

ABSTRACT
High fracture rate and high circulating levels of the Wnt inhibitor, sclerostin, have been reported in diabetic patients. We studied the effects of Wnt signaling activation on bone health in a mouse model of insulin‐deficient diabetes. We introduced the sclerostin‐resistant Lrp5 A214V mutation, associated with high bone mass, in mice carrying the Ins2 Akita mutation (Akita), which results in loss of beta cells, insulin deficiency, and diabetes in males. Akita mice accrue less trabecular bone mass with age relative to wild type (WT). Double heterozygous Lrp5 A214V/Akita mutants have high trabecular bone mass and cortical thickness relative to WT animals, as do Lrp5 A214V single mutants. Likewise, the Lrp5 A214V mutation prevents deterioration of biomechanical properties occurring in Akita mice. Notably, Lrp5 A214V/Akita mice develop fasting hyperglycemia and glucose intolerance with a delay relative to Akita mice (7 to 8 vs. 5 to 6 weeks, respectively), despite lack of insulin production in both groups by 6 weeks of age. Although insulin sensitivity is partially preserved in double heterozygous Lrp5 A214V/Akita relative to Akita mutants up to 30 weeks of age, insulin‐dependent phosphorylated protein kinase B (pAKT) activation in vitro is not altered by the Lrp5 A214V mutation. Although white adipose tissue depots are equally reduced in both compound and Akita mice, the Lrp5 A214V mutation prevents brown adipose tissue whitening that occurs in Akita mice. Thus, hyperactivation of Lrp5‐dependent signaling fully protects bone mass and strength in prolonged hyperglycemia and improves peripheral glucose metabolism in an insulin independent manner. Wnt signaling activation represents an ideal therapeutic approach for diabetic patients at high risk of fracture. © 2021 The Authors. Journal of Bone and Mineral Research published by Wiley Periodicals LLC on behalf of American Society for Bone and Mineral Research (ASBMR).
Keywords: Lrp5, WNT SIGNALING, BONE, DIABETES, BROWN ADIPOSE TISSUE
Introduction
The incidence of type 1 diabetes (T1D) and type 2 diabetes (T2D) is increasing worldwide, whereas improvements in diabetes treatment have contributed to an exponential increase in the number of fragility fractures in these patients. Indeed, low‐trauma fractures now substantially contribute to deterioration of quality of life and mortality in diabetic patients, in addition to vascular complications.( 1, 2 ) However, the mechanisms by which diabetes exposes patients to high risk of low‐trauma fractures remain only partly known. The modestly lower bone mineral density (BMD) cannot entirely explain the several‐fold higher fracture rate in T1D patients relative to age‐matched controls,( 3 ) and BMD is even higher than normal in T2D, after adjustment for body size.( 4, 5 ) Low bone turnover is common among T2D and T1D, although histologic studies in T1D are not all consistent with decreased bone formation.( 6, 7 ) Subtle defects in bone microarchitecture and increased accumulation of advanced glycosylation end‐products (AGEs) and pentosidine, which alter bone stiffness, have been reported in T1D subjects.( 8 ) Abnormal accumulation of AGEs in bone has also been observed in a rat model of T1D.( 9 ) High glucose concentration in extracellular fluids and AGEs can increase expression of the Wnt inhibitor, sclerostin, and decrease receptor activator of NF‐κB ligand (RANKL) production in vitro, potentially explaining the low bone turnover in T1D.( 10 ) Consistent with these observations, we have recently shown that circulating glycated hemoglobin is positively associated with increased fracture rate in T1D patients.( 11 )
Canonical Wnt signaling is a key modulator of bone homeostasis. In humans, gain‐of‐function mutations of the LRP5 Wnt co‐receptor gene are associated with high bone mass.( 12, 13 ) Likewise, loss‐of‐function mutations of SOST, the gene encoding sclerostin, cause sclerosteosis or van Buchem disease, both high bone mass disorders.( 14, 15 ) Importantly, an anti‐sclerostin antibody, romosozumab, has been recently approved for use in the United States and other countries for increasing bone mass and reducing fracture risk in osteoporosis.( 16 ) Mounting evidence suggests that the Wnt signaling pathway, and sclerostin in particular, might be implicated in the mechanisms leading to low bone formation and suboptimal bone quality of diabetes patients. Increased circulating sclerostin has been observed in subjects with T1D( 17 ) and T2D, where it is inversely correlated with bone turnover markers, but is positively associated with BMD.( 18, 19, 20, 21, 22 ). Indeed, we have recently found that SOST expression is increased in bone of T2D patients.( 23 ) Preclinical studies further suggest that Wnt signaling is involved in glucose metabolism. Mice with a null mutation of the Lrp5 gene are osteopenic and glucose intolerant.( 24, 25 ) By contrast, Sost knockout mice have higher insulin sensitivity and lower adiposity than normal littermates,( 26 ) in addition to high bone mass.( 27 )
Based on this background, we used mouse genetic models to ask whether activating Wnt signaling may counteract the bone and metabolic abnormalities that develop in insulin‐dependent diabetes. Results confirm that a sclerostin‐insensitive Lrp5 mutant leads to high bone mass accrual and improved bone strength in mice with prolonged, severe hyperglycemia. Importantly, the gain‐of‐function Lrp5 mutation delays the onset of hyperglycemia and prevents whitening of brown adipose tissue that occurs in insulin‐deficient mice. Our studies suggest that therapies based on Wnt activation may be beneficial to both bone and glucose homeostasis.
Materials and Methods
Animals
Mice carrying the germline Lrp5 A214V mutant allele, which results in high bone mass (HBM) phenotype,( 28 ) were a generous gift from Dr. Matthew L. Warman (Harvard University, Boston, MA, USA). The Lrp5 A214V mutation renders the receptor insensitive to antagonists, particularly sclerostin, which consequently hyperactivates Wnt signaling.( 29 ) Ins2 Akita (Akita) mice were purchased from Jackson Laboratories (Stock number 003548; Bar Harbor, ME, USA). These mice carry one mutant Ins2 allele that causes insulin protein misfolding, cellular stress, and eventual apoptosis of beta cells, thereby leading to spontaneous hyperglycemia by 4 to 5 weeks of age in males.( 30 ) Heterozygous Lrp5 +/A214V (HBM) mice were mated with Ins2 Akita mice, to obtain, in one generation, double heterozygous Lrp5 +/A214V;Ins2 Akita (HBM/Akita) mice, as well as single heterozygous and wild‐type (WT) mice. All the mouse lines used in this project were bred in a C57BL/6J background, and littermates were used as controls. Mice were weaned at postnatal day 28 (P28), fed a regular chow and housed in a room maintained at constant temperature (25°C) on a 12‐h light and 12‐h dark cycle. Genotyping for Lrp5 was performed via polymerase chain reaction (PCR) on genomic DNA extracted from mouse tails using the HotSHOT method,( 31 ) and primers to detect the WT and Lrp5 A214V alleles, as described.( 28 ) Genotyping for the Ins2 Akita allele was performed by TransnetYX, Inc. (Cordova, TN, USA). Tissues for postmortem analysis (adipose tissue depots, pancreas) were collected immediately after euthanasia. Brown adipose tissue (BAT) was extracted from the intrascapular region; white adipose tissue was harvested from gonadal/inguinal and retroperitoneal areas (gWAT, rWAT, respectively). Tissues were snap frozen in liquid nitrogen until processing. All animal procedures were performed in accordance with procedures approved by the Institutional Animal Care and Use Committee of Washington University in St. Louis (Protocols number 20140279, 20170095, and amendments).
Body composition analysis
To determine whole‐body composition, dual‐energy x‐ray absorptiometry (DXA) was performed using a Faxitron UltraFocus100 scanner (Faxitron Bioptics, LLC, Tucson, AZ, USA) at different ages. Mice were anesthetized by 2% isoflurane inhalation, a concentration that has no impact on metabolic parameters,( 32 ) and placed in a prostrate position on an imaging‐positioning tray. Whole‐body scans were made excluding head and tail. Analyses included whole‐body BMD, bone mineral content (BMC), percent body fat, and lean mass. The instrument was calibrated daily before use, and one investigator (Giulia Leanza) performed all scans.
BAT histology
The entire BAT depot was excised from the intrascapular region of 30‐week‐old mice, fixed in ethanol, and processed for paraffin inclusion. Sections were stained with hematoxylin and eosin and imaged by bright‐field microscopy (NanoZoomer; Hamamatsu Photonics K.K., Hamamatsu, Japan). Sections from four blocks, each from a different mouse, were analyzed per genotype.
Bone microarchitecture
For in vivo longitudinal analysis of bone microarchitecture, mice were scanned at 6 and 20 weeks of age at the proximal tibia by micro–computed tomography (μCT) (VIVA CT40; SCANCO Medical AG, Brüttisellen, Switzerland) as described.( 33 ) Before scanning, the leg was fully extended, while the mouse was kept in a mask to ensure the continuous flow of 2% isoflurane during the entire procedure. The leg was immobilized in the scanner using a custom designed fixture, so as to minimize any variability due to leg repositioning. Microstructural analysis of trabecular and cortical bone was also performed as described.( 33 ) For ex vivo μCT analysis, tibias or femurs were stored at −20°C until use, then placed in 2% agarose gel and scanned using a μCT‐40 system (SCANCO Medical AG) as described.( 34 )
Bone biomechanics
For assessment of bone strength, dissected tibias were tested in three‐point bending to failure or fracture using methods described.( 33 ) Briefly, specimens were stabilized over two supports placed 7 mm apart in an Instron 8841 apparatus (Instron, Norwood, MA, USA). A loading force was applied in the anteroposterior direction midway between the two supports by a displacement ramp at a rate of 0.03 mm/s. Force and displacement data were collected at 100 Hz (Labview 5.0; National Instruments, Austin, TX, USA), and test curves were analyzed as described.( 35 )
Glucose metabolism
Capillary blood was obtained by cutting 1 to 2 mm of tissue from the tail tip with sharp scissors, and glucose measurements were performed using the On‐call Express Blood Glucose Meter (ACON Laboratories, San Diego, CA, USA). Because the detection limit of this device is 600 mg/dl, blood glucose above this level was recorded as 600 mg/dl but considered to be a lower limit of the true value.( 36 ) Intraperitoneal glucose tolerance test (ipGTT) was performed after 6 h morning or 14 h overnight fasting on conscious mice. Glucose sampling was obtained at baseline and 15, 30, 45, 60, 90, and 120 min after an intraperitoneal glucose injection (1.5 g/kg dextrose in 50% solution).( 37 ) Insulin tolerance test (ITT) was performed at 7 weeks of age after 6 h morning fasting. Insulin (0.5 U/kg body weight) was administered intraperitoneally( 38 ); blood glucose was obtained at baseline and 15, 30, 45, 60, 90, and 120 min after insulin injection.
