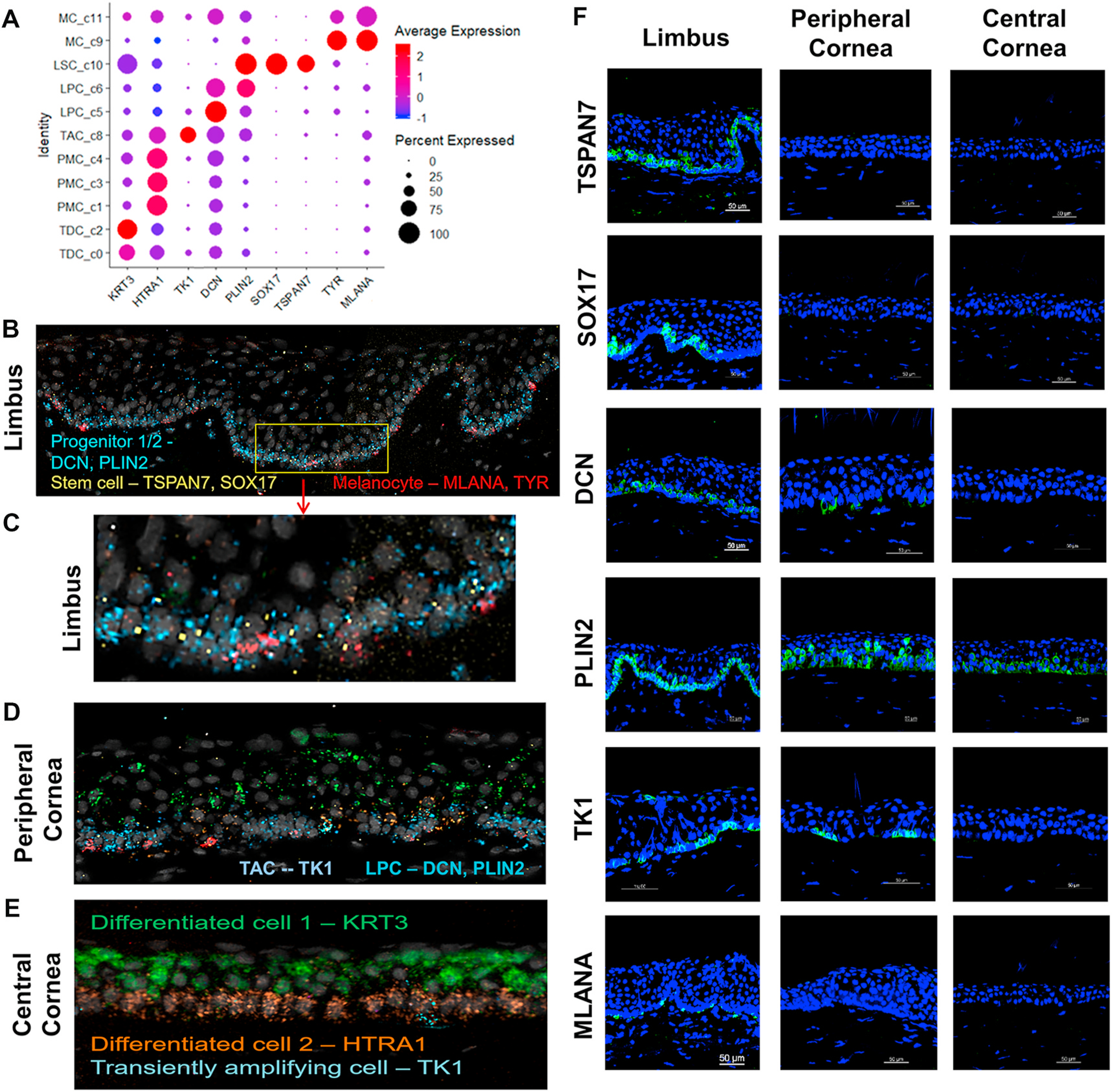
Validation of cell type-specific marker genes in five sub cell types at transcript and protein levels using RNAscope and immunofluorescent staining. A. Dot plot shows the expression of 9 marker genes selected from five cell-types. B-E. RNAscope in-situ validation shows the spatial expression of the transcripts of cell type-specific markers in three different regions of corneal tissues. Progenitor cells on limbus were identified by DCN and PLIN2 in blue, Stem cells by TSPAN7 and SOX17 in yellow, and Melanocytes by MLANA and TYR in red (B,C). On peripheral cornea more cell types were observed by known markers such as TK1 for TAC, DCN and PLIN2 for LPC and TDC for KRT3 (D). On central cornea, KRT3 for TDC and HTRA1 for PMC were expressed (E). F. Immunofluorescent staining shows the spatial localization of these marker proteins at limbus, peripheral, and central cornea.
Using the RNAscope HiPlex assay [61], we examined the spatial localization of all 9 marker transcript expressions mentioned above simultaneously in human corneal limbal tissue. As shown in Fig. 6B and C, TSPAN7+ and SOX17+ cells were sparsely distributed at the limbal basal epithelial layer, but not at the corneal epithelium, which is consistent with the idea that these cells are LSCs. DCN+ and PLIN2+ cells were identified at the same basal layer and likely represent limbal progenitor cells in agreement with their features and locations within the tissue. Moreover, we observed that TK1+ cells, which we identified as TACs, were sparsely distributed at the basal epithelium of peripheral cornea (Fig. 6D). HTRA1+ cells, which indicate PMCs, were dominantly distributed at suprabasal epithelial layers, while KRT3+ cells, indicating TDCs, were enriched at superficial layers of the central cornea (Fig. 6E). Interestingly, MLANA+ and TYR + melanocytes were sparsely located at the limbal basal layer where LSC resides (Fig. 6B and C).
