

Abstract
Hereditary disorders are frequently caused by genetic variants that affect pre‐messenger RNA splicing. Though genetic variants in the canonical splice motifs are almost always disrupting splicing, the pathogenicity of variants in the noncanonical splice sites (NCSS) and deep intronic (DI) regions are difficult to predict. Multiple splice prediction tools have been developed for this purpose, with the latest tools employing deep learning algorithms. We benchmarked established and deep learning splice prediction tools on published gold standard sets of 71 NCSS and 81 DI variants in the ABCA4 gene and 61 NCSS variants in the MYBPC3 gene with functional assessment in midigene and minigene splice assays. The selection of splice prediction tools included CADD, DSSP, GeneSplicer, MaxEntScan, MMSplice, NNSPLICE, SPIDEX, SpliceAI, SpliceRover, and SpliceSiteFinder‐like. The best‐performing splice prediction tool for the different variants was SpliceRover for ABCA4 NCSS variants, SpliceAI for ABCA4 DI variants, and the Alamut 3/4 consensus approach (GeneSplicer, MaxEntScacn, NNSPLICE and SpliceSiteFinder‐like) for NCSS variants in MYBPC3 based on the area under the receiver operator curve. Overall, the performance in a real‐time clinical setting is much more modest than reported by the developers of the tools.
Keywords: ABCA4, deep learning, MYBPC3, RNA splicing, splice prediction tools, variant effect prediction
The choice of splice prediction tool depends on the dataset. However, deep learning tools show promising results for variants tested in midigene splice assays.
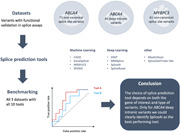
1. INTRODUCTION
An estimated 50% of pathogenic variants result in aberrant splicing (López‐Bigas et al., 2005; Pan et al., 2008). Genetic variants may affect all sequence elements required for correct splicing, including the three core elements, which are recognized by the spliceosome: The canonical 5ʹ splice donor site (SDS), the canonical 3ʹ splice acceptor site (SAS), and the branchpoint. Both the SDS and SAS contain conserved dinucleotides. At the SDS, the most commonly encountered dinucleotide is a GT and at the SAS invariably an AG. Alternative dinucleotides for the SDS are known, of which GC with a frequency of 1% is the most common one (Sheth et al., 2006). In contrast with the SDS and SAS, the branchpoint motif is less conserved (Will & Lührmann, 2011). The noncanonical sequences around the canonical splice sites are part of the splice site consensus and therefore also conserved. The noncanonical sequences at the SAS are located from 14 to 3 nucleotides (nt) upstream and 2 nt downstream, that is, in the exon. For the SDS, these are the last 2 nt of the exon and positions 3 to 6 downstream. In addition to the three main core elements, other cis‐acting elements, such as intronic and exonic splicing enhancers and silencers, are involved in splicing (Albert et al., 2018; Glisovic et al., 2008).
Variants affecting canonical sequences are considered to have a major effect, where the relevant exon is skipped and even skipping of neighboring exons can be observed. In the presence of alternative splice sites in or outside of the exon, partial exon skipping or exon elongation also have been observed (Fadaie et al., 2019; Fang et al., 2001; Khan et al., 2020; Labonne et al., 2016; Ramalho et al., 2003; Sangermano et al., 2018; Symoens et al., 2011). Variants in the noncanonical splicing motifs are referred to as noncanonical splice site (NCSS) variants. Disrupting NCSS variants usually affects splicing by weakening the existing splice site (Bradley et al., 2005; Shaw et al., 2003), and occasionally by creating a new splice site (Fadaie et al., 2019). On the contrary, deep‐intronic (DI) variants are known to both create or strengthen cryptic splice sites (Fadaie et al., 2019; Khan et al., 2020; Sangermano et al., 2018; Sobczyńska‐Tomaszewska et al., 2013; Sun & Chasin, 2000). In general, if DI variants alter splicing, it is through pseudo‐exon inclusion into the messenger RNA due to the creation of a cryptic SAS or SDS when an appropriate naturally existing SAS or SDS, respectively, is present (Dhir & Buratti, 2010; Romano et al., 2013).
To determine the impact of a putative pathogenic variant or variant of unknown significance (VUS) on splicing, in silico splice prediction tools may be employed. The available tools make use of three different algorithms: Motif‐based algorithms, classical machine learning algorithms, and deep learning algorithms. Whereas classical machine learning algorithms rely on preselected features, novel deep learning tools show promising improvements in the field of in silico splice prediction (Cheng et al., 2019; Louadi et al. 2019; Naito, 2019), as they learn informative features from the data. Deep learning algorithms may capture more complex information, such as the distance between different sequence motifs, structural motifs, and nonlinear relationships. They may also capture the joint effects of the SDS and SAS, explaining splice site interdependence (Hefferon et al., 2002; Khan et al., 2020; Ohno et al., 2018). Most in silico splice prediction tools are trained and evaluated on RNA‐seq data, achieving high scores for accuracy and precision. The high precision, however, often cannot be reproduced in diagnostics. The reported area under the precision‐recall curve for SpliceAI for instance is 0.98 (Jaganathan et al., 2019), whereas SpliceAI demonstrated lower performance in small clinical real‐time test sets (Ellingford et al., 2019; Wai et al., 2020).
In the past, nondeep learning tools have been compared to each other (Jian et al., 2014; Moles‐Fernández et al., 2018), whereas more recently, one deep learning tool has been compared to nondeep learning tools, in which case the deep learning tool has shown to be more accurate in its predictions (Ellingford et al., 2019; Jaganathan et al., 2019; Jian et al., 2014; Ohno et al., 2018). Currently, there is no study comparing different deep learning splice prediction tools on a clinically relevant set of variants. Therefore, in this study, we compared the motif‐based algorithm SpliceSiteFinder‐like (Shapiro & Senapathy, 1987), the interaction‐based algorithm MaxEntScan (Yeo & Burge, 2004), the classical machine learning tools CADD (Rentzsch et al., 2019), GeneSplicer (Pertea, 2001), NNSPLICE (Reese, 1997), and SPIDEX (Xiong et al., 2015) and the deep learning tools DSSP (Naito, 2019), MMSplice (Cheng et al., 2019), SpliceAI (Jaganathan et al., 2019) and SpliceRover (Zuallaert et al., 2018). A motivation for this selection is given in Section 2. The comparison was done on two of the largest, high‐confidence sets of variants that are rare, potentially clinically relevant, and for which the effect of splicing has been functionally assessed using mini‐ or midigene assays.
The variants are located in genes coding for adenosine triphosphate‐binding cassette subfamily A member 4 (ABCA4) and myosin‐binding protein C (MYBPC3). The ABCA4 protein is expressed in the retina where it removes retinaldehyde from the photoreceptor cells (Molday et al., 2000; Sun & Nathans, 1997). Biallelic pathogenic variants in ABCA4 cause Stargardt disease (STGD1), which displays a spectrum of retinal phenotypes encompassing early‐onset, classical, and late‐onset STGD1, depending on the severity of the two alleles (Allikmets et al., 1997; Cremers et al., 1998, 2020; Maugeri et al., 2000). MYBPC3 is involved in muscle contraction in heart muscle cells, and defects are associated with cardiomyopathy (Marston et al., 2009; van Dijk et al., 2009).
