

Summary
Whole metagenomic shotgun (WMS) sequencing has dramatically enhanced our ability to study microbial genomics. The possibility to unveil the genetic makeup of bacteria that cannot be easily isolated has significantly expanded our microbiological horizon. Here, we report an approach aimed at uncovering novel bacterial species by the use of targeted WMS sequencing. Employing in silico data retrieved from metabolic modelling to formulate a chemically defined medium (CDM), we were able to isolate and subsequently sequence the genomes of six putative novel species of bacteria from the gut of non‐human primates.
Introduction
A plethora of microbial species reside in the gastrointestinal tract of animals, and represent a complex microbial consortium also known as the gut microbiota (Lozupone et al., 2012; Milani et al., 2017b). The identified microbial groups in this environment encompass representatives of every domain of life, i.e., Archaea, Bacteria and Eukarya (Lozupone et al., 2012; Milani et al., 2017b). Within these domains, a complex dynamic drives the gut microbiota homeostasis, which may involve viruses, microbe‐microbe and host–microbe interactions (Clemente et al., 2012; Bokulich et al., 2016; Milani et al., 2017b). In recent years, various approaches have been employed to study the gut microbiota composition. Classical microbiology procedures, such as culturomic methods, allow the isolation and identification of microbial taxa that are culturable. PCR and sequence‐based approaches, such as 16S rRNA gene‐based or internal transcribed spacer (ITS) microbial profiling, unveil the detailed composition of the microbial gut consortia, demonstrating the existence of microorganisms that remain uncharacterized because they appear recalcitrant to in vitro cultivation (Ellegaard and Engel, 2016; Milani et al., 2020). Recently, whole metagenome shotgun (WMS) sequencing techniques have revolutionized the study of microbial genomics, allowing the reconstruction of genomes belonging to microbial species that have escaped isolation employing classical culturomic approaches, and such sequences have for this reason been referred to as microbial dark matter (Rinke et al., 2013).
Among microorganisms that reside in the animal gut, members of the genus Bifidobacterium represent an important and extensively studied bacterial component due to the generally accepted important role they play as part of the early gut microbiota during the very first stages of life of mammals (O'Callaghan and van Sinderen, 2016; Turroni et al., 2018; Mancabelli et al., 2020; Turroni et al., 2020). In the last 3 years, a large number of novel species of the genus Bifidobacterium have been described, resulting in a genus that currently constitutes 94 different (sub)species (Duranti et al., 2017; Lugli et al., 2018b; Modesto et al., 2018a; Modesto et al., 2018c; Modesto et al., 2018b; Duranti et al., 2019; Modesto et al., 2019b; Modesto et al., 2019a; Duranti et al., 2020; Modesto et al., 2020b; Modesto et al., 2020a; Neuzil‐Bunesova et al., 2020; Neuzil‐Bunesova et al., 2021). The majority of these most recently discovered novel bifidobacterial species have been isolated from mammals, in particular from faecal samples of primates, such as species of the genus Callimico, Callithrix, Saguinus and Saimiri (Duranti et al., 2017; Lugli et al., 2018b; Modesto et al., 2018a; Modesto et al., 2018c; Modesto et al., 2018b; Duranti et al., 2019; Modesto et al., 2019a; Duranti et al., 2020; Modesto et al., 2020a; Neuzil‐Bunesova et al., 2021). Nonetheless, a number of studies suggest that novel bifidobacterial species resident in the gut of Mammalia are yet to be discovered, thereby representing part of the microbial dark matter of the mammalian gut (Milani et al., 2017a; Alessandri et al., 2020; Lugli et al., 2020b).
Recently, using a WMS approach, the reconstruction of genome sequences belonging to novel bifidobacterial species allowed their isolation based on predicted metabolic properties (Lugli et al., 2019b). The in silico prediction allowed selection of novel species due to specific carbohydrate substrates that specifically support their growth, resulting in isolation and subsequent genome sequencing of these microorganisms (Lugli et al., 2019b). Notwithstanding, the WMS approach's main limitation is the ability to retrieve genetic information of low abundance microorganisms since assembling data requires at least a 5× read coverage. While taxonomic classification of low abundance sequences may still allow compositional analysis (Hillmann et al., 2018), associated microbial genome reconstruction attempts are likely to result in an inconsistent genome assembly (Malmstrom and Eloe‐Fadrosh, 2019). Therefore, gathering sufficient genetic data belonging to low abundant microorganisms through a targeted WMS approach is crucial to obtain an informative genome assembly (Cohrs et al., 2017; Lugli et al., 2017a; Vezzulli et al., 2017; Clark et al., 2018). Accordingly, targeted bifidobacterial DNA amplification from mammalian faecal samples allowed an up to 26,500‐fold enrichment of DNA belonging to this genus (Lugli et al., 2019a). The subsequent assembly of sequenced DNA resulted in the reconstruction of genomes belonging to the Bifidobacterium adolescentis and Bifidobacterium longum species employing probes designed on genome sequences of 62 species of the genus Bifidobacterium (Lugli et al., 2019a).
The current study was aimed at exploring the dark matter of bifidobacterial communities by applying targeted WMS sequencing to reconstruct genomes of putative novel species of the Bifidobacterium genus, followed by in silico prediction of their nutritional requirements by means of metabolic modelling, which in turn facilitated their cultivation and isolation.
Results and discussion
Targeted genome sequencing of bifidobacteria
In order to discover putative novel bifidobacterial species, faecal material of non‐human primates was selected as an appropriate source since it had recently been shown to represent an important reservoir of bifidobacterial diversity (Duranti et al., 2017; Lugli et al., 2018b; Modesto et al., 2018a; Modesto et al., 2018c; Modesto et al., 2018b; Duranti et al., 2019; Modesto et al., 2019a; Duranti et al., 2020; Modesto et al., 2020a; Neuzil‐Bunesova et al., 2021). Consequently, the examined samples were selected based on a previous study investigating the co‐phylogeny of primate‐associated bifidobacteria (Lugli et al., 2020b). Six non‐human primate faecal samples containing a very high abundance of putative novel bifidobacterial species, as identified by means of an ITS bifidobacterial profiling approach, were thus selected (Lugli et al., 2020b) (Table 1). Notably, the high level of putative novel species denotes relative abundances based on the sole identification of bifidobacteria, thus representing a fraction of the overall microbial composition of these samples. In detail, samples were collected from six different monkey species: Callithrix pygmaea (CaPy), Leontopithecus chrysomelas (LeCh), Leontopithecus rosalia (LeRo), Mico argentatus (MiAr), Saguinus imperator (SaIm) and Saguinus oedipus (SaOe). Since our interest was focused on the targeted sequencing of novel bifidobacterial species, a specific protocol was developed as part of this study to enrich DNA belonging to novel members of this genus employing probes previously used to enrich bifidobacterial DNA from mammalian faecal samples (Lugli et al., 2019a) (see Methods) (Fig. S1).
Table 1.
