

Abstract
The redox chemistry of the heterobimetallic triple‐decker complexes [(Cp*Fe)(Cp′′′Co)(μ,η5:η4‐E5)] (E=P (1), As (2), Cp*=1,2,3,4,5‐pentamethyl‐cyclopentadienyl, Cp′′′=1,2,4‐tri‐tertbutyl‐cyclopentadienyl) and [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)] (E=P (10), As (11)) was investigated. Compound 1 and 2 could be oxidized to the monocations 3 and 4 and further to the dications 5 and 6, while the initially folded cyclo‐E5 ligand planarizes upon oxidation. The reduction leads to an opposite change in the geometry of the middle deck, which is now folded stronger into the direction of the other metal fragment (formation of monoanions 7 and 8). For the arsenic compound 8, a different behavior is found since a fragmentation into an As6 (9) and As3 ligand complex occurs. The Co and Ni triple‐decker complexes 10 and 11 can be oxidized initially to the heterometallic monocations 12 and 13, which are not stable in solution and convert selectively into the homometallic nickel complexes 14 and 15 and the cobalt complexes 16 and 17. This behavior was further proven by the oxidation of [(Cp′′′Co)(Cp′′Ni)(μ,η3:η2‐P3)] (19, Cp′′=1,3‐di‐tertbutyl‐cyclopentadienyl) comprising two different Cp ligands. The transfer of {CpRM} fragments can be suppressed when a {W(CO)5} unit is coordinated to the P3 ligand (20) prior to the oxidation and the mixed cobalt and nickel cation 21 can be isolated. The reduction of 10 and 11 yields the heterometallic monoanions 22 and 23, where no transfer of the {CpRM} fragments is observed.
Keywords: arsenic, heterometallic complexes• phosphorus, redox chemistry, transfer reactions
The redox chemistry of heterobimetallic triple‐decker complexes (Fe/Co and Co/Ni) with polypnictogen ligands (E=P, As) was studied first by cyclic voltammetry and then experimentally. The complexes respond to the addition and withdrawal of electrons either by the rearrangement of the triple‐decker geometry, by the fragmentation of the En ligand and/or by the exchange of {Cp′′′M} fragments.
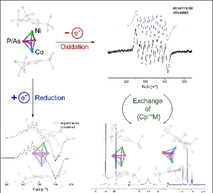
Introduction
Elemental modifications of sulfur, phosphorus or arsenic can be reduced leading to a degradation under bond cleavage to yield S6 2−, P3− or As3−, respectively.[1] On the other hand, oxidation reactions can cause the formation of new element‐element bonds. While for the oxidation of S8 to S8 2+ a novel S−S bond is formed, the oxidation of white phosphorus (P4) is accompanied by an aggregation to form P9 +.[2] A similar reactivity can be observed for cyclic phosphines or arsines such as tBu4E4 (E=P, As). Both can be reduced by degradation to tBu2E2 2− using for example elemental potassium.[3] On the other hand, organometallic complexes such as ferrocene reveal a versatile redox chemistry. The parent ferrocene has played an important role in recent research,[4] not only as the redox couple [Cp2Fe]/[Cp2Fe]+, which is widely used as a reference system for electrochemical investigation methods such as cyclic voltammetry.[5] Moreover, ferrocene‐containing fragments are commonly used as molecular switches in redox switchable catalysis (RSC).[6] For some systems, more redox states are needed and therefore more than one ferrocene unit was incorporated e. g. in macrocycles.[7] According to the isolobal relationship between CH and P or As, the CH units in C5H5 ligand can partially be replaced by pnictogen atoms revealing ligands as for instance phospholes (C4H4P, C2H2P3)[8, 9, 10] or arsoles (C4H4As, C2H2As3),[8, 10, 11] or cyclo‐E5 (polypnictogen) ligands as for instance in the pentaphospha‐ and pentaarsaferrocene [Cp*Fe(η5‐E5)] (E=P, As).[12, 13] The latter reveal a distinct redox chemistry,[14] however, most of these processes are irreversible and proceed via dimerization and fragmentation processes. A similar behavior was observed for the sandwich complexes [Cp′′′Co(η4‐P4)][15] and [(Cp′′′Ni(η3‐P3)][16] and the tetrahedrane compound [{CpMo(CO)2}2(μ,η2:η2‐E2)][17] (E=P, As, Sb, Bi). These complexes respond to redox reactions with either dimerization (e. g. to [(Cp′′′Co)2(μ,η3:η3‐P8)]2−(A), Scheme 1) or a fragmentation and rearrangement takes places (e. g. to [(Cp′′′Ni)2(μ,η2:η2‐P8)]2− (B), Scheme 1).
Scheme 1.
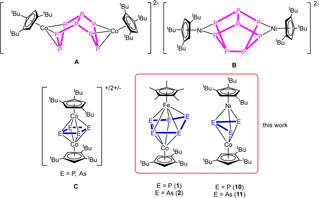
Selected examples of oxidized and reduced polypnictogen ligand complexes heterobimetallic triple‐decker complexes [(Cp*Fe)(Cp′′′Co)(μ,η5:η4‐E5)] (E=P (1), As (2)) and [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)] (E=P (10), As (11)).
On the other hand, going from sandwich complexes to homometallic polypnictogen triple‐decker complexes, a fully reversible redox chemistry over two oxidation states as e. g. in [(CpRMo)2(μ,η6:η6‐E6)] (E=P, As)[18] or even over four and five redox stages takes place, which was found for the cobalt complex [(Cp′′′Co)2(μ,η2:η2‐E2)] (E=P, As).[19] Oxidation and reduction of the latter two induce a cyclization of the initially separated E2 units to form a cyclo‐E4 ligand in [(Cp′′′Co)2(μ,η4:η4‐E4)]+/2+/− (C, Scheme 1).
Intrigued by the diversity of the structural changes and/or rearrangement processes upon oxidation and/or reduction of the homometallic polypnictogen ligand complexes, we were interested in investigating how the heterobimetallic polypnictogen triple‐decker complexes would respond to the withdrawal or addition of electrons. Would a simple rearrangement of the En ligand, a release of a {CpRM} fragment and/or an aggregation to larger polypnictogen ligands take place to form multimetallic cluster complexes? Would the redox processes be reversible and would there be a difference between phosphorus‐ and arsenic‐containing derivatives, considering the in general lower E−E bond energy for arsenic? Therefore, we were intrigued to investigate the redox chemistry of the mixed iron/cobalt triple‐decker complexes [(Cp*Fe)(Cp′′′Co)(μ,η5:η4‐E5)] (E=P (1), As (2)) and the mixed cobalt/nickel triple‐decker complexes [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)] (E=P (10), As (11) to explore the response of these complex systems within the redox behavior.
Results and Discussion
The heterobimetallic triple‐decker complexes [(Cp*Fe)(Cp′′′Co)(μ,η5:η4‐E5)] (E=P (1), As (2)) can be easily synthesized by the reaction of the cobalt toluene complex [(Cp′′′Co)2(μ,η4:η4‐C7H8)] with [Cp*Fe(η5‐E5)] (E=P, As)] in gram scale.[20, 21] To obtain a first overview if redox processes are accessible, cyclic voltammetry measurements were performed revealing several redox processes (cf. Supporting Information). Both compounds can be oxidized using one equivalent of the Ag(I) salt containing the weakly coordinating anion [FAl]− ([FAl{OC6F10(C6F5)}3]−) leading to the isostructural compounds [(Cp*Fe)(Cp′′′Co)(μ,η5:η5‐E5)][FAl] (E=P (3), As (4)) which can be isolated in crystalline yields of 55 and 63 %, respectively (Scheme 2). Compound 3 can be obtained as green, 4 as brown air‐ and moisture‐sensitive solids. Crystals suitable for single crystal X‐ray structure analysis were obtained from concentrated solutions in CH2Cl2 layered with hexane at ‐30 °C. The structures in the solid state (Figure 1) reveal dinuclear triple‐decker complexes with a slightly folded cyclo‐E5 ligand as middle deck while the ligand planarized compared to the starting materials. The E5 ligand coordinates to both iron and cobalt fragments in an η5 fashion, while E1 is bent out of the plane (fold angle E2‐E4‐E5‐E1) by approx. 9.4° (3) and 13.6° (4), respectively (35.0° in 1, 37.9° in 2). All E−E bonds are shortened compared to 1 and 2. For 3, very similar P‐P distances between 2.1214(9) and 2.1601(9) Å are present, while 1 shows three shorter distances (P1‐P2 2.1459(10) Å, P3‐P4 2.1283(10) Å, P1‐P5 2.1498(10) Å) and two longer ones (P2‐P3 2.2244(11) Å, P4‐P5 2.2407(10) Å). For the analog arsenic compound 4, the situation is similar. All As−As distances are in the range between 2.3224(8) and 2.3619(8) Å and shortened compared to 2, while in 2, three shorter distances (As1‐As2 2.3682(5) Å, As3‐As4 2.3401(5) Å, As1‐As5 2.3716(6) Å) and two longer ones (As2‐As3 2.4627(5) Å, As4‐As5 2.4383(6) Å) are present. Compounds 3 and 4 are paramagnetic. The 1H NMR spectrum of 3 in CD2Cl2 reveals four strongly broadened and shifted signals centered at δ=5.66, 3.76, 3.29 and −65.22 ppm with an integral ratio of 15 : 9 : 18 : 2, while the signal at 5.66 ppm can be assigned to the Cp* ligand, and the other three singlets correspond to the Cp′′′ ligand. The 31P{1H} NMR spectrum shows one broad singlet at δ=275.1 ppm. The 1H NMR spectrum of 4 in CD2Cl2 reveals three strongly broadened and shifted signals centered at δ=3.84, 2.74 and −1.09 ppm with an integral ratio of 9 : 18 : 15, while the signals at 3.84 and 2.74 ppm can be assigned to the tBu groups of the Cp′′′ ligands and the singlet at −1.09 ppm of the Cp* ligand. No signal for the C−H groups of the Cp′′′ ligand can be detected between +125 and −125 ppm. The Evans NMR spectrum shows an effective magnetic moment of 2.04 μB for 3 and 1.90 μB for 4 corresponding to about one unpaired electron each. The EPR spectrum of 3 at 77 K (frozen o‐C6H4F2 solution) exhibits a strong rhombic resonance with hyperfine coupling to Co (g1 =2.155, g2 =2.035, g3 =1.948, A1 Co=290, A2 Co=55, A3 Co=120 MHz, Figure 2). DFT calculations reveal that the spin density is mainly located on the Co1 atom with some minor participation of P1 and Fe1 (cf. Figure 2) For 4, the EPR spectra reveal rhombic resonances similar to those of 3. The calculations show that the spin density is more equally distributed over Fe, Co and As1 and, compared to 3, the signal is more asymmetric with the g values being more different (77 K, solid: g1 =2.235, g2 =1.952, g3 =1.896, Figure 2). The presence of the hyperfine coupling in the EPR spectrum of 3 and the pronounced rhombic signal in 4 indicates a rather metal centered oxidation.
