

Abstract
Background
The coagulation factors (F)V and VIII are homologous proteins that support hemostasis through their regulation of FX activity. Hemophilia A (HA) patients have reduced FVIII activity and a prolonged bleeding time that is corrected through the administration of exogenous FVIII. Around one‐third of severe HA patients develop FVIII neutralizing antibodies, known as “inhibitors,” which neutralize FVIII activity and preclude them from further FVIII therapy.
Objectives
We hypothesized that, based on the degree of homology between FV and FVIII (~40%), FVIII‐neutralizing antibodies could cross react with FV. To test this hypothesis, a panel of recombinant, patient‐derived, FVIII‐neutralizing antibodies were screened for cross‐reactivity against FV.
Methods
Factor V and FVIII activity was measured using one‐stage clotting assays; structural analysis was carried out using a structural approach.
Results
We detected FV neutralizing activity with the anti‐FVIII A2 domain antibody NB11B2. Because this antibody was derived from an HA inhibitor patient, FV‐neutralizing activity was then evaluated in a number of HA inhibitor patient plasma samples; nine alloimmune samples had FV‐neutralizing activity whereas no FV neutralizing activity was found in the two autoimmune samples available. We next examined the degree of surface homology between FV and FVIII and found that structural similarity could explain the cross reactivity of the anti‐A2 antibody and likely accounts for the cross reactivity we observed in patient samples.
Conclusions
Although this novel observation is of interest, further work will be needed to determine whether FV neutralization in HA patient samples contributes to their bleeding diathesis.
Keywords: coagulation, coagulation factor V, coagulation factor VIII, FVIII inhibitors, hemostasis
Essentials.
The hypothesis that structural homology between FV and FVIII could lead to cross reactivity was tested.
Recombinant FVIII neutralizing antibodies (inhibitors) were screened for FV neutralizing activity.
Recombinant FVIII inhibitors neutralize FV; FV neutralization was also observed in FVIII inhibitor patient samples.
Structural similarity between FV and FVIII underpins cross reactivity and is measurable in FVIII inhibitor patient samples.
1. INTRODUCTION
Coagulation factors VIII (FVIII) and V (FV) share significant similarity with regard to structure and function. They are both large, Ca2+‐dependent plasma glycoproteins secreted as single inactive polypeptide chains that function as nonenzymic cofactors for serine proteases. FV and FVIII are proteolytically cleaved into heavy and light chains that are noncovalently associated by a Ca2+ ion. A summary of the similarities between FV and FVIII (and the related ceruloplasmin) is in Table 1.
TABLE 1.
Comparison of coagulation FV and FVIII and the related ceruloplasmin
| Feature | FV | FVIII | Ceruloplasmin |
|---|---|---|---|
| Synthesis (as inactive precursor) | Liver > endothelial cells > megakaryocytes ‐ also in platelet alpha granules | Liver > endothelial cells > megakaryocytes (also in platelet alpha granules) | Liver |
| Plasma concentration | 10 µg/ml (18) |
0.1–0.2 µg/ml (19) 0.3–2.6 nM (20) |
200–350 µg/ml |
| Structure | A1 ‐A2‐B‐A3‐C1‐C2 | A1‐A2‐B‐A3‐B‐C1‐C2 | A1‐A2‐A3 |
| Size |
Heavy chain = 709 aa, 150 kDa Light chain = 651 aa, 74 kDa |
Heavy chain = 740 aa Light chain = 684 aa |
2‐1065aa |
| Cations | Ca2+, Zn2+ | Ca2+, Zn2+ | Six Cu2+ per monomer |
| Proteolysis | Thrombin (FXa and plasmin) cleaves FV | Thrombin sites 391/392, 759/760, 1332/1333b, 1667/1668b, 1708/1709 | Nonea |
| Main function | FV increases FXa activity towards prothrombin (300 000‐fold over FXa alone) to increase the efficiency of thrombin generation | FVIII increases FIXa activity ~10 000‐fold | Copper carrying, iron metabolism (ferroxidase) |
| Sulfation | Essential for efficient proteolytic cleavage by thrombin and activation | Sulfation at Tyr1699 is essential for VWF binding | Nonea |
| Inactivation | aPC | aPC | |
| Signal peptides | 28 aa | 19 aa | 19 aa |
| Glycosylation | Extensive N‐ and O‐linked glycosylation | Extensive N‐ and O‐linked glycosylation | Six N‐linked modification |
| Disulfide bonds | 167–193, 248–329, 500–526, 603–684, 1725–1751, 1907–2061, 2066–2221 | 172–198, 267–348, 547–573, 649–730, 1851–1877, 1918–1922, 2040–2188, 2193–2345 | 174–200, 276–537, 534–560, 637–718, 874–900 |
Abbreviations: >(i.e., x > y) indicates that the level of protein expression in x is greater than y; aPC, activated protein C; F, factor.
