

Abstract
Background
We classified non‐demented European Prevention of Alzheimer's Dementia (EPAD) participants through the amyloid/tau/neurodegeneration (ATN) scheme and assessed their neuropsychological and imaging profiles.
Materials and methods
From 1500 EPAD participants, 312 were excluded. Cerebrospinal fluid cut‐offs of 1000 pg/mL for amyloid beta (Aß)1‐42 and 27 pg/mL for p‐tau181 were validated using Gaussian mixture models. Given strong correlation of p‐tau and t‐tau (R2 = 0.98, P < 0.001), neurodegeneration was defined by age‐adjusted hippocampal volume. Multinomial regressions were used to test whether neuropsychological tests and regional brain volumes could distinguish ATN stages.
Results
Age was 65 ± 7 years, with 58% females and 38% apolipoprotein E (APOE) ε4 carriers; 57.1% were A–T–N–, 32.5% were in the Alzheimer's disease (AD) continuum, and 10.4% suspected non‐Alzheimer's pathology. Age and cerebrovascular burden progressed with biomarker positivity (P < 0.001). Cognitive dysfunction appeared with T+. Paradoxically higher regional gray matter volumes were observed in A+T–N– compared to A–T–N– (P < 0.001).
Discussion
In non‐demented individuals along the AD continuum, p‐tau drives cognitive dysfunction. Memory and language domains are affected in the earliest stages.
Keywords: Alzheimer's disease (AD), amyloid beta, amyloid/tau/neurodegeneration (ATN) staging, cognition, European Prevention of Alzheimer's Dementia (EPAD), magnetic resonance imaging (MRI), neurodegeneration, neuroimaging, tau
1. INTRODUCTION
Finding disease‐modifying therapies for Alzheimer's disease (AD), the most prevalent cause of dementia, is an international priority. The most effective approach may be to slow or prevent AD progression prior to dementia. Secondary prevention strategies aim to identify individuals with evidence of AD pathology but who have not (yet) developed symptoms, that is, at a preclinical stage.1
To provide a reference framework, an update on the biological definition of AD has been published, describing the disease solely in terms of biomarkers.2 In the amyloid/tau/neurodegeneration (ATN) framework, the AD continuum is defined by the deposition of amyloid beta (Aβ) plaques in the brain. As the presence of amyloid seems to be necessary but not sufficient for the development of AD dementia,3 a more fine‐grained description of the disease can be obtained through classification in terms of the additional presence of tau pathology and neurodegeneration. Clinical characterization of these subgroups has been limited. In addition, there is limited consensus on the operationalization of these criteria.
To apply the ATN criteria, possibly for selection of at‐risk individuals into trials, cut‐off values must be defined. However, the definition of cut‐offs in a non‐demented population such as ours has proven challenging. Cut‐offs are typically defined using populations of symptomatic individuals, excluding the long pre‐symptomatic phase of AD.4, 5 This has led to variability in proposed cut‐off values, ranging from 880 to 1100 pg/mL for cerebrospinal fluid (CSF) Aβ1‐426, 7, 8 and from 19 to 27 pg/mL for CSF phosphorylated tau (p‐tau) using the Roche Elecsys assays.6, 9 Differences in cut‐offs for CSF Aβ1‐42 may also be related to the use of different pre‐analytical protocols for CSF handling.10 Similarly, cut‐off values for AD‐specific neurodegeneration (N) markers vary considerably.11 Jack et al. proposed the use of either CSF total tau (t‐tau) or magnetic resonance imaging (MRI) measures of atrophy as proxies for neurodegeneration.12 One possible way to define AD‐specific neurodegeneration is based on hippocampal volume (HCV),13 as there is evidence that the hippocampus is one of the earliest structures affected in AD and undergoes disproportionate atrophy.14 HCV can be assessed using visual rating scales,15 or quantified through segmentation.16
The aim of the current study is to investigate the effect of ATN staging on neuropsychological profiles and atrophy patterns within the European Prevention of Alzheimer's Dementia (EPAD) cohort.1, 17
2. MATERIALS AND METHODS
2.1. Study participants
We included baseline data of the first 1500 participants consented in the EPAD cohort from 21 different European sites (Figure S1 in supporting information).1, 17 The study protocol has been reported elsewhere.17 Participants ≥50 years of age, with at least 7 years of education, and a study partner were included. Exclusion criteria were the presence of conditions associated with neurodegeneration or affecting cognition, Clinical Dementia Rating (CDR) ≥1, contraindications to MRI or lumbar puncture, and cancer or history of cancer in the preceding 5 years. After obtaining written informed consent, a screening visit was conducted to check whether the study participants fulfilled these criteria,18, 19 and 144 participants were excluded (Figure S2 in supporting information). Demographic details were collected. Each participant underwent clinical and neurological assessment, brain MRI, a lumbar puncture to evaluate biomarkers in the CSF, and neuropsychological assessment. Participants with missing CSF or MRI data were excluded (n = 168; Figure S2). The total remaining sample included 1188 study participants.
HIGHLIGHTS
Cut‐off values for cerebrospinal fluid (CSF) amyloid and tau were confirmed in a non‐demented population.
CSF t‐tau may not be optimal to define neurodegeneration in non‐demented cohorts.
Cognitive performance drops when CSF p‐tau reaches abnormal levels.
Cerebrovascular burden increases along the Alzheimer's disease continuum.
Brain regional volumes might show bi‐directional changes in A+T–N–.
RESEARCH IN CONTEXT
Systematic Review: The amyloid/tau/neurodegeneration (ATN) framework provides a biological definition of Alzheimer's disease (AD)‐related biomarker changes, independent of cognitive performance. To understand their significance in a non‐demented population, we operationalized the ATN classification in the European Prevention of Alzheimer's Dementia (EPAD) cohort and examined the associated neuropsychological and radiological profiles.
Interpretation: Data‐driven models confirm that the proposed cerebrospinal fluid cut‐offs values for amyloid beta (Aß)42 and p‐tau181 are valid in a non‐demented population. Moreover, our results show that (1) t‐tau is not appropriate for staging N; (2 cognitive dysfunction, especially within the delayed memory domain, coincides with T positivity; (3) cerebrovascular burden parallels ATN biomarker progression, and (4) regional atrophy measures show paradoxical higher volumes in the basal forebrain, putamen, postcentral, and middle occipital gyri, early along the AD continuum and atrophy afterward.
Future directions: In a non‐demented population, ATN profiles convey important neuropsychological and structural information that may help identify subjects for enrolment in secondary prevention trials.
2.2. Cerebrospinal fluid
CSF was obtained using a harmonized pre‐analytical protocol. Analyses were performed using the fully automatized Roche Elecsys System in a single laboratory (University of Gothenburg).17 Concentrations of Aβ1‐42, phosphorylated tau (p‐tau181), and total tau (t‐tau) were determined according to the manufacturer's instructions.
2.3. Neuropsychological evaluation
The EPAD Neuropsychological Examination (ENE) battery covers relevant cognitive domains and was collected with standardized procedures on a tablet.20, 21 Tests performed included the Mini‐Mental State Examination (MMSE),22 CDR,23 Advanced Instrumental Activities of Daily Living (AIADL), Geriatric Depression Scale (GDS),24 the Repeatable Battery for the Assessment of Neuropsychological Status (RBANS),25 the Four Mountains Test (4MT),26 and the Virtual Reality Supermarket Trolley test (VRST).27 More information on the neuropsychological tests is reported in the supporting information.
2.4. MRI scans
Brain MRI scans were performed with standardized acquisition protocols, including 3D‐T1, 3D‐FLAIR, 2D‐T2, and 2D‐T2* sequences. The images were centrally evaluated by experienced raters, blind to the clinical and neuropsychological data. Visual assessment of the scans included white matter hyperintensities,28, 29 perivascular spaces (PVS),30, 31 microbleeds (CMBs),30 medial temporal lobe atrophy (MTA),32 and posterior cortical atrophy (PCA;33 supporting information). Regional gray matter volumes were determined on 3D‐T1 weighted images using a segmentation process based on atlas‐propagation with the Learning Embeddings for Atlas Propagation (LEAP) framework.34
2.5. Biomarkers cut‐offs
Following the recently published research framework,3 we classified participants into different groups based on the presence (+) or absence (–) of abnormal CSF Aβ1‐42 values (labeled “A”), p‐tau181 (labeled “T”), and neurodegeneration or neuronal injury (labeled “N”).
We referred to CSF Aβ1‐42 levels < 1000 pg/mL to define A+ and CSF p‐tau181 > 27 pg/mL to define T+, as suggested previously.7, 10, 11 These CSF cut‐offs were validated for our sample using Gaussian mixture models (GMMs). GMM analysis implements an expectation maximization algorithm assuming that data points are generated from a mixture of Gaussian distributions, which were labeled as “normal,” “pathological,” or “noise” based on visual inspection. The intersection point between the normal and the pathological identified components were used as cut‐off points. The results of GMM analysis for Aβ1‐42 and p‐tau181 are shown in Figure 1. The three components identified in Aβ1‐42 were visually inspected and classified as normal, pathological, and noise distributions. The intersection point between the normal and the pathological components corresponded to 1007 pg/mL, close to the reference published cut‐point of 1000 pg/mL (Figure 1A). GMM analysis for p‐tau revealed two components, whose intersection point was located at 27 pg/mL (Figure 1B). Thus, we considered both the published cut‐offs valid for our sample.
