

Abstract
Aims
The aim of this study was to identify risk variants and haplotypes that impair dihydropyrimidine dehydrogenase (DPD) activity and are, therefore, candidate risk variants for severe toxicity to 5‐fluorouracil (5‐FU) chemotherapy.
Methods
Plasma dihydrouracil/uracil (UH2/U) ratios were measured as a population marker for DPD activity in a total of 1382 subjects from 4 independent studies. Genotype and haplotype correlations with UH2/U ratios were assessed.
Results
Significantly lower UH2/U ratios (panova < 2 × 10−16) were observed in carriers of the 4 well‐studied 5‐FU toxicity risk variants with mean differences (MD) of −43.7% for DPYD c.1905 + 1G > A (rs3918290), −46.0% for DPYD c.1679T > G (rs55886062), −37.1%, for DPYD c.2846A > T (rs67376798), and −13.2% for DPYD c.1129‐5923C > G (rs75017182). An additional variant, DPYD c.496A > G (rs2297595), was also associated with lower UH2/U ratios (P < .0001, MD: −12.6%). A haplotype analysis was performed for variants in linkage disequilibrium with c.496A > G, which consisted of the common variant c.85T > C (rs1801265) and the risk variant c.1129‐5923C > G. Both haplotypes carrying c.496A > G were associated with decreased UH2/U ratios (H3, P = .003, MD: −9.6%; H5, P = .002, MD: −16.9%). A haplotype carrying only the variant c.85T > C (H2) was associated with elevated ratios (P = .004, MD: +8.6%).
Conclusions
Based on our data, DPYD‐c.496A > G is a strong candidate risk allele for 5‐FU toxicity. Our data suggest that DPYD‐c.85T > C might be protective; however, the deleterious impacts of the linked alleles c.496A > G and c.1129‐5923C > G likely limit this effect in patients. The possible protective effect of c.85T > C and linkage disequilibrium with c.496A > G and c.1129‐5923C > G may have hampered prior association studies and should be considered in future clinical studies.
Keywords: 5‐fluorouracil, chemotherapy, dihydropyrimidine dehydrogenase, DPYD, haplotype, pharmacogenetics, uracil
What is already known about this subject
Rare enzyme impairing variants in the DPYD gene are predictive biomarkers for 5‐FU related toxicities.
However, not all cases can be explained by these rare variants.
Two common DPYD variants (c.85T > C and c.496A > G) are controversially discussed for their impact on DPD phenotype and role in 5‐FU toxicity.
What this study adds
Our study found that the effects of c.85T > C and c.496A > G depend on haplotype structure.
We observed an enzyme activity enhancing effect for c.85C, whereas c.496G was associated with impaired activity.
Multi‐locus effects within DPYD may evolve as a marker for prediction of 5‐FU related toxicities.
1. INTRODUCTION
The uracil catabolizing enzyme dihydropyrimidine dehydrogenase (DPD), which is encoded by the DPYD gene, is crucial for the catabolism of the fluoropyrimidine (FP) drug 5‐fluorouracil (5‐FU). Cancer patients with impaired DPD activity are at higher risk of developing severe 5‐FU related toxicities compared to patients with normal DPD function. Such impairment in DPD activity can result from genetic variation in DPYD.1 Currently, 4 DPYD risk variants (c.1905 + 1G > A, rs3918290; c.1679T > G, rs55886062; c.2846A > T, rs67376798; and c.1129‐5923C > G, rs75017182, which is tagged by c.1236G > A/HapB3) are considered clinically relevant markers for predicting 5‐FU‐related toxicities pretherapy.2, 3 While these 4 DPYD risk variants are clinically important, they account for only a fraction of toxicity cases.2 Other variants and haplotypes of the polymorphic DPYD gene remain to be evaluated for their effect on the DPD phenotype, as well as their clinical relevance.
The impact of certain additional DPYD variants is not as straightforward and requires further investigation to reconcile disparate results. For example, DPYD c.85T > C (rs1801265) and c.496A > G (rs2297595) are both exonic single nucleotide polymorphisms (SNPs) that lead to amino acid changes in the DPD protein (p.C29R and p.M166V, respectively). The c.85T > C variant was described as deleterious upon discovery because it was initially observed in DPD deficient patients.4 Recombinant p.C29R‐containing DPD showed impaired protein function when expressed in Escherichia coli.4 However, later studies failed to corroborate those conclusions.5 Clinical studies also report disparate results in regard to the role of c.85T > C in 5‐FU related toxicities. Two recent studies suggest a protective effect for the c.85C allele, which suggests that DPD activity might be higher in carriers of c.85C.6, 7 However, other studies failed to show a protective effect8, 9, 10 or suggested association with increased toxicity risk,11, 12 which, could not be replicated.12 Similar discrepancies have been reported for c.496A > G: Some studies suggest that c.496G is linked to FP toxicitiy,8, 9, 13 while others fail to demonstrate an association.6, 7, 14 Another study has suggested a protective effect for the variant.15 In vitro studies have similarly yielded inconclusive data pertaining to the effect of these variants on DPD function.16, 17, 18 Guidelines from the Clinical Pharmacogenetics Implementation Consortium (CPIC) have graded both variants as normal‐function alleles due to the lack of clear evidence linking these variants with 5‐FU toxicity.2 Our previous study suggested linkage disequilibrium (LD) between c.85T > C and c.496A > G19; however, detailed multi‐SNP analyses of these variants have not been reported.
In the present study, endogenous plasma dihydrouracil/uracil (UH2/U) ratios were used as a surrogate marker for systemic DPD activity. Previous studies have suggested that carriers of deleterious DPYD variants display significantly reduced steady‐state plasma UH2/U ratios, consistent with lower systemic DPD activity.7, 20 However, high interindividual variability and small population sizes limited the statistical power of these studies.7, 20 The goals of the present study were to establish population‐level reference values for UH2/U ratios in carriers of deleterious DPYD variants and to identify correlations between UH2/U ratios and multimarker DPYD haplotypes linked to c.85T > C and c.496A > G variants within in a large population.
