

Abstract
Background
Arterial and venous thrombosis are both common in antiphospholipid syndrome (APS). Recent studies have shown that anti‐factor Xa (FXa) therapy in APS patients leads to a greater number of patients with arterial thrombosis than with warfarin. We hypothesize that this may be due to the lowering of prothrombin levels by warfarin.
Objectives
To investigate whether antiprothrombin antibodies induce platelet aggregation and to identify the platelet receptors involved. A second aim was to investigate the effect of reduced prothrombin levels on antiprothrombin antibody‐induced platelet aggregation.
Methods
Enzyme‐linked immunosorbent assays were performed to measure binding of antiprothrombin antibodies to prothrombin fragment 1+2 and prothrombin. Platelet aggregation assays in washed platelets were performed. FcγRIIA was immunoprecipitated and tyrosine‐phosphorylated FcγRIIA was measured by western blot.
Results
The antiprothrombin antibodies 28F4 and 3B1 had lupus anticoagulant (LAC) activity and caused platelet aggregation in the presence of Ca2+ and prothrombin. Antiprothrombin antibodies without LAC activity did not activate platelets. Inhibition of Syk and Src kinases and FcγRIIA blocked platelet aggregation. Fab and F(ab’)2 fragments of 28F4 were unable to induce platelet aggregation. Immunoprecipitations showed that whole 28F4 immunoglobulin G induced tyrosine phosphorylation of FcγRIIA. Platelet aggregation was significantly reduced when prothrombin levels were reduced from 1 µM to 0.2 µM.
Conclusions
Antiprothrombin antibodies with LAC activity are able to activate platelets via FcγRIIA. Decreased prothrombin levels resulted in less antiprothrombin antibody‐mediated platelet aggregation. This may explain the lower incidence of arterial thrombosis in patients treated with warfarin than with anti‐FXa therapy.
Keywords: antiphospholipid syndrome, antiprothrombin antibodies, DOACs, thrombosis, vitamin K antagonists
Essentials.
Arterial and venous thrombosis are both common in antiphospholipid syndrome and this is linked to the presence of antiprothrombin antibodies.
Antiprothrombin antibodies with lupus anticoagulant activity are able to activate platelets via the FcγRIIA receptor.
Decreased prothrombin levels resulted in less antiprothrombin antibody‐mediated platelet aggregation.
The reduction in antiprothrombin levels by warfarin may explain the lower incidence of arterial thrombosis in patients than with anti‐FXa therapy.
1. INTRODUCTION
Antiphospholipid syndrome (APS) is characterized by thrombosis and/or pregnancy complications due to the persistent presence of antiphospholipid antibodies.1 Laboratory criteria for classification of APS include detection of lupus anticoagulant (LAC), anti‐β2‐glycoprotein I (aβ2GPI) and anticardiolipin (aCL) immunoglobulin G (IgG)/M antibodies.1 Clinical criteria for classification of thrombotic APS include venous, arterial, and small vessel thrombosis. The updated European League Against Rheumatism recommendations on managing thrombotic APS patients recommend long‐term vitamin K antagonists as the standard of care.2
Direct oral anticoagulants (DOACs) such as rivaroxaban, a direct factor Xa (FXa) inhibitor, are currently prescribed to patients with venous thromboembolism and atrial fibrillation. The advantage of DOACs is that they are given at a fixed dose and do not need laboratory monitoring. Interestingly, two trials reported predominantly arterial thrombosis in patients treated with rivaroxaban with a high rate of stroke.3, 4 No cases of stroke were reported in patients treated with warfarin.3, 4 Recent professional guidance statements have been issued regarding the use of DOACs in APS patients and recommended against DOACs in triple positive APS patients (defined by the presence of LAC, aβ2GPI, and aCL antibodies) and in arterial thrombosis.5
Blood platelets play a key role in arterial thrombosis.6 Antibodies against β2GPI are known to activate platelets via glycoprotein (GP) Ibα and apolipoprotein E receptor 2.7, 8 Triple‐positive APS patients have the highest risk of thrombotic complications, and these patients usually have both aβ2GPI antibodies and anti‐phosphatidylserine/prothrombin (aPS/PT) antibodies.9 A systematic review identified aPS/PT antibodies as a risk factor for arterial thrombosis.10 Still, experimental data on aPS/PT antibodies and platelet aggregation are lacking.