Scheme 2.
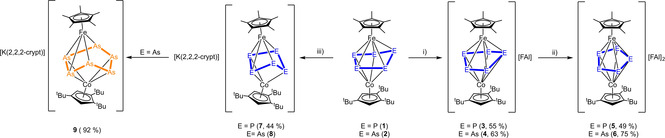
Reaction of [(Cp*Fe)(Cp′′′Co)(μ,η5:η4‐E5)] (E=P (1), As (2)) with: i) Ag[FAl] in CH2Cl2 at room temperature; ii) Ag[FAl] in o‐C6H4F2 at room temperature and iii) KC8 in the presence of 2,2,2‐crpytand in dme.
Figure 1.
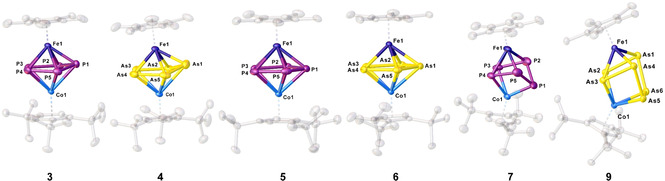
Molecular structures of 3–7 and 9 in the solid state. Thermal ellipsoids are drawn with 50 % probability level. Hydrogen atoms, solvent molecules, anions and cations, respectively, are omitted for clarity.
Figure 2.
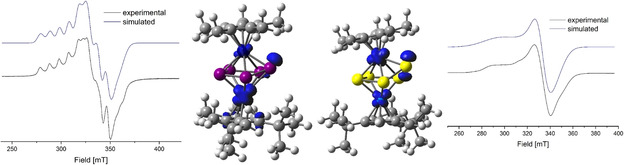
Experimental (black) and simulated (blue) X‐band EPR spectra of 3 (left) and 4 (right) in frozen o‐C6H4F2 solution and the iso‐surfaces of the calculated spin density (BP86/def2‐TZVP level of theory) in 3 (left middle) and 4 (right middle).
Using two equivalents of Ag[FAl] in the oxidation of 1 and 2, respectively, gives the dicationic compounds [(Cp*Fe)(Cp′′′Co)(μ,η5:η5‐E5)][FAl]2 (E=P (5), As (6)) in crystalline yields of 49 and 75 %, respectively (Scheme 2). Both compounds can be obtained as air‐ and moisture‐sensitive brown solids. Crystals suitable for X‐ray single crystal structure analysis were obtained from concentrated solutions in o‐C6H4F2 layered with hexane (5) and toluene (6) and stored at −30 °C, respectively. The structures in the solid state (Figure 1) reveal dinuclear triple‐decker complexes with a planar cyclo‐E5 ligand as middle deck. In the compounds 5 and 6, the cyclo‐E5 ligand coordinates to both the iron and cobalt fragments in an η5 fashion and is planar, while it is slightly folded in the monocations 3 and 4 or strongly folded in the related starting materials 1 and 2. For 5, all P‐P distances are between 2.1425(16) and 2.1521(16) Å and are in the range between P‐P single and double bonds.[22, 23] They are only slightly longer than in [Cp*Fe(η5‐P5)] showing distances in the range of 2.1158(15) and 2.1268(16) Å.[24] For 6, the As−As distances are in the range of 2.3390(6) and 2.3515(8) Å and also between single and double bonds.[22, 23] Compared to [Cp*Fe(η5‐As5)] the As−As bonds are only slightly elongated (2.317(1)–2.330(1) Å).[25] Interestingly, the diamagnetic dicationic compounds 5 and 6 are isostructural and isoelectronic to the monocationic homo‐ and heterobimetallic complexes [(CpRM)(CpR′M′)(μ,η5:η5‐E5)]+ (M=Fe, Ru, M′=Fe, Ru, E=P, As).[12, 26] The 1H NMR spectrum of 5 in o‐C6H4F2 with C6D6 capillary shows four singlets centered at δ=3.69, 1.49, 1.39 and 1.21 ppm with an integral ratio of 2 : 18 : 9 : 15. In the 31P{1H} spectrum of 5, a sharp singlet at δ=52.7 ppm is monitored. In the 1H NMR spectrum of 6, four singlets centered at δ=4.20, 1.25, 1.18 and 0.86 ppm with an integral ratio of 2 : 18 : 9 : 15 are detected.
Therefore, the oxidation of 1 and 2 leads to a successive planarization of the folded cyclo‐E5 ligand resulting in a completely planar ligand in 5 and 6, which is accompanied by an overall significant shortening of all E‐E distances. All oxidation reactions are fully reversible upon addition of KC8.
The question arises how 1 and 2 will respond to a chemical reduction. Compound 1 can be reduced using potassium graphite in the presence of 2,2,2‐cryptand to give [K(2,2,2‐crypt][(Cp*Fe)(Cp′′′Co)(μ,η4:η3‐P5)] (7) in crystalline yields of 44 % (Scheme 2). Compound 7 can be isolated as an extremely air‐ and moisture‐sensitive dark green solid. Crystals of 7 suitable for single crystal X‐ray structure analysis were obtained from a concentrated solution in thf layered with hexane at −30 °C. The structure of 7 in the solid state (Figure 1) reveals a dinuclear triple‐decker complex with a strongly folded cyclo‐P5 middle deck. The P5 ligand coordinates to the iron fragment in an η4 and to the cobalt fragment in an η3 fashion. One P atom (P1) is bent out of the plane by approx. 44.5°. All P‐P distances are in the range of single bonds. Compound 7 is paramagnetic. The 1H NMR spectrum in thf‐d8 shows three broad and strongly shifted signals for the tBu and Me groups of the Cp ligands, while no signal for the H atoms directly attached to the Cp′′′ ligand can be detected. The effective magnetic moment was determined to be 2.01 μB by the Evans method corresponding to about one unpaired electron. The EPR spectrum shows an isotropic resonance with g values near to 2.0 indicating one unpaired electron (293 K, solid, giso =2.060, cf. Supporting Information), while the spectra of 3 and 4 show rhombic resonances.
The folding of the cyclo‐P5 ligand in 1 is clearly dependent on the oxidation state. The oxidation to the mono cation 3 and the dication 5 leads to a successive planarization of the P5 ligand, while the reduction inverts the folding and the P1 atom turns from the coordination to Fe1 to Co1 accompanied by an increase of the folding angle. For the reduction of the arsenic complex 2, potassium graphite in the presence of 2,2,2‐cryptand was used (Scheme 2). Regardless of numerous attempts, it was not possible to obtain crystals of the analog arsenic complex [K(2,2,2‐crypt)][(Cp*Fe)(Cp′′′Co)(μ,η4:η3‐As5)] (8) yielding always only brown oily residues. But the presence of 8 was proved by paramagnetic signals in the 1H NMR spectrum of the reaction mixture (mixture of 8, 9 and unknown compound, cf. Supporting Information) and a strong isotropic resonance in the EPR spectra of the precipitated solid of the reaction mixture with g values near to 2 (77 K, solid: g iso=2.123, r.t, solid: g iso=2.109). Leaving the Schlenk flasks with the oily residues of the crystallization at room temperature for a few weeks, a large amount of crystalline [K(2,2,2‐crypt)][(Cp*Fe)(Cp′′′Co)(μ,η4:η4‐As6)] (9) could be obtained in isolated yields of 92 % (Scheme 2). The formation of 9 is reproducible. The 1H NMR spectrum of the mother liquor reveals [(Cp*Fe)(Cp′′′Co)(μ,η3:η3‐As3)] as a side product.[21] The solid‐state structure of 9 is shown in Figure 1. The molecular structure reveals a dinuclear triple‐decker complex with a newly formed bent cyclo‐As6 ligand as middle deck. All As−As bonds except As2‐As3 are in the range of single bonds (2.3441(5)–2.4420(5) Å). The As2‐As3 distance of 2.6907(5) Å displays a strongly elongated As−As single bond indicated by a WBI of 0.52.[23] The structural motif of 9 is reminiscent of the isoelectronic cobalt complex [(Cp+Co)2(μ,η4:η4‐As6)],[27] however, in 9 as heterobinuclear complex. The 1H NMR spectrum of 9 in thf‐d8 shows four singlets centered at δ=4.14, 1.61, 1.53 and 1.35 ppm with an integral ratio of 2 : 15 : 18 : 9.
While the reduction of 1 to 7 is fully reversible upon addition of AgBF4, this is not the case for the arsenic derivate 2 due to the fragmentation of the As5 ligand to yield 9.