2. METHODS
2.1. Study populations
Four independent study cohorts were evaluated. For 2 cohorts, previously published UH2/U ratio data were used. These include data from 320 healthy blood donors, referred to herein as Sistonen et al.20 and pretreatment data from 550 cancer patients, referred to as Meulendijks et al.21 For the third cohort, plasma UH2/U ratios were measured in 204 subjects from the Mayo Clinic Biobank. Details of this cohort, which is referred to as Nie et al., have been previously reported.22 For the fourth cohort, plasma samples from 308 healthy blood donors were collected in 2017 at the Regional Blood Transfusion Service of the Swiss Red Cross, Bern, Switzerland. This previously unpublished cohort is referred to herein as Hamzic et al. The Nie et al.22 cohort was enriched for carriers of DPYD risk variants (c.1905 + 1G > A, c.1679T > G, c.2846A > T, and c.1129‐5923C > G) as previously detailed22; therefore, it is understood that any assessments of allele frequency within this population would be biased in that regard. Specimens from the Sistonen et al.20 and Hamzic et al. cohorts were obtained without intentional enrichment for specific genotypes and, therefore, can be considered representative of the populations from which they were derived. The Meulendijks et al. population21 was prescreened for c.1905 + 1G > A carriers, which were excluded. Genotype data for c.85T > C and c.496A > G were not available from the Meulendijks et al. cohort.21 All cohorts had information on uracil and dihydrouracil levels, age, sex and DPYD risk variants. Information about ethnic background was only available for the Meulendijks et al. study,21 in which ~95% of patients were of Caucasian origin. The Sistonen et al.20 and Hamzic et al. cohorts were collected from Swiss blood donors, which are expected to be predominantly of Caucasian origin. The population in Nie et al.22 was restricted to individuals that self‐declared race as white (Figure S1). For all contributing studies, authors stated that they obtained appropriate institutional review board approval or followed the principles outlined in the Declaration of Helsinki for all human or animal experimental investigations. In addition, for investigations involving human subjects, informed consent has been obtained from the participants involved.
2.1.1. Sample processing
The blood samples from Hamzic et al. were collected on ice and processed within 2 hours of collection. Plasma was isolated from whole blood collected in EDTA tubes and stored at −80°C. DNA was extracted from buffy coat layers using the Qiagen DNA Blood Mini Kit. DNA concentration and quality were measured using a NanoDrop 1000 spectrophotometer. DNA samples were stored at −20°C prior to genetic analyses. Plasma samples from Nie et al.22 were isolated within 2 hours of collection and stored as described above for Hamzic et al. DNA from Nie et al. was prepared as previously detailed.22 For the Sistonen et al. and Meulendijks et al. cohorts,20, 21 only available data were used; therefore, no additional sample processing was required for these cohorts. However, for each of these cohorts,20 , 21 the blood samples were processed within 1 hour of collection. An overview of the sample processing for each individual cohort is available in Figure S1.
2.2. Quantitation of metabolites in plasma
In the Hamzic et al. and Nie et al.22 cohorts, endogenous plasma uracil and dihydrouracil levels were measured using a liquid chromatography–tandem mass spectrometry method previously detailed by Büchel et al.23 The mass spectrometric analysis was performed by multiple reaction monitoring on a Sciex QTrap 5500 mass spectrometer.
2.3. SNP genotyping
The Hamzic et al. cohort was genotyped for DPYD variants (c.1905 + 1G > A, c.1679T > G, c.2846A > T, c.1129‐5923C > G) using previously validated TaqMan assays (Thermo Fisher Scientific) on a Quantstudio 6 in 384‐well format. All DPYD risk variants were validated by Sanger sequencing. The DPYD variant c.85T > C (rs1801265) was genotyped using custom‐designed KASP genotyping assays (KASP, Biosearch Technologies). Genotyping assays for c.85T > C and c.496A > G were retrospectively validated with previously genotyped samples from Sistonen et al.20 and Sanger sequencing. Genotype information on DPYD risk variants was previously reported for Nie et al.22; c.85T > C and c.496A > G were newly genotyped for this study using custom‐designed rhAMP SNP Assays (Integrated DNA Technologies) using a LightCycler 480 System.
2.4. Data and statistical analysis
The data and statistical analysis comply with the recommendations on experimental design and analysis in pharmacology.24
2.5. Model building and single marker analysis
All statistical analyses were performed using R version 3.6.1 (R Foundation for Statistical Computing, Vienna, Austria) on R Studio v1.2.5001 (R‐studio Inc., Boston, MA, USA). To analyse the association of DPYD variants with UH2/U‐ratios, a linear mixed model was used to account for potential study cohort effects. UH2/U‐ratios were log2‐transformed to account for non‐normal distributions (Figure S1). Age and sex were tested independently for association with UH2/U‐ratios and were included in models analyzing the effect of genetic variants if P < .2.25 For genetic association testing, DPYD variants and sex were treated as fixed effects, whereas the study cohort was coded as a blocking factor and included as a random effect variable in the model. Linear mixed modelling was performed using the lmerTest package26 and the lmer function27 in R. In more detail, analysis of associations of DPYD risk variants with UH2/U ratios included all 4 DPYD risk variants (c.1905 + 1G > A, c.1679T > G, c.2846A > T, and c.1129‐5923C > G) in 1 linear mixed model with sex (female) as a fixed co‐factor and cohort as a random factor: lmer (log2ratio ~ risk_name + female + (1|cohort), data=df). The model assessing associations of common DPYD polymorphisms with UH2/U ratios was performed individually for each variant (c.85T > C and c.496A > G) and adjusted for DPYD risk variant carrier status (cofactor: risk): lmer (log2ratio ~ c.85T.C + female + risk + (1|cohort), data=df) and lmer (log2ratio ~ c.496A.G + female + risk + (1|cohort), data=df), respectively.
2.6. Haplotype analysis
LD between individual DPYD variants was calculated with the genetics package in R.28 Variants, which were significantly linked consistently in all investigated cohorts, were used for haplotype analysis. The samples from Meulendijks et al.21 were excluded for this analysis because c.85T > C and c.496A > G genotypes were not available. The haplo.glm function of the haplo.stats package in R was used for haplotype inference.29, 30 This method is permissive for ambiguous haplotypes and allows multivariate analysis (model was adjusted to sex and DPYD risk variant carrier status). To account for ambiguity in haplotype inference, the posterior probabilities of the haplotypes were used as weights for the regression coefficient. To compare haplotype frequencies in different populations, phase 3 data from the 1000 Genomes Project, accessed through the API of LDLink (https://ldlink.nci.nih.gov/), was assessed using the LDhap function.31 Circular bar plots were generated using ggplot2 in R‐Studio.32
2.7. Correction for multiple testing
We set the threshold for statistical significance in the linkage analysis as P < .0083 (Bonferroni correction n = 6, α = .05). Consistent significant LD was noted between 3 (c.85T > C, c.496A > G, and c.1129‐5923C > G) of the 6 investigated variants. To account for this partial genetic correlation, we used a threshold of P < .0125 for the single‐marker analysis (Bonferroni correction n = 4, α = .05). A threshold of P < .01 was used for haplotype analyses because 5 haplotypes were tested for association with UH2/U ratios (Bonferroni correction n = 5, α = .05).