We further investigated the spatial localization of these new markers at the protein level with fresh donor corneal tissues using immunofluorescent staining. To validate LSC (C10), TSPAN7 and SOX17 proteins were observed to be exclusively localized at the limbal basal layer while the suprabasal and superficial limbal cells and full layers of the peripheral and central corneal epithelium were totally negative (Fig. 6F). This localization pattern is consistent with the RNAscope results, suggesting that these genes may serve as the new markers exclusive for LSCs.
The immunoreactivity against DCN and PLIN2 was localized not only at limbal basal epithelial cells, but also at partial peripheral corneal basal cells, indicating a distinguishable pattern from the LSC markers. Interestingly, the localization of these two proteins was different in the corneal epithelium. DCN protein for C5 was much less and weaker in peripheral cornea and non-existent in the central cornea (Fig. 6F). In contrast, PLIN2 for C6 was localized much more with strong intensity in peripheral cornea while weaker expression by central corneal epithelial cells. The difference may suggest that C5 is an early stage of LPC while C6 is a later stage of LPC.
TK1, a new marker related to cell proliferation for C8 was sparsely located on the basal layer of limbus and peripheral cornea (Fig. 6F), indicating that C8 uniquely represents TACs, the intermediate progenitor cell developed from LSCs. C9/11 were validated as melanocytes sparsely located in the limbal basal epithelium by MLANA antibody (Fig. 6F).
Altogether, we have validated different cell types based on the spatial location of new markers at transcript and protein levels on human corneal tissue, and the results are consistent with LSC concept and differentiation model.
Functional validation of new marker genes for LSC regeneration capacity
Human limbal epithelial cells (HLECs) in vitro culture models [32, 35] were used for functional validation of these new marker genes regarding expression, production, and regulation related to cell growth, proliferation, and tissue regeneration. Using an in vitro epithelial regeneration model, we observed that the potential LSC markers, TSPAN7 and SOX17, were rarely expressed by a few small cells in HLECs, but became dramatically stimulated during the epithelial regeneration process after wound (Fig. 7A and B).
Fig. 7.
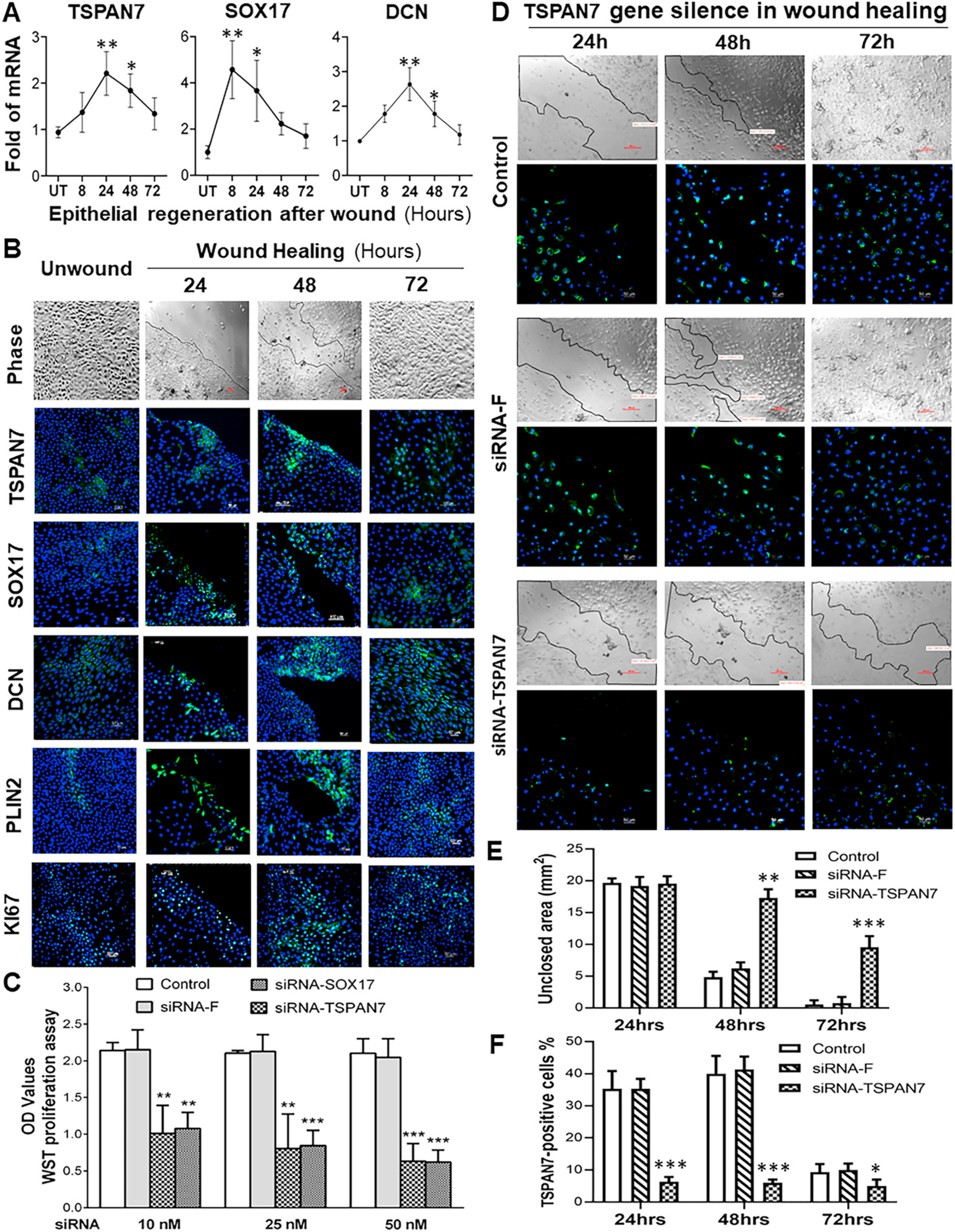
Functional validation of new markers for LSC in vitro culture models of epithelial regeneration and RNA interference. A. The stimulated mRNA expression of TSPAN7, SOX17 and DCN in 8–72 h after wound in human limbal epithelial cultures, evaluated by RT-qPCR. B. Representative images show phase and immunofluorescent staining of 5 markers in cultures after wound in 24–72 h. C. Blockade of TSPAN7 and SOX17 by their specific siRNAs respectively suppressed the cell proliferation of primary HLECs in comparison to the normal controls, as measured by WST assay. D. Representative images of phase and immunofluorescent staining show that siRNA interference significantly blocked the activation of TSPAN7 and inhibited epithelial regeneration after wound at 24–72 h. E. The quantification plot of the unclosed areas (mm2) for images in D. F. The quantification plot of the TSPAN7-positive cells (%) for images in D. Results shown are the Mean ± SD. *P < 0.05, **P < 0.01 and ***P < 0.001; n = 4, compared with controls.