The second scope of this work was the investigation of the redox behavior of the mixed cobalt and nickel triple decker complexes [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)] (E=P (10), As (11). Both compounds can be easily prepared from the reaction of [Cp′′′Ni(η3‐E3)] (E=P, As) with the cobalt toluene complex [(Cp′′′Co)2(μ,η4:η4‐C7H8)] in gram scale. The synthesis of 10 was already reported[20] and the arsenic derivative 11 was prepared analogously (cf. experimental section). To obtain a first insight into possible redox processes of 10 and 11, cyclic voltammetric measurements were conducted, which reveal several possible redox processes (cf. Supporting Information). The oxidation of 10 and 11 with one equivalent Ag[FAl] leads to a color change of dark brown to red‐brown within one hour (10) and to an immediate color change from dark green to brown (11), respectively. The 1H NMR spectra in CD2Cl2 of both reaction mixtures after one hour reveal the formation of paramagnetic species, which is in agreement with the abstraction of one electron by the oxidation. Stirring the mixtures overnight and recording another 1H NMR spectrum after one day shows the formation of two new diamagnetic species each while the initial paramagnetic compounds have disappeared completely. In the 31P{1H} NMR spectrum of the reaction mixture of 10 with Ag[FAl] in CD2Cl2, no signals could be detected after one hour, but, after one day, a broad singlet centered at δ=140.2 ppm and a set of a doublet (δ=789.0 ppm) and a triplet (δ=−218.0 ppm) with a 1 JPP coupling constant of 361 Hz and an integral ratio of 2 : 1 could be observed. The first species could be identified as [(Cp′′′Ni)2(μ,η3:η3‐P3)][FAl] (14) (cf. Scheme 3), which had already been reported, however with [GaCl4]− as a counterion.[28] The mixtures from the reaction of 10 and 11 with Ag[FAl], respectively, were layered with hexane and left at −30 °C for crystallization. After a few days, red blocks of [(Cp′′′Co)2(μ,η3:η3‐P3)][FAl] (16) and brown blocks of [(Cp′′′Co)2(μ,η3:η3‐As3)][FAl] (17) could be isolated in 65 % and 17 % yields, respectively (Scheme 3). The 1H and 31P{1H} NMR spectra of the mother liquors show each an enrichment of the nickel complexes [(Cp′′′Ni)2(μ,η3:η3‐P3)][FAl] (14) and [(Cp′′′Ni)2(μ,η3:η3‐As3)][FAl] (15). The identity of 14 was proven by comparison with the NMR spectra of [(Cp′′′Ni)2(μ,η3:η3‐P3)][GaCl4],[28] and analytically pure [(Cp′′′Ni)2(μ,η3:η3‐As3)][FAl] (15) could be obtained by the oxidation of [Cp′′′Ni(η3‐As3)] with [FcDiac][FAl] more selectively (cf. experimental section). Obviously, a strong tendency of a rearrangement to homometallic triple‐decker complexes is present. Before these rearrangement processes take place, the oxidation of 10 and 11 to the heterometallic monocations [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)][FAl] (E=P (12), As (13)) displays the first step. Compounds 12 and 13 could not be crystallized due to their further transformation to 14/16 and 15/17 in solution, respectively (Scheme 3). But the reaction can be stopped by precipitation with hexane after a few minutes. The obtained solids contain 12 and 13 as major compounds (determined by NMR) and were further investigated by EPR spectroscopy. The X‐band EPR spectrum of a freshly prepared CH2Cl2 solution (see Figure 3) of 12 at room temperature shows a rhombic resonance (g1 =2.023, g2 =2.031, g3 =2.014) with a well resolved hyperfine coupling to cobalt (A1 Co=60, A2 Co=64, A3 Co=63 MHz), indicating that the oxidation occurred on the metal centers. The EPR spectrum in frozen solution (77 K) shows one broad resonance while the coupling is not resolved (cf. Supporting Information). The solid compound 12 is EPR‐silent at room temperature and 77 K. For the arsenic derivative 13, a resonance can be detected for the solid and of the frozen CH2Cl2 solution only at 77 K. This resonance at 77 K shows a rhombic character (g1 =2.155, g2 =2.071, g3 =2.016) without any hyperfine couplings. Since the molecular structures could not be determined by XRD, DFT calculations (BP86/def2‐TZVP level of theory) were performed starting either on the strongly bent geometry of the cobalt complexes 16/17 or from the more planar geometry of the nickel complexes 14/15. The bent isomers 12‐bent/13‐bent are energetically favored by 20.3 and 3.5 kJ/mol, respectively, compared to their linear isomers 12‐linear/13‐linear. The spin density of both isomers differs significantly, being neither mainly located on the nickel atom (12‐bent/13‐bent) nor on the cobalt atom (12‐linear/13‐linear) (cf. Figure 3 and S83). In agreement with the EPR data of 12, the linear isomer seems to be predominant in solution, albeit energetically slightly disfavored. In the case of the arsenic derivative 13, the bent isomer seems to be predominant in frozen solution as well as in the solid state due to the absence of a hyperfine coupling to cobalt and the presence of a rhombic signal shape (the free electron is preferably located on a metal center). Since 12 and 13 could not be obtained as analytically pure compounds, no Evans NMR experiments were conducted. The further rearrangement of the heterometallic cation 12/13 to the homometallic nickel (14/15) and cobalt (16/17) cations is exothermic by 95.5 and 73.9 kJ/mol (related to 12‐bent/13‐bent), respectively.
Scheme 3.
Reaction of [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)] (E=P (10), As (11) with i) Ag[FAl] in CH2Cl2, ii) leaving the reaction mixture at room temperature overnight and iii) potassium graphite in dme in the presence of 2,2,2‐cryptand.
Figure 3.
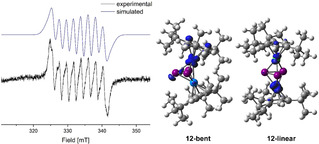
X‐band EPR spectrum of 12 in CH2Cl2 at room temperature (experimental (black) and simulated (blue)) and the isosurfaces of the calculated spin densities in 12‐bent and 12‐linear.
As already mentioned, leaving the reaction mixtures of the oxidation experiments of 10 and 11 with Ag[FAl] for crystallization yields single crystals of [(Cp′′′Co)2(μ,η3:η3‐E3)][FAl] (E=P (16), As (17)) suitable for X‐ray diffractions. The structures in the solid state are depicted in Figure 4. The molecular structure reveals a strongly bent triple‐decker complex with an E3 chain ligand coordinated to two {Cp′′′Co} fragments. The E3 ligand shows two short distances (P1‐P2 2.1532(12) Å, P2‐P3 2.1516(21) Å, As1‐As2 2.3770(5) Å, As2‐As3 2.3638(5) Å being in the range between single[23] and double[22] bonds, which is also in agreement with the calculated WBIs of 1.07/1.07 (16) and 1.02/1.02 (17). The outer E atoms E1 and E3 are 3.2571(11) Å (16) and 3.5176(6) Å (17) away from each other and clearly represent no bond (WBIs of 0.17 (16) and 0.17 (17)). The Co⋅⋅⋅Co distances of 2.6213(6) Å (16) and 2.6827(6) Å (17) are quite short and an interaction could not be dismissed, supported by the calculated WBIs of 0.18 (16) and 0.17 (17), respectively. Compound 13 and 15 are isoelectronic and isostructural to the starting materials 10 and 11, while 17 is isoelectronic and isostructural to the mixed iron and cobalt complex [(Cp*Fe)(Cp′′′Co)(μ,η3:η3‐As3)].[21] Single crystals of the nickel complexes [(Cp′′′Ni)2(μ,η3:η3‐E3)][FAl] (E=P (14), As (15)) as the second product were not obtained from these reaction mixtures. Since the phosphorus derivative was already reported,[28] only the so far unknown arsenic complex 15 will be discussed here. Single crystals of 15 were obtained from a concentrated solution of the reaction mixture of the oxidation of [Cp′′′Ni(η3‐As3)] with [FcDiac][FAl] in CH2Cl2 layered with pentane at room temperature. The structure in the solid state (Figure 4) shows a triple‐decker complex with an As3 ligand coordinated to two {Cp′′′Ni} fragments. The As3 ligand shows two shorter distance (As1‐As2 2.4179(4) Å, As2‐As3 2.4294(3) Å) and one long distance (As1‐As3 2.8730(4) Å). The short distance is in the range between As−As single and double bonds (calculated WBIs of 1.02 and 1.03) and the long distance is too long to be considered as a bond, but since the distance is still below the sum of van der Waals radii,[29] a weak interaction could not be dismissed, which is underlined by the calculated WBI of 0.36. The Ni atoms are 3.5434(5) Å apart from each other, which is clearly too long for an interaction (WBI of 0.05).
Figure 4.
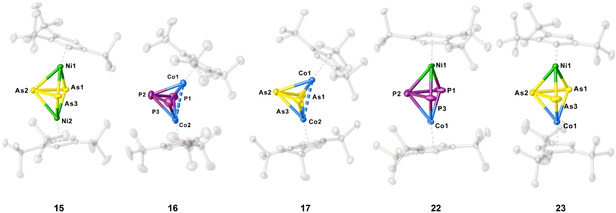
Molecular structures of 15, 16, 17, 22 and 23 in the solid state. Thermal ellipsoids are drawn with 50 % probability level. Hydrogen atoms, solvent molecules, anions and cations, respectively, are omitted for clarity.
To obtain further proof of the transfer of the {Cp′′′M} fragments during the oxidation process, [(Cp′′′Co)(Cp′′Ni)(μ,η3:η2‐P3)] (18) with two different Cp ligands was synthesized by the reaction of [Cp′′Ni(η3‐P3)] with [(Cp′′′Co)2(μ,η4:η4‐C7H8)]. Interestingly, the change from Cp′′′ to Cp′′ leads to a significant change in the geometry of the heterometallic triple‐decker complex in the solid state (Figure 5). Due to the lower steric demand of the Cp′′ ligand, the geometry changes from a slightly bent triple‐decker complex such as in 10 to a structural motif where the {Cp′′Ni} fragment is almost in the plane of the P3 ligand (torsion angle of only 7.5°).
Figure 5.
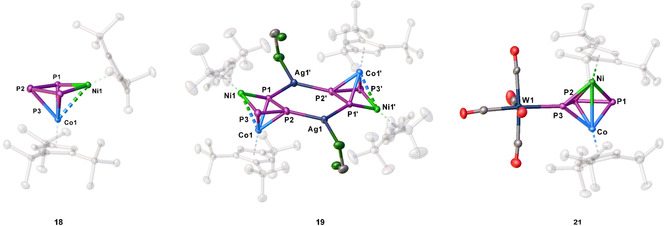
Molecular structures of 18, 19 and 21 in the solid state. Thermal ellipsoids are drawn with 50 % probability level. Hydrogen atoms, solvent molecules and anions, respectively, are omitted for clarity
This geometry is energetically favored by 33.5 kJ/mol towards the stretched triple‐decker geometry observed for 10 (cf. Figure S86). In the 31P{1H} NMR spectrum of 18 in C6D6, only the bent isomer can be detected in solution, indicated by a doublet centered at δ=356.0 and a triplet centered at δ=148.5 ppm with a 1 JPP coupling constant of 360 Hz. In contrast, for 10, the bent isomer is also energetically favored by 22.1 kJ/mol, but both can be detected in solution in a solvent‐ and temperature‐dependent equilibrium.[20] Due to the isolobal analogy between {CpRNi} and P, complex 18 is isolobal to the cyclo‐P4 complex [Cp′′′Co(η4‐P4)].[30] Contrary to expectations, the reaction of 18 with Ag[FAl] does not lead to an oxidized complex but instead to the coordination compound [(Cp′′′Co)(Cp′′Ni)(μ4,η3:η2:η1:η1‐P3){Ag(CH2Cl2)}]2[FAl]2 (19) (Scheme 4). Crystals suitable for X‐ray single crystal structure analysis could be obtained from a concentrated solution in CH2Cl2 layered with hexane at room temperature. The structure in the solid state (Figure 5) shows a dimeric structural motif with a central six‐membered ring consisting of two Ag atoms saturated each by a CH2Cl2 molecule and two P atoms of each molecule 18. Initially targeting to obtain the analog copper coordination compound, we reacted 18 with [Cu(MeCN)3.5][FAl] (Scheme 4). Interestingly, a short time after the addition of the copper salt to 18, a metal mirror was observed on the glass wall and, after workup, single crystals of 16 could be obtained, revealing clearly the transfer of {CpRM} fragments during the oxidation process. It was not possible to identify further Cp′′Ni‐containing products. The question arises as to whether the transfer of the metal fragments can be hindered. One possibility would be to coordinate an innocent organometallic fragment to the Pn ligand prior to oxidation. For this reason, we used the {W(CO)5}‐stabilized complex [(Cp′′′Co)(Cp′′′Ni)(μ3,η3:η2:η1‐P3){W(CO5)}] (20)[20] and reacted it with Ag[FAl]. Directly after addition of the silver salt, a metal mirror was observed and a 1H NMR spectrum in CD2Cl2 was recorded showing the formation of new paramagnetic species. After stirring for one day, another NMR spectrum was recorded showing that the paramagnetic species was still present, not having disappeared as observed for the oxidation of 10 and 11, respectively. After workup and crystallization, single crystals of [(Cp′′′Co)(Cp′′′Ni)(μ3,η3:η3:η1‐P3){W(CO)5}][FAl] (21) (Scheme 5)were obtained. The structure in the solid state reveals a strongly bent triple‐decker geometry where one of the outer P atoms is coordinated to a {W(CO)5} fragment. Since both metal fragments contain the same Cp′′′ ligand, a mixed occupancy could not be dismissed. The paramagnetism of 21 is in agreement with two different metal atoms. The X‐band EPR spectra of the obtained crystals shows a rhombic resonance at room temperature and 77 K, the latter being simulated (cf. Figure S12, g1 =2.060, g2 =2.007, g3 =1.984, giso =2.017). The 1H NMR spectrum in CD2Cl2 reveals three very broad signals centered at δ=5.73, 2.01 and 1.74 ppm.
