

Summary
Programmed cell death (apoptosis) is an integral part of tissue homeostasis in complex organisms, allowing for tissue turnover, repair, and renewal while simultaneously inhibiting the release of self antigens and danger signals from apoptotic cell-derived constituents that can result in immune activation, inflammation, and autoimmunity. Unlike cells in culture, the physiological fate of cells that die by apoptosis in vivo is their rapid recognition and engulfment by phagocytic cells (a process called efferocytosis). To this end, apoptotic cells express specific eat-me signals, such as externalized Phosphatidylserine (PS), that are recognized in a specific context by receptors to initiate signaling pathways for engulfment. The importance of carefully regulated recognition and clearance pathways is evident in the spectrum of inflammatory and autoimmune disorders caused by defects in PS receptors and signaling molecules. However, in recent years, several additional cell death pathways have emerged, including immunogenic cell death, necroptosis, pyroptosis, and netosis, that interweave different cell death pathways with distinct innate and adaptive responses from classical apoptosis that can shape long-term host immunity. In this review, we discuss the role of different cell death pathways in terms of their immune potential outcomes specifically resulting in specific cell corpse/phagocyte interactions (phagocytic synapses) that impinge on host immunity, with a main emphasis on tolerance and cancer immunotherapy.
Keywords: Cell death, tolerance, phosphatidylserine, tumor immunity, immunogenic cell death
Introduction
Apoptosis, also called programmed cell death, is an evolutionarily conserved and highly regulated cell death modality. In both developing and adult tissues, apoptosis allows for the continuous removal of cells that are aged, genetically damaged, infected with pathogens, or self-reactive, and critical for self-renewal and the maintenance of tissue homeostasis in metazoans (1–4). Unlike cells in tissue culture that progress to late apoptosis and produce advanced features of the apoptotic program, the final fate of apoptotic cell death under physiological conditions is their rapid removal by neighboring or recruited phagocytic cells (a term called efferocytosis to distinguish the engulfment of apoptotic cells from other phagocytic processes) (5, 6). Efferocytosis is achieved by an intricate collaborative array of eat-me signals on the apoptotic cells and recognition receptors on the phagocyte that, when intact, achieves efficient clearance that promotes self-tolerance and resolution of inflammation (7, 8). However, when clearance fails, or is delayed, or when cells under alternative cell death modalities induced by stress, dying cells externalize or release distinct intracellular constituents and itineraries that can be recognized as danger signals that feature death-associated inflammation, including the production of self-reactive immune cells that depending on context, can induce autoimmunity and, if properly controlled, host anti-tumor immunity (9–12). Indeed, over the past decade, a broader definition of the immune consequences from dying cells has emerged that reflects a most active and emerging field of immunology. In this review, we highlight some of the historical milestones in the field of efferocytosis, with a main focus on how apoptosis and subsequent efferocytosis impinge on innate and cognate immune responses in cancer. We begin with recognition of the early genetics of the cell death and engulfment that propagated the field to its current status.
Historical milestones: Genetics in Caenorhabditis elegans (C. elegans) provided an impetus to study the biology of efferocytosis
By the early 1990’s, the field of Cell Death was in a rapid expansionary phase following the identification of a series of Cell Death Defective genes (commonly abbreviated CED genes) in C. elegans, a genetically tractable model organism characterized by massive cell death during embryonic development (13–16). These studies, pioneered by H. Robert Horvitz and his colleagues, and culminating in a shared Nobel prize in 2002 for elucidating the genetics of the cell death program (17), helped establish a central dogma of apoptotic cell death in which apoptosis is regulated by a set of genes; affirmatively regulated by CED3 (Caspase) and CED4 (Apoptosis Protease activating Factor-1), and negatively regulated by CED9 (homologous Bcl-2 family protein) (15, 18–20). Subsequent biochemical studies in vertebrate systems showed that the apoptotic gene products, along with Cytochrome-c, comprised a protein interactome called the apoptosome, a complex quaternary structure assembled intracellularly in responses to both intrinsic and extrinsic cell death stimulus (21).
Perhaps initially less universally recognized, but conceptually of equal importance, the genetic studies in C elegans also identified a second set of cell death defective genes (indeed a larger array of genes) comprising CED1, CED2, CED5, CED6, CED7, CED8, CED10, and CED12 that regulated the engulfment of apoptotic corpses (22, 23) (Fig. 1). Worms harboring CED mutants for engulfment genes generally had normal physiological apoptosis (although in some mutants, cell death was delayed suggesting engulfment can regulate the commitment to apoptosis (24)), but cell corpses remained or fragmented in tissues that could be observed by differential interference contrast microscopy. Importantly, the identification of CED genes that abrogated efferocytosis unequivocally demonstrated that clearance (like apoptosis) was genetically programmed in multicellular organisms, ensuring the rapid and decisive detection and removal of cell corpses by neighboring viable cells (13).
Figure 1. Genetic regulation of cell death and engulfment pathways in C.elegans.
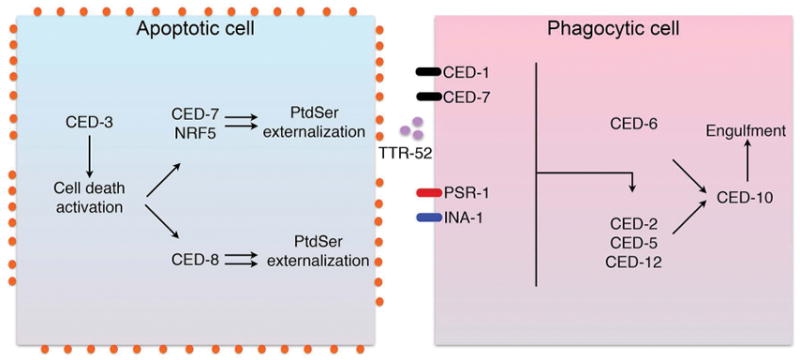
During apoptotic cell (left panel), caspase (CED-3) activation leads to a cell death cascade that includes pathways leading to CED7 and CED8-mediated externalization of PS. PS externalization on dying cells is recognized by a series of PS receptors and PS bridging molecules that detect the dying cells, that in turn engage signaling pathways involving CED-2, CED-5 and CED12 that lead to CED-10 activation, for subsequent engulfment and degradation of the apoptotic cell (right panel).
Genetic complementation and mapping studies revealed that the aforementioned genes that regulated engulfment encoded two evolutionarily conserved modules (25), the first defined by a complementation group involving CED2 (Crk), CED5 (DOCK180), CED12 (ELMO), CED10 (Rac1) (26), and a second complementation group comprised of CED1 (CD91/Scavenger receptor from endothelial cells (SREC)-like protein (27), CED6 (GULP) (28), and CED7 (ATP binding cassette ABC1 transporter) (29). CED8 (Xkr8), the last of the engulfment-defective genes to be molecularly characterized, encoded a lipid scramblase, subsequently shown to be involved in the externalization of phosphatidylserine (PS) (30, 31), and as discussed below, a signal important for the recognition and internalization of apoptotic cells. Analogous to the apoptosis pathways involving CED4, CED3, and CED9 module, the engulfment pathways could also be mapped by genetic rescue experiments; For example, a CED2 or CED5 loss of function mutant could be rescued by a CED10 gain of function mutant. Moreover, while studies identified two complementation groups, later studies showed that CED10 gain of function mutants could rescue many of the other CED mutants (including CED2, CED5, CED12, CED6 and CED7), implicating the Rho family GTPase, Rac1, as a downstream master switch signal for the clearance of apoptotic cells in multicellular organisms (Fig. 1) (25).
However, unlike the CED3, CED4, CED9, apoptosome complex, that provided a paradigm for a novel biology involving a structured protease platform, the discovery of the CED2/CED5/CED12/CED10 switch had already been discovered and characterized in the context of another biology, namely from previous work in retroviral oncogenesis, and how signal transduction is achieved by modular domains and protein-protein interactions. Most notably, CED2/Crk had been initially discovered as a transforming gene encoded by avian retrovirus CT10, an oncogenic variant of Crk II in which viral Gag sequences were fused to sequences homologous to the regulatory region of Src kinases, the co-called Src homology 2 (SH2) and Src homology 3 (SH3) domains (32–34).
Indeed, earlier biochemical and functional studies with v-Crk showed that the Crk SH2 domain bound specific tyrosine phosphorylated cytoskeletal proteins and focal adhesion proteins (35), and simultaneous, via its SH3 domain, to proline-rich elements in DOCK180 (Downstream of Crk) which represents that mammalian homology of CED5 (36, 37). DOCK180, in turn, represents an atypical guanine nucleotide exchange factor (GNEF), that instead of catalyzing GDP/GTP exchange via a PH/Dbl motif, forms an unusual bipartite GNEF with ELMO/CED12 via another SH3/PxxP mediated interaction (38, 39). Together Crk/DOCK180/ELMO form a ternary complex that localizes to tyrosine phosphorylated cytoskeletal proteins, thereby exchanging GDP for GTP on Rac1 (the mammalian homolog of CED10) in response to upstream receptor activation or cytoskeletal reorganization at the plasma membrane. These data indicate that CED2 (Crk II), DOCK180 (CED5), CED12 (ELMO) and CED1 (Rac1) have overlapping functions with respect to cell motility and efferocytosis, which is likely regulated by both the nature of the extracellular ligands (deposition of extracellular matrix versus apoptotic cells) as well as the nature of the upstream receptors.
The identification of CED2/Crk II, a SH2 domain-containing pTyr sensor and central modulator of efferocytosis, by inference, suggested that upstream receptors, either directly or indirectly, would impinge on tyrosine phosphorylation-dependent efferocytosis. Indeed, exposure of cells to general tyrosine kinase inhibitors effectively blocked efferocytosis in both professional and non-professional phagocytes, and several receptors for apoptotic cells have been linked to the Crk II/DOCK180/Rac1 module (40). These include αvβ5 integrin/MFG-E8 in DCs and epithelial cells (41), α-INA-1-β-PAT3 in C. elegans (42) and αPS3/βv in Drosophila (the sole integrin in Drosophila) (43, 44). However, it is also clear that not all efferocytosis receptors utilize the aforementioned modules the same way, and there are variations in the modes of activation of CED10, for example, Bai1 can employ CED12/CED5 independent of CED2, and TIM-4 uses Vav1 rather than DOCK180/ELMO to activate CED10 (45) (Fig. 2).
Figure 2. Detection of PS via PS receptors, and signaling via Crk, DOCK180, Rac1 in phagocytic cells.
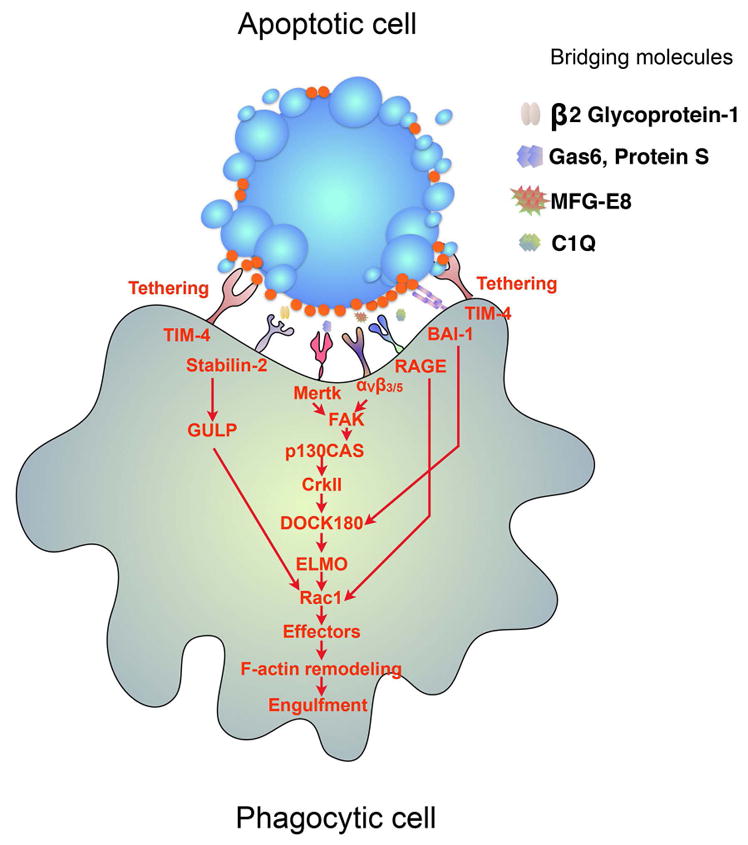
Apoptotic cells express “eat-me” signals, such as externalized PS in response to apoptotic stimuli. Externalized PS, in turn, is recognized directly by a variety of PS receptors expressed on phagocytes and bridging molecules such C1q, MFG-E8, Gas6, and Pros1. Many PS receptors and PS bridging molecules, when engaged bv apoptotic cells, subsequently lead to activation of an evolutionarily conserved Crk-DOCK-Rac1 pathway.
A second complementation group in C. elegans, CED1, CED7, and CED6, comprises a receptor-initiated cascade, and like CED2/CED5/CED10 module, is highly evolutionarily conserved and also regulated by post-receptor tyrosine phosphorylation-dependent signaling (22). In Drosophila and mammals, the homologs/orthologs of CED1 are Draper and Jedi/MEGF10, respectively, and all these gene products contain multiple EGF repeats in the extracellular domain, a single trans-membrane domain, and an NpXY phosphotyrosine-binding domain that binds CED6/Gulp (46). The intracellular domains of Jedi and MEGF10 also contain ITAM motifs that directly interact with Syk to modulate phagocytosis (47). In the case for CED6, this molecule can also interact with Dynamin and Rap7, suggesting a link between internalization and delivery to a degradation compartment (48).
