

Abstract
Aim
Phase 2b study to assess efficacy, safety, thrombogenicity, immunogenicity and tolerability with 28 days of daily dosing of subcutaneous (SQ) dalcinonacog alfa as prophylaxis for haemophilia B (HB).
Methods
Adult males with a confirmed diagnosis of congenital HB (factor IX [FIX] activity <2%) received daily dalcinonacog alfa 100 IU/kg SQ until day 28. The primary efficacy endpoint was the number of participants who achieved a steady‐state FIX activity level ≥12%. Tolerability, thrombogenicity and immunogenicity were study safety endpoints.
Results
Of 6 participants who received study drug, one discontinued the study on day 7 due to injection‐site reactions (ISR). Of the 5 participants completing the study, FIX activity level exceeded 12% in 3 participants at day 7, increasing to 4 participants on days 14, 21 and 28 and all 5 at day 29. Pharmacokinetic findings (including mean alpha and beta half‐life of 5.3 days and 3.9 days, respectively, and mean residence time of 6.2 days) supported prolonged effects. Thrombogenicity markers remained normal throughout prophylactic injections or showed some initial increases followed by decreases with continued dosing. Two participants had anti‐drug antibodies to dalcinonacog alfa at study end, none had neutralizing antibody. Two participants had ISR, both resolved. Reports of redness, swelling, tenderness or pain among the first 3 participants prompted dose‐splitting for the last 3 participants, leading to fewer ISR.
Conclusion
Subcutaneous dalcinonacog alfa is effective in raising FIX levels into the mild haemophilia range, comparable to intravenous extended half‐life FIX clotting factors.
Keywords: clinical trial, dalcinonacog alfa, factor IX, haemophilia B, immunogenicity, prophylaxis, subcutaneous
1. INTRODUCTION
Haemophilia B (HB) is a bleeding disorder caused by factor IX (FIX) deficiency because of an X chromosome mutation,1 characterized by frequent and spontaneous bleeding into joints, muscles and body cavities. The current globally accepted standard of care is intravenous (IV) FIX replacement therapy administered prophylactically, at regular intervals, to prevent the onset of bleeding episodes.2, 3, 4, 5 Intravenous infusions are a significant barrier to prophylaxis adherence, especially in paediatric patients and adults with poor venous access.6, 7, 8 Extended half‐life recombinant FIX (rFIX) products that allow for weekly to every‐2‐week dosing have been introduced in recent years, conferring a 4‐ to 6‐fold increase in half‐life and reducing infusions by 60% relative to standard half‐life products but have had variable bleed prevention.9 Subcutaneous (SQ) administration of rFIX with a large volume of distribution would provide major advantages over IV administration by improving quality of life and reducing healthcare costs.10
Dalcinonacog alfa is a novel rFIX variant developed using a rational design approach with three‐point substitutions (Arg318Tyr; Arg338Glu; Thr343Arg) that confer a 3‐fold higher catalytic speed of factor X activation than that of wild‐type FIX of factor X, 10‐fold affinity for activated factor VIII (FVIIIa), and 15‐fold resistance to inhibition by antithrombin III.11, 12 Pharmacokinetic (PK) observations in murine models and early clinical studies collectively support that the potency of dalcinonacog alfa is >20‐fold that of recombinant wild‐type FIX dosed at the same mass, thereby allowing for higher activity at equal dosing.13, 14 Dalcinonacog alfa was evaluated in a Phase 1/2, open‐label, multicentre, dose‐escalation study in patients with previously treated HB.14 In a cohort receiving SQ dosing, activity levels after 6 daily doses in 5 participants reached a median of 15.7%, with 4 of these participants having FIX levels >12%. Median half‐life was 63.2 h. Initial IV dosing was also shown to increase SQ dalcinonacog alfa bioavailability by increasing extravascular compartment saturation, resulting in activity levels greater than those expected from simple addition of SQ to IV activity.
Here we report the findings of a Phase 2b study of SQ administration of dalcinonacog alfa in adults with congenital HB, primarily designed to assess efficacy (based on the proportion of participants achieving steady‐state FIX activity levels ≥12%), thrombogenicity, immunogenicity and tolerability with 28 days of daily dosing.