FIGURE 1.
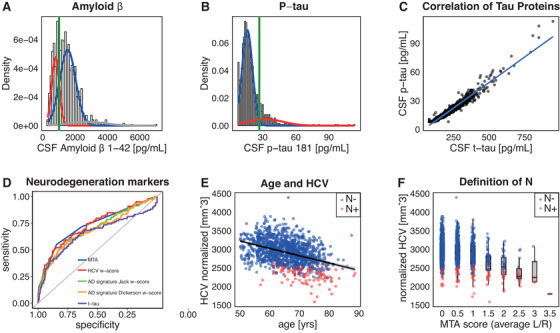
Gaussian mixture models of cerebrospinal fluid (CSF) amyloid beta (Aß)1‐42 (A) and p‐tau181 (B; Roche Elecsys assay). Pathological component is indicated in red, normal in blue, other components in gray; the green line indicates cut‐off values at the intersection point of the two components. CSF Aß1‐42 cut‐off of 1000 pg/mL and p‐tau181 cut‐off of 27 pg/mL were confirmed. C, Correlation between tau proteins (Pearson's correlation coefficient = 0.976, P‐value < 0.001). D, Receiver operating characteristic (ROC) curves distinguishing A– Clinical Dementia Rating (CDR) = 0 from A+ CRR = 0.5 groups with different neurodegeneration markers. E, Relation between age and hippocampal volume (HCV). F, Concordance between medial temporal atrophy (MTA) scores and normalized HCV. The left/right scores MTA and HCV were averaged. Neurodegeneration was defined on the base of age‐adjusted normalized HCV w‐scores
Potential neurodegeneration (N) markers were t‐tau, MTA, HCV, and volume of AD signature regions as defined by Jack et al. (enthorinal, inferior temporal, middle temporal, and fusiform cortices)34 and by Dickerson et al. (medial and inferior temporal cortices, temporal pole, angular gyrus, superior frontal gyrus, superior parietal lobule, supramarginal gyrus, and inferior frontal sulcus).35 HCV and AD signature region volumes were divided by a normalization factor approximated for original brain size. We compared how these potential neurodegeneration markers could distinguish A– individuals with CDR = 0 from A+ individuals with CDR = 0.5 by generating receiver operating characteristic (ROC) curves, as suggested in a recent work.11 The highest area under the curve (AUC) was used to select the optimal marker for N definition.
A strong linear relationship was observed between p‐tau and t‐tau (r = 0.98, P < 0.001), suggesting that these measures could not be used independently for T and N staging, respectively, in our population (Figure 1C). As HCV and AD signature regions have a known relationship with age,3 we used age‐adjusted w‐scores to account for this effect. We confirmed a HCV decrease with age in our data (r = –0.39, P < 0.001; Figure 2E) and then used age‐adjusted w‐scores to disentangle neurodenegeneration from normal aging36 (Figures 2E, 2F).
FIGURE 2.
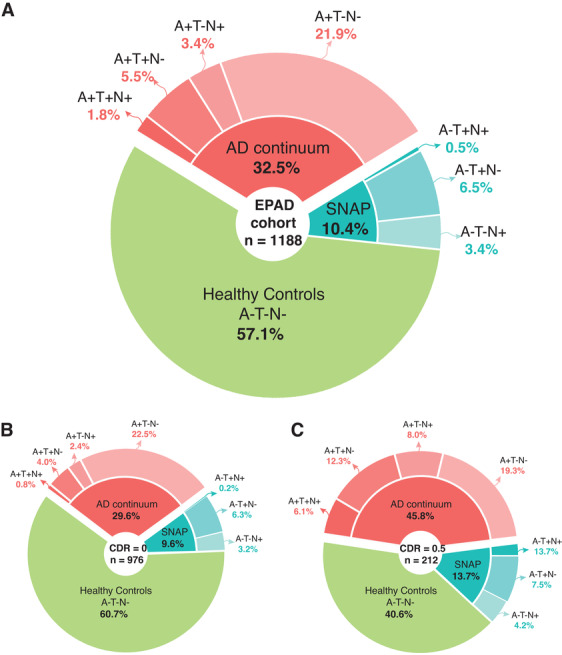
Classification of the first European Prevention of Alzheimer's Dementia (EPAD) study participants according to the amyloid/tau/neurodegeneration (ATN) classification scheme. We applied the ATN classification scheme using the following cut‐offs: A+ if participants demonstrated cerebrospinal fluid (CSF) amyloid beta (Aß)42 levels < 1000 pg/mL (A– otherwise); T+ if they showed CSF p‐tau levels > 27 pg/mL (T– otherwise); N+ if they had an age adjusted hippocampal volume w‐score < 1.5 standard deviation from the mean (N– otherwise). Out of 1500 screened participant, n = 312 individuals were excluded from analyses because of they either did not fulfil EPAD eligibility criteria (n = 144) or they had missing CSF or magnetic resonance imaging (MRI) data (n = 168), reducing the total sample to n = 1188. A, Of these, 678 participants were classified as healthy controls (A–T–N–). Individuals with amyloid positivity (A+) were classified as in the AD continuum (n = 387 [25.7%], of which 261 [21.9%] A+T–N–; 40 [3.4%] A+T–N+; 65 [5.5%] A+T+N–; 21 [1.8%] A+T+N+). Finally, individuals negative to amyloid but positive to other biomarkers were defined as suspected non‐Alzheimer's disease pathologic change (SNAP; of which 40 [3.4%] A–T–N+; 77 [6.5%] A–T+N–; 6 [0.5%] A–T+N+). We then proceeded to stratify these groups on the base of Clinical Dementa Rating (CDR) global score. B, A total of 975 study participants had a CDR score of 0. Of these, 592 (60.7%) were A–T–N–, 289 (29.6%) were in the AD spectrum (of which 219 [22.5%] A+T–N–; 23 [2.4%] A+T–N+; 39 [4.0%] A+T+N‐; 8 [0.8%] A+T+N+), and 94 (9.6%) were classified as SNAP (of which 31 [3.2%] A–T–N+; 61 [6.3%] A‐T+N‐; 2 [0.2%] A‐T+N+). C, Out of the remaining 212 study participants with a CDR score of 0.5, 86 (40.6%) were A–T–N‐, 97 (45.8%) were in the AD spectrum (of which 41 [19.3%] A+T–N–; 17 [8.0%] A+T–N+; 26 [12.3%] A+T+N–; 13 [6.1%] A+T+N+), and 29 (13.7%) were classified as SNAP (of which 9 [4.2%] A–T–N+; 16 [7.5%] A–T+N–; 4 [1.9%] A–T+N+)
Results of the ROC curve analysis for the N marker definition are shown in Figure 1D. ROC curves distinguishing A– individuals with CDR = 0 from A+ individuals with CDR = 0.5 performed best for HCV (AUC = 0.74; 95% confidence interval [CI]: 0.68–0.80) and MTA (AUC = 0.73; 95% CI: 0.67–0.79), followed by AD signature regions as defined by Jack et al.34 (AUC = 0.72; 95% CI: 0.66–0.78) and by Dickerson et al.35 (AUC = 0.70; 95% CI: 0.64–0.76), and finally by t‐tau (AUC = 0.65; 95% CI: 0.58–0.72; Figure 1D). Because HCV was normally distributed, precluding the use of GMM, N+ was defined for individuals with HCV w‐score < –1.5 standard deviations from the mean.
In line with Jack et al.2, we defined A–T–N– as the reference group, A+ individuals with or without positivity for T and N as in the AD continuum, and subjects with positivity for T and/or N but not for A as suspected non‐AD pathologic change (SNAP). Within the AD continuum, A+T–N– were identified as a group with AD pathological changes, A+T+N‐ and A+T+N+ as groups with AD, and A+T‐N+ as AD and concomitant suspected non‐AD pathologic change (atypical AD).
2.6. Statistical analyses
The ATN groups were compared based on demographic, clinical, neuropsychological, and radiological characteristics with nonparametric tests, that is, Kruskal–Wallis test for continuous variables or Chi‐square tests for categorical variables. As the objective of EPAD is to identify subjects at risk of AD dementia, SNAP and A+T‐N+ (atypical AD) groups were excluded from further analyses.
Based on the results of the Kruskal–Wallis tests, we selected as potentially interesting the RBANS outcomes that could distinguish among groups with a P‐value < 0.001, and we proceeded to build three multinomial logistic regression models to examine the predictive value of RBANS scores for classification into the ATN groups. Dependent variables were the ATN groups and predictors were RBANS total scale for model 1, significant RBANS index scores (attention, immediate memory, delayed memory) for model 2, and significant RBANS subtests (coding, figure recall, list learning, list recall, list recognition, semantic fluency, story memory, story recall) for model 3. All models were adjusted for age, sex, years of education, family history of dementia, and site of data collection.