Circulating hormones
Blood samples were drawn from the tail vein in heparinized capillary tubes during an ipGTT at time 0, immediately before dextrose injection, and 30 min after injection. Serum insulin, glucagon, and C‐peptide were measured using Millipore Sigma's MILLIPLEX MAP Mouse Metabolic Hormone Magnetic Bead Panel ‐ Metabolism Multiplex Assay MMHMAG‐44K (Millipore Sigma, Burlington, MA, USA), containing beads for insulin, glucagon, and C‐peptide 2 (Washington University Immunoassay Core, St. Louis, MO, USA). Insulin‐like growth factor 1 (IGF‐1) was measured using mouse IGF‐1 (BR55) LXSAMSM 1plex (R&D Systems, Minneapolis, MN, USA) by the Washington University Immunoassay Core. Glu, Gla13, and total mouse osteocalcin were measured using three specific enzyme‐linked immunosorbent assays (ELISAs), as described( 39 ) by two co‐authors (Mathieu Ferron, Céline Schott). Briefly, 96‐well ELISA plates (R&D Systems) were coated with anti‐Glu‐osteocalcin, anti‐Gla13‐osteocalcin, or anti‐Mid‐osteocalcin antibodies diluted in coating buffer (Immunochemistry Technologies, Bloomington, MN, USA) and incubated overnight at room temperature. Then, the plates were washed and blocked with assay diluent (0.1% Tween and 3% bovine serum albumin [BSA] in phosphate buffered saline [PBS]) for 4 h at room temperature. Afterward, standards or serum samples was added to assay diluent and plates were sealed and incubated overnight at 4°C. The Gla13 and total osteocalcin (OCN) (Mid) ELISA were run using synthetic 3XGla‐OCN (Bio‐Synthesis Inc., Lewisville, TX, USA) as standard, whereas Glu‐OCN ELISA was run using bacterial produced Glu‐OCN as standard. After five washes, horseradish peroxidase–conjugated anti‐CT‐osteocalcin (osteocalcin C‐terminus) antibody was added to each well and placed on a shaker for 1 h at room temperature. After further washes, the plates were incubated with the substrate, tetramethylbenzidine (Pierce, Rockford, IL, USA) for 15 min. The reaction was stopped with 100 μl of HCl 1M and detected by colorimetry at 450 nm wavelength in a microplate reader. Polynomial second‐order standard curves were used to determine the concentration of Glu (undercarboxylated), Gla13 (carboxylated), and total osteocalcin. Glu13 was calculated by subtracting Gla13 from total osteocalcin.
Whole‐pancreas insulin content
After euthanasia, the pancreas was isolated, weighted (wet weight), then minced and homogenized using a mechanical homogenizer in acid/ethanol extraction buffer (77% 100 proof ethanol, 1.5% HCl; 1 ml/100 mg tissue). The extract was incubated overnight at 4°C and centrifuged at 400g for 30 min at 4°C. Supernatants were stored at −20°C until analysis. Insulin content was determined by ELISAs at 1:5000 dilution, as detailed.( 36 )
Adipocyte cultures
Adipocyte cultures were prepared from external ear stromal cells following an established procedure.( 40 ) External ears were collected and cut in small pieces in ice‐cold Hank's balanced salt solution (HBSS) containing antibiotics; incubated in 2 mg/ml collagenase I (Worthington Biochemical Corp., Lakewood, NJ, USA) for 1 h at 37°C, filtered through a 70‐μm cell strainer (BD Biosciences, San Jose, CA, USA) and pelleted by centrifugation at 189 relative centrifugation force (RCF) (1300 revolutions per minute (rpm)) for 10 min. Cells were re‐suspended, seeded, and subcultured in Dulbecco's modified Eagle medium (DMEM)/Ham's F‐12 Nutrient Mixture (F12) containing 15% fetal bovine serum (FBS) and 10 ng/ml fibroblast growth factor (FGF). Confluent cultures were incubated in adipogenic medium (5 μg/ml insulin, 1μM dexamethasone, 500μM isobutyl‐methylxanthine, 5μM rosiglitazone; Sigma, St. Louis, MO, USA) for 2 days; switched to DMED/F12 with 10% FBS, 5 μg/ml insulin and 5μM rosiglitazone for 2 days; then cultured in DMEM/F12 with 10% FBS and visually monitored until accumulation of lipid droplets. For the signaling experiments, cells were incubated in serum‐free media for 2 h before exposure to insulin.
Immunoblots
Protein extracts were prepared by lysing cells cultured in six‐well plates with radioimmunoprecipitation assay (RIPA) buffer (50mM Tris, 150mM NaCl, 1mM ethylenediamine tetraacetic acid [EDTA], 0.5% NaDOAc, 0.1% sodium dodecylsulfate [SDS], and 1.0% NP‐40) plus a phosphatase and protease inhibitor cocktail (Xpert P3200‐001 and P3100‐001; GenDEPOT, Katy, TX, USA). Protein concentration was determined by the Bio‐Rad method (DC Protein Assay), and equal amounts of proteins were subjected to SDS–polyacrylamide gel electrophoresis (SDS‐PAGE) gels (8–15%). Proteins were transferred onto polyvinylidene fluoride (PVDF) membranes and incubated overnight with primary antibody at 4°C, followed by a 1‐h incubation with secondary goat anti‐mouse or goat anti‐rabbit Alexa‐Fluor 680 (Molecular Probes, Eugene, OR, USA), or donkey anti‐goat 780 (LI‐COR Biosciences, Lincoln, NE, USA) antibody. Primary antibodies include total protein kinase B (Akt), phospho‐Akt (Ser473) (Cell Signaling Technology, Beverly, MA, USA) and β‐actin (Santa Cruz Biotechnology, Santa Cruz, CA, USA). The results were visualized using Li‐Cor Odyssey Infrared Imaging System (LI‐COR Biosciences).
Data presentation and statistical analysis
Group data are presented in box‐plots with median and interquartile range; whiskers represent maximum and minimum values. When appropriate, arithmetic mean is shown as plus sign within the box‐plot. Unless otherwise noted, repeated measures are plotted as median and interquartile range. Differences between groups were assessed using one‐way analysis of variance (ANOVA), and repeated measures were analyzed by two‐way ANOVA or mixed‐effects models, in cases of missing data points, followed by Tukey's tests to adjust p values for multiple comparisons. Data were managed in Microsoft Excel (Microsoft Corp., Redmond, WA, USA), plotted, and analyzed using Prism 8.0 and 9.1 (GraphPad Software, San Diego, CA, USA).
Results
The Lrp5A214V mutation results in high whole‐body bone mass and prevents BAT whitening, but not loss of WAT in the Akita background
Validating the Lrp5 A214V model, whole‐body BMD and BMC measured by DXA were significantly higher in the HBM groups relative to either the Akita or control groups up to at least 26 weeks of age (Figure 1A,B ), whereas body weight and length were not different among the four experimental groups (Figure 1, Supplemental Figure S1 A,B). Aside a slightly lower BMD at 6 weeks in HBM/Akita mice, there were no differences in whole‐body bone mass between double and single HBM mutants. Likewise, analysis of whole‐body fat and lean mass by DXA showed no significant differences among groups at the different time points (Figure 1C,D ). However, direct postmortem assessment of fat depots revealed significantly lower gonadal and retroperitoneal WAT accumulation in both Akita and double mutants relative to the other two groups (Figure 2 A, Figure 1, Supplemental Figure S1 C). By contrast, BAT mass was significantly higher in HBM/Akita mice relative to both WT and Akita groups; median BAT mass was also higher in HBM than in WT, but the groups did not significantly differ (Figure 2B ). Histological sections of BAT revealed denser cellularity and reduced number of lipid droplet‐containing cells in HBM and HBM/Akita (Figure 2D,E ), relative to WT BAT (Figure 2C ). By contrast, BAT isolated from Akita mice was almost entirely constituted by lipid containing adipocytes (Figure 2F ). Thus, the HBM mutation increases BAT mass and prevents BAT whitening in Akita mice.
Fig. 1.