The overall aim was to investigate whether antiprothrombin antibodies stimulate platelet activation and to identify the underlying mechanism. This included investigation of the effect of reduced prothrombin levels on platelet aggregation as warfarin inhibits the vitamin K–dependent synthesis of biologically active prothrombin, whereas DOACs do not.11 Decreased levels of prothrombin may explain why warfarin‐treated APS patients are protected against arterial thrombosis, whereas those treated with rivaroxaban are not.
2. MATERIAL AND METHODS
2.1. Reagents
Purified human prothrombin was from Synapse Research Institute (Maastricht, The Netherlands) and rivaroxaban (Xarelto) was from Bayer Schering Pharma AG. Anti‐FcγRIIA monoclonal antibody (mAb) IV.3 was purified in the laboratory from a hybridoma.12 The antiprothrombin mAb 28F4 is a mouse IgG antibody, as described previously.13, 14 Antiprothrombin fragment 1+2 mAbs 3B1, 6A3, 11H2, and 8H11 were developed and produced according to standard procedures.15 LAC was measured by activated partial thromboplastin time (Diagnostica Stago), dilute Russell Viper Venom Time (Diagnostica Stago), and Ecarin Clotting Time (ECT; Diagnostic Reagents). The Pierce Classic IP Kit from Thermo Fisher Scientific was used for immunoprecipitation. High‐binding enzyme‐linked immunosorbent assay (ELISA) plates from Costar were used for the ELISAs. Sheep anti‐human prothrombin IgG horseradish peroxidase–labeled antibody was from Affinity Biologicals, Inc., prothrombin fragment 1+2 was from Haematologic Technologies, Inc. All other reagents are previously described or are from Sigma‐Aldrich.16
2.2. Preparation of washed platelets
Blood was drawn by venipuncture into 4% sodium citrate and then, within 5 min of blood draw, mixed with 10% acidic citrate dextrose (120 mM sodium citrate, 110 mM glucose, 80 mM citric acid) from consenting, self‐reported healthy volunteers. Platelet‐rich plasma was obtained by centrifugation at 200g for 20 min at room temperature. Washed platelets were obtained by further centrifugation of platelet‐rich plasma at 1000g for 10 min in the presence of 0.2 μg/ml prostacyclin and resuspended in modified‐Tyrode's‐HEPES buffer (134 mM NaCl, 0.34 mM Na2HPO4, 2.9 mM KCl, 12 mM NaHCO3, 20 mM HEPES, 5 mM glucose, 1 mM MgCl2; pH 7.3), as previously described.16 Platelets were used at 2 × 108/ml and 4 × 108/ml for aggregation studies and immunoprecipitation, respectively.
2.3. Fab and F(ab′)2 antibody fragments
Fab fragments from 28F4 were generated by 4‐h incubation of 5 mg/ml antibody with immobilized Ficin in the presence of 25 mM cysteine (Pierce, ThermoFisher) at 37°C. F(ab′)2 fragments from 28F4 were generated by 24‐h incubation of 5 mg/ml antibody with immobilized Ficin in the presence of 4 mM cysteine (Pierce) at 37°C. Preparations were then applied to an immobilized protein A column (Pierce) followed by dialysis to remove any remaining cysteine. The purity of the Fab and F(ab′)2 fragments was verified by sodium dodecyl sulphate–polyacrylamide gel electrophoresis.