Uniprot.
Activating cleavages.
Hemophilia A (HA) is a rare bleeding disorder that can be congenital or acquired. HA patients lack functional FVIII and are given exogenous FVIII products to restore hemostasis. A complication of congenital hemophilia is the alloimmune response to FVIII biotherapeutics and the development and circulation of FVIII‐neutralizing antibodies that preclude these “inhibitor” patients from further treatment with FVIII. Because of the structural similarity between FVIII and FV, we hypothesized that there may exist some cross reactivity between the FVIII‐neutralizing antibodies in HA inhibitor patient and FV.
A recombinant anti‐FVIII antibody panel, described previously,1 was used to test the novel research hypothesis that FVIII‐neutralizing antibodies can cross react with and neutralize FV owing to the high degree of homology that exists between these two proteins.
2. MATERIALS AND METHODS
2.1. Recombinant FVIII antibodies
In brief, the variable domains of heavy or light chain of patient‐derived FVIII‐neutralizing antibodies were cloned into an IgG4 framework and expressed in Chinese hamster ovary cells as recombinant antibodies. A full characterization of these antibodies has been described previously.1
2.2. Patient samples
Patient plasma samples from congenital (alloimmune) and acquired (autoimmune) patients were sourced from the archive at the Oxford Haemophilia Centre.
2.3. Measurement of FV activity by prothrombin time (PT) assay
Recombinant antibodies were diluted in TBS buffer to 10× final concentration before being added (1/10) to FVIII‐deficient plasma (F8DP, Siemens), which served as the FV source, and incubated for 2 h at 37°C. Prothrombin time was measured on an ACL Top 550 blood analyser. Siemens Innovin reagent was reconstituted and stored according to the manufacturer’s instructions. Patient plasma samples were either diluted in 4% human serum albumin or used neat, added 50:50 with F8DP (Siemens) as the source of FV, and incubated for 2 h at 37°C. Patient samples were then assessed for PT using Siemens Innovin reagent and FV‐deficient plasma (Siemens) on a Sysmex CS5100 blood analyzer. This in vitro assay, like the one‐stage clotting assay used to measure FVIII activity, is a turbidimetric endpoint assay where the time taken to clot is converted to % FV activity using a standard curve.
2.4. Calculating Bethesda titer
The FVIII‐neutralizing activity of FVIII‐neutralizing antibodies is reported in Bethesda units (BU)/ml (Bethesda titre) using the equation that follows. It is important that one chooses the dilution factor of patient plasma that gives closest to a 50% reduction in residual FVIII activity (ideally between 25% and 75%) to calculate the Bethesda titer. The higher the Bethesda titer, the more potent the FVIII neutralizing activity is.
2.5. FV modelling
A structure for FV was modelled using the RaptorX software.2, 3, 4 The heavy and light chains were processed separately, and, among the RaptorX database, chains A and B of pdb (Protein Data Bank) 4BDV5 were identified as suitable templates for the homology modelling, respectively. This confirms the similarity between FV and FVIII because pdb 4BDV is the crystal structure of a truncated B‐domain human FVIII. Modeller 9.246 was used to finally build the three‐dimensional models for FV light and heavy chains. FV structures were then aligned to pdb 4BDV using the MultiSeq environment,7 as implemented in VMD.8 More specifically, the STAMP algorithm9 was used for alignment, and the root mean square deviation (RMSD) and QH (QH ranges from 0 to 1 where QH = 1 refers to identical proteins) 10 values were used as indicators of structural similarity. FV pdb files are provided as Appendices S1 and S2.
2.6. Statistical analysis
The data in Figure 1A were analyzed by two‐way analysis of variance using multiple comparisons analysis with Tukey correction; p values of <.001 are shown with a double asterisk, whereas <.05 are shown with a single asterisk.
FIGURE 1.