2. MATERIAL AND METHODS
2.1. Study design
This was a single centre, open‐label Phase 2b study of SQ prophylaxis with dalcinonacog alfa in adults with HB. The screening period duration was up to 4 weeks, and SQ prophylaxis with dalcinonacog alfa was given over a 28‐day treatment period, with a goal of achieving steady‐state levels ≥12% FIX activity. The study (NCT03995784) was conducted from 18 June 2019 until 20 March 2020 in accordance with the Declaration of Helsinki and was approved by the independent ethics committee/institutional review board at the study site. All patients provided informed consent prior to enrolment.
Dalcinonacog alfa is produced by a recombinant Chinese hamster ovary cell clone in suspension culture that co‐expresses rFIX and recombinant human wt‐furin, which is extensively characterized. Stored cell banks are free of human blood or plasma products. No additives of animal or human origin are used during the cell culture, purification and formulation processes. Recombinant FIX is purified by a four‐step chromatography purification process. Dalcinonacog alfa is formulated as a sterile, non‐pyrogenic, lyophilized powder preparation intended to be reconstituted for administration by IV infusion or SQ injection. Each eligible participant in the current study received an IV dose of 50 IU/kg to increase the saturation of collagen IV and maintain higher FIX levels until steady‐state levels are reached, followed 35 (±5) min later by SQ dosing of 100 IU/kg. Daily SQ doses of 100 IU/kg were administered until day 28 (28 total SQ doses). Half‐life was calculated from FIX levels on days 29–33 (after administration of the last SQ dose). An end‐of‐study visit occurred 30 (±2) days after the last dose of study drug (Figure 1). Participants were instructed to use their currently prescribed FIX replacement product and regimen for treatment of any spontaneous or traumatic bleed while on study drug, with immediate reporting to the clinical investigative team for further measures, including decisions regarding continuation of daily study drug administration. There were no concomitant medication restrictions.
FIGURE 1.

Study schema
Study drug administration was to be interrupted for a thrombotic event, clinical evidence of inhibitor formation, laboratory results suggesting a high titre antibody may have been developing, or trough activity levels >80% where subsequent dosing was determined in consultation with the Sponsor. If interruption had occurred, study duration may have been extended to include a full dosing schedule. If an urgent need for a surgical procedure or an event required extended hospitalization (>48 h), a FIX activity level was to be urgently obtained and measured to determine the need for any additional treatment and/or whether the event required study drug interruption.
2.2. Patient population
Eligible patients included adult males (age ≥18 years) with a confirmed diagnosis of congenital HB (FIX activity <2%) at a single clinical investigation site in South Africa. Patients with a history or family history of FIX inhibitors, or with positive antibody testing at screening, absolute CD4 count <200 cells/μl, platelet count <100,000, compromised hepatic or renal function, FIX gene mutation 128G>A, history of other clinically relevant coagulation disorders, or advanced atherosclerotic disease, known deep‐vein thrombosis, or high risk of venous thromboembolism were excluded from participation. Treatment in a clinical trial within the previous 30 days or 3 half‐lives (whichever was greater) and current immunomodulatory therapy were also exclusion criteria.
2.3. Assessments
The primary objective was to determine the number of participants who achieved a steady‐state FIX activity level ≥12% with daily dosing. Secondary endpoints included occurrences of clinically significant thrombogenicity marker levels resulting from SQ administration of dalcinonacog alfa and a confirmed antibody response with high titre to dalcinonacog alfa (and whether it was inhibitory and cross‐reactive to BeneFIX), as well as pharmacokinetic variables, immunogenicity and safety. On day 1, FIX activity levels, coagulation assays, thrombogenicity markers and safety assessments were performed at pre‐IV dose and repeated 35 (±5) min later prior to the first SQ dose. Subsequent assessments were performed predose on days 2, 3, 7, 14, 21 and 28 (treatment period) and on days 29–33 (washout period), which included measurement of daily FIX activity levels, unless FIX activity level was known to be <5% as measured by local laboratory.