Similarly, we compared regional volumes across the ATN groups with Kruskal–Wallis tests. For brain regions detecting differences among A–T–N– and the different stages of the AD continuum with a P‐value < 0.001, we built a multinomial regression model to test whether such volumes could distinguish among the different ATN stages, after adjusting for age, sex, family history of dementia, head size, and site of data collection. We limited the interpretation of the model to findings with a P‐value < 0.01. The analyses were performed using R version 3.6.0 (https://www.R‐project.org/).
3. RESULTS
3.1. Prevalence of ATN stages
The reference group (A–T–N–) comprised 678 individuals (57.1%); 387 (32.5%) participants had abnormal amyloid, 169 (14.2%) abnormal p‐tau, and 107 (9.0%) abnormal HCV, based on a priori defined cut‐off values for the CSF markers6, 9, 10 and on age‐adjusted HCV w‐score. Individuals classified as SNAP were 10.4%, showing positivity to T (n = 77, 6.5%), N (n = 40, 3.4%), or both (n = 6, 0.5%; Figure 2A). Of those in the AD continuum, 261 (21.9%) were classified as A+T–N–, 65 (5.5%) A+T+N–, 40 (3.4%) A+T–N+, and 21 (2.1%) A+T+N+. Individuals with a CDR = 0.5 (n = 212) showed increased positivity to biomarkers (Figures 2B, 2C).
3.2. ATN stages
We report in Table 1 an overview of our data from A–T–N– across subjects in the ATN continuum. More than 99% of our study population was White. Male‐to‐female ratio was slightly unbalanced, with a predominance of males in the A+T+N+ group. There were no differences in years of education or handedness among the ATN groups. Age significantly increased progressing in positivity along the ATN stages (P < 0.001). Prevalence of apolipoprotein E (APOE) ε4 carriers also increased with biomarker positivity along the AD continuum, being 29.8% for the A–T–N–, 46.8% for the A+T–N–, 66.7% for the A+T+N–, and 75.0% for the A+T+N+ group. Groups showed significant differences in MMSE, CDR, AIADL, and RBANS total score (P < 0.001). Cognitive domains that showed significant differences were RBANS composite scores on attention, delayed memory, and immediate memory (P < 0.001). PCA increased along the AD continuum although being generally low (P < 0.001). Finally, increasing cerebrovascular burden, as measured with Fazekas scale rating white matter hyperintensities and Potter scale rating PVS, was shown progressing along the AD continuum (P < 0.001).
TABLE 1.
Descriptive characteristics of the study population by ATN staging
| A–T–N– (n = 678) | A+T–N– (n = 261) | A+T+N– (n = 65) | A+T–N+ (n = 40) | A+T+N+ (n = 21) | Total (n = 1065) | P | |
|---|---|---|---|---|---|---|---|
| Demographics | |||||||
| Age | 64.3 ± 6.7 | 65.2 ± 7.1 | 70.1 ± 5.5 | 70.5 ± 6.7 | 75.1 ± 5.1 | 65.3 ± 7.0 | **<0.001 |
| Sex, male | 269 (39.7%) | 112 (42.9%) | 25 (38.5%) | 29 (72.5%) | 16 (76.2%) | 451 (42.3%) | **<0.001 |
| Education (years) | 14.6 ± 3.6 | 14.6 ± 3.8 | 13.7 ± 3.6 | 15.4 ± 3.6 | 14.2 (5.1) | 14.5 ± 3.7 | 0.252 |
| Handedness, right hand | 630 (93.5%) | 244 (93.8%) | 61 (93.8%) | 40 (100.0%) | 19 (90.5%) | 994 (93.8%) | 0.532 |
| Family history of dementia, positive | 472 (69.6%) | 174 (66.7%) | 50 (76.9%) | 30 (75.0%) | 14 (66.7%) | 740 (69.5%) | 0.504 |
| APOE ε4, carrier | 191 (29.8%) | 111 (46.6%) | 42 (66.7%) | 18 (48.6%) | 15 (75.0) | 377 (37.7%) | **<0.001 |
| Neuropsychological tests | |||||||
| MMSE | 28.8 ± 1.4 | 28.8 ± 1.5 | 27.8 ± 2.0 | 28 ± 1.9 | 26.5 (3.9) | 28.7 (1.6) | **<0.001 |
| CDR global score, 0.5 | 86 (12.7%) | 41 (15.7%) | 26 (40.0%) | 17 (42.5%) | 13 (61.9) | 183 (17.2%) | **<0.001 |
| AIADL total score | 0.31 ± 0.93 | 0.91 ± 3.11 | 2.25 ± 4.67 | 1.65 ± 2.82 | 5.75 (10.26) | 0.74 ± 2.72 | **<0.001 |
| GDS | 4.6 ± 4.4 | 5.2 ± 5.2 | 5.0 (4.4) | 5.2 ± 4.4 | 5.6 (4.3) | 4.8 ± 4.6 | 0.401 |
| VR Supermarket Trolley Task | 9.6 ± 3.6 | 10.2 ± 3.6 | 8.9 ± 3.8 | 8.0 ± 4.1 | 8.2 ± 4.2 | 9.6 ± 3.6 | *0.001 |
| Four Mountains Task | 9.7 ± 2.4 | 9.4 ± 2.7 | 7.9 ± 2.1 | 8.6 ± 3.4 | 7.3 ± 1.9 | 9.5 ± 2.5 | * < 0.001 |
| RBANS total scale | 105.5 ± 12.4 | 104.7 ± 12.4 | 95.3 ± 16.2 | 98.4 ± 14.1 | 89.7 ± 14.8 | 104.1 ± 13.2 | **<0.001 |
| RBANS domains | |||||||
| RBANS attention index | 101.2 ± 15.6 | 99.9 ± 16.3 | 92.3 ± 15.8 | 93.3 ± 16.3 | 90.2 ± 15.5 | 99.8 ± 16.1 | **<0.001 |
| RBANS delayed memory index | 104.4 ± 11.8 | 103.6 ± 13.5 | 94.5 ± 21.1 | 98.4 ± 14.6 | 82.5 ± 25.3 | 103.0 ± 14.0 | **<0.001 |
| RBANS immediate memory index | 106.4 ± 12.6 | 105.8 ± 13.4 | 96.7 ± 18.2 | 98.6 ± 14.1 | 84.5 ± 19.3 | 104.9 ± 14.0 | **<0.001 |
| RBANS language index | 99.5 ± 10.0 | 99. ± 10.6 | 94.7 ± 12.1 | 98.7 ± 8.2 | 95.1 ± 9.1 | 99.0 ± 10.3 | 0.003 |
| RBANS visuo‐constructional index | 108.9 ± 15.8 | 109.1 ± 14.5 | 104.2 ± 14.7 | 105.1 ± 18.3 | 108.4 ± 15.4 | 108.5 ± 15.5 | 0.100 |
| RBANS subtests | |||||||
| RBANS coding | 46.5 ± 10.5 | 44.4 ± 11.1 | 38.0 ± 11.4 | 38.0 ± 12.8 | 33.5 ± 11.3 | 44.9 ± 11.2 | **<0.001 |
| RBANS digit span | 10.1 ± 2.4 | 10.3 ± 2.6 | 9.5 ± 2.5 | 9.4 ± 2.5 | 9.1 ± 2.2 | 10.1 ± 2.4 | *0.024 |
| RBANS figure copy | 18.4 ± 2.3 | 18.3 ± 2.1 | 17.9 ± 2.1 | 17.8 ± 2.5 | 18.1 ± 2.3 | 18.3 ± 2.2 | 0.234 |
| RBANS figure recall | 14.3 ± 3.6 | 14.4. ± 3.7 | 11.8 ± 5.3 | 12.1 ± 4.3 | 8.7 ± 5.5 | 14.0 ± 3.9 | **<0.001 |
| RBANS list learning | 29.1 ± 4.3 | 28.8 ± 4.4 | 25.5 ± 6.3 | 25.0 ± 5.6 | 19.8 ± 6.7 | 28.5 ± 4.9 | **<0.001 |
| RBANS line orientation | 18.0 ± 2.4 | 18.3 ± 1.8 | 17.5 ± 2.3 | 17.6 ± 2.3 | 17.7 ± 2.7 | 18.0 ± 2.3 | *0.059 |
| RBANS list recall | 6.6 ± 2.1 | 6.1 ± 2.5 | 4.8 ± 2.8 | 4.5 ± 2.5 | 3.1 ± 2.8 | 6.2 ± 2.4 | **<0.001 |
| RBANS list recognition | 19.4 ± 1.0 | 19.3 ± 1.1 | 18.6 ± 2.1 | 19.1 ± 1.2 | 17.6 ± 2.7 | 19.3 ± 1.2 | **<0.001 |
| RBANS picture naming | 9.8 ± 0.9 | 9.8 ± 0.4 | 9.7 ± 0.7 | 9.9 ± 0.3 | 9.9 ± 0.3 | 9.8 ± 0.8 | 0.355 |
| RBANS semantic fluency | 20.6 ± 5.1 | 20.0 ± 5.7 | 17.6 ± 5.6 | 18.7 ± 4.9 | 16.2 ± 4.2 | 20.1 ± 5.3 | **<0.001 |
| RBANS story memory | 18.8 ± 3.0 | 18.8 ± 3.2 | 16.7 ± 4.4 | 17.6 ± 3.2 | 13.7 ± 4.3 | 18.5 ± 3.3 | **<0.001 |
| RBANS story recall | 9.4 ± 1.7 | 9.2 ± 1.9 | 7.7 ± 3.2 | 8.9 ± 2.3 | 5.6 ± 3.7 | 9.1 ± 2.0 | **<0.001 |
| Radiological visual rating scales | |||||||
| Fazekas score deep | 0.79 ± 0.67 | 0.92 ± 0.73 | 1.22 ± 0.80 | 1.20 ± 0.82 | 1.33 ± 0.80 | 0.88 ± 0.72 | **<0.001 |
| Fazekas score periventricular | 0.42 ± 0.65 | 0.52 ± 0.69 | 0.86 ± 0.79 | 0.90 ± 0.93 | 1.19 ± 0.93 | 0.51 ± 0.71 | **<0.001 |
| ePVS basal ganglia | 1.06 ± 0.44 | 1.16 ± 0.50 | 1.34 ± 0.67 | 1.20 ± 0.56 | 1.29 ± 0.64 | 1.11 ± 0.49 | **<0.001 |
| ePVS centrum semiovale | 1.27 ± 0.78 | 1.34 ± 0.85 | 1.85 ± 0.97 | 1.65 ± 0.92 | 1.81 ± 0.93 | 1.35 ± 0.84 | **<0.001 |