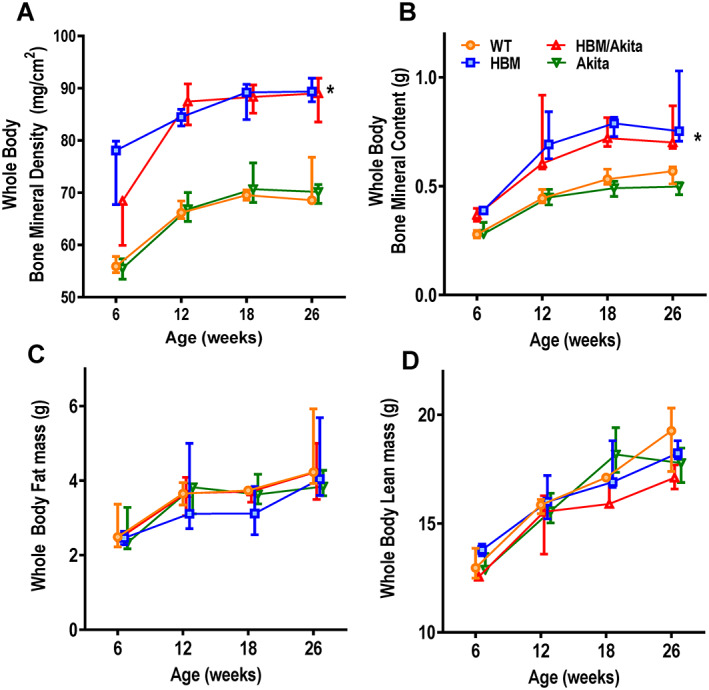
Body composition analysis. (A) Whole‐body bone mineral density, (B) bone mineral content, (C) fat mass, and (D) lean mass measured by DXA at different ages in the different mutants. n = 3–7 at 6 weeks, 6–11 at 12 weeks, n = 3–6 at 18 weeks, n = 5–7 at 26 weeks. Asterisk indicates p < .001 for HBM and HBM/Akita versus WT and Akita at all time points, except for HBM/Akita at 6 weeks, when p > .10 versus all other groups (adjusted p values by Tukey's multiple comparison test after two‐way ANOVA). Abbreviations: ANOVA, analysis of variance; DXA, dual‐energy x‐ray absorptiometry; HBM, high bone mass; WT, wild‐type.
Fig. 2.
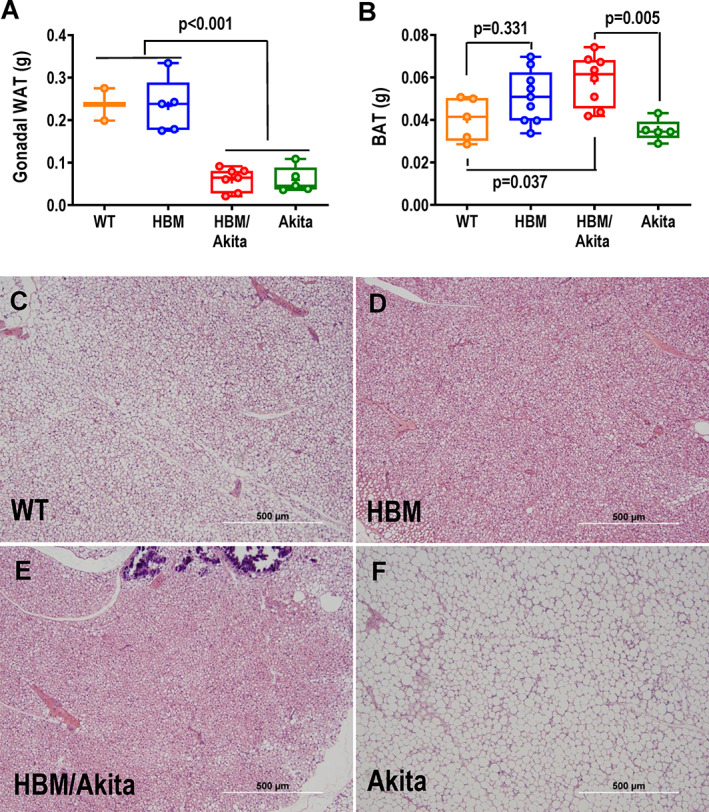
Fat tissue mass and histology. (A) Gonadal WAT, and (B) suprascapular BAT mass determined postmortem at 30 weeks of age. Brackets indicate adjusted p values (Tukey's multiple comparison test) after one‐way ANOVA; in A, brackets indicate comparison of all groups on one side with all groups on the other side. (C–F) H&E‐stained sections of BAT representative of the four genotype groups at 30 weeks of age. Abbreviations: ANOVA, analysis of variance; BAT, brown adipose tissue; H&E, hematoxylin and eosin; WAT, white adipose tissue.
The Lrp5A214V mutation improves bone microarchitecture and preserves bone strength in the Akita background
In vivo analysis of bone microarchitecture by μCT confirmed higher volumetric trabecular bone volume/total volume (BV/TV) and BMD in both HBM and HBM/Akita mutants relative to WT and Akita at 6 and 20 weeks of age (Figure 3A–C ). Of note, the difference between WT and HBM groups was significant only at 20 weeks, reflecting increasing BV/TV and BMD with age in HBM and HBM/Akita mice, relative to minimal changes in WT and Akita mice (Figure 3B,C ). Accordingly, trabecular thickness was higher and trabecular spacing lower in both HBM groups relative to WT and Akita (Figure 3, Supplemental Figure S2A,B ). Also, trabecular thickness increased with age in both HBM groups, without changes in trabecular spacing, consistent with bone mass accrual with age in these groups. Again, only minor age‐related trends were observed in trabecular thickness and spacing in the WT and Akita groups (Figure 3, Supplemental Figure 2A,B ). As for trabecular bone, cortical thickness and cortical bone area were also higher in both HBM mutants relative to both WT and Akita groups at both ages and increased in all group with age (Figure 3 D, Figure 3, Supplemental Figure S2C ). Total cortical area, representing bone cross‐sectional size, was higher in HBM than in WT at 6 weeks, but it was not different between HBM/Akita and Akita mutants; however, both HBM groups did not experience the decline in total cortical area that occurred in WT and Akita, resulting in larger cross‐sectional area in the HBM mutants at 20 weeks (Figure 3E ). Consistent with increased cortical thickness, medullary area decreased with age in all groups, without evident genotype effect (Figure 3, Supplemental Figure S2D ). Tissue mineral density, which represents the degree of mineralization, was higher in trabecular bone of both HBM groups at 6 weeks, but this difference disappeared at 20 weeks; and both trabecular and cortical tissue mineral density increased with age in all groups rather uniformly (Figure 3, Supplemental Figure S2E,F ).
Fig. 3.
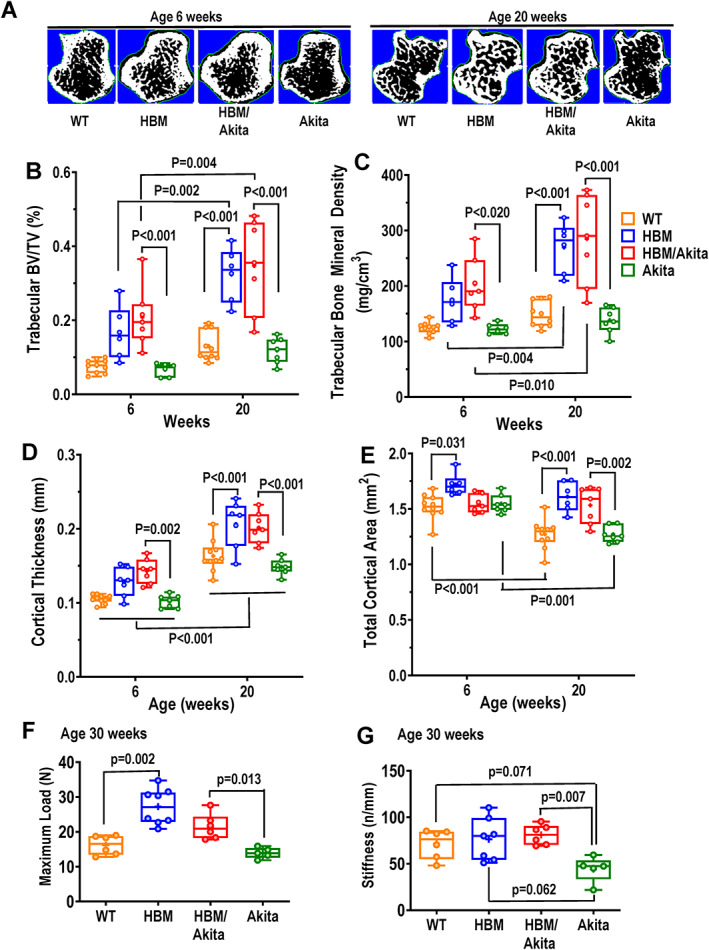
Bone microarchitecture and biomechanics. (A) Cross‐sections of proximal tibias from in vivo μCT scans representative of the four experimental groups at 6 and 20 weeks of age. (B) Volumetric trabecular BV/TV, (C) trabecular bone mineral density, (D) cortical thickness, and (E) total cortical area in the four genotype groups at two different ages. (F) Maximum load, and (G) stiffness derived from three‐point bending analysis of femurs at 30 weeks of age. Brackets indicate adjusted p values (Tukey's multiple comparison test) after two‐way ANOVA (B–E), or one‐way ANOVA (F,G). Brackets on multiple groups (D) indicate pairwise comparison of each genotype group at 6 and 20 weeks. Pairwise comparisons with p > .10 are not indicated. Abbreviations: μCT, micro–computed tomography; ANOVA, analysis of variance; BV/TV, bone volume/total volume.
We then assessed bone biomechanical properties in femurs from a subset of mice followed until age 30 weeks. As anticipated, maximum load in a three‐point bending was significantly higher in HBM relative to WT bones, and in HBM/Akita relative to Akita mice, whereas no difference was observed between Akita and WT mice (Figure 3F ). On the other hand, bone stiffness was significantly lower in Akita compared to all other groups, although the difference was below the significance threshold only relative to HBM/Akita double mutants, most likely because of low power (Figure 3G ). Microarchitectural parameters determined ex vivo in these 30‐week‐old mice subjected to bone strength testing confirmed high volumetric BV/TV and BMD, cortical thickness and bone area in HBM and HBM/Akita relative to the other groups, but no significant differences were noted between Akita and WT (Figure 3, Supplemental Figure S3A–D ).