Bifidobacterium species distribution among samples.
| Sample | LeCh | LeRo | MiAr | CaPy | SaOe | SaIm |
|---|---|---|---|---|---|---|
| Scientific name | Leontopithecus chrysomelas | Leontopithecus rosalia | Mico argentatus | Callithrix pygmaea | Saguinus Oedipus | Saguinus imperator |
| Putative bifidobacterial novel species (ITS) | 72% | 65% | 73% | 91% | 74% | 46% |
| Amplified bifidobacterial abundance (targeted WMS) | 100% | 98% | 100% | 98% | 31% | 95% |
| Bifidobacterial relative abundance (targeted WMS) | ||||||
| Bifidobacterium adolescentis | 5.3% | 3.1% | 0.0% | 0.0% | 2.4% | 0.0% |
| Bifidobacterium aerophilum | 2.8% | 4.1% | 2.8% | 0.0% | 0.0% | 3.9% |
| Bifidobacterium aesculapii | 0.7% | 0.0% | 0.7% | 0.0% | 0.0% | 0.0% |
| Bifidobacterium animalis | 0.0% | 0.8% | 0.0% | 0.0% | 0.0% | 0.0% |
| Bifidobacterium avesanii | 0.0% | 0.0% | 0.0% | 0.8% | 0.0% | 0.0% |
| Bifidobacterium biavatii | 7.7% | 4.7% | 2.8% | 0.0% | 0.7% | 4.6% |
| Bifidobacterium callimiconis | 1.9% | 0.0% | 1.1% | 0.0% | 0.0% | 0.6% |
| Bifidobacterium callitrichidarum | 5.2% | 6.9% | 1.8% | 0.0% | 2.7% | 3.8% |
| Bifidobacterium callitrichos | 3.2% | 1.0% | 1.7% | 0.9% | 2.0% | 3.9% |
| Bifidobacterium catenulatum | 0.0% | 0.7% | 0.0% | 0.0% | 0.0% | 0.0% |
| Bifidobacterium felsineum | 0.9% | 0.0% | 0.0% | 1.1% | 0.0% | 0.7% |
| Bifidobacterium goeldii | 1.9% | 5.5% | 0.0% | 2.5% | 0.0% | 0.0% |
| Bifidobacterium imperatoris | 4.0% | 1.6% | 8.6% | 0.0% | 1.6% | 7.4% |
| Bifidobacterium longum | 0.0% | 1.0% | 1.0% | 0.0% | 0.0% | 0.9% |
| Bifidobacterium margollesii | 0.0% | 0.0% | 0.0% | 1.1% | 0.0% | 0.0% |
| Bifidobacterium myosotis | 0.0% | 0.0% | 0.0% | 45.2% | 0.0% | 0.0% |
| Bifidobacterium parmae | 6.7% | 2.9% | 8.3% | 2.2% | 0.7% | 1.8% |
| Bifidobacterium primatium | 0.7% | 0.6% | 0.9% | 0.0% | 0.0% | 0.0% |
| Bifidobacterium pseudocatenulatum | 1.0% | 10.9% | 0.0% | 0.0% | 0.0% | 0.0% |
| Bifidobacterium ramosum | 2.3% | 4.2% | 2.5% | 0.0% | 0.0% | 4.0% |
| Bifidobacterium reuteri | 0.0% | 0.0% | 0.0% | 1.6% | 0.0% | 0.0% |
| Bifidobacterium rousetti | 3.8% | 1.2% | 2.0% | 4.4% | 2.8% | 4.8% |
| Bifidobacterium saguini | 2.9% | 5.0% | 9.9% | 0.0% | 3.1% | 21.1% |
| Bifidobacterium stellenboschense | 7.8% | 5.5% | 13.6% | 1.8% | 0.9% | 3.7% |
| Bifidobacterium unknown species | 40.2% | 37.1% | 39.9% | 18.1% | 13.3% | 29.4% |
| Bifidobacterium vansinderenii | 0.8% | 1.6% | 2.6% | 0.0% | 1.1% | 4.0% |
| Bifidobacterium vespertilionis | 0.0% | 0.0% | 0.0% | 18.1% | 0.0% | 0.0% |
The Bifidobacterium‐targeted WMS approach on the latter samples produced approximately 130 million of paired‐end reads with an average length of 150 bp. Taxonomic classification of sequenced reads, using an enhanced version of the METAnnotatorX pipeline (Milani et al., 2018), revealed that all sequences were predicted to be of bifidobacterial origin in samples LeCh and MiAr (Table 1). Furthermore, 98% of sequenced DNA of samples LeRo and CaPy was shown to belong to bifidobacterial genomes, while targeted sequencing of sample SaIm and SaOe indicated that 95% and 31% of the obtained sequences originated from bifidobacteria, respectively (Table 1). Detailed taxonomical classification of short‐read sequences revealed that, depending on the analysed samples, between 7% and 40% of the deduced reads belonged to putative novel bifidobacterial species (Table 1). These percentages were determined using an updated microbial database encompassing each chromosomal sequence retrieved from the NCBI genome database, including all 94 bifidobacterial subspecies identified to date, supporting the notion that DNA of putative novel bifidobacterial species was not lost in the bifidobacterial DNA enrichment process.
Genome reconstruction of members of the genus Bifidobacterium
Genomic data sets retrieved from targeted WMS sequencing were then assembled to rebuild the bifidobacterial DNA genomic structure (Milani et al., 2018). Assembled contigs longer than 5000 nucleotides were taxonomically classified, distinguishing the genetic material of known bacterial species from that of unknown/putative novel bacterial species. The updated microbial database mentioned above was also employed in the assembled contigs' taxonomic classification to cover the bifidobacterial biodiversity available to date, encompassing 94 subspecies of the genus. Using this procedure, a genetic amount of 160 Kb to 5.4 Mb belonging to putative novel bifidobacterial species was reconstructed from the enriched sequence reads of the assessed samples, resulting in assembled contigs with a total length of 11.6 Mb, which was predicted to represented genomic fragments of as yet unclassified bifidobacteria (Fig. 1). Samples LeCh, MiAr and LeRo were shown to correspond to enriched sequences with a percentage of putative novel bifidobacterial DNA above 30%. The most abundant species reconstructed in the process were bifidobacterial gut commensals of primates, such as Bifidobacterium saguini, Bifidobacterium pseudocatenulatum, Bifidobacterium vansinderenii, Bifidobacterium adolescentis and Bifidobacterium myosotis (Lugli et al., 2020b) (Fig. 1).
Fig. 1.