Scheme 4.
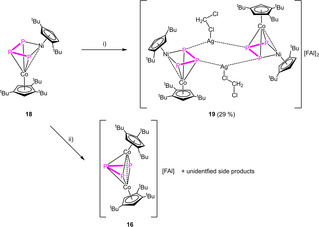
Reaction of 18 with: i) Ag[FAl] and ii) [Cu(MeCN)3.5][FAl] in CH2Cl2 at room temperature.
Scheme 5.
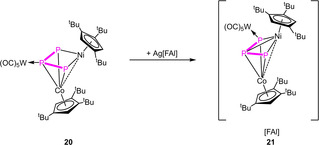
Oxidation of 20 with Ag[FAl] in CH2Cl2.
As already mentioned, the cyclic voltammograms of both 10 and 11 reveal possible reduction processes. The reduction of 10 and 11 yields [K(2,2,2‐crypt)][(Cp′′′Co)(Cp′′′Ni)(μ3,η3:η3‐E3)] (E=P (22), As (23), Scheme 3) in crystalline yields of 76 % and 73 %, respectively. Crystals suitable for X‐ray single crystal structure analysis could be obtained from a concentrated solution in thf layered with hexane at room temperature. The molecular structure in the solid state (Figure 4) shows a triple‐decker complex with both Cp′′′ ligands being almost parallel to each other (Cp′′′NiCoCp′′′ angle of 174.1° (22) and 174.9° (23)). The E3 middle deck shows two shorter E−E bonds (P1‐P2 2.1792(10) Å, P2‐P3 2.172(1) Å, As1‐As2 2.362(12) Å, As2‐As3 2.4327(7) Å) and two longer E⋅⋅⋅E contacts (P1‐P3 2.5537(11) Å, As1‐As3 2.7799)10) Å). The short distances are in the range between single and double bonds, indicated by the calculated WBIs of 1.04/1.08 for 22 and 0.99/1.02 for 23. The long distances are too long to be considered as bonds, but an interaction should be present, indicated by the WBIs of 0.45 (22) and 0.68 (23). Compounds 22 and 23 are both paramagnetic, which agrees with the addition of one electron by the reduction process and no rearrangement as in the oxidations mentioned above took place. The related 1H NMR spectra in thf‐d8 reveal several broad and partially strongly shifted signals. The magnetic moments in solution were determined by the Evans method to be 1.92 (22) and 1.22 (23) μB corresponding to roughly one unpaired electron each. At room temperature, the X‐band EPR spectra of solid 22 show only a very weak resonance and, at 77 K, a strong resonance with a rhombic character. In contrast, the spectra of a thf solution of 22 show at room temperature a resonance with a poorly resolved hyperfine coupling and, at 77 K, a strong resonance with a hyperfine coupling to cobalt and phosphorus (Figure 6). The latter was simulated (g1 =2.143, g2 =2.120, g3 =1.980, giso =2.081, A1 Co=149 MHz, A2 Co=50 MHz, A3 Co=43 MHz, A1 P=14 MHz, A2 P=36 MHz, A3 P=42 MHz). Compound 23 is EPR‐silent at room temperature. The X‐band EPR spectrum in a frozen thf solution at 77 K shows a rhombic resonance with a poorly resolved hyperfine coupling to cobalt, while the spectrum of solid 23 shows a rhombic signal without a resolved coupling (Figure 6, g1 =2.214, g2 =2.142, g3 =2.039, giso =2.146). DFT calculations (BP86/def2‐TZVP level of theory) reveal that the spin density is mainly located on the Co and only slightly on the Ni atom. In the case of 22, also an additional small contribution of the P3 ligand is observed. The reduction of 10 and 11 to 22 and 23 is fully reversible upon addition of AgBF4.
Figure 6.
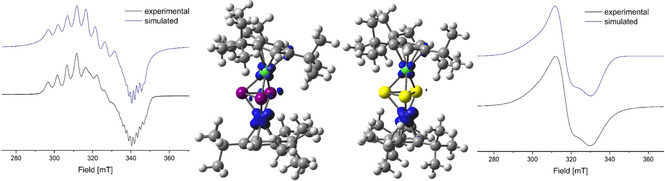
Experimental (black) and simulated (blue) X‐band EPR spectra of 22 (left) in frozen thf solution and 23 (right) in the solid state and the iso‐surfaces of the calculated spin density (BP86/def2‐TZVP level of theory) in 22 (left middle) and 23 (right middle).
Conclusion
It was shown that heterobimetallic triple‐decker complexes possessing a polypnictogen ligand as middle deck show a versatile redox chemistry. They respond to the redox reaction either with slight changes in the geometry, in the fragmentation of the En ligand or in the exchange of {Cp′′′M} fragments under formation of homometallic complexes. The iron and cobalt triple‐decker complexes [(Cp*Fe)(Cp′′′Co)(μ,η5:η4‐E5)] (E=P (1), As (2)) can be oxidized twice and reduced once, although the reduction of 2 is accompanied by a fragmentation of the As5 ligand into As6 and As3‐containing species. The folding of the cyclo‐E5 ligand is strongly dependent on the oxidation state. While oxidation leads to a successive planarization of the middle decks, reduction increases the fold angle and the coordination of the bent E atom changes from iron to cobalt. While in this case the outcome of the reduction is dependent on the pnictogen atom, this does not apply for the cobalt nickel triple‐decker complexes [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐E3)] (E=P (10), As (11). Both can be reduced to 22 and 21, while the oxidation to the heterometallic cations 12 and 13 does not display the end of the reaction, but a strong tendency to rearrange to the homo‐metallic cobalt complexes 16 and 17 and the nickel complexes 14 and 15 is observed. The transfer of {Cp′′′M} fragments was further investigated by EPR and NMR spectroscopy. Directly after the reaction, paramagnetic species (12 and 13) are present, which completely convert within one day into the homometallic diamagnetic compounds (14‐17). Since both metal fragments contain the same Cp′′′ ligand, the analog starting material with two different Cp ligands was prepared and analog reactions performed, revealing the same transfer of {Cp′′′M} fragments upon oxidation. This transfer can be remediated when an innocent ligand ({W(CO)5} is coordinated prior to the oxidation. The reversibility of the mentioned redox processes is very good for the mixed iron and cobalt complexes 1 and 2 (except reduction of 2) and comparable with the homometallic triple‐decker complexes mentioned in the introduction. For the mixed cobalt and nickel complexes 10 and 11, only the reduction is reversible, while the oxidation is accompanied by an exchange of the {Cp′′′M} fragments to form homometallic species.
Experimental Section
Synthetic procedures: All manipulations were performed under an atmosphere of dry argon using standard glove‐box and Schlenk techniques. All solvents were degassed and purified by standard procedures. The compounds [Cp′′′Ni(CO)]2,[31] [(Cp*Fe)(Cp′′′Co)(μ,η5:η4‐E5)] (E=P (1), As (2)),[20, 21] [KC8],[32] Ag[FAl],[33] [Cu(MeCN)3.5][FAl],[34] [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐P3)] (10),[20] [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η2:η1‐P3){W(CO)5}] (20),[20] [Cp′′Ni(η3‐P3)],[35] [(Cp′′′Co)2(η4:η4‐C7H8)].[36] 18‐c‐6 and 2,2,2‐cryptand were purchased commercially. The NMR spectra were recorded with a Bruker Avance 400 spectrometer (1H: 400.13 MHz, 31P: 161.976 MHz). The chemical shifts are given in ppm referenced to external SiMe4 (1H) and H3PO4 (31P). Elemental analyses were determined with an Elementar Vario EL III apparatus. The X‐Band EPR measurements were carried out with a MiniScope MS400 device with a frequency of 9.44 GHz and a rectangular resonator TE102 of the company Magnettech GmbH. The LIFDI‐MS spectra were recorded with a Finnigan MAT 95 mass spectrometer. The ESI‐MS spectra were acquired on a ThermoQuest Finnigan MAT TSQ 7000 mass spectrometer.
Synthesis of [Cp′′′Ni(η3‐As3)]: A freshly prepared solution of yellow arsenic in boiling xylene (starting from 5 g grey arsenic) was reacted with [(Cp′′′Ni(CO)]2 (800 mg, 6.248 mmol) under reflux for three hours. Five batches were combined and worked up together. The solvent was removed in vacuo, the residue dissolved in CH2Cl2 and SiO2 was added. The preabsorbed reaction mixture was purified by column chromatography (SiO2, hexane, 15×4.5 cm). Using hexane, an intense red fraction was eluted. The solvent was removed in vacuo and the oily residue dissolved in CH2Cl2 and precipitated with MeCN yielding a red powder. Yield: 3.70 g (57 %). 1H NMR (CD2Cl2, 25 °C): δ [ppm]=5.45 (s, 2H, C5 H 2 tBu3), 1.28 (s, 18H, C5H2 t Bu 3), 1.10 (s, 9H, C5H2 t Bu3 ). FD‐MS (toluene): m/z=1250.0 (53 %, [(Cp′′′Ni)3As5]),), 882.0 (100 %, [(Cp′′′Ni)2As4]),), 807.0 (63 %, [(Cp′′′Ni)2As3]), 515.9( 41 %, [M]+). EA C17H29NiAs3: calc [%]: C 39.50; H 5.66; found [%]: C 39.15; H 5.27.