2.4. Lupus anticoagulant testing
Normal pooled plasma was incubated with 50 µg/ml mAb for 10 min at 37°C before LAC testing. LAC was performed according to the SSC guidelines and manufacturer's instructions for ECT were followed.17, 18
2.5. ELISA
The mAb 28F4 (5 µg/ml) was coated onto an ELISA plate overnight at 4°C. Blocking buffer consisted of 5% bovine serum albumin and 5 mM CaCl2 in Tris‐buffered saline (pH 7.6). Washing buffer was 5 mM CaCl2 and 0.1% Tween in Tris‐buffered saline (pH 7.6). Increasing concentrations of antigen (prothrombin fragment 1+2, purified prothrombin, or platelet‐poor plasma) was incubated for 1 h at room temperature. Sheep anti‐human prothrombin IgG horseradish peroxidase‐labeled antibody (1:1000) was used as secondary antibody followed by staining with tetra methyl benzidine (ThermoFisher). The reaction was stopped with 2 M H2SO4 and absorption was measured at 405 nm.
2.6. Platelet aggregation assay
Platelet aggregation was assessed by light transmission aggregometry in a PAP‐8E (Bio/Data Corporation) aggregometer for up to 30 min. Washed platelets were incubated with aPT antibodies for 5 min with an antiprothrombin antibody (50 μg/ml) followed by CaCl2 (1 mM, 3 mM, or 10 mM) and purified human prothrombin (1 µM). In some studies, platelets were preincubated with eptifibatide (9 µM), vorapaxar (1 µM) + BMS‐986120 (1 µM), indomethacin (10 µM), ticagrelor (10 µM), PRT‐060318 (5 µM), dasatinib (10 µM), mAb IV.3 (10 µg/ml), or rivaroxaban (200, 400, 800, or 1000 nM) for 10 min.
2.7. Immunoprecipitation and western blotting
Washed platelets were pretreated with 9 μM eptifibatide to block integrin αIIbβ3. Cross‐linking of IV.3 IgG (10 µg/ml) with 10 µg/ml anti‐Fc (Biolegend) served as a positive control. Platelets were stimulated at 37°C with stirring at 1200 rpm on a PAP‐8E aggregometer. Reactions were terminated by addition of ice‐cold lysis buffer (0.025 M Tris, 0.15 M NaCl, 0.001 M EDTA, 1% NP‐40, 5% glycerol, pH 7.4). Lysates were incubated overnight with mAb IV.3 to form immune complexes. Precipitated proteins were separated by reducing sodium dodecyl sulphate–polyacrylamide gel electrophoresis, electro‐transferred, and Western blotted with the anti‐phosphotyrosine antibody, clone 4G10.
2.8. Ethics
Ethical approval for collecting blood healthy volunteers was granted by Birmingham University Internal Ethical Review (ERN_11‐0175).
2.9. Statistical analysis
All data are presented as mean ± standard deviation (SD). A p‐value <.05 was considered statistically significant. Statistical analysis was performed using Wilcoxon signed‐rank test, Kruskal‐Wallis test with Dunn's multiple comparison test and Mann‐Whitney U test as stated. All statistical analyses were performed using GraphPad Prism 7 (GraphPad Software Inc.).
3. RESULTS
3.1. Lupus anticoagulant activity
The five antiprothrombin mAbs (28F4, 3B1, 6A3, 11H2, and 8H11) were raised against F1+2, the activation peptide of prothrombin. Of these mAbs, only 28F4 and 3B1 caused LAC activity in the dilute Russell Viper Venom Time and activated partial thromboplastin time tests. The other three mAbs (11H2, 6A3, and 8H11) had no effect on the clotting time (Table S1). The ECT reagent converts prothrombin into thrombin, independently of phospholipids. The mAbs 28F4 and 3B1 did not prolong the ECT and did not inhibit thrombin activity when tested with a fluorescent substrate. However, mAb 28F4 did bind to prothrombin fragment 1+2, purified prothrombin, and native prothrombin in plasma in an ELISA assay (Figure S1).