| ePVS perivascular midbrain | 0.56 ± 0.50 | 0.54 ± 0.50 | 0.58 ± 0.50 | 0.65 ± 0.48 | 0.71 ± 0.46 | 0.56 ± 0.50 | 0.424 |
| CMBs, present | 86 (12.8%) | 34 (13.0%) | 15 (23.1%) | 8 (20.0%) | 5 (23.8%) | 148 (14.0%) | 0.079 |
| MTA average | 0.29 ± 0.45 | 0.48 ± 0.53 | 0.58 ± 0.70 | 1.12 ± 1.03 | 1.02 ± 0.81 | 0.40 ± 0.56 | **<0.001 |
| PCA | 0.49 ± 0.61 | 0.51 ± 0.59 | 0.63 ± 0.67 | 0.72 ± 0.85 | 1.05 ± 0.67 | 0.52 ± 0.63 | **<0.001 |
| Volumetric MRI analyses | |||||||
| Brain volume [cm3] | 1341 ± 61.3 | 1329 ± 69.5 | 1319 ± 72.5 | 1249 ± 64.1 | 1229 ± 56.4 | 1331 ± 67.9 | **<0.001 |
| Hippocampal volume [mm3] | 2992 ± 299.1 | 2919 ± 292.4 | 2921 ± 319.3 | 2330 ± 168.4 | 2258 ± 248.8 | 2930 ± 333.3 | **<0.001 |
Notes: Values are expressed as mean ± standard deviation for continuous variables and number (percentage) for dichotomous variables. Significance value was set at P‐value < 0.05 and P‐values were reported as follows.
P < 0.05.
P < 0.01.
Abbreviations: AIADL, Advanced Instrumental Activities of Daily Living; APOE, apolipoprotein E; CDR, Clinical Dementia Rating; CMBs, cerebral microbleeds; ePVS, enlarged perivascular spaces; GDS, Geriatric Depression Scale; MMSE, Mini‐Mental State Examination; MTA, medial temporal atrophy; PCA, posterior cortical atrophy; RBANS, Repeatable Battery for the Assessment of Neuropsychological Status; VR, virtual reality.
Comparison of demographics, clinical, neuropsychological, and radiological data among the A–T–N– subjects in the AD continuum and SNAP is reported in Table S1 in supporting information. Compared to A–T–N–, SNAP were older, had a higher male prevalence, lower CDR and RBANS total scores, and higher scores on the AIADL questionnaire. They had similar scores to individuals in the AD continuum at RBANS domains and subtests, with a lower performance compared to A–T–N– (P‐value < 0.01 for attention, delayed, and immediate memory). Their vascular burden was similar to A–T–N–, while their atrophy profiles were similar to subjects in the AD continuum, showing increased neurodegeneration compared to A–T–N– (P‐value < 0.001).
3.3. Relationship between cognitive performance and ATN stages
The results of multinomial regression analyses predicting ATN stage based on ENE battery results are presented in Table 2. RBANS total scale significantly distinguished A–T–N– from A+T+N–, A+T–N+, and A+T+N+ stages (P‐values < 0.01); between individuals with only amyloid pathology (A+T–N–) and A+T+N–, A+T–N+, and A+T+N+ stages (P‐values < 0.01); and between A+T–N+ and A+T+N+ (P‐value < 0.05); but not between A–T–N– and individuals with only amyloid pathology (A+T–N–), or between A+T+N– and A+T–N+ (model 1).
TABLE 2.
Multinomial regression with RBANS total score (model 1) and domains (model 2)
| Ref. group: A–T–N– | A+T–N– | A+T+N– | A+T–N+ | A+T+N+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | |
| Model 1: Multinomial regression with RBANS total score | ||||||||||||
| RBANS total | –0.005 | 0.995 | 0.979–1.011 | –0.004 ** | 0.956 | 0.931–0.982 | –0.039 ** | 0.962 | 0.937–0.988 | –0.086 ** | 0.918 | 0.890 – 0.946 |
| Model 2: Multinomial regression with RBANS cognitive domains | ||||||||||||
| RBANS attention | –0.003 | 0.997 | 0.984–1.010 | –0.014 | 0.986 | 0.963–1.009 | ‐0‐.025 * | 0.975 | 0.952–0.999 | –0.022 | 0.979 | 0.949 – 1.009 |
| RBANS immediate memory | –0.002 | 0.998 | 0.979–1.017 | –0.023 | 0.978 | 0.949–1.007 | –0.015 | 0.985 | 0.956–1.015 | –0.046 ** | 0.955 | 0.923 – 0.988 |
| RBANS delayed memory | 0.001 | 1.001 | 0.983 – 1.019 | –0.017 | 0.982 | 0.957 – 1.009 | –0.015 | 0.989 | 0.960–1.017 | –0.037 * | 0.963 | 0.936 – 0.992 |
| RBANS language | 0.001 | 1.001 | 0.984 – 1.016 | –0.014 | 0.985 | 0.957 – 1.014 | 0.043 * | 1.044 | 1.002–1.0876 | 0.037 | 1.038 | 0.977 – 1.103 |
| RBANS visuo‐construction | 0.003 | 1.003 | 0.992 – 1.014 | 0.001 | 1.001 | 0.984 – 1.019 | –0.013 | 0.987 | 0.965–.009 | 0.0159 | 1.016 | 0.983 – 1.050 |
| Ref. group: A+T–N– | A–T–N– | A+T+N– | A+T‐N+ | A+T+N+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | |
| Model 1: Multinomial regression with RBANS total score | ||||||||||||
| RBANS total | –0.039 ** | 0.961 | 0.934 – 0.989 | –0.036 * | 0.964 | 0.938–0.991 | ‐0.081 ** | 0.923 | 0.894 – 0.952 | |||
| Model 2: Multinomial regression with RBANS cognitive domains | ||||||||||||
| RBANS attention | –0.011 | 0.989 | 0.964 – 1.014 | –0‐.023 | 0.977 | 0.953–1.001 | –0.019 | 0.981 | 0.951 – 1.013 | |||
| RBANS immediate memory | –0.021 | 0.980 | 0.949 – 1.011 | –0.014 | 0.985 | 0.955–1.017 | –0.044 * | 0.957 | 0.923 – 0.993 | |||
| RBANS delayed memory | –0.018 | 0.982 | 0.954 – 1.011 | –0.010 | 0.991 | 0.961–1.021 | –0.038 * | 0.962 | 0.933 – 0.993 | |||
| RBANS language | –0.015 | 0.985 | 0.955 – 1.016 | 0.043 * | 1.044 | 1.002–1.104 | 0.037 | 1.038 | 0.976 – 1.104 | |||
| RBANS visuo‐construction | –0.001 | 0.998 | 0.980 – 1.017 | –0.016 | 0.984 | 0.961–1.007 | 0.013 | 1.013 | 0.980 – 1.047 | |||
| Ref. group: A+T+N– | A–T–N– | A+T–N– | A+T‐N+ | A+T+N+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | |
| Model 1: Multinomial regression with RBANS total score | ||||||||||||
| RBANS total | 0.013 | 0.101 | 0.982 – 1.046 | –0.041 * | 0.960 | 0.928 – 0.993 | ||||||
| Model 2: Multinomial regression with RBANS cognitive domains | ||||||||||||
| RBANS attention | –0.010 | 0.990 | 0.962 – 1.019 | –0.007 | 0.993 | 0.958 – 1.028 | ||||||
| RBANS immediate memory | –0.009 | 1.008 | 0.973 –1.045 | –0.023 | 0.977 | 0.940 – 1.016 | ||||||
| RBANS delayed memory | 0.010 | 1.011 | 0.978 – 1.044 | –0.020 | 0.980 | 0.948 – 1.013 | ||||||
| RBANS language | 0.058 * | 1.060 | 1.011 – 1.111 | 0.052 | 1.053 | 0.989 – 1.123 | ||||||
| RBANS visuo‐construction | –0.014 | 0.985 | 0.960 – 1.012 | 0.014 | 1.015 | 0.980 – 1.051 | ||||||
| Ref. group: A+T‐N+ | A–T–N– | A+T–N– | A+T+N‐ | A+T+N+ | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | ß | OR | 95% CI for OR | |
| Model 1: Multinomial regression with RBANS total score | ||||||||||||
| RBANS total | –0.047 * | 0.954 | 0.916 – 0.994 | |||||||||
| Model 2: Multinomial regression with RBANS cognitive domains | ||||||||||||
| RBANS attention | 0.012 | 1.012 | 0.971 –1.055 | |||||||||
| RBANS immediate memory | –0.024 | 0.977 | 0.930 – 1.025 | |||||||||
| RBANS delayed memory | –0.037 | 0.964 | 0.925 – 1.005 | |||||||||
| RBANS language | –0.006 | 0.994 | 0916 – 0.999 | |||||||||
| RBANS visuo‐construction | 0.029 | 1.030 | 0.928 – 1.069 | |||||||||
NOTES: All models were corrected for age, sex, years of education, family history of dementia, and site of data acquisition. Significance value was set at P‐value < 0.05 and P‐values were reported as follows.