2. METHODS
2.1. Datasets
Seventy‐one ABCA4 NCSS variants, 81 ABCA4 DI variants, and 61 MYBPC3 NCSS variants with functional validation from LOVD, ClinVar, and ExAC were selected (Table S1). The selection and splice assays were already performed, and details can be found in the references listed in Table S1. Only variants that may disrupt splicing by affecting noncanonical splice sites or that may create novel splice sites in deep intronic regions are included.
The selection criterion for functional validation of the ABCA4 variants was a 2% difference in splice score for at least two of the Alamut programs (SpliceSiteFinder‐like, MaxEntScan, NNSPLICE, GeneSplicer, and Human Splicing Finder) and relative strength of at least 75% for novel splice sites, which has shown to include most of the disruptive variants in the past (Khan et al., 2020; Sangermano et al., 2019). To assess the pathogenicity of putative causative ABCA4 variants, splice assays were performed using midigenes as previously described (Fadaie et al., 2019; Khan et al., 2020; Sangermano et al., 2018). In short, midigenes contain multiple exons and introns to mimic a natural genomic context for testing the effect of variants on splicing. A construct containing the wild type is then compared to a construct, including the mutant variant, which is introduced by site‐directed mutagenesis. After independent transfection into HEK293T cells, ABCA4 transcripts were amplified using reverse‐transcription polymerase chain reaction (RT‐PCR) and separated on a 2% agarose gel to determine the percentage of mutant RNA in comparison with the control line. ABCA4 variants with more than 20% mutant RNA were classified as splice altering (Sangermano et al., 2018). For MYBPC3 variants, the selection criterion was a lower MaxEntScan score than the score of the reference nucleotide. Selected variants were assessed in minigenes that contained a CMV promoter and a 500‐base pair oligonucleotide with the relevant intron flanked by exon fragments (Ito et al., 2017). Computational quantification of RT‐PCR transcripts with a significant difference (p < .001, two‐sided Fisher's exact test) between wild type and the mutant transcript was performed, and variants with a significant difference were classified as splice altering. Both the ABCA4 and MYBPC3 data sets were aligned to the human genome reference GRCh37/hg19 assembly.
2.2. In silico splice prediction tools
In silico splice prediction tools were selected based on the following criteria:
-
‐
The tool is freely available.
-
‐
The tool can be applied to a variant in either variant or sequence format.
-
‐
The tool either uses deep learning or is widely applied in routine diagnostics.
-
‐
The tool can predict a score for most of the variants in the data set.
An overview of all in silico prediction tools and their characteristics is provided in Table 1. Tools were grouped into the categories classical machine learning, deep learning, and others based on their underlying algorithm. Deep learning is a part of machine learning. They differ in the way they define their features. In machine learning, the features are defined by the user before the training of the model. In deep learning, the features are defined during the training of the model. Deep learning, therefore, offers possibilities to capture more complex features, but the included feature definition also contributes to the black‐box character of deep learning.
Table 1.
Overview of the most important properties of the different splice prediction tools
| Tool | Approach | Algorithm | Score range | Characteristic | Training data | Input data | Nucleotide positions | Interface | Year |
|---|---|---|---|---|---|---|---|---|---|
| CADD | Support vector machine with linear kernel | ML | – | Integrates more than 60 genomic features into a single score | 13,141,299 SNVs, 627,071 insertions and 926,968 deletions from simulated and observed variants | VCF file | – | Website | 2014 |
| DSSP | CNN with long short‐term memory | DL | 0–1 | Individual prediction for SDS and SAS | HS3D | 140 nt sequence with consensus sequence the middle | 140 nt | Python script | 2018 |
| GeneSplicer | Decision tree and Markov model | ML | 0–15 | Markov model captures additional dependencies among neighboring bases at splice sites | 1323 plant genes and 1115 human genes | FASTA sequence | Up to 80 nt on both sites of splice site | Alamut | 2001 |
| MaxEntScan | Maximum entropy |
Other |
0–12 | Use of different constraints sorted by the effect on entropy, only second‐order dependencies | 1821 nonredundant transcripts with 12,715 introns | 9‐mer FASTA sequence | 9 nt at SAS, 23 nt at SDS | Alamut | 2004 |
| MMSplice | Individual modules scoring exon, intron, and splice sites | DL |
– |
Predicts quantitative physical measures of splicing | Vex‐seq + GENCODE | VCF file | All nucleotides in intron, exon, intron structure | Python package | 2018 |
| NNSPLICE | Hidden Markov model and neural network | ML | 0–1 | Captures pairwise correlations between adjacent nucleotides | 285 multiple‐exon human DNA sequences from GenBank | FASTA sequence | −7 to +8 at SAS, −21 to +20 at SDS | Alamut | 1997 |
| SPIDEX | Bayesian modeling | ML | 0–1 | Tissue‐specific PSI values | Illumina Human Body Map 2.0 project | VCF file | Depending on features, up to 2000 nt in introns and 300 nt in exons | Txt file with precomputed values | 2015 |
| SpliceAI | Deep learning with ResNet blocks | DL | 0–1 | Predicts nucleosome positioning from sequence | GENCODE | VCF file | 10,000 nt | Python package | 2019 |
| SpliceRover | CNN | DL | 0–1 | Identifies regions/structures of interest by normalizing contribution scores, and individual models for SDS and SAS | Human and plant | FASTA sequence | Minimal 400 nt | Website | 2018 |
| SpliceSiteFinder‐like | Position weight matrices | Other | 0–100 | – | – | – | – | Alamut | 1987 |
Abbreviations: CNN, convolutional neural network; DL, deep learning; ML, machine learning; nt, nucleotides; SAS, splice acceptor site; SDS, splice donor site; VCF, variant call format.
Delta scores according to formula (1) were calculated for tools that provided a separate score for wild type and variant sequences. The absolute value of the score was used for tools that returned negative values to only compare the magnitude of splice change:
| (1) |
The commonly applied tools GeneSplicer, MaxEntScan, NNSPLICE, and SpliceSiteFinder‐like were accessed from Alamut Visual Software version 2.13 (SOPHiA GENETICS). Missing values, given the default settings of the Alamut tools, likely do not result in a change compared to wild type and are unlikely to affect splicing. Therefore, we replaced them with zero. When multiple splice sites close to the investigated variant were scored, the score for the canonical splice site was chosen for NCSS variants, and the score for the novel created/strengthened splice site was chosen for DI variants.
The other tools were accessed separately from either a website, available scripts or files with precomputed scores. Tools accessed via a website were CADD v1.6 and SpliceRover. For CADD, a variant call format (VCF) file with the variants was uploaded to the website, and raw scores were obtained. SpliceRover required a FASTA sequence with a minimal length of 400 nt. Thus, we included 410‐nt long sequences around the variant of interest as input. For 11 variants that caused an error message, we used a different input length to obtain a score (ABCA4: 1000 nt for c.769−605T>C, c.769−1778T>C, c.302+628C>T, and 750 for c.769−788A>T; MYBPC3: 1000 for c.3815−10T>G, c.2906−12C>T, c.1928−11G>A, c.1625−8C>G, c.1227−9C>A, c.1091−8G>A and 750 for c.906−8T>C). Python scripts were available for DSSP, SpliceAI and MMSplice. DSSP required input sequences of 140 nt with the SAS dinucleotide at positions 69 and 70 or the SDS dinucleotide at positions 71 and 72. Donor and acceptor sequences were processed with separate Python scripts available on the DSSP GitHub (https://github.com/DSSP-github/DSSP). SpliceAI v1.3.1 was applied to a VCF file. MMSplice v2.0.0 was also applied to VCF files but returned multiple scores for most variants. The maximum absolute delta logit PSI score across all exons was chosen as the primary score. A file with precomputed scores was available for SPIDEX v1.0. The data and all analysis scripts can be found at https://github.com/cmbi/Benchmarking_splice_prediction_tools.