The Lrp5A214V mutation delays the onset of hyperglycemia and glucose intolerance in Akita mice
We monitored glucose homeostasis to validate the Akita model and correlate with the skeletal phenotype. Although blood glucose in WT and HBM mice was normal and stable over the entire observation period (18 weeks), Akita mice, as expected, developed hyperglycemia (blood glucose ≥300 mg/dl) between 4 and 6 weeks of age on standard diet. Compound HBM/Akita mice became hyperglycemic by 7 weeks of age in average, and the degree of hyperglycemia was slightly milder than Akita mice, with significantly lower random blood glucose at multiple time points within the first 12 weeks of age (Figure 4A ). Accordingly, the proportion of HBM/Akita mice with random blood glucose ≥300 mg/dl at 6 weeks was 30%, compared with 90% of Akita mice, whereas all HBM/Akita mice were hyperglycemic by 12 weeks (Figure 4B ). Such difference was even clearer when blood glucose was measured after an overnight fast; although all Akita mice were frankly hyperglycemic relative to the other groups at 6 weeks of age, the HBM/Akita double mutants showed normal or slightly elevated overnight fasting blood glucose compared to the nondiabetic groups at this age (Figure 4C ). Thus, the HBM mutation delays the onset of hyperglycemia in Akita mice.
Fig. 4.
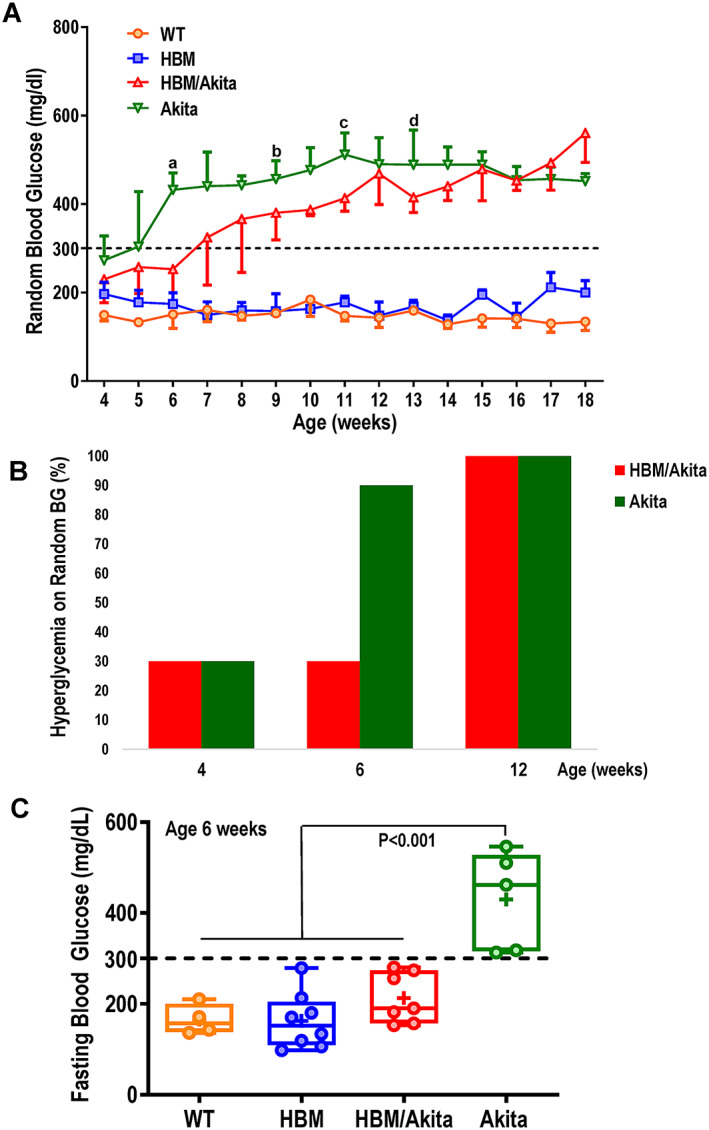
Blood glucose. (A) Random blood glucose (median and interquartile range) measured weekly in the four genotype groups (n = 4–16 per group at each time point). a p = .025, b p = .095, c p = .007, d p = .052 versus HBM/Akita (adjusted p values by Tukey's multiple comparison test after mixed‐effect analysis). Akita higher than WT and HBM at all time points (p < .01). (B) Percentage of mice with hyperglycemia (random glucose>300 mg/dl) in HBM and Akita groups at 4, 6, and 12 weeks of age. (C) Fasting blood glucose at 6 weeks of age (n = 4–8/genotype). Brackets indicate adjusted p values (Tukey's multiple comparison test) after one‐way ANOVA. Abbreviations: ANOVA, analysis of variance; HBM, high bone mass; WT, wild‐type.
ipGTT at 6 weeks of age confirmed reduced glucose tolerance in Akita relative to WT and HBM mice. Glucose tolerance was more variable but significantly better in HBM/Akita relative to Akita mice, although the double mutants remained hyperglycemic up to 2 hours after glucose load (Figure 5A ). By contrast, at 8 weeks of age glucose tolerance was equally compromised in both HBM/Akita and Akita mice (Figure 5B ), consistent with fasting hyperglycemia in both groups at this age (Figure 4A ). Comparison of areas under the curve (AUC) for the ipGTT confirmed delayed development of glucose intolerance in the double HBM/Akita mutants, revealing a larger spread of individual data points in both Akita and double mutants relative to the other two groups at 6 weeks (Figure 5C), but tighter clustering toward higher AUC values at 8 weeks (Figure 5D ). Notably, at 6 weeks, more than half of HBM/Akita had ipGTT AUC within the normal range, suggesting preserved glucose tolerance in a large proportion of double mutants at this age (Figure 5C ). In these experiments, HBM mice, which are heterozygous for the Lrp5 A214V mutation, exhibited normal glucose tolerance (Figure 5A,B ). However, homozygous Lrp5 A214V/A214V mutants showed significantly lower blood glucose relative to WT littermates 15 and 30 min after a glucose load (Figure 5, Supplemental Figure S4). The HBM group showed an intermediate response to glucose, suggesting that stronger Lrp5‐mediated signaling may favor glucose utilization in otherwise healthy conditions.
Fig. 5.
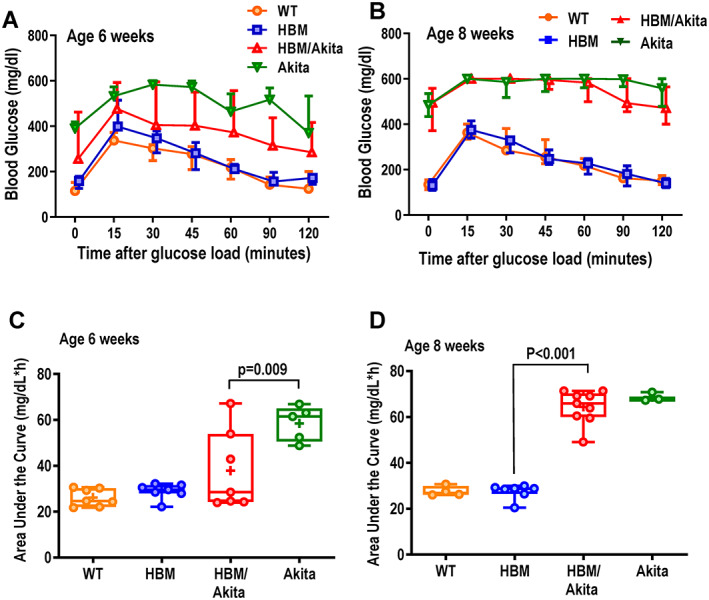
ipGTT. (A,B) Blood glucose before and after an intraperitoneal glucose load (1.5 g/kg) in the four genotype groups at age 6 (WT, HBM, and HBM/Akita, n = 7; Akita, n = 5) and 8 weeks (WT, Akita, n = 4; HBM, n = 7; HBM/Akita, n = 9). (At 6 weeks: p > .10 at all time points for HBM/Akita relative to all the other groups; p < .01 for Akita relative to WT and HBM [except 120 min, when p = .018, and p = .027, respectively]; at 8 weeks: p < .001 for Akita and HBM/Akita vs. WT and HBM at all time points [two‐way ANOVA and Tukey's multiple comparison test]). (C,D) Area under the curve for blood glucose during the ipGTT at 6 and 8 weeks of age. Brackets indicate adjusted p values (Tukey's multiple comparison test) after one‐way ANOVA. Pairwise comparisons with p > .10 are not indicated. Abbreviations: ANOVA, analysis of variance; HBM, high bone mass; ipGTT, intraperitoneal glucose tolerance test; WT, wild‐type.
The Lrp5A214V mutant ameliorates insulin responsiveness in Akita mice without altering insulin production or signaling and independently of osteocalcin
We then performed intraperitoneal ITT (ipITT) in the four groups at 7 weeks, when most of the double mutant mice are not yet hyperglycemic, and at 30 weeks, after a prolonged diabetic status. At 7 weeks, HBM/Akita mice, though already borderline hyperglycemic, exhibited a strong response to insulin with a fast decrease in blood glucose, which remained within or just above the normal range for at least 2 hours after insulin administration. Although hyperglycemic throughout the test, Akita mice also responded to insulin with a significant decline in blood glucose at 60 min, suggesting that they are not insulin insensitive at this age (Figure 6A ). At 30 weeks of age, Akita mice showed essentially no response to insulin, consistent with the notion that prolonged hyperglycemia eventually impairs insulin sensitivity. By contrast, insulin was able to decrease blood glucose by more than 30% in the HBM/Akita, even though these mice remained frankly hyperglycemic (Figure 6B ). AUC analysis confirmed that at 7 weeks the Akita group was significantly less responsive to insulin relative to all the other groups, including the HBM/Akita group where a wider spread of data points was observed (Figure 6C ). At 30 weeks both Akita and double mutants were significantly less insulin sensitive relative to the non‐Akita groups (Figure 6D ).