Dalcinonacog alfa activity measurement variability between platforms and reagents has been published, including data that the chromogenic assay should not be used.15 FIX activity was assayed at Haemtech Biopharma Services (Essex Junction, Vermont, USA), with an activated partial thromboplastin time (aPTT) FIX one‐stage clotting assay on an ACL‐TOP instrument (Instrumentation Laboratory) applying the recommended SynthASil® (Instrumentation Laboratory) reagents and calibrators. Clearance, volume of distribution at steady‐state and half‐life (t1/2) (alpha and beta phase) were determined from day 28 and subsequent samples during washout. Pharmacodynamic (PD) assessments included the results of specific coagulation assays (prothrombin time and aPTT) and thrombogenicity biomarkers: fibrinogen (HemosIL Fibrinogen‐C), D‐dimer (HemosIL D‐Dimer HS), prothrombin fragments 1 + 2 [F1 + 2] (Siemens Enzygnost F1 + 2 kit) and thrombin‐antithrombin [TAT] (Siemens Enzygnost TAT kit) were performed at the central laboratory. Anti‐drug antibodies (ADA) to dalcinonacog alfa and BeneFIX were detected through a direct binding enzyme‐linked immunosorbent assay (ELISA). ADA titres were compared to the assay cut point that was set to 5% false positive for screening, and 1% false positive for confirmation, and then, titres were obtained if confirmed. Neutralizing antibodies (NAb) to dalcinonacog alfa and BeneFIX were sought through the Nijmegen modified Bethesda assay. Safety was assessed on the basis of adverse events (AEs), haematology and chemistry, electrocardiograms, physical examinations, vital signs, occurrence of a confirmed antibody response to dalcinonacog alfa or occurrence of thrombotic events.
2.4. Statistical analysis
As this study was not inferential, no formal sample size calculation was required; however, 6 participants were sufficient to provide guidance on the dose range required to achieve steady‐state activity ≥12% and starting SQ dose for subsequent studies. Continuous and categorical variables were summarized descriptively.
All analyses were based on available data, with no imputation of missing data. The analysis sets included a safety population (all participants who received ≥1 dose of study drug), PK population (any participant who was administered ≥1 week of daily SQ dosing and had FIX activity values analyzed at ≥4 time points at day 28 and after) and PD population (any participant who was administered ≥1 week of daily SQ dosing and who had ≥1 non‐missing PD parameter). In the event that thrombogenicity markers were below the limit of quantification, all analyses were performed with the imputed midpoint of the potential values for this observation. For coagulation assays, the limit of quantification was used for the analysis.
Pharmacokinetic analysis was performed using compartmental and non‐compartmental analysis with Demitasse 2000 (programme created by M Lee), and other statistical outputs were produced using SAS statistical package version 9.4 or higher. A semiparametric model described by Lee et al16 was used to calculate the equilibration (alpha phase) and terminal (beta phase) t1/2. Other parameters were calculated using a standard non‐compartmental approach based on the trapezoidal rule.
All AE were coded using MedDRA Version 20.1. A treatment‐emergent AE (TEAE) was defined as an AE that either began after the first dose of study drug or worsened after the first dose of study drug, but not later than the date of last dose +30 days.
3. RESULTS
3.1. Study patient population
Of 11 patients screened, 4 did not meet the eligibility criteria and 1 withdrew consent before receiving any study medication. A total of 6 participants received study drug (0.72–0.9 ml SQ), comprising the safety population. One of these participants, who received 0.83 ml SQ injections, discontinued the study on day 7 due to injection‐site reactions (ISR) on days 1, 2 and 3, but not days 4, 5 or 6, resulting in 5 participants in the PK and PD populations. The other 5 participants completed treatment and the study.
Demographics for the 6 treated participants are presented in Table 1. The median age was 33 years. Overall, the median number (range) of bleeds reported within the prior 6 months and prior 50 days upon entering the study were 1.5 (0–8) and 0.5 (0–2), respectively.
TABLE 1.