2.8. Nomenclature of targets and ligands
Key protein targets and ligands in this article are hyperlinked to corresponding entries in http://www.guidetopharmacology.org, and are permanently archived in the Concise Guide to PHARMACOLOGY 2019/20.33
3. RESULTS
3.1. Population characteristics
This study included data from 4 cohorts. Study characteristics are summarized in Table 1. Data on DPYD variants c.85T > C, c.496A > G, and c.1905 + 1G > A were available for 3 of the 4 cohorts (Table 2). Notably, the distribution and levels of UH2/U ratios measured in healthy volunteers (Sistonen et al.20 and Hamzic et al.) were comparable to the pretherapeutic UH2/U levels measured in cancer patients (Meulendijks et al.).21 The median UH2/U ratio was lower in Nie et al.22 compared to the other 3 cohorts (Table 1, Figure S1), which was accounted for in subsequent analyses by including study cohort as factor in the multivariate regression. Differences in age and sex distributions were noted among the cohorts, which was attributed to differences in study design. In the univariate model, we observed a P‐value below the predefined model‐inclusion threshold value of 0.2 for sex but not for age; therefore, sex was included as a factor in the multivariate model.
TABLE 1.
Study characteristics
| Characteristics | Sistonen et al.20 (n = 320) | Hamzic et al. (n = 308) | Meulendijks et al.21 (n = 550) | Nie et al.22 (n = 204) |
|---|---|---|---|---|
| Study cohort | Healthy volunteers | Healthy volunteers | Cancer patients | Biobank samples |
| Median age (y) | 46 | 50 | 59 | 61 |
| Male (%) | 228 (71%) | 204 (66%) | 232 (42%) | 84 (41%) |
| Female (%) | 92 (29%) | 104 (34%) | 318 (58%) | 120 (59%) |
| Median UH2/U ratio (ng/mL) | 11.4 | 11.2 | 11.2 | 8.7 |
UH2/U, dihydrouracil/uracil
TABLE 2.
Variants in DPYD are associated with altered UH2/ dihydrouracil/uracil ratios
| Allele frequencies | |||||||||
|---|---|---|---|---|---|---|---|---|---|
| Genetic variant | P‐valuea | Log2 βa | % change in ratiosa | Combined cohortb | Sistonen et al.20 | Hamzic et al. | Meulendijks et al.21 | Nie et al.22, c | dbSNPd (EUR) |
| c.1129‐5923C > G (rs75017182) | .0003 | −0.20 | −13.2% | 3.66% | 1.56% | 2.11% | 2.00% | 13.72% | 2.39% |
| c.1679T > G (rs55886062) | 9.2 × 10 −7 | −0.89 | −46.0% | 0.29% | 0.32% | 0.32% | 0.18% | 0.49% | 0.06% |
| c.1905 + 1G > A (rs3918290) | 1.5 × 10 −9 | −0.83 | −43.7% | 0.54% | 0.16% | 0.16% | 0b | 3.18% | 0.50% |
| c.2846A > T (rs67376798) | 8.0 × 10 −10 | −0.67 | −37.1% | 0.83% | 0.31% | 0.48% | 0.54% | 2.90% | 0.42% |
| c.85T > C (rs1801265) | .067 | +0.06 | +2.4% | 24.63%b | 22.50% | 23.00% | NAb | 30.10% | 21.79% |
| c.496A > G (rs2297595) | 8.7 × 10 −6 | −0.20 | −12.6% | 10.81%b | 12.50% | 11.50% | NAb | 7.10% | 11.93% |
P‐values and β‐coefficients were calculated in the complete cohort using a multivariate model with sex, study cohort, and DPYD risk variants as independent variables; P‐values < .01 are in bold, and % change in ratios is given per allele. DPYD risk variants have been included in the same linear mixed model using an ANOVA‐based approach (P anova = 2 × 10−16). For c.85T > C and c.496A > G, individual linear mixed models were performed including DPYD risk status as a co‐factor.
The study from Meulendijks et al.21 excluded carriers of c.1905 + 1G > A in their study and was not genotyped for c.85T > C and c.496A > G; the complete study population size for c.85T > C and c.496A > G is n = 832.
The cohort of Nie et al.22 was enriched for DPYD risk variant carriers (c.1129‐5923C > G,c.1679T > G, c.1905 + 1G > A, and c.2846A > T).
European population (EUR), https://www.ncbi.nlm.nih.gov/snp.
3.2. The impact of known toxicity‐associated DPYD variants on plasma UH2/U ratios
To determine the extent to which known toxicity‐associated variants impact DPD function in vivo, we investigated the effects of c.1905 + 1G > A, c.1679T > G, c.2846A > T, and c.1129‐5923C > G on plasma UH2/U ratios. As expected, all 4 risk variants were individually associated with lower UH2/U ratios (Figure 1 and Table 2; P anova < 2.0 × 10−16), confirming impaired enzyme activity for carriers of these variants. As expected, a stronger mean decrease in UH2/U ratios was observed in individuals that were heterozygous for a nonfunctional variant, c.1905 + 1G > A or c.1679T > G, compared to heterozygous carriers of a decreased function variant, c.2846A > T or c.1129‐5923C > G (Figure 1 and Table 2). No compound heterozygous or homozygous risk variant carriers were observed in any of the 4 cohorts. At the level of individual cohorts, we observed, in general, similar effect sizes (i.e. β‐coefficients) for the associations of DPYD variants with UH2/U ratios, indicating that our results were not likely to have been driven by a single study (Figure S3).
FIGURE 1.