While the above-mentioned genetic studies in C. elegans provided a conceptual framework for how efferocytosis is organized in metazoans, and a specific example for how phagocytic cells employ an evolutionarily conserved actin cytoskeletal module for clearance, only in more recent years has information emerged with respect to how apoptotic cells reciprocally provide eat me signals for their engulfment. In this respect, two of the engulfment defective genes, namely CED7 and CED8, encode an ATP binding cassette (ABCA1) transporter (CED7) (49) and a lipid scramblase (CED8) (31), respectively, that in part function in the externalization of PS (50), and emblematic eat me signal that is recognized by PS receptors. In the case for the CED1, CED7, CED6 recognition pathway, independent studies by Wang et al and Mapes et al found that CED1 indirectly interacts with a PS bridging factors identified as Transthyretin-like protein 52 (TTR-52) that binds both PS and CED1 (51, 52). The role for CED7 in this pathway is less demonstrable, although it appears important for both the externalization of PS, possibly in collaboration with secreted lipid transfer/LPS-binding family protein NRF-5, as well as for CED1-mediated corpse clearance (53). More recent studies by Conradt and colleagues, also in the C. elegans model, showed that CED3 (caspase) activity on the dying cells was required for subsequent clustering of CED1 on neighboring efferocytic cells, suggesting that a gradient of Caspase activity exists and can be detected by receptors on neighboring cells through the externalization of PS (54). As such, the previous reconciliation that CED1 recognizes recognizing PS in a complex with opsonins such as TTR-52 supports a model whereby caspase activation (in the dying cells) induces PS externalization, and that PS clustering may be a driving signal for activation of PS receptors on the efferocytosing cell (Fig. 3).
Figure 3. Model for the differential engulfment outcomes resulting from PS externalization via caspase activation (Xkr8/ATP11C) and via cellular stress (TMEM16F).
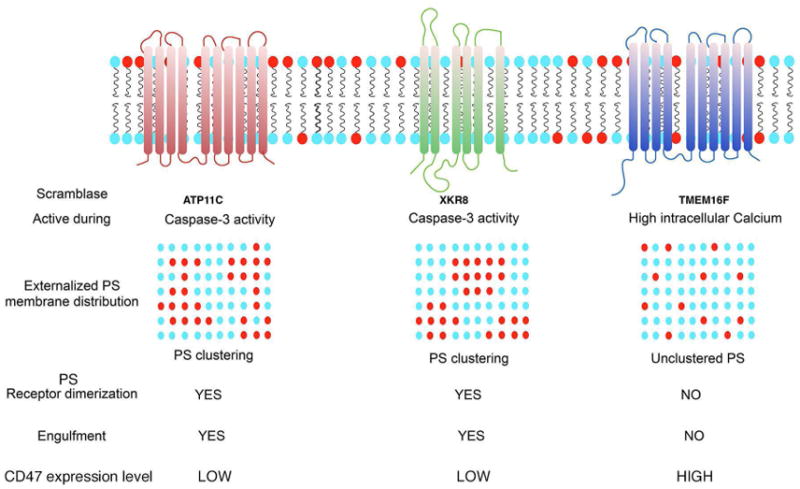
As developed in the text, PS is externalized via the activity of distinct classes of lipid transporters during apoptosis versus cellular stress. During apoptosis, PS becomes externalized concomitant with caspase activation by the inactivation of ATP11C (flippase), and the concomitant activation of Xkr8 (scramblase), leading to irreversible PS externalization. By contrast, during cellular stress, activation of calcium-dependent TMEM16F (scramblase) leads to transient and reversible externalization of PS. In addition to the issue of reversibility of PS externalization, other factors, such as the down-regulation of the don’t-eat me receptor CD47, and the lateral movement and clustering of PS, likely contribute differential signaling of externalized PS have been implicated in the differential fates of externalized PS (see ref 59, 69).
Further genetic evidence supporting externalized PS as a pre-eminent eat-me signal, as well as a conceptual argument for the centrality of PS externalization in clearance, emerged via the recent molecular characterization of CED8/Xkr8 as an evolutionarily conserved PS scramblase in worms and mammals (30, 50). Functionally, the CED8/Xkr8 gene product comprises a 10 trans-membrane domain spanning plasma membrane protein, is typically inactive as a scramblase in native cells, but becomes active following caspase 3/7-dependent cleavage during apoptosis (55). Truncated Xkr8, in turn, promotes a conformational induced oligomerization process allowing for the subsequent vertical transfer lipids from the inner to the outer membrane to break membrane asymmetry (55). In mammals, simultaneous caspase 3/7-dependent inactivation of ATP11C (an ATP-dependent flippase) appears to functionally synergize with Xkr8 by preventing the re-transfer of PS from the outer to the inner membrane, and allowing PS externalization to be more irreversible (56) (Fig. 3). Further down-regulation of don’t-eat me signals (i.e CD47) may sustain the PS/PSR interactions, that inhibits phagocytosis by binding to its receptor, signal regulatory protein-α(SIRPα) (57). The importance of CD47 as a don’t eat-me signal is exemplified by the fact that CD47-deficient red cells are more rapidly engulfed by splenic macrophages than native CD47+ erythrocytes (58). Although more research is needed to understand the exact relationships between PS externalization and CD47 down-regulation, in several cancer cells, particularly circulating leukemia stem cells and blast cells, overexpress CD47 on their surface (59, 60), which ultimately may interfere with their ability to be engulfed by phagocytes, even after apoptosis.
However, unlike the irreversible externalization of PS initiated by caspase activation, apoptosis, and efferocytosis, it is important to emphasize that PS can also be externalized on a variety of viable cells, including activated T cells (61) (62), mast cells (63), neutrophils (64), myoblasts (65), and platelets (66), due in part to rises in intracellular calcium (66–68). These data clearly indicate that externalization of PS, per se, is not sufficient for efferocytosis, but likely depends on the topology of PS in the membrane, including the lateral mobility of PS in apoptotic membranes, as has been suggested by Herrmann and others (69–71) (Fig. 3). In addition, unlike the situation during apoptosis, reversible externalization of PS does not lead to inactivation of flippases or down-regulation of don’t eat me signals, and may therefore mainly serve as nucleation sites for the recruitment PS binding proteins such as clotting factors, but not sufficient for efferocytosis (67). One of the best studied of the calcium activated scramblases is TMEM16F, that becomes activated in platelets due to elevated intracellular calcium concentrations, and in doing so, undergoes a calcium induced conformational change and homo-dimerization that allows for the vertical movement of PS across the lipid bilayer and the outward translocation of phospholipids (72). Expression of a constitutively active mutant of TMEM16F leads to constitutively externalized PS, although these cells are not engulfed when cultured with phagocytes (67), and recent studies by Nagata and colleagues have identified specific scrambling domains required for translocation (72). Interestingly, loss of function mutations of in TMEM16F (or genetic knockouts) lead to Scott Syndrome, a mild bleeding disorder characterized by the lack of PS externalization and coincident lack of recruitment of clotting factors to initiate the coagulation process (68).
PS/PS-receptor interactions and the regulation of efferocytosis in higher metazoans
The aforementioned studies in the C. elegans genetic models provided a critical framework for efferocytosis, that involved (i) caspase mediated PS externalization (clustering) (Xkr8/CED8), (ii) recognition of externalized PS by directly binding PS receptors and bridging molecules (TTR52/CED1)/CED7, and (iii) linkage of post-receptor signaling pathways to an evolutionarily conserved adaptor proteins regulated by tyrosine phosphorylation to activate Rac1 (CED2->CED5->CED12->CED10). Notably, however, the model in C. elegans was somewhat underwhelming in predicting the subsequent complexity of PS receptors and bridging molecules in higher metazoans. Nonetheless, the redistribution and externalization of PS on aged red cell membranes and apoptotic cells, and its role in phagocytosis has been appreciated since the early 1980s, clearly before the above-mentioned genetic studies in C. elegans, by early pioneering observations by Schriot and colleagues describing externalized PS as a facilitator for the clearance of aged red blood cells (73, 74), and later studies by Fadok and Henson showing that exposure of PS on apoptotic lymphocytes trigged their phagocytosis by macrophages (75).
Since the initial reports describing the cloning and characterization of the first PS receptor, called PSR-1, involved in the clearance of apoptotic cells (76, 77), the repertoire of PS receptors and PS bridging molecules implicated in apoptotic cell clearance has steadily increased. Indeed, over the past decade, PS has been shown to bind to phagocytes directly via various PS receptors that include CD300 family proteins (CD300b and CD300f), Stabilin-2 family members (also called hyaluronic acid receptor for endocytosis/HARE), T Cell Immunoglobulin mucin 1 (TIM-1, and family members TIM-3 and TIM-4), Brain-specific angiogenesis inhibitor-1 (BAI1) and Receptor for advanced glycation end products (RAGE), each of which employ different PS binding mechanisms and ligand-binding domains to mediate the interaction (78, 79). Adding complexity, in addition to the receptors that recognize PS directly, PS-mediated efferocytosis involves soluble bridging molecules that recognize PS and indirectly target phagocytic receptors. Included in this class are Milk Fat Globule EGF Factor 8 (MFG-E8, also called Lactadherin), Developmental Endothelial Locus 1 (Del-1), Growth arrest specific factor 6 (Gas6), Protein S, β2GPI, and C1q. Each of these molecules possesses cis-acting bifunctional domains that (i) interact with PS on the apoptotic cells and (ii) receptors on phagocytic cells (8). For example, MFG-E8 and Del-1, secreted from macrophages and tumor cells, binds PS via its C1 and C2 domains, and interacts simultaneously with αvβ5 and αvβ3 integrins through the RGD motif in the EGF domain, stimulating engulfment (80, 81). In an analogous topological arrangement, endogenous proteins Gas6 and Pros1 employ a vitamin K modified Gla domain to bind externalized PS, and laminin-like globular domains to bind Tyro3, Axl, and Mertk (TAM receptors) (82). It is clear that, despite their common biological function, PS receptors are disparate in their structural arrangement and sequence homology across member. PS receptors often are regulated by different transcriptional and post-translational mechanisms, and in many cases have unique expression patterns that add complexity to their biology (83).
Finally, in addition to receptors or soluble proteins alluded to above that recognize PS when presented as an external signal, several scavenger receptors can broadly recognize the negative charge PS, adding both diversity and complexity to PS receptor signaling and function (45). For example, CD36, CD91 (also called LDL-receptor related protein; LRP, the homolog of CED1) and CD68 have been implicated in the recognition and clearance of apoptotic cells, although as noted above, these receptors likely also recognize additional chemical modalities on the apoptotic cells, such as modified carbohydrates, acetylated components of the glycocalyx, and altered sialic acid residues (so-called apoptotic cell associated molecular patterns; ACAMPs) (3, 45). Interestingly, there have also been reports that scavenger receptors, and possibly even conventional PS receptors, preferentially recognize oxidized PS (oxPS), whereby the acyl chains of PS become oxidized during apoptosis. For example, CD36, CD68, as well as CD91 have been reported to preferentially interact with oxPS compared to non-oxidized PS as scavenger receptors, and several PS binding opsonins, such as MFG-E8 and C1q, may also bind with higher affinity towards oxPS than non-oxidized PS (84).
Mechanistically, the interaction of PS receptors and scavenger receptors with oxidized PS may have functional consequences with respect to how externalized PS on apopototic cells, but not non-apoptotic cells, acts as a signal for efferocytosis. In this capacity, interesting studies by Kagan and colleagues showed that cytochrome c can acquire a gain-of-function peroxidase activity once released from the mitochondria (85, 86). According to this idea, cytochrome c released from the mitochondria during mitochondrial outer membrane permeabilization (MOMP) would serve two interrelated functions, first as a central component of the apoptosome, and second, to concomitantly catalyze the oxidation of PS to ensure that apoptotic cells are swiftly and decisively cleared by phagocytes. Such a mechanism might add a level of molecular assurance that activation of caspases on the dying cell is efficiently coupled the externalization of PS by Xkr8. Along these lines, as the fundamental mechanisms of lipid trans-bilayer movements become better understood at the molecular level, it would also be interesting to determine whether oxPS is also preferentially scrambled by Xkr8.
While the repertoire of PS receptors has grown in diversity and complexity compared to the biology of PS receptors in the worm, many of the known PS receptors and PS bridging molecules signal through CED2 (Crk), CED5 (DOCK180), CED12 (Elmo), CED10 (Rac1) or CED6 (Gulp), CED10 (Rac1), and ultimately require Rac1 (Fig. 2). The universality of the PS->PSR->PSR-post-receptor signaling itinerary in both worms and mammals is conceptually important, and highlights the centrality of PS as an efferocytic signal in multicellular organisms.
PS/PS-receptor interactions and the regulation of immune homeostasis
The expansion in the numbers and complexity of PS receptors and PS bridging molecules in higher metazoans might simply reflect the increased complexity of efferocytosis in complex metazoans. However, it is also plausible (and likely) that the increased diversity and repertoire of PS receptors (as well as other efferocytosis receptors) functions to broaden the regulation of innate and adaptive immune responses in these higher organisms. Nematodes have a simplified and primitive host defense systems against pathogens, do not express macrophages, and express a single TLR (Tol-1), but do not express MyD88 or NF-κB (87), such that clearance failure in worms is not associated with a notable inflammatory response.
