

To the Editor:
Sickle cell disease (SCD) is a severe genetic disorder that impacts approximately 20 million people worldwide.1 The causative β6 Glu‐Val substitution is a gain of function mutation; in the deoxygenated state, the mutant protein, Hb S, can form polymers, leading to red blood cell sickling and precipitating downstream consequences, including vaso‐occlusion (pain crisis), hemolytic anemia, stroke and related pathophysiology.2, 3 Polymerization is exponentially dependent on deoxy Hb S concentration.4 Thus, relatively small changes in deoxyHb S concentration will significantly impact polymerization, red blood cell sickling, and ultimately the clinical course of the disease. Pharmacologic evidence for the benefit of reducing the concentration of deoxyHb S arises from studies of covalent modification of Hb S, where stabilization of the oxygenated conformation increases Hb O2 affinity, reduces RBC sickling, extends RBC half‐life, and ultimately reduces the frequency of vaso‐occlusive crisis (VOC).5, 6, 7, 8 In clinical trials, ex vivo carbamylation of patient blood led to improvements in hemolytic anemia; treated patients exhibited a 2.7 g/dl increase in hemoglobin, a 58% decrease in reticulocytes, a 65% decrease in irreversibly sickled cells, and an 80% decrease in frequency of painful crises.8 Subsequent oxyHb S stabilizing molecules were developed based on the observation that benzaldehyde derivatives formed stable, covalent Schiff bases with hemoglobin. The most advanced of this class of molecules is the covalent compound Voxelotor (GBT 440, Oxbryta), which was approved by the FDA in 2019 for the treatment of SCD. In the pivotal study, 59% of patients in the higher dose group (1500 mg/day) had increases of 1 g/dl or greater in hemoglobin, with a mean hemoglobin modification of 26.5%.9 While the impact of covalent Hb S modifiers on hemolytic anemia is well established, the rise in hemoglobin observed for voxelotor falls short of the effects observed by ex vivo carbamylation, and was not accompanied by a significant effect on the frequency of VOC. This suggests that the therapeutic potential of hemoglobin modification has not been fully realized.
PF‐7059013 is a non‐covalent modifier of hemoglobin that stabilizes the oxygenated state (see Gopalsamy et al.). Here we present the impact this molecule has on a well‐established mouse model of sickle cell disease. Treatment with PF‐7059013 demonstrated robust changes in key markers of hemolytic anemia in the Townes mouse model,10 suggesting it has the potential to be a potent and efficacious therapy for SCD.
PF‐07059013 was orally administered to Townes SCD model mice twice daily at a dose of 200 mg/kg for 15 days. This dose was selected as it was expected to result in approximately 25% hemoglobin coverage.11 At 30 minutes post the initial dose, total blood concentrations for individual animals were 2–4 mM, consistent with the low total blood clearance observed in the single dose administration studies. These values translate to approximately 40%–60% hemoglobin coverage, based on measured hemoglobin concentrations. The high total blood concentrations observed following the initial dose were maintained for the duration the 15‐day dose period (Supporting Information S1).
Animals treated with PF‐07059013 show a significant stabilization of the oxygenated state. The average p50 decreased by 53.7% (±21.2%) in the treated group, relative to vehicle, and the average p20, a more sensitive marker of compound occupancy, decreased by 84.4% (±2.6%) in the treated group relative to vehicle (Supporting Information S1). As expected from the large shifts in oxygen affinity, blood from animals in the treated group showed significant reductions in RBC sickling. Under stringent hypoxic conditions, treatment with PF‐07059013 resulted in a 37.8% (±9%) decrease in RBC sickling (Figure 1(A)).
FIGURE 1.