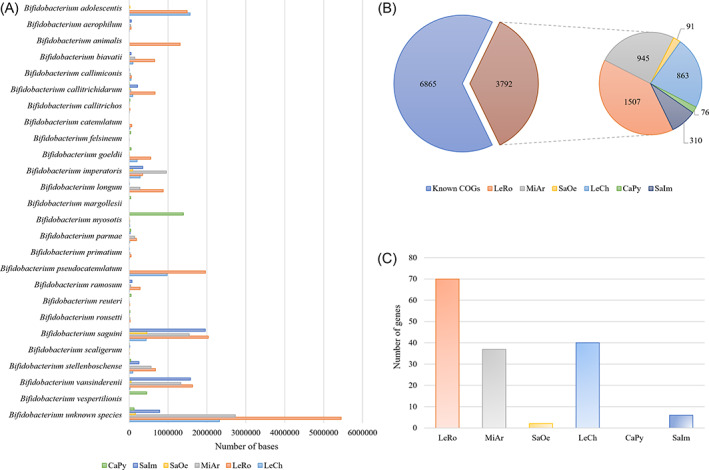
Identification of unique genomic signatures of putative novel strains belonging to the genus Bifidobacterium. Panel A displays the relative abundance of the reconstructed bifidobacterial genomic material at species level obtained from CaPy, SaIm, SaOc, MiAr, LeRo and LeCh samples. The x‐axis shows the number of base pairs (bp) assigned to each species. Only species that display at least 30 kb of the total amount of the assembled data were included in the histogram. Panel B exhibits the distribution of unique COGs identified in putative novel bifidobacterial species. Panel C shows the number of unique genes of putative novel bifidobacterial species correlated with the degradation of carbon sources in each sample. [Color figure can be viewed at wileyonlinelibrary.com]
Unique features of unknown bifidobacterial species
Reconstructed genome sequences belonging to members of the genus Bifidobacterium were investigated to discern apparently unique genetic signatures that do not belong to known species. Predicted genes were assigned to two groups for each sample: genes belonging to known species were assigned to one group, while genes associated with putative novel bifidobacterial species were assigned to the other group. Following this, a pangenome analysis was undertaken to determine putative orthologous genes between these two bifidobacterial gene groups. This analysis resulted in the identification of clusters of orthologous genes (COGs) in each sample, thereby allowing the clustering of genes that are shared between groups and genes present in a single group. Specifically, a total of 3792 COGs were collected among analysed samples, representing genes identified only in novel bifidobacterial species (Fig. 1). Sample LeRo, followed by samples MiAr and LeCh possessed the highest number of COGs related to putative novel bifidobacterial taxa, 1509, 945 and 863, respectively (Fig. 1).
Metabolic modelling of the bifidobacterial dark matter
Those unique genes, belonging to putative novel bifidobacterial species, were used to access the metabolic signature of the bifidobacterial dark matter. Combining a screening of 15 databases of protein domains, motifs and folds in deduced protein structures, we identified 155 genes, whose protein products were correlated with the metabolism of various carbon sources (Fig. 1 and Table S1). The most informative results revealed that putative novel bifidobacteria carried genes encoding proteins with predicted activities such as α‐l‐arabinofuranosidase, α‐mannosidase, α‐rhamnosidase, endo‐α‐N‐acetylgalactosaminidase, β‐galactosidase, β‐mannosidase, pullulanase and additional glycosyl hydrolases (GHs) especially belonging to families GH31 and GH43 (Table S1). Notably, such an enzymatic repertoire is predicted to catalyse the hydrolysis of arabinans, (arabino)xylans and glycans containing N‐acetyl galactosamine, galactose, mannose and/or rhamnose, such as N‐ and O‐glycans. Thus, this prediction guided us to select specific substrates to support growth of bifidobacteria corresponding to genetic dark matter identified in these samples. In this context, d‐galactose, d‐mannose, l‐rhamnose, d‐xylose and pullulan were selected as suitable substrates for the isolation of putative novel bifidobacterial species.
Targeted culturomics of bifidobacteria
To retrieve bifidobacterial strains associated with the primate gut, several cultivation attempts were performed. For this purpose, aliquots of faecal samples from CaPy, LeCh, LeRo, MiAr, SaIm and SaO were added to a chemically defined medium (CDM), containing particular glycans that were selected based on the predicted metabolic modelling reconstruction of the shotgun metagenomics data (see above). These cultivation experiments using specific carbohydrates allowed growth of 27 phenotypically distinct bacterial isolates. These isolates were genotypically characterized by the amplification and sequencing of their ITS sequences (Milani et al., 2014), which were then compared to a previously described ITS bifidobacterial database (Lugli et al., 2020a) updated with all bifidobacterial species classified to date in order to identify strains that do not belong to previously characterized bifidobacterial species, i.e., showing an ITS sequence identity lower than 99% (Milani et al., 2014; Lugli et al., 2019b). This approach resulted in the identification of 10 bifidobacterial isolates that appear to belong to novel species (Table 2). Additional growth experiments were performed on selective enriched media to highlight the growth ability of such isolated strains (see Supporting Information and Fig. S2).
Table 2.
Bifidobacterial strain selection.
| Strain | Host | ITS sequence identity | Predicted species (ITS)a |
|---|---|---|---|
| CP1 | Callithrix pygmaea | 98% | Bifidobacterium callimiconis LMG 30938 |
| CP2 | Callithrix pygmaea | 81% | Bifidobacterium catulorum DSM 103154 |
| CP3 | Callithrix pygmaea | 80% | Bifidobacterium dentium DSM 20436 |
| CP4 | Callithrix pygmaea | 90% | Bifidobacterium imperatoris LMG 30297 |
| LC1 | Leontopithecus chrysomelas | 100% | Bifidobacterium imperatoris LMG 30297 |
| LC2 | Leontopithecus chrysomelas | 99% | Bifidobacterium parmae LMG 30295 |
| LC3 | Leontopithecus chrysomelas | 100% | Bifidobacterium pseudocatenulatum DSM 20439 |
| LC4 | Leontopithecus chrysomelas | 100% | Bifidobacterium pseudocatenulatum DSM 20439 |
| LC5 | Leontopithecus chrysomelas | 100% | Bifidobacterium pseudocatenulatum DSM 20439 |
| LC6 | Leontopithecus chrysomelas | 94% | Bifidobacterium saguini DSM 23967 |
| LR1 | Leontopithecus rosalia | 99% | Bifidobacterium longum NCC 2705 |
| LR2 | Leontopithecus rosalia | 99% | Bifidobacterium parmae LMG 30295 |
| LR3 | Leontopithecus rosalia | 99% | Bifidobacterium pseudocatenulatum DSM 20439 |
| LR4 | Leontopithecus rosalia | 99% | Bifidobacterium pseudocatenulatum DSM 20439 |
| LR5 | Leontopithecus rosalia | 99% | Bifidobacterium vansinderenii LMG 30126 |
| MA1 | Mico argentatus | 85% | Bifidobacterium biavatii DSM 23969 |
| MA2 | Mico argentatus | 82% | Bifidobacterium callitrichos DSM 23973 |
| MA3 | Mico argentatus | 100% | Bifidobacterium dentium DSM 20436 |
| MA4 | Mico argentatus | 100% | Bifidobacterium parmae LMG 30295 |
| SI1 | Saguinus imperator | 99% | Bifidobacterium saguini DSM 23967 |
| SI2 | Saguinus imperator | 99% | Bifidobacterium vansinderenii LMG 30126 |
| SO1 | Saguinus Oedipus | 93% | Bifidobacterium adolescentis ATCC 15703 |
| SO2 | Saguinus Oedipus | 72% | Bifidobacterium callitrichos DSM 23973 |
| SO3 | Saguinus Oedipus | 100% | Bifidobacterium felsineum DSM 103139 |
| SO4 | Saguinus Oedipus | 85% | Bifidobacterium imperatoris LMG 30297 |
| SO5 | Saguinus Oedipus | 100% | Bifidobacterium lemurum DSM 28807 |
| SO6 | Saguinus Oedipus | 100% | Bifidobacterium longum DSM 20088 |
based on the database hit. Reliability may vary on the base of the ITS sequence identity.