In higher mammals, the expansion in PS receptors, as well as other receptors that recognize the surface of the apoptotic cells, provides a higher level of discrimination of the complex lipidome and proteome of the apoptotic cell surface, and subsequently, the ability to make an appropriate post-signaling cascades in order to mount meaningful immune responses. Therefore, while many PS receptors (and other apoptotic cell receptors) are studied as individual receptors that aims to understand specific post-receptor outcomes, it may be more meaningful to use systems biology approaches to study PS receptors as, physiologically, many PS receptors are likely activated at the same time, inducing complex pleotropic outcomes and function cooperatively and synergize in signaling. Examples of this type of cooperation include cooperation between MFG-E8 and Gas6, via their signaling receptors, αvβ5 integrin and Mertk, that crosstalk in order to activate Rac1 (88). Similar examples have been described for Stabilin-2 and αϖβ5 (89), TIM-4 and integrin (90), C1q and Mertk (91), BAI and TIM-4 (92) as well as PS receptors with other cytokine receptors such as Axl with INFAR (93) and Axl with LRP-1 and RANBP9 (94). In several of these scenario’s, cooperatively can be supported by loss of function genetic knockouts (for example for resident macrophages, combined loss of function of Mertk and Tim4 have maximal inhibitory effects on efferocytosis (95)). However, such higher order signaling of different PS receptors is under-appreciated functionally, and the idea of a phagocytic synapse, akin the immunological synapse for T cell signaling, is still generally depicted only in models. Since the surface of the apoptotic likely engages multiple receptors simultaneously, systems biology approaches to probe the collective activation of a series of receptors is likely to provide important information as to how apoptotic cells signal.
Indeed, the notion that multiple phagocytic receptors, including one or more PS receptors, act coordinately to influence complex immune outcomes during homeostatic efferocytosis is consistent with the equally complex actions of apoptotic cells, that include the suppression of pro-inflammatory cytokines such as IL-1β, TNF-α, and IL-17, and the production of anti-inflammatory cytokines and resolving factors that include IL-10, TGF-β, PAF, and PEG2 (3, 96) (70, 97), which can further polarize immune subsets towards M2 “wound-healing” macrophages, immature DCs, and T regulatory cells. Furthermore, apoptotic cells can also influence the potency and activity of soluble cytokines such as IL-4 and IL-13 that render the expression of anti-inflammatory/tissue-repair genes (98), and the half-life of cytokines and interferons (99). Moreover, macrophages have also been observed to crosstalk to non-professional phagocytes, for example via the production of IGF-1, to dampen inflammatory responses on epithelial cells (100).
While presently it is not clear how individual PS receptors, or specific combinations of PS receptors and other efferocytosis receptors promote cytokine production or the polarization of professional phagocytes towards non-activating subtypes (although some PS receptors, such as TAMs and TIMs possess ITIM inhibitory motifs or have been linked to inhibitory pathways (101, 102) (103)), the importance of PS receptors in immune homeostasis is clearly evident in the spectrum of inflammatory and autoimmune disorders caused by defects in PS receptors and their signaling molecules (70). Genetic ablation of PS receptors, including members of the TAM family, members of the TIM family, SCARF1, CD300, MFG-E8, and C1q display phenotypic outcomes that manifest increased deposition of apoptotic cells in vivo, increased production of inflammatory cytokines, and often auto-immunity (78, 79). Mechanistically, it is well established that non-cleared apoptotic cell or their remnants release intracellular constituents as self-antigens in peripheral tissues, secondary lymphoid tissues, or germinal centers and can release intracellular components as danger signals (9). Under these conditions, auto- reactive intracellular constituents such as self-nucleic acids and modified histones are released as danger signals from un-cleared cells, which can lead to auto-antibody production and a pathology highly reminiscent of systemic lupus erythematosis (SLE) (11, 104). On the other hand, interesting studies from Ravichandran and colleagues showed that boosting cell clearance in vivo, by overexpressing the PS receptor BAI in colonic epithelial cells in a gain-of-function capacity, could improve disease outcome in a colitis models by dampening inflammatory cytokines (105). It will be of interest to determine whether these observations can be phenocopied as a therapeutic strategy in inflammatory conditions, for example using agonistic antibodies towards specific PS receptors.
Externalized PS is dys-regulated in the tumor microenvironment
As noted above, unlike the scenario in the PS-Receptor knockout mice, it is rare to detect Annexin V-positive apoptotic cells in vivo, even in tissues such as the spleen and thymus where is the rapid cell turnover, implying that clearance is profoundly proficient. By contrast, in the tumor microenvironment (as well as in stress or during viral and pathogen infections), constitutive PS exposures persist in tissues, imposing a significant barrier to mount a host anti-tumor immune response (70).
The mechanism(s) by which PS becomes constitutively externalized in the tumor microenvironment is complex and likely multifactorial, although at least three mechanisms appear to cooperate in order to create a PS-positive immunosuppressed environmental milieu. These include (i) the high apoptotic indexes of cancers, (ii) the stressed endothelium and stress tumor cells, and (iii) PS + microparticles such as exosomes and microvesicles. In the case for solid tumors with a high apoptotic index, if tumor cells die by classic apoptosis, followed by Xkr8-mediated PS externalization, the typically beneficial tolerogenic signals of apoptotic cell death (including the polarization of M2 macrophages, development of T regulatory cells, and suppression of antigen presenting cells) can impose a significant barrier to achieve host immunity and actively drive immune evasion (70). Moreover, if chemotherapies and targeted oncogene therapies induce classic apoptosis, as opposed to immunogenic cell death (see below) these strategies could further increase the PS burden in the tumor micro-environment, and inadvertently contribute to immune escape mechanisms.
A second contributing factor to the constitutive elevation of PS in the tumor microenvironment arises from the stressed vascular endothelial cell and metabolically stressed viable tumor cells (106). In addition to dying tumor cells, the harsh microenvironment of the tumor can lead to metabolic stress in both the vasculature and the tumor cells, leading to elevated intracellular calcium and constitutive activation of calcium-activated PS scramblases such as TMEM16F and related members (72). Whether activation of stress-activated PS scramblases is a general hallmark of cancers, for example according to Hanahan and Weinberg criterions, remain to be tested (107), clearly experimentally models that knockout of scramblases in cancer cells as experimental strategies, or development of blocking antibodies to TMEM16 or Xkr8, will be important experiments and might shed light on these questions.
Finally, a significant source PS in the tumor microenvironment is derived from PS-positive microvesicles and PS-positive exosomes, which increase both the source and surface area of externalized PS (108, 109). In the case for activated cells, such as platelets and endothelial cells that are activated by elevation in intracellular calcium, interesting studies by Fjuii et al showed that activated TMEM16F, and possibly other TMEM members that support Ca2+-dependent phospholipid scrambling, not only support PS externalization, but also support the release of PS-positive microparticles (110). This observation, combined with recent observations by Schroit and colleagues showing that tumor exosomes, but not exosomes form non-transformed cells are PS positive (111), suggest that PS positive exosomes not only contribute significantly to the constitutive elevation of PS in the tumor microenvironment, but also may have diagnostic value to predict tumor status in patients (112).
Development of PS targeting biologicals and Mabs to neutralize externalized PS
The aforementioned discussion that PS is constitutively externalized in cancers, but not naïve and native tissues, implicates a conceptual strategy that externalized PS may represent a fortuitous target in cancer biology, by interfering with the immunosuppressive signaling properties of PS. Two of the better studied therapeutic strategies of this class include recombinant Annexin V (AnxV) proteins and the PS targeting monoclonal antibody developed by Thorpe and colleagues, and subsequently tested by Peregrine Pharmaceuticals. In the case for AnxV, early studies by Herrmann and colleagues showed that Anx5 binds with high affinity to externalized PS on dying cells associated with tumor models, can subsequently induce improved host anti-tumor immunity, presumably by blocking the immunosuppressive of PS as developed above (113–116). More recently, systemic administration of a series of PS targeting monoclonal antibodies have also supported PS targeting as a baseline general strategy for cancer (117–120). Since the pre-clinical studies performed by Thorpe and colleagues over 10 years ago in mice, several company-sponsored and investigator-sponsored trials have been designed and reported with PS targeting antibodies (70), several of which showed enhanced levels of CD4+ and CD8+ tumor-infiltrating lymphocytes and improved anti-tumor responses (121). Further mechanistic studies with AnxV and PS targeting Mabs, and other PS targeting agents, should be of interest, particularly to better understand whether these strategies block and mask PS, akin to the phenotype of PS receptor KO’s, hence delaying clearance pathways and favoring the indirect release of operational danger or stress signals which can activate immunogenic signals in the tumor microenvironment (Table 1).
Table 1. Summary of current PtdSer -targeting modalities and their current stage of development.
Listed are current PtdSer -targeting biological and Mabs under development and their current stage in clinical application. Presently, the β2GPI-Binding Mab, Bavituximab (Peregrine Pharmaceuticals), is the only PtdSer -targeting agent in clinical trials, however there are many other promising therapeutics in preclinical stages. Notably, these therapies are not restricted to Mabs and in fact represent several unique PS-binding materials including: cationic small molecules, fusion proteins, nanoparticles, nanovesicles, and peptide-peptoid hybrids. Further studies comparing affinities and specificities towards PtdSer, and their applicability in driving immunogenic signals in cancer is an important future goal.
| Name | PS Targeting Strategy | Development Stage | Noteworthy Publications |
|---|---|---|---|
| Bavituximab | β2GPI-binding Mab | Phase III NSCLC Phase III Metastatic Breast Cancer | DeRose et al, 2011 ref. 18 |
| ch1N11 | β2GPI-binding Mab | Preclinical | Gray et al, 2016 Freimark et al, 2016 ref. 122, 123 |
| 11.31 | PS-direct binding Mab | Preclinical | Moody et al, 2010 Kasikara et al, 2017 ref. 129, 176 |
| PGN635 F(ab’)2-SPIO | β2GPI-binding F(ab’)2 fused to superparamagnetic iron oxide (SPIO) nanoparticle. | Preclinical | Zhang et al, 2014 ref. 171 |
| SapC-DOPS | Nanovesicles targeting membranes exposing PS. | Preclinical | Davis et al, 2016 ref. 172 |
| mCTH-ANXA1/5 | Annexin A1 and Annexin A5 fused to cystathione gamma-lyase. Used in combo with rapamycin and /or cyclophosphamide. | Preclinical | Krais et al, 2017 ref. 173 |
| GW4869 | Cationic small molecule that binds to externalized PS. | Preclinical | Vuckovic et al, 2017 ref. 174 |
| PPS1, PPS1D1 | Peptide-Peptoid Hybrids that bind to externalized PS on cancer cells. | Preclinical | Martharage et al, 2015 ref. 175 |
Due to the earlier success of the pre-clinical and phase I and phase II human trials led to a larger phase III clinical trial called SUNRISE (Stimulating ImmUne RespoNse thRough BavItuximab in a PhaSe III Lung Cancer Study), a randomized, double bind, placebo-controlled registration trial by Peregrine for previously-treated locally advanced or metastatic non-squamous non-small cell lung cancer (NSCLC). Bavituximab is a chimeric monoclonal constructed from the Fv region of murine antibody 3G4 (used successfully in many pre-clinical studies) fused to the Fc of a human IgG. Disappointingly, in February of 2016, the SUNRISE trail was discontinued following an interim analysis showing that bavituximab plus docetaxel group did not show a sufficient improvement in overall survival as compared to docetaxel monotherapy group.
While the bavituximab plus docetaxel studies had a disappointing outcome, clearly more mechanistic research will be required to evaluate the mechanisms of action of PS targeting antibodies, as well as increase the repertoire of combinatorial agents that may maximize therapeutic efficacy. For example, in follow to the SUNRISE trial, additional biomarkers have been identified, including high levels of beta-2 glycoprotein and serum IFN-γ that appear to segregate with strong patient responders. Moreover, in a recent follow up, it has been observed bavituximab-treated patients that continued on to checkpoint therapeutics, such as anti-PD1/PDL1, have significantly improved outcomes compared to the docetaxel arm, suggesting that previous exposure to bavituximab may have elevated the patient’s immune thresholds for the subsequent exposure to anti-PD1. Clearly, future studies aimed to understand this biology are meritorious of further investigation. Additional studies investigating the combined efforts of bavituximab and checkpoints, empirically, to reverse the immunosuppressive phenotypes of solid cancers will be great interest.
In support of this model, several recent studies support the idea that PS targeting Mabs can act synergistically with other immune checkpoint inhibitors. These studies utilize a PS-targeting chimeric ch1N11, a mouse IgG2a-κ antibody with human variable heavy and light chain regions. Ch1N11 binds to β2GPI complexed to PS and is considered a “pre-clinical equivalent to Bavituximab”. Recently, it has been primarily used to conduct mechanistic studies involving immune checkpoint combinations. In the study by Gray and colleagues, these investigators found that ch1N11, in addition to anti-PD1 showed synergistic effects with respect to tumor shrinkage, metastasis, and at the molecular and cellular level, with skewed immune responses (increased CD8+ and CD3+ T cells) and inflammatory cytokines (122). Furthermore, Freimark et al showed that ch1N11 enhanced CTLA-4 and PD-1 antibody antitumor activity in Melanoma (123). In this study, ch1N11 combo therapies with either anti-CTLA-4 or anti-PD1 significantly increased checkpoint inhibition in comparison to the corresponding mono-therapies, resulting in an elevated infiltration of CD4+ and CD8+ lymphocytes. Moreover, in these combination groups, there were elevated ratios of CD8+ T-cells to myeloid-derived suppressor cells (MDSCs) and regulatory T cells (Tregs) within tumors. Accompanying these results were also increased levels of cells producing pro-inflammatory cytokines such as IL-2, IFN-γ, and TNF-α. Taken together, recent data strongly suggest that cocktails containing immune-checkpoint inhibitors and PS-antibodies may be most effective route of therapy for non-immunogenic tumors.