MMSplice and Spidex could not calculate a score for more than half of the ABCA4 DI variants, and we, therefore, excluded these tools completely for the analysis of DI variants. MMSplice only considers variants located within 300 nt of the SDS or SAS. SPIDEX scores were retrieved from files with precomputed values, which did not include DI variants.
2.3. Classification metrics and receiver operator curve
The accuracy, sensitivity, specificity, positive predictive value (PPV), negative predictive value (NPV), and Matthews correlation coefficient (MCC) were calculated for each data set. The formulas of the applied classification metrics are provided in Table S2. In addition to the standard statistical measures, MCC was used. The MCC is best suited for unbalanced data sets, whereas other metrics are influenced by the size of the positive and negative groups. A consensus of the Alamut tools (GeneSplicer, MaxEntScan, NNSPLICE, and SpliceSiteFinder‐like) is frequently considered in diagnostics. Therefore, an Alamut consensus with 3/4 tools was included in the assessment. Sklearn 0.19.2 for Python was used to calculate the area under the curve (AUC) and the optimal cutoff to separate the true positives and true negatives for each prediction tool.
3. RESULTS
3.1. Variants
Seventy‐one ABCA4 NCSS variants, 81 ABCA4 DI variants, and 61 MYBPC3 NCSS variants were evaluated with a selection of splice prediction tools (Table 1). All variants were taken from previously published data sets (Bauwens et al., 2019; Braun et al., 2013; Fadaie et al., 2019; Ito et al., 2017; Khan et al., 2019, 2020; Sangermano et al. 2018, 2019; Zernant et al., 2014) and either might weaken an existing noncanonical splice site or create a novel splice site in deep intronic regions. The number of variants that alter splicing and variants that have no effect on splicing is provided in Figure 1a. Ninety percent (64 out of 71) of ABCA4 NCSS variants altered splicing, whereas 74% (60 out of 81) of ABCA4 DI variants had no effect on splicing. MYBPC3 NCSS variants showed a more even distribution with 56% (34 out of 61) splice‐altering variants. For all three data sets, more variants were located near the SDS than the SAS (Figure 1b). Figure 1c and d show the distribution of variants around the SAS and SDS, respectively, for all NCSS variants. On the donor site, most splice‐altering variants were located at the last exonic position, and on the acceptor site, most splice‐altering variants were located at the first and second exonic position.
Figure 1.
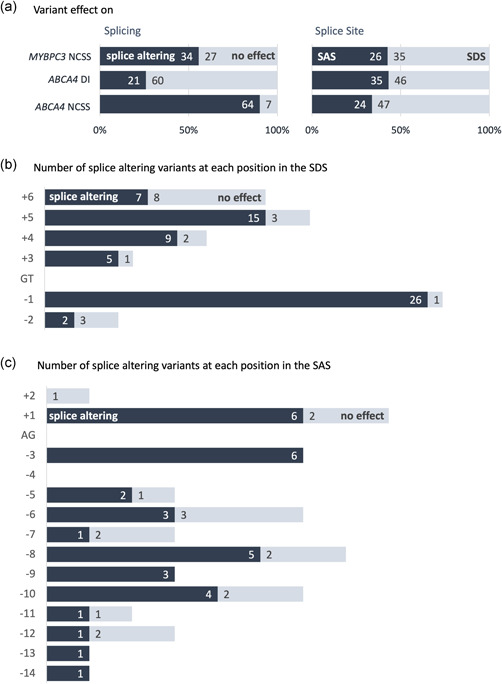
Variant effect on splicing and splice site. (a) Distribution of splice‐altering variants and distribution of variants that affected either the splice acceptor site (SAS) or splice donor site (SDS) in the ABCA4 NCSS, ABCA4 DI, and MYBPC3 NCSS data set. (b, c) Plot of the number splice‐altering and nonsplice‐altering NCSS variants present at the SDS (+3 to +6, panel b) and SAS (−14 to −3, panel c) and the first or last two nucleotides of the exon and the number of variants found to affect splicing
3.2. Receiver operator curve (ROC) and area under the curve (AUC)
The ROC curves with AUCs of the five best‐performing tools for each data set are provided in Figure 2a–c. For ABCA4 NCSS variants those tools were SpliceRover, SpliceAI, DSSP, Spidex and SpliceSiteFinder‐like. The best‐performing tools for the ABCA4 DI variants were SpliceAI, SpliceRover, GeneSplicer, NNSPLICE, and MaxEntScan. For MYBPC3 NCSS variants, Alamut 3/4, SpliceSiteFinder‐like, NNSPLICE, MMSplice and MaxEntScan achieved the highest AUC. ROC curves including all tools are provided in Figure S1.
Figure 2.
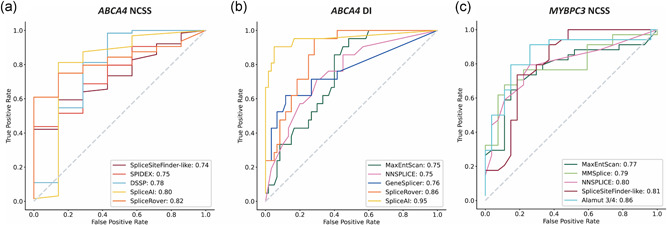
Receiver operator curve (ROC) and area under the curve (AUC) for the five splice prediction tools with the highest AUC for each data set. ROC curves for (a) ABCA4 NCSS variants, (b) ABCA4 DI variants, and (c) MYBPC3 NCSS variants. The AUC values are given in the insets
3.3.
3.3.1.
Figure 3 compares the tools of the three different categories based on their AUC value for the three different data sets. The tools with the highest AUC value for each category were SpliceAI for deep learning, NNSPLICE for machine learning, and the Alamut 3/4 consensus approach and MaxEntScan for the other category. For the ABCA4 NCSS variants, the deep learning tools SpliceAI, SpliceRover and DSSP had the highest AUC. SpliceAI outperformed all other tools on the ABCA4 DI variants. For the MYBPC3 dataset, the Alamut tools GeneSplicer, NaxEntScan, NNSPLICE and SpliceSiteFinder‐like, and MMSplice achieve the highest AUC.
Figure 3.

Comparison of the area under the curve (AUC) for all tools in the three different data sets. In addition to the individual tools, the Alamut 3/4 consensus was included. The best tool for each category is highlighted in dark blue. For the other category, both the Alamut 3/4 consensus approach and MaxEntScan showed comparable high AUC values and are, therefore, highlighted
3.4. Comparison of thresholds
The ROC was used to determine the best threshold for each data set to classify the variants as splice‐altering or nonsplice‐altering. Table 2 shows the comparison of the thresholds identified with the ROC curve with the predefined threshold for the different tools suggested by the developers. The best threshold to maximize the number of true positives and true negatives depended highly on the data set. For MaxEntScan and NNSPLICE the best thresholds were higher than the predefined threshold, whereas the best thresholds for DSSP, MMSplice, SPIDEX and SpliceAI were lower. SpliceSiteFinder‐like thresholds were higher than the predefined threshold for ABCA4 DI variants and MYBPC3 variants, and the threshold for ABCA4 NCSS variants was lower. The thresholds for CADD were difficult to compare to the predefined thresholds because these utilize a threshold depending on the location of the variant. GeneSplicer and SpliceRover have no predefined threshold. Three tools, MMSplice, SpliceAI and SpliceSiteFinder‐like, showed thresholds close to the predefined threshold.