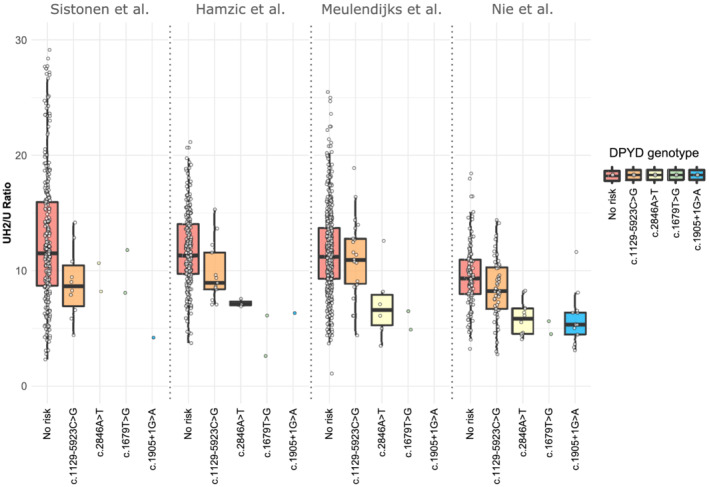
Distribution of dihydrouracil/uracil (UH2/U) ratios according to DPYD risk genotype. Boxplots represent UH2/U ratios according to study cohorts and DPYD risk variant carriers in different colours. From left to right, the populations carrying: none of the 4 risk variants (red), c.1129‐5923G (orange), c.2846 T (yellow), c.1679G (green), and c.1905 + 1A (blue). All DPYD risk variant carriers were heterozygous for the mutation. The study from Meulendijks et al.21 excluded carriers of c.1905 + 1G > A in their study. The Nie et al.22 cohort was enriched for DPYD risk variant carriers (c.1129‐5923C > G, c.1679T > G, c.1905 + 1G > A, and c.2846A > T). Each variant was significantly associated with decreased ratios (Table 2). The boxes represent the first and third quartile, and the black bar represents the median. The whiskers represent 1.5× of the interquartile range
3.3. The effects of c.85T > C and c.496A > G on DPD activity depend on haplotype structure
We previously reported LD between c.85T > C and c.496A > G.19 In the present study, we observed statistically significant LD between c.85T > C, c.496A > G, and c.1129‐5923C > G (Figure S2). Notably, statistically significant linkage between c.85C/c.496G and between c.85C/c.1129‐5923G was consistently observed in all cohorts (P < .0083). The linkage between c.496G and c.1129‐5923G was only significant in the combined cohort, probably due to a lack of statistical power in the other cohorts. The D′‐values observed in the combined cohort were comparable to the values reported for the European population in the LDLink database,31 i.e. 0.66 vs. 0.69 for c.85C/c.496G, 0.90 vs. 0.89 for c.85C/c.1129‐5923G, and 0.99 vs. 1.0 for c.496G/c.1129‐5923G (Figure S2). In contrast, the R 2‐values are slightly different for c.85C/c.496G and c.85C/c.1129‐5923G compared to the values reported in the LDLink database31: 0.16 vs. 0.24 and 0.12 vs. 0.07, respectively. This can be explained by the above‐mentioned enrichment of c.1129‐5923G‐ and other DPYD‐risk variant carriers in the Nie et al. cohort.22 In more detail, this enrichment led to allele frequency differences between the combined cohort and the reference European population, as shown in Table 2, affecting the R 2‐values. No significant LD was detected between the 3 rare DPYD risk variants c.1679T > G, c.1905 + 1G > A and c.2846A > T and all other variants (Figure S2).
At the single variant level, c.496A > G was significantly associated with lower UH2/U ratios (Table 2 and Figure S4). The c.85T > C variant did not show significant association with altered UH2/U ratios (Table 2 and Figure S4).
Because of the LD observed between c.85T > C, c.496A > G and c.1129‐5923C > G, as well as the discordant evidence in the literature concerning potential contributions of c.85T > C and c.496A > G to FP toxicity risk, we sought to perform expanded haplotype analyses on these 3 variants within our study cohorts. Haplotype phasing for most samples could be directly observed based on genotypes (n = 670, 81%). For the remaining samples (n = 162, 19%), paired diplotypes were inferred computationally. Posterior probabilities >94.5% were observed for 160 (99%) of the 162 inferred haplotype pairs. The 2 remaining haplotype pairs (1%) had posterior probabilities of 80.0%. The most frequent haplotype was termed H1 and consisted of all wild‐type alleles (Table 3). The H1 haplotype was therefore considered the base haplotype to which other haplotypes were compared. The c.85T > C variant was present in 3 haplotypes (H2–H4), and the c.496A > G variant was present in haplotypes H3 and H5. The c.1129‐5923C > G variant was present in 2 haplotypes, H4 and H6.
TABLE 3.
Association of c.85T > C and c.496A > G with UH2/U ratios depends on haplotype structure
| Haplotype frequencies | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Haplotype | DPYD c.85T > C | DPYD c.496A > G | DPYD c.1129‐5923C > G | P‐valuea | Log2 βa | % change in ratiosa | Combined cohortb | Sistonen et al.20 | Hamzic et al. | Nie et al.22 | LD LINKc |
| (%/n) | (%) | (%) | (%) | (%) | |||||||
| H1 | T | A | C | Base haplotype | 72.2/1201 | 74.3 | 73.9 | 66.8 | 75.6 | ||
| H2 | C | A | C | .004 | 0.119 | 8.6% | 12.2/203 | 11.8 | 12.4 | 12.4 | 10.2 |
| H3 | C | G | C | .003 | −0.146 | −9.6% | 8.1/135 | 9.1 | 9.1 | 4.9 | 9.1 |
| H4 | C | A | G | <.0001 | −0.266 | −16.8% | 4.4/73 | 1.4 | 1.7 | 12.8 | 2.2 |
| H5 | T | G | C | .002 | −0.267 | −16.9% | 2.7/45 | 3.2 | 2.5 | 2.2 | 2.9 |
| H6 | T | A | G | .765 | −0.069 | −4.7% | 0.4/7 | 0.0 | 0.4 | 0.9 | 0.0 |
Minor alleles are in bold and underlined.
β‐coefficients and P‐values were calculated in the complete cohort using a multivariate model with sex, study cohort and DPYD risk variants as independent variables; P‐values < .01 are in bold, and % change in ratios is given per allele.
The observed haplotype frequency does not reflect true population frequencies because the Nie et al. cohort is enriched for DPYD risk variants. Total sample size n = 1664 haplotypes.
Frequencies for the European population obtained from LDhap Tool, https://ldlink.nci.nih.gov/.
Of the haplotypes containing c.85T > C, H3 and H4 were associated with lower UH2/U ratios, whereas H2, which only contained c.85T > C, was associated with elevated UH2/U ratios (Table 3). Both c.496A > G‐containing haplotypes, H3 and H5, were associated with significantly lower UH2/U ratios. A stronger effect size was observed for H5, which only contained c.496A > G, compared to H3, which also contained c.85T > C. Haplotype H4, which contained both c.1129‐5923C > G and c.85T > C, was associated with significantly decreased UH2/U ratios. The rarest haplotype, H6, contained only c.1129‐5923C > G and was the only haplotype with UH2/U ratios that were not significantly different from H1, which was probably due to its low frequency. Overall, our results indicate that within haplotypes, c.496A > G and c.1129‐5923C > G impair DPD activity. In contrast, c.85T > C tends to increase DPD activity. Combinations of offsetting variables tended to moderate the effects, particularly as observed for haplotypes containing c.496A > G; the carrier frequency of H6 was too low to directly investigate this phenomenon in c.1129‐5923C > G‐containing haplotypes. Similar to our observations for single‐variant analyses, our results do not appear to be driven by a single study because β‐coefficients were similar between populations (Figure S3 C).
It is noted that the Nie et al. cohort22 was intentionally enriched for carriers of c.1129‐5923C > G to address the original hypothesis of that study. Furthermore, the Sistonen et al.20 and Hamzic et al. populations were both collected within Switzerland. Therefore, to determine the relevance of these haplotypes in additional global populations, genotype data was retrieved from the 1000 Genomes Project34 (Figure 2). Estimated haplotype frequencies varied greatly among 1000 Genomes populations. The most common haplotype, H1, showed the highest frequency in East Asians, whereas haplotype H2 was most frequent in Africans. Both of these haplotypes were similarly distributed over sub‐populations within the East Asian and African superpopulations. The rarer haplotypes, H3, H4, H5 and H6, showed comparably high variability in frequency. Interestingly, haplotype H4 was only detected in American, South Asian and European populations, whereas haplotype H5 was more widely distributed globally. The rarest haplotype, H6, was only observed in 4 subpopulations and was completely absent from the East Asian population.