Targeting PS receptors in the tumor microenvironment
In addition to the targeting of PS in the tumor microenvironment, emerging studies also suggest that targeting subsets of PS receptors, mainly the TIM and TAM receptors, might also have therapeutic value in cancer immunotherapy. In the case for TIMs, most notably TIM-3, these receptors mediate immune tolerance in mouse and human models, and while not clear how they transmit inhibitory signals from PS, targeting TIMs appears promising for the improvement of current immunotherapies (124).
The TAM receptors (Tyro-3, Axl, and Mertk) are also emerging as interesting targets in cancer immunology, particularly in their capacity to act as myeloid checkpoint inhibitors. As such, all three TAMs can be sporadically up-regulated on several tumor types and subsets and promote oncogenic signaling, they are also expressed on myeloid derived cells where they act as PS sensors and inhibitory receptors for externalized PS (82, 125). Curiously, it is also apparent that TAMs are differentially expressed on different myeloid subsets within the tumor microenvironment, for example resident and infiltrating macrophages express Mertk, while immature DC’s express Axl. Moreover, TAMs are regulated differentially, whereby Mertk is up-regulated by dexamethasone and tolerogenic conditions, while Axl, appears to be reciprocally regulated with respect to Mertk in the tumor microenvironment, and for example, is up-regulated by pro-inflammatory components such as poly-I:C, LPS, and other danger signals (126, 127). In this scenario, blocking the activation of Mertk on macrophages under tolerogenic conditions, while preserving Axl-mediated efferocytosis on DCs might have therapeutic values in models of immunogenic death. Indeed, support of this model, interesting studies by Cook and colleagues showed that transplantation of Mertk (−/−) bone marrow, but not WT bone marrow, into lethally-irradiated MMTV-PyVmT mice decreased tumor growth and cytokine production by tumor CD11+ cells as well as improved host anti-tumor immunity, including the appearance of tumor specific T cells with anti-tumor activity (128). Together, these studies suggest that antagonistic Mertk antibodies (via their role as PS receptors) may be attractive therapeutic targets in cancer (Fig. 4).
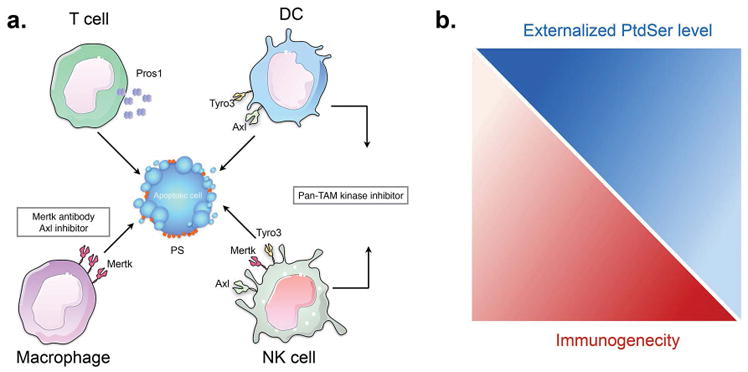
Figure 4. PS receptors (Tyro3, Axl, and Mertk) differentially impact the tumor microenvironment via the interaction of apoptotic cells.
(A) The expression of TAMs and their ligands (Gas6 and Pros1) on infiltrating tumor-associated immune subsets may act as immune checkpoint inhibitors that promote tolerogenic signals in the tumor microenvironment. Examples include; Mertk expressed on macrophages, Tyro3 and Axl expressed on DCs, and Tyro3, Axl, and Mertk expressed on NK cells, and Pros1 expressed on activated T cells. In recent years, the development of TAM antagonists are being assessed as myeloid checkpoint inhibitors in cancer. In some scenario’s, inhibition of Mertk on macrophages might be expected to skew efferocytosis towards antigen presenting cells (DCs) that establish tumor immunity. In other cases, pan TAM inhibitors might stimulate global immunogenic outcomes by blocking multiple inhibitory signals. (B). Externalized PS is a double-edge sword in the tumor microenvironment. In this model, tumors with high deposition of externalized PS will have low immunogenicity, and vice versa, such that PS targeting antibodies are expected to have important therapeutic value in cancer immunotherapy
Moreover, analogous to the above-mentioned strategies to combine PS targeting antibodies anti-PD1 or anti-PDL1 antibodies, combinatorial strategies that combine TAM inhibitors with other checkpoint inhibitors may also be meritorious of further development. In support of this model, we have recently shown that PS (apoptotic cells) promotes TAM-mediated up-regulation of PDL1 on several tumor cell lines (129). Further studies showing combined action of generalized TAM inhibitors with anti-PD1 in non-small cell lung cancer, or TAM inhibitors with anti-PD1 head and neck cancers supports this notion (130, 131). Currently, there are three clinical trials involving and Axl tyrosine kinase inhibitor, BGB324 (BerGenBio), along with anti-PD1 monoclonal antibody Pembrolizumab (Keytruda, Merck) for the treatment of NSCLC, AML, and Melanoma (BergenBio). Furthermore, there are numerous TAM therapeutics in preclinical studies that have potential to synergize with other immune checkpoint molecule therapies.
Tolerogenic versus Inflammatory Cell Death Pathways
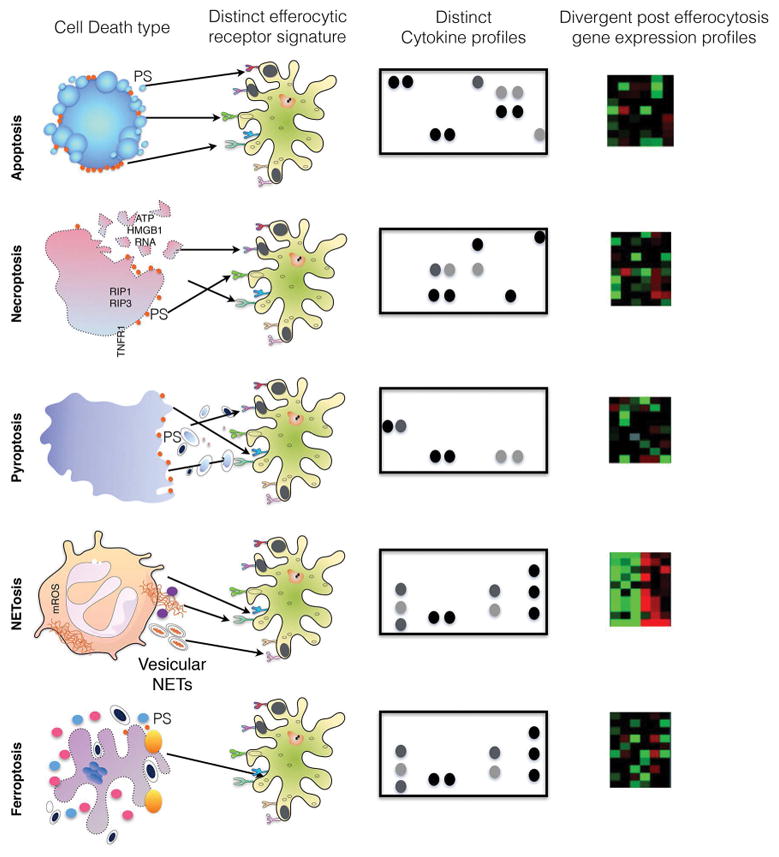
As described above, classical caspase-mediated apoptosis, induced by chemotherapeutics or oncogene-targeted therapeutics, and the subsequent tolerogenic signals manifested by the clearance of PS-positive apoptotic cells by PS receptors would likely produce poorly immunogenic tumors, expected to evade rejection by the host immune system. By contrast, over the past several years, alternative apoptotic death mechanisms and numerous non-apoptotic cell death pathways have been described (i.e. necroptosis, pyroptosis, ferroptosis, NETosis, programmed necrosis)(132–138), often these forms of cell death are defined by morphological features and criteria, associated with different biomarkers and cell surface molecules and neo-antigens associated with the membrane (139). In turn, specific alterations on the surface of the dying cells are expected to have unique “interactomes” on the surface of the phagocyte (akin to a specific phagocytic synapse) in order to alter immunological consequences at the level of professional phagocytes as well as non-professional phagocytes (Fig. 5). In this capacity, the constellation of receptors engaged on the phagocyte serves as an immune surveillance system to “autopsy” the history of the dying cell. Depending on the specific lipidome and proteome of the apoptotic cell surface, specific information would be provided, whether the cell died a natural or unnatural death, and whether it was stressed, transformed, or virally infected. Subsequently, mediated by the changes in gene expression and cytokines produced, and the processing of antigens derived from the apoptotic cells, both innate and adaptive immune responses are expected to be regulated by apoptotic cells.
Figure 5. Decoding cell death modules by phagocytic cells into efferocytic synapses.
Cells that have undergone cell death through various mechanisms include apoptosis, necroptosis, pyroptosis, NETosis, and ferroptosis. These different death modalities produce distinct proteomic and lipidomic surface components that in turn engage specific repertoires of surface receptors on the engulfing cells, akin to specific phagocytic synapses. Intuitively, these ligand receptor interactions will produce signature gene expression and cytokine profiles. Such systems biology platforms may help better define different cell death platforms and how they influence the tumor microenvironment.
However, the aforementioned definition of tolerogenic and inflammatory cell death has been challenged, spearheaded by important studies by Albert et al, showing that apoptotic influenza infected cells, when challenged to DCs in the presence of a danger signal, could cross present internalized antigens on MHC Class I molecules to CD8+ T cells (140). More recently, elegant work pioneered by Kroemer and colleagues identified two morphological equivalent, but immunological distinct forms of apoptosis, described as immunogenic cell death (ICD) and classic (non-immunogenic) cell death (141). Unlike tolerogenic death, ICD is defined by the spatiotemporal externalization or release of dander signals of damage-associated molecular patterns (DAMPs) that increase the immunogenicity of caspase-activated dying cancer cells that lead to a durable anti-tumor response (142, 143). The first inducer of ICD was the anthrocycline drug, doxorubicin, but other inducers include oxaliplatin, ionizing radiation, cardiac glycosides, and photodynamic therapies with Hypericin and many others have been identified (144).
Mechanistically, several conditions must be met to define tumor cell death as ICD. In this capacity, ICD has been shown to depend on the activation of endoplasmic reticulum (ER) stress as well as induction of reactive oxygen species (ROS), which induce the redistribution of a series of “lock- and-key” molecular determinants on the surface of the apoptotic cell, that when engulfed by DCs, can act as danger signals to cross-present tumor antigens to anti-tumor T cells to mount an anti-tumor response (145–147). Then list of danger signals important for ICD include (i) the surface exposure of calreticulin (CRT) (148), (ii) the surface exposure of heat shock proteins 70 and 90 (HSP70 and HSP90) (149), (iii) the secretion of ATP (150), and (iv) the release of high mobility group box 1 (HMGB1) (151). Functionally, CRT acts as a danger signal and appears traffics apoptotic cargo into a cross- presentation competent itinerary via its receptor CD91, HMGB1 and HSP70/90 bind TLR’s and appear to lead to activation and maturation of DCs as a pre-requisite of cross-presentation, and ATP appears to be required not only to recruit DCs to the vicinity of apoptotic tumor cells via purinoreceptor-1 (P2Y2) receptors, but also to activate the inflammasome and induce DC maturation and prime DC for anti-tumor adaptive immunity (152, 153).
In addition to their role in ICD, both CRT and HMGB1 have been shown to interact with PS, and therefore it is possible that in addition to their role in inflammatory signaling, exposure of these molecules masks that tolerogenic signals of PS-mediated efferocytosis (154–156). Consistent with this idea HMGB1 can inhibit PS-mediated efferocytosis in apoptotic neutrophils (155). Whether CRT, which binds PS through its C-terminal acidic region (156), also blocks PS signaling awaits further investigation. Similarly, studies combining ICD inducers with PS targeting antibodies or AnxV might also have combinatorial value to skew immune responses in favor immunogenicity.
Role of intrinsic oncogenic pathways in immune escape
While the aforementioned discussion suggests that distinct forms of death reflect the nature of the cell-death inducing signals, for example anthracyclins induce immunogenic death, and docetaxol induces tolerogenic cell death, an important and emerging idea in the cell death field is that certain oncogenic pathways may drive intrinsic immune escape pathways. Indeed, of the multiple ways by which tumor maintains and grows in presence of hostile host immune system is by devising means to evade host immune response(157, 158). While expression of immune checkpoints (such as PD-L1) that block the cytotoxic immune cells including CD8+ T cells and decrease of antigen presentation, by lowering MHC-I expression, have emerged one of the common mechanisms by which tumor cells evade immune response, the molecular basis of these processes were still largely unknown until recently. New evidences link a select list of oncogenes, that are frequently up-regulated in gain-of-function manner, to immune response to tumors.