Factor V cross reactivity can be detected with the A2‐specific, patient‐derived NB11B2 as well as patient samples. (A) Recombinant FVIII‐neutralizing antibodies were incubated with FV in F8DP for 2 h at 37°C before being assessed for prothrombin time on an ACL Top 550 blood analyzer (Werfen). Patient samples, diluted in 4% human serum albumin, were incubated with an equal volume of F8DP for 2 h at 37°C before FV activity (prothrombin time) or FVIII activity (chromogenic assay) was measured. Residual FV activity is shown in panel B, transformed data in panel C, and bar chart summaries in panel D. (B–D) FVIII activity is shown in black, FV activity is shown in red. (E–F) correlation between the strength of FV and FVIII neutralizing activity. F, factor
3. RESULTS AND DISCUSSION
3.1. Recombinant, patient‐derived FVIII neutralizing antibodies inhibit FV
To begin, PT was used to measure and quantify FV activity. As shown in Figure 1A, NB11B2 reduced FV activity at 1 µM and 0.3 µM, indicating that cross reactivity of this antibody with FV is possible. NB11B2 is an anti‐A2 domain antibody originally derived from an HA patient antibody isolated more than a decade ago.11 Although it is interesting that recombinant NB11B2 can neutralize FV activity when spiked into F8DP, whether FV neutralization would have been detectable in plasma samples from this patient is unknown; NB11B2 has an IC50 of 0.0001 µM for FVIII, so this interaction would likely dominate in vivo. To investigate FV neutralization in FVIII inhibitor patient samples, PT was initially measured in three alloimmune HA inhibitor patient plasma samples. These samples had FVIII Bethesda titers of 6 BU/ml, 11 BU/ml, and 115 BU/ml (the latter diluted to ~30 BU/ml), designated A, B, and C, respectively. NB11B2 (at a Bethesda titer of ~50 BU/ml) was included as a positive control; concentration‐dependent prolongation of PT (presented as % residual FV activity) was detected in all three patient samples (Figure 1B [raw data] and Figure 1C [transformed data]). To quantify the FV‐neutralizing activity, an FV Bethesda titer was calculated using the equation that is used to calculate FVIII Bethesda titers, substituting % residual FV activity for % residual FVIII activity. The FV Bethesda titer of A was 0.6 FV BU/ml, B had FV Bethesda titer of 0.2 FV BU/ml, and C diluted to 30 FVIII BU/ml had a FV Bethesda titer of 0.3 BU/ml. It was surprising to note that FV‐neutralizing activity was detectable in all three patient samples, and we decided to assess additional samples, including two derived from acquired HA inhibitor patients. As shown in Figure 1D, mild to moderate FV‐neutralizing activity was detected in all nine alloimmune HA inhibitor patients (solid bars), with FV Bethesda titers ranging from 0.1 to 1.2 FVBU/ml; no FV‐neutralizing activity was detectable in the acquired (autoimmune) samples (hashed bars). It was somewhat surprising that the strength of FV‐neutralizing activity did not correlate with the strength of the FVIII‐neutralizing activity (Figure 1E, F; R = 0.1125, slope not significantly different to zero). These novel findings suggest FVIII‐neutralizing antibodies can cross react with FV in vivo, but whether it has a significant impact on hemostasis in HA patients remains to be determined. The PT assay measures clotting time initiated by the extrinsic coagulation pathway. Although not a direct measure of FV activity, the number of molecules that could underpin the observations in this study are limited to tissue factor, FVII, FX, and FV. This leads us to conclude that the increase in PT observed with the FVIII‐neutralizing antibodies is likely due to FV neutralization, owing to the considerable homology between FV and FVIII. To gain further insight into these experimental findings, we next evaluated whether there was a structural basis for our observations. X‐ray crystallography data are available for FVIII but the structure for FV has only been partially solved. A structural approach was therefore used to generate a novel FV structure that could be used to compare the regions of FV and FVIII that correspond to the epitopes of the recombinant FVIII‐neutralizing antibodies used in this study (Figure 1A).