Synthesis of [(Cp*Fe)(Cp′′′Co)(μ,η5:η5‐P5)][FAl] (3): A solution of Ag[FAl] (233.8 mg, 0.157 mmol, 1 eq) in CH2Cl2 was added to a stirred solution of 1 (100 mg, 0.157 mmol, 1 eq) in CH2Cl2 at room temperature, while the color changed from dark red to brown‐green and a dark precipitate (Ag0) was obtained. After stirring for 1 h, the reaction mixture was filtered over diatomaceous earth. The solvent of the green solution was reduced, layered with hexane and stored at −30 °C. After a few days, 3 could be obtained as green plates. The supernatant was decanted off, washed with hexane and dried in vacuo. Yield: 174 mg (55 %). 1H NMR (CD2Cl2, 25 °C): δ [ppm]=5.66 (br, ω 1/2=13 Hz, 15H, C5 Me5 ), 3.76 (br, ω 1/2=45 Hz 9H, C5H2 t Bu 3), 3.29 (br, ω 1/2=42 Hz 18H, C5H2 t Bu 3), −65.22 (br, ω 1/2=622 Hz, 2H, C5 H 2 tBu3). 19F{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=−112.6 (d, J F‐F=285 Hz, 6F), −117.1 (d, J F‐F=279 Hz, 6F), −121.8 (d, J F‐F=279 Hz, 3F), −127.9 (s, 6F), −130.6 (d, J F‐F=274 Hz, 6F), −137.2 (d, J F‐F=276 Hz, 6F), −141.2 (d, J F‐F=274 Hz, 3F), −154.3 (t, J F‐F=22 Hz, 6F), −165.0 (t, J F‐F=18 Hz, 6F), −172.0 (s, AlF, 1F). 31P{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=275.1 (br, ω 1/2=2600 Hz, P 5). 31P NMR (CD2Cl2, 25 °C): δ [ppm]=275.1 (br, ω 1/2=2600 Hz, P 5). Evans‐NMR (CD2Cl2, 25 °C): μeff =2.04 μB corresponding to 1.27 unpaired electrons. ESI‐MS (CH2Cl2): m/z=638.1 (100 %, [M]+), 607.1 (1 %, [M−P]+). EA C63H44O3AlF46P5FeCo: calc [%]: C 37.47; H 2.20; found [%]: C 37.46; H 2.31. X‐band EPR (77 K, frozen CH2Cl2 solution) g1 =2.155, g2 =2.035, g3 =1.948, A1 Co=290.125, A2 Co=55.125, A3 Co=120.125 MHz, (77 K, solid) (overall shape similar to (77 K, liquid), but coupling not resolved).
Synthesis of [(Cp*Fe)(Cp′′′Co)(μ,η5:η5‐As5)][FAl] (4): A solution of Ag[FAl] (173.5 mg, 0.1165 mmol, 1 eq) in CH2Cl2 was added to a stirred solution of 2 (100 mg, 0.1165 mmol, 1 eq) in CH2Cl2 at room temperature, while the color changed from dark green to brown‐green and a dark precipitate (Ag0) was obtained. After stirring for 1 h, the reaction mixture was filtered over diatomaceous earth. The solvent of the brown‐green solution was reduced, layered with hexane and stored at −30 °C. After a few days, 4 could be obtained as brown plates. The supernatant was decanted off, washed with hexane and dried in vacuo. Yield: 165 mg (63 %). 1H NMR (CD2Cl2, 25 °C): δ [ppm]=3.84 (br, ω 1/2=167 Hz, 9H, C5H2 t Bu 3), 2.74 (br, ω 1/2=119 Hz, 18H, C5H2 t Bu 3), −1.09 (br, ω 1/2=40 Hz, 15H, C5 Me 5). 19F{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=−112.6 (d, J F‐F=285 Hz, 6F), −117.1 (d, J F‐F=279 Hz, 6F), −121.8 (d, J F‐F=279 Hz, 3F), −127.9 (s, 6F), −130.6 (d, J F‐F=274 Hz, 6F), −137.2 (d, J F‐F=276 Hz, 6F), −141.2 (d, J F‐F=274 Hz, 3F), −154.3 (t, J F‐F=22 Hz, 6F), −165.0 (t, J F‐F=18 Hz, 6F), −172.0 (s, AlF, 1F). Evans‐NMR (CD2Cl2, 25 °C): μeff =1.90 μB corresponding to 1.14 unpaired electrons. ESI‐MS (CH2Cl2): m/z=857.9 (100 %, [M]+), 782.9 (67 %, [M−As]+), 708.0 (13 %, [M‐2As]+). EA C63H44O3AlF46As5FeCo: calc [%]: C 33.79; H 1.98; found [%]: C 34.06; H 2.01. X‐band EPR (77 K, solid) g1 =2.235, g2 =1.952, g3 =1.896, (77 K, frozen CH2Cl2 solution) g iso=2.04, (r.t, CH2Cl2 solution) g iso=2.06, (r.t, solid) g iso=2.04.
Synthesis of [(Cp*Fe)(Cp′′′Co)(μ,η5:η5‐P5)][FAl]2 (5): A solution of Ag[FAl] (238 mg, 0.160 mmol, 2.05 eq) in o‐C6H4F2 was added to a stirred solution of 1 (50 mg, 0.078 mmol, 1 eq) in o‐C6H4F2 at room temperature, while the color changed from dark red to brown and a dark precipitate (Ag0) was obtained. After stirring for 1 h, the reaction mixture was filtered over diatomaceous earth. The solvent of the brown solution was reduced, layered with toluene and stored at −30 °C. After a few days, 5 could be obtained as brown blocks. The supernatant was decanted off, washed with hexane and dried in vacuo. Yield: 131 mg (49 %). 1H NMR (CD2Cl2, 25 °C): δ [ppm]=4.69 (s, 2H, C5 H 2 tBu3), 1.49 (s, 18H, C5H2 t Bu 3), 1.39 (s, 9H, C5H2 t Bu3 ), 1.21 (s, 15H, C5 Me 5). 19F{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=−112.6 (d, J F‐F=285 Hz, 6F), −117.1 (d, J F‐F=279 Hz, 6F), −121.8 (d, J F‐F=279 Hz, 3F), −127.9 (s, 6F), −130.6 (d, J F‐F=274 Hz, 6F), −137.2 (d, J F‐F=276 Hz, 6F), −141.2 (d, J F‐F=274 Hz, 3F), −154.3 (t, J F‐F=22 Hz, 6F), −165.0 (t, J F‐F=18 Hz, 6F), −172.0 (s, AlF, 1F). 31P{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=54.7 (s, P 5). 31P NMR (CD2Cl2, 25 °C): δ [ppm]=54.7 (s, P 5). ESI‐MS (CH2Cl2): m/z=638.1 (100 %, [M]+). ESI‐MS (o‐C6H4F2): m/z=638.1 (100 %, [M]+), 319.0 (7 %, [M]2+). EA C99H44O6Al2F92P5FeCo x C6H4F2: calc [%]: C 35.88; H 1.37; found [%]: C 35.58; H 1.42.
Synthesis of [(Cp*Fe)(Cp′′′Co)(μ,η5:η5‐As5)][FAl]2 (6): A solution of Ag[FAl] (375 mg, 0.2518 mmol, 2 eq) in o‐C6H4F2 was added to a stirred solution of 2 (108 mg, 0.1259 mmol, 1 eq) in o‐C6H4F2 at room temperature, while the color changed from dark green to brown and a dark precipitate (Ag0) was obtained. After stirring for 4 h, the reaction mixture was filtered over diatomaceous earth. The solvent of the brown solution was reduced, layered with toluene and stored at −30 °C. After a few days, 6 could be obtained as brown blocks. The supernatant was decanted off, washed with hexane and dried in vacuo. Yield: 344 mg (75 %). 1H NMR (o‐C6H4F2 + C6D6 capillary, 25 °C): δ [ppm]=4.20 (s, 2H, C5 H 2 tBu3), 1.25 (s, 18H, C5H2 t Bu 3), 1.18 (s, 9H, C5H2 t Bu 3), 0.86 (s, 15H, C5 Me 5). 19F{1H} NMR (o‐C6H4F2 + C6D6 capillary, 25 °C): δ [ppm]=−112.6 (d, J F‐F=285 Hz, 6F), −117.1 (d, J F‐F=279 Hz, 6F), −121.8 (d, J F‐F=279 Hz, 3F), −127.9 (s, 6F), −130.6 (d, J F‐F=274 Hz, 6F), −137.2 (d, J F‐F=276 Hz, 6F), −141.2 (d, J F‐F=274 Hz, 3F), −154.3 (t, J F‐F=22 Hz, 6F), −165.0 (t, J F‐F=18 Hz, 6F), −172.0 (s, AlF, 1F). ESI‐MS (o‐C6H4F2): m/z=857.8 (56 %, [M]+), 428.9 (100 %, [M]2+). EA C99H44O6Al2F92P5FeCo: calc [%]: C 32.84; H 1.22; found [%]: C 32.55; H 1.46.
Synthesis of [K(2,2,2‐crypt)][(Cp*Fe)(Cp′′′Co)(μ,η4:η3‐P5)] (7): A suspension of KC8 (25 mg, 0.188, 1. 2 eq) in dme was added to a stirred solution of 1 (100 mg, 0.1567 mmol, 1 eq) and 2,2,2‐crypt (59 mg, 0.1567 mmol, 1 eq) in dme, while the color changed from dark red to brown green. After stirring for 3 h it was filtered over diatomaceous earth. The solvent was reduced and layered with hexane at room temperature. 7 could be obtained as dark green blocks. The supernatant was decanted off and the crystals dried in vacuo. Yield: 88 mg (44 %) 1H NMR (thf‐d8, 25 °C): δ [ppm]=6.24 (br, 9H, C5H2 t Bu 3), 4.15 (br, 15H, C5 Me 5), 3.70 (br, 12H, crypt), 3.66 (br, 12H, cypt), 3.44 (br, 18H, C5H2 t Bu 3), 2.66 (br, 12 H, crypt). EA C45H80O6N2P5FeCoK: calc [%]: C 51.29; H 7.65; N 2.66; found [%]: C 51.21; H 7.35; N 2.57. Evans‐NMR (thf‐d8, 25 °C): μeff =2.01 μB corresponding to 1.24 unpaired electrons. X‐band EPR (77 K, solid) g iso=2.047, (77 K, frozen dme solution) g iso=2.041, (r.t, dme solution) g iso=2.078, (r.t, solid) g iso=2.060.