The role of PI3K-AKT-mTOR pathway is well established in the regulation of cell proliferation survival and growth. While most of the published data suggest a cell intrinsic role this pathway in regulation of tumor cell growth and proliferation, recent evidences unfold a cell extrinsic view of this pathway in control of tumor immunity. In a mice model of lung cancer, targeting PI3K-AKT-mTOR pathway by rapamycin was found to be associated with fewer immunosuppressive FoxP3+ regulatory T cells (Treg) cells, indicating a link between this pathway and immunosuppression(159). Mechanistically, it was found that oncogenic activation of PI3K-AKT-mTOR pathway regulates PD-L1 (an important immune checkpoint) expression on non-small cell lung cancer cells. Consistently, when combined with mTOR targeting rapamycin, anti-PD1 antibody provided a synergistic benefit in tumor regression, decrease of Tregs and increase of tumor infiltrating CD8+ T cells.
Another mechanism by which mTOR signaling plays a role in immune evasion in mammary tumor cells is by modulation of inter-tumoral MDSC infiltration. shRNA screens, unbiased genomic analysis of human breast tumors established an unconventional role of mTOR signaling in accumulation of tumor promoting MDSC by driving of G-CSF expression(160). Although mTOR plays an important role in T cell activation and hence is not a good target for inducing immunogenicity, these studies have provided an important mechanistic insight into the possible reasons of inter-tumoral MDSC heterogeneity and variable response to immune checkpoint inhibitors.
These results were further affirmed by another observation that PTEN (a negative regulator of PI3K-AKT pathway) loss in melanoma patients correlates with less T cell infiltration in primary tumor and poor efficacy of anti-PD1 therapy(161). Similarly, loss of PTEN renders the tumor immune microenvironment immunosuppressive by skewing the cytokines milieu. Knockdown of PTEN by shRNA resulted in increased PD-L1 in human breast cancer cells indicating a direct mechanistic link between PTEN loss and immunosuppression(162). In the PTEN null melanoma tumors, targeting the PI3K-AKT pathway by pharmacological inhibition provides enhances the tumor immunogenicity and provides therapeutic synergy with anti-PD1(163).
Myc, an important transcription factor that is commonly dysregulated in many human cancers, is an important regulator is tumor immunity. By ChIP analysis in human melanoma cells and tetracycline-off mice model (wherein tetracycline controls Myc expression), it was shown that Myc expression in melanoma can control tumor immunity by transcriptional control of PD-L1 and CD47, two immune checkpoint molecules(164). By regulation of don’t-find-me and don’t-eat-me signals Myc is implicated for the first time in tumor immunity. Therapeutic agents that target Myc activation or expression in cancers with overexpressed Myc, may enhance the sustained anti-tumor host immune response.
In non-small cell lung cancer, oncogenic drivers and their genetic basis of immune evasion are becoming clearer with recent reports. Oncogenic EGFR mutation a common feature in NSCLC is shown to drive oncogenic transformation and PD-L1 expression in lung cancer cells(165, 166). Another oncoprotein formed in NSCLC due to fusion of two individual genes (EML4 and ALK) known as EM4-ALK fusion protein have been reported to drive PD-L1(167). Moreover, in NSCLC patients there is a clear co-relation between PD-L1 levels and EM4-ALK protein expression. Consistent with these observations, ALK specific inhibitor attenuates the PD-L1 expression in lung cancer cells. Interestingly, EM4-ALK induced PD-L1 expression is attenuated by pharmacological inhibition of MEK-ERK and PI3K-AKT pathways indicating an intricate link between these three pathways in regulation of tumor immunity by PD1-PD-L1 axis(163, 167).
Similarly, in human melanoma cells, targeting BRAF-MAPK signaling pathway by RNAi or pharmacological inhibitors of MEK alleviates tumor immune evasion by suppressing the immunosuppressive cytokines IL-6, IL-10 and VEGF levels(168). This indicates the role of constitutive MAPK signaling in immune evasion by human melanomas. In an important landmark study, perturbation of tumor cell intrinsic signaling by b catenin pathway was shown to be important regulator of tumor immunity by CD8+ T cells in melanoma(169). By comparative gene expression profiling between T-cell non-inflamed and inflamed cohorts of metastatic human cutaneous melanoma samples, active beta catenin signaling was found to be active in non-T cell inflamed cohort. Further experiments using genetically engineered BRAF V600E/ Pten−/− mice showed that activated WNT/B-catenin pathway lead to suppression of recruitment of dermal CD103+ DC and T cell infiltration due to defective T cell priming. Finally, B-catenin target gene expression inversely correlates with intratumoral CD8+ and DC cell populations.
Lastly, another cell intrinsic mechanism by which tumor cells evade immune response is by manipulating antigen presentation by dendritic cells and macrophages that is key for cytotoxic T cells to recognize and eliminate the tumor cells. Tumor cells evade this process of immune recognition by lowering the expression of MHC I on their cell surface that is recognized by CD8+ T cytotoxic cells. Human kinase regulation plays a major role in regulating the MHC I levels and thus the antigen presentations in multiple tumor types(170). A recent RNAi based screen of 526 human kinases and further validation by pharmacological inhibitors revealed an important role for MEK1, RET and EGFR for regulation of MHC-I expression and antigen presentation in human mesothelioma and lung cancer cells. Consistently, activation of MAPK or EGFR pathway caused down-regulation of MHC-I.
Summary and Conclusions
Over the past two decades, the biology of efferocytosis, like that of the biology of apoptosis and immunogenic cell death, has made great strides, whereby many of the eat-me signals and receptors for apoptotic cells have been identified and molecularly characterized. However, there are several challenges ahead that require further investigation. First, recent studies indicate that efferocytosis receptors, both for tolerogenic and ICD, do not act alone but rather cooperate and synergize to decode the spatial array of eat me signals defined by different forms of cell death. Presently, the exact nature of these receptor interactions are not known (akin to efferocytic synapses with different co-stimulatory and co-inhibitory modules) and therefore it is also not known if and how receptors traffic apoptotic cells to distinct intracellular itineraries, as well as how they cooperate to drive complex immune outcomes. Clever designs of global systems biology approaches and next-generation sequencing will be required to fully understand these processes. Second, recent studies focusing on immunogenic cells death, and targeting PS and PS receptors suggest that efferocytosis pathways may be experimentally manipulated, for example to regulate tolerance in autoimmunity and cancer vaccines with ICD. Finally, recent studies suggest that specific oncogenic pathways, such as FAK, Erk, and Myc, can intrinsically effect immunosuppressive pathways. Clearly, the goals in cancer immunology will be to optimize immunogenicity over tolerance for the design of new chemotherapeutic strategies into multimodal therapeutic protocols.
Acknowledgments
We would like to thank members of the Birge laboratory, particularly Canan Kasikara and Viral Davra for helpful discussions. This work was supported in part form Grant NIH CA 165077 as well as a grant New Jersey Health Foundation to RBB. We acknowledge Rutgers Society of Research Scholars award to Sushil Kumar.
Footnotes
Disclosure of potential conflicts of interest:
No potential conflicts of interest were disclosed by the authors.
References
- 1.Elliott MR, Ravichandran KS. Clearance of apoptotic cells: implications in health and disease. J Cell Biol. 2010;189:1059–1070. doi: 10.1083/jcb.201004096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Savill J, Dransfield I, Gregory C, Haslett C. A blast from the past: clearance of apoptotic cells regulates immune responses. Nat Rev Immunol. 2002;2:965–975. doi: 10.1038/nri957. [DOI] [PubMed] [Google Scholar]
- 3.Savill J, Fadok V. Corpse clearance defines the meaning of cell death. Nature. 2000;407:784–788. doi: 10.1038/35037722. [DOI] [PubMed] [Google Scholar]
- 4.Voll RE, Herrmann M, Roth EA, et al. Immunosuppressive effects of apoptotic cells. Nature. 1997;390:350–351. doi: 10.1038/37022. [DOI] [PubMed] [Google Scholar]
- 5.Duvall E, Wyllie AH, Morris RG. Macrophage recognition of cells undergoing programmed cell death (apoptosis) Immunology. 1985;56:351–358. [PMC free article] [PubMed] [Google Scholar]
- 6.deCathelineau AM, Henson PM. The final step in programmed cell death: phagocytes carry apoptotic cells to the grave. Essays Biochem. 2003;39:105–117. doi: 10.1042/bse0390105. [DOI] [PubMed] [Google Scholar]
- 7.Fadok VA, Bratton DL, Konowal A, et al. Macrophages that have ingested apoptotic cells in vitro inhibit proinflammatory cytokine production through autocrine/paracrine mechanisms involving TGF-beta, PGE2, and PAF. J Clin Invest. 1998;101:890–898. doi: 10.1172/JCI1112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hawkins LA, Devitt A. Current understanding of the mechanisms for clearance of apoptotic cells-a fine balance. J Cell Death. 2013;6:57–68. doi: 10.4137/JCD.S11037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Baumann I, Kolowos W, Voll RE, et al. Impaired uptake of apoptotic cells into tingible body macrophages in germinal centers of patients with systemic lupus erythematosus. Arthritis Rheum. 2002;46:191–201. doi: 10.1002/1529-0131(200201)46:1<191::AID-ART10027>3.0.CO;2-K. [DOI] [PubMed] [Google Scholar]
- 10.Mackiewicz M, Huppi K, Pitt JJ, et al. Identification of the receptor tyrosine kinase AXL in breast cancer as a target for the human miR-34a microRNA. Breast Cancer Res Treat. 2011;130:663–679. doi: 10.1007/s10549-011-1690-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Munoz LE, Lauber K, Schiller M, et al. The role of defective clearance of apoptotic cells in systemic autoimmunity. Nat Rev Rheumatol. 2010;6:280–289. doi: 10.1038/nrrheum.2010.46. [DOI] [PubMed] [Google Scholar]
- 12.Shao WH, Cohen PL. Disturbances of apoptotic cell clearance in systemic lupus erythematosus. Arthritis Res Ther. 2011;13:202. doi: 10.1186/ar3206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hengartner MO, Horvitz HR. The ins and outs of programmed cell death during C. elegans development. Philos Trans R Soc Lond B Biol Sci. 1994;345:243–246. doi: 10.1098/rstb.1994.0100. [DOI] [PubMed] [Google Scholar]
- 14.Yuan J, Horvitz HR. A first insight into the molecular mechanisms of apoptosis. Cell. 2004;116:S53–56. 51–S59. doi: 10.1016/s0092-8674(04)00028-5. [DOI] [PubMed] [Google Scholar]
- 15.Yuan J, Shaham S, Ledoux S, et al. The C. elegans cell death gene ced-3 encodes a protein similar to mammalian interleukin-1 beta-converting enzyme. Cell. 1993;75:641–652. doi: 10.1016/0092-8674(93)90485-9. [DOI] [PubMed] [Google Scholar]
- 16.Yuan JY, Horvitz HR. The Caenorhabditis elegans genes ced-3 and ced-4 act cell autonomously to cause programmed cell death. Dev Biol. 1990;138:33–41. doi: 10.1016/0012-1606(90)90174-h. [DOI] [PubMed] [Google Scholar]
- 17.Horvitz HR. Worms, life, and death (Nobel lecture) Chembiochem. 2003;4:697–711. doi: 10.1002/cbic.200300614. [DOI] [PubMed] [Google Scholar]
- 18.Hengartner MO, Ellis RE, Horvitz HR. Caenorhabditis elegans gene ced-9 protects cells from programmed cell death. Nature. 1992;356:494–499. doi: 10.1038/356494a0. [DOI] [PubMed] [Google Scholar]
- 19.Hengartner MO, Horvitz HR. C. elegans cell survival gene ced-9 encodes a functional homolog of the mammalian proto-oncogene bcl-2. Cell. 1994;76:665–676. doi: 10.1016/0092-8674(94)90506-1. [DOI] [PubMed] [Google Scholar]
- 20.Yuan J, Horvitz HR. The Caenorhabditis elegans cell death gene ced-4 encodes a novel protein and is expressed during the period of extensive programmed cell death. Development. 1992;116:309–320. doi: 10.1242/dev.116.2.309. [DOI] [PubMed] [Google Scholar]