3.2. Structural similarity between FVIII and FV underpins FV cross reactivity
Using the protein structure prediction server RaptorX,2, 3, 4 which selects structural templates for a “query” protein sequence (in this case, FV) from an extensive database of experimentally solved structures in an unbiased manner, a novel FV structure was generated. Theoretical FVa structures had been generated previously and were based on experimentally derived data for ceruloplasmin A domains and the FV C2 domain.12, 13 More recently, the crystal structure of truncated B domain‐deleted human FVIII has been solved (PDB 4BDV5); the RaptorX server selected PBD 4BDV as a template for the FV model in an entirely unbiased manner. Indicators of the final model quality, including alignment score and p value, were favorable (Figure 2). This novel structure was then used to evaluate the degree of structural similarity between FV and FVIII to see whether it provided a rational basis for the cross reactivity that was observed in patient samples. The model was also used to evaluate the degree of similarity between epitopes to which the recombinant antibodies bind, as the epitopes are known. The RaptorX‐generated FV structure was aligned with FVIII (4BDV) using the STAMP algorithm (Figure 2A) and the QH parameter used to identify areas of similarity (blue – QH = 1) and dissimilarity (red – QH <0.3; Figure 2B). FV surface representations are also shown in Figure 2C (front) and Figure 2D (back) to highlight regions of similarity (gray) and dissimilarity (red) to FVIII. The prevalence of gray visually confirms the high degree of similarity between FV and FVIII and quantification using the RMSD and QH confirmed the model was of good quality; structural conservation between FV and FVIII is 87%–88%. This degree of similarity supports the level of cross reactivity that was observed in patient samples. Furthermore, the epitope reported for the parent antibody from which NB11B2 was derived11 maps broadly to FVIII residues 379–456 (A2 domain, Figure 2E), which display significant similarity in this FV model, and likely account for its cross reactivity with FV.
FIGURE 2.
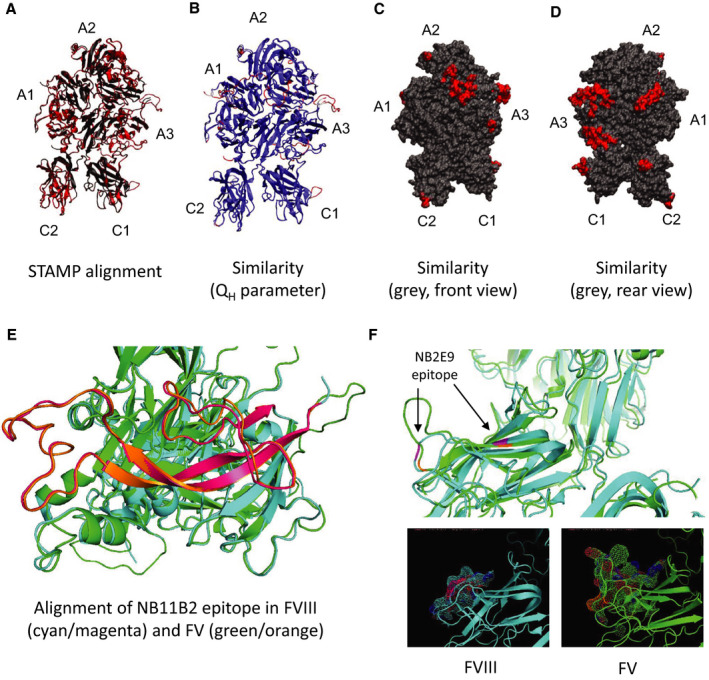
Structural similarity between FVIII and FV. (A) FV‐FVIII alignment using the STAMP algorithm,9 which works by minimizing the Cα distance between the aligned residues from each molecule (FV in red, FVIII in black). A score of 468.7 (range, 1–709) and a p value of 1.314e−40 was obtained for the FV heavy chains modelled against the chain A of pdb 4BDV, indicative of a good quality model (p values <10−3–10−4 are generally desired). The only exception was Pro686‐Arg737, which was omitted from the final structure because of poor fit. FV light chains modelled against chain B of pdb 4BDV had a score of 575.5 (range, 1–651) and the p value was 4.652e−43, which again was indicative of good‐quality modelling across the entire domain. (B) Alignment of FVIII and FV colored according to the QH (QH is a measure of similarity and ranges from 0 to 1 where QH = 1 refers to identical proteins) parameter, a measure of similarity between the aligned structures. QH ranges from 0 to 1 where QH =1 refers to identical proteins.10 Blue areas indicate structural similarity (QH =1) and areas of poor structural similarity (QH <0.3) are shown in red. Average values of QH of 0.8731 and 0.882 were obtained for the heavy and light chains, respectively. The RMSD between the two structures was 0.7135 nm for the heavy chains and 0.6236 nm for the light chains. (C–D) Surface representation of FV, where the areas showing poor similarity with FVIII are colored in red. (C) is a front view, while (D) is a rear view. (E) Residues implicated in FVIII binding for the parent antibody of NB11B2. The A2 domain is shown in cyan for FVIII and green for FV, and the residues involved in antibody binding are shown in magenta for FVIII and orange for FV. (F) Residues implicated in FVIII binding for the parent antibody of NB2E9. The C1 domain is shown in cyan for FVIII and green for FV, and the residues involved in antibody binding are shown in magenta for FVIII and orange for FV. F, factor; pdb, Protein Data Bank; RMSD, root mean square deviation