P < 0.05.
P < 0.01.
Abbreviations: CI, confidence interval; OR, odds ratio; RBANS, Repeatable Battery for the Assessment of Neuropsychological Status; VR, virtual reality.
In the second model, language could discriminate A+T–N+ from A–T–N–, A+T–N–, A+T+N– (P‐values < 0.05), but not from A+T+N+. Moreover, A+T–N+ performed significantly worse than A–T–N– at attention tasks (P‐value < 0.05). Immediate and delayed memory could discriminate A+T+N+ from those with no tau pathology or neurodegeneration, that is, A–T–N– and A+T–N– (P‐values < 0.05).
The third model demonstrated that the coding task was able to differentiate between A–T–N– and A+T+N– and A+T‐N+ (P‐value < 0.05), while list learning could discriminate between A–T–N– and A+T+N+ and between A+T–N– and A+T+N+ (P‐values < 0.05; Table S3 in supporting information). Age was associated with ATN stages in all models except while trying to discriminate between A–T–N– and individuals with only amyloid pathology (A+T–N–; not shown).
3.4. Regional brain volumes across the ATN stages
The results of the exploratory tests to distinguish ATN stages based on regional brain volumes are summarized in Table S4 in supporting information. Basal forebrain, precentral gyrus, middle cingulate gyrus, postcentral gyrus, middle occipital gyrus, amygdala, enthorinal cortex, and deep gray matter regions (nucleus accumbens, putamen) differed among groups with a P‐value ≤0.001. The results of the regression models distinguishing ATN stages based on regional volumetric measures are reported in Table 3. Compared to A–T–N–, the A+T–N– demonstrated higher volumes in the basal forebrain, postcentral gyrus, middle occipital gyrus, and putamen and lower volumes of enthorinal cortex and nucleus accumbens (P‐value < 0.01). The A+T+N– group had higher volumes in pre‐ and postcentral gyri compared to A–T–N (P‐value < 0.05), and lower volumes of the amygdala (P‐value < 0.001), enthorinal cortex, and nucleus accumbens (P‐value < 0.05). Finally, both A+T–N+ and A+T+N+ demonstrated lower volumes than A–T–N– and A+T–N– in the amygdala, enthorinal cortex, and nucleus accumbens (P‐value < 0.01), but these values were not significantly different between A+T–N+ and A+T+N– or between A+T+N+ and A+T–N+. A lower volume of the middle cingulate gyrus was reported in A+T+N+ compared to A+T+N– and A+T–N+ (P‐value < 0.05). The only region that was significantly different between A+T–N+ and A+T+N– was the middle occipital gyrus, showing a lower volume in A+T–N+ (Figure 3).
TABLE 3.
Results of multinomial regression models distinguishing ATN stages on the base of regional volumes. Reference (ref.) group is indicated at the top of the table
| Ref. group: A–T–N– | A+T–N– | A+T+N– | A+T–N+ | A+T+N+ | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | |
| Basal forebrain | 0.0019 | 0.0006 | 2.9199 | **0.0035 | –0.0006 | 0.0013 | –0.4552 | 0.6490 | –0.0034 | 0.0018 | –1.8584 | 0.0631 | –0.0062 | 0.0028 | –2.2163 | *0.02667 |
| Precentral gyrus | 0.0001 | 0.0001 | 1.7138 | 0.0866 | 0.0003 | 0.0001 | 2.3901 | *0.0168 | 0.0001 | 0.0001 | 0.7718 | 0.4402 | 0.0000 | 0.0002 | 0.1412 | 0.8877 |
| middle cingulate gyrus | 0.0004 | 0.0002 | 2.5897 | 0.0096 | 0.0005 | 0.0003 | 1.6198 | 0.1053 | 0.0003 | 0.0003 | 0.8713 | 0.3836 | –0.0010 | 0.0005 | –1.9986 | *0.04565 |
| postcentral gyrus | 0.0004 | 0.0001 | 4.7520 | **<0.0001 | 0.0003 | 0.0002 | 2.0679 | *0.0386 | –0.0002 | 0.0002 | –0.9109 | 0.3623 | 0.0000 | 0.0003 | 0.0040 | 0.9968 |
| middle occipital gyrus | 0.0004 | 0.0002 | 2.4888 | *0.0128 | 0.0000 | 0.0003 | 0.1590 | 0.8737 | 0.0003 | 0.0004 | 0.9349 | 0.3498 | –0.0006 | 0.0005 | –1.2101 | 0.2262 |
| Amygdala | –0.0009 | 0.0005 | –1.8602 | 0.0629 | –0.0028 | 0.0008 | –3.5115 | **0.0005 | –0.0045 | 0.0009 | –4.9321 | **<0.0001 | –0.0060 | 0.0012 | –4.9969 | **<0.0001 |
| enthorinal cortex | –0.0007 | 0.0003 | –2.8579 | **0.0043 | –0.0010 | 0.0004 | –2.3067 | *0.0211 | –0.0021 | 0.0005 | –4.1523 | **<0.0001 | –0.0030 | 0.0007 | –4.4222 | **<0.0001 |
| nucleus accumbens | –0.0068 | 0.0023 | –2.9848 | **0.0028 | –0.0032 | 0.0015 | –2.0747 | *0.0380 | –0.0072 | 0.0018 | –3.9510 | **<0.0001 | –0.0068 | 0.0023 | –2.9848 | **0.0028 |
| putamen | 0.0003 | 0.0001 | 3.9221 | **0.0001 | 0.0003 | 0.0001 | 2.1428 | *0.0321 | 0.0000 | 0.0002 | –0.0604 | 0.9518 | –0.0001 | 0.0003 | –0.4570 | 0.6477 |
| Ref. group: A+T–N– | A–T–N– | A+T+N– | A+T–N+ | A+T+N+ | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | |
| basal forebrain | –0.0025 | 0.0013 | –1.8520 | 0.0640 | –0.0052 | 0.0018 | –2.8364 | **0.0046 | –0.0081 | 0.0028 | –2.8617 | **0.0042 | ||||
| precentral gyrus | 0.0002 | 0.0001 | 1.3535 | 0.1759 | 0.0000 | 0.0002 | –0.0498 | 0.9603 | –0.0001 | 0.0002 | –0.4140 | 0.6789 | ||||
| middle cingulate gyrus | 0.0001 | 0.0003 | 0.1808 | 0.8565 | –0.0001 | 0.0003 | –0.3948 | 0.6930 | –0.0014 | 0.0005 | –2.7642 | **0.0057 | ||||
| postcentral gyrus | –0.0001 | 0.0002 | –0.5258 | 0.5990 | –0.0006 | 0.0002 | –2.8829 | **0.0039 | –0.0004 | 0.0003 | –1.4437 | 0.1488 | ||||
| middle occipital gyrus | –0.0004 | 0.0003 | –1.1370 | 0.2555 | –0.0001 | 0.0004 | –0.1764 | 0.8600 | –0.0011 | 0.0005 | –1.9504 | 0.0511 | ||||
| amygdala | –0.0019 | 0.0009 | –2.2142 | *0.02682 | –0.0036 | 0.0009 | –3.7754 | **0.0002 | –0.0051 | 0.0012 | –4.1312 | **<0.0001 | ||||
| enthorinal cortex | –0.0003 | 0.0005 | –0.6053 | 0.5450 | –0.0014 | 0.0005 | –2.6838 | **0.0073 | –0.0023 | 0.0007 | –3.3165 | **0.0009 | ||||
| nucleus accumbens | –0.0022 | 0.0016 | –1.3238 | 0.1856 | –0.0062 | 0.0019 | –3.2726 | **0.0011 | –0.0058 | 0.0023 | –2.4778 | *0.0132 | ||||
| putamen | 0.0000 | 0.0002 | –0.1635 | 0.8701 | –0.0003 | 0.0002 | –1.7736 | 0.0761 | –0.0005 | 0.0003 | –1.6631 | 0.0963 | ||||
| Ref. group: A+T+N– | A–T–N– | A+T–N– | A+T–N+ | A+T+N+ | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | |
| basal forebrain | –0.0028 | 0.0021 | –1.3166 | 0.1880 | –0.0056 | –0.0030 | 1.8900 | 0.0588 | ||||||||
| precentral gyrus | –0.0002 | 0.0002 | –1.0367 | 0.2999 | –0.0003 | 0.0002 | –1.1383 | 0.2550 | ||||||||
| middle cingulate gyrus | –0.0002 | 0.0004 | –0.4650 | 0.6420 | –0.0015 | 0.0006 | –2.6465 | **0.0081 | ||||||||