Fig. 6.
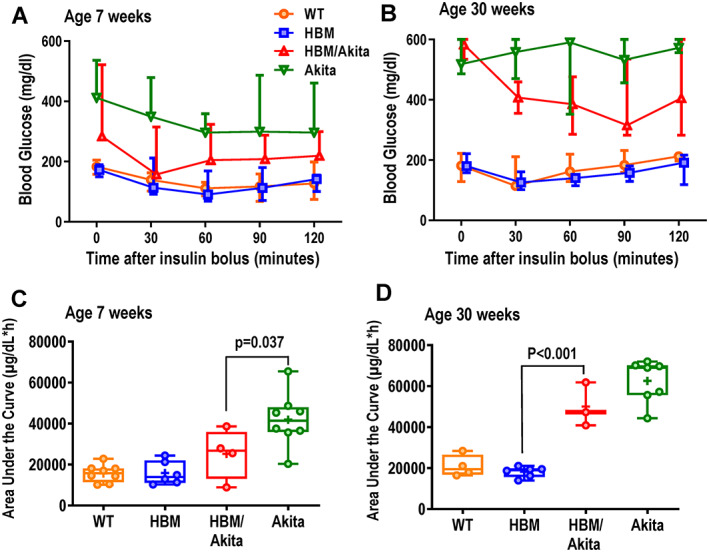
ipITT. (A,B) Blood glucose before and after intraperitoneal injection of insulin (0.5 U/kg) in the four genotype groups at age 7 weeks (WT, Akita, n = 8; HBM, n = 6; Akita, HBM/Akita, n = 4) and 30 weeks (WT, n = 3; HBM, n = 6; Akita, n = 7; HBM/Akita, n = 3). (At 6 weeks: p > .10 at all time points for HBM/Akita relative to all the other groups; p < .01 for Akita relative to WT and HBM at time points; at 8 weeks: p < .001 for Akita and HBM/Akita vs. WT and HBM at all time‐points [two‐way ANOVA and Tukey's multiple comparison test]). (C,D) Area under the curve for blood glucose during the ITT at 7 and 30 weeks of age. Brackets indicate adjusted p values (Tukey's multiple comparison test) after one‐way ANOVA. Pairwise comparisons with p > .10 are not indicated. Abbreviations: ANOVA, analysis of variance; HBM, high bone mass; ipITT, intraperitoneal insulin tolerance test; WT, wild‐type.
To obtain insights on the potential mechanisms by which onset of hyperglycemia is delayed in HBM/Akita mutants, we first measured glucose regulating hormones at 6 weeks of age, when hyperglycemia has developed in Akita, but not in the double HBM/Akita mutants (Figure 4B ). Median fasting serum insulin was lower in HBM relative to WT mice, but statistical analysis showed no differences. As expected, serum insulin increased in both groups 30 min after a glucose load. Conversely, both Akita and HBM/Akita mice showed undetectable serum insulin at time 0 or 30 min after glucose load (Figure 7A ). Serum C‐peptide followed a similar pattern, with overlapping baseline values in HBM and WT groups, trends to increase after glucose load, and very low levels in both Akita groups at time 0 and 30 min after glucose load (Figure 7B ). Circulating glucagon was very low in WT and HBM groups, but it was higher in Akita mice before the glucose load, consistent with their hyperglycemia. Notably, serum glucagon was widely variable among the HBM/Akita and Akita groups, with about half of the animals showing frankly elevated levels and a decrease after glucose load only in HBM/Akita mice (Figure 7C ). Two other circulating glucose regulating factors were determined. Serum IGF‐1 was highly variable in WT and HBM groups, with a decrease after glucose load in WT; and it was barely detectable in both Akita and HBM/Akita groups (Figure 7D ). Baseline total osteocalcin, measured at age 28 to 30 weeks, was lower in both Akita and HBM/Akita mice relative to WT and HBM groups (Figure 7E ); and Glu13 (undercarboxylated) osteocalcin, which has been proposed as a regulator of glucose homeostasis,( 41 ) followed a similar trend (Figure 7F ), although higher variability precluded rejection of the hypothesis zero of no differences among groups (F = 2.594; p = .083). Gla13 (carboxylated) osteocalcin was not different among groups (Supplemental Figure S4B ). Of note, similar levels of total and Glu13 osteocalcin were observed in WT and HBM mice.
Fig. 7.
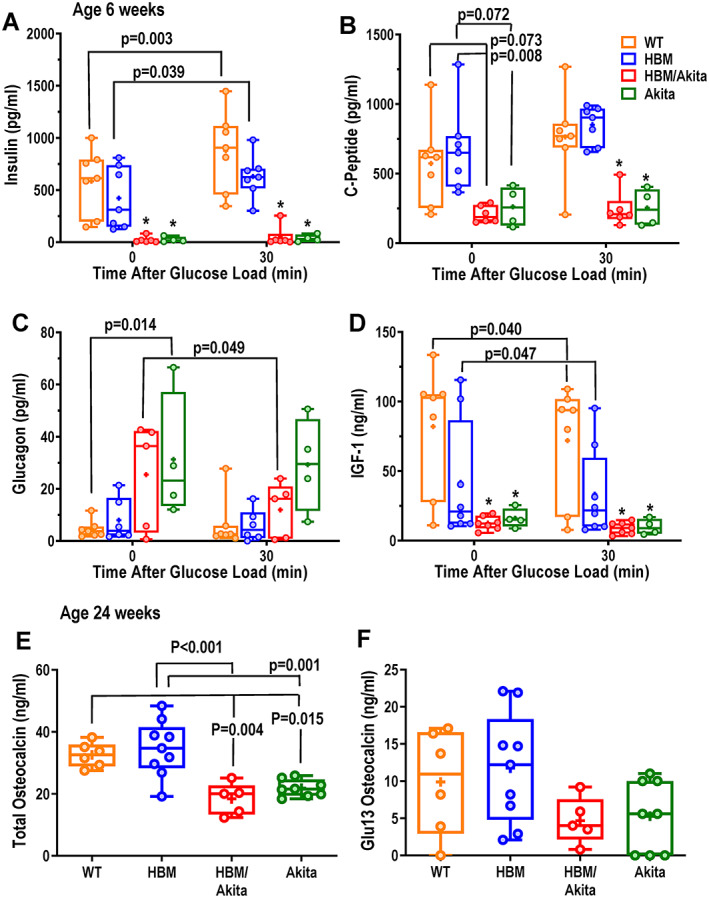
Glucose regulating hormones and osteocalcin. (A) Serum insulin, (B) C‐peptide, (C) glucagon and (D) IGF‐1 measured before (time 0) and 30 min after a glucose load (i.p. dextrose, 1.5 mg/g) in 6‐week‐old mice. (E) Serum total and (F) decarboxylated (Glu13) osteocalcin in the four genotype groups at age 28–30 weeks. Brackets indicate adjusted p values (Tukey's multiple comparison test) after two‐way ANOVA; *p < .01 versus WT and HBM at either time point. Pairwise comparisons with p > .10 are not indicated. Abbreviations: ANOVA, analysis of variance; HBM, high bone mass; IGF‐1, insulin‐like growth factor 1; WT, wild‐type.
Because the Lrp5 A214V mutant is globally expressed, we asked whether it may affect insulin production by pancreatic beta cells. Direct measurement of total insulin content in pancreas at 6 weeks of age confirmed barely detectable insulin in any of the Akita or HBM/Akita double mutant mice at this age, further validating the Akita model; notably, pancreatic insulin content was significantly lower in HBM compared to WT mice (Figure 8A ). Finally, we tested insulin responsiveness in extraskeletal and bone cells using pAkt as a readout. In adipocyte cultures obtained from ear mesenchymal stem cells (EMSCs) grown in adipogenic medium, insulin rapidly stimulated Akt phosphorylation to the same extent in both WT and HBM cells, as evidenced by the appearance of strong pAkt bands at 5 and 30 min after insulin exposure in immunoblots, without changes in total Akt abundance (Figure 8B ). Identical results were obtained in three replicate experiments using cells from different animals (Figure 8C , Supplemental Figure S5A,B ). Consistently, bone marrow stromal cells (BMSCs) from either WT or HBM mice showed similar pAkt activation upon insulin exposure, though to a less extent than EMSC‐derived adipocytes (Supplemental Figure 5C ).
Fig. 8.