As shown in Fig. 7A with RT-qPCR results, their mRNA expression increased and reached the peak levels at 8–24 h during epithelial cell migration and growth period, and then decreased to near the normal levels when the wound areas were closured at 48–72 h. Immunofluorescent staining confirmed the stimulated patterns of these markers at the protein level. As shown in Fig. 7B, the increase of TSPAN7+ and SOX17+ cells with stronger immunofluorescent intensity were observed at wounding edges and reached peak levels at 24–48 h. However, when epithelial regeneration was near complete at 48–72 h, the number of positively stained cells and intensity were reduced. Activation of TSPAN7 and SOX17 markers was accompanied by stimulation of progenitor cell markers DCN and PLIN2, as well as proliferation marker KI67. The results provide evidence that these new markers represent LSCs with a capacity to regenerate lost cells and repair the corneal epithelial tissues, the most important functional feature of stem cells [8].
To confirm the functional role of the new markers in maintaining the stem cell properties, RNA interference by siRNA was performed. Interestingly, we observed that the blockade of TSPAN7 and SOX17 by their specific siRNAs respectively suppressed the cell proliferation of primary HLECs in comparison with the normal controls, as measured by WST assay (Fig. 7C). The blockade of TSPAN7 expression by siRNA significantly inhibited epithelial regeneration after wound, as evaluated the unclosed areas by phase images and TSPAN7-positive cells by immunofluorescent staining with quantification (Fig. 7D–F).
Novel signaling pathways and transcription factors were revealed to maintain LSC stemness and drive LSC differentiation
To characterize genetic regulatory networks (GRNs) along the LSC differentiation trajectory, we applied SCENIC analysis [31], which revealed 51 highly expressed transcription factors (TFs) in our dataset (Fig. 8A). These TFs are found to participate in several pathways that maintain and regulate the stemness, differentiation and regeneration, such as Wnt/β-catenin, PI3K/Akt, Notch, cell cycle, senescence, and Hippo signaling pathways [62–66].
Fig. 8.
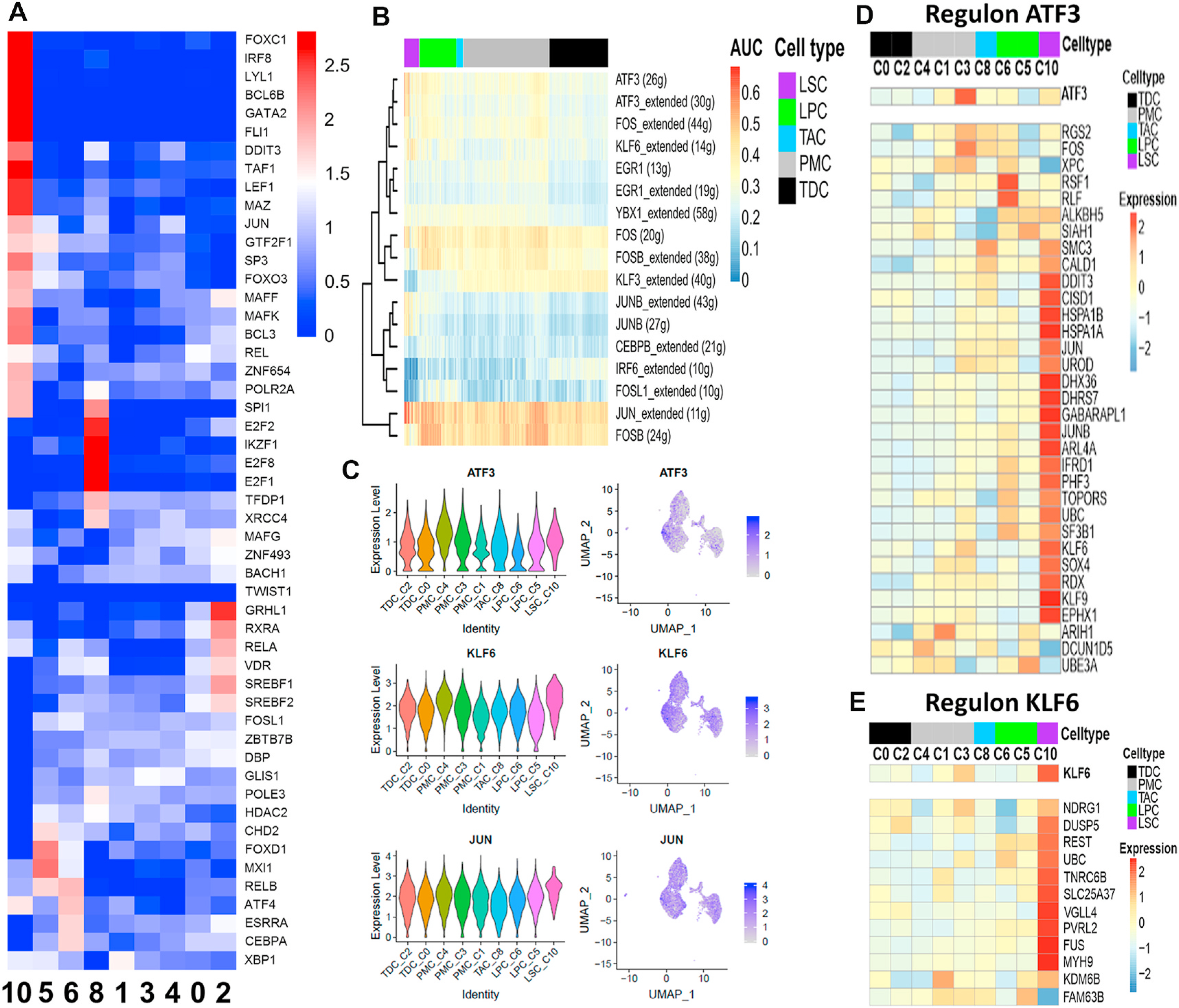
Gene regulatory networks using SCENIC. A. Heatmap for expression of transcription factors (TFs) across clusters found by SCENIC. B. Heatmap of AUC values across the cells indicating the activity of a regulon in a cell using AUCell used as a statistical method based on the "Area Under the Curve" (AUC). SCENIC was performed on 708 pseudo cells which were averaged by 20 cells from LPC, TAC, PMC, and TDC. LSCs were used without averaging values due to the low number of cells. C. Violin and feature plot of regulator genes enriched in LSC. D, E. Heatmap of the expression profile of ATF3 and KLF6 regulons with target genes having binding motifs on their regulatory regions. Color code represents each cell type. Expression values were centered and z scaled by row.