Reaction of [(Cp*Fe)(Cp′′′Co)(μ,η5:η4‐As5)] (2) with KC8 : Compound 2 (100 mg, 0.1166 mmol, 1 eq), KC8 (19 mg, 0.1399, 1.2 eq) and 2,2,2‐crypt (43.9 mg, 0.1166 mmol, 1 eq) were weighed in together and dimethoxyethane was added, while the color changed from dark green to brown. After stirring for 5 minutes, it was filtered over diatomaceous earth and the solvent was reduced. An excess of hexane was used for precipitation. The microcrystalline solid was washed with hexane (slight brown to green color) and dried in vacuo. The 1H NMR spectrum in thf‐d8 reveals the formation of 9 beside the paramagnetic compound [K(2,2,2‐crypt)][(Cp*Fe)(Cp′′′Co)(μ,η4:η3‐As5)] (8) (cf. Figure S24). X‐Band EPR measurement of the obtained solid reveals a strong isotropic resonance. Compound 8: X‐band EPR (77 K, solid) g iso=2.123, (r.t, solid) g iso=2.109.
Synthesis of [K(2,2,2‐crypt)][(Cp*Fe)(Cp′′′Co)(μ,η4:η4‐As6)] (9): Compound 2 (500 mg, 0.5827 mmol, 1 eq), KC8 (88 mg, 0.651, 1.12 eq) and 2,2,2‐crypt (219.5 mg, 0.5827 mmol, 1 eq) were weighed in together and dimethoxyethane was added. After stirring overnight (color change from green to brown), it was filtered over diatomaceous earth. The solvent was reduced and layered with hexane at room temperature. After a few days, an oily solid was observed at the Schlenk wall. Leaving it for a few weeks, crystalline solid (needles) of 9 were obtained. The supernatant was decanted off, washed with hexane and the crystals dried in vacuo. The 1H NMR spectrum of the brown‐green mother liquor reveals [(Cp*Fe)(Cp′′′Co)(μ,η3:η3‐As3)][21] as a major compound. Yield: 481 mg (92 %). 1H NMR (thf‐d8, 25 °C): δ [ppm]=4.14 (s, 2H, C5 H 2 tBu3), 3.61 (s, 12H, C18 H 36N2O6), 3.57 (overlapped with solvent signal, C18 H 36N2O6), 2.56 (t, 3 JHH =4.4 Hz, 12H, C18 H 36N2O6), 1.61 (s, 15H, C5 Me 5), 1.53 (s, 18H, C5H2 t Bu 3), 1. 35 (s, 9H, C5H2 t Bu 3). ESI‐MS (dme): m/z=932.9 (100 %, [M]−). EA C45H80O6N2As6FeCoK: calc [%]: C 40.08; H 5.98; N 2.08; found [%]: C 40.60; H 6.12; N 1.94.
Synthesis of [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐As3)] (11): A solution of [(Cp′′′Co)2(μ,η4:η4‐C7H8) (985 mg, 1.455 mmol, 1 eq) in hexane was added to a stirred solution of [Cp′′′Ni(η3‐As3)] (1.50 mg, 2.91 mmol, 2 eq) in hexane at room temperature, while the color changed to green. After stirring for one hour, the solvent was removed in vacuo. The residue was dissolved in CH2Cl2 and layered with MeCN and stored at room temperature. After a few days, 11 can be obtained as green needles. The supernatant was decanted off and the remaining crystals dried in vacuo. Yield: 2.05 g (85 %). 1 H NMR (C6D6, 25 °C): δ [ppm]=5.14 (s, 2H, C5 H 2 tBu3), 4.90 (s, 2H, C5 H 2 tBu3), 1.43 (s, 18H, C5H2 t Bu 3), 1.34 (s, 18H, C5H2 t Bu 3), 1.06 (s, 9H, C5H2 t Bu 3), 0.96 (s, 9H, C5H2 t Bu 3). LIFDI‐MS (toluene): m/z =808.0 (100 %, [M]+). EA C34H58CoNiAs3: calc [%]: C 50.46; H 7.22; found [%]: C 50.07; H 6.84.
Reaction of [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐P3)] (10) with Ag[FAl]: A solution of Ag[FAl] (220 mg, 0.148 mmol, 1 eq) in CH2Cl2 was added to a stirred solution of 10 (100 mg, 0.148 mmol, 1 eq) in CH2Cl2 at −80 °C. The reaction mixture was warmed to room temperature, while a color change from brown to red‐brown and a dark precipitate (Ag0) was observed. The desired [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐P3)][FAl] (12) was precipitated by addition of hexane, the solvent was decanted off and the residue dried in vacuo. The obtained solid was investigated by NMR and EPR spectroscopy, revealing the paramagnetic character of 12. 1H NMR (CD2Cl2, 25 °C): δ [ppm]=3.65 (br, ω 1/2=127 Hz, 27H, C5H2 t Bu 3), 2.96 (br, ω 1/2=53 Hz, 18H, C5H2 t Bu 3), 2.49 (br, ω 1/2=48 Hz, 9H, C5H2 t Bu 3). X‐band EPR (r.t, solution) g iso=2.023, g 1=2.023, g 2=2.031, g 3=2.014, A 1 Co=59.56, A 2 Co=63.97, A 3 Co=62.83 MHz; (77 K, frozen CH2Cl2 solution) g iso=2.025.
Oxidation of [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐As3)] (11): A solution of Ag[FAl] (92 mg, 0.062 mmol, 1 eq) in CH2Cl2 was added to a stirred solution of 11 (50 mg, 0.062 mmol, 1 eq) in CH2Cl2 at −80 °C, while the color changed from green to red‐brown. The reaction mixture was warmed to room temperature and a dark precipitate (Ag0) was observed. The desired [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐As3)][FAl] (13) was precipitated by addition of hexane, the solvent was decanted off and the residue dried in vacuo. The obtained solid was investigated by 1H NMR and EPR spectroscopy, revealing the paramagnetic character of 13. The residual solid was again dissolved in CH2Cl2 and stirred for five days. The solvent was removed in vacuo and an 1H NMR spectrum in CD2Cl2 was recorded, revealing the formation of the two diamagnetic species [(Cp′′′Co)2(μ,η3:η3‐As3)][FAl] (17) and [(Cp′′′Ni)2(μ,η3:η3‐As3)][FAl] (15). Very few single crystals of 17 suitable for X‐ray diffractions were obtained by layering a concentrated solution of the reaction mixture in o‐C6H4F2 with pentane at −30 °C. Analytically pure 15 can be obtained by the oxidation of [Cp′′′Ni(η3‐As3)] with diacetyl‐ferrocenium (vide infra). Analytical data of 13: 1H NMR (CD2Cl2, 25 °C): δ [ppm]=3.04 (br, 18H, C5H2 t Bu 3), 2.99 (br,9H, C5H2 t Bu 3), 2.76 (br, 18H, C5H2 t Bu 3), 2.25 (br, 18H, C5H2 t Bu 3), −23.51 (br, 2H, C5 H 2 tBu3). X‐band EPR (77 K, solid) g1 =2.155, g2 =2.071, g3 =2.016, (77 K, frozen CH2Cl2 solution) giso =2.070.
Oxidation of [Cp′′′Ni(η3‐As3)]: Addition of a blueish green solution of [FcDiac][FAl] (83 mg, 0.05 mmol, 0.5 eq.) in CH2Cl2 to a dark red solution of Cp′′′Ni(η3‐As3) (52 mg, 0.1 mmol, 1 eq.) in CH2Cl2 at −80 °C afforded a rapid color change to dark brown. The reaction mixture was left to warm to room temperature and stirred for another 2 h. Afterwards pentane was added to precipitate 15 as a brown powder. The supernatant was decanted off, the solid washed two times with toluene and another two times with pentane. The residue was dissolved in CH2Cl2 and layered with pentane. After a few days, 15 could be obtained as brown blocks. The supernatant was decanted off and the crystals washed with hexane and dried in vacuo. Yield: 55 mg (50 %). 1H NMR (CD2Cl2, 25 °C): δ [ppm]=5.09 (s, 2H, C5 H 2 tBu3), 1.35 (s, 18H, C5H2 t Bu 3), 0.96 (s, 9H, C5H2 t Bu 3). ESI‐MS (CH2Cl2): m/z=841.1 (0.1 %, [M+H2O+O]), 807.1 (0.6 %, [M]+), 373.2 (24 %, [Cp′′′Ni(MeCN)2]), 332.2 (100 %, [Cp′′′NiMeCN]). EA C70H58O3Ni2As3AlF46: calc [%]: C 38.39; H 2.67; found [%]: C 38.61; H 2.70.
Synthesis of [(Cp′′′Co)2(μ,η3:η3‐P3)][FAl] (16): A solution of Ag[FAl] (220 mg, 0.148 mmol, 1 eq) in CH2Cl2 was added to a stirred solution of 10 (100 mg, 0.148 mmol, 1 eq) in CH2Cl2 at room temperature, while the color changed from brown to red‐brown and a dark precipitate (Ag0) was obtained. The reaction was completed by stirring for 20 h (dark red color, shorter reaction times do not lead to a full conversion). The reaction mixture was filtered over diatomaceous earth. The 1H and 31P{1H} NMR spectra in CD2Cl2 reveals the formation of [(Cp′′′Co)2(μ,η3:η3‐P3)][FAl] (16) and [(Cp′′′Ni)2(μ,η3:η3‐P3)][FAl] (14).[28] The solvent of the red solution was reduced, layered with toluene and stored at −30 °C. After a few days, 16 can be obtained as red rods. The supernatant was decanted off, washed with hexane and dried in vacuo. Yield: 86 mg (65 %). 1 H NMR (CD2Cl2, 25 °C): δ [ppm]=4.89 (s, 4H, C5 H 2 tBu3), 1.41 (s, 36H, C5H2 t Bu 3), 1.23 (s, 18H, C5H2 t Bu 3). 31 P{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=789.0 (d, 1 JPP =361 Hz, 2P, P 3), −218.0 (t, 1 JPP =361 Hz, 1P, P 3). 31 P NMR (CD2Cl2, 25 °C): δ [ppm]=789.0 (d, 1 JPP =361 Hz, 2P, P 3), −218.0 (t, 1 JPP =361 Hz, 1P, P 3). 19 F{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=‐112.6 (d, J F‐F=285 Hz, 6F), −117.1 (d, J F‐F=279 Hz, 6F), −121.8 (d, J F‐F=279 Hz, 3F), −127.9 (s, 6F), −130.6 (d, J F‐F=274 Hz, 6F), −137.2 (d, J F‐F=276 Hz, 6F), −141.2 (d, J F‐F=274 Hz, 3F), −154.3 (t, J F‐F=22 Hz, 6F), −165.0 (t, J F‐F=18 Hz, 6F), −172.0 (s, AlF, 1F). ESI‐MS (CH2Cl2): m/z=677.2 (100 %, [M]+). EA C70H58O3AlF46Co2P3: calc [%]: C 40.83; H 2.84; found [%]: C 40.91; H 3.12.