Genome sequencing and comparative analyses
Putative novel bifidobacterial strains were subjected to whole genome shotgun sequencing revealing genome sizes ranging from 2.7 to 3.4 Mb obtained as a result of a genome coverage that ranged from 53‐ to 137‐fold (Table 3). The average nucleotide identity (ANI) analysis of the decoded bifidobacterial genomes with all known bifidobacterial (sub)species revealed that six strains displayed ANI values below 94% (Table 3). In contrast, strains CP1, CP3, CP4 and SO2 belonged to Bifidobacterium callimiconis, Bifidobacterium vespertilionis, Bifidobacterium felsineum and Bifidobacterium simiarum species, respectively. Thus, based on the notion that bacterial strains displaying an ANI value < 95% are considered to belong to distinct species, isolates CP2, LC6, MA1, MA2, SO1 and SO4 are assigned as novel species of the genus Bifidobacterium (Richter and Rossello‐Mora, 2009; Lugli et al., 2014; Lugli et al., 2017b; Lugli et al., 2018a).
Table 3.
General genetic features.
| Biological origin | Average Coverage | Number of assembled contigs | Genome length | Average GC percentage | Number of predicted ORFs | tRNA | rRNAa | ANI value | |
|---|---|---|---|---|---|---|---|---|---|
| CP1 | Callithrix pygmaea | 111 | 16 | 2,896,801 | 62.36 | 2188 | 58 | 4 | 97.9% Bifidobacterium callimiconis LMG 30938T |
| CP2 | Callithrix pygmaea | 59 | 26 | 2,790,418 | 65.91 | 2115 | 57 | 2 | 87.4% Bifidobacterium platyrrhinorum DSM 106029T |
| CP3 | Callithrix pygmaea | 57 | 51 | 3,034,123 | 64.22 | 2328 | 57 | 3 | 97.9% Bifidobacterium vespertilionis DSM 106025T |
| CP4 | Callithrix pygmaea | 61 | 36 | 2,679,727 | 57.53 | 2196 | 54 | 2 | 98.6% Bifidobacterium felsineum DSM 103139T |
| LC6 | Leontopithecus chrysomelas | 105 | 19 | 2,697,321 | 57.97 | 2096 | 56 | 3 | 86.7% Bifidobacterium imperatoris LMG 30297T |
| MA1 | Mico argentatus | 92 | 133 | 3,348,570 | 64.36 | 2628 | 68 | 3 | 90.8% Bifidobacterium biavatii DSM 23969T |
| MA2 | Mico argentatus | 53 | 25 | 2,771,886 | 65.97 | 2210 | 63 | 3 | 89.1% Bifidobacterium rousetti DSM 106027T |
| SO1 | Saguinus Oedipus | 132 | 52 | 3,401,546 | 62.36 | 2807 | 97 | 3 | 89.1% Bifidobacterium aerophilum DSM 100689T |
| SO2 | Saguinus Oedipus | 137 | 37 | 2,799,858 | 63.68 | 2172 | 57 | 3 | 98.1% Bifidobacterium simiarum DSM 103153T |
| SO4 | Saguinus Oedipus | 94 | 37 | 2,912,164 | 62.98 | 2265 | 58 | 4 | 93.9% Bifidobacterium callitrichidarum DSM 103152T |
Predicted number of rRNA loci.
Shotgun reconstructions of these six novel bifidobacterial species allowed us to compare the obtained genome sequences with those predicted to belong to unknown species in the original metagenomic data sets. Sequence alignments involving contigs longer than 1000 nucleotides retrieved from targeted WMS sequencing and the six individual bacterial genome sequences showed a conspicuous portion of dark matter with identity values above 99.95% (Fig. S3). Specifically, 69.2% and 50.9% of the chromosome length of candidate LC6 and MA2 was previously assembled by the targeted WMS sequencing (Fig. S3). Following, 31.2% and 19.7% of MA1 and CP4 was reconstructed in the corresponding metagenomic data set. Besides, the genome sequences of the remaining two candidate novel bifidobacteria, represented by SO1 and SO4, were retrieved at low abundance in their corresponding metagenomic data sets, revealing that their genomic repertoire was insignificant when formulating the CDM. Furthermore, a comparison between the reconstructed genes of the six novel bifidobacterial species in respect to the genomic DNA sequences used to design the probes allowed to estimate an average of 10.6% of unknown genes retrieved through targeted genome sequencing (Table S2).
Phylogenomic inference of novel isolated strains
A phylogenomic investigation involving a genome‐wide approach was then performed employing the collected data of the 10 isolated bifidobacterial strains. A core‐genome‐based phylogenomic tree was build using the predicted proteome of 94 bifidobacterial type strains combined with that of the 10 newly isolated strains. Predicted 41,591 clusters of orthologous genes identified by comparative genomics analysis of the 104 bifidobacterial strains allowed the identification of a core genome of 162 shared COGs. Paralogues identified in COGs were discarded, resulting in 130 core protein‐encoding sequences, homologues of which are present in a single copy in each genome. Concatenation of these protein sequences was used to build a Bifidobacterium phylogenomic tree, unveiling the novel species position within the Bifidobacterium genus phylogeny (Fig. 2). Strains CP2, LC6, MA2 and SO4 clustered in the B. longum phylogenetic group (Lugli et al., 2014; Lugli et al., 2017b; Lugli et al., 2018a), while strains MA1 and SO1 grouped together with members of the Bifidobacterium bifidum group (Lugli et al., 2014; Lugli et al., 2017b; Lugli et al., 2018a). Altogether, the phylogenomic inference revealed that each of the sequenced bifidobacterial strain shares a core tree branch with the predicted closest species identified in the ANI analysis (Table 1). The morphology of the six novel isolated strains has been reported in Fig. S4.
Fig. 2.
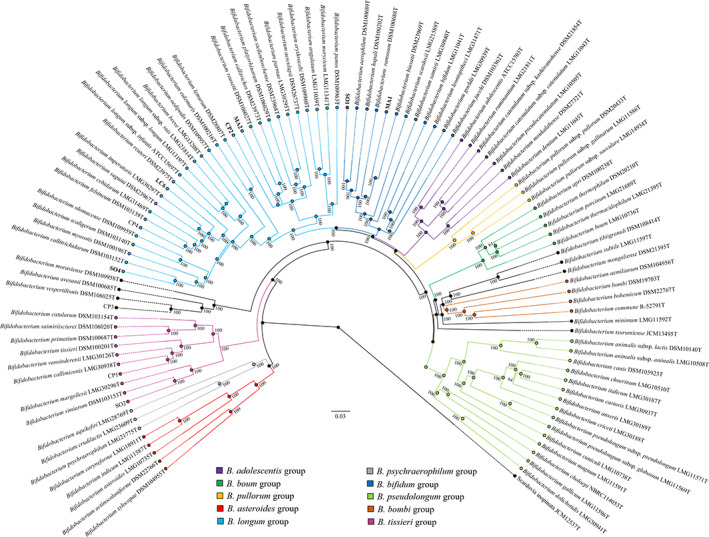
Phylogenomic tree of the genus Bifidobacterium based on the concatenation of 130 core protein sequences from genomes of 10 novel strains isolated in this study and the 94 type strains of the genus Bifidobacterium. Different colours show the division into 10 phylogenetic groups, of which B. longum and B. bifidum groups are populated by the novel strains (highlighted in bold). The phylogenetic tree was constructed by the Neighbour‐joining method, with the genome sequence of Scardovia inopinata JCM 12537 as outgroup. Bootstrap percentages above 50 are shown at node points, based on 1000 replicates of the phylogenetic tree. [Color figure can be viewed at wileyonlinelibrary.com]
Conclusions
To discover and isolate novel bifidobacterial species, we examined six faecal samples of non‐human primates that were suspected to harbour novel species of the genus (Lugli et al., 2020b). Using a targeted WMS approach, we were able to reconstruct a large portion of such unknown bifidobacterial species, thereby obtaining insights into their metabolic abilities. Accordingly, the genetic makeup identified prior to cultivation attempts guided us in selecting suitable growth substrates for a culturomics approach, which successfully retrieved six novel species of the genus, increasing our knowledge of bifidobacterial biodiversity and reducing our ignorance of metagenomic dark matter. The proposed approach should in principle be applicable to many bacterial genera of which genome sequences of known species have previously been decoded.