Among the highest expressed TFs from C10 in red/pink color on the heatmap (Fig. 8A), FOXC1 is a DNA-binding TF that plays a role in a broad range of cellular and developmental processes in eye, bones, cardiovascular, kidney and skin, and BCL6B is important for spermatogonial stem cell self-renewal and survival, both of which require PI3K/AKT signaling pathway [62–64]. LEF1, DDIT3, TAF1, SP3 and POLR2A are regulators for cell cycle and senescence via Wnt and Hippo pathways [65,66]. Interestingly, SPI1, LYL1, MAFK, FOXO3 and GATA2 are involved in the maintenance, differentiation, and development of hematopoietic stem cells [67,68]. All these TFs, known and unknown, may provide new opportunities to explore novel signaling pathways for human LSC.
Seventeen regulons that show distinct patterns across different cell clusters were revealed (Fig. 8B). Several regulons such as ATF3, KLF6, JUN, and YBX1 are enriched in LSC (Fig. 8B–E), implying a critical role in the maintenance of stem cells. For examples, ATF3 (activating transcription factor 3) is proposed as a novel marker for ependymal and spinal stem cells and plays an important role in regulating cell cycle and apoptosis. Very recently, ATF3 is found to maintain the quiescence in the stem cell-enriched limbal basal cells [21], protect the retinal ganglion cells, and promote optic nerve regeneration [69]. KLF6 is a key factor in regulating cell proliferation, apoptosis, rat pituitary stem/progenitor cells, and mouse ES cell differentiation [70]. These regulons open a new window to explore the novel pathways for LSC maintenance and differentiation.
Discussion
Human LSCs have been widely accepted to reside at basal layer of limbal epithelium and well characterized regarding to the undifferentiated status, quiescent slow cycling with high proliferation potential, self-renewal and tissue regeneration capacity. However, Current knowledge on human limbus and LSC concept is mostly based on broad investigations at the tissue and bulk cell population levels. The heterogeneity of human limbal basal epithelium is still a mystery and the definitive molecular markers of LSCs remains elusive [11,16,17].
The basal layer of limbal epithelium has been known to harbor the stem cells or stem/progenitor cells, but not the differentiated mature epithelial cells, which are localized at the superficial layers of limbus and the most layers of central cornea. The intermediate progenitor TACs are located in the basal layer of cornea. The compartment localization of these cell types has provided a valuable model to study the properties of LSC in adult stem cell biology. Using the scRNA-seq approach with a total of 16,360 single cells isolated from the human basal limbus, we have revealed 12 cell clusters in the heterogeneous limbal basal epithelium. Among them, 9 clusters fall into 5 major cell types including LSC, LPC, TAC, PMC and TDC, spanning all stages during LSC differentiation.