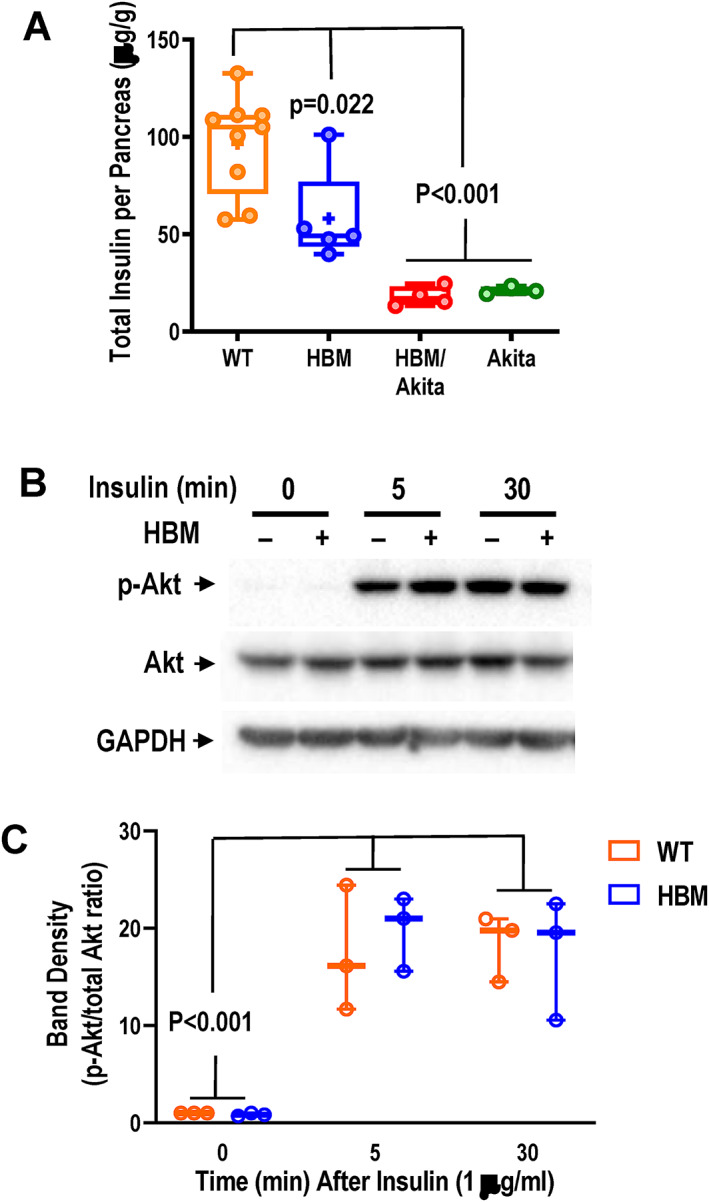
Insulin production and signaling. (A) Total pancreatic insulin content assessed by ELISA in whole pancreas homogenates of 6‐week‐old mice. Brackets indicate adjusted p values (Tukey's multiple comparison test) after one‐way ANOVA. (B) Immunoblots of whole‐cell lysates from ear mesenchymal stem cell–derived adipocyte cultures before and after 5 or 30 min of exposure to 1 μg/ml of insulin. (C) Densitometric analysis of immunoblot bands shown in B and Supplemental Figure S5A,B , expressed as pAKT/total Akt ratio. Brackets indicate adjusted p‐values (Tukey's multiple comparison test) after one‐way ANOVA (time, F = 33.82, p < .001; genotype, F = 0.057, p = .815; interaction, F = 0.252, p = .781. Abbreviations: ANOVA, analysis of variance; ELISA, enzyme‐linked immunosorbent assay; pAKT, phosphorylated protein kinase B.
Discussion
Our study demonstrates that the Lrp5 A214V mutation results in high bone mass and improves bone microarchitecture and bone strength despite prolonged insulin‐deficient diabetes in mice. We also show that this mutation delays the onset of diabetes and ameliorates glucose tolerance, primarily via insulin‐independent mechanisms. Thus, our work suggests that therapeutic agents based on Wnt signaling activation should be useful in improving bone fragility in patients with diabetes and may have the added benefit of also improving energy metabolism in early stages of the disease.
Despite the severe metabolic imbalance, we found no detectable postnatal developmental or growth defects in Akita mice, at least up to age 20 weeks. This is consistent with previous observations,( 42, 43 ) although an age‐related reduction in body weight has been reported in older Akita mice.( 44, 45 ) We did not detect differences in whole‐body bone mass or microarchitectural abnormalities in Akita relative to WT mice. Indeed, bone mass in the HBM/Akita compound mutants was as high as in the HBM mutants, and trabecular and cortical bone continued to accrue through at least 20 weeks of age just like in the HBM single mutants. However, others have shown that the effect of hyperglycemia on whole‐body bone density is slow and may become evident only at 1 year of age.( 46 ) Lack of densitometric or microstructural abnormalities may be considered a limitation of the Akita model; and lack of insulitis is also a difference with the human disease. On the other hand, with 100% penetrance in males and longtime survival, Akita mice represent an excellent platform to study the effect of prolonged hyperglycemia and insulin deficiency on bone. Indeed, we did detect decreased stiffness in 30‐week‐old Akita bones, suggesting that prolonged hyperglycemia alters bone material properties rather than bone mass, microarchitecture, or mineralization, probably by abnormal glycosylation of matrix proteins.( 47, 48 ) More to the point, conserved bone stiffness and improved bone strength in HBM/Akita relative to Akita mice suggest that the Lrp5 A214V mutation fully overrides any effects of prolonged hyperglycemia and insulin deficiency on bone mass and strength. Intriguingly, high bone mass accrual with the Lrp5 A214V mutation in the Akita background occurs with lower circulating osteocalcin, a marker of bone formation. Such observation is consistent with accumulated clinical evidence that bone formation is reduced in T1DM( 7 ); it also implies that the Lrp5 A214V mutation is more effective in decreasing bone resorption than in counteracting the effect of hyperglycemia on bone formation, to account for the high bone mass. Consistent with this conclusion, in clinical trials the anti‐sclerostin antibody, romosozumab transiently stimulates bone formation and reduces bone resorption, but continuous bone mass accrual with prolonged treatment occurs with reduced bone formation and resorption.( 16 )
Other mouse genetic models demonstrate a positive action of Wnt signaling on glucose metabolism. Sost knockout mice have improved glucose tolerance and insulin sensitivity and reduced white adipose depots, leading to the suggestion of an endocrine function of sclerostin, which is assumed to be produced only in bone.( 26 ) By contrast, Lrp5 knockout mice have markedly impaired glucose tolerance and decreased glucose‐induced insulin secretion.( 25 ) As expected, Lrp5 A214V and Sost −/− mice share many similarities in glucose homeostasis, because both mutations result in enhanced Wnt signaling. Glucose tolerance is improved in both, although this is evident only with homozygous Lrp5 A214V/A214V mice in a nondiabetic background. Insulin sensitivity is improved in Sost −/− mice, and the Lrp5 A214V mutation also improves insulin sensitivity in a diabetic context, even though it does not affect insulin signaling in bone cells. Furthermore, the reduced pancreatic insulin content in Lrp5 A214V mutants is consistent with reduced beta cell area and size observed in Sost −/− mice.( 26 ) The reason for reduced insulin content in Wnt signaling gain‐of‐function mutants remains elusive. Intriguingly, insulin‐dependent glucose uptake is increased in Sost −/− mice,( 26 ) suggesting lower insulin requirement. There are also differences between the two models; unlike the reduced WAT depots in Sost −/− mice, the Lrp5 A214V mutation does not alter body adiposity. Further, Akt activation in response to insulin is not altered in cultured Lrp5 A214V adipocytes and BMSC relative to WT cells, whereas enhanced insulin signaling was reported in Sost knockout cells.( 26 ) Lack of effect of Lrp5 A214V on WAT mass may be explained by recent findings that sclerostin favors adiposity via binding to Lrp4( 49 ); thus, this endocrine action of sclerostin may not require canonical, Lrp5‐mediated Wnt signaling. Of note, the Lrp5 A214V mutation renders the receptor sclerostin‐insensitive,( 28 ) but not capable of ligand‐independent signaling; therefore, our data would support the hypothesis that improved glucose metabolism is related to resistance to the inhibitory action of sclerostin in different peripheral tissues, thus reinforcing the notion that sclerostin acts in an endocrine fashion to regulate glucose metabolism and adiposity.
Although insulin‐stimulated glucose utilization is improved in HBM/Akita mice, delayed onset of diabetes cannot be mediated by increased insulin production or sensitivity, because not only is insulin abundance reduced in the pancreas of HBM mice, but HBM/Akita mutants remain able to respond to glucose load for 2 to 3 weeks after their beta cell functionality is completely lost. Thus, an insulin‐independent mechanism allows Lrp5 A214V mutants to support glucose homeostasis for the first few weeks after insulin failure, though such mechanism is obviously not sufficient to prevent insulin‐deficient diabetes. Undercarboxylated osteocalcin can function as a glucose regulatory factor,( 50, 51 ) and circulating osteocalcin has been reported elevated in subjects with gain‐of‐function LRP5 mutations.( 12 ) Although we have not measured osteocalcin at the onset of the metabolic abnormalities, the lower‐than‐normal osteocalcin in HBM/Akita mice is the opposite of what would be expected if osteocalcin were the mechanism of improved glucose tolerance in these mice. Likewise, circulating undercarboxylated osteocalcin and adiponectin are normal in Sost −/− mice.( 27 ) The surprisingly larger BAT depots in HBM/Akita mutants, in the face of decreased WAT, and more so, prevention of BAT whitening by the Lrp5 A214V mutation in the Akita background, may hold the key to this unexpected finding. Transplantation of normal BAT into streptozotocin‐induced diabetic mice restores euglycemia and normal glucose tolerance, and reduces inflammation, independently of insulin.( 52 ) Increased production of IGF‐1 was proposed as a mechanism; however, IGF‐1 was normal in our Lrp5 A214V mutants, even after glucose load, and it was undetectable in both Akita and double mutants. In Sost −/− mice, BAT mass is normal, although genes associated with adipocyte browning or beiging, such as Ucp‐1 are upregulated in WAT.( 27 ) Therefore, it is possible that Wnt signaling may enhance the release of other adipokines or “batokines” with a positive effect on glucose metabolism.
In summary, this study demonstrates that Wnt signaling may provide a common thread between bone and energy metabolism. Activated Wnt signaling improves bone mass, microarchitecture and strength in insulin‐deficient diabetes and has positive effects on glucose homeostasis. Diabetes leads to cardiovascular complications, and because there are concerns that romosozumab, an anti‐sclerostin antibody, also increases the risk of cardiovascular events, sclerostin inhibition should be considered in subjects with diabetes without complications.