- 21.Zhou M, Li Y, Hu Q, et al. Atomic structure of the apoptosome: mechanism of cytochrome c- and dATP-mediated activation of Apaf-1. Genes Dev. 2015;29:2349–2361. doi: 10.1101/gad.272278.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ellis RE, Jacobson DM, Horvitz HR. Genes required for the engulfment of cell corpses during programmed cell death in Caenorhabditis elegans. Genetics. 1991;129:79–94. doi: 10.1093/genetics/129.1.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Reddien PW, Horvitz HR. The engulfment process of programmed cell death in caenorhabditis elegans. Annu Rev Cell Dev Biol. 2004;20:193–221. doi: 10.1146/annurev.cellbio.20.022003.114619. [DOI] [PubMed] [Google Scholar]
- 24.Reddien PW, Cameron S, Horvitz HR. Phagocytosis promotes programmed cell death in C. elegans. Nature. 2001;412:198–202. doi: 10.1038/35084096. [DOI] [PubMed] [Google Scholar]
- 25.Kinchen JM, Cabello J, Klingele D, et al. Two pathways converge at CED-10 to mediate actin rearrangement and corpse removal in C. elegans. Nature. 2005;434:93–99. doi: 10.1038/nature03263. [DOI] [PubMed] [Google Scholar]
- 26.Reddien PW, Horvitz HR. CED-2/CrkII and CED-10/Rac control phagocytosis and cell migration in Caenorhabditis elegans. Nat Cell Biol. 2000;2:131–136. doi: 10.1038/35004000. [DOI] [PubMed] [Google Scholar]
- 27.Zhou Z, Hartwieg E, Horvitz HR. CED-1 is a transmembrane receptor that mediates cell corpse engulfment in C. elegans. Cell. 2001;104:43–56. doi: 10.1016/s0092-8674(01)00190-8. [DOI] [PubMed] [Google Scholar]
- 28.Liu QA, Hengartner MO. Candidate adaptor protein CED-6 promotes the engulfment of apoptotic cells in C. elegans. Cell. 1998;93:961–972. doi: 10.1016/s0092-8674(00)81202-7. [DOI] [PubMed] [Google Scholar]
- 29.Wu YC, Horvitz HR. The C. elegans cell corpse engulfment gene ced-7 encodes a protein similar to ABC transporters. Cell. 1998;93:951–960. doi: 10.1016/s0092-8674(00)81201-5. [DOI] [PubMed] [Google Scholar]
- 30.Suzuki J, Denning DP, Imanishi E, et al. Xk-related protein 8 and CED-8 promote phosphatidylserine exposure in apoptotic cells. Science. 2013;341:403–406. doi: 10.1126/science.1236758. [DOI] [PubMed] [Google Scholar]
- 31.Suzuki J, Imanishi E, Nagata S. Exposure of phosphatidylserine by Xk-related protein family members during apoptosis. J Biol Chem. 2014;289:30257–30267. doi: 10.1074/jbc.M114.583419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Matsuda M, Mayer BJ, Fukui Y, Hanafusa H. Binding of transforming protein, P47gag-crk, to a broad range of phosphotyrosine-containing proteins. Science. 1990;248:1537–1539. doi: 10.1126/science.1694307. [DOI] [PubMed] [Google Scholar]
- 33.Mayer BJ, Hamaguchi M, Hanafusa H. A novel viral oncogene with structural similarity to phospholipase C. Nature. 1988;332:272–275. doi: 10.1038/332272a0. [DOI] [PubMed] [Google Scholar]
- 34.Reichman CT, Mayer BJ, Keshav S, Hanafusa H. The product of the cellular crk gene consists primarily of SH2 and SH3 regions. Cell Growth Differ. 1992;3:451–460. [PubMed] [Google Scholar]
- 35.Birge RB, Fajardo JE, Reichman C, et al. Identification and characterization of a high-affinity interaction between v-Crk and tyrosine-phosphorylated paxillin in CT10-transformed fibroblasts. Mol Cell Biol. 1993;13:4648–4656. doi: 10.1128/mcb.13.8.4648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hasegawa H, Kiyokawa E, Tanaka S, et al. DOCK180, a major CRK-binding protein, alters cell morphology upon translocation to the cell membrane. Mol Cell Biol. 1996;16:1770–1776. doi: 10.1128/mcb.16.4.1770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Matsuda M, Hashimoto Y, Muroya K, et al. CRK protein binds to two guanine nucleotide-releasing proteins for the Ras family and modulates nerve growth factor-induced activation of Ras in PC12 cells. Mol Cell Biol. 1994;14:5495–5500. doi: 10.1128/mcb.14.8.5495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Brugnera E, Haney L, Grimsley C, et al. Unconventional Rac-GEF activity is mediated through the Dock180-ELMO complex. Nat Cell Biol. 2002;4:574–582. doi: 10.1038/ncb824. [DOI] [PubMed] [Google Scholar]
- 39.Grimsley CM, Kinchen JM, Tosello-Trampont AC, et al. Dock180 and ELMO1 proteins cooperate to promote evolutionarily conserved Rac-dependent cell migration. J Biol Chem. 2004;279:6087–6097. doi: 10.1074/jbc.M307087200. [DOI] [PubMed] [Google Scholar]
- 40.Albert ML, Kim JI, Birge RB. alphavbeta5 integrin recruits the CrkII-Dock180-rac1 complex for phagocytosis of apoptotic cells. Nat Cell Biol. 2000;2:899–905. doi: 10.1038/35046549. [DOI] [PubMed] [Google Scholar]
- 41.Akakura S, Singh S, Spataro M, et al. The opsonin MFG-E8 is a ligand for the alphavbeta5 integrin and triggers DOCK180-dependent Rac1 activation for the phagocytosis of apoptotic cells. Exp Cell Res. 2004;292:403–416. doi: 10.1016/j.yexcr.2003.09.011. [DOI] [PubMed] [Google Scholar]
- 42.Hsu TY, Wu YC. Engulfment of apoptotic cells in C. elegans is mediated by integrin alpha/SRC signaling. Curr Biol. 2010;20:477–486. doi: 10.1016/j.cub.2010.01.062. [DOI] [PubMed] [Google Scholar]
- 43.Nonaka S, Nagaosa K, Mori T, et al. Integrin alphaPS3/betanu-mediated phagocytosis of apoptotic cells and bacteria in Drosophila. J Biol Chem. 2013;288:10374–10380. doi: 10.1074/jbc.M113.451427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Tung TT, Nagaosa K, Fujita Y, et al. Phosphatidylserine recognition and induction of apoptotic cell clearance by Drosophila engulfment receptor Draper. J Biochem. 2013;153:483–491. doi: 10.1093/jb/mvt014. [DOI] [PubMed] [Google Scholar]
- 45.Penberthy KK, Ravichandran KS. Apoptotic cell recognition receptors and scavenger receptors. Immunol Rev. 2016;269:44–59. doi: 10.1111/imr.12376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Wu HH, Bellmunt E, Scheib JL, et al. Glial precursors clear sensory neuron corpses during development via Jedi-1, an engulfment receptor. Nat Neurosci. 2009;12:1534–1541. doi: 10.1038/nn.2446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Scheib JL, Sullivan CS, Carter BD. Jedi-1 and MEGF10 signal engulfment of apoptotic neurons through the tyrosine kinase Syk. J Neurosci. 2012;32:13022–13031. doi: 10.1523/JNEUROSCI.6350-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yu X, Lu N, Zhou Z. Phagocytic receptor CED-1 initiates a signaling pathway for degrading engulfed apoptotic cells. PLoS Biol. 2008;6:e61. doi: 10.1371/journal.pbio.0060061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hamon Y, Broccardo C, Chambenoit O, et al. ABC1 promotes engulfment of apoptotic cells and transbilayer redistribution of phosphatidylserine. Nat Cell Biol. 2000;2:399–406. doi: 10.1038/35017029. [DOI] [PubMed] [Google Scholar]
- 50.Chen YZ, Mapes J, Lee ES, et al. Caspase-mediated activation of Caenorhabditis elegans CED-8 promotes apoptosis and phosphatidylserine externalization. Nat Commun. 2013;4:2726. doi: 10.1038/ncomms3726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Mapes J, Chen YZ, Kim A, et al. CED-1, CED-7, and TTR-52 regulate surface phosphatidylserine expression on apoptotic and phagocytic cells. Curr Biol. 2012;22:1267–1275. doi: 10.1016/j.cub.2012.05.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang X, Li W, Zhao D, et al. Caenorhabditis elegans transthyretin-like protein TTR-52 mediates recognition of apoptotic cells by the CED-1 phagocyte receptor. Nat Cell Biol. 2010;12:655–664. doi: 10.1038/ncb2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Zhang Y, Wang H, Kage-Nakadai E, et al. C. elegans secreted lipid-binding protein NRF-5 mediates PS appearance on phagocytes for cell corpse engulfment. Curr Biol. 2012;22:1276–1284. doi: 10.1016/j.cub.2012.06.004. [DOI] [PubMed] [Google Scholar]
- 54.Conradt B, Wu YC, Xue D. Programmed Cell Death During Caenorhabditis elegans Development. Genetics. 2016;203:1533–1562. doi: 10.1534/genetics.115.186247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Suzuki J, Imanishi E, Nagata S. Xkr8 phospholipid scrambling complex in apoptotic phosphatidylserine exposure. Proc Natl Acad Sci U S A. 2016;113:9509–9514. doi: 10.1073/pnas.1610403113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Segawa K, Kurata S, Yanagihashi Y, et al. Caspase-mediated cleavage of phospholipid flippase for apoptotic phosphatidylserine exposure. Science. 2014;344:1164–1168. doi: 10.1126/science.1252809. [DOI] [PubMed] [Google Scholar]
- 57.Barclay AN, Van den Berg TK. The interaction between signal regulatory protein alpha (SIRPalpha) and CD47: structure, function, and therapeutic target. Annu Rev Immunol. 2014;32:25–50. doi: 10.1146/annurev-immunol-032713-120142. [DOI] [PubMed] [Google Scholar]
- 58.Oldenborg PA, Zheleznyak A, Fang YF, et al. Role of CD47 as a marker of self on red blood cells. Science. 2000;288:2051–2054. doi: 10.1126/science.288.5473.2051. [DOI] [PubMed] [Google Scholar]
- 59.Jaiswal S, Jamieson CH, Pang WW, et al. CD47 is upregulated on circulating hematopoietic stem cells and leukemia cells to avoid phagocytosis. Cell. 2009;138:271–285. doi: 10.1016/j.cell.2009.05.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Majeti R, Chao MP, Alizadeh AA, et al. CD47 is an adverse prognostic factor and therapeutic antibody target on human acute myeloid leukemia stem cells. Cell. 2009;138:286–299. doi: 10.1016/j.cell.2009.05.045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Elliott JI, Surprenant A, Marelli-Berg FM, et al. Membrane phosphatidylserine distribution as a non-apoptotic signalling mechanism in lymphocytes. Nat Cell Biol. 2005;7:808–816. doi: 10.1038/ncb1279. [DOI] [PubMed] [Google Scholar]
- 62.Carrera Silva EA, Chan PY, Joannas L, et al. T cell-derived protein S engages TAM receptor signaling in dendritic cells to control the magnitude of the immune response. Immunity. 2013;39:160–170. doi: 10.1016/j.immuni.2013.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martin S, Pombo I, Poncet P, et al. Immunologic stimulation of mast cells leads to the reversible exposure of phosphatidylserine in the absence of apoptosis. Int Arch Allergy Immunol. 2000;123:249–258. doi: 10.1159/000024451. [DOI] [PubMed] [Google Scholar]
- 64.Frasch SC, Henson PM, Nagaosa K, et al. Phospholipid flip-flop and phospholipid scramblase 1 (PLSCR1) co-localize to uropod rafts in formylated Met-Leu-Phe-stimulated neutrophils. J Biol Chem. 2004;279:17625–17633. doi: 10.1074/jbc.M313414200. [DOI] [PubMed] [Google Scholar]
- 65.Hochreiter-Hufford AE, Lee CS, Kinchen JM, et al. Phosphatidylserine receptor BAI1 and apoptotic cells as new promoters of myoblast fusion. Nature. 2013;497:263–267. doi: 10.1038/nature12135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Yang H, Kim A, David T, et al. TMEM16F forms a Ca2+-activated cation channel required for lipid scrambling in platelets during blood coagulation. Cell. 2012;151:111–122. doi: 10.1016/j.cell.2012.07.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Segawa K, Suzuki J, Nagata S. Constitutive exposure of phosphatidylserine on viable cells. Proc Natl Acad Sci U S A. 2011;108:19246–19251. doi: 10.1073/pnas.1114799108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Suzuki J, Umeda M, Sims PJ, Nagata S. Calcium-dependent phospholipid scrambling by TMEM16F. Nature. 2010;468:834–838. doi: 10.1038/nature09583. [DOI] [PubMed] [Google Scholar]
- 69.Biermann M, Maueroder C, Brauner JM, et al. Surface code--biophysical signals for apoptotic cell clearance. Phys Biol. 2013;10:065007. doi: 10.1088/1478-3975/10/6/065007. [DOI] [PubMed] [Google Scholar]
- 70.Birge RB, Boeltz S, Kumar S, et al. Phosphatidylserine is a global immunosuppressive signal in efferocytosis, infectious disease, and cancer. Cell Death Differ. 2016 doi: 10.1038/cdd.2016.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ishii H, Mori T, Shiratsuchi A, et al. Distinct localization of lipid rafts and externalized phosphatidylserine at the surface of apoptotic cells. Biochem Biophys Res Commun. 2005;327:94–99. doi: 10.1016/j.bbrc.2004.11.135. [DOI] [PubMed] [Google Scholar]
