

Abstract
Prevention of hypertriglyceridemia is one of the biomedical targets in Glycogen Storage Disease type Ia (GSD Ia) patients, yet it is unclear how hypoglycemia links to plasma triglyceride (TG) levels. We analyzed whole‐body TG metabolism in normoglycemic (fed) and hypoglycemic (fasted) hepatocyte‐specific glucose‐6‐phosphatase deficient (L‐G6pc −/−) mice. De novo fatty acid synthesis contributed substantially to hepatic TG accumulation in normoglycemic L‐G6pc −/− mice. In hypoglycemic conditions, enhanced adipose tissue lipolysis was the main driver of liver steatosis, supported by elevated free fatty acid concentrations in GSD Ia mice and GSD Ia patients. Plasma very‐low‐density lipoprotein (VLDL) levels were increased in GSD Ia patients and in normoglycemic L‐G6pc −/− mice, and further elevated in hypoglycemic L‐G6pc −/− mice. VLDL‐TG secretion rates were doubled in normo‐ and hypoglycemic L‐G6pc −/− mice, while VLDL‐TG catabolism was selectively inhibited in hypoglycemic L‐G6pc −/− mice. In conclusion, fasting‐induced hypoglycemia in L‐G6pc −/− mice promotes adipose tissue lipolysis and arrests VLDL catabolism. This mechanism likely contributes to aggravated liver steatosis and dyslipidemia in GSD Ia patients with poor glycemic control and may explain clinical heterogeneity in hypertriglyceridemia between GSD Ia patients.
Keywords: Glycogen Storage Disease type Ia, hepatic steatosis, hypertriglyceridemia, metabolic control, translational research

Synopsis.
This work shows that fasted hepatocyte‐specific G6pc knockout mice show a concomitant increase in VLDL‐TG production and reduction in VLDL catabolism, resulting in more pronounced hypertriglyceridemia in hypoglycemic Glycogen Storage Disease type la.
1. INTRODUCTION
Glycogen Storage Disease type Ia (GSD Ia) is an inborn error of carbohydrate metabolism caused by a deficiency of the catalytic subunit (G6PC) of the glucose‐6‐phosphatase (G6Pase) complex G6PC, selectively expressed in liver, kidney, and intestine, which catalyzes the final step in gluconeogenesis and glycogenolysis by hydrolyzing glucose‐6‐phosphate to glucose and is the key enzyme for glucose homeostasis during fasting.1
GSD Ia patients clinically present with severe fasting intolerance and hepatomegaly, biochemically characterized by nonketotic hypoglycemia, hyperlactacidemia, hyperuricemia, hypercholesterolemia, hepatic steatosis, and hypertriglyceridemia.2, 3 Currently, dietary management still is the cornerstone of treatment for GSD patients to maintain normoglycemia and prevent secondary metabolic derangement (such as hyperlipidemia) as well as long‐term complications. Therefore, GSD Ia patients have to adhere to a strict diet, consisting of frequent meals with restriction of simple sugars, relatively small doses of uncooked cornstarch (UCCS) during the day and either UCCS doses or continuous (naso)gastric drip feeding through the night.3, 4, 5 Although strict dietary management and compliance have significantly improved outcomes,6, 7, 8 long‐term complications of GSD Ia,4, 7, 9, 10 among which the development of liver adenomas in young adulthood,9, 10, 11, 12 still occur.
Triglyceride (TG) concentrations are currently regarded as an important biomarker for metabolic control in GSD Ia patients.4, 13, 14 Importantly, “Uncertainties regarding optimal metabolic control both clinically and biochemically (like lactate, ketones, and/or lipids) in hepatic GSDs” is among the top research priorities that were recently established by the international GSD priority setting partnership.15 We previously reported large phenotypic heterogeneity in TG levels between GSD Ia patients (16, 17); however, the mechanisms underlying (variations in) hypertriglyceridemia in GSD Ia are as yet incompletely understood. Such insight requires an in‐depth analysis of the different physiological pathways involved in systemic TG metabolism. Mouse models for GSD Ia used in preclinical studies are commonly investigated under specific, controlled fasting conditions. Such studies at one hand limit transability of preclinical findings as dietary management in GSD Ia patients involves frequent intake of small meals, while they do not accommodate the extreme ends of the glycemic condition that GSD Ia patients face, that is, normoglycemia and severe hypoglycemia.