FIGURE 2.
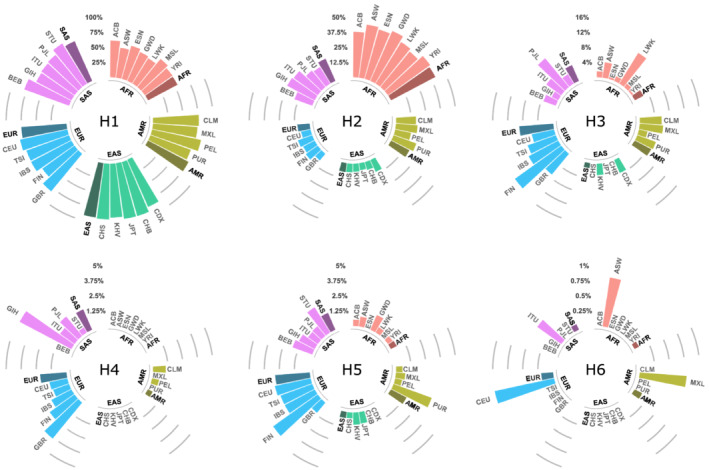
Haplotype frequencies across different world populations (1000 Genomes Project). Populations are clustered in superpopulations and visualized in different colours. Individual bars represent haplotype frequencies of populations with the corresponding population code. The shaded bars represent the average haplotype frequency in the superpopulation. The haplotype code H1–H6 refers to the corresponding haplotype, as defined in our study (Table 3). Haplotype data are based on the phase 3 analysis of the 1000 Genomes Project, including a total of n = 2504 genomes.34 Superpopulation code: AFR: African; AMR: Ad Mixed American; EAS: East Asian; EUR: European; SAS: South Asian. Population code: ACB: African Caribbeans in Barbados; ASW: Americans of African Ancestry in SW USA; BEB: Bengali from Bangladesh; GBR: British in England and Scotland; CDX: Chinese Dai in Xishuangbanna, China; CLM: Colombians from Medellin, Colombia; ESN: Esan in Nigeria; FIN: Finnish in Finland; GWD: Gambian in Western Divisions in the Gambia; GIH: Gujarati Indian from Houston, Texas; CHB: Han Chinese in Beijing, China; IBS: Iberian Population in Spain; ITU: Indian Telugu from the UK; JPT: Japanese in Tokyo, Japan; KHV: Kinh in Ho Chi Minh City, Vietnam; LWK: Luhya in Webuye, Kenya; MSL: Mende in Sierra Leone; MXL: Mexican Ancestry from Los Angeles, USA; PEL: Peruvians from Lima, Peru; PUR: Puerto Ricans from Puerto Rico; PJL: Punjabi from Lahore, Pakistan; CHS: Southern Han Chinese; STU: Sri Lankan Tamil from the UK; TSI: Toscani in Italia; CEU: Utah Residents (CEPH) with Northern and Western European Ancestry; YRI: Yoruba in Ibadan, Nigeria; AFR: African; AMR: Ad Mixed American; EAS: East Asian; EUR: European; SAS: South Asian
4. DISCUSSION
Using plasma UH2/U ratios as a surrogate marker for systemic DPD activity at a population level, we identified DPYD c.496A > G as a strong candidate risk variant for 5‐FU toxicity that correlated with reduced UH2/U in both single marker and haplotype tests. The level of change associated with c.496A > G was similar to that for the well‐characterized 5‐FU toxicity risk variant c.1129‐5923C > G. Our data also suggest that c.85T > C increases DPD enzyme activity and, therefore, might be protective against 5‐FU toxicity. LD between c.85T > C and both c.496A > G and c.1129‐5923C > G was also observed, which could moderate the impact of individual SNPs depending on the specific haplotype structure, indicating that haplotype‐based tests consisting of these 3 variants could greatly improve predictive tests for 5‐FU‐associated adverse events.
In the present study, all 4 commonly tested 5‐FU toxicity risk variants in DPYD, c.1905 + 1G > A, c.1679T > G, c.2846A > T and c.1129‐5923C > G, were strongly associated with decreased UH2/U ratios in single variant tests (Table 2). Previous reports have shown that DPD function is differentially impaired by each of the 4 variants, with c.1905 + 1G > A completely ablating function and c.1679T > G being strongly deleterious to function, whereas c.2846A > T and c.1129‐5923C > G are only partially deleterious to function.2, 18, 22 Consistent with these previous reports, we noted larger effect sizes in carriers of c.1905 + 1G > A and c.1679T > G compared to carriers of 2846A > T and c.1129‐5923C > G (Table 2). These data indicate that, although UH2/U has been shown to have limited utility as a predictive biomarker of 5‐FU toxicity at the individual patient level,7, 20, 21, 35 given a sufficiently large sample size, endogenous plasma UH2/U ratios can be used as a surrogate marker for systemic DPD activity in correlative studies.
Within our study cohort, we noted evidence for linkage between the toxicity‐associated variant c.1129‐5923C > G and the variants c.85T > C and c.496A > G. Previous studies have reported unclear associations with 5‐FU toxicity risk for c.85T > C and c.496A > G, with various studies reporting contradictory results.6, 7, 8, 9, 10, 11, 12, 13, 14, 15 At the single variant level, both c.496A > G and c.1129‐5923C > G were significantly associated with lower UH2/U ratios, and c.85T > C did not show evidence for association (Table 2). The most common haplotype containing c.496A > G (H3) displayed a modest but significant reduction in UH2/U ratios compared to the base haplotype (H1; −9.6%, P = .003; Table 3). Notably, H3 also contained c.85T > C. Compared to H3, the haplotype containing only c.85T > C (H2) showed a markedly increased UH2/U ratio that was significantly higher than the base haplotype (H1; +8.6%, P = .004). UH2/U ratios were 18.2% lower in H3 compared to H2, which is similar to the 16.9% reduction observed for the haplotype containing only c.496A > G compared to the base haplotype (H5; P = .002). The similar effect size for H5 and the most common c.1129‐5923C > G‐containing haplotype (H4) suggests that H5 might be similarly predictive of 5‐FU toxicity risk. Further investigation of the clinical importance of haplotypes containing c.496A > G in the clinical trial setting is needed to fully establish correlations with 5‐FU toxicity risk.