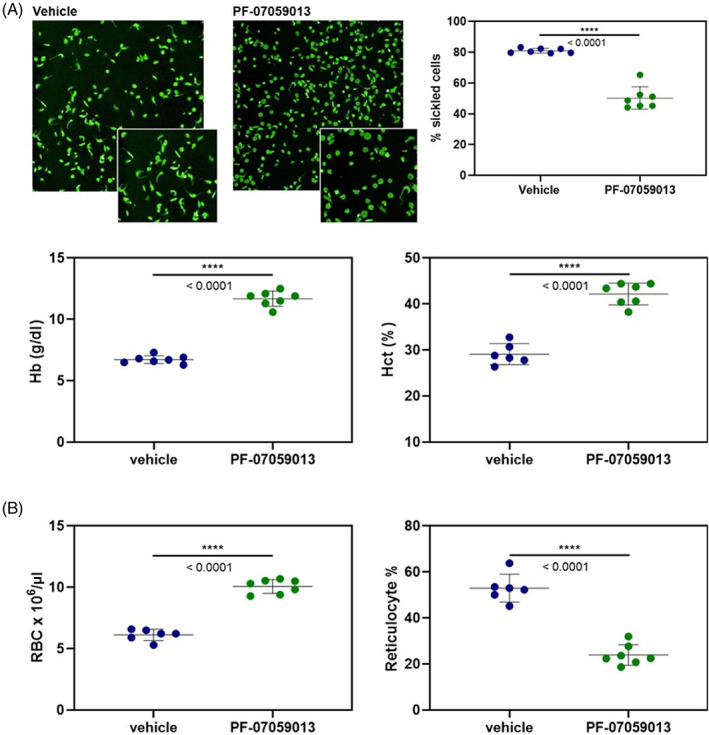
(A) Left: Representative images of red blood cells from vehicle treated and PFE‐07059013 treated animals, following 4 h of hypoxic exposure. Inset: Expansion of section of depicted field to highlight differences in number of sickled cells. Right: Individual values for RBC sickling for vehicle treated and PF‐07059013 treated mice. The treated animals showed a robust and statistically significant reduction in RBC sickling. (B) Markers of hemolytic anemia measured for vehicle treated (blue) and PF‐07059013 treated (green) mice. Note, PF‐07059013 treated animals showed increases in RBC count, hemoglobin (Hb), and hematocrit (Hct), indicating a substantial reduction in hemolysis. PF‐07059013 treated animals also showed a significant decrease in reticulocytes, suggesting decreased hematopoiesis in the PF‐07059013 animals, due to the decreased hemolysis
Consistent with reduction of RBC sickling, following 15 days of dosing, mice treated with PF‐07059013 showed significant improvement in markers of hemolytic anemia. PF‐07059013 treated animals showed a 42.4% (±4.2%) increase in hemoglobin, with a mean increase in hemoglobin of 5 g/dl, as well as a 30.9% (±0.7%) increase in hematocrit, and a 39.2 (±9.3%) increase in red blood cells relative to vehicle. All of the changes were statistically significant, and the increases restored the hemoglobin, hematocrit, and RBC counts of the treated group to values similar to wild type (C7BL/6) mice (Figure 1(B)). In addition, treatment with PF‐07059013 resulted in a 54.7% (±2.4%) decrease in reticulocytes (Figure 1(B)). Note, PF‐07059013 achieves consistently high levels of hemoglobin occupancy in in vivo studies using the Townes SCD murine model, which were sustained for the duration of dosing. Taken together, these results indicate that a non‐covalent molecule has the potential to be efficacious for the treatment of sickle cell disease, and that the presence of a reactive aldehyde is not a requirement for potency.