First, we defined the 9 clusters into 5 major groups of cell types based on their expression patterns of 36 genes associated with SC properties and cell cycle status (Fig. 3). Interestingly, the highly differentiated cell clusters do not express the putative epithelial SC markers; and vice versa. Then we performed trajectory analysis to confirm the cell lineages among these cell types in the limbal basal epithelium by Monocle 3 and CytoTRACE [26–29]. Both analyses show the differentiation of the cells along the trajectory in the order of clusters 10, 5, 6, 8, 1, 3, 4, 0, and 2 (Fig. 4A–F). Functional PCA analysis further showed that the top DEGs in these 9 clusters follow a significant trend along this trajectory (Fig. 4G–I).
These 5 major cell types were further validated by the spatial localization of 9 new marker genes at transcript and protein levels in human corneal limbal tissue using the RNAscope HiPlex and antibody staining (Fig. 6). For example, TSPAN7+ and SOX17+ cells are sparsely distributed only at the limbal basal epithelial layer, but not at the corneal epithelium, indicating putative LSCs. DCN+ and PLIN2+ cells are enriched at the basal layer of the limbal and peripheral cornea, serving as LPC. TK1+ cells, identified for TAC, are sparsely distributed at the basal epithelium of the peripheral cornea. HTRA1+ cells as PMCs were dominantly distributed at suprabasal epithelial layers, while KRT3+ cells, indicating TDCs, were enriched at superficial layers of the central cornea. In addition, the differential distribution patterns of DCN and PLIN2 further distinguishes C5 and C6 as early and later LPC, respectively.
Altogether, these findings demonstrate that five cell types, LSC, LPC, TAC, PMC and TDC, representing major stages and trajectories of LSC stemness and differentiation, are localized in the heterogeneous limbal basal epithelium. The findings are beyond the traditional concept that these different cell types are spatially located in different compartments from limbus to central cornea. These features reveal new insight into how LSCs make themselves ready to meet the requirement of rapid turnover of corneal epithelial tissue.
The definitive molecular markers of LSCs remain elusive although many have been proposed in the past three decades [11,16,17]. This scRNA-seq dataset identified C10, one of the smallest clusters, as LSCs based on the stemness characteristics. The cells in C10 are quiescent at the undifferentiated state enriched with all 12 marker genes of putative epithelial SCs. C10 contains 69 cells, accounting for 0.4% of the 16,360 single cells. The estimated number of LSCs is approximately ~320 per human corneal limbus based on the isolated limbal basal cell number averaged at 80,000 per cornea in this study.
From LSC (C10) enriched top DEGs (Fig. 5, Supplementary Table 5) we identified exclusively expressed genes that have not been previously reported to link the LSC properties. In this study, we validated that TSPAN7+ and SOX17+ cells are sparsely distributed only at the limbal basal epithelial layer (Fig. 6). Using an in vitro epithelial regeneration model, we observed that TSPAN7 and SOX17 were rarely expressed by a few small cells in human limbal epithelial cells (HLECs), but dramatically stimulated at mRNA and protein levels by wounding, especially during cell migration and growth period (Fig. 7A and B). Activation of TSPAN7 and SOX17 was followed by stimulation of progenitor cell markers DCN and PLIN2, as well as proliferation marker KI67 (Fig. 7B). Furthermore, the blockade of TSPAN7 and SOX17 by RNA interference with specific siRNAs suppressed cell proliferation and significantly inhibited epithelial regeneration after wound (Fig. 7C–F).
Altogether, the validation results provide evidence that TSPAN7 and SOX17 serve as new markers for identifying LSCs with a capacity to regenerate lost cells and repair the corneal epithelial tissues, the most important feature of adult stem cells [8].
In conclusion, this human corneal scRNA-Seq identifies the LSC population in the limbal basal epithelium with multiple new marker genes discovered. Two novel genes TSPAN7 and SOX17 are identified as LSC markers based on their exclusively spatial localization in limbal basal epithelium and functional role in tissue regeneration. Interestingly, all major cell types mapping the trajectory of LSC differentiation are uncovered in the heterogeneous limbal basal epithelium. The transcription factor networks linking novel signaling pathways to maintain LSC stemness are revealed for further studies.
Supplementary Material
Acknowledgements
This work is supported by grants from National Institutes of Health (NIH), National Eye Institute (NEI), EY023598 (DQL), EY011915 (SCP), EY018571 (RC), EY022356 (RC), and EY002520 (Core Grant for Vision Research), National Science Foundation (NSF) (DMS-1620957 to H.M.), Lions Foundation for Sight, Research to Prevent Blindness, the Oshman Foundation and the William Stamps Farish Fund. Single-cell RNA-Seq was performed at the Single Cell Genomics Core at BCM partially supported by National Institutes of Health (NIH) shared instrument grants (S10OD018033, S10OD023469 to RC).
We thank Drs. Marshall Bowes Hamill and Alice Y. Matoba for thier kind support and the Lions Eye Bank of Texas for providing human corneoscleral tissues.
Footnotes
Declaration of competing interest
The authors declare no competing interests.
Appendix A. Supplementary data
Supplementary data to this article can be found online at https://doi.org/10.1016/j.jtos.2020.12.004.