3.2. Platelet aggregation
Adding mAbs 28F4 and 3B1, but not 11H2, 6A3, and 8H11, resulted in aggregation of washed platelets in the presence of prothrombin and 10 mM Ca2+ (Figure 1A), indicating a correlation between LAC activity and platelet aggregation. mAb 28F4 induced aggregation in washed platelets in the presence but not absence of Ca2+ (data not shown) or prothrombin (Figure S2). The 28F4 also induced platelet aggregation in the presence lower Ca2+ concentrations (1 mM Ca2+ and 3 mM Ca2+; Figure S3). 28F4 was the most potent at inducing platelet aggregation and LAC activity. Preincubation of washed platelets with 1 µg/ml 28F4 resulted in partial aggregation. Full platelet aggregation was achieved with 10 µg/ml 28F4. Addition of 200–1000 nM rivaroxaban to washed platelets did not influence the aggregation profile.
FIGURE 1.

The mAbs 28F4 and 3B1 induce platelet aggregation in the presence of Ca2+ and prothrombin. (A) Representative traces of light transmission aggregometry using washed platelets at a concentration of 2 × 108/ml. Aliquots of platelet suspension were stimulated with 50 µg/ml of mAbs 28F4, 3B1, 6A3, 11H2, or 8H11 in the presence of 10 mM Ca2+ and 1 µM prothrombin (n = 3). (B) Quantification of platelet aggregation induced by increasing doses of mAb 28F4. Platelet aggregation was measured as increase of light transmission. Results are shown as mean ± SD (n = 3). Statistical significance was analyzed using the Kruskal‐Wallis test. *p < .05. mAbs, monoclonal antibodies; NS, not significant
3.3. Platelet signalling
Inhibitors were used to investigate the mechanism involved in antiprothrombin‐induced platelet aggregation to low (10 µg/ml) and high (50 µg/ml) concentrations of mAb 28F4. Pretreatment of platelets with the αIIbβ3 antagonist eptifibatide inhibited platelet aggregation to both concentrations of mAb 28F4 (Table 1). Indomethacin and ticagrelor, which block cyclooxygenase and the P2Y12 receptor, respectively, attenuated but did not block platelet aggregation. Platelet aggregation induced by mAb 28F4 was not altered in the combined presence of the PAR‐1 and PAR‐4 antagonists, vorapaxar, and BMS‐986120, respectively. The same concentrations of the two antagonists blocked thrombin mediated platelet aggregation (data not shown). The Syk and Src inhibitors, PRT‐060318 and dasatinib, respectively, blocked aggregation to both concentrations of mAb 28F4, which indicates involvement of signalling via receptors containing an immunoreceptor tyrosine‐based activation motif (ITAM). Three ITAM‐containing receptors of human platelets are known including: C‐type lectin‐like receptor 2, GPVI, and FcγRIIA (Fc receptor for IgG).
TABLE 1.
ITAM signaling is involved in antiprothrombin induced platelet aggregation
| 28F4 (10 µg/ml) | 28F4 (50 µg/ml) | |
|---|---|---|
| % Aggregation ± SD | % Aggregation ± SD | |
| Vehicle | 65.4 ± 10.2 | 70.0 ± 8.1 |
| Eptifibatide (9 µM) | 4.5 ± 1.4 | 18.0 ± 9.2 |
| Indomethacin (10 µM) | 22.3 ± 11.2 | 38.9 ± 18.2 |
| PRT‐06318 (5 µM) | 2.5 ± 2.8 | 3.9 ± 1.8 |
| Ticagrelor (10 µM) | 11.0 ± 6.0 | 19.6 ± 15.7 |
| Dasatinib (10 µM) | 2.7 ± 1.7 | 4.2 ± 2.6 |
| Vorapaxar (1 µM) + BMS‐986120 (1 µM) | 65.8 ± 2.3 | 74.6 ± 4.1 |
Low dose of mAb 28F4 (10 µg/ml) and high dose of mAb 28F4 (50 µg/ml) was used to assess the effect of platelet antagonists. Platelet aggregation was measured as increase of light transmission. Results are shown as mean ± standard deviation (SD) (n = 3).