- 72.Gyobu S, Ishihara K, Suzuki J, et al. Characterization of the scrambling domain of the TMEM16 family. Proc Natl Acad Sci U S A. 2017;114:6274–6279. doi: 10.1073/pnas.1703391114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schroit AJ, Madsen JW, Tanaka Y. In vivo recognition and clearance of red blood cells containing phosphatidylserine in their plasma membranes. J Biol Chem. 1985;260:5131–5138. [PubMed] [Google Scholar]
- 74.Schroit AJ, Tanaka Y, Madsen J, Fidler IJ. The recognition of red blood cells by macrophages: role of phosphatidylserine and possible implications of membrane phospholipid asymmetry. Biol Cell. 1984;51:227–238. doi: 10.1111/j.1768-322x.1984.tb00303.x. [DOI] [PubMed] [Google Scholar]
- 75.Fadok VA, Voelker DR, Campbell PA, et al. Exposure of phosphatidylserine on the surface of apoptotic lymphocytes triggers specific recognition and removal by macrophages. J Immunol. 1992;148:2207–2216. [PubMed] [Google Scholar]
- 76.Fadok VA, Bratton DL, Rose DM, et al. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature. 2000;405:85–90. doi: 10.1038/35011084. [DOI] [PubMed] [Google Scholar]
- 77.Wang X, Wu YC, Fadok VA, et al. Cell corpse engulfment mediated by C. elegans phosphatidylserine receptor through CED-5 and CED-12. Science. 2003;302:1563–1566. doi: 10.1126/science.1087641. [DOI] [PubMed] [Google Scholar]
- 78.Arandjelovic S, Ravichandran KS. Phagocytosis of apoptotic cells in homeostasis. Nat Immunol. 2015;16:907–917. doi: 10.1038/ni.3253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kimani SG, Geng K, Kasikara C, et al. Contribution of Defective PS Recognition and Efferocytosis to Chronic Inflammation and Autoimmunity. Front Immunol. 2014;5:566. doi: 10.3389/fimmu.2014.00566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hanayama R, Tanaka M, Miwa K, et al. Identification of a factor that links apoptotic cells to phagocytes. Nature. 2002;417:182–187. doi: 10.1038/417182a. [DOI] [PubMed] [Google Scholar]
- 81.Yoshida H, Kawane K, Koike M, et al. Phosphatidylserine-dependent engulfment by macrophages of nuclei from erythroid precursor cells. Nature. 2005;437:754–758. doi: 10.1038/nature03964. [DOI] [PubMed] [Google Scholar]
- 82.Lemke G. Biology of the TAM receptors. Cold Spring Harb Perspect Biol. 2013;5:a009076. doi: 10.1101/cshperspect.a009076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Davra V, Kimani SG, Calianese D, Birge RB. Ligand Activation of TAM Family Receptors-Implications for Tumor Biology and Therapeutic Response. Cancers (Basel) 2016:8. doi: 10.3390/cancers8120107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Tyurin VA, Balasubramanian K, Winnica D, et al. Oxidatively modified phosphatidylserines on the surface of apoptotic cells are essential phagocytic ‘eat-me’ signals: cleavage and inhibition of phagocytosis by Lp-PLA2. Cell Death Differ. 2014;21:825–835. doi: 10.1038/cdd.2014.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kagan VE, Borisenko GG, Serinkan BF, et al. Appetizing rancidity of apoptotic cells for macrophages: oxidation, externalization, and recognition of phosphatidylserine. Am J Physiol Lung Cell Mol Physiol. 2003;285:L1–17. doi: 10.1152/ajplung.00365.2002. [DOI] [PubMed] [Google Scholar]
- 86.Kagan VE, Gleiss B, Tyurina YY, et al. A role for oxidative stress in apoptosis: oxidation and externalization of phosphatidylserine is required for macrophage clearance of cells undergoing Fas-mediated apoptosis. J Immunol. 2002;169:487–499. doi: 10.4049/jimmunol.169.1.487. [DOI] [PubMed] [Google Scholar]
- 87.Irazoqui JE, Urbach JM, Ausubel FM. Evolution of host innate defence: insights from Caenorhabditis elegans and primitive invertebrates. Nat Rev Immunol. 2010;10:47–58. doi: 10.1038/nri2689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Wu Y, Singh S, Georgescu MM, Birge RB. A role for Mer tyrosine kinase in alphavbeta5 integrin-mediated phagocytosis of apoptotic cells. J Cell Sci. 2005;118:539–553. doi: 10.1242/jcs.01632. [DOI] [PubMed] [Google Scholar]
- 89.Kim S, Park SY, Kim SY, et al. Cross talk between engulfment receptors stabilin-2 and integrin alphavbeta5 orchestrates engulfment of phosphatidylserine-exposed erythrocytes. Mol Cell Biol. 2012;32:2698–2708. doi: 10.1128/MCB.06743-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Flannagan RS, Canton J, Furuya W, et al. The phosphatidylserine receptor TIM4 utilizes integrins as coreceptors to effect phagocytosis. Mol Biol Cell. 2014;25:1511–1522. doi: 10.1091/mbc.E13-04-0212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Galvan MD, Foreman DB, Zeng E, et al. Complement component C1q regulates macrophage expression of Mer tyrosine kinase to promote clearance of apoptotic cells. J Immunol. 2012;188:3716–3723. doi: 10.4049/jimmunol.1102920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Mazaheri F, Breus O, Durdu S, et al. Distinct roles for BAI1 and TIM-4 in the engulfment of dying neurons by microglia. Nat Commun. 2014;5:4046. doi: 10.1038/ncomms5046. [DOI] [PubMed] [Google Scholar]
- 93.Rothlin CV, Ghosh S, Zuniga EI, et al. TAM receptors are pleiotropic inhibitors of the innate immune response. Cell. 2007;131:1124–1136. doi: 10.1016/j.cell.2007.10.034. [DOI] [PubMed] [Google Scholar]
- 94.Subramanian M, Hayes CD, Thome JJ, et al. An AXL/LRP-1/RANBP9 complex mediates DC efferocytosis and antigen cross-presentation in vivo. J Clin Invest. 2014;124:1296–1308. doi: 10.1172/JCI72051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Nishi C, Toda S, Segawa K, Nagata S. Tim4- and MerTK-mediated engulfment of apoptotic cells by mouse resident peritoneal macrophages. Mol Cell Biol. 2014;34:1512–1520. doi: 10.1128/MCB.01394-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Huynh ML, Fadok VA, Henson PM. Phosphatidylserine-dependent ingestion of apoptotic cells promotes TGF-beta1 secretion and the resolution of inflammation. J Clin Invest. 2002;109:41–50. doi: 10.1172/JCI11638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Birge RB, Ucker DS. Innate apoptotic immunity: the calming touch of death. Cell Death Differ. 2008;15:1096–1102. doi: 10.1038/cdd.2008.58. [DOI] [PubMed] [Google Scholar]
- 98.Bosurgi L, Cao YG, Cabeza-Cabrerizo M, et al. Macrophage function in tissue repair and remodeling requires IL-4 or IL-13 with apoptotic cells. Science. 2017;356:1072–1076. doi: 10.1126/science.aai8132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Oyler-Yaniv J, Oyler-Yaniv A, Shakiba M, et al. Catch and Release of Cytokines Mediated by Tumor Phosphatidylserine Converts Transient Exposure into Long-Lived Inflammation. Mol Cell. 2017;66:635–647. e637. doi: 10.1016/j.molcel.2017.05.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Han CZ, Juncadella IJ, Kinchen JM, et al. Macrophages redirect phagocytosis by non-professional phagocytes and influence inflammation. Nature. 2016;539:570–574. doi: 10.1038/nature20141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Du W, Yang M, Turner A, et al. TIM-3 as a Target for Cancer Immunotherapy and Mechanisms of Action. Int J Mol Sci. 2017:18. doi: 10.3390/ijms18030645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rothlin CV, Carrera-Silva EA, Bosurgi L, Ghosh S. TAM receptor signaling in immune homeostasis. Annu Rev Immunol. 2015;33:355–391. doi: 10.1146/annurev-immunol-032414-112103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Lee J, Su EW, Zhu C, et al. Phosphotyrosine-dependent coupling of Tim-3 to T-cell receptor signaling pathways. Mol Cell Biol. 2011;31:3963–3974. doi: 10.1128/MCB.05297-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Gaipl US, Munoz LE, Grossmayer G, et al. Clearance deficiency and systemic lupus erythematosus (SLE) J Autoimmun. 2007;28:114–121. doi: 10.1016/j.jaut.2007.02.005. [DOI] [PubMed] [Google Scholar]
- 105.Lee CS, Penberthy KK, Wheeler KM, et al. Boosting Apoptotic Cell Clearance by Colonic Epithelial Cells Attenuates Inflammation In Vivo. Immunity. 2016;44:807–820. doi: 10.1016/j.immuni.2016.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Ran S, Downes A, Thorpe PE. Increased exposure of anionic phospholipids on the surface of tumor blood vessels. Cancer Res. 2002;62:6132–6140. [PubMed] [Google Scholar]
- 107.Hanahan D, Weinberg RA. Hallmarks of cancer: the next generation. Cell. 2011;144:646–674. doi: 10.1016/j.cell.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 108.Lima LG, Chammas R, Monteiro RQ, et al. Tumor-derived microvesicles modulate the establishment of metastatic melanoma in a phosphatidylserine-dependent manner. Cancer Lett. 2009;283:168–175. doi: 10.1016/j.canlet.2009.03.041. [DOI] [PubMed] [Google Scholar]
- 109.Lima LG, Oliveira AS, Campos LC, et al. Malignant transformation in melanocytes is associated with increased production of procoagulant microvesicles. Thromb Haemost. 2011;106:712–723. doi: 10.1160/TH11-03-0143. [DOI] [PubMed] [Google Scholar]
- 110.Fujii T, Sakata A, Nishimura S, et al. TMEM16F is required for phosphatidylserine exposure and microparticle release in activated mouse platelets. Proc Natl Acad Sci U S A. 2015;112:12800–12805. doi: 10.1073/pnas.1516594112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Lea J, Sharma R, Yang F, et al. Detection of phosphatidylserine-positive exosomes as a diagnostic marker for ovarian malignancies: a proof of concept study. Oncotarget. 2017;8:14395–14407. doi: 10.18632/oncotarget.14795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Sharma R, Huang X, Brekken RA, Schroit AJ. Detection of phosphatidylserine-positive exosomes for the diagnosis of early-stage malignancies. Br J Cancer. 2017 doi: 10.1038/bjc.2017.183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bondanza A, Zimmermann VS, Rovere-Querini P, et al. Inhibition of phosphatidylserine recognition heightens the immunogenicity of irradiated lymphoma cells in vivo. J Exp Med. 2004;200:1157–1165. doi: 10.1084/jem.20040327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Frey B, Schildkopf P, Rodel F, et al. AnnexinA5 renders dead tumor cells immunogenic--implications for multimodal cancer therapies. J Immunotoxicol. 2009;6:209–216. doi: 10.3109/15476910903204058. [DOI] [PubMed] [Google Scholar]
- 115.Munoz LE, Frey B, Pausch F, et al. The role of annexin A5 in the modulation of the immune response against dying and dead cells. Curr Med Chem. 2007;14:271–277. doi: 10.2174/092986707779941131. [DOI] [PubMed] [Google Scholar]
- 116.Stach CM, Turnay X, Voll RE, et al. Treatment with annexin V increases immunogenicity of apoptotic human T-cells in Balb/c mice. Cell Death Differ. 2000;7:911–915. doi: 10.1038/sj.cdd.4400715. [DOI] [PubMed] [Google Scholar]
- 117.Beck AW, Luster TA, Miller AF, et al. Combination of a monoclonal anti-phosphatidylserine antibody with gemcitabine strongly inhibits the growth and metastasis of orthotopic pancreatic tumors in mice. Int J Cancer. 2006;118:2639–2643. doi: 10.1002/ijc.21684. [DOI] [PubMed] [Google Scholar]
- 118.DeRose P, Thorpe PE, Gerber DE. Development of bavituximab, a vascular targeting agent with immune-modulating properties, for lung cancer treatment. Immunotherapy. 2011;3:933–944. doi: 10.2217/imt.11.87. [DOI] [PubMed] [Google Scholar]
- 119.He J, Yin Y, Luster TA, et al. Antiphosphatidylserine antibody combined with irradiation damages tumor blood vessels and induces tumor immunity in a rat model of glioblastoma. Clin Cancer Res. 2009;15:6871–6880. doi: 10.1158/1078-0432.CCR-09-1499. [DOI] [PubMed] [Google Scholar]
- 120.Huang X, Bennett M, Thorpe PE. A monoclonal antibody that binds anionic phospholipids on tumor blood vessels enhances the antitumor effect of docetaxel on human breast tumors in mice. Cancer Res. 2005;65:4408–4416. doi: 10.1158/0008-5472.CAN-05-0031. [DOI] [PubMed] [Google Scholar]
- 121.Yin Y, Huang X, Lynn KD, Thorpe PE. Phosphatidylserine-targeting antibody induces M1 macrophage polarization and promotes myeloid-derived suppressor cell differentiation. Cancer Immunol Res. 2013;1:256–268. doi: 10.1158/2326-6066.CIR-13-0073. [DOI] [PubMed] [Google Scholar]