References
- [1].Schermer A, Galvin S, Sun TT. Differentiation-related expression of a major 64k corneal keratin in vivo and in culture suggests limbal location of corneal epithelial stem cells. J Cell Biol 1986;103:49–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Tseng SC. Concept and application of limbal stem cells. Eye 1989;3(Pt 2):141–57. [DOI] [PubMed] [Google Scholar]
- [3].Cotsarelis G, Cheng SZ, Dong G, Sun TT, Lavker RM. Existence of slow-cycling limbal epithelial basal cells that can be preferentially stimulated to proliferate: implications on epithelial stem cells. Cell 1989;57:201–9. [DOI] [PubMed] [Google Scholar]
- [4].Dua HS, Saini JS, Azuara-Blanco A, Gupta P. Limbal stem cell deficiency: concept, aetiology, clinical presentation, diagnosis and management. Indian JOphthalmol 2000;48:83–92. [PubMed] [Google Scholar]
- [5].Gheorghe A, Pop M, Mrini F, Barac R, Vargau I. Ocular surface reconstruction in limbal stem cell deficiency. Rom J Ophthalmol 2016;60:2–5. [PMC free article] [PubMed] [Google Scholar]
- [6].Vazirani J, Nair D, Shanbhag S, Wurity S, Ranjan A, Sangwan V. Limbal stem cell deficiency-demography and underlying causes. Am J Ophthalmol 2018;188: 99–103. [DOI] [PubMed] [Google Scholar]
- [7].Ramaesh K, Ramaesh T, Dutton GN, Dhillon B. Evolving concepts on the pathogenic mechanisms of aniridia related keratopathy. IntJBiochemCell Biol 2005;37:547–57. [DOI] [PubMed] [Google Scholar]
- [8].Post Y, Clevers H. Defining adult stem cell function at its simplest: the ability to replace lost cells through mitosis. Cell Stem Cell 2019;25:174–83. [DOI] [PubMed] [Google Scholar]
- [9].Vascotto SG, Griffith M. Localization of candidate stem and progenitor cell markers within the human cornea, limbus, and bulbar conjunctiva in vivo and in cell culture. Anat Rec A Discov Mol Cell Evol Biol 2006;288:921–31. [DOI] [PubMed] [Google Scholar]
- [10].Gonzalez G, Sasamoto Y, Ksander BR, Frank MH, Frank NY. Limbal stem cells: identity, developmental origin, and therapeutic potential7. Wiley Interdiscip Rev Dev Biol; 2018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Lavker RM, Tseng SC, Sun TT. Corneal epithelial stem cells at the limbus: looking at some old problems from a new angle. ExpEye Res 2004;78:433–46. [DOI] [PubMed] [Google Scholar]
- [12].Li DQ, Wang Z, Yoon KC, Bian F. Characterization, isolation, expansion and clinical therapy of human corneal epithelial stem/progenitor cells. J Stem Cell 2014;9: 79–91. [PubMed] [Google Scholar]
- [13].Luznik Z, Bertolin M, Breda C, Ferrari B, Barbaro V, Schollmayer P, et al. Preservation of ocular epithelial limbal stem cells: the new frontier in regenerative medicine. Adv Exp Med Biol 2016;951:179–89. [DOI] [PubMed] [Google Scholar]
- [14].Pellegrini G, Golisano O, Paterna P, Lambiase A, Bonini S, Rama P, et al. Location and clonal analysis of stem cells and their differentiated progeny in the human ocular surface. JCell Biol 1999;145:769–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Zhou M, Li XM, Lavker RM. Transcriptional profiling of enriched populations of stem cells versus transient amplifying cells. A comparison of limbal and corneal epithelial basal cells. J Biol Chem 2006;281:19600–9. [DOI] [PubMed] [Google Scholar]
- [16].Joe AW, Yeung SN. Concise review: identifying limbal stem cells: classical concepts and new challenges. Stem Cells Transl Med 2014;3:318–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Guo ZH, Zhang W, Jia YYS, Liu QX, Li ZF, Lin JS. An insight into the difficulties in the discovery of specific biomarkers of limbal stem cells. Int J Mol Sci 2018;19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Shekhar K, Lapan SW, Whitney IE, Tran NM, Macosko EZ, Kowalczyk M, et al. Comprehensive classification of retinal bipolar neurons by single-cell transcriptomics. Cell 2016;166:1308–1323 e30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Plass M, Solana J, Wolf FA, Ayoub S, Misios A, Glazar P, et al. Cell type atlas and lineage tree of a whole complex animal by single-cell transcriptomics. Science 2018;360. [DOI] [PubMed] [Google Scholar]
- [20].Tasic B. Single cell transcriptomics in neuroscience: cell classification and beyond. Curr Opin Neurobiol 2018;50:242–9. [DOI] [PubMed] [Google Scholar]
- [21].Kaplan N, Wang J, Wray B, Patel P, Yang W, Peng H, et al. Single-cell rna transcriptome helps define the limbal/corneal epithelial stem/early transit amplifying cells and how autophagy affects this population. Invest Ophthalmol Vis Sci 2019;60:3570–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Li D-Q, Chen Z, Song XJ, de Paiva CS, Kim HS, Pflugfelder SC. Partial enrichment of a population of human limbal epithelial cells with putative stem cell properties based on collagen type iv adhesiveness. ExpEye Res 2005;80:581–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Butler A, Hoffman P, Smibert P, Papalexi E, Satija R. Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 2018;36:411–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, et al. Single-cell rna-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res 2015;25:1860–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Tirosh I, Izar B, Prakadan SM, Wadsworth MH, Treacy D, Trombetta JJ, et al. Dissecting the multicellular ecosystem of metastatic melanoma by single-cell rna-seq. Science 2016;352:189–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods 2017;14:979–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Qiu X, Hill A, Packer J, Lin D, Ma YA, Trapnell C. Single-cell mrna quantification and differential analysis with census. Nat Methods 2017;14:309–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Gulati GS, Sikandar SS, Wesche DJ, Manjunath A, Bharadwaj A, Berger MJ, et al. Single-cell transcriptional diversity is a hallmark of developmental potential. Science 2020;367:405–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Sang P, Wang L, Cao J. Parametric functional principal component analysis. Biometrics 2017;73:802–10. [DOI] [PubMed] [Google Scholar]
- [31].Aibar S, Gonzalez-Blas CB, Moerman T, Huynh-Thu VA, Imrichova H, Hulselmans G, et al. Scenic: single-cell regulatory network inference and clustering. Nat Methods 2017;14:1083–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Lu R, Qu Y, Ge J, Zhang L, Su Z, Pflugfelder SC, et al. Transcription factor tcf4 maintains the properties of human corneal epithelial stem cells. Stem Cell 2012;30: 753–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].de Paiva CS, Chen Z, Corrales RM, Pflugfelder SC, Li D-Q. Abcg2 transporter identifies a population of clonogenic human limbal epithelial cells. Stem Cell 2005; 23:63–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Chen Z, de Paiva CS, Luo L, Kretzer FL, Pflugfelder SC, Li D-Q. Characterization of putative stem cell phenotype in human limbal epithelia. Stem Cell 2004;22: 355–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Qu Y, Chi W, Hua X, Deng R, Li J, Liu Z, et al. Unique expression pattern and functional role of periostin in human limbal stem cells. PloS One 2015;10: e0117139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Bian F, Liu W, Yoon KC, Lu R, Zhou N, Ma P, et al. Molecular signatures and biological pathway profiles of human corneal epithelial progenitor cells. Int J Biochem Cell Biol 2010;42:1142–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zieske JD. Perpetuation of stem cells in the eye. Eye 1994;8(Pt 2):163–9. [DOI] [PubMed] [Google Scholar]
- [38].Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive integration of single-cell data. Cell 2019;177:1888–1902 e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Kao WW, Liu CY, Converse RL, Shiraishi A, Kao CW, Ishizaki M, et al. Keratin 12-deficient mice have fragile corneal epithelia. Invest OphthalmolVisSci 1996;37: 2572–84. [PubMed] [Google Scholar]
- [40].Tong L, Corrales RM, Chen Z, Villarreal AL, De Paiva CS, Beuerman R, et al. Expression and regulation of cornified envelope proteins in human corneal epithelium. Invest Ophthalmol Vis Sci 2006;47:1938–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Chen Z, Evans WH, Pflugfelder SC, Li D-Q. Gap junction protein connexin 43 serves as a negative marker for a stem cell-containing population of human limbal epithelial cells. Stem Cell 2006;24:1265–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Joyce NC, Meklir B, Joyce SJ, Zieske JD. Cell cycle protein expression and proliferative status in human corneal cells. Invest OphthalmolVisSci 1996;37: 645–55. [PubMed] [Google Scholar]
- [43].Lehrer MS, Sun TT, Lavker RM. Strategies of epithelial repair: modulation of stem cell and transit amplifying cell proliferation. JCell Sci 1998;111(Pt 19):2867–75. [DOI] [PubMed] [Google Scholar]
- [44].Lu R, Bian F, Zhang X, Qi H, Chuang EY, Pflugfelder SC, et al. The beta-catenin/tcf4/survivin signaling maintains a less differentiated phenotype and high proliferative capacity of human corneal epithelial progenitor cells. Int J Biochem Cell Biol 2011;43:751–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ksander BR, Kolovou PE, Wilson BJ, Saab KR, Guo Q, Ma J, et al. Abcb5 is a limbal stem cell gene required for corneal development and repair. Nature 2014;511: 353–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Watanabe K, Nishida K, Yamato M, Umemoto T, Sumide T, Yamamoto K, et al. Human limbal epithelium contains side population cells expressing the atp-binding cassette transporter abcg2. FEBS Lett 2004;565:6–10. [DOI] [PubMed] [Google Scholar]
- [47].Stepp MA. Corneal integrins and their functions. Exp Eye Res 2006;83:3–15. [DOI] [PubMed] [Google Scholar]
- [48].Pajoohesh-Ganji A, Stepp MA. In search of markers for the stem cells of the corneal epithelium. Biol Cell 2005;97:265–76. [DOI] [PubMed] [Google Scholar]
- [49].Szklarczyk D, Franceschini A, Wyder S, Forslund K, Heller D, Huerta-Cepas J, et al. String v10: protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res 2014;43:D447–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bohrer C, Pfurr S, Mammadzada K, Schildge S, Plappert L, Hils M, et al. The balance of id3 and e47 determines neural stem/precursor cell differentiation into astrocytes. EMBO J 2015;34:2804–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Gadomski S, Singh SK, Singh S, Sarkar T, Klarmann KD, Berenschot M, et al. Id1 and id3 maintain steady-state hematopoiesis by promoting sinusoidal endothelial cell survival and regeneration. Cell Rep 2020;31:107572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Karlsson G, Rorby E, Pina C, Soneji S, Reckzeh K, Miharada K, et al. The tetraspanin cd9 affords high-purity capture of all murine hematopoietic stem cells. Cell Rep 2013;4:642–8. [DOI] [PubMed] [Google Scholar]