3.4. Involvement of FcγRIIA
Of the three platelet ITAM‐containing receptors, FcγRIIA receptor was felt to be the most likely to be involved in the response to the antiprothrombin antibodies because it is a low affinity receptor for the Fc domain. Tyrosine phosphorylation of FcγRIIA was induced by mAb 28F4 as measured by immunoprecipitation and western blotting (Figure 2). Crosslinked IV.3 (α‐FcγRIIA antibody) was used as a positive control. As expected, rhodocytin and CRP, known stimulators of C‐type lectin‐like receptor 2 and GPVI, respectively, did not result in tyrosine phosphorylation of FcγRIIA. Non‐crosslinked mAb IV.3 showed a trend toward inhibiting 28F4‐ and 3B1‐induced platelet aggregation (Figure 3 and not shown). Fab and F(ab′)2 antibody fragments of mAb 28F4 were unable to induce platelet aggregation (not shown) confirming the critical role of the Fc domain.
FIGURE 2.
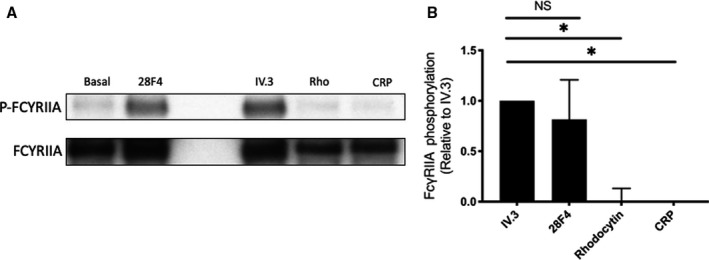
FcγRIIA is tyrosine‐phosphorylated in the presence of mAb 28F4. (A) Representative blot of five experiments. (B) Mean data of signal for p‐FcγRIIA. The signal obtained from crosslinked mAb IV.3 was normalized against total FcγRIIA and set as a reference (1 AU). Differences were analyzed with the Kruskal‐Wallis test. Results are shown as mean ± SD with n = 5. *p < .05. mAb, monoclonal antibody; NS, not significant
FIGURE 3.

Antiprothrombin‐induced platelet aggregation can be blocked by mAb IV.3 (anti‐FcγRIIA) antibody. Aliquots of platelet suspension were preincubated with mAb IV.3 (10 µg/ml) followed by stimulation using mAbs 28F4 (50 µg/ml) or 3B1 (50 µg/ml). Platelet aggregation was measured as increase of light transmission. Results are shown as mean ± SD (n = 5). Statistical significance was analyzed using the Mann‐Whitney U test (*p < .05). mAb, monoclonal antibody; SD, standard deviation
3.5. The link between prothrombin levels and platelet aggregation
We investigated whether prothrombin levels affected mAb 28F4‐induced platelet aggregation as a possible explanation for anti‐FXa therapy failure in patients with APS. Platelet aggregation was assessed in washed platelets in the presence of 50 µg/ml 28F4 with two concentrations of prothrombin. Aggregation was found to be attenuated by 59% when prothrombin levels were reduced from 1 µM to 0.2 µM, suggesting a direct link between prothrombin levels and platelet aggregation (Figure 4).
FIGURE 4.

Decreased prothrombin levels results in attenuated platelet aggregation. (A) Representative traces of light transmission aggregometry using washed platelets at a concentration of 2 x 108/ml. Aliquots of platelet suspension were preincubated with 0.2 μM prothrombin or 1 μM prothrombin, followed by stimulation using 50 μg/ml mAb 28F4. (B) Quantification of platelet aggregation in the presence of two concentrations of prothrombin. Platelet aggregation was measured as increase of light transmission. Results are shown as mean ± SD (n = 5). Statistical significance was analyzed using the Mann‐Whitney U test (*p < 0.05). SD, standard deviation
4. DISCUSSION
In this study, we show that (1) antiprothrombin antibodies with LAC activity, but not those without LAC activity, are able to activate platelets via the FcγRIIA receptor; and (2) decreased prothrombin levels result in less antiprothrombin antibody‐mediated platelet aggregation. These results suggest an important role of antiprothrombin antibodies with LAC activity in platelet activation. We speculate that the LAC activity is required for binding of the prothrombin/antiprothrombin complex to the platelet surface leading to binding of the Fc portion of the antiprothrombin antibodies to FcγRIIA and platelet activation.