| postcentral gyrus | –0.0005 | 0.0002 | –2.1051 | *0.0353 | –0.0003 | 0.0003 | –1.0610 | 0.2887 | ||||||||
| middle occipital gyrus | 0.0003 | 0.0004 | 0.6618 | 0.5081 | –0.0007 | 0.0006 | –1.1858 | 0.2357 | ||||||||
| amygdala | –0.0017 | 0.0011 | –1.5714 | 0.1161 | –0.0032 | 0.0013 | –2.4326 | *0.0150 | ||||||||
| enthorinal cortex | –0.0011 | 0.0006 | –1.8579 | 0.0632 | –0.0020 | 0.0008 | –2.6803 | **0.0074 | ||||||||
| nucleus accumbens | –0.0041 | 0.0022 | –1.8866 | 0.0592 | –0.0036 | 0.0025 | –1.4462 | 0.1481 | ||||||||
| putamen | –0.0003 | 0.0002 | –1.4316 | 0.1523 | –0.0004 | 0.0003 | –1.4751 | 0.1402 | ||||||||
| Ref. group: A+T‐N+ | A‐T‐N‐ | A+T‐N‐ | A+T‐N+ | A+T+N+ | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | Estimate | Std. Error | z‐value | Pr(> |z|) | |
| basal forebrain | –0.0028 | 0.0031 | –0.9134 | 0.3611 | ||||||||||||
| precentral gyrus | –0.0001 | 0.0002 | –0.3384 | 0.7351 | ||||||||||||
| middle cingulate gyrus | –0.0013 | 0.0006 | –2.3099 | *0.0209 | ||||||||||||
| postcentral gyrus | 0.0002 | 0.0003 | 0.5570 | 0.5776 | ||||||||||||
| middle occipital gyrus | –0.0010 | 0.0006 | –1.6270 | 0.1037 | ||||||||||||
| amygdala | –0.0015 | 0.0013 | –1.1463 | 0.2517 | ||||||||||||
| enthorinal cortex | –0.0009 | 0.0008 | –1.1340 | 0.2568 | ||||||||||||
| nucleus accumbens | 0.0004 | 0.0026 | 0.1629 | 0.8706 | ||||||||||||
| putamen | –0.0001 | 0.0003 | –0.3601 | 0.7188 | ||||||||||||
Notes: Significant results are indicated in dark blue if they indicate a higher volume (i.e., positive regression coefficient) and in light blue if they have a lower volume (i.e., negative regression coefficient). All the analyses were corrected for age, sex, family history of dementia, brain size, and site of data collection. Significance value was set at ‐Pvalue < 0.05 and P‐values were reported as follows.
P < 0.05.
P < 0.01.
FIGURE 3.
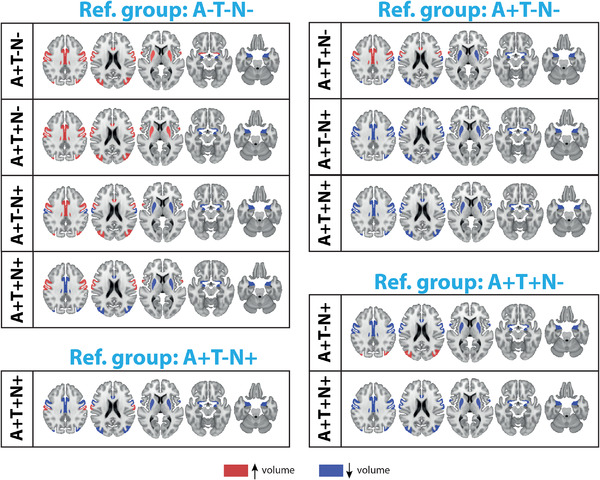
Regional volumes able to distinguish between the different amyloid/tau/neurodegeneration (ATN) stages (P‐value < 0.05). The areas indicated in red show a higher volume, while those in blue show a lower volume compared to the reference (ref.) group. Reference group is indicated at the top of the image. The regression coefficients and P‐values are reported in Table 3
4. DISCUSSION
The goal of EPAD study was to create a deeply phenotyped cohort of non‐demented subjects to select potential participants for secondary AD prevention trials.17 When classifying our dataset according to the ATN criteria,2 the portion of potential trial‐ready subjects, that is, individuals in the AD continuum, was 32.5%, of which 21.9% were A+T–N–, 3.4% A+T–N+ (atypical AD), 5.5% A+T+N–, and 1.8% A+T+N+. Brain regional volumetric differences among A–T–N– and A+T–N– were seen in the basal forebrain, postcentral and middle occipital gyri, amygdala, entorhinal cortex, nucleus accumbens, and putamen, while differences further along the AD continuum were detected by RBANS neuropsychological tests, especially in the memory domain. SNAP showed neuropsychological and neurodegeneration profiles similar to individuals in the AD continuum, but demonstrated a higher male prevalence, lower prevalence of APOE ε4 carriers, and lower cerebrovascular burden (similar to A–T–N–).
The percentage of EPAD participants in the AD continuum was lower than other AD cohorts that are partially population‐based such as the Australian Imaging, Biomarker & Lifestyle Flagship Study of Ageing (AIBL),37 or the Alzheimer's Disease Neuroimaging Initiative (ADNI;38 EPAD: 32.5%, AIBL: 45.2%, ADNI: 38.4%; Table S2 in supporting information). These differences might be explained by the younger age (average age EPAD: 65 years; AIBL: 72 years; ADNI: 74 years) and better cognitive status (EPAD CDR = 0.5: 17.9%; AIBL mild cognitive impairment [MCI]: 18.5%, ADNI MCI: 60.8%) of EPAD participants. EPAD recruited a high portion of A–T–N– (57.1%); still, these individuals often have a positive family history for AD, part of the prescreening process in EPAD,17, 18, 19 which might imply a greater chance of developing AD pathology, and that the distribution of ATN groups may shift upon longitudinal follow‐up. This is currently being studied. The prevalence of SNAP was 10.4%, generally lower than other cohorts, showing that EPAD is successful in filtering out these subjects in the pre‐screening phase.
We confirmed the CSF cut‐offs of Aβ1‐42 and p‐tau181,8 obtaining values in line with literature findings.5, 6, 7, 9, 39 The prevalence distribution across ATN stages in our study deviated from that reported by Weigand at al. on the ADNI population, especially with regard to tau positivity.40 Such differences might be explained by the different composition of the cohorts as well as the different methods used to define tau positivity, that is, CSF p‐tau in our case, positron emission tomography (PET) 18F AV1451 in the report of Weigand et al. In fact, a high rate of discordance between fluid and imaging biomarkers has been reported, suggesting that they capture different aspects of tau pathology.41
The definition of neurodegeneration remains more contentious, as the guidelines propose to use CSF t‐tau, MRI data, or fluorodeoxyglucose (FDG)‐PET (the latter of which was not available within EPAD and thus not explored). According to our data, the high correlation between p‐tau and t‐tau does not support the use of t‐tau for defining N+ within a non‐demented population. We chose to define N based on the hippocampus volume, a structure that shows high sensitivity to neurodegeneration in AD.13, 14 We used HCV rather than MTA, proposed elsewhere,13 providing finer granularity and greater data variance (being a parametric as opposed to a ordinal measure) while keeping in mind that MTA scores showed a high correlation with HCV values. The cut‐off used relates well with the clinically used average MTA score42 (Figure 2F). This definition of N had of course an impact on the percentage of N+ individuals.36 Nevertheless, these cut‐offs remain to be validated with longitudinal data.