Comparing the activity of early covalent modifiers in the Townes model with PF‐07059013 is not possible, as the development of many of those molecules predated the development of the transgenic mouse models. However, it is possible to compare the performance of PF‐07059013 in the Townes SCD mouse with previously published Voxelotor pre‐clinical data in the Townes model. Oksenberg et al. report that twice‐daily oral dosing of Townes SCD mice with Voxelotor/GBT 440 at 100 or 150 mg/kg for 9–12 days resulted in hemoglobin occupancies ranging from 11%–39.7%.12 The pharmacodynamic effects of Voxelotor were strongly correlated with the degree of hemoglobin occupancy, as only the animals attaining >30% occupancy (4/14) showed changes in reticulocyte count or red blood cell half‐life relative to vehicle. Thus, PF‐07059013 achieved high degrees of hemoglobin coverage upon twice‐daily oral dosing at 200 mg/kg for 15 days. As PF‐07059013 binds ditopically to Hb (two compound: one tetramer), the dose is comparable to the doses used in the Voxelotor animal studies,12 as PF‐07059013 requires twice as much compound to achieve the same hemoglobin occupancy percentages. In contrast with Voxelotor preclinical studies12 all of the animals dosed with PF‐07059013 (n = 7) achieved >40% hemoglobin occupancy. The consistently high occupancy level across all animals leads to a uniform improvement in markers of hemolytic anemia. Similarly, all PF‐07059013 treated animals showed decreases in RBC sickling, ranging from 35.3%–45.5%. The observed decrease in RBC sickling with PF‐07059013 treatment is consistent with the decrease observed for Voxelotor in Townes SCD mice that had high Hb occupancy.12
The role of increases in hemoglobin in the overall pathology of sickle cell disease, particularly as it relates to VOC, is not completely understood. In the Voxelotor pivotal trial, 59% of patients treated with 1500 mg/day experienced hemoglobin increases of 1 g/dl or greater (average = 1.1 g/dl), and showed reductions in reticulocytes and bilirubin, consistent with improvements in hemolytic anemia, following 24 weeks of dosing.9 Post‐hoc analysis of the pivotal trial results indicated that patients who achieved a hemoglobin level of 10 g/dl or greater had reduced incidence of VOC (50/179), with the greatest benefit observed in the small group of patients that had hemoglobin levels of 12 g/dl or greater (10/179).13 These data are consistent with the observation that increased hemoglobin can lead to reductions in VOC, provided RBC sickling is sufficiently impeded; Diedrich et al. demonstrated a substantial reduction in VOC frequency following weekly extracorporeal carbamylation.8 After 3 months of treatment, hemoglobin had increased by an average of 2.7 g/dl to an average of 8.8 g/dl and occurrences of VOC decreased by 80%.8 An increase in hemoglobin alone is likely not sufficient, as SCD patients undergoing exchange transfusions still experience VOC.14 Ex vivo carbamylation was most efficacious when hemoglobin occupancy was above 35%, suggesting achieving and maintaining high levels of hemoglobin occupancy may be crucial for making the maximum reduction in RBC sickling, reducing hemolysis, and increasing hemoglobin. These data indicate that it is possible to correlate the hemoglobin increase mediated by stabilization of the oxygenated state to resolution of VOC, and further suggest that the size of the increase in hemoglobin may be an important influencer of other clinical outcomes. Based in part on the magnitude and consistency of the response in the Townes SCD mouse model presented here, clinical studies of PF‐07059013 are currently underway.
CONFLICT OF INTEREST
All authors listed were employees of Pfizer Inc and declare no conflict of interest.
AUTHOR CONTRIBUTIONS
Kelly M. Knee: designed research, performed research, analyzed data, wrote the paper. Reema Jasuja: designed research, performed research, analyzed data. Amey Barakat: performed research, analyzed data. Dharani Rao: performed research, analyzed data. Zane Wenzel: performed research, analyzed data. Jayasankar Jasti: performed research, analyzed data. Jonathan Novak: performed research, analyzed data. Kevin Beaumont: designed research, analyzed data. David W. Piotrowski: designed research, analyzed data. Phil Jeffery: designed research, analyzed data. Christine Bulawa: designed research, analyzed data. John E. Murphy: analyzed data, designed research. Jay M. Janz: performed research, designed research, analyzed data, wrote the paper.