While c.85T > C was not significantly associated with altered UH2/U ratios at the single variant level (Table 2), haplotypes that contain c.85T > C tended to have higher UH2/U levels than matched haplotypes without the variant (Table 3). H2, which contains only c.85T > C, showed significantly higher UH2/U than the base haplotype containing no variants (H1; 8.6% higher ratio; P = .004). As discussed above, a similar effect was noted in haplotypes containing c.496A > G, with H3 showing a less severe reduction in UH2/U ratio from the base haplotype than H5 (−9.6% compared to −16.9%). Given this, we would expect c.85T > C to also modulate the effect of c.1129‐5923C > G; however, there was an inadequate number of carriers of the recombinant haplotype (H6) to test this hypothesis (Table 3). Overall, these data indicate that c.85T > C might be associated with a modest increase in DPD activity. This conclusion is consistent with a previous report in which recombinant DPD protein containing p.C29R (the translated product of the c.85T > C variant) had elevated enzyme activity compared to wild‐type DPD.17 Collectively, these data suggest that in single variant analyses, the increase in UH2/U associated with c.85T > C was likely masked because of linkage with c.496A > G and c.1129‐5923C > G. This also suggests that the deleterious effects of c.496A > G and c.1129‐5923C > G might be stronger than the enhancing effect of c.85T > C.
Consistent with our findings, similar effects were noted in a study in which haplotypes containing c.496A > G, but not c.85T > C or c.1129‐5923C > G, were enriched in patients with severe FP toxicity.15 Furthermore, carriers of the haplotype consisting of just c.85T > C, corresponding to haplotype H2 in the present study, were enriched in the population that did not experience toxicity.15 Association of c.496A > G with FP toxicity was also suggested in another study, in which linkage between variants was assessed, but haplotype association was not tested.9 An additional study36 provides suggestive data that the H3 haplotype (Table 3) might contribute to reduced DPD function. While a direct haplotype assessment was not performed, individuals carrying both c.85T > C and c.496A > G, which would presumably be enriched for H3 carriers based on haplotype frequencies, exhibited significantly impaired DPD function.36
While our results suggest an impact of c.85T > C and c.496A > G on DPD activity, we cannot unambiguously conclude that the observed changes in UH2/U ratios are due to altered protein activity and not changes in gene expression. GTEx37 data indicate that c.85T > C is associated with higher DPYD gene expression in certain tissues, suggesting that altered regulation might contribute to the observed phenotype (Figure S5). We also note that GTEx37 data do not link c.496A > G with expression. Given the relative commonality of these variants and LD with the toxicity‐associated variant rs75017182 (discussed above), future studies are warranted to investigate the mechanism(s) through which these variants alter DPD function.
High variability in haplotype frequencies was noted for global populations in the 1000 Genomes Project34 data (Figure 2). Two haplotypes that showed similar frequencies and effect sizes in our study (H4 and H5; Table 3) showed varied global distributions. H4 was most common in European and South Asian populations, but it was absent from African and East Asian populations. H5 was present in all superpopulations, with higher frequencies than H4 in African, American and East Asian populations (Figure 2). Based on global frequencies, haplotype H5, which contains only c.496A > G, could potentially be more important for the prediction of FP‐related toxicities on the global scale than H4, the primary haplotype containing the risk variant c.1129‐5923C > G. Additionally, the H2 haplotype (suggestive for higher DPD activity) showed the highest frequency in African populations (Figure 2). It is noted that the deleterious DPYD variant rs11523289838 (c.557A > G, p.Y186C) is in strong LD with c.85T > C (D′ = 1.00, data from 1000 Genomes Project). Additional studies are needed to evaluate the effect of these and other haplotypes on FP toxicity risk in varied racial/ethnic populations.
Collectively, our results highlight the importance of careful multivariant haplotype assessments in pharmacogenomic studies, even if evidence suggests that individual variants might only confer a weak or non‐significant effect. Given the potential multilocus impact of c.85T > C and c.496A > G on DPD phenotype identified in this study, the further evaluation of these variants and associated haplotypes in the context of clinical outcomes using sufficiently sized patient populations is urgently needed.
COMPETING INTERESTS
The authors have no other relevant affiliations or financial involvement with any organization or entity with a financial interest in or financial conflict with the subject matter or materials discussed in the manuscript apart from those disclosed. No writing assistance was utilized in the production of this manuscript.
CONTRIBUTORS
C.R.L., S.H., S.O. and U.A. were involved in the conception and design of the study and drafted the article. S.H. and D.S. organized and carried out the genetic and mass spectrometric analyses. C.R.L., S.H., S.O., R.D., C.N. and U.A. were involved in the analysis and interpretation of data. D.M., S.F., M.W. and S.S. provided additional samples, data, infrastructure, and/or expertise in statistical modelling and mass spectrometry. All authors were involved in the critical revision of the manuscript and gave final approval of the version to be published. Financial support for this study was provided by a research grant from the Swiss National Science Foundation (163205) to C.R. Largiadèr.
Supporting information
FIGURE S1 Violin and QQ plots of UH2/U ratio distribution and characteristics in the individual cohorts
FIGURE S2 Genetic linkage of DPYD variants in individual cohorts, the combined cohort and the reference EUR‐population retrieved from the LDlink database (last accessed on 16 November 2020). Notably, statistically significant linkage between c.85C/c.496G and between c.85C/c.1129‐5923G, was consistently observed in all cohorts (P < .0083).
FIGURE S3 Effect of DPYD variants and haplotypes on UH2/U ratios in individual cohorts. ns: P > .05; *: P ≤ .05; **: P ≤ .01; ***: P ≤ .001; ****: P ≤ .0001. (A) DPYD risk variants, (B) c.85T > C and c.496A > G and C haplotypes
FIGURE S4 Effect of c.85T > C and c.496A > G on UH2/U ratios. Boxplots according to study cohorts and DPYD variant on the x‐axis. (A) c.85T > C genotype is grouped in different colours. From left to right: TT (red), TC (green), CC (blue); (B) c.496A > G from left to right: AA (red), AG (green), GG (blue).
FIGURE S5DPYD c.85T > C is significantly associated with DPYD gene expression. Statistically significant effects are reported for skin and oesophagus mucosa (skin: P = 9.6 × 10−8, m‐value = 1; oesophagus mucosa: P = 3.8 × 10−8, m‐value = 1). The liver, which is the main site of DPYD expression, is weakly associated (P = .02, m‐value = 0.623) with increased expression for the c.85C allele. However, the liver is under‐represented with n = 208 samples compared to skin (n = 517) and oesophagus mucosa (n = 497). This graph was obtained from the GTEx Portal Website (https://www.gtexportal.org/home/) on 04 April 2020.
FIGURE S6 Effect of c.85T > C and c.496A > G on UH2/U ratios. Boxplots according to study cohorts and DPYD variant on the x‐axis. (A) c.85T > C genotype is grouped in different colours. From left to right: TT (red), TC (green), CC (blue); (B) c.496A > G from left to right: AA (red), AG (green), GG (blue).