Other brain areas also show significant atrophy in AD.14, 43 Our data suggested that PCA, although generally low, was higher with increasing biomarker positivity from A–T–N– along the AD continuum. Moreover, the middle cingulate gyrus was the only region able to distinguish between A+T–N+ and A+T+N+ stages, which otherwise showed very similar imaging profiles. The importance of such regions in distinguishing the ATN stages might be related to the age of our sample, as parietal atrophy seems to be specific for young‐onset AD. Interestingly, A+T–N– showed higher regional volumes compared to A–T–N– in the basal forebrain, postcentral and middle occipital gyri, and putamen (Table 3). This is in line with other literature findings44 and might be due to microglia activation leading to inflammation45 or to leakage of the blood‐brain barrier.46
The amygdala and entorhinal cortex also could distinguish among ATN stages, showing a lower volume in subjects with higher biomarker positivity, in contrast with one study showing higher volume in A+ individuals.47 These differences might be partially explained by the different definition in amyloid positivity.
Neuropsychological findings demonstrated that one or a few tests are insufficient for an exhaustive profiling of individuals within the AD continuum, and that composite scores help in detecting initial subtle changes in cognitive performance.48 Moreover, our data support the line of evidence claiming that neurofibrillary tangles deposition is a prerequisite for cognitive dysfunction. Indeed, RBANS total score could differentiate between A–T–N– and T+ individuals independent of their N status, but not between A–T–N– and A+T–N–, or between A+T+N– and A+T+N+, although some reports in the literature show that early cognitive dysfunction in A+ individuals might be detected.49 Moreover, the SNAP group (mostly T+) showed neuropsychological profiles similar to individuals in the AD continuum, confirming that T might drive such changes. Our findings are in line with literature, as individuals with only amyloid pathology are expected to be at risk of cognitive decline but the timeline remains unknown.3
The relationship between RBANS total score and ATN groups seems to be driven by memory tasks in individuals with neurodegeneration, although A+T‐N+ could be best distinguished from other groups—but not from A+T+N+—on the base of language tasks. It is not trivial to detect insidious cognitive dysfunction due to early AD pathological changes, thus the RBANS battery seems appropriate for the EPAD cohort. Although RBANS tests are not corrected for education, this was irrelevant for our study as the years of education were high and approximately the same for all groups (on average 14 years), probably explaining why our sample scored higher compared to the general population.
Our data confirmed that cerebrovascular disorders co‐occur with AD pathology. Although EPAD protocol restricted eligibility to individuals with no major cerebrovascular pathology (Figure S2), the degree of small vessel disease was higher in A+ individuals. In line with this, SNAP showed a lower vascular burden compared to subjects in the AD continuum. The cross‐sectional nature and the design of this study do not allow for any causal inference regarding the link between cerebrovascular disorders and AD pathology nor with regard to the extent of the impact of concomitant cerebrovascular pathology on the cognitive performance of individuals along the AD continuum.50
Other than cerebrovascular disorders, we were able to confirm several well‐known risk factors of AD‐related pathology, including APOE ε4 genotype, positive family history of dementia, and age. Specifically, based on the prevalence of APOE ε4 carriers per ATN stage, we can confirm that APOE ε4 seems to be a major driver of AD pathology, in line with current evidence.51
Some methodological concerns should be addressed. EPAD is a multicenter study, using slightly different recruitment strategies per center. As in other cohorts, our sample is not totally representative of the general population, missing minorities such as individuals with lower education and non‐Whites. Cut‐offs for ATN staging need to be validated with longitudinal data and with other non‐demented populations, investigating progression in cognitive decline for each stage. Nevertheless, we believe that this study sets firm bases for recruitment into clinical trials beyond simple neuropsychological performance using multimodal biomarkers, including CSF‐based biomarkers, advanced neuropsychological findings, and radiological outcomes.
To conclude, our findings show that, in a non‐demented population, t‐tau is not appropriate for describing neurodegeneration and cognitive dysfunction, especially in the memory domain, appears concomitantly to positivity to p‐tau. Moreover, bi‐directional regional volumetric changes might be observed in the brain, possibly reflecting neurodegenerative and inflammatory processes. Finally, cerebrovascular burden increases together with biomarker positivity along the AD continuum.
AUTHOR CONTRIBUTIONS
SI: data collection, study design, statistical analyses and data interpretation, manuscript writing; CdB: data collection, study design and coordination, manuscript writing, supervision of project; LM: data interpretation, manuscript writing; IV: statistical analyses, data interpretation; CdP: data collection and interpretation; PJV: study design, data interpretation, supervision of project; FB: study design, data interpretation, supervision of project. All authors critically revised the manuscript and approved the final version.
ETHICS APPROVAL AND CONSENT TO PARTICIPATE
The study was approved by the ethical committees of all participating centers. All study participants provided written informed consent.
AVAILABILITY OF DATA AND MATERIALS
Data can be shared upon request.
CONFLICTS OF INTEREST
The authors declare that they have no competing interests.
Supporting information
SUPPORTING INFORMATION
ACKNOWLEDGMENTS
This publication is part of the EPAD LCS (European Prevention of Alzheimer's Dementia Longitudinal Cohort Study). The authors would like to express their most sincere gratitude to the EPAD LCS participants, without whom this research would have not been possible.
Ingala S, De Boer C, Masselink LA, et al. Application of the ATN classification scheme in a population without dementia: Findings from the EPAD cohort. Alzheimer's Dement. 2021;17:1189–1204. 10.1002/alz.12292
Funding information
This work has received support from the EU/EFPIA Innovative Medicines Initiative Joint Undertaking EPAD grant agreement n◦ 115736.AMW, VW, and FB have received funding from the European Union's Horizon 2020 research and innovation program under grant agreement No. 666992. JDG holds a “Ramón y Cajal” fellowship (RYC‐2013‐13054). FB is supported by the NIHR UCLH biomedical research center. WvdF and FB are recipients of a JPND grand for E‐DADS (project number 733051106). Research of the Alzheimer Center Amsterdam is part of the neurodegeneration research program of Amsterdam Neuroscience. The Alzheimer Center Amsterdam is supported by Stichting Alzheimer Nederland and Stichting VUmc fonds. WF is recipient of ZonMW Memorabel (ABIDE; project no 733050201), a project in the context of the Dutch Deltaplan Dementie.
REFERENCES
- 1.Ritchie CW, Molinuevo JL, Truyen L, Satlin A, Van der Geyten S, Lovestone S. Development of interventions for the secondary prevention of Alzheimer's dementia: the European Prevention of Alzheimer's Dementia (EPAD) project. The Lancet Psychiatry. 2016;3:179‐186. [DOI] [PubMed] [Google Scholar]
- 2.Jack CR, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimer's Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.ten Kate M, Ingala S, Schwarz AJ, et al. Secondary prevention of Alzheimer's dementia: neuroimaging contributions. Alzheimers Res Ther. 2018;10:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Shaw LM, Waligorska T, Fields L, et al. Derivation of cutoffs for the Elecsys® amyloid β (1‐42) assay in Alzheimer's disease. Alzheimer's Dement Diagnosis. Assess Dis Monit. 2018;10:698‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Vogelgsang J, Wedekind D, Bouter C, Klafki HW, Wiltfang J. Reproducibility of Alzheimer's Disease Cerebrospinal Fluid‐Biomarker Measurements under Clinical Routine Conditions. J Alzheimers Dis. 2018;62:203‐212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schindler SE, Gray JD, Gordon BA, et al. Cerebrospinal fluid biomarkers measured by Elecsys assays compared to amyloid imaging. Alzheimer's Dement. 2018;14:1460‐1469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shaw LM, Waligorska T, Fields L, et al. Derivation of cutoffs for the Elecsys((R)) amyloid beta (1‐42) assay in Alzheimer's disease. Alzheimers Dement (Amst). 2018;10:698‐705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐beta PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimers Dement. 2018;14:1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Willemse EAJ, van Maurik IS, Tijms BM, et al. Diagnostic performance of Elecsys immunoassays for cerebrospinal fluid Alzheimer's disease biomarkers in a nonacademic, multicenter memory clinic cohort: the ABIDE project. Alzheimers Dement (Amst). 2018;10:563‐572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Hansson O, Seibyl J, Stomrud E, et al. CSF biomarkers of Alzheimer's disease concord with amyloid‐β PET and predict clinical progression: a study of fully automated immunoassays in BioFINDER and ADNI cohorts. Alzheimer's Dement. 2018;14:1470‐1481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mattsson‐Carlgren N, Leuzy A, Janelidze S, et al. The implications of different approaches to define AT(N) in Alzheimer disease. Neurology. 2020;94:e2233‐e2244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Jack CR Jr, Bennett DA, Blennow K, et al. NIA‐AA Research Framework: toward a biological definition of Alzheimer's disease. Alzheimers Dement. 2018;14:535‐562. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Altomare D, De Wilde A, Ossenkoppele R, et al. Applying the ATN scheme in a memory clinic population: the ABIDE project. Neurology. 2019;93:E1635‐E1646. [DOI] [PubMed] [Google Scholar]