Demographic characteristics of study participants
| Parameter | N = 6 |
|---|---|
| Age (y) | |
| Mean ± standard deviation | 34.5 ± 11.5 |
| Median (minimum, maximum) | 33 (19, 53) |
| Race n (%) | |
| Black or African American | 5 (83.3) |
| White (not Hispanic or Latino) | 1 (16.7) |
| Weight (kg) | |
| Median (minimum, maximum) | 62.8 (57.8, 72.7) |
| Body mass index (kg/m2) | |
| Median (minimum, maximum) | 21.6 (19, 26.7) |
Median (IQR) study drug exposure was 28 days (2.0), ranging from 6 days (n = 1) to 29 days (n = 2), and mean compliance was 99.4%.
3.2. Primary efficacy
All participants achieved FIX activity levels >12% during treatment (Figure 2). FIX activity level >12% was achieved by 3 of 5 participants at day 7; 4 of 5 participants on days 14, 21 and 28; and all 5 participants at day 29 (Figure 2). No bleeding events occurred during the study.
FIGURE 2.

Factor IX activity levels with daily subcutaneous dalcinonacog alfa dosing for the six study participants
3.3. Pharmacokinetic parameters
Pharmacokinetic parameters are summarized in Table 2. Mean alpha and beta t1/2 were 5.3 days and 3.9 days, respectively, and the mean residence time was 6.2 days, indicating a prolonged effect with SQ administration of dalcinonacog alfa.
TABLE 2.
Summary of pharmacokinetic parameters (pharmacokinetic population) n = 5
| Mean (SD) |
CV (%) |
Median (IQR) |
Minimum, Maximum |
GM (CV%) |
|
|---|---|---|---|---|---|
| Maximum concentration (IU/dl) during SQ dosing | 20.7 (5.6) | 26.9 | 22.6 (8.6) | 13.9, 26.8 | 20.1 (29.2) |
| Half‐Life‐1(alpha) (day) | 5.3 (3.0) | 57.3 | 5.3 (4.3) | 3.2, 7.5 | 4.9 (66.8) |
| Half‐Life‐1(beta) (day) | 3.9 (1.1) | 29.3 | 4.2 (1.7) | 2.5, 5.1 | 3.7 (32.1) |
| Mean residence time (day) | 6.2 (1.6) | 26.1 | 6.7 (2.3) | 4.3, 8.1 | 6.0 (27.7) |
| AUC infinity observation (IU/dl × day) | 126.0 (26.8) | 21.3 | 122.2 (10.9) | 99.1, 170.5 | 124.0 (20.2) |
| AUC to last non‐zero concentration (IU/dl × day) | 75.4 (17.2) | 22.9 | 71.9 (26.0) | 58.0, 98.5 | 73.9 (22.9) |
| Volume of distribution at steady‐state observed (dl/kg) | 5.0 (1.5) | 30.8 | 4.1 (2.8) | 3.8, 6.7 | 4.8 (30.7) |
| Clearance (dl/day/kg) | 0.82 (0.15) | 18.7 | 0.82 (0.08) | 0.59, 1.0 | 0.81 (20.17) |
Abbreviations: AUC, area under curve; CV, coefficient of variation; GM, geometric mean; IQR, interquartile range; SD, standard deviation; SQ, subcutaneous.
3.4. Coagulation and thrombogenicity markers
No thrombotic events occurred during the study. Changes in thrombogenicity markers are shown in Figure 3. D‐dimer values showed an increase from baseline throughout the treatment period but decreased as dosing continued. Prothrombin fragment F1 + 2 values were elevated for 2 participants, but all values were within the normal range after day 14 of dosing. Fibrinogen values remained normal throughout the treatment period. TAT values were elevated in 3 participants during the first 14 days and then normalized. TAT elevation at a single time point (day 28) in one participant and an elevation at day 33 in another were believed to be artefactual. There was no correlation at any given time point for abnormal D‐dimer, F1 + 2 or TAT values for any individual participant or the study population, indicating that these were sporadic changes and did not indicate evidence for thrombogenicity. There was no correlation between ISR and thrombogenicity markers.
FIGURE 3.