Table 2.
Comparison of the optimal thresholds for each data set with the suggested threshold by the developers
| Tool | ABCA4 NCSS | ABCA4 DI | MYBPC3 NCSS | Suggested threshold |
|---|---|---|---|---|
| CADD | 2.66 | 0.24 | 2.09 | 5ʹ extended: 7.39, 3ʹ intronic: 0.0964, exonic: 0.39 |
| DSSP | 0.01 | 0.13 | 0.01 | 0.30 |
| GeneSplicer | 0.18 | 0.05 | 0.21 | – |
| MaxEntScan | 0.26 | 0.31 | 0.24 | 0.10 |
| MMSplice | 1.42 | – | 1.37 | 2 |
| NNSPLICE | 0.13 | 0.40 | 0.30 | 0.05 |
| Spidex | 0.86 | – | 1.72 | 5 |
| SpliceAI | 0.19 | 0.18 | 0.11 | 0.20 |
| SpliceRover | 0.18 | 0.26 | 0.10 | – |
| SpliceSiteFinder‐like | 0.01 | 0.12 | 0.09 | 0.05 |
3.5. Performance assessment of the splice prediction tools
The accuracy, sensitivity, specificity, PPV, NPV, and MCC for each data set, as defined in Table S2, are provided in Tables 3, 4, 5. For the ABCA4 NCSS variants, the PPV was above 90% for all tools and the NPV was below 30% for all tools. This can be explained by the imbalance in the variants as the majority of the variants in this data set affected splicing. The highest MCC, a measure that is optimal for unbalanced test data sets, was found for SpliceAI and SpliceRover. For ABCA4 DI variants, the tools with the lowest MCC, and also PPV and specificity, corresponded to the tools with the lowest AUC (SpliceSiteFinder‐like and CADD). SpliceAI showed the highest accuracy, PPV, sensitivity, specificity, NPV, and MCC. For the MYBPC3 NCSS data set, all tools showed a reasonable performace based on both the AUC and MCC. The tool with the highest AUC was the Alamut 3/4 consensus approach, followed by GeneSplicer, MMSplice and SpliceSiteFinder‐like.
Table 3.
Confusion matrix and statistical measures of the ABCA4 NCSS variants
| Tool | Missing values | TP | FP | TN | FN | Accuracy (%) | PPV (%) | Sensitivity (%) | Specificity (%) | NPV (%) | MCC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Alamut Consensus 3/4 | 0 | 35 | 2 | 5 | 29 | 56 | 95 | 55 | 71 | 15 | 0.16 |
| CADD | 0 | 40 | 3 | 4 | 24 | 62 | 93 | 63 | 57 | 14 | 0.12 |
| DSSP | 0 | 51 | 2 | 5 | 13 | 79 | 96 | 80 | 71 | 28 | 0.35 |
| GeneSplicer | 0 | 31 | 3 | 4 | 33 | 49 | 91 | 48 | 57 | 11 | 0.03 |
| MaxEntScan | 0 | 40 | 2 | 5 | 24 | 63 | 95 | 63 | 71 | 17 | 0.21 |
| MMSplice | 0 | 43 | 2 | 5 | 21 | 68 | 96 | 67 | 71 | 19 | 0.24 |
| NNSPLICE | 0 | 42 | 2 | 5 | 22 | 66 | 95 | 66 | 71 | 19 | 0.23 |
| Spidex | 5 | 43 | 2 | 5 | 21 | 68 | 96 | 67 | 71 | 19 | 0.24 |
| SpliceAI | 0 | 50 | 1 | 6 | 14 | 79 | 98 | 78 | 86 | 30 | 0.42 |
| SpliceRover | 0 | 48 | 1 | 6 | 16 | 76 | 98 | 75 | 86 | 27 | 0.39 |
| SpliceSiteFinder‐like | 0 | 40 | 2 | 5 | 24 | 63 | 95 | 63 | 71 | 17 | 0.21 |
Abbreviations: FN, false negatives; FP, false positives; MCC, Mathew's correlation coefficient; NPV, negative predictive value; PPV, positive predictive value; TN, true negatives; TP, true positives.
Table 4.
Confusion matrix and statistical measures of the ABCA4 DI variants
| Tool | Missing values | TP | FP | TN | FN | Accuracy (%) | PPV (%) | Sensitivity (%) | Specificity (%) | NPV (%) | MCC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Alamut Consensus 3/4 | 0 | 11 | 14 | 46 | 10 | 70 | 44 | 52 | 77 | 82 | 0.28 |
| CADD | 0 | 12 | 24 | 36 | 9 | 59 | 33 | 57 | 60 | 80 | 0.15 |
| DSSP | 0 | 13 | 19 | 41 | 8 | 67 | 41 | 62 | 68 | 84 | 0.27 |
| GeneSplicer | 0 | 14 | 16 | 44 | 7 | 72 | 47 | 67 | 73 | 86 | 0.36 |
| MaxEntScan | 0 | 13 | 21 | 39 | 8 | 64 | 38 | 62 | 65 | 83 | 0.24 |
| NNSPLICE | 0 | 14 | 17 | 43 | 7 | 70 | 45 | 67 | 72 | 86 | 0.35 |
| SpliceAI | 0 | 19 | 3 | 57 | 2 | 94 | 86 | 90 | 95 | 97 | 0.84 |
| SpliceRover | 0 | 15 | 14 | 46 | 6 | 75 | 52 | 71 | 77 | 88 | 0.44 |
| SpliceSiteFinder‐like | 0 | 11 | 27 | 33 | 10 | 54 | 29 | 52 | 55 | 77 | 0.06 |
Abbreviations: FN, false negatives; FP, false positives; MCC, Mathew's correlation coefficient; NPV, negative predictive value; PPV, positive predictive value; TN, true negatives; TP, true positives.
Table 5.
Confusion matrix and statistical measures of the MYBPC3 NCSS variants
| Tool | Missing values | TP | FP | TN | FN | Accuracy (%) | PPV (%) | Sensitivity (%) | Specificity (%) | NPV (%) | MCC |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Alamut Consensus 3/4 | 0 | 23 | 4 | 23 | 11 | 75 | 85 | 68 | 85 | 68 | 0.53 |
| CADD | 0 | 21 | 8 | 19 | 13 | 66 | 72 | 62 | 70 | 59 | 0.32 |
| DSSP | 0 | 22 | 10 | 17 | 12 | 64 | 69 | 65 | 63 | 59 | 0.28 |
| GeneSplicer | 0 | 25 | 6 | 21 | 9 | 75 | 81 | 74 | 78 | 70 | 0.51 |
| MaxEntScan | 0 | 24 | 6 |
21 |
10 | 74 | 80 | 71 | 78 | 68 | 0.48 |
| MMSplice | 0 | 25 | 6 | 21 | 9 | 75 | 81 | 74 | 78 | 70 | 0.51 |
| NNSPLICE | 0 | 23 | 6 | 21 | 11 | 72 | 79 | 68 | 78 | 66 | 0.45 |
| Spidex | 3 | 20 | 9 | 18 | 14 | 62 | 69 | 59 | 67 | 56 | 0.25 |
| SpliceAI | 0 | 22 | 8 | 19 | 12 | 67 | 73 | 65 | 70 | 61 | 0.35 |
| SpliceRover | 0 | 22 | 9 | 18 | 12 | 66 | 71 | 65 | 67 | 60 | 0.31 |
| SpliceSiteFinder‐like | 0 | 25 | 6 | 21 | 9 | 75 | 81 | 74 | 78 | 70 | 0.51 |
Abbreviations: FN, false negatives; FP, false positives; MCC, Mathew's correlation coefficient; NPV, negative predictive value; PPV, positive predictive value; TN, true negatives; TP, true positives.