Supporting information
Appendix S1. Supporting information
ACKNOWLEDGMENTS
The authors wish to thank Dr. Carlo Brugnara (Boston Children's Hospital) for assistance in obtaining SCD patient blood, and for valuable project discussions. We thank Dr. John Kelly (Northeastern University Co‐op program), Joseph Nneji (Northeastern University Co‐op program), and Victoria Ball (Northeastern University Co‐op Program) for assistance in hemoglobin purification. We thank Jazmyne Lopez (Pfizer Occupational Health and Wellness) for coordinating healthy human blood sample collection, and Dr. David Karanian and Dr. Jamie DaSilva (Pfizer Drug Safety Research and Development) for providing whole blood from relevant toxicology species.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
REFERENCES
- 1.Modell B, Darlison M. Global epidemiology of haemoglobin disorders and derived service indicators. Bull World Health Organ. 2008;86(6):480‐487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kato G, Gladwin MT, Steinberg MH. Deconstructing sickle cell disease: reappraisal of the role of hemolysis in the development of clinical subphenotypes. Blood Rev. 2007;21(1):37‐47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Rees D, Williams TN, Gladwin MT. Sickle‐cell disease. Lancet. 2010;376:2018‐2031. [DOI] [PubMed] [Google Scholar]
- 4.Ferrone F. Kinetics of sickle hemoglobin polymerization II. A double nucleation mechanism. J Mol Biol. 1985;183:611‐631. [DOI] [PubMed] [Google Scholar]
- 5.Cerami A, Manning JM. Potassium cyanate as an inhibitor of the sickling of erythrocytes in vitro. Proc Natl Acad Sci U S A. 1971;68(6):1180‐1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Njikam N, Jones WM, Nigen AM, Gillette PN, Williams RC, Manning JM. Carbamylation of the chains of hemoglobin S by cyanate in vitro and in vivo. J Biol Chem. 1973;248(23):8052‐8056. [PubMed] [Google Scholar]
- 7.Gillette P, Manning JM, Cerami A. Increased survival of sickle cell erythrocytes after treatment in vitro with sodium cyanate. Proc Natl Acad Sci U S A. 1971;68(11):2791‐2793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Diederich D, Trueworth RC, Gill P, Cader M, Larsen WE. Hematologic and clinical responses in patients with sickle cell anemia after chronic extracorporeal red cell carbamylation. J Clin Investig. 1976;58:542‐653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Vichinsky E, Hoppe CC, Ataga KI, et al. A phase 3 randomized trial of Voxelotor in sickle cell disease. N Engl J Med. 2019;381(6):509‐510. [DOI] [PubMed] [Google Scholar]
- 10.Ryan T, Ciavatta DJ, Townes TM. Knockout‐transgenic mouse model of sickle cell disease. Science. 1997;278:873‐876. [DOI] [PubMed] [Google Scholar]
- 11.Gopalsamy A, Aulabaugh AE, Barakat A, et al. PF‐07059013: a non‐covalent modulator of hemoglobin for treatment of sickle cell disease. J Med Chem. 2021;64:326‐342. [DOI] [PubMed] [Google Scholar]
- 12.Oksenberg D, Dufu K, Patel MP, et al. GBT440 increases haemoglobin oxygen affinity, reduces sickling and prolongs RBC half‐life in a murine model of sickle cell disease. Br J Hematol. 2016;175:141‐153. [DOI] [PubMed] [Google Scholar]
- 13.Vichinsky E, Gordeuk VR, Telfer P, et al. Higher hemoglobin levels achieved with Voxelotor are associated with lower vaso occlusive crisis incidence: 72 week analysis from the HOPE study [abstract]. Blood. 2020;136:31‐32. [Google Scholar]
- 14.Swerdlow P. Red cell exchange in sickle cell disease. Hematology Am Soc Educ Program. 2006;1:48‐53. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Appendix S1. Supporting information
Data Availability Statement
The data that support the findings of this study are available from the corresponding author upon reasonable request.