- 14.Pini L, Pievani M, Bocchetta M, et al. Brain atrophy in Alzheimer's Disease and aging. Ageing Res Rev. 2016;30:25‐48. [DOI] [PubMed] [Google Scholar]
- 15.Persson K, Barca ML, Cavallin L, et al. Comparison of automated volumetry of the hippocampus using NeuroQuant(R) and visual assessment of the medial temporal lobe in Alzheimer's disease. Acta Radiol. 2018;59:997‐1001. [DOI] [PubMed] [Google Scholar]
- 16.Clerx L, van Rossum IA, Burns L, et al. Measurements of medial temporal lobe atrophy for prediction of Alzheimer's disease in subjects with mild cognitive impairment. Neurobiol Aging. 2013;34:2003‐2013. [DOI] [PubMed] [Google Scholar]
- 17.Solomon A, Kivipelto M, Molinuevo JL, Tom B, Ritchie CW. European Prevention of Alzheimer's Dementia Longitudinal Cohort Study (EPAD LCS): study protocol. BMJ Open. 2019;8:e021017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Vermunt L, Veal CD, ter Meulen L, et al. European Prevention of Alzheimer's Dementia Registry: recruitment and prescreening approach for a longitudinal cohort and prevention trials. Alzheimer's Dement. 2018;14:837‐842. [DOI] [PubMed] [Google Scholar]
- 19.Vermunt L, Muniz‐Terrera G, Ter Meulen L, et al. Prescreening for European Prevention of Alzheimer Dementia (EPAD) trial‐ready cohort: impact of AD risk factors and recruitment settings. Alzheimer's Res Ther. 2020;12:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Ritchie K, Ropacki M, Albala B, et al. Recommended cognitive outcomes in preclinical Alzheimer ’ s disease : consensus statement from the European Prevention of Alzheimer ’ s Dementia project. Alzheimer's Dement. 2017;13:186‐195. [DOI] [PubMed] [Google Scholar]
- 21.Muniz Terrera G, Harrison JE, Ritchie CW, Ritchie K. Cognitive Functions as Predictors of Alzheimer's Disease Biomarker Status in the European Prevention of Alzheimer's Dementia Cohort. J Alzheimer's Dis. 2020;74:1203‐1210. [DOI] [PubMed] [Google Scholar]
- 22.Folstein MF, Robins LN, Helzer JE. The Mini‐Mental State Examination. Arch Gen Psychiatry. 1983;40:812. [DOI] [PubMed] [Google Scholar]
- 23.Morris JC. The Clinical Dementia Rating (CDR). Neurology. 1993;43:2412‐2412‐a. [DOI] [PubMed] [Google Scholar]
- 24.Yesavage JA, Brink TL, Rose TL, et al. Development and validation of a geriatric depression screening scale: a preliminary report. J Psychiatr Res. 1982;17:37‐49. [DOI] [PubMed] [Google Scholar]
- 25.Randolph C, Tierney MC, Mohr E, Chase TN. The Repeatable Battery for the Assessment of Neuropsychological Status (RBANS): preliminary clinical validity. J Clin Exp Neuropsychol. 1998;20:310‐319. [DOI] [PubMed] [Google Scholar]
- 26.Hartley T, Bird CM, Chan D, et al. The hippocampus is required for short‐term topographical memory in humans. Hippocampus. 2007;17:34‐48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Tu S, Wong S, Hodges JR, Irish M, Piguet O, Hornberger M. Lost in spatial translation ‐ A novel tool to objectively assess spatial disorientation in Alzheimer's disease and frontotemporal dementia. Cortex. 2015;67:83‐94. [DOI] [PubMed] [Google Scholar]
- 28.Fazekas F, Chawluk JB, Alavi A, Hurtig HI, Zimmerman RA. MR signal abnormalities at 1.5 T in Alzheimer's dementia and normal aging. Am J Roentgenol. 1987;149:351‐356. [DOI] [PubMed] [Google Scholar]
- 29.Wardlaw JM, Smith EE, Biessels GJ, et al. Position Paper Neuroimaging standards for research into small vessel disease and its contribution to ageing and neurodegeneration. Lancet Neurol. 2013;12:822‐838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cordonnier C, Potter GM, Jackson CA, et al. Improving interrater agreement about brain microbleeds: development of the Brain Observer MicroBleed Scale (BOMBS). Stroke. 2009;40:94‐99. [DOI] [PubMed] [Google Scholar]
- 31.Potter GM, Chappell FM, Morris Z, Wardlaw JM. Cerebral perivascular spaces visible on magnetic resonance imaging: development of a qualitative rating scale and its observer reliability. Cerebrovasc Dis. 2015;39:224‐231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Scheltens P, Leys D, Barkhof F, et al. Atrophy of medial temporal lobes on MRI in “probable” Alzheimer's disease and normal ageing: diagnostic value and neuropsychological correlates. J Neurol Neurosurg Psychiatry. 1992;55:967‐972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Koedam ELGE, Lehmann M, Van Der Flier WM, et al. Visual assessment of posterior atrophy development of a MRI rating scale. Eur Radiol. 2011;21:2618‐2625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jack CR, Wiste HJ, Weigand SD, et al. Defining imaging biomarker cut points for brain aging and Alzheimer's disease. Alzheimer's Dement. 2017;13:205‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Dickerson BC, Stoub TR, Shah RC, et al. Alzheimer‐signature MRI biomarker predicts AD dementia in cognitively normal adults. Neurology. 2011;76:1395‐1402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jack CR, Petersen RC, Xu YC, et al. Prediction of AD with MRI‐based hippocampal volume in mild cognitive impairment. Neurology. 1999;52:1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Burnham S, Bourgeat P, Doré V, et al. IMPLEMENTING THE ATN CLASSIFICATION IN AIBL. Alzheimer's Dement. 2017;13:P1511. [Google Scholar]
- 38.Guo T, Landau SM, Jagust WJ. Longitudinal amyloid, neurodegeneration and cognition in pre‐dementia elderly individuals with different ATN profiles. Alzheimer's Dement J Alzheimer's Assoc. 2019;15:P1485. [Google Scholar]
- 39.Milà‐Alomà M, Sánchez‐Benavides G, Grau‐Rivera O, et al. Prevalence of amyloid‐β and tau pathology in middle‐aged cognitively unimpaired individuals: the alfa study. Alzheimer's Dement. 2019;15:P1341‐P1342. [Google Scholar]
- 40.Weigand AJ, Bangen KJ, Thomas KR, et al. Is tau in the absence of amyloid on the Alzheimer's continuum?: a study of discordant PET positivity. Brain Commun. 2020;2:1‐18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.La Joie R, Bejanin A, Fagan AM, et al. Associations between [18F]AV1451 tau PET and CSF measures of tau pathology in a clinical sample. Neurology. 2018;90:E282‐E290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rhodius‐Meester HFM, Benedictus MR, Wattjes MP, et al. MRI visual ratings of brain atrophy and white matter hyperintensities across the spectrum of cognitive decline are differently affected by age and diagnosis. Front Aging Neurosci. 2017;9:1‐12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Tondelli M, Wilcock GK, Nichelli P, De Jager CA, Jenkinson M, Zamboni G. Structural MRI changes detectable up to ten years before clinical Alzheimer's disease. Neurobiol Aging. 2012;33:825. e25‐36. [DOI] [PubMed] [Google Scholar]
- 44.Femminella GD, Dani M, Wood M, et al. Microglial activation in early Alzheimer trajectory is associated with higher gray matter volume. Neurology. 2019;92:E1331‐E1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keren‐Shaul H, Spinrad A, Weiner A, et al. A Unique Microglia Type Associated with Restricting Development of Alzheimer's Disease. Cell. 2017;169:1276‐1290.e17. [DOI] [PubMed] [Google Scholar]
- 46.Montagne A, Nation DA, Sagare AP, et al. APOE4 leads to blood–brain barrier dysfunction predicting cognitive decline. Nature. 2020;581:71‐76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chételat G, Villemagne VL, Pike KE, et al. Larger temporal volume in elderly with high versus low beta‐amyloid deposition. Brain. 2010;133:3349‐3358. [DOI] [PubMed] [Google Scholar]
- 48.Jonaitis EM, Koscik RL, Clark LR, et al. Measuring longitudinal cognition: individual tests versus composites. Alzheimer's Dement Diagnosis. Assess Dis Monit. 2019;11:74‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hedden T, Oh H, Younger AP, Patel TA. Meta‐analysis of amyloid‐cognition relations in cognitively normal older adults. Neurology. 2013;80:1341‐1348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Carmichael O, Schwarz C, Drucker D, et al. Longitudinal changes in white matter disease and cognition in the first year of the Alzheimer disease neuroimaging initiative. Arch Neurol. 2010;67:1370‐1378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Hunsberger HC, Pinky PD, Smith W, Suppiramaniam V, Reed MN. The role of APOE4 in Alzheimer's disease: strategies for future therapeutic interventions. Neuronal Signal. 2019;3:1‐15. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
SUPPORTING INFORMATION
Data Availability Statement
Data can be shared upon request.