ACKNOWLEDGEMENTS
The authors thank Daniel Schärer and Gisela Andrey‐Zürcher for technical assistance. The authors also thank Claudia Bühler for technical assistance regarding mass spectrometry. The Regional Blood Transfusion Service of the Swiss Red Cross Bern is acknowledged for collecting blood samples of the healthy volunteers, and the Liquid Biobank Bern (www.biobankbern.ch) for sample storage and processing. The authors also thank The Mayo Clinic Center for Individualized Medicine for the use of biospecimens from the Mayo Clinic Biobank.
Hamzic S, Schärer D, Offer SM, et al. Haplotype structure defines effects of common DPYD variants c.85T > C (rs1801265) and c.496A > G (rs2297595) on dihydropyrimidine dehydrogenase activity: Implication for 5‐fluorouracil toxicity. Br J Clin Pharmacol. 2021;87:3234–3243. 10.1111/bcp.14742
Principal Investigator: We conducted an observational study; therefore, no interventions were performed and/or no substance were administered to human subjects/patients for this study. The study was approved from the Swiss ethics committee: Req‐2017‐00033. Prof. Carlo Largiadèr ensured that the necessary consents for the data and analysis of the samples were obtained.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author, upon reasonable request.
REFERENCES
- 1.Amstutz U, Froehlich TK, Largiadèr CR. Dihydropyrimidine dehydrogenase gene as a major predictor of severe 5‐fluorouracil toxicity. Pharmacogenomics. 2011;12(9):1321‐1336. [DOI] [PubMed] [Google Scholar]
- 2.Amstutz U, Henricks LM, Offer SM, et al. Clinical Pharmacogenetics Implementation Consortium (CPIC) Guideline for Dihydropyrimidine Dehydrogenase Genotype and Fluoropyrimidine Dosing: 2017 Update. Clin Pharmacol Ther. 2018;103(2):210‐216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Henricks LM, Lunenburg CATC, de Man FM, et al. DPYD genotype‐guided dose individualisation of fluoropyrimidine therapy in patients with cancer: a prospective safety analysis. Lancet Oncol. 2018;19(11):1459‐1467. [DOI] [PubMed] [Google Scholar]
- 4.Vreken P, Van Kuilenburg ABP, Meinsma R, Van Gennip AH. Identification of novel point mutations in the dihydropyrimidine dehydrogenase gene. J Inherit Metab Dis. 1997;20(3):335‐338. [DOI] [PubMed] [Google Scholar]
- 5.van Kuilenburg ABP, De Abreu RA, van Gennip AH. Pharmacogenetic and clinical aspects of dihydropyrimidine dehydrogenase deficiency. Ann Clin Biochem. 2003;40(1):41‐45. [DOI] [PubMed] [Google Scholar]
- 6.Madi A, Fisher D, Maughan TS, et al. Pharmacogenetic analyses of 2183 patients with advanced colorectal cancer; potential role for common dihydropyrimidine dehydrogenase variants in toxicity to chemotherapy. Eur J Cancer. 2018;102:31‐39. [DOI] [PubMed] [Google Scholar]
- 7.Etienne‐Grimaldi M‐C, Boyer JC, Béroud C, et al. New advances in DPYD genotype and risk of severe toxicity under capecitabine. PLoS One. 2017;12(5):e0175998. 10.1371/journal.pone.0175998 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Ruzzo A, Graziano F, Galli F, et al. Dihydropyrimidine dehydrogenase pharmacogenetics for predicting fluoropyrimidine‐related toxicity in the randomised, phase III adjuvant TOSCA trial in high‐risk colon cancer patients. Br J Cancer. 2017;117(9):1269‐1277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Gross E, Busse B, Riemenschneider M, et al. Strong Association of a Common Dihydropyrimidine Dehydrogenase Gene Polymorphism with Fluoropyrimidine‐Related Toxicity in Cancer Patients. PLoS One. 2008;3:1‐7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Milano G. Highlight on DPYD gene polymorphisms and treatment by capecitabine*. Scand J Clin Lab Invest. 2016;76(sup245):S30‐S33. [DOI] [PubMed] [Google Scholar]
- 11.Khushman M, Patel GK, Hosein PJ, et al. Germline pharmacogenomics of DPYD*9A (c.85T>C) variant in patients with gastrointestinal malignancies treated with fluoropyrimidines. J Gastrointest Oncol. 2018;9(3):416‐424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Maharjan AS, McMillin GA, Patel GK, et al. The Prevalence of DPYD*9A (c.85T>C) Genotype and the Genotype‐Phenotype Correlation in Patients with Gastrointestinal Malignancies Treated With Fluoropyrimidines: Updated Analysis. Clin Colorectal Cancer. 2019;18(3):e280‐e286. [DOI] [PubMed] [Google Scholar]
- 13.Falvella FS, Cheli S, Martinetti A, et al. DPD and UGT1A1 deficiency in colorectal cancer patients receiving triplet chemotherapy with fluoropyrimidines, oxaliplatin and irinotecan. Br J Clin Pharmacol. 2015;80(3):581‐588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Loganayagam A, Arenas Hernandez M, Corrigan A, et al. Pharmacogenetic variants in the DPYD, TYMS, CDA and MTHFR genes are clinically significant predictors of fluoropyrimidine toxicity. Br J Cancer. 2013;108(12):2505‐2515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Kleibl Z, Fidlerova J, Kleiblova P, et al. Influence of dihydropyrimidine dehydrogenase gene (DPYD) coding sequence variants on the development of fluoropyrimidine‐related toxicity in patients with high‐grade toxicity and patients with excellent tolerance of fluoropyrimidine‐based chemotherapy. Neoplasma. 2009;56(4):303‐316. [DOI] [PubMed] [Google Scholar]
- 16.van Kuilenburg ABP, Meijer J, Tanck MWT, et al. Phenotypic and clinical implications of variants in the dihydropyrimidine dehydrogenase gene. Biochim Biophys Acta ‐ Mol Basis Dis. 2016;1862(4):754‐762. [DOI] [PubMed] [Google Scholar]
- 17.Offer SM, Wegner NJ, Fossum C, Wang K, Diasio RB. Phenotypic profiling of DPYD variations relevant to 5‐fluorouracil sensitivity using real‐time cellular analysis and in vitro measurement of enzyme activity. Cancer Res. 2013;73(6):1958‐1968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Offer SM, Fossum CC, Wegner NJ. Comparative Functional Analysis of DPYD Variants of Potential Clinical Relevance to Dihydropyrimidine Dehydrogenase Activity Comparative Functional Analysis of DPYD Variants of Potential Clinical Relevance to Dihydropyrimidine Dehydrogenase Activity. Cancer Res. 2014;74:2545‐2554. 10.1158/0008-5472.CAN-13-2482 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Amstutz U, Farese S, Aebi S, Largiadèr CR. Dihydropyrimidine dehydrogenase gene variation and severe 5‐fluorouracil toxicity: a haplotype assessment. Pharmacogenomics. 2009;10(6):931‐944. [DOI] [PubMed] [Google Scholar]