Experimental procedures
Design of myBaits® probe for targeted WMS sequencing
Probes used in this study were built on the basis of the chromosomal DNA sequence of 62 bifidobacterial type strains. Genome sequences of the selected strains were sent to Arbor Biosciences (Ann Arbor, MI, USA), where the probes were designed to provide a custom myBaits® kit for DNA enrichment of members of the genus Bifidobacterium (myBaits® WGE Custom Cat. No. 302416). In this context, biotinylated RNA baits were produced randomly by using the chromosomal DNA sequence provided. This approach allowed to cover the complete genome sequences of the 62 bifidobacterial type strains (Fig. S1). Thus, more than 20,000 baits were manufactured representing portions of the chromosomal DNA, including both coding and non‐coding sequences. Following this process, 16 reactions were provided, allowing bulk enrichment of genome‐wide endogenous DNA from complex metagenomic samples such as environmental DNA. Accordingly, DNA from faecal samples of primates was used in the targeted WMS approach combining one of the 16 reagent kit.
Microbial DNA extraction
Microbial DNA was extracted using the QIAmp DNA Stool Mini Kit following the manufacturer's instructions (Qiagen, Hilden, Germany) from faecal samples of primates collected from previous studies (Milani et al., 2017a; Lugli et al., 2020b). DNA concentration and purity of each sample was then investigated employing a Picodrop microtiter Spectrophotometer (Picodrop, Hinxton, UK).
Targeted DNA sequencing of novel bifidobacterial species
Capture of bifidobacterial DNA was performed in solution using the custom MyBaits® kit according to the manufacturer's protocol (Hybridization Capture for Targeted NGS Manual Version 4.01). To enrich the DNA of unknown bifidobacterial species, a temperature of 63 °C was used for DNA hybridization and over 40 h of actual hybridization. According to the manufacturer's instructions, DNA library preparation was performed using the Nextera XT DNA sample preparation kit (Illumina, San Diego, CA, USA). One ng input DNA from each sample was used for library preparation. The isolated DNA underwent fragmentation, adapter ligation and amplification. Illumina libraries were pooled equimolarly, denatured and diluted to a concentration of 1.5 pM. Sequencing was performed on a NextSeq 550 instrument (Illumina) using a 2× 150 bp High Output sequencing kit and a deliberate spiking of 1% PhiX control library.
Taxonomic classification of the reads and WMS assembly
To analyse high‐quality sequenced data only, each dataset was subjected to a filtering step removing low quality reads (minimum mean quality score 20, window size 5, quality threshold 25 and minimum length 100) using the fastq‐mcf script (https://github.com/ExpressionAnalysis/ea-utils/blob/wiki/FastqMcf.md). Filtered reads were then collected and taxonomically classified through the METAnnotatorX pipeline (Milani et al., 2018), using the up to date genome RefSeq database retrieved from NCBI. Filtered reads were then subjected to whole metagenome assembly using Spades v3.14 (Antipov et al., 2016) with default parameters and the metagenomic flag option (−meta) together with k‐mer sizes of 21, 33, 55 and 77. As mentioned above for the short reads, reconstructed contig sequences were taxonomically classified based on their sequence identity using megablast (Chen et al., 2015). In all, the METAnnotatorX pipeline was employed for various purposes, from read filtering to taxonomic classification of the assembled contigs (Milani et al., 2018).
Comparative genomics
Pangenome calculations were performed using the pan‐genome analysis pipeline PGAP (Zhao et al., 2012). Predicted proteomes were screened for orthologues between groups using BLAST analysis (cutoff, E‐value of < 1 × 10%–5% and 50% identity across at least 80% of either protein sequence) (Altschul et al., 1990). The resulting output was clustered into protein families through MCL (graph theory‐based Markov clustering algorithm) using the gene family method. Using this approach, unique protein families encoded by unknown bifidobacterial species were identified. The presence of functional domains of unique genes predicted to belong to unknown bifidobacterial species was performed using InterProScan (Jones et al., 2014). Queried databases were CDD, Pfam, TIGRFAM, Gene3D, PANTHER, SUPERFAMILY, PRINTS, ProSitePatterns, PIRSF, Hamap, Coils, SMART, ProSiteProfiles, SFLD and MobiDBLite.
Bifidobacterial isolation
One gram of a faecal sample was mixed with 9 ml of phosphate‐buffered saline (PBS), pH 6.5. Serial dilutions and subsequent platings were performed using a combination of five carbon sources. The employed growth medium consisted of a chemically defined medium (CDM) with the addition of 50 μg/ml mupirocin (Delchimica, Italy), 0.05% (wt/vol) l‐cysteine hydrochloride and 1% (wt/vol) of each of the five carbohydrates, i.e., d‐galactose, d‐mannose, l‐rhamnose, d‐xylose and pullulan. CDM contains (per litre of distilled water) 4.0 g of sodium acetate; 1.0 g of tri‐ammonium citrate; 2.0 g of KH2PO4; 2.0 g of K2HPO4; 0.5 g of MgSO4.7H2O; 0.05 g of MnSO4.H2O; 0.02 g of FeSO4.7H2O; 0.2 g of CaCl2; 20 mg of adenine; 40 mg of xanthine; 0.4 g of cysteine; 0.3 g of aspartic acid; 0.3 g of glutamic acid; 0.2 g of each the following amino acids: alanine, arginine, glycine, histidine, isoleucine, leucine, lysine, methionine, phenylalanine, proline, serine, threonine, tryptophan, tyrosine and valine; 0.5 g of orotic acid; 0.5 mg of p‐aminobenzoic acid; 0.5 mg of folic acid, 2.0 mg of nicotinic acid; 2.0 mg of Ca‐pantothenate; 1.0 mg of biotin; 2.0 mg of pyridoxal; 2.0 mg of riboflavin; and 1.0 mg of vitamin B12. The medium was sterilized by filtration (0.22 μm). Agar plates were incubated for 48 h at 37°C in a chamber (Concept 400; Ruskin) with an anaerobic atmosphere (2.99% H2, 17.01% CO2 and 80% N2). Morphologically distinct colonies that developed on CDM plates were randomly picked and re‐streaked to isolate purified bacterial strains.