Synthesis of [(Cp′′′Co)2(μ,η3:η3‐As3)][FAl] (17):A solution of Ag[FAl] (184 mg, 0.1235 mmol, 1 eq) in CH2Cl2 was added to a stirred solution of 11 (100 mg, 0.1235 mmol, 1 eq) in CH2Cl2, while the color changed immediately from dark green to brown. The reaction mixture was stirred overnight and filtered over diatomaceous earth. The solvent was reduced, layered with hexane and stored at −30 °C. After a few days, 17 could be obtained as brown blocks, with 15 remaining in the mother liquor. The mother liquor was decanted off, the obtained crystals washed with hexane and dried in vacuo. Yield: 45 mg (17 %).Analytical data of 17: 1H NMR (CD2Cl2, 25 °C): δ [ppm]=4.79 (s, 2H, C5 H 2 tBu3), 1.35 (s, 18H, C5H2 t Bu 3), 1.15 (s, 9H, C5H2 t Bu 3). 19F{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=−112.6 (d, J F‐F=285 Hz, 6F), −117.1 (d, J F‐F=279 Hz, 6F), −121.8 (d, J F‐F=279 Hz, 3F), −127.9 (s, 6F), −130.6 (d, J F‐F=274 Hz, 6F), −137.2 (d, J F‐F=276 Hz, 6F), −141.2 (d, J F‐F=274 Hz, 3F), −154.3 (t, J F‐F=22 Hz, 6F), −165.0 (t, J F‐F=18 Hz, 6F), ‐172.0 (s, AlF, 1F).ESI‐MS (CH2Cl2): m/z=809.09 (100 %, [M]+). EA C70H58O3AlF46Co2As3x(C6H4F2)0.5: calc [%]: C 39.01; H 2.69; found [%]: C 38.89; H 2.62.
Synthesis of [(Cp′′′Co)(Cp′′Ni)(μ,η3:η2‐P3)] (18): A solution of [(Cp′′′Co)2(μ,η4:η4‐C7H8) (680 mg, 1.00 mmol, 1 eq) in toluene was added to a stirred solution of [Cp′′Ni(η3‐P3)] (658 mg, 2.00 mmol, 2 eq) in toluene at room temperature. After stirring for one hour, the solvent was removed in vacuo. The residue was dissolved in CH2Cl2 and layered with MeCN and stored at −30 °C. After a few days, 18 was obtained as brown needles. The supernatant was decanted off and the remaining crystals dried in vacuo. Yield: 730 mg (59 %). 1H NMR (C6D6, 25 °C): δ [ppm]=4.96 (s, 1H, C5 H 3 tBu2), 4.81 (s, 2H, C5 H 3 tBu2), 4.78 (s, 2H, C5 H 2 tBu3), 1.39 (s, 18H, C5H2 t Bu 3), 1.26 (s, 9H, C5H2 t Bu 3), 1.23 (s, 18H, C5H3 t Bu 2). 31P{1H} NMR (C6D6, 25 °C): δ [ppm]=375.0 (d, 1 JPP =360 Hz, 2P, P 3), 148.5 (t, 1 JPP =360 Hz, 1P, P 3). LIFDI‐MS (toluene): m/z=620.1 (100 %, [M]+). EA C30H50CoNiP3: calc [%]: C 57.99; H 8.11; found [%]: C 57.63; H 7.97.
Synthesis of [(Cp′′′Co)(Cp′′Ni)(μ4,η3:η2:η1:η1‐P3)Ag(CH2Cl2)]2[FAl]2 (19): A solution of Ag[FAl] (240 mg, 0.161 mmol, 1 eq) in CH2Cl2 was added to a stirred solution of 18 (100 mg, 0.161 mmol, 1 eq) in CH2Cl2, while the color changed to red‐brown. After stirring for two hours, the volume of the solvent was reduced and layered with hexane. After storage at room temperature for a few days, 19 could be obtained as brown blocks. The supernatant was decanted off and the remaining crystals dried in vacuo. Yield: 120 mg (29 %). 1H NMR (CD2Cl2, 25 °C): δ [ppm]=4.93 (br, 3H, C5 H 3 tBu2), 4.17 (br, 2H, C5 H 2 tBu3), 1.51 (s, 9H, C5H2 t Bu 3/C5H3 t Bu 2), 1.49 (s, 9H, C5H2 t Bu 3/C5H3 t Bu 2), 1.43 (s, 9H, C5H2 t Bu 3/C5H3 t Bu 2), 1.22 (s, 18H, C5H2 t Bu 3/C5H3 t Bu 2). 31P{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=334.2 (br), 321.9 (br), 125.9 (br), 110.8 (br). ESI‐MS (CH2Cl2): m/z=1477.5 (100 %, [M+F]+). EA C67H52O3CoNiP3AgAlCl2F46×1.4 CH2Cl2: calc [%]: C 35.42; H 2.39; found [%]: C 35.28; H 2.30.
Synthesis of [(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3:η1‐P3){W(CO)5}][FAl] (21):A solution of Ag[FAl] (149 mg, 0.10 mmol, 1 eq) in CH2Cl2 was added to a stirred solution of 20 (100 mg, 0.10 mmol, 1 eq) in CH2Cl2 and stirred overnight, while the color changed from red to brown and a dark precipitate (Ag0) was observed. The reaction mixture was filtered over diatomaceous earth. The solvent was removed in vacuo. The residue was washed with hexane (slightly brown), dissolved in CH2Cl2, layered with hexane and stored at room temperature. After a few days, few crystals of 21 could be obtained in form of brown blocks. 1H NMR (CD2Cl2, 25 °C): δ [ppm]=5.73 (very broad, C5 H 2 tBu3), 2.01 (br, 36H, C5H2 t Bu 3), 1.74 (br, 18H, C5H2 t Bu 3). 19F{1H} NMR (CD2Cl2, 25 °C): δ [ppm]=−112.6 (d, J F‐F=285 Hz, 6F), −117.1 (d, J F‐F=279 Hz, 6F), −121.8 (d, J F‐F=279 Hz, 3F), −127.9 (s, 6F), −130.6 (d, J F‐F=274 Hz, 6F), −137.2 (d, J F‐F=276 Hz, 6F), −141.2 (d, J F‐F=274 Hz, 3F), −154.3 (t, J F‐F=22 Hz, 6F), −165.0 (t, J F‐F=18 Hz, 6F), ‐172.0 (s, AlF, 1F). X‐band EPR (293 K, solid) rhombic spectrum similar to the spectrum at 77 K, no simulation performed; (77 K, solid) g iso=2.017, g 1=2.060, g 2=2.007, g 3=1.984.
Synthesis of [K(2,2,2‐crpyt)][(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐P3)] (22): A suspension of KC8 (25 mg, 0.18 mmol, 1.22 eq) in dimethoxyethane was added to a stirred solution of 10 (100 mg, 0.1476 mmol, 1 eq) and 2,2,2‐cryptand (56 mg, 0.1476 mmol, 1 eq) in dimethoxyethane at −50 °C, while the color changed from brown to dark brown. The reaction mixture was allowed to reach room temperature overnight. The reaction mixture was filtered over diatomaceous earth, the solvent removed in vacuo and washed with hexane. The residue was dissolved in thf, layered with hexane and stored at room temperature. After a few days, 20 could be obtained as brown plates. The supernatant was decanted off, washed with hexane and dried in vacuo. Yield: 123 mg (76 %). 1H NMR (thf‐d8, 25 °C): δ [ppm]=3.65 (s, C18 H 36N2O6), 3.61 (s, C18 H 36N2O6), 2.78 (br, C5H2 t Bu 3),2.62 (s, C18 H 36N2O6), 2.33 (br, C5H2 t Bu 3), 1.63 (br, C5H2 t Bu 3). Evans‐NMR (thf‐d8, 25 °C): μeff =1.92 μB; corresponding to roughly one unpaired electron. ESI‐MS (dme): m/z=676.2 (39 %, [M]−), 385.1 (100 %, [M−Cp‘‘‘Ni]−). EA C52H94O6N2KCoNiP3: calc [%]: C 57.14; H 8.67, N 2.56; found [%]: C 57.22; H 8.50, N 2.52. X‐band EPR (293 K, solid) giso =2.079, (77 K, solid) giso =2.061, (293 K, solution) rhombic spectrum with fine couplings, badly resolved, no simulation performed, (77 K, liquid) g iso=2.081, g 1=2.143, g 2=2.120, g 3=1.980, A 1 Co=148.53, A 2 Co=50.32, A 3 Co=42.72, A 1 P=13.63, A 2 P=35.94, A 3 P=42.13 MHz).
Synthesis of [K(2,2,2‐crypt)][(Cp′′′Co)(Cp′′′Ni)(μ,η3:η3‐As3)] (23): A suspension of KC8 (25 mg, 0.1900 mmol, 1.5 eq) in dimethoxyethane was added to a stirred solution of 16 (100 mg, 0.1236 mmol, 1 eq) and 2,2,2‐cryptand (47 mg, 0.1236 mmol, 1 eq) in dimethoxyethane at room temperature, while the color changed from green to red‐brown. After one hour, the reaction mixture was filtered over diatomaceous earth, the solvent removed in vacuo and washed with hexane. The residue was dissolved in thf, layered with hexane and stored at room temperature. After a few days, 23 could be obtained as brown blocks. The supernatant was decanted off, washed with hexane and dried in vacuo. Yield: 110 mg (73 %). 1H NMR (thf‐d8, 25 °C): δ [ppm]=3.61 (s, C18 H 36N2O6), 3.57 (s, overlapped with solvent signal, C18 H 36N2O6), 3.16 (br, C5H2 t Bu 3),2.59 (br, C18 H 36N2O6+C5H2 t Bu 3), 1.41 (br, C5H2 t Bu 3). Evans‐NMR (thf‐d8, 25 °C): μeff =1.22 μB; corresponding to roughly one unpaired electron. ESI‐MS (dme): no fragments can be assigned properly. EA C52H94N2O6KCoNiAs3: calc [%]: C 50.99; H 7.74; N 2.29; found [%]: C 51.22; H 7.21, N 2.23. 76. X‐band EPR (77 K, solid) giso =2.141, g 1=2.214, g 2=2.142, g 3=2.039, (77 K, solution) rhombic spectrum with fine couplings, badly resolved, no simulation performed.