4. DISCUSSION
Increasing use of whole genome and whole exome sequencing in routine diagnostics requires in silico splice prediction tools to select likely pathogenic variants for further testing. To date, there are studies evaluating single splice prediction tools, but none comparing multiple deep learning tools. This study benchmarked selected established and deep learning in silico splice prediction tools based on multiple classification metrics on two of the largest sets of variants for which the effect of splicing is functionally assessed using mini‐ or midigene assays. The data showed that SpliceAI, the Alamut 3/4 consensus approach, NNSPLICE, and MaxEntScan perform well on all data sets based on the AUC. Though for ABCA4 variants tools of the deep learning category showed the highest AUC values, the Alamut tools and MMSplice performed best on the MYBPC3 variants. Additionally, this study demonstrated that the choice of the best splice prediction tool may depend on the gene of interest and the type of splice‐altering variants. Only for ABCA4 DI variants we could clearly identify SpliceAI as the best performing tool.
We included NCSS and DI variants in the ABCA4 gene and NCSS variants in the MYBPC3 gene. There was no single best‐performing splice prediction tool for all three data sets. This may be explained in several ways. ABCA4 and MYBPC3 are expressed in a tissue‐specific manner, with high expression in the retina and heart muscle, respectively. The representation of splice patterns in these tissues in the data used for training different deep learning algorithms may affect its performance. None of the tools included retina tissue in its training data, as far as we can judge. Moreover, most splice prediction tools focus on the area around the canonical splice sites and were not trained on DI variants, which explains their lower performance on the DI data set. Another reason for differences in performance may lie in the selection criteria used to functionally assess the ABCA4 and MYBPC3 variants. MYBPC3 variants were selected for functional validation based on MaxEntScan scores, and ABCA4 variants were selected when they showed a difference in splice score for at least two of the Alamut programs (including Human Splicing Finder) and/or a delta score of at least 2%. This may lead to a positive bias in the performance assessment for the tools that were used to select the variants, but we find the opposite, where Alamut 3/4 performs best on the MYBPC3 data, and MaxEntScan performs relatively well on the ABCA4 data set. Yet another source of difference in performance can be found in the functional assays used for their evaluation; ABCA4 variants were tested in midigenes and MYBPC3 variants in minigenes. In most cases, minigenes and midigenes result in the same transcripts but when the flanking exons of the minigene vector are stronger than the ones in the gene of interest, they can cause artifacts. Ideally, we would have used only data sets tested in midigenes; however, the data was not available. The different splice assays might also explain why SpliceAI performed better on the ABCA4 data sets than on the MYBPC3 data set.
The performance measures of splice prediction tools need to be carefully chosen, in particular when there is an imbalance in the number of splice‐altering and nonsplice‐altering variants. In the ABCA4 NCSS data set, most variants affected splicing, whereas most ABCA4 DI variants had no effect on splicing. The MYBPC3 data set contained about the same number of splice and nonsplice‐altering variants. Imbalance in the data set influences most classification metrics. If the positive (splice‐altering) and negative class (nonsplice‐altering) are interchanged during the calculation of the metric, the metric changes. The only metric not influenced by class imbalance is MCC and we regard this as the preferred measure in the current setting.
Our results are consistent with previous studies that included a smaller number of splice prediction tools. Wai et al. (2020) compared Alamut, Human Splicing Finder, and SpliceAI on 257 VUSs (NCSS and DI) from blood RNA samples showing that SpliceAI outperformed the other tools with an AUC of 0.951. A second study by Ellingford et al. (2019) compared SpliceAI, SPIDEX, S‐CAP, CADD, and MaxEntScan first in a real‐time assessment of 21 variants and then in variant prioritization of nearly 3000 variants. The real‐time assessment showed that SpliceAI and MaxEntScan achieved a good performance. In the variant prioritization of the large cohort only SpliceAI, Spidex, and CADD are compared. Here, SpliceAI showed the highest AUC (0.96). Our AUC values for SpliceAI were 0.80 (ABCA4 NCSS), 0.95 (ABCA4 DI), and 0.72 (MYBPC3 NCSS). Especially the AUCs of the NCSS data sets are lower than the AUC found in the two other studies. There can be multiple explanations for this. First, our data sets are smaller, making the right prediction for each individual variant more important. Second, we used variants located in only one gene, whereas the abovementioned studies used variants in a variety of genes. This could indicate that for genes with tissue‐specific expression the available splice prediction tools are not specialized enough, for reasons explained above. Third, we evaluated tools based on functional assessment with midi‐ or minigenes assays, which currently represent the best medium‐throughput tools. Still, also this experimental set‐up has limitations since the splice assays were performed in human kidney cells. This means that tissue‐specific splicing events may be missed. For ABCA4 it is known that variants can lead to tissue‐specific pseudo‐exon inclusion (Albert et al., 2018). Another limitation is that the percentage mutant RNA of the ABCA4 variants is determined based on RT‐PCR products visualized on agarose gels. RT‐PCR has a bias toward smaller segments, and this can lead to incorrect classification of the variants. A better alternative would be to use RNA‐sequencing, which captures bigger segments as well as smaller segments.
A general observation made in our benchmark study is that the prediction of the in silico tools on a set of clinically relevant variants varies considerably from the performance described in the original paper. SpliceAI, for instance, achieves an area under the precision‐recall curve (PR‐AUC) of 0.98 on RNA‐seq data (Jaganathan et al., 2019). For our data sets, the PR‐AUC is 0.93 for ABCA4 NCSS variants, 0.91 for ABCA4 DI variants, and 0.74 for MYBPC3 NCSS variants. The higher performance observed by the authors can be explained by the use of an RNA‐seq dataset. Large population‐based RNA‐seq data sets like GTEx (Lonsdale et al., 2013) may contain non‐pathogenic rare splice‐altering variants (Mertes et al., 2021) and have an extreme skew toward variants with a neutral effect on splicing. This may introduce biases that make their results poorly generalizable to variants encountered in a clinical setting with a higher prior likelihood for affecting splicing. Moreover, circularity, that is, incomplete independence of the variants used for training and testing, may result in overestimation of the performance of the model (Grimm et al., 2015). This is why it is important to use a truly independent set of clinically relevant variants to evaluate the performance of the splice prediction tools. Additionally, it is important to use the right evaluation metrics to compare different algorithms. As shown for the ABCA4 variants, imbalance in the data set influences the classification metrics and, therefore, also the comparison. The precision‐recall curve uses the PPV and sensitivity to calculate the AUC. The imbalance in the data set has an influence on both metrics, which makes it difficult to compare highly imbalanced data sets based on the PR‐AUC.
To conclude, there are a variety of different splice prediction tools available. It is not easy to choose which tool to use because different tools may perform better in different contexts. The best‐performing tools make use of different algorithms, deep learning (SpliceAI), machine learning (NNSPLICE), and interactions (MaxEntScan). Deep learning has the possibility to improve splice prediction but is not a guarantee for success: For both ABCA4 datasets the deep learning tools showed promosing results, while for the MYBPC3 dataset the more traditional Alamut tools achieved better results. Only for ABCA4 DI the deep learning tool SpliceAI clearly outperformed all other tools. In the end, it is always a trade‐off between knowing the features in traditional machine learning, and thereby also limiting the complexity and numbers of features, and the possibility to capture more complex features in deep learning but having a black‐box algorithm.