Sequencing of bifidobacterial ITS
Selected isolates were subjected to DNA isolation and characterized using an ITS sequencing approach. Cells from 10 ml of an overnight culture were harvested by centrifugation at 6000 rpm for 8 min. Obtained cell pellets were used for DNA extraction using the GenElute™ Bacterial Genomic DNA kit (Sigma‐Aldrich, Darmstadt, Germany) following the manufacturer's guidelines. Internal transcribed spacer (ITS) sequences were amplified from extracted DNA using primer pair Bif23S_ITS (5′‐AGATGTTTCACTTCCCTGCG‐3′) and Bif16S_ITS (5′‐CCTTGTACACACCGCCCG‐3′). Nucleotide sequencing of the ITS region was performed by Eurofins Mix2Seq Kit service (Eurofins Genomics, Germany) using 16S_bif‐SEQ1 (5′‐CGTCAAGTCATGAAAGTGGG‐3′). Finally, ITS sequences were compared to a publicly available database composed of an exhaustive collection of bifidobacterial ITS sequences (http://probiogenomics.unipr.it/pbi/) using the BLAST tool (Altschul et al., 1990).
Bifidobacterial genome sequencing
The genome sequences of Bifidobacterium strains were determined by GenProbio srl (Parma, Italy) using a MiSeq platform (Illumina, UK). A genome library was generated using the TruSeq Nano DNA kit following a specified protocol (part no. 15041110 rev. D). The generated library samples were then loaded into a 600‐cycle flow cell version 3 (Illumina). Paired fastq files of shotgun genomics were used as input for SPAdes assembler v3.14 (Antipov et al., 2016). De novo genomic assemblies were performed using default parameters enabling the flag option –isolates coupled with a list of k‐mer sizes of 21, 33, 55, 77, 99 and 127. ORFs of each assembled genome were predicted with Prodigal (Hyatt et al., 2010) and annotated utilizing the MEGAnnotator pipeline (Lugli et al., 2016).
Phylogenomic analysis of novel Bifidobacterium species
A pangenome calculation was performed including 94 Bifidobacterium type strains genomes as well as the genomes of the novel strains identified in this study (Lugli et al., 2018a). Using this approach, unique protein families encoded by the analysed Bifidobacterium genomes were identified, allowing the prediction of the core genome of the Bifidobacterium genus. The concatenated core genome sequences of the Bifidobacterium genus were then aligned using MAFFT (Multiple Alignment using Fast Fourier Transform) (Katoh and Standley, 2013), and the corresponding phylogenomic tree was constructed using the neighbour‐joining method in Clustal W version 2.1 (Larkin et al., 2007). The core genome tree was built using FigTree (http://tree.bio.ed.ac.uk/software/figtree/). For each genome pair, a value for the average nucleotide identity (ANI) was calculated using FastANI (Jain et al., 2018). Previous Bifidobacterium‐based phylogenomic studies identified an ANI threshold of about 94% to discriminate between species (Lugli et al., 2014; Lugli et al., 2017b; Lugli et al., 2018a).
Phenotypic characterization
The morphology of novel bifidobacterial taxa was determined using phase‐contrast microscopy after incubating each strain under anaerobic conditions at 37°C for 24 h.
Conflict of interest
The authors declare that they have no competing interests.
Author contributions
G.A.L. performed bioinformatics and statistical analyses and wrote the manuscript; G.A. performed the in vitro analyses and wrote the manuscript; C.M. validate the bioinformatics analyses and edited the manuscript; A.V. and C.A. performed the ITS profiling and shotgun metagenomics sequencing; F.F., C.T. and L.M. validate the bioinformatics analyses; A.M., L.R., D.V.S and F.T. supervised the project and edited the manuscript; M.V. supervised the project and designed the study.
Supporting information
Fig. S1. Schematic representation of the targeted bifidobacterial WMS approach.
Fig. S2. Carbohydrate growth assays of the isolated bifidobacterial stains. The heat map illustrates the average optical densities (OD600) of three independent replicates for each isolated strain at two different time points, 24 and 48 h.
Fig. S3. Alignment of genome sequences of bifidobacterial isolates against contigs longer than 1000 nucleotides from targeted WMS sequencing. Panel a to panel l report the sequence alignment of strain LC6, MA2, MA1, SO4, SO2, SO1, CP3, CP2, CP1 and CP4.
Fig. S4. CP2, LC6, MA1, MA2, SO1 and SO4 cellular morphologies as determined by the use of phase‐contrast microscopy. Bar, 10 μm.
Table S1. Metabolic profile of the bifidobacterial dark matter.
Table S2. Unknown genes retrieved between reconstructed genomes.
Table S3. Metabolic profile of the isolated bifidobacterial strains.
Acknowledgements
We thank GenProbio srl for the financial support of the Laboratory of Probiogenomics. Part of this research is conducted using the High Performance Computing (HPC) facility of the University of Parma. D.v.S. is a member of The APC Microbiome Institute funded by Science Foundation Ireland (SFI), through the Irish Government's National Development Plan (Grant numbers SFI/12/RC/2273a and SFI/12/RC/2273b). This work was financially supported by a PostDoc fellowship (Bando Ricerca Finalizzata) to G.A. F.T. is funded by Italian Ministry of Health through the Bando Ricerca Finalizzata (Grant Number GR‐2018‐12365988).
Data availability
Raw sequences of shotgun metagenomics experiments are accessible through SRA study BioProject PRJNA698773. Ten genome sequences have also been deposited in the GenBank database under the BioProject PRJNA698773.