Antiprothrombin antibodies with LAC activity are associated with APC resistance.19 We were able to demonstrate platelet aggregation caused by antiprothrombin antibodies with LAC activity, indicating that antiprothrombin antibodies with LAC activity are potentially pathogenic. Antiprothrombin antibodies can be detected by coating prothrombin on irradiated ELISA plates or using phosphatidylserine–prothrombin complex as antigen.10 The presence of aPS/PT antibodies is considered to be a stronger predictor of thrombosis than antiprothrombin antibodies.10 However, we demonstrated binding of mAb 28F4 to prothrombin in the absence of phosphatidylserine, by ELISA. A recent study showed that at least two subpopulations of aPS/PT antibodies exist; aPS/PT antibodies that bind to the “open” conformation of prothrombin and aPS/PT antibodies able to react with “closed” prothrombin.20 There were, however, no different pathological effects found between the two subpopulations of aPS/PT antibodies. In contrast, we were able to distinguish between antiprothrombin antibodies that were able to induce platelet aggregation and those that did not by measuring LAC activity. From the five antiprothrombin antibodies investigated, only mAbs 28F4 and 3B1 were able to induce platelet aggregation and LAC activity. Although mAb 28F4 is able to bind closed prothrombin, thrombin activity was not inhibited, which could be explained by the affinity of the investigated mAbs toward the activation peptide of prothrombin, fragment 1+2, and not thrombin. These data indicate that LAC activity determines the ability of antiprothrombin antibodies to induce platelet aggregation.
Data on the role of antiprothrombin antibodies in platelet activation are limited, probably because platelet aggregation studies are performed in citrated plasma and prothrombin needs Ca2+ to bind to platelets.21 Although platelet function assays were not performed, one study showed the potency of mAb 28F4 to induce platelet activation by measuring thromboxane B2 levels, phospholipid‐related platelet procoagulant activity, and thrombin generation.19 The platelet‐dependent effect of mAb 28F4 was found to be reduced in the presence of an integrin αIIbβ3 inhibitor, but not with IV.3.19 The inability of showing involvement of the FcγRIIA receptor might be due to the low concentration of mAb IV.3 used in their study.19 We have showed involvement of the FcγRIIA receptor in mAb 28F4 induced platelet activation in three ways: (1) inhibited platelet aggregation by IV.3; (2) tyrosine phosphorylation of the FcγRIIA receptor; and (3) the inability of Fab and F(ab′)2 fragments of mAb 28F4 to aggregate platelets. In this respect, the mechanism of platelet activation by aPT antibodies mimics another autoimmune disease; heparin‐induced thrombocytopenia and thrombosis.22 The presence of Ca2+ proved to be essential for platelet aggregation induced by antiprothrombin antibodies, suggesting Ca2+‐dependent binding of antiprothrombin‐prothrombin immune complexes to negatively charged phospholipids expressed by platelets. Here, we show a major role for FcγRIIA in antiprothrombin‐induced platelet aggregation.
The trial of rivaroxaban in antiphospholipid syndrome trial included APS patients with exclusively triple positive antiphospholipid (LAC, aCL and aβ2GPI antibodies) in which rivaroxaban was compared with warfarin therapy.3 Although aPS/PT antibodies were not measured in the trial, aPS/PT antibodies have shown to correlate well with triple positivity in other studies.20, 23 We found that platelet aggregation was attenuated when prothrombin levels were reduced. The link between prothrombin levels and platelet aggregation may explain the failure of anti‐FXa therapy in patients with APS as rivaroxaban does not affect prothrombin levels in plasma, whereas vitamin K antagonists (e.g., warfarin) reduce active prothrombin (antigen) levels.
There are several limitations in our study. We used mAbs that recognize the prothrombin F1+2 fragment, but we did not further specify the site of recognition. In addition, we did not confirm our findings with patient antibodies and did not evaluate antiprothrombin‐induced platelet activation in patients receiving vitamin K antagonists or rivaroxaban therapy. Additional data are needed on the specification of antiprothrombin antibodies and their effect on platelets.