- 122.Gray MJ, Gong J, Hatch MM, et al. Phosphatidylserine-targeting antibodies augment the anti-tumorigenic activity of anti-PD-1 therapy by enhancing immune activation and downregulating pro-oncogenic factors induced by T-cell checkpoint inhibition in murine triple-negative breast cancers. Breast Cancer Res. 2016;18:50. doi: 10.1186/s13058-016-0708-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Freimark BD, Gong J, Ye D, et al. Antibody-Mediated Phosphatidylserine Blockade Enhances the Antitumor Responses to CTLA-4 and PD-1 Antibodies in Melanoma. Cancer Immunol Res. 2016;4:531–540. doi: 10.1158/2326-6066.CIR-15-0250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Hahn AW, Gill DM, Pal SK, Agarwal N. The future of immune checkpoint cancer therapy after PD-1 and CTLA-4. Immunotherapy. 2017;9:681–692. doi: 10.2217/imt-2017-0024. [DOI] [PubMed] [Google Scholar]
- 125.Graham DK, DeRyckere D, Davies KD, Earp HS. The TAM family: phosphatidylserine sensing receptor tyrosine kinases gone awry in cancer. Nat Rev Cancer. 2014;14:769–785. doi: 10.1038/nrc3847. [DOI] [PubMed] [Google Scholar]
- 126.Lemke G. Phosphatidylserine Is the Signal for TAM Receptors and Their Ligands. Trends Biochem Sci. 2017 doi: 10.1016/j.tibs.2017.06.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Zagorska A, Traves PG, Lew ED, et al. Diversification of TAM receptor tyrosine kinase function. Nat Immunol. 2014;15:920–928. doi: 10.1038/ni.2986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Cook RS, Jacobsen KM, Wofford AM, et al. MerTK inhibition in tumor leukocytes decreases tumor growth and metastasis. J Clin Invest. 2013;123:3231–3242. doi: 10.1172/JCI67655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Kasikara C, Kumar S, Kimani S, et al. Phosphatidylserine Sensing by TAM Receptors Regulates AKT-dependent Chemoresistance and PD-L1 Expression. Mol Cancer Res. 2017 doi: 10.1158/1541-7786.MCR-16-0350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.von Massenhausen A, Bragelmann J, Billig H, et al. Implication of the Receptor Tyrosine Kinase AXL in Head and Neck Cancer Progression. Int J Mol Sci. 2016:18. doi: 10.3390/ijms18010007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Vouri M, An Q, Birt M, et al. Small molecule inhibition of Axl receptor tyrosine kinase potently suppresses multiple malignant properties of glioma cells. Oncotarget. 2015;6:16183–16197. doi: 10.18632/oncotarget.3952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Abiko K, Mandai M, Hamanishi J, et al. PD-L1 on tumor cells is induced in ascites and promotes peritoneal dissemination of ovarian cancer through CTL dysfunction. Clin Cancer Res. 2013;19:1363–1374. doi: 10.1158/1078-0432.CCR-12-2199. [DOI] [PubMed] [Google Scholar]
- 133.Galluzzi L, Buque A, Kepp O, et al. Immunogenic cell death in cancer and infectious disease. Nat Rev Immunol. 2017;17:97–111. doi: 10.1038/nri.2016.107. [DOI] [PubMed] [Google Scholar]
- 134.Jorgensen I, Rayamajhi M, Miao EA. Programmed cell death as a defence against infection. Nat Rev Immunol. 2017;17:151–164. doi: 10.1038/nri.2016.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Nagata S, Tanaka M. Programmed cell death and the immune system. Nat Rev Immunol. 2017;17:333–340. doi: 10.1038/nri.2016.153. [DOI] [PubMed] [Google Scholar]
- 136.Segawa K, Nagata S. An Apoptotic ‘Eat Me’ Signal: Phosphatidylserine Exposure. Trends Cell Biol. 2015;25:639–650. doi: 10.1016/j.tcb.2015.08.003. [DOI] [PubMed] [Google Scholar]
- 137.Toda S, Nishi C, Yanagihashi Y, et al. Clearance of Apoptotic Cells and Pyrenocytes. Curr Top Dev Biol. 2015;114:267–295. doi: 10.1016/bs.ctdb.2015.07.017. [DOI] [PubMed] [Google Scholar]
- 138.Yang WS, Stockwell BR. Ferroptosis: Death by Lipid Peroxidation. Trends Cell Biol. 2016;26:165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kroemer G, Galluzzi L, Vandenabeele P, et al. Classification of cell death: recommendations of the Nomenclature Committee on Cell Death 2009. Cell Death Differ. 2009;16:3–11. doi: 10.1038/cdd.2008.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Albert ML, Sauter B, Bhardwaj N. Dendritic cells acquire antigen from apoptotic cells and induce class I-restricted CTLs. Nature. 1998;392:86–89. doi: 10.1038/32183. [DOI] [PubMed] [Google Scholar]
- 141.Kroemer G, Galluzzi L, Kepp O, Zitvogel L. Immunogenic cell death in cancer therapy. Annu Rev Immunol. 2013;31:51–72. doi: 10.1146/annurev-immunol-032712-100008. [DOI] [PubMed] [Google Scholar]
- 142.Dudek AM, Garg AD, Krysko DV, et al. Inducers of immunogenic cancer cell death. Cytokine Growth Factor Rev. 2013;24:319–333. doi: 10.1016/j.cytogfr.2013.01.005. [DOI] [PubMed] [Google Scholar]
- 143.Kepp O, Tesniere A, Zitvogel L, Kroemer G. The immunogenicity of tumor cell death. Curr Opin Oncol. 2009;21:71–76. doi: 10.1097/CCO.0b013e32831bc375. [DOI] [PubMed] [Google Scholar]
- 144.Garg AD, Galluzzi L, Apetoh L, et al. Molecular and Translational Classifications of DAMPs in Immunogenic Cell Death. Front Immunol. 2015;6:588. doi: 10.3389/fimmu.2015.00588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Garg AD, Krysko DV, Vandenabeele P, Agostinis P. The emergence of phox-ER stress induced immunogenic apoptosis. Oncoimmunology. 2012;1:786–788. doi: 10.4161/onci.19750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Kepp O, Menger L, Vacchelli E, et al. Crosstalk between ER stress and immunogenic cell death. Cytokine Growth Factor Rev. 2013;24:311–318. doi: 10.1016/j.cytogfr.2013.05.001. [DOI] [PubMed] [Google Scholar]
- 147.Panaretakis T, Kepp O, Brockmeier U, et al. Mechanisms of pre-apoptotic calreticulin exposure in immunogenic cell death. EMBO J. 2009;28:578–590. doi: 10.1038/emboj.2009.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Obeid M, Tesniere A, Ghiringhelli F, et al. Calreticulin exposure dictates the immunogenicity of cancer cell death. Nat Med. 2007;13:54–61. doi: 10.1038/nm1523. [DOI] [PubMed] [Google Scholar]
- 149.Spisek R, Charalambous A, Mazumder A, et al. Bortezomib enhances dendritic cell (DC)-mediated induction of immunity to human myeloma via exposure of cell surface heat shock protein 90 on dying tumor cells: therapeutic implications. Blood. 2007;109:4839–4845. doi: 10.1182/blood-2006-10-054221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Ghiringhelli F, Apetoh L, Tesniere A, et al. Activation of the NLRP3 inflammasome in dendritic cells induces IL-1beta-dependent adaptive immunity against tumors. Nat Med. 2009;15:1170–1178. doi: 10.1038/nm.2028. [DOI] [PubMed] [Google Scholar]
- 151.Apetoh L, Ghiringhelli F, Tesniere A, et al. The interaction between HMGB1 and TLR4 dictates the outcome of anticancer chemotherapy and radiotherapy. Immunol Rev. 2007;220:47–59. doi: 10.1111/j.1600-065X.2007.00573.x. [DOI] [PubMed] [Google Scholar]
- 152.Aymeric L, Apetoh L, Ghiringhelli F, et al. Tumor cell death and ATP release prime dendritic cells and efficient anticancer immunity. Cancer Res. 2010;70:855–858. doi: 10.1158/0008-5472.CAN-09-3566. [DOI] [PubMed] [Google Scholar]
- 153.Locher C, Conforti R, Aymeric L, et al. Desirable cell death during anticancer chemotherapy. Ann N Y Acad Sci. 2010;1209:99–108. doi: 10.1111/j.1749-6632.2010.05763.x. [DOI] [PubMed] [Google Scholar]
- 154.Davis K, Banerjee S, Friggeri A, et al. Poly(ADP-ribosyl)ation of high mobility group box 1 (HMGB1) protein enhances inhibition of efferocytosis. Mol Med. 2012;18:359–369. doi: 10.2119/molmed.2011.00203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Liu G, Wang J, Park YJ, et al. High mobility group protein-1 inhibits phagocytosis of apoptotic neutrophils through binding to phosphatidylserine. J Immunol. 2008;181:4240–4246. doi: 10.4049/jimmunol.181.6.4240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Wijeyesakere SJ, Bedi SK, Huynh D, Raghavan M. The C-Terminal Acidic Region of Calreticulin Mediates Phosphatidylserine Binding and Apoptotic Cell Phagocytosis. J Immunol. 2016;196:3896–3909. doi: 10.4049/jimmunol.1502122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Gubin MM, Zhang X, Schuster H, et al. Checkpoint blockade cancer immunotherapy targets tumour-specific mutant antigens. Nature. 2014;515:577–581. doi: 10.1038/nature13988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Vanneman M, Dranoff G. Combining immunotherapy and targeted therapies in cancer treatment. Nat Rev Cancer. 2012;12:237–251. doi: 10.1038/nrc3237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Welte T, Kim IS, Tian L, et al. Oncogenic mTOR signalling recruits myeloid-derived suppressor cells to promote tumour initiation. Nat Cell Biol. 2016;18:632–644. doi: 10.1038/ncb3355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Lastwika KJ, Wilson W, 3rd, Li QK, et al. Control of PD-L1 Expression by Oncogenic Activation of the AKT-mTOR Pathway in Non-Small Cell Lung Cancer. Cancer Res. 2016;76:227–238. doi: 10.1158/0008-5472.CAN-14-3362. [DOI] [PubMed] [Google Scholar]
- 161.Dong Y, Richards JA, Gupta R, et al. PTEN functions as a melanoma tumor suppressor by promoting host immune response. Oncogene. 2014;33:4632–4642. doi: 10.1038/onc.2013.409. [DOI] [PubMed] [Google Scholar]
- 162.Mittendorf EA, Philips AV, Meric-Bernstam F, et al. PD-L1 Expression in Triple Negative Breast Cancer. Cancer immunology research. 2014;2:361–370. doi: 10.1158/2326-6066.CIR-13-0127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Peng W, Chen JQ, Liu C, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov. 2016;6:202–216. doi: 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Casey SC, Tong L, Li Y, et al. MYC regulates the antitumor immune response through CD47 and PD-L1. Science. 2016;352:227–231. doi: 10.1126/science.aac9935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Chen N, Fang W, Zhan J, et al. Upregulation of PD-L1 by EGFR Activation Mediates the Immune Escape in EGFR-Driven NSCLC: Implication for Optional Immune Targeted Therapy for NSCLC Patients with EGFR Mutation. J Thorac Oncol. 2015;10:910–923. doi: 10.1097/JTO.0000000000000500. [DOI] [PubMed] [Google Scholar]
- 166.Tang Y, Fang W, Zhang Y, et al. The association between PD-L1 and EGFR status and the prognostic value of PD-L1 in advanced non-small cell lung cancer patients treated with EGFR-TKIs. Oncotarget. 2015;6:14209–14219. doi: 10.18632/oncotarget.3694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Ota K, Azuma K, Kawahara A, et al. Induction of PD-L1 Expression by the EML4-ALK Oncoprotein and Downstream Signaling Pathways in Non-Small Cell Lung Cancer. Clin Cancer Res. 2015;21:4014–4021. doi: 10.1158/1078-0432.CCR-15-0016. [DOI] [PubMed] [Google Scholar]
- 168.Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–1656. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Spranger S, Bao R, Gajewski TF. Melanoma-intrinsic beta-catenin signalling prevents anti-tumour immunity. Nature. 2015;523:231–235. doi: 10.1038/nature14404. [DOI] [PubMed] [Google Scholar]
- 170.Brea EJ, Oh CY, Manchado E, et al. Kinase Regulation of Human MHC Class I Molecule Expression on Cancer Cells. Cancer Immunol Res. 2016;4:936–947. doi: 10.1158/2326-6066.CIR-16-0177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Zhang L, Zhou H, Belzile O, et al. Phosphatidylserine-targeted bimodal liposomal nanoparticles for in vivo imaging of breast cancer in mice. J Control Release. 2014;183:114–123. doi: 10.1016/j.jconrel.2014.03.043. [DOI] [PubMed] [Google Scholar]
- 172.Davis HW, Hussain N, Qi X. Detection of cancer cells using SapC-DOPS nanovesicles. Mol Cancer. 2016;15:33. doi: 10.1186/s12943-016-0519-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Krais JJ, Virani N, McKernan PH, et al. Antitumor Synergism and Enhanced Survival with a Tumor Vasculature-Targeted Enzyme Prodrug System, Rapamycin, and Cyclophosphamide. Mol Cancer Ther. 2017 doi: 10.1158/1535-7163.MCT-16-0263. [DOI] [PubMed] [Google Scholar]
- 174.Vuckovic S, Vandyke K, Rickards DA, et al. The cationic small molecule GW4869 is cytotoxic to high phosphatidylserine-expressing myeloma cells. Br J Haematol. 2017;177:423–440. doi: 10.1111/bjh.14561. [DOI] [PubMed] [Google Scholar]
- 175.Matharage JM, Minna JD, Brekken RA, Udugamasooriya DG. Unbiased Selection of Peptide-Peptoid Hybrids Specific for Lung Cancer Compared to Normal Lung Epithelial Cells. ACS Chem Biol. 2015;10:2891–2899. doi: 10.1021/acschembio.5b00592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 176.Moody MA, Liao HX, Alam SM, et al. Anti-phospholipid human monoclonal antibodies inhibit CCR5-tropic HIV-1 and induce beta-chemokines. J Exp Med. 2010;207:763–776. doi: 10.1084/jem.20091281. [DOI] [PMC free article] [PubMed] [Google Scholar]