Disclosures
All authors have nothing to disclose.
Peer review
The peer review history for this article is available at https://publons.com/publon/10.1002/jbmr.4303.
Supporting information
Supplemental Figure S1 Post‐natal Growth and WAT Mass. (A) Body weight, and (B) length at different ages in the different mutants. N = 11–16 at 4 weeks and 4–6 at 18 weeks. p > .05 by 2‐ way ANOVA for genotype effect in both measures. (C) Retroperitoneal white adipose tissue (WAT) mass determined post‐mortem at 30 weeks of age. Brackets represent adjusted p‐values (Tukey's multiple comparison test) after one‐way ANOVA, comparing all groups on one side with all groups on the other side.
Supplemental Figure S2. Bone Microarchitecture in Vivo. (A) Trabecular thickness, (B) trabecular spacing, (C) cortical bone area, (D) cortical medullary area, (E) trabecular tissue mineral density, and (D) cortical tissue mineral density assessed by in vivo μCT in the 4 genotype groups at 6 and 20 weeks of age. Brackets represent adjusted p‐values (Tukey's multiple comparison test) after two‐way ANOVA. Brackets on multiple groups (C‐F) indicate pairwise comparison of each genotype group at 6 and 20 weeks. Pairwise comparisons with p > .10 are not indicated.
Supplemental Figure S3. Bone Microarchitecture Post‐Mortem. (A) Volumetric trabecular bone volume/total volume (BV/TV), (B) trabecular bone mineral density, (C) cortical thickness, and (D) cortical bone area in the 4 genotype groups at 30 weeks of age measured by μCT. Brackets represent adjusted p‐values (Tukey's multiple comparison test) after one‐way ANOVA..
Supplemental Figure S4. Intraperitoneal Glucose Tolerance Test and Serum Osteocalcin. (A) Blood glucose before and after an intraperitoneal glucose load (1.5 mg/kg) at age 10–12 weeks (n = 7–10). *p = .0192 for WT vs. Lrp5A214V/A214V; two‐way ANOVA and Bonferroni post‐hoc test. (B) Serum Gla13 (carboxylated) osteocalcin in ***‐week‐old mice. F = 1.257, p = .312 (ANOVA).
Supplemental Figure S5. Insulin Signaling. (A, B) Two replicates of immunoblots of whole cell lysates from external ear‐derived adipocyte cultures before and after 5 or 30 minutes of exposure to 1 μg/mL of insulin. Quantitative band densitometry is shown in Figure 8. (C) Immunoblot of bone marrow stromal cell lysates before and after 5 or 30 minutes of exposure to 20 μg/mL of insulin.
Appendix S1. Supporting Information.
Acknowledgments
This work was supported by a grant from Società Italiana per l'Osteoporosi e le Malattie del Metabolismo Minerale e Scheletrico (SIOMMMS; to Rocky Strollo), from Fondazione Diabete Ricerca‐AMD and Società Italiana di Diabetologia (AMD‐SID; to Giulia Leanza), and by a grant from the Barnes‐Jewish Foundation (to Roberto Civitelli), and National Institutes of Health (NIH) DK098584 (to Maria S. Remedi). Further support was provided by National Institutes of Health (NIH) P30 AR057235 (Core Center for Musculoskeletal Biology and Medicine). We are grateful to Yung Kim and Michael Brodt for their assistance with the bone biomechanics experiments and analysis.
Author contributions: Giulia Leanza: Conceptualization; data curation; funding acquisition; investigation; methodology; project administration; writing‐original draft. Francesca Fontana: Conceptualization; data curation; formal analysis; investigation; methodology; supervision. Seung‐Yon Lee: Data curation; formal analysis; investigation; methodology. Maria Remedi: Data curation; formal analysis; investigation; supervision. Celine Schott: Data curation; investigation. Mathieu Ferron: Data curation; methodology; supervision; writing‐review & editing. Malcolm Hamilton‐Hall: Data curation; formal analysis; visualization. Yael Alippe: Data curation; investigation; methodology. Rocky Strollo: Conceptualization; funding acquisition; supervision. Nicola Napoli: Conceptualization; data curation; funding acquisition; project administration; supervision; writing‐review & editing. Roberto Civitelli: Conceptualization; data curation; formal analysis; funding acquisition; project administration; supervision; writing‐original draft; writing‐review & editing.
Data availability statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.
References
- 1.Sellmeyer DE, Civitelli R, Hofbauer LC, Khosla S, Lecka‐Czernik B, Schwartz AV. Skeletal metabolism, fracture risk, and fracture outcomes in type 1 and type 2 diabetes. Diabetes. 2016;65(7):1757‐1766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Napoli N, Strollo R, Paladini A, Briganti SI, Pozzilli P, Epstein S. The alliance of mesenchymal stem cells, bone, and diabetes. Int J Endocrinol. 2014;2014:690783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Weber DR, Haynes K, Leonard MB, Willi SM, Denburg MR. Type 1 diabetes is associated with an increased risk of fracture across the life span: a population‐based cohort study using The Health Improvement Network (THIN). Diabetes Care. 2015;38(10):1913‐1920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Napoli N, Conte C, Pedone C, et al. Effect of insulin resistance on BMD and fracture risk in older adults. J Clin Endocrinol Metab. 2019;104(8):3303‐3310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schwartz AV, Vittinghoff E, Bauer DC, et al. Association of BMD and FRAX score with risk of fracture in older adults with type 2 diabetes. JAMA. 2011;305(21):2184‐2192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Armas LA, Akhter MP, Drincic A, Recker RR. Trabecular bone histomorphometry in humans with type 1 diabetes mellitus. Bone. 2012;50(1):91‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Starup‐Linde J, Vestergaard P. Biochemical bone turnover markers in diabetes mellitus ‐ a systematic review. Bone. 2016;82:69‐78. [DOI] [PubMed] [Google Scholar]
- 8.Farlay D, Armas LA, Gineyts E, Akhter MP, Recker RR, Boivin G. Nonenzymatic glycation and degree of mineralization are higher in bone from fractured patients with type 1 diabetes mellitus. J Bone Miner Res. 2016;31(1):190‐195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Saito M, Fujii K, Mori Y, Marumo K. Role of collagen enzymatic and glycation induced cross‐links as a determinant of bone quality in spontaneously diabetic WBN/Kob rats. Osteoporos Int. 2006;17(10):1514‐1523. [DOI] [PubMed] [Google Scholar]
- 10.Tanaka K, Yamaguchi T, Kanazawa I, Sugimoto T. Effects of high glucose and advanced glycation end products on the expressions of sclerostin and RANKL as well as apoptosis in osteocyte‐like MLO‐Y4‐A2 cells. Biochem Biophys Res Commun. 2015;461(2):193‐199. [DOI] [PubMed] [Google Scholar]
- 11.Leanza G, Maddaloni E, Pitocco D, et al. Risk factors for fragility fractures in type 1 diabetes. Bone. 2019;125:194‐199. [DOI] [PubMed] [Google Scholar]
- 12.Boyden LM, Mao J, Belsky J, et al. High bone density due to a mutation in LDL‐receptor‐related protein 5. N Engl J Med. 2002;346(20):1513‐1521. [DOI] [PubMed] [Google Scholar]
- 13.Little RD, Carulli JP, Del Mastro RG, et al. A mutation in the LDL receptor‐related protein 5 gene results in the autosomal dominant high‐bone‐mass trait. Am J Hum Genet. 2002;70(1):11‐19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Napoli N, Chandran M, Pierroz DD, et al. Mechanisms of diabetes mellitus‐induced bone fragility. Nat Rev Endocrinol. 2017;13(4):208‐219. [DOI] [PubMed] [Google Scholar]
- 15.Niziolek PJ, Farmer TL, Cui Y, Turner CH, Warman ML, Robling AG. High‐bone‐mass‐producing mutations in the Wnt signaling pathway result in distinct skeletal phenotypes. Bone. 2011;49(5):1010‐1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.McClung MR, Grauer A, Boonen S, et al. Romosozumab in postmenopausal women with low bone mineral density. N Engl J Med. 2014;370(5):412‐420. [DOI] [PubMed] [Google Scholar]
- 17.Hygum K, Starup‐Linde J, Harslof T, Vestergaard P, Langdahl BL. Mechanisms in endocrinology: diabetes mellitus, a state of low bone turnover ‐ a systematic review and meta‐analysis. Eur J Endocrinol. 2017;176(3):R137‐R157. [DOI] [PubMed] [Google Scholar]
- 18.Garcia‐Martin A, Rozas‐Moreno P, Reyes‐Garcia R, et al. Circulating levels of sclerostin are increased in patients with type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(1):234‐241. [DOI] [PubMed] [Google Scholar]
- 19.Gaudio A, Privitera F, Battaglia K, et al. Sclerostin levels associated with inhibition of the Wnt/beta‐catenin signaling and reduced bone turnover in type 2 diabetes mellitus. J Clin Endocrinol Metab. 2012;97(10):3744‐3750. [DOI] [PubMed] [Google Scholar]
- 20.Hie M, Iitsuka N, Otsuka T, Tsukamoto I. Insulin‐dependent diabetes mellitus decreases osteoblastogenesis associated with the inhibition of Wnt signaling through increased expression of Sost and Dkk1 and inhibition of Akt activation. Int J Mol Med. 2011;28(3):455‐462. [DOI] [PubMed] [Google Scholar]
- 21.Portal‐Nunez S, Lozano D, de Castro LF, de Gortazar AR, Nogues X, Esbrit P. Alterations of the Wnt/beta‐catenin pathway and its target genes for the N‐ and C‐terminal domains of parathyroid hormone‐related protein in bone from diabetic mice. FEBS Lett. 2010;584(14):3095‐3100. [DOI] [PubMed] [Google Scholar]