With regard to the recombinant antibodies that lacked cross reactivity, their epitopes lie in areas of the FVIII molecules that lack similarity to FV (regions of FV showing the lowest similarity to FVIII are summarized in Table 2). For example, the FVIII C1 “spike” K2092‐F2093 epitope for NB3314 is absent from our FV model (it maps into residues Y1984‐L1985 in FV). Furthermore, the lack of FV neutralization by NB2C11 also fits with this model; X‐ray crystallography studies identified the C2 domain spike M2199‐F2200 as supporting the FVIII‐antibody interaction and in FV, the corresponding C2 spike instead comprises two sequential tryptophan (W2091‐W2092, which also lie in a region of high dissimilarity; see Table 2) residues,15 explaining the lack of cross reactivity. For NB41, its epitope was mapped to the FVIII a3‐A3‐C1 region and defined as R1803‐K181816 and in an area described as dissimilar to FV (residues K1679‐E1696 of FV in this model; see Table 2). Finally, NB2E9 is derived from a patient that neutralized wild‐type exogenous FVIII, but not their endogenous FVIII, which had a mutation at R2150.17 A second residue, E2066, is also implicated in binding of this antibody and is positioned in a solvent‐facing C1 loop (2064TKEPFSW2070). In this FV model, the corresponding R2150 aligns well (to R2042 in FV), but the residue corresponding to E2066 (E1951 in FV) resides in a much larger loop (1949SVEKLAAEFASKPW1962) that takes up considerably more space (Figure 2F) and may occlude R2042 (corresponding to R2150 in FVIII), accounting for the lack of neutralization. To summarize, by using this model to systematically evaluate the recombinant FVIII‐neutralizing antibody epitopes, it was possible to identify a structural basis for the experimental observations of this study. NB11B2 is the only recombinant antibody in this panel that increased PT and whose epitope could be mapped to an area of significant similarity, indicating a structural basis for the cross reactivity observed.
TABLE 2.
Regions of dissimilarity between an experimentally derived FVIII structure and modelled FV structure
| Heavy Chain (FV Against 4BDV) | Light Chain (FV Against 4BDV) |
|---|---|
| R46‐T59, turn/coil, A1 domain | S1574‐G1577, coil, A3 domain |
| Q226‐S228, turn, A1 domain | F1596‐D1612, turn/coil, A3 domain |
| K332‐H346, coil, A1‐A2 domain | K1679‐E1696, turn/coil, A3 domain |
| E458‐N460, alpha‐helix, A2 domain | K1787‐K1800, coil, A3 domain |
| K527‐I542, coil, A2 domain | K1952‐S1959, turn/coil, C1 domain |
| W2091‐G2093, turn/coil, C2 domain | |
| K2170‐V2174, turn, C2 domain |
Residue numbering and secondary structure pattern refer to FV.
Abbreviation: F, factor.
4. CONCLUSION
We report here for the first time that FVIII‐neutralizing antibodies from alloimmune (but not autoimmune) HA inhibitor patients exhibit FV‐neutralizing activity in the PT assay. Using a computationally derived structure for FV, structural characteristics were correlated with experimental data for the recombinant antibodies and a structural basis was found for these observations. Although these findings are novel and interesting, their bearing on the clinical presentation of HA patients remain to be seen: just because there is cross reactivity with FV does not mean that it will contribute to the clinical phenotype of the patient or necessitate any changes to how HA inhibitor patients are managed and treated. However, FV cross reactivity could account for disharmony between Bethesda titers and bleeding phenotype in some patients.
CONFLICT OF INTEREST
The authors declare no competing interests.