WEB RESOURCES
https://github.com/cmbi/Benchmarking_splice_prediction_tools
https://github.com/DSSP-github/DSSP
https://github.com/gagneurlab/MMSplice_MTSplice
https://github.com/Illumina/SpliceAI
https://cadd.gs.washington.edu/score
http://bioit2.irc.ugent.be/rover/splicerover
http://www.openbioinformatics.org/annovar/spidex_download_form.php
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
Supporting information
Supporting information.
Supporting information.
ACKNOWLEDGMENTS
The work of Mubeen Khan was supported by RetinaUK, grant no. GR591 (to Frans P. M. Cremers).
Riepe, T. V., Khan, M., Roosing, S., Cremers, F. P. M., & 't Hoen, P. A. C. (2021). Benchmarking deep learning splice prediction tools using functional splice assays. Human Mutation, 42, 799–810. 10.1002/humu.24212
DATA AVAILABILITY STATEMENT
Variants used for the study are publicly available online at LOVD (https://www.lovd.nl/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and ExAC (https://exac.broadinstitute.org/).
REFERENCES
- Albert, S., Garanto, A., Sangermano, R., Khan, M., Bax, N. M., Hoyng, C. B., Zernant, J., Lee, W., Allikmets, R., Collin, R. W. J., & Cremers, F. P. M. (2018). Identification and rescue of splice defects caused by two neighboring deep‐intronic ABCA4 mutations underlying Stargardt disease. American Journal of Human Genetics, 102(4), 517–527. 10.1016/j.ajhg.2018.02.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allikmets, R., Singh, N., Sun, H., Shroyer, N. F., Hutchinson, A., Chidambaram, A., Gerrard, B., Baird, L., Stauffer, D., Peiffer, A., Rattner, A., Smallwood, P., Li, Y., Anderson, K. L., Lupski, J. R., Nathans, J., Leppert, M., Dean, M., & Lewis, R. A. (1997). A photoreceptor cell‐specific ATP‐binding transporter gene (ABCR) is mutated in recessive Stargardt macular dystrophy. Nature Genetics, 15(3), 236–246. 10.1038/ng0397-236 [DOI] [PubMed] [Google Scholar]
- Bauwens, M., Garanto, A., Sangermano, R., Naessens, S., Weisschuh, N., de Zaeytijd, J., Khan, M., Sadler, F., Balikova, I., van Cauwenbergh, C., Rosseel, T, Bauwens, J., de Leeneer, K., de Jaegere, S., van Laethem, T., de Vries, M., Carss, K., Arno, G., Fakin, A., … de Baere, E. (2019). ABCA4‐associated disease as a model for missing heritability in autosomal recessive disorders: Novel noncoding splice, cis‐regulatory, structural, and recurrent hypomorphic variants. Genetics in Medicine, 21(8), 1761–1771. 10.1038/s41436-018-0420-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bradley, K. J., Cavaco, B. M., Bowl, M. R., Harding, B., Young, A., & Thakker, R. V. (2005). Utilisation of a cryptic non‐canonical donor splice site of the gene encoding PARAFIBROMIN is associated with familial isolated primary hyperparathyroidism. Journal of Medical Genetics, 42(8), e51. 10.1136/jmg.2005.032201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Braun, T. A., Mullins, R. F., Wagner, A. H., Andorf, J. L., Johnston, R. M., Bakall, B. B., Deluca, A. P., Fishman, G. A., Lam, B. L., Weleber, R. G., Cideciyan, A. V., Jacobson, S. G., Sheffield, V. C., Tucker, B. A., & Stone, E. M. (2013). Non‐exomic and synonymous variants in ABCA4 are an important cause of Stargardt disease. Human Molecular Genetics, 22(25), 5136–5145. 10.1093/hmg/ddt367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng, J., Nguyen, T. Y. D., Cygan, K. J., Çelik, M. H., Fairbrother, W. G., Avsec, Ž., & Gagneur, J. (2019). MMSplice: Modular modeling improves the predictions of genetic variant effects on splicing. Genome Biology, 20(1), 48. 10.1186/s13059-019-1653-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremers, F. P. M., Lee, W., Collin, R. W. J., & Allikmets, R. (2020). Clinical spectrum, genetic complexity and therapeutic approaches for retinal disease caused by ABCA4 mutations. Progress in Retinal and Eye Research, 79, 100861. 10.1016/j.preteyeres.2020.100861 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cremers, F. P. M., Van De Pol, D. J. R., Van Driel, M., Den Hollander, A. I., van Haren, F. J. J., Knoers, N. V. A. M., Tijmes, N., Bergen, A. A., Rohrschneider, K., Blankenagel, A., Pinckers, A. J., Deutman, A. F., & Hoyng, C. B. (1998). Autosomal recessive retinitis pigmentosa and cone‐rod dystrophy caused by splice site mutations in the Stargardt's disease gene ABCR. Human Molecular Genetics, 7(3), 355–362. 10.1093/hmg/7.3.355 [DOI] [PubMed] [Google Scholar]
- Dhir, A., & Buratti, E. (2010). Alternative splicing: Role of pseudoexons in human disease and potential therapeutic strategies: Minireview. FEBS Journal, 277(4), 841–855. 10.1111/j.1742-4658.2009.07520.x [DOI] [PubMed] [Google Scholar]
- van Dijk, S. J., Dooijes, D., Remedios, C., Dos, Michels, M., Lamers, J. M. J., Winegrad, S., Schlossarek, S., Carrier, L., ten Cate, F. J., Stienen, G. J. M., & van Velden, J. D. (2009). Cardiac myosin‐binding protein C mutations and hypertrophic ardiomyopathy haploinsufficiency, deranged phosphorylation, and cardiomyocyte dysfunction. Circulation, 119(11), 1473–1483. 10.1161/CIRCULATIONAHA.108.838672 [DOI] [PubMed] [Google Scholar]
- Ellingford, J. M., Thomas, H. B., Rowlands, C., Arno, G., Beaman, G., Gomes‐Silva, B., Gossan, N., Hardcastle, C., Campbell, C., Webb, K., O'Callaghan, C., Hirst, R. A., Ramsden, S., Jones, E., Clayton‐Smith, J., Webster, A. R., Genomics England Research Consortium , O'Keefe, R. T., & Black, G. C. (2019). Functional and in‐silico interrogation of rare genomic variants impacting RNA splicing for the diagnosis of genomic disorders. BioRxiv, 781088. 10.1101/781088 [DOI] [Google Scholar]
- Fadaie, Z., Khan, M., Del Pozo‐Valero, M., Cornelis, S. S., Ayuso, C., Cremers, F. P. M., Roosing, S., & The ABCA4 Study Group . (2019). Identification of splice defects due to noncanonical splice site or deep‐intronic variants in ABCA4. Human Mutation, 40(12), 2365–2376. 10.1002/humu.23890 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang, L. J., Simard, M. J., Vidaud, D., Assouline, B., Lemieux, B., Vidaud, M., Chabot, B., & Thirion, J. P. (2001). A novel mutation in the neurofibromatosis type 1 (NF1) gene promotes skipping of two exons by preventing exon definition. Journal of Molecular Biology, 307(5), 1261–1270. 10.1006/jmbi.2001.4561 [DOI] [PubMed] [Google Scholar]