- 20.Sistonen J, Büchel B, Froehlich TK, et al. Predicting 5‐fluorouracil toxicity: DPD genotype and 5,6‐dihydrouracil:uracil ratio. Pharmacogenomics. 2014;15(13):1653‐1666. [DOI] [PubMed] [Google Scholar]
- 21.Meulendijks D, Henricks LM, Jacobs BAW, et al. Pretreatment serum uracil concentration as a predictor of severe and fatal fluoropyrimidine‐associated toxicity. Br J Cancer. 2017;116(11):1415‐1424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Nie Q, Shrestha S, Tapper EE, et al. Quantitative Contribution of rs75017182 to Dihydropyrimidine Dehydrogenase mRNA Splicing and Enzyme Activity. Clin Pharmacol Ther. 2017;102(4):662‐670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Büchel B, Rhyn P, Schürch S, Bühr C, Amstutz U, R. Largiadèr C. LC‐MS/MS method for simultaneous analysis of uracil, 5,6‐dihydrouracil, 5‐fluorouracil and 5‐fluoro‐5,6‐dihydrouracil in human plasma for therapeutic drug monitoring and toxicity prediction in cancer patients. Biomed Chromatogr. 2013;27(1):7‐16. [DOI] [PubMed] [Google Scholar]
- 24.Curtis MJ, Alexander S, Cirino G, et al. Experimental design and analysis and their reporting II: updated and simplified guidance for authors and peer reviewers. Br J Pharmacol. 2018;175(7):987‐993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Pardoe I. Applied Regression Modeling. Second ed. Hoboken, New Jersey: John Wiley & Sons, Inc.; 2013. 10.1002/9781118345054 [DOI] [Google Scholar]
- 26.Kuznetsova A, Brockhoff PB, Christensen RH. B lmerTest Package: Tests in Linear Mixed Effects Models. J Stat Softw. 2017;82:1‐26. [Google Scholar]
- 27.Bates D, Mächler M, Bolker BM, Walker SC. Fitting linear mixed‐effects models using lme4. J Stat Softw. 2015;67:1‐48. [Google Scholar]
- 28.Warnes G, Gorjanc G, Leisch F, Man M. Genetics: Population genetics. R package version 1.3. 8.1. 2. R Foundation for Statistical Computing. 2019.
- 29.Schaid DJ, Rowland CM, Tines DE, Jacobson RM, Poland GA. Score tests for association between traits and haplotypes when linkage phase is ambiguous. Am J Hum Genet. 2002;70(2):425‐434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Sinnwell JP, Schaid DJ. Haplo Stats (Version 1.6.0): Statistical Methods for Haplotypes When Linkage Phase Is Ambiguous. Rochester, MN, USA: Mayo Clinic Division of Health Sciences Research; 2013. [Google Scholar]
- 31.LDlink|An Interactive Web Tool for Exploring Linkage Disequilibrium in Population Groups. Retrieve from: http://analysistools.nci.nih.gov/LDlink/ (Accessed: 3rd February 2016)
- 32.Wickham H. ggplot2: Elegant Graphics for Data Analysis. New York: Springer‐Verlag; 2016. [Google Scholar]
- 33.Alexander SPH, Fabbro D, Kelly E, et al. The Concise Guide to PHARMACOLOGY 2019/20: Enzymes. Br J Pharmacol. 2019;176:S297‐S396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Auton A, Abecasis GR, Altshuler DM, et al. A global reference for human genetic variation. Nature. 2015;526(7571):68‐74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Meulendijks D, Cats A, Beijnen JH, Schellens JHM. Improving safety of fluoropyrimidine chemotherapy by individualizing treatment based on dihydropyrimidine dehydrogenase activity – Ready for clinical practice ? Cancer Treat Rev. 2016;50:23‐34. [DOI] [PubMed] [Google Scholar]
- 36.Gentile G, Botticelli A, Lionetto L, et al. Genotype‐phenotype correlations in 5‐fluorouracil metabolism: a candidate DPYD haplotype to improve toxicity prediction. Pharmacogenomics J. 2015;16:320‐325. 10.1038/tpj.2015.56 [DOI] [PubMed] [Google Scholar]
- 37.Lonsdale J, Thomas J, Salvatore M, et al. The Genotype‐Tissue Expression (GTEx) project. Nat Genet. 2013;45(6):580‐585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Offer SM, Lee AM, Mattison LK, Fossum C, Wegner NJ, Diasio RB. A DPYD variant (Y186C) in individuals of african ancestry is associated with reduced DPD enzyme activity. Clin Pharmacol Ther. 2013;94(1):158‐166. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
FIGURE S1 Violin and QQ plots of UH2/U ratio distribution and characteristics in the individual cohorts
FIGURE S2 Genetic linkage of DPYD variants in individual cohorts, the combined cohort and the reference EUR‐population retrieved from the LDlink database (last accessed on 16 November 2020). Notably, statistically significant linkage between c.85C/c.496G and between c.85C/c.1129‐5923G, was consistently observed in all cohorts (P < .0083).
FIGURE S3 Effect of DPYD variants and haplotypes on UH2/U ratios in individual cohorts. ns: P > .05; *: P ≤ .05; **: P ≤ .01; ***: P ≤ .001; ****: P ≤ .0001. (A) DPYD risk variants, (B) c.85T > C and c.496A > G and C haplotypes
FIGURE S4 Effect of c.85T > C and c.496A > G on UH2/U ratios. Boxplots according to study cohorts and DPYD variant on the x‐axis. (A) c.85T > C genotype is grouped in different colours. From left to right: TT (red), TC (green), CC (blue); (B) c.496A > G from left to right: AA (red), AG (green), GG (blue).
FIGURE S5DPYD c.85T > C is significantly associated with DPYD gene expression. Statistically significant effects are reported for skin and oesophagus mucosa (skin: P = 9.6 × 10−8, m‐value = 1; oesophagus mucosa: P = 3.8 × 10−8, m‐value = 1). The liver, which is the main site of DPYD expression, is weakly associated (P = .02, m‐value = 0.623) with increased expression for the c.85C allele. However, the liver is under‐represented with n = 208 samples compared to skin (n = 517) and oesophagus mucosa (n = 497). This graph was obtained from the GTEx Portal Website (https://www.gtexportal.org/home/) on 04 April 2020.
FIGURE S6 Effect of c.85T > C and c.496A > G on UH2/U ratios. Boxplots according to study cohorts and DPYD variant on the x‐axis. (A) c.85T > C genotype is grouped in different colours. From left to right: TT (red), TC (green), CC (blue); (B) c.496A > G from left to right: AA (red), AG (green), GG (blue).
Data Availability Statement
The data that support the findings of this study are available from the corresponding author, upon reasonable request.