- 22.Gennari L, Merlotti D, Valenti R, et al. Circulating sclerostin levels and bone turnover in type 1 and type 2 diabetes. J Clin Endocrinol Metab. 2012;97(5):1737‐1744. [DOI] [PubMed] [Google Scholar]
- 23.Piccoli A, Cannata F, Strollo R, et al. Sclerostin regulation, microarchitecture, and advanced glycation end‐products in the bone of elderly women with type 2 diabetes. J Bone Miner Res. 2020;35(12):2415‐2422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kato M, Patel MS, Levasseur R, et al. Cbfa1‐independent decrease in osteoblast proliferation, osteopenia, and persistent embryonic eye vascularization in mice deficient in Lrp5, a Wnt coreceptor. J Cell Biol. 2002;157(2):303‐314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Fujino T, Asaba H, Kang MJ, et al. Low‐density lipoprotein receptor‐related protein 5 (LRP5) is essential for normal cholesterol metabolism and glucose‐induced insulin secretion. Proc Natl Acad Sci U S A. 2003;100(1):229‐234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Kim SP, Frey JL, Li Z, et al. Sclerostin influences body composition by regulating catabolic and anabolic metabolism in adipocytes. Proc Natl Acad Sci U S A. 2017;114(52):E11238‐E11247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Li X, Ominsky MS, Niu QT, et al. Targeted deletion of the sclerostin gene in mice results in increased bone formation and bone strength. J Bone Miner Res. 2008;23(6):860‐869. [DOI] [PubMed] [Google Scholar]
- 28.Cui Y, Niziolek PJ, MacDonald BT, et al. Lrp5 functions in bone to regulate bone mass. Nat Med. 2011;17(6):684‐691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Niziolek PJ, MacDonald BT, Kedlaya R, et al. High bone mass‐causing mutant LRP5 receptors are resistant to endogenous inhibitors in vivo. J Bone Miner Res. 2015;30(10):1822‐1830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yoshioka M, Kayo T, Ikeda T, Koizumi A. A novel locus, Mody4, distal to D7Mit189 on chromosome 7 determines early‐onset NIDDM in nonobese C57BL/6 (Akita) mutant mice. Diabetes. 1997;46(5):887‐894. [DOI] [PubMed] [Google Scholar]
- 31.Wang MW, Wei S, Faccio R, et al. The HIV protease inhibitor ritonavir blocks osteoclastogenesis and function by impairing RANKL‐induced signaling. J Clin Invest. 2004;114(2):206‐213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Szczesny G, Veihelmann A, Massberg S, Nolte D, Messmer K. Long‐term anaesthesia using inhalatory isoflurane in different strains of mice‐the haemodynamic effects. Lab Anim. 2004;38(1):64‐69. [DOI] [PubMed] [Google Scholar]
- 33.Grimston SK, Goldberg DB, Watkins M, Brodt MD, Silva MJ, Civitelli R. Connexin43 deficiency reduces the sensitivity of cortical bone to the effects of muscle paralysis. J Bone Miner Res. 2011;26(9):2151‐2160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Di Benedetto A, Watkins M, Grimston S, et al. N‐cadherin and cadherin 11 modulate postnatal bone growth and osteoblast differentiation by distinct mechanisms. J Cell Sci. 2010;123(Pt 15):2640‐2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Silva MJ, Ulrich SR. In vitro sodium fluoride exposure decreases torsional and bending strength and increases ductility of mouse femora. J Biomech. 2000;33(2):231‐234. [DOI] [PubMed] [Google Scholar]
- 36.Wang Z, York NW, Nichols CG, Remedi MS. Pancreatic beta cell dedifferentiation in diabetes and redifferentiation following insulin therapy. Cell Metab. 2014;19(5):872‐882. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ayala JE, Samuel VT, Morton GJ, et al. Standard operating procedures for describing and performing metabolic tests of glucose homeostasis in mice. Dis Model Mech. 2010;3(9‐10):525‐534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Remedi MS, Friedman JB, Nichols CG. Diabetes induced by gain‐of‐function mutations in the Kir6.1 subunit of the KATP channel. J Gen Physiol. 2017;149(1):75‐84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ferron M, Wei J, Yoshizawa T, Ducy P, Karsenty G. An ELISA‐based method to quantify osteocalcin carboxylation in mice. Biochem Biophys Res Commun. 2010;397(4):691‐696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Rim JS, Mynatt RL, Gawronska‐Kozak B. Mesenchymal stem cells from the outer ear: a novel adult stem cell model system for the study of adipogenesis. FASEB J. 2005;19(9):1205‐1207. [DOI] [PubMed] [Google Scholar]
- 41.Ferron M, Wei J, Yoshizawa T, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell. 2010;142(2):296‐308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maresch CC, Stute DC, Ludlow H, et al. Hyperglycemia is associated with reduced testicular function and activin dysregulation in the Ins2(Akita+/−) mouse model of type 1 diabetes. Mol Cell Endocrinol. 2017;446:91‐101. [DOI] [PubMed] [Google Scholar]
- 43.Han Z, Guo J, Conley SM, Naash MI. Retinal angiogenesis in the Ins2(Akita) mouse model of diabetic retinopathy. Invest Ophthalmol Vis Sci. 2013;54(1):574‐584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Coe LM, Zhang J, McCabe LR. Both spontaneous Ins2(+/−) and streptozotocin‐induced type I diabetes cause bone loss in young mice. J Cell Physiol. 2013;228(4):689‐695. [DOI] [PubMed] [Google Scholar]
- 45.Vastani N, Guenther F, Gentry C, et al. Impaired nociception in the diabetic Ins2(+/Akita) mouse. Diabetes. 2018;67(8):1650‐1662. [DOI] [PubMed] [Google Scholar]
- 46.Botolin S, McCabe LR. Bone loss and increased bone adiposity in spontaneous and pharmacologically induced diabetic mice. Endocrinology. 2007;148(1):198‐205. [DOI] [PubMed] [Google Scholar]
- 47.Rubin MR, Paschalis EP, Poundarik A, et al. Advanced glycation endproducts and bone material properties in type 1 diabetic mice. PLoS One. 2016;11(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Picke AK, Campbell G, Napoli N, Hofbauer LC, Rauner M. Update on the impact of type 2 diabetes mellitus on bone metabolism and material properties. Endocr Connect. 2019;8(3):R55‐R70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kim SP, Da H, Li Z, et al. Lrp4 expression by adipocytes and osteoblasts differentially impacts sclerostin's endocrine effects on body composition and glucose metabolism. J Biol Chem. 2019;294(17):6899‐6911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Wei J, Ferron M, Clarke CJ, et al. Bone‐specific insulin resistance disrupts whole‐body glucose homeostasis via decreased osteocalcin activation. J Clin Invest. 2014;124(4):1‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Lacombe J, Al Rifai O, Loter L, et al. Measurement of bioactive osteocalcin in humans using a novel immunoassay reveals association with glucose metabolism and beta‐cell function. Am J Physiol Endocrinol Metab. 2020;318(3):E381‐E391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gunawardana SC, Piston DW. Reversal of type 1 diabetes in mice by brown adipose tissue transplant. Diabetes. 2012;61(3):674‐682. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Figure S1 Post‐natal Growth and WAT Mass. (A) Body weight, and (B) length at different ages in the different mutants. N = 11–16 at 4 weeks and 4–6 at 18 weeks. p > .05 by 2‐ way ANOVA for genotype effect in both measures. (C) Retroperitoneal white adipose tissue (WAT) mass determined post‐mortem at 30 weeks of age. Brackets represent adjusted p‐values (Tukey's multiple comparison test) after one‐way ANOVA, comparing all groups on one side with all groups on the other side.
Supplemental Figure S2. Bone Microarchitecture in Vivo. (A) Trabecular thickness, (B) trabecular spacing, (C) cortical bone area, (D) cortical medullary area, (E) trabecular tissue mineral density, and (D) cortical tissue mineral density assessed by in vivo μCT in the 4 genotype groups at 6 and 20 weeks of age. Brackets represent adjusted p‐values (Tukey's multiple comparison test) after two‐way ANOVA. Brackets on multiple groups (C‐F) indicate pairwise comparison of each genotype group at 6 and 20 weeks. Pairwise comparisons with p > .10 are not indicated.
Supplemental Figure S3. Bone Microarchitecture Post‐Mortem. (A) Volumetric trabecular bone volume/total volume (BV/TV), (B) trabecular bone mineral density, (C) cortical thickness, and (D) cortical bone area in the 4 genotype groups at 30 weeks of age measured by μCT. Brackets represent adjusted p‐values (Tukey's multiple comparison test) after one‐way ANOVA..
Supplemental Figure S4. Intraperitoneal Glucose Tolerance Test and Serum Osteocalcin. (A) Blood glucose before and after an intraperitoneal glucose load (1.5 mg/kg) at age 10–12 weeks (n = 7–10). *p = .0192 for WT vs. Lrp5A214V/A214V; two‐way ANOVA and Bonferroni post‐hoc test. (B) Serum Gla13 (carboxylated) osteocalcin in ***‐week‐old mice. F = 1.257, p = .312 (ANOVA).
Supplemental Figure S5. Insulin Signaling. (A, B) Two replicates of immunoblots of whole cell lysates from external ear‐derived adipocyte cultures before and after 5 or 30 minutes of exposure to 1 μg/mL of insulin. Quantitative band densitometry is shown in Figure 8. (C) Immunoblot of bone marrow stromal cell lysates before and after 5 or 30 minutes of exposure to 20 μg/mL of insulin.
Appendix S1. Supporting Information.
Data Availability Statement
The data that support the findings of this study are available from the corresponding authors upon reasonable request.