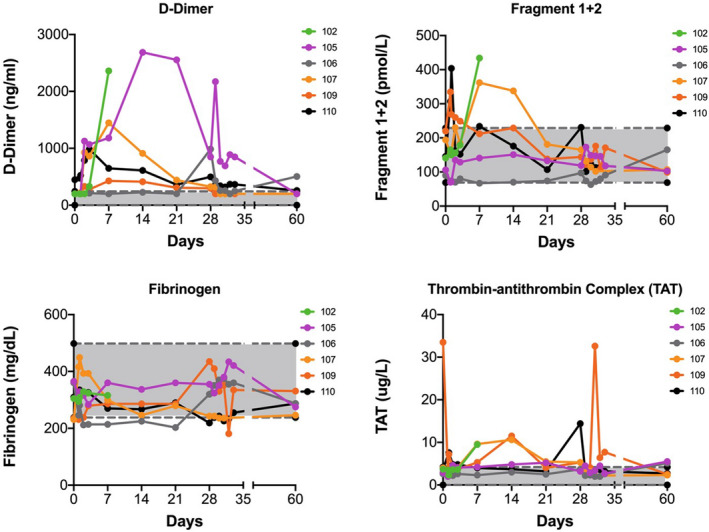
D‐dimer, prothrombin fragments 1 + 2, fibrinogen and thrombin‐antithrombin results with daily subcutaneous dalcinonacog alfa dosing
3.5. Immunogenicity
No NAb to dalcinonacog alfa were detected throughout the study period. Two participants had ADA to dalcinonacog alfa at study end. One participant had titres of 156 and 182 (repeat assay) with negative NAb (Bethesda <0.48). The participant's BeneFIX end‐of‐study ADA titre was 112, increasing from 62 at screening. This participant had negative ADA specimens at intervening time points, reflecting that these titres were close to the assay cut‐off. Another participant had an increase in titre from baseline 235 to 1103 at study end with negative NAb (Bethesda <0.48). The participant's screening BeneFIX ADA titre was 224, increasing to 298 at day 28.
3.6. Safety
Two participants reported TEAE. Both were ISR and considered study drug related and were of moderate severity. One of these participants experienced reactions that included localized bleeding following the first, second and third injections, and the other developed haematomata on the abdomen at the injection site. Outcomes for both TEAE were considered resolved; however, the events in the former participant led to study discontinuation on day 7.
Overall, among the first 3 participants enrolled, ISR of redness, swelling, tenderness or pain occurred. Subsequently, the last 3 participants enrolled split the dose of 100 IU/kg into 2 equal volume adjacent injections: 1 had no ISR, 1 reported pain with the IV and first SQ injection only, and 1 had sporadic reports of pain on day 7 and pain and redness on days 17 and 18.
There were no serious TEAE, deaths or adverse or abnormal findings reported for clinical laboratory, chemistry, vital signs or other safety measurements during the study.
4. DISCUSSION
This open‐label Phase 2b study explored the efficacy, PK, PD and safety of SQ dalcinonacog alfa over a 4‐week treatment period in achieving steady‐state FIX activity levels ≥12% when dosed daily. The premise was that a potent novel FIX variant administered SQ could provide therapeutic levels of FIX without the development of ADA. The primary efficacy endpoint in this study was met. With 28 days of daily dosing of SQ dalcinonacog alfa, all participants achieved steady‐state FIX activity levels >12% and no bleeding events occurred. Washout PK data collectively support a prolonged half‐life and mean residence time with SQ administration. Dalcinonacog alfa was also safe and well tolerated, and no participant developed NAb.
Overall, dalcinonacog alfa appeared to be an effective preventive treatment for the study duration, based on its ability to achieve FIX activity levels ≥12% in all participants. Steady‐state levels of FIX were documented after 2 weeks of dosing, and by day 29, or earlier, the mean FIX activity was 19.4%, exceeding the 12% goal. The mean beta t1/2 of 3.9 days was consistent with the prior Phase 1/2 study of dalcinonacog alfa where SQ beta t1/2 was 66‒103 h.14 On day 1, participant 109 received a higher IV dose than allowed per protocol (150 IU/kg); however, the IV dose would have been eliminated within 6 days and therefore did not affect any later results. No bleeding events or thrombotic events were reported during the treatment or washout period. In addition to no reported blood clotting events, no relevant alterations in blood clotting markers were observed. Sporadic increases in biomarker D‐dimer decreased with continued dosing and were not synchronous with changes in F1 + 2 or TAT.