AUTHOR CONTRIBUTIONS
Carmen H. Coxon carried out and directed the majority of work and wrote the manuscript. James Beavis oversaw and facilitated clinical laboratory experiments. Andrea Arsiccio developed the factor V model. Sanj Raut supported and facilitated the research and contributed to writing of the manuscript. All authors critically reviewed the manuscript and approved the final version.
Supporting information
App S1
App S2
ACKNOWLEDGMENTS
The authors thank Dr. Nicola Curry for her support of this work and for her intellectual input with questions relating to the clinical management of hemophilia A patients.
Manuscript Handled by: Robert Gosselin
Final decision: Robert Gosselin, 16 April 2021
REFERENCES
- 1.Coxon CH, Yu X, Beavis J, et al. Characterisation and application of recombinant FVIII‐neutralising antibodies from haemophilia A inhibitor patients. Brit J Haematol. 2020. 10.1111/bjh.17227 (Epub ahead of print). [DOI] [PubMed] [Google Scholar]
- 2.Peng J, Xu J. Low‐homology protein threading. Bioinformatics. 2010;26(12):i294‐i300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ma J, Peng J, Wang S, Xu J. A conditional neural fields model for protein threading. Bioinformatics. 2012;28(12):i59‐i66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ma J, Wang S, Zhao F, Xu J. Protein threading using context‐specific alignment potential. Bioinformatics (Oxford, England). 2013;29(13):i257‐i265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Svensson LA, Thim L, Olsen OH, Nicolaisen EM. Evaluation of the metal binding sites in a recombinant coagulation factor VIII identifies two sites with unique metal binding properties. Biol Chem. 2013;394(6):761. [DOI] [PubMed] [Google Scholar]
- 6.Eswar N, Webb B, Marti‐Renom MA, et al. Comparative protein structure modeling using modeller. Curr Protoc Bioinformatics. 2006;15(1):5.6.1‐5.6.30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Roberts E, Eargle J, Wright D, Luthey‐Schulten Z. MultiSeq: unifying sequence and structure data for evolutionary analysis. BMC Bioinformatics. 2006;7(1):382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Humphrey W, Dalke A, Schulten K. VMD: visual molecular dynamics. J Mol Graph. 1996;14(1):33‐38. [DOI] [PubMed] [Google Scholar]
- 9.Russell RB, Barton GJ. Multiple protein sequence alignment from tertiary structure comparison: assignment of global and residue confidence levels. Proteins. 1992;14(2):309‐323. [DOI] [PubMed] [Google Scholar]
- 10.O'Donoghue P, Luthey‐Schulten Z. On the evolution of structure in aminoacyl‐tRNA synthetases. Microbiol Mol Biol Rev. 2003;67(4):550‐573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Jacquemin M, Gilles JG, Saint‐Remy J‐M. Antibodies binding to the A2 domain of FVIII and inhibiting coagulation activity. United States Patent. 2010;Patent;No. US7858089B2.
- 12.Pellequer JL, Gale AJ, Getzoff ED, Griffin JH. Three‐dimensional model of coagulation factor Va bound to activated protein C. Thromb Haemost. 2000;84(5):849‐857. [PubMed] [Google Scholar]
- 13.Orban T, Kalafatis M, Gogonea V. Completed three‐dimensional model of human coagulation factor Va. Molecular dynamics simulations and structural analyses. Biochemistry‐Us. 2005;44(39):13082‐13090. [DOI] [PubMed] [Google Scholar]
- 14.Bloem E, Van den Biggelaar M, Wroblewska A, et al. Factor VIII C1‐domain spikes 2092–2093 and 2158–2159 comprise regions that modulate cofactor function and cellular uptake. J Thromb Haemost. 2013;11:211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Gilbert GE, Novakovic VA, Kaufman RJ, Miao HZ, Pipe SW. Conservative mutations in the C2 domains of factor VIII and factor V alter phospholipid binding and cofactor activity. Blood. 2012;120(9):1923‐1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bovenschen N, Boertjes RC, van Stempvoort G, et al. Low density lipoprotein receptor‐related protein and factor IXa share structural requirements for binding to the A3 domain of coagulation factor VIII. J Biol Chem. 2003;278(11):9370‐9377. [DOI] [PubMed] [Google Scholar]
- 17.Jacquemin M, Benhida A, Peerlinck K, et al. A human antibody directed to the factor VIII C1 domain inhibits factor VIII cofactor activity and binding to von Willebrand factor. Blood. 2000;95(1):156‐163. [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
App S1
App S2