- Glisovic, T., Bachorik, J. L., Yong, J., & Dreyfuss, G. (2008). RNA‐binding proteins and post‐transcriptional gene regulation. FEBS Letters, 582(14), 1977–1986. 10.1016/j.febslet.2008.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grimm, D. G., Azencott, C. A., Aicheler, F., Gieraths, U., Macarthur, D. G., Samocha, K. E., Cooper, D. N., Stenson, P. D., Daly, M. J., Smoller, J. W., Duncan, L.E., & Borgwardt, K. M. (2015). The evaluation of tools used to predict the impact of missense variants is hindered by two types of circularity. Human Mutation, 36(5), 513–523. 10.1002/humu.22768 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hefferon, T. W., Broackes‐Carter, F. C., Harris, A., & Cutting, G. R. (2002). Atypical 5′ splice sites cause CFTR exon 9 to be vulnerable to skipping. American Journal of Human Genetics, 71(2), 294–303. 10.1086/341664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito, K., Patel, P. N., Gorham, J. M., McDonough, B., DePalma, S. R., Adler, E. E., Seidman, J. G., Lam, L., MacRae, C. A., Mohiuddin, S. M., Fatkin, D., & Seidman, C. E. (2017). Identification of pathogenic gene mutations in LMNA and MYBPC3 that alter RNA splicing. Proceedings of the National Academy of Sciences of the United States of America, 114(29), 7689–7694. 10.1073/pnas.1707741114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaganathan, K., Kyriazopoulou Panagiotopoulou, S., McRae, J. F., Darbandi, S. F., Knowles, D., Li, Y. I., Farh, K. K. H., Kosmicki, J. A., Arbelaez, J., Cui, W., Schwartz, G. B., Chow, E. D., Kanterakis, E., Gao, H., Kia, A., Batzoglou, S., Sanders, S. J., & Farh, K.‐H. (2019). Predicting splicing from primary sequence with deep learning. Cell, 176(3), 535–548. 10.1016/j.cell.2018.12.015 [DOI] [PubMed] [Google Scholar]
- Jian, X., Boerwinkle, E., & Liu, X. (2014). In silico tools for splicing defect prediction: A survey from the viewpoint of end users. Genetics in Medicine, 16(7), 497–503. 10.1038/gim.2013.176 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M., Cornelis, S. S., Pozo‐Valero, M. D., Whelan, L., Runhart, E. H., Mishra, K., Bults, F., Al Swaiti, Y., Al Talbishi, A., de Baere, E., Banfi, S., Banin, E., Bauwens, M., Ben‐Yosef, T., Boon, C. J. F., van den Born, I., Defoort, S., Devos, A., Dockery, A., … Cremers, F. P. M. (2020). Resolving the dark matter of ABCA4 for 1054 Stargardt disease probands through integrated genomics and transcriptomics. Genetics in Medicine, 22(7), 1235–1246. 10.1038/s41436-020-0787-4 [DOI] [PubMed] [Google Scholar]
- Khan, M., Cornelis, S. S., Sangermano, R., Post, I. J. M., Groesbeek, A. J., Amsu, J., Gilissen, C., Garanto, A., Collin, R. W. J., & Cremers, F. P. M. (2020). In or out? New insights on exon recognition through splice‐site interdependency. International Journal of Molecular Sciences, 21(7):2300. 10.3390/ijms21072300 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan, M., Fadaie, Z., Cornelis, S. S., Cremers, F. P. M., & Roosing, S. (2019). Identification and Analysis of Genes Associated with Inherited Retinal Diseases. In Methods in Molecular Biology (1834, pp. 3–27). Humana Press Inc. 10.1007/978-1-4939-8669-9_1 [DOI] [PubMed] [Google Scholar]
- Labonne, J. D. J., Chung, M. J., Jones, J. R., Anand, P., Wenzel, W., Iacoboni, D., Layman, L. C., & Kim, H. G. (2016). Concomitant partial exon skipping by a unique missense mutation of RPS6KA3 causes Coffin‐Lowry syndrome. Gene, 575(1), 42–47. 10.1016/j.gene.2015.08.032 [DOI] [PubMed] [Google Scholar]
- Lonsdale, J., Thomas, J., Salvatore, M., Phillips, R., Lo, E., Shad, S., Moore, H. F., Walters, G., Garcia, F., Young, N., Foster, B., Moser, M., Karasik, E., Gillard, B, Ramsey, K., Sullivan, S., Bridge, J., Magazine, H, Syron, J., … Hasz, R. (2013). The Genotype‐Tissue Expression (GTEx) project. Nature Genetics, 45(6), 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- López‐Bigas, N., Audit, B., Ouzounis, C., Parra, G., & Guigó, R. (2005). Are splicing mutations the most frequent cause of hereditary disease? FEBS Letters, 579(9), 1900–1903. 10.1016/j.febslet.2005.02.047 [DOI] [PubMed] [Google Scholar]
- Louadi, Z., Oubounyt, M., Tayara, H., & … Chong, K. T. (2019). Deep splicing code: Classifying alternative splicing events using deep learning. Genes, 34(8), 330–333. 10.3390/genes10080587 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marston, S., Copeland, O., Jacques, A., Livesey, K., Tsang, V., McKenna, W. J., Jalilzadeh, S., Carballo, S., Redwood, C, & Watkins, H. (2009). Evidence from human myectomy samples that MYBPC3 mutations cause hypertrophic cardiomyopathy through haploinsufficiency. Circulation Research, 105(3), 219–222. 10.1161/CIRCRESAHA.109.202440 [DOI] [PubMed] [Google Scholar]
- Maugeri, A., Klevering, B. J., Rohrschneider, K., Blankenagel, A., Brunner, H. G., Deutman, A. F., Hoyng, C. B., & Cremers, F. P. M. (2000). Mutations in the ABCA4 (ABCR) gene are the major cause of autosomal recessive cone‐rod dystrophy. American Journal of Human Genetics, 67(4), 960–966. 10.1086/303079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mertes, C., Scheller, I. F., Yépez, V. A., Çelik, M. H., Liang, Y., Kremer, L. S., Gusic, M., Prokisch, H., & Gagneur, J. (2021). Detection of aberrant splicing events in RNA‐seq data using FRASER. Nature Communications, 12(1), 1–13. 10.1038/s41467-020-20573-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molday, L. L., Rabin, A. R., & Molday, R. S. (2000). ABCR expression in foveal cone photoreceptors and its role in Stargardt macular dystrophy. Nature Genetics, 25(3), 257–258. 10.1038/77004 [DOI] [PubMed] [Google Scholar]
- Moles‐Fernández, A., Duran‐Lozano, L., Montalban, G., Bonache, S., López‐Perolio, I., Menéndez, M., Santamariña, M., Behar, R., Blanco, A., Carrasco, E., López‐Fernández, A., Stjepanovic, N., Balmaña, J., Capellá, G., Pineda, M., Vega, A, Lázaro, C., de la Hoya, M., Diez, O, & Gutiérrez‐Enríquez, S. (2018). Computational tools for splicing defect prediction in breast/ovarian cancer genes: How efficient are they at predicting RNA alterations? Frontiers in Genetics, 9, 366. 10.3389/fgene.2018.00366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Naito, T. (2019). Predicting the impact of single nucleotide variants on splicing via sequence‐based deep neural networks and genomic features. Human Mutation, 40(9), 1261–1269. 10.1002/humu.23794 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohno, K., Takeda, J. I., & Masuda, A. (2018). Rules and tools to predict the splicing effects of exonic and intronic mutations. Wiley Interdisciplinary Reviews: RNA, 9(1), 1451. 10.1002/wrna.1451 [DOI] [PubMed] [Google Scholar]
- Pan, Q., Shai, O., Lee, L. J., Frey, B. J., & Blencowe, B. J. (2008). Deep surveying of alternative splicing complexity in the human transcriptome by high‐throughput sequencing. Nature Genetics, 40(12), 1413–1415. 10.1038/ng.259 [DOI] [PubMed] [Google Scholar]