- [53].Zeng A, Li H, Guo L, Gao X, McKinney S, Wang Y, et al. Prospectively isolated tetraspanin(+) neoblasts are adult pluripotent stem cells underlying planaria regeneration. Cell 2018;173:1593–1608 e20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Kashef J, Diana T, Oelgeschlager M, Nazarenko I. Expression of the tetraspanin family members tspan3, tspan4, tspan5 and tspan7 during xenopus laevis embryonic development. Gene Expr Patterns 2013;13:1–11. [DOI] [PubMed] [Google Scholar]
- [55].He S, Kim I, Lim MS, Morrison SJ. Sox17 expression confers self-renewal potential and fetal stem cell characteristics upon adult hematopoietic progenitors. Genes Dev 2011;25:1613–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Wang P, Rodriguez RT, Wang J, Ghodasara A, Kim SK. Targeting sox17 in human embryonic stem cells creates unique strategies for isolating and analyzing developing endoderm. Cell Stem Cell 2011;8:335–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Engert S, Burtscher I, Liao WP, Dulev S, Schotta G, Lickert H. Wnt/beta-catenin signalling regulates sox17 expression and is essential for organizer and endoderm formation in the mouse. Development 2013;140:3128–38. [DOI] [PubMed] [Google Scholar]
- [58].Miyachi K, Yamada T, Kawagishi-Hotta M, Hasebe Y, Date Y, Hasegawa S, et al. Extracellular proteoglycan decorin maintains human hair follicle stem cells. J Dermatol 2018;45:1403–10. [DOI] [PubMed] [Google Scholar]
- [59].Liu X, Lu X, Song K, Blackman MR. Natural functions of plin2 mediating wnt/licl signaling and glycogen synthase kinase 3 (gsk3)/gsk3 substrate-related effects are modulated by lipid. Mol Cell Biol 2016;36:421–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Jiang HP, Serrero G. Isolation and characterization of a full-length cdna coding for an adipose differentiation-related protein. Proc Natl Acad Sci U S A 1992;89: 7856–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Chan S, Filezac de L’Etang A, Rangell L, Caplazi P, Lowe JB, Romeo V. A method for manual and automated multiplex rnascope in situ hybridization and immunocytochemistry on cytospin samples. PloS One 2018;13:e0207619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].Oatley JM, Avarbock MR, Telaranta AI, Fearon DT, Brinster RL. Identifying genes important for spermatogonial stem cell self-renewal and survival. Proc. Natl. Acad. Sci. U.S.A 2006;103:9524–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Omatsu Y, Seike M, Sugiyama T, Kume T, Nagasawa T. Foxc1 is a critical regulator of haematopoietic stem/progenitor cell niche formation. Nature 2014;508:536–40. [DOI] [PubMed] [Google Scholar]
- [64].Lay K, Kume T, Fuchs E. Foxc1 maintains the hair follicle stem cell niche and governs stem cell quiescence to preserve long-term tissue-regenerating potential. Proc Natl Acad Sci U S A 2016;113:E1506–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Armenteros T, Andreu Z, Hortiguela R, Lie DC, Mira H. Bmp and wnt signalling cooperate through lef1 in the neuronal specification of adult hippocampal neural stem and progenitor cells. Sci Rep 2018;8:9241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Petersson M, Brylka H, Kraus A, John S, Rappl G, Schettina P, et al. Tcf/lef1 activity controls establishment of diverse stem and progenitor cell compartments in mouse epidermis. EMBO J 2011;30:3004–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Bergiers I, Andrews T, Vargel Bolukbasi O, Buness A, Janosz E, Lopez-Anguita N, et al. Single-cell transcriptomics reveals a new dynamical function of transcription factors during embryonic hematopoiesis. Elife 2018;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Kauts ML, De Leo B, Rodriguez-Seoane C, Ronn R, Glykofrydis F, Maglitto A, et al. Rapid mast cell generation from gata2 reporter pluripotent stem cells. Stem Cell Reports 2018;11:1009–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Kole C, Brommer B, Nakaya N, Sengupta M, Bonet-Ponce L, Zhao T, et al. Activating transcription factor 3 (atf3) protects retinal ganglion cells and promotes functional preservation after optic nerve crush. Invest Ophthalmol Vis Sci 2020;61: 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Ueharu H, Higuchi M, Nishimura N, Yoshida S, Shibuya S, Sensui K, et al. Expression of kruppel-like factor 6, klf6, in rat pituitary stem/progenitor cells and its regulation of the prrx2 gene. J Reprod Dev 2014;60:304–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All raw data are available throughout Gene Expression Omnibus (GEO) with accession number GSE153515 (https://urldefense.proofpoint.com/v2/url?u=https-3A__www.ncbi.nlm.nih.gov_geo_query_acc.cgi-3Facc-3DGSE153515&d=DwIBAg&c=ZQs-KZ8oxEw0p81sqgiaRA&r=0ttAf5n03KatecVFHf58mw&m=FGyZl_yfIX4uoDe0fAXWlLVMKCgHTkLuKKcbI1dzx4U&s=7x8PSA8eY8Wg4vi2IoVrwtJZW5xNkg_qk92jJJI6wzk&e=). R scripts used to analyze are freely available at the github (https://github.com/sangbaekim/10xRNAseq_LSC).