References
- Alessandri, G., Milani, C., Mancabelli, L., Longhi, G., Anzalone, R., Lugli, G.A., et al. (2020) Deciphering the bifidobacterial populations within the canine and feline gut microbiota. Appl Environ Microbiol 86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altschul, S.F., Gish, W., Miller, W., Myers, E.W., and Lipman, D.J. (1990) Basic local alignment search tool. J Mol Biol 215: 403–410. [DOI] [PubMed] [Google Scholar]
- Antipov, D., Korobeynikov, A., McLean, J.S., and Pevzner, P.A. (2016) hybridSPAdes: an algorithm for hybrid assembly of short and long reads. Bioinformatics 32: 1009–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokulich, N.A., Chung, J., Battaglia, T., Henderson, N., Jay, M., Li, H., et al. (2016) Antibiotics, birth mode, and diet shape microbiome maturation during early life. Sci Transl Med 8: 343ra382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen, Y., Ye, W., Zhang, Y., and Xu, Y. (2015) High speed BLASTN: an accelerated MegaBLAST search tool. Nucleic Acids Res 43: 7762–7768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark, S.A., Doyle, R., Lucidarme, J., Borrow, R., and Breuer, J. (2018) Targeted DNA enrichment and whole genome sequencing of Neisseria meningitidis directly from clinical specimens. Int J Med Microbiol 308: 256–262. [DOI] [PubMed] [Google Scholar]
- Clemente, J.C., Ursell, L.K., Parfrey, L.W., and Knight, R. (2012) The impact of the gut microbiota on human health: an integrative view. Cell 148: 1258–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohrs, R.J., Lee, K.S., Beach, A., Sanford, B., Baird, N.L., Como, C., et al. (2017) Targeted genome sequencing reveals varicella‐zoster virus open Reading frame 12 deletion. J Virol 91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duranti, S., Mangifesta, M., Lugli, G.A., Turroni, F., Anzalone, R., Milani, C., et al. (2017) Bifidobacterium vansinderenii sp. nov., isolated from faeces of emperor tamarin (Saguinus imperator). Int J Syst Evol Microbiol 67: 3987–3995. [DOI] [PubMed] [Google Scholar]
- Duranti, S., Lugli, G.A., Napoli, S., Anzalone, R., Milani, C., Mancabelli, L., et al. (2019) Characterization of the phylogenetic diversity of five novel species belonging to the genus Bifidobacterium: Bifidobacterium castoris sp. nov., Bifidobacterium callimiconis sp. nov., Bifidobacterium goeldii sp. nov., Bifidobacterium samirii sp. nov. and Bifidobacterium dolichotidis sp. nov. Int J Syst Evol Microbiol 69: 1288–1298. [DOI] [PubMed] [Google Scholar]
- Duranti, S., Lugli, G.A., Viappiani, A., Mancabelli, L., Alessandri, G., Anzalone, R., et al. (2020) Characterization of the phylogenetic diversity of two novel species belonging to the genus Bifidobacterium: Bifidobacterium cebidarum sp. nov. and Bifidobacterium leontopitheci sp. nov. Int J Syst Evol Microbiol 70: 2288–2297. [DOI] [PubMed] [Google Scholar]
- Ellegaard, K.M., and Engel, P. (2016) Beyond 16S rRNA community profiling: intra‐species diversity in the gut microbiota. Front Microbiol 7: 1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hillmann, B., Al‐Ghalith, G.A., Shields‐Cutler, R.R., Zhu, Q., Gohl, D.M., Beckman, K.B., et al. (2018) Evaluating the information content of shallow shotgun metagenomics. mSystems 3: e00069‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hyatt, D., Chen, G.L., Locascio, P.F., Land, M.L., Larimer, F.W., and Hauser, L.J. (2010) Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11: 119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jain, C., Rodriguez, R.L., Phillippy, A.M., Konstantinidis, K.T., and Aluru, S. (2018) High throughput ANI analysis of 90K prokaryotic genomes reveals clear species boundaries. Nat Commun 9: 5114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones, P., Binns, D., Chang, H.Y., Fraser, M., Li, W., McAnulla, C., et al. (2014) InterProScan 5: genome‐scale protein function classification. Bioinformatics 30: 1236–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh, K., and Standley, D.M. (2013) MAFFT multiple sequence alignment software version 7: improvements in performance and usability. Mol Biol Evol 30: 772–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Larkin, M.A., Blackshields, G., Brown, N.P., Chenna, R., McGettigan, P.A., McWilliam, H., et al. (2007) Clustal W and Clustal X version 2.0. Bioinformatics 23: 2947–2948. [DOI] [PubMed] [Google Scholar]
- Lozupone, C.A., Stombaugh, J.I., Gordon, J.I., Jansson, J.K., and Knight, R. (2012) Diversity, stability and resilience of the human gut microbiota. Nature 489: 220–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A., Milani, C., Mancabelli, L., van Sinderen, D., and Ventura, M. (2016) MEGAnnotator: a user‐friendly pipeline for microbial genomes assembly and annotation. FEMS Microbiol Lett 363: fnw049. [DOI] [PubMed] [Google Scholar]
- Lugli, G.A., Duranti, S., Milani, C., Mancabelli, L., Turroni, F., Sinderen, D.V., and Ventura, M. (2019a) Uncovering Bifidobacteria via targeted sequencing of the mammalian gut microbiota. Microorganisms 7: 535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A., Milani, C., Mancabelli, L., Turroni, F., Ferrario, C., Duranti, S., et al. (2017a) Ancient bacteria of the Otzi's microbiome: a genomic tale from the copper age. Microbiome 5: 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A., Milani, C., Duranti, S., Mancabelli, L., Mangifesta, M., Turroni, F., et al. (2018a) Tracking the taxonomy of the genus bifidobacterium based on a phylogenomic approach. Appl Environ Microbiol 84: e02249‐17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A., Milani, C., Duranti, S., Alessandri, G., Turroni, F., Mancabelli, L., et al. (2019b) Isolation of novel gut bifidobacteria using a combination of metagenomic and cultivation approaches. Genome Biol 20: 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A., Tarracchini, C., Alessandri, G., Milani, C., Mancabelli, L., Turroni, F., et al. (2020a) Decoding the genomic variability among members of the Bifidobacterium dentium species. Microorganisms 8: 1720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A., Mangifesta, M., Duranti, S., Anzalone, R., Milani, C., Mancabelli, L., et al. (2018b) Phylogenetic classification of six novel species belonging to the genus Bifidobacterium comprising Bifidobacterium anseris sp. nov., Bifidobacterium criceti sp. nov., Bifidobacterium imperatoris sp. nov., Bifidobacterium italicum sp. nov., Bifidobacterium margollesii sp. nov. and Bifidobacterium parmae sp. nov. Syst Appl Microbiol 41: 173–183. [DOI] [PubMed] [Google Scholar]
- Lugli, G.A., Milani, C., Turroni, F., Duranti, S., Ferrario, C., Viappiani, A., et al. (2014) Investigation of the evolutionary development of the genus Bifidobacterium by comparative genomics. Appl Environ Microbiol 80: 6383–6394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A., Milani, C., Turroni, F., Duranti, S., Mancabelli, L., Mangifesta, M., et al. (2017b) Comparative genomic and phylogenomic analyses of the Bifidobacteriaceae family. BMC Genomics 18: 568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugli, G.A., Alessandri, G., Milani, C., Mancabelli, L., Ruiz, L., Fontana, F., et al. (2020b) Evolutionary development and co‐phylogeny of primate‐associated bifidobacteria. Environ Microbiol 22: 3375–3393. [DOI] [PubMed] [Google Scholar]