- Pertea, M. (2001). GeneSplicer: A new computational method for splice site prediction. Nucleic Acids Research, 29(5), 1185–1190. 10.1093/nar/29.5.1185 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramalho, A. S., Beck, S., Penque, D., Gonska, T., Seydewitz, H. H., Mall, M., & Amaral, M. D. (2003). Transcript analysis of the cystic fibrosis splicing mutation 1525‐1G>A shows use of multiple alternative splicing sites and suggests a putative role of exonic splicing enhancers. Journal of Medical Genetics, 40(7), e88. 10.1136/jmg.40.7.e88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reese, M. G. (1997). Improved splice site detection in Genie. Journal of Computational Biology, 4(3), 311–323. 10.1089/cmb.1997.4.311 [DOI] [PubMed] [Google Scholar]
- Rentzsch, P., Witten, D., Cooper, G. M., Shendure, J., & Kircher, M. (2019). CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Research, 47(D1), D886–D894. 10.1093/nar/gky1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romano, M., Buratti, E., & Baralle, D. (2013). Role of pseudoexons and pseudointrons in human cancer. International Journal of Cell Biology, 2013, 810572. 10.1155/2013/810572 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangermano, R., Garanto, A., Khan, M., Runhart, E. H., Bauwens, M., Bax, N. M., van den Born, L. I., Khan, M. I., Cornelis, S. S., Verheij, J. B. G. M., Pott, J.‐W. R., Thiadens, A. A. H. J., Klaver, C. C. W., Puech, B., Meunier, I., Naessens, S., Arno, G., Fakin, A., … Cremers, F. P. M. (2019). Deep‐intronic ABCA4 variants explain missing heritability in Stargardt disease and allow correction of splice defects by antisense oligonucleotides. Genetics in Medicine, 21(8), 1751–1760. 10.1038/s41436-018-0414-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sangermano, R., Khan, M., Cornelis, S. S., Richelle, V., Albert, S., Garanto, A., Elmelik, D., Qamar, R., Lugtenberg, D., van den Born, L. I., Collin, R. W. J., & Cremers, F. P. M. (2018). ABCA4 midigenes reveal the full splice spectrum of all reported noncanonical splice site variants in Stargardt disease. Genome Research, 28(1), 100–110. 10.1101/gr.226621.117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shapiro, M. B., & Senapathy, P. (1987). RNA splice junctions of different classes of eukaryotes: Sequence statistics and functional implications in gene expression. Nucleic Acids Research, 15(17), 7155–7174. 10.1093/nar/15.17.7155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaw, M. A., Brunetti‐Pierri, N., Kádasi, L., Kovácová, V., Van Maldergem, L., de Brasi, D., Gécz, J., & Salerno, M. (2003). Identification of three novel SEDL mutations, including mutation in the rare, non‐canonical splice site of exon 4. Clinical Genetics, 64(3), 235–242. 10.1034/j.1399-0004.2003.00132.x [DOI] [PubMed] [Google Scholar]
- Sheth, N., Roca, X., Hastings, M. L., Roeder, T., Krainer, A. R., & Sachidanandam, R. (2006). Comprehensive splice‐site analysis using comparative genomics. Nucleic Acids Research, 34(14), 3955–3967. 10.1093/nar/gkl556 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sobczyńska‐Tomaszewska, A., Ołtarzewski, M., Czerska, K., Wertheim‐Tysarowska, K., Sands, D., Walkowiak, J., Bal, J., & Mazurczak, T. (2013). Newborn screening for cystic fibrosis: Polish 4 years’ experience with CFTR sequencing strategy. European Journal of Human Genetics, 21(4), 391–396. 10.1038/ejhg.2012.180 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H., & Chasin, L. A. (2000). Multiple splicing defects in an intronic false exon. Molecular and Cellular Biology, 20(17), 6414–6425. 10.1128/mcb.20.17.6414-6425.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun, H., & Nathans, J. (1997). Stargardt's ABCR is localized to the disc membrane of retinal rod outer segments. Nature Genetics, 17(1), 15–16. 10.1038/ng0997-15 [DOI] [PubMed] [Google Scholar]
- Symoens, S., Malfait, F., Vlummens, P., Hermanns‐Lê, T., Syx, D., & de Paepe, A. (2011). A novel splice variant in the N‐propeptide of COL5A1 causes an EDS phenotype with severe kyphoscoliosis and eye involvement. PLOS One, 6(5), e20121. 10.1371/journal.pone.0020121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai, H. A., Lord, J., Lyon, M., Gunning, A., Kelly, H., Cibin, P., Seaby, E. G., Spiers‐Fitzgerald, K., Lye, J., Ellard, S., Thomas, N. S., Bunyan, D. J., Douglas, A. G. L., Baralle, D., & Splicing and disease working group (2020). Blood RNA analysis can increase clinical diagnostic rate and resolve variants of uncertain significance. Genetics in Medicine, 22(6), 1005–1014. 10.1038/s41436-020-0766-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Will, C. L., & Lührmann, R. (2011). Spliceosome structure and function. Cold Spring Harbor Perspectives in Biology, 3(7), 1–2. 10.1101/cshperspect.a003707 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong, H. Y., Alipanahi, B., Lee, L. J., Bretschneider, H., Merico, D., Yuen, R. K. C., Hua, Y., Gueroussov, S., Najafabadi, H. S., Hughes, T. R., Morris, Q., Barash, Y., Krainer, A. R., Jojic, N., Scherer, S. W., Blencowe, B. J., & Frey, B. J. (2015). The human splicing code reveals new insights into the genetic determinants of disease. Science, 347(6218), 1254806. 10.1126/science.1254806 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yeo, G., & Burge, C. B. (2004). Maximum entropy modeling of short sequence motifs with applications to RNA splicing signals. Journal of Computational Biology, 11(2–3), 377–394. 10.1089/1066527041410418 [DOI] [PubMed] [Google Scholar]
- Zernant, J., Xie, Y. A., Ayuso, C., Riveiro‐Alvarez, R., Lopez‐Martinez, M. A., Simonelli, F., Allikmets, R., Testa, F., Gorin, M. B., Strom, S. P., Bertelsen, M., Rosenberg, T., Boone, P. M., Yuan, B., Ayyagari, R., Nagy, P. L., Tsang, S. H., Gouras, P., Collison, F. T., … Fishman, G. A. (2014). Analysis of the ABCA4 genomic locus in Stargardt disease. Human Molecular Genetics, 23(25), 6797–6806. 10.1093/hmg/ddu396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuallaert, J., Godin, F., Kim, M., Soete, A., Saeys, Y., & de Neve, W. (2018). Splicerover: Interpretable convolutional neural networks for improved splice site prediction. Bioinformatics, 34(24), 4180–4188. 10.1093/bioinformatics/bty497 [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting information.
Supporting information.
Data Availability Statement
Variants used for the study are publicly available online at LOVD (https://www.lovd.nl/), ClinVar (https://www.ncbi.nlm.nih.gov/clinvar/), and ExAC (https://exac.broadinstitute.org/).