Crystallographic data: Deposition numbers 2064647 (3), 2064648 (4), 2064649 (5), 2064650 (6), 2064651 (7), 2064652 (9), 2064653 (11), 2064654 (15), 2064655 (16), 2064656 (17), 2064657 (18), 2064658 (19), 2064659 (21), 2064660 (22), 2064661 (23) and 2064662 ([Cp′′′Ni(η3‐As3)]) contain the supplementary crystallographic data for this paper. These data are provided free of charge by the joint Cambridge Crystallographic Data Centre and Fachinformationszentrum Karlsruhe Access Structures service.
Conflict of interest
The authors declare no conflict of interest.
Supporting information
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary
Acknowledgements
This work was supported by the Deutsche Forschungsgemeinschaft within the project Sche 384/33‐2 and 384/36‐1. M. P. is grateful to the Fonds der Chemischen Industrie for a PhD fellowship and S. R. is grateful to the Studienstiftung des Deutschen Volkes for a PhD fellowship. Open access funding enabled and organized by Projekt DEAL.
M. Piesch, S. Reichl, C. Riesinger, M. Seidl, G. Balazs, M. Scheer, Chem. Eur. J. 2021, 27, 9129.
Dedicated to Professor Andreas Hirsch on the occasion of his 60th birthday
Contributor Information
Dr. Martin Piesch, https://www.uni‐regensburg.de/chemie‐pharmazie/anorganische‐chemie‐scheer/
Prof. Dr. Manfred Scheer, Email: manfred.scheer@ur.de.
References
- 1.
- 1a.Gnutzmann G., Wilhelm Dorn F., Klemm W., Z. Anorg. Allg. Chem. 1961, 309, 210–225; [Google Scholar]
- 1b.Delamar M., J. Electroanal. Chem. Interfacial Electrochem. 1975, 63, 339–349; [Google Scholar]
- 1c.Brauer G., Zintl E., Z. physik. Chem. 1937, B37, 323–352. [Google Scholar]
- 2.
- 2a.Köchner T., Engesser T. A., Scherer H., Plattner D. A., Steffani A., Krossing I., Angew. Chem. Int. Ed. 2012, 51, 6529–6531; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2012, 124, 6635–6637; [Google Scholar]
- 2b.Gillespie R. J., Passmore J., Ummat P. K., Vaidya O. C., Inorg. Chem. 1971, 10, 1327–1332. [Google Scholar]
- 3.
- 3a.Tzschach A., Kiesel V., J. Prakt. Chem. 1971, 313, 259–264; [Google Scholar]
- 3b.Baudler M., Gruner C., Fürstenberg G., Kloth B., Saykowski F., Özer U., Z. Anorg. Allg. Chem. 1978, 446, 169–176. [Google Scholar]
- 4.Astruc D., Eur. J. Inorg. Chem. 2017, 2017, 6–29. [Google Scholar]
- 5.Gritzner G., Kuta J., Pure Appl. Chem. 1984, 56, 461–466. [Google Scholar]
- 6.
- 6a.Arumugam K., Varnado C. D., Sproules S., Lynch V. M., Bielawski C. W., Chem. Eur. J. 2013, 19, 10866–10875; [DOI] [PubMed] [Google Scholar]
- 6b.Allgeier A. M., Mirkin C. A., Angew. Chem. Int. Ed. 1998, 37, 894–908; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 1998, 110, 936–952. [Google Scholar]
- 7.Neumann P., Dib H., Caminade A.-M., Hey-Hawkins E., Angew. Chem. Int. Ed. 2015, 54, 311–314; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2015, 127, 316–319. [Google Scholar]
- 8.Turbervill R. S. P., Jupp A. R., McCullough P. S. B., Ergöçmen D., Goicoechea J. M., Organometallics 2013, 32, 2234–2244. [Google Scholar]
- 9.
- 9a.Nief F., Ricard L., Organometallics 2001, 20, 3884–3890; [Google Scholar]
- 9b.Du S., Chai Z., Hu J., Zhang W.-X., Xi Z., Chin. J. Org. Chem. 2019, 39, 2338; [Google Scholar]
- 9c.Du S., Yin J., Chi Y., Xu L., Zhang W.-X., Angew. Chem. Int. Ed. 2017, 56, 15886–15890; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 16102–16106; [Google Scholar]
- 9d.Du S., Yang J., Hu J., Chai Z., Luo G., Luo Y., Zhang W.-X., Xi Z., J. Am. Chem. Soc. 2019, 141, 6843–6847. [DOI] [PubMed] [Google Scholar]
- 10.Turbervill R. S. P., Goicoechea J. M., Chem. Commun. 2012, 48, 6100–6102. [DOI] [PubMed] [Google Scholar]
- 11.Ashe A. J., Mahmoud S., Elschenbroich C., Wünsch M., Angew. Chem. Int. Ed. Engl. 1987, 26, 229–230; [Google Scholar]; Angew. Chem. 1987, 99, 249–250. [Google Scholar]
- 12.Scherer O. J., Blath C., Wolmershäuser G., J. Organomet. Chem. 1990, 387, C21-C24. [Google Scholar]
- 13.Scherer O. J., Brück T., Angew. Chem. Int. Ed. Engl. 1987, 26, 59; [Google Scholar]; Angew. Chem. 1987, 99, 59. [Google Scholar]
- 14.
- 14a.Butovskiy M. V., Balázs G., Bodensteiner M., Peresypkina E. V., Virovets A. V., Sutter J., Scheer M., Angew. Chem. Int. Ed. 2013, 52, 2972–2976; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2013, 125, 3045–3049; [Google Scholar]
- 14b.Schmidt M., Konieczny D., Peresypkina E. V., Virovets A. V., Balázs G., Bodensteiner M., Riedlberger F., Krauss H., Scheer M., Angew. Chem. Int. Ed. 2017, 56, 7307–7311; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 7413–7417. [Google Scholar]
- 15.Piesch M., Seidl M., Scheer M., Chem. Sci. 2020, 11, 6745–6751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Mädl E., Balázs G., Peresypkina E. V., Scheer M., Angew. Chem. Int. Ed. 2016, 55, 7702–7707; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2016, 128, 7833–7838. [Google Scholar]
- 17.Dütsch L., Fleischmann M., Welsch S., Balázs G., Kremer W., Scheer M., Angew. Chem. Int. Ed. 2018, 57, 3256–3261; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2018, 130, 3311–3317. [Google Scholar]
- 18.Fleischmann M., Dielmann F., Balázs G., Scheer M., Chem. Eur. J. 2016, 22, 15248–15251. [DOI] [PubMed] [Google Scholar]
- 19.Piesch M., Graßl C., Scheer M., Angew. Chem. Int. Ed. 2020, 59, 7154–7160; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2020, 132, 7220–7227. [Google Scholar]
- 20.Piesch M., Dielmann F., Reichl S., Scheer M., Chem. Eur. J. 2020, 26, 1518–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Piesch M., Scheer M., Organometallics 2020, 39, 4247–4252. [Google Scholar]
- 22.Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 12770–12779. [DOI] [PubMed] [Google Scholar]
- 23.Pyykkö P., Atsumi M., Chem. Eur. J. 2009, 15, 186–197. [DOI] [PubMed] [Google Scholar]
- 24.Scheer M., Gregoriades L. J., Virovets A. V., Kunz W., Neueder R., Krossing I., Angew. Chem. Int. Ed. 2006, 45, 5689–5693; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2006, 118, 5818–5822. [Google Scholar]
- 25.Fleischmann M., Welsch S., Krauss H., Schmidt M., Bodensteiner M., Peresypkina E. V., Sierka M., Gröger C., Scheer M., Chem. Eur. J. 2014, 20, 3759–3768. [DOI] [PubMed] [Google Scholar]
- 26.
- 26a.Kudinov A. R., Loginov D. A., Starikova Z. A., Petrovskii P. V., Corsini M., Zanello P., Eur. J. Inorg. Chem. 2002, 2002, 3018–3027; [Google Scholar]
- 26b.Scherer O. J., Brück T., Wolmershäuser G., Chem. Ber. 1989, 122, 2049–2054; [Google Scholar]
- 26c.Schmidt M., Seitz A. E., Eckhardt M., Balázs G., Peresypkina E. V., Virovets A. V., Riedlberger F., Bodensteiner M., Zolnhofer E. M., Meyer K. et al., J. Am. Chem. Soc. 2017, 139, 13981–13984. [DOI] [PubMed] [Google Scholar]
- 27.Scherer O. J., Pfeiffer K., Heckmann G., Wolmershäuser G., J. Organomet. Chem. 1992, 425, 141–149. [Google Scholar]
- 28.Scheer M., Riesinger C., Duetsch L., Balazs G., Bodensteiner M., Chem. Eur. J. 2020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Alvarez S., Dalton Trans. 2013, 42, 8617–8636. [DOI] [PubMed] [Google Scholar]
- 30.Dielmann F., Timoshkin A., Piesch M., Balázs G., Scheer M., Angew. Chem. Int. Ed. 2017, 56, 1671–1675; [DOI] [PMC free article] [PubMed] [Google Scholar]; Angew. Chem. 2017, 129, 1693–1698. [Google Scholar]
- 31.Sitzmann H., Wolmershäuser G., Z. Naturforsch. B 1995, 50, 750–756. [Google Scholar]
- 32.Lalancette J.-M., Rollin G., Dumas P., Can. J. Chem. 1972, 50, 3058–3062. [Google Scholar]
- 33.Köchner T., Trapp N., Engesser T. A., Lehner A. J., Röhr C., Riedel S., Knapp C., Scherer H., Krossing I., Angew. Chem. Int. Ed. 2011, 50, 11253–11256; [DOI] [PubMed] [Google Scholar]; Angew. Chem. 2011, 123, 11449–11452. [Google Scholar]
- 34.Elsayed Moussa M., Piesch M., Fleischmann M., Schreiner A., Seidl M., Scheer M., Dalton Trans. 2018, 47, 16031–16035. [DOI] [PubMed] [Google Scholar]
- 35.Scherer O. J., Dave T., Braun J., Wolmershäuser G., J. Organomet. Chem. 1988, 350, C20-C24. [Google Scholar]
- 36.Schneider J. J., Wolf D., Janiak C., Heinemann O., Rust J., Krüger C., Chem. Eur. J. 1998, 4, 1982–1991. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
As a service to our authors and readers, this journal provides supporting information supplied by the authors. Such materials are peer reviewed and may be re‐organized for online delivery, but are not copy‐edited or typeset. Technical support issues arising from supporting information (other than missing files) should be addressed to the authors.
Supplementary