- Malmstrom, R.R., and Eloe‐Fadrosh, E.A. (2019) Advancing genome‐resolved metagenomics beyond the shotgun. mSystems 4: e00118‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancabelli, L., Tarracchini, C., Milani, C., Lugli, G.A., Fontana, F., Turroni, F., et al. (2020) Multi‐population cohort meta‐analysis of human intestinal microbiota in early life reveals the existence of infant community state types (ICSTs). Comput Struct Biotechnol J 18: 2480–2493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani, C., Lugli, G.A., Turroni, F., Mancabelli, L., Duranti, S., Viappiani, A., et al. (2014) Evaluation of bifidobacterial community composition in the human gut by means of a targeted amplicon sequencing (ITS) protocol. FEMS Microbiol Ecol 90: 493–503. [DOI] [PubMed] [Google Scholar]
- Milani, C., Mangifesta, M., Mancabelli, L., Lugli, G.A., James, K., Duranti, S., et al. (2017a) Unveiling bifidobacterial biogeography across the mammalian branch of the tree of life. ISME J 11: 2834–2847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani, C., Casey, E., Lugli, G.A., Moore, R., Kaczorowska, J., Feehily, C., et al. (2018) Tracing mother‐infant transmission of bacteriophages by means of a novel analytical tool for shotgun metagenomic datasets: METAnnotatorX. Microbiome 6: 145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani, C., Duranti, S., Bottacini, F., Casey, E., Turroni, F., Mahony, J., et al. (2017b) The first microbial colonizers of the human gut: composition, activities, and health implications of the infant gut microbiota. Microbiol Mol Biol Rev 81: e00036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani, C., Alessandri, G., Mangifesta, M., Mancabelli, L., Lugli, G.A., Fontana, F., et al. (2020) Untangling species‐level composition of complex bacterial communities through a novel metagenomic approach. mSystems 5: e00404‐20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Modesto, M., Michelini, S., Oki, K., Biavati, B., Watanabe, K., and Mattarelli, P. (2018a) Bifidobacterium catulorum sp. nov., a novel taxon from the faeces of the baby common marmoset (Callithrix jacchus). Int J Syst Evol Microbiol 68: 575–581. [DOI] [PubMed] [Google Scholar]
- Modesto, M., Puglisi, E., Bonetti, A., Michelini, S., Spiezio, C., Sandri, C., et al. (2018b) Bifidobacterium primatium sp. nov., Bifidobacterium scaligerum sp. nov., Bifidobacterium felsineum sp. nov. and Bifidobacterium simiarum sp. nov.: four novel taxa isolated from the faeces of the cotton top tamarin (Saguinus oedipus) and the emperor tamarin (Saguinus imperator). Syst Appl Microbiol 41: 593–603. [DOI] [PubMed] [Google Scholar]
- Modesto, M., Watanabe, K., Arita, M., Satti, M., Oki, K., Sciavilla, P., et al. (2019a) Bifidobacterium jacchi sp. nov., isolated from the faeces of a baby common marmoset (Callithrix jacchus). Int J Syst Evol Microbiol 69: 2477–2485. [DOI] [PubMed] [Google Scholar]
- Modesto, M., Michelini, S., Sansosti, M.C., De Filippo, C., Cavalieri, D., Qvirist, L., et al. (2018c) Bifidobacterium callitrichidarum sp. nov. from the faeces of the emperor tamarin (Saguinus imperator). Int J Syst Evol Microbiol 68: 141–148. [DOI] [PubMed] [Google Scholar]
- Modesto, M., Satti, M., Watanabe, K., Scarafile, D., Huang, C.H., Liou, J.S., et al. (2020a) Phylogenetic characterization of two novel species of the genus Bifidobacterium: Bifidobacterium saimiriisciurei sp. nov. and Bifidobacterium platyrrhinorum sp. nov. Syst Appl Microbiol 43: 126111. [DOI] [PubMed] [Google Scholar]
- Modesto, M., Satti, M., Watanabe, K., Huang, C.H., Liou, J.S., Tamura, T., et al. (2020b) Bifidobacteria in two‐toed sloths (Choloepus didactylus): phylogenetic characterization of the novel taxon Bifidobacterium choloepi sp. nov. Int J Syst Evol Microbiol 70: 6115–6125. [DOI] [PubMed] [Google Scholar]
- Modesto, M., Satti, M., Watanabe, K., Puglisi, E., Morelli, L., Huang, C.H., et al. (2019b) Characterization of Bifidobacterium species in feaces of the Egyptian fruit bat: description of B. vespertilionis sp. nov. and B. rousetti sp. nov. Syst Appl Microbiol 42: 126017. [DOI] [PubMed] [Google Scholar]
- Neuzil‐Bunesova, V., Lugli, G.A., Modrackova, N., Vlkova, E., Bolechova, P., Burtscher, J., et al. (2021) Five novel bifidobacterial species isolated from faeces of primates in two Czech zoos: Bifidobacterium erythrocebi sp. nov., Bifidobacterium moraviense sp. nov., Bifidobacterium oedipodis sp. nov., Bifidobacterium olomucense sp. nov. and Bifidobacterium panos sp. nov. Int J Syst Evol Microbiol 71: 1–12. [DOI] [PubMed] [Google Scholar]
- Neuzil‐Bunesova, V., Lugli, G.A., Modrackova, N., Makovska, M., Mrazek, J., Mekadim, C., et al. (2020) Bifidobacterium canis sp. nov., a novel member of the Bifidobacterium pseudolongum phylogenetic group isolated from faeces of a dog (Canis lupus f. familiaris). Int J Syst Evol Microbiol 70: 5040–5047. [DOI] [PubMed] [Google Scholar]
- O'Callaghan, A., and van Sinderen, D. (2016) Bifidobacteria and their role as members of the human gut microbiota. Front Microbiol 7: 925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Richter, M., and Rossello‐Mora, R. (2009) Shifting the genomic gold standard for the prokaryotic species definition. Proc Natl Acad Sci U S A 106: 19126–19131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rinke, C., Schwientek, P., Sczyrba, A., Ivanova, N.N., Anderson, I.J., Cheng, J.F., et al. (2013) Insights into the phylogeny and coding potential of microbial dark matter. Nature 499: 431–437. [DOI] [PubMed] [Google Scholar]
- Turroni, F., Milani, C., Duranti, S., Ferrario, C., Lugli, G.A., Mancabelli, L., et al. (2018) Bifidobacteria and the infant gut: an example of co‐evolution and natural selection. Cell Mol Life Sci 75: 103–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turroni, F., Milani, C., Duranti, S., Lugli, G.A., Bernasconi, S., Margolles, A., et al. (2020) The infant gut microbiome as a microbial organ influencing host well‐being. Ital J Pediatr 46: 16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vezzulli, L., Grande, C., Tassistro, G., Brettar, I., Hofle, M.G., Pereira, R.P., et al. (2017) Whole‐genome enrichment provides deep insights into vibrio cholerae metagenome from an African River. Microb Ecol 73: 734–738. [DOI] [PubMed] [Google Scholar]
- Zhao, Y., Wu, J., Yang, J., Sun, S., Xiao, J., and Yu, J. (2012) PGAP: pan‐genomes analysis pipeline. Bioinformatics 28: 416–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Schematic representation of the targeted bifidobacterial WMS approach.
Fig. S2. Carbohydrate growth assays of the isolated bifidobacterial stains. The heat map illustrates the average optical densities (OD600) of three independent replicates for each isolated strain at two different time points, 24 and 48 h.
Fig. S3. Alignment of genome sequences of bifidobacterial isolates against contigs longer than 1000 nucleotides from targeted WMS sequencing. Panel a to panel l report the sequence alignment of strain LC6, MA2, MA1, SO4, SO2, SO1, CP3, CP2, CP1 and CP4.
Fig. S4. CP2, LC6, MA1, MA2, SO1 and SO4 cellular morphologies as determined by the use of phase‐contrast microscopy. Bar, 10 μm.
Table S1. Metabolic profile of the bifidobacterial dark matter.
Table S2. Unknown genes retrieved between reconstructed genomes.
Table S3. Metabolic profile of the isolated bifidobacterial strains.
Data Availability Statement
Raw sequences of shotgun metagenomics experiments are accessible through SRA study BioProject PRJNA698773. Ten genome sequences have also been deposited in the GenBank database under the BioProject PRJNA698773.