Approximately 150 SQ injections were administered during this study, with only 2 participants developing moderate ISR, both resolving without sequelae (although 1 participant discontinued on day 7 of the study after ISR from the first 3 doses). The last 3 subjects (with split dosing) had no, or only sporadic, mild ISR. In the Phase 1/2 study, mild pain, erythema and redness were reported at the injection site in initial injections of a greater dose (140 IU/kg) but not at later injections. One participant reported their AE to be moderate for the first 2 injections and mild thereafter.14 No NAb were detected. As NAb typically emerge within 10 exposure days (ED) and the majority within 20 ED, this study reinforces the conclusion of analyses that have collectively found that dalcinonacog alfa is no more immunogenic than wild‐type FIX, but longer periods of observation are desirable. In the Phase 1/2 trial of 2 participants who received dalcinonacog alfa IV followed by dalcinonacog alfa SQ, both developed NAb (transiently in 1 participant) that did not inhibit BeneFIX and safely returned to wild‐type FIX treatment.14 The most likely cause was the human leucocyte antigen (HLA) signature of these 2 participants, who were cousins and shared a rare gene mutation of the propeptide region (1 participant had all 3 HLA types predictive for immunogenicity and the other had 1 HLA type).17
It is important to highlight that any molecular modifications of therapeutic proteins, such as FIX, may create a non‐self‐epitope which can stimulate innate immune responses. Consequently, such alterations could trigger the development of ADA, with or without neutralizing activity, hypersensitivity reactions, or breakdown of immune tolerance to the endogenous protein. NAb may, therefore, be reported following administration of novel therapies. However, antibodies to a therapeutic protein may not have clinical effects as shown by Whelan et al, who found that approximately 30% of haemophilia A patients have antibodies binding to FVIII without any effect on treatment outcome.18 We found that low‐titre ADA to FIX that were not neutralizing were detected during screening, at baseline or during the safety follow‐up period in 2 treated subjects, with approximately 150 ED collectively recorded by participants. Immunogenicity should continue to be monitored in future studies with dalcinonacog alfa.
We acknowledge the small sample size of 6 participants and additional study design limitations, including that this was a single centre study that evaluated only 28 days of dosing. A Phase 2 study, with an increased number of participants, longer duration of dosing and exploration of less frequent dosing regimens, is warranted.
These results support SQ dalcinonacog alfa reaching high levels of FIX and continuous protection because of stable levels and high volume of distribution, as seen with wild‐type‐FIX compared with extended half‐life alternatives. These attributes could provide efficacious prophylaxis for patients with HB.
CONFLICT OF INTEREST
JM has received research grants from Bayer, Biogen, Biomarin, CSL, Novo Nordisk, Sobi, Roche and Unique; has served as a member of the scientific advisory committee of Amgen, Bayer, Biotest, Biogen, Baxalta, CSL Behring, Catalyst Biosciences, Novo Nordisk, Roche and Spark; and has served as a member of the speaker bureau of Alnylam, Bayer, Biotest, Biogen, Novo Nordisk, Pfizer, Sobi, Shire, Roche, ISTH and WFH. HL and FDG are employees and stockholders of Catalyst Biosciences. ML has acted as a paid consultant to Catalyst Biosciences.
AUTHOR CONTRIBUTIONS
JM performed the research, HL designed the research study, FDG conducted the trial operational execution, ML analysed the data, and both JM and HL wrote the paper.
ACKNOWLEDGEMENTS
The study was funded by Catalyst Biosciences. Editorial support was provided by Laurie Orloski, PharmD, of InSeption Group and funded by Catalyst Biosciences, and by Angie Dale, PhD, of Catalyst Biosciences.