Here, we have shown that only antiprothrombin antibodies with LAC activity are able to activate platelets via the FcγRIIA receptor. Decreased prothrombin levels resulted in attenuated platelet aggregation, which might be an explanation for the low prevalence of arterial thrombosis in APS patients treated with warfarin compared with rivaroxaban therapy.
CONFLICT OF INTEREST
Drs. Chayoua and de Laat are employees of Synapse Research Institute, part of Diagnostica Stago SAS. Dr. de Groot is an advisor of Synapse Research Institute. Dr. Watson holds a BHF Chair (03/003). The other authors state that they have no relevant conflict of interest.
AUTHOR CONTRIBUTIONS
Walid Chayoua, Philip G. de Groot, Phillip L. R. Nicolson, and Stephen P. Watson designed the study. Walid Chayoua, Phillip L. R. Nicolson, Caroline Kardeby, and Lourdes Garcia‐Quintanilla performed the measurements. LAC was measured under supervision of Joost C. M. Meijers and Katrien M. J. Devreese. Walid Chayoua, Phillip L. R. Nicolson, Stephen P. Watson, Bas de Laat, and Philip G. de Groot interpreted data, performed statistical analyses, and wrote the manuscript. Joost C. M. Meijers, Caroline Kardeby, Lourdes Garcia‐Quintanilla, and Katrien M. J. Devreese critically reviewed the manuscript.
Supporting information
Figure S1
Figure S2
{kind=link}
Figure S3
{kind=link}
Table S1
ACKNOWLEDGMENTS
The authors thank Ying Di for her technical assistance. W.C. was supported by a grant (HS‐BAFTA, Harry Struijker‐Boudier Award For Talented Academics) issued by the Cardiovascular Research Institute Maastricht (CARIM).
Manuscript handled by: Wolfgang Bergmeier
Final decision: Wolfgang Bergmeier, 15 March 2021
REFERENCES
- 1.Miyakis S, Lockshin MD, Atsumi T, et al. International consensus statement on an update of the classification criteria for definite antiphospholipid syndrome (APS). J Thromb Haemost. 2006;4:295‐306. [DOI] [PubMed] [Google Scholar]
- 2.Tektonidou MG, Andreoli L, Limper M, et al. EULAR recommendations for the management of antiphospholipid syndrome in adults. Ann Rheum Dis. 2019;78:1296‐1304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pengo V, Denas G, Zoppellaro G, et al. Rivaroxaban vs warfarin in high‐risk patients with antiphospholipid syndrome. Blood. 2018;132:1365‐1371. [DOI] [PubMed] [Google Scholar]
- 4.Ordi‐Ros J, Sáez‐Comet L, Pérez‐Conesa M, et al. Rivaroxaban versus vitamin K antagonist in antiphospholipid syndrome: a randomized noninferiority trial. Ann Intern Med. 2019;10:685‐694. [DOI] [PubMed] [Google Scholar]
- 5.Zuily S, Cohen H, Isenberg D, et al. Use of direct oral anticoagulants in patients with thrombotic antiphospholipid syndrome: Guidance from the Scientific and Standardization Committee of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2020;18:2126‐2137. [DOI] [PubMed] [Google Scholar]
- 6.Koupenova M, Kehrel BE, Corkrey HA, Freedman JE. Thrombosis and platelets: an update. Eur Heart J. 2017;11:785‐791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Urbanus RT, Pennings MTT, Derksen RHWM, Groot PGD. Platelet activation by dimeric β2‐glycoprotein I requires signaling via both glycoprotein Ibα and apolipoprotein E receptor 2′. J Thromb Haemost. 2008;6:1405‐1412. [DOI] [PubMed] [Google Scholar]
- 8.Shi T, Giannakopoulos B, Yan X, et al. Anti–β2‐glycoprotein I antibodies in complex with β2‐glycoprotein I can activate platelets in a dysregulated manner via glycoprotein Ib‐IX‐V. Arthritis Rheum. 2006;54:2558‐2567. [DOI] [PubMed] [Google Scholar]