Mahlangu J, Levy H, Lee M, Del Greco F. Efficacy and safety of subcutaneous prophylaxis with dalcinonacog alfa in adults with haemophilia B. Haemophilia. 2021;27:574–580. 10.1111/hae.14315
DATA AVAILABILITY STATEMENT
Data available on request from the authors: The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Mannucci PM, Tuddenham EG. The hemophilias−from royal genes to gene therapy. N Engl J Med. 2001;344(23):1773‐1779. [DOI] [PubMed] [Google Scholar]
- 2.Giangrande P. Haemophilia B: Christmas disease. Expert Opin Pharmacother. 2005;6(9):1517‐1524. [DOI] [PubMed] [Google Scholar]
- 3.Fischer K, Van Den Berg M. Prophylaxis for severe haemophilia: clinical and economical issues. Haemophilia. 2003;9(4):376‐381. [DOI] [PubMed] [Google Scholar]
- 4.Ljung RC. Prophylactic infusion regimens in the management of hemophilia. Thromb Haemost. 1999;82(2):525‐530. [PubMed] [Google Scholar]
- 5.Manco‐Johnson MJ, Abshire TC, Shapiro AD, et al. Prophylaxis versus episodic treatment to prevent joint disease in boys with severe hemophilia. N Engl J Med. 2007;357(6):535‐544. [DOI] [PubMed] [Google Scholar]
- 6.Blanchette P, Rivard G, Israels S, et al. A survey of factor prophylaxis in the Canadian haemophilia A population. Haemophilia. 2004;10(6):679‐683. [DOI] [PubMed] [Google Scholar]
- 7.De Moerloose P, Urbancik W, Van Den Berg HM, Richards M. A survey of adherence to haemophilia therapy in six European countries: results and recommendations. Haemophilia. 2008;14(5):931‐938. [DOI] [PubMed] [Google Scholar]
- 8.Hacker MR, Geraghty S, Manco‐Johnson M. Barriers to compliance with prophylaxis therapy in haemophilia. Haemophilia. 2001;7(4):392‐396. [DOI] [PubMed] [Google Scholar]
- 9.Mannucci PM. Hemophilia therapy: the future has begun. Haematologica. 2020;105(3):545‐553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stoner KL, Harder H, Fallowfield LJ, Jenkins VA. Intravenous versus subcutaneous drug administration. Which do patients prefer? A systematic review. Patient. 2015;8(2):145‐153. [DOI] [PubMed] [Google Scholar]
- 11.Nichols TC, Levy H, Merricks EP, Raymer RA, Lee ML. Preclinical evaluation of a next‐generation, subcutaneously administered, coagulation factor IX variant, dalcinonacog alfa. PLoS One. 2020;15(10):e0240896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Madison E, Thanos C, Blouse G, et al. CB‐FIX: an improved second generation FIX drug candidate [abstract]. J Thromb Haemost. 2015;13(Suppl 2):209‐210. [Google Scholar]
- 13.Hong S‐B, Levy H, Jung JY, et al. Pharmacokinetics of subcutaneously administered CB2679d/ISU304 in wild‐type and hemophilia B mice. Blood. 2016;128(22):1389. [Google Scholar]
- 14.You CW, Hong SB, Kim S, et al. Safety, pharmacokinetics, and pharmacodynamics of a next‐generation subcutaneously administered coagulation factor IX variant, dalcinonacog alfa, in previously treated hemophilia B patients. J Thromb Haemost. 2021;19(4):967‐975. [DOI] [PubMed] [Google Scholar]
- 15.Williams SC, Gray E. Activity measurements of dalcinonacog alfa. Haemophilia. 2020;26(2):346‐353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee ML, Wai‐Yin P, Kingdon HS. A two‐phase linear regression model for biologic half‐life data. J Lab Clin Med. 1990;115(6):745‐748. [PubMed] [Google Scholar]
- 17.Blouse GE. A Comprehensive in silico and in vitro immunogenicity risk assessment of dalcinonacog alfa shows no increased risk compared with wild‐type FIX. Presented at the Second Annual 2019 Hemophilia Drug Development (HDD) Summit; August 21, 2019; Boston, MA. [Google Scholar]
- 18.Whelan SF, Hofbauer CJ, Horling FM, et al. Distinct characteristics of antibody responses against factor VIII in healthy individuals and in different cohorts of hemophilia A patients. Blood. 2013;121(6):1039‐1048. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
Data available on request from the authors: The data that support the findings of this study are available from the corresponding author upon reasonable request.