- 9.Litvinova E, Darnige L, Kirilovsky A, Burnel Y, de Luna G, Dragon‐Durey M‐A. Prevalence and significance of non‐conventional antiphospholipid antibodies in patients with clinical APS criteria. Front Immunol. 2018;9:2971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sciascia S, Sanna G, Murru V, Roccatello D, Khamashta MA, Bertolaccini ML. Antiprothrombin (aPT) and anti‐phosphatidylserine/prothrombin (aPS/PT) antibodies and the risk of thrombosis in the antiphospholipid syndrome: a systematic review. Thromb Haemost. 2014;111:354‐364. [DOI] [PubMed] [Google Scholar]
- 11.Horton JD, Bushwick BM. Warfarin therapy: evolving strategies in anticoagulation. Am Fam Physician. 1999;59:635‐646. [PubMed] [Google Scholar]
- 12.Arman M, Krauel K, Tilley DO, et al. Amplification of bacteria‐induced platelet activation is triggered by FcγRIIA, integrin αIIbβ3, and platelet factor 4. Blood. 2014;123:3166‐3174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Arnout J, Vanrusselt M, Nevens C, Smans K, Wittevrongel C, Vermylen J. Some murine monoclonal antibodies against human prothrombin have lupus anticoagulant activity. Thromb Haemost 1999; Suppl August: 65.10456456 [Google Scholar]
- 14.Querrec AL, Arnout J, Arnoux D, et al. Quantification of lupus anticoagulants in clinical samples using anti‐ β2GP1 and antiprothrombin monoclonal antibodies. Thromb Haemost. 2001;86:584‐589. [PubMed] [Google Scholar]
- 15.Köhler G, Milstein C. Continuous cultures of fused cells secreting antibody of predefined specificity. Nature. 1975;256:495‐497. [DOI] [PubMed] [Google Scholar]
- 16.Nicolson PLR, Hughes CE, Watson S, et al. Inhibition of Btk by Btk‐specific concentrations of ibrutinib and acalabrutinib delays but does not block platelet aggregation mediated by glycoprotein VI. Haematologica. 2018;103:2097‐2108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Devreese KMJ, de Groot PG, De Laat B, et al. Guidance from the scientific and standardization committee for lupus anticoagulant/antiphospholipid antibodies of the International Society on Thrombosis and Haemostasis. J Thromb Haemost. 2020;18:2828‐2839. [DOI] [PubMed] [Google Scholar]
- 18.Moore GW, Culhane AP, Maloney JC, Archer RA, Breen KA, Hunt BJ. Taipan snake venom time coupled with ecarin time enhances lupus anticoagulant detection in nonanticoagulated patients. Blood Coagul Fibrinolysis. 2016;27:477‐480. [DOI] [PubMed] [Google Scholar]
- 19.Membre A, Wahl D, Latger‐Cannard V, et al. The effect of platelet activation on the hypercoagulability induced by murine monoclonal antiphospholipid antibodies. Haematologica. 2008;93:566‐573. [DOI] [PubMed] [Google Scholar]
- 20.Chinnaraj M, Planer W, Pengo V, Pozzi N. Discovery and characterization of 2 novel subpopulations of aPS/PT antibodies in patients at high risk of thrombosis. Blood Adv. 2019;3:1738‐1749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Palatinus AA, Ahuja KDK, Adams MJ. Effects of antiphospholipid antibodies on in vitro platelet aggregation. Clin Appl Thromb Hemost. 2012;18:59‐65. [DOI] [PubMed] [Google Scholar]
- 22.Warkentin TE, Greinacher A. Management of heparin‐induced thrombocytopenia. Curr Opin Hematol. 2016;23:462‐470. [DOI] [PubMed] [Google Scholar]
- 23.Pengo V, Banzato A, Bison E, et al. Laboratory testing for antiphospholipid syndrome. Int J Lab Hematol. 2016;38(Suppl 1):27‐31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1
Figure S2
Figure S3
Table S1