Because hypertriglyceridemia and poor glycemic control are associated with long‐term complications such as liver adenoma progression,14, 18, 19, 20 detailed understanding of the mechanistic link between glycemic control and hyperlipidemia is of great importance to further improve and personalize GSD Ia patient care. Previous work in tissue‐specific G6pc knockout mice have shown that hepatocyte‐specific deficiency of G6PC is the major determinant of hypoglycemia and hypertriglyceridemia in GSD Ia.21 In the current study, we therefore performed a systematic analysis of whole‐body TG metabolism in normoglycemic (fed) and in hypoglycemic (fasted) hepatocyte‐specific G6pc knockout (L‐G6pc −/−) mice. Our preclinical findings indicate that severe hypertriglyceridemia in GSD Ia patients with poor glycemic control is most likely due to impaired catabolism of TG‐rich lipoproteins.21
2. RESULTS
2.1. Hyperlipidemia and hepatic steatosis are aggravated in hypoglycemic L‐G6pc −/− mice
Fast protein liquid chromatography (FPLC) analysis revealed that excess TGs and cholesterol in serum of normoglycemic GSD Ia patients were mainly associated with VLDL/chylomicrons (Figure 1A, Table S1). Since hypoglycemia and hyperlipidemia in GSD Ia are caused by the loss of G6PC activity,21 we characterized TG metabolism in L‐G6pc −/− mice. Consistent with observations in patients, plasma TG and cholesterol levels were increased in fed (normoglycemic) L‐G6pc −/− mice as compared to wildtype (L‐G6pc +/+) littermates (Figure 1B, Table 1). Overnight fasting resulted in hypoglycemia in L‐G6pc −/− mice with additional increases in plasma TG levels (Figure 1B, Table 1). Under both conditions, L‐G6pc −/− mice exhibited increased lipid accumulation as compared to wild‐type controls (Figure 1C,E), due to the hepatic accumulation of TGs and cholesterol esters (CEs; Figures 1E and S1A,B). Similar to what was observed for plasma TG levels, the largest increase in hepatic TG and CE content was observed in hypoglycemic L‐G6pc −/− mice (Figures 1C,E and S1B). Hepatic G6P and glycogen contents were most markedly increased in hypoglycemic L‐G6pc −/− mice as compared to wild‐type controls (Table 1). Exacerbated liver steatosis of hypoglycemic L‐G6pc −/− mice was associated with an increase in hepatic lipid droplet area (Figure 1D). Oleate (C18:1) was the most abundant hepatic fatty acid in both normo‐ and hypoglycemic L‐G6pc −/− mice, with 3‐fold increases in its content in hypoglycemic compared to normoglycemic L‐G6pc −/− mice (Figure 1F, Table S2), reflecting the higher degree of TG and CE accumulation under hypoglycemic conditions. Linoleate (C18:2), an essential fatty acid that cannot be synthesized de novo, was selectively increased in hypoglycemic L‐G6pc −/− mice, while hepatic palmitate (C16:0) content was similar in L‐G6pc +/+ mice and L‐G6pc −/− mice (Figure 1F). These findings suggest that part of the hepatic fatty acids accumulated in hypoglycemic L‐G6pc −/− mice is derived from extrahepatic sources, and not from de novo lipogenesis.
FIGURE 1.
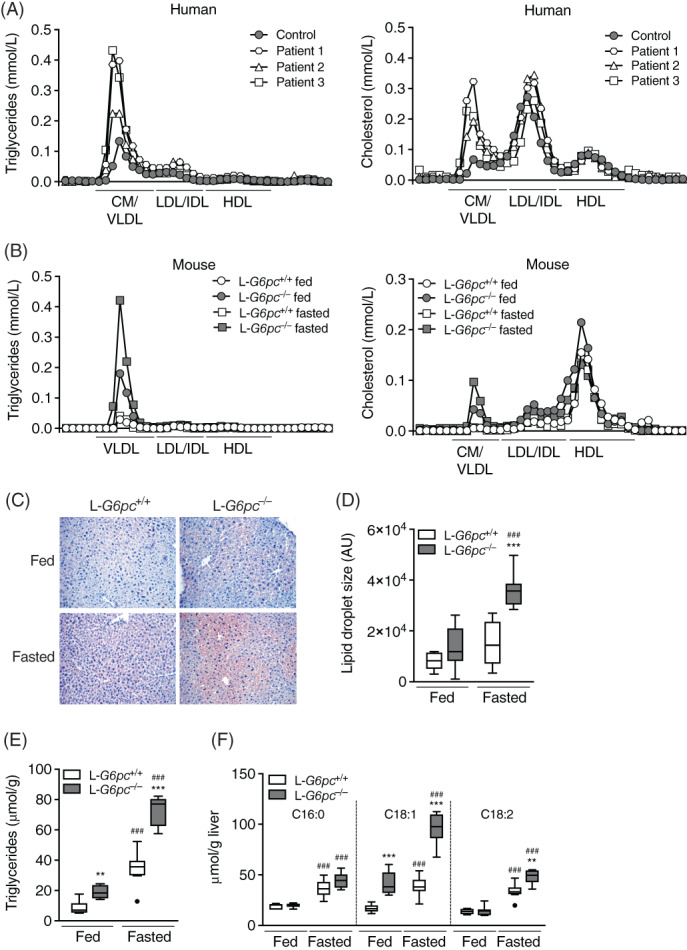
Hyperlipidemia and hepatic steatosis are aggravated in hypoglycemic L‐G6pc −/− mice. Triglyceride and total cholesterol concentrations in lipoprotein profiles of (A) GSD Ia patients (n = 3) and healthy control subject (n = 1) and (B) L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 1). C, Representative pictures of Oil‐red‐O staining and (D) quantification of lipid droplet area in livers of L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 6‐8). E, Hepatic triglyceride levels and (F) absolute values of hepatic total fatty acids in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 6‐8). GSD Ia, Glycogen Storage Disease type Ia
TABLE 1.
Plasma and liver characteristics in male L‐G6pc −/− mice and wild‐type littermates in fed state or after an overnight fast
| Parameter | L‐G6pc +/+ fed | L‐G6pc −/− fed | P value | L‐G6pc +/+ fasted | L‐G6pc −/− fasted | P value |
|---|---|---|---|---|---|---|
| Median (range) | Median (range) | Median (range) | Median (range) | |||
| Plasma | ||||||
| Glucose (mmol/L) | 8.6 (6.8‐9.5) | 6.5 (5.6‐9.9) | .126 | 5.0 (3.7‐8.4) | 2.1 (1.1‐2.4) | <.001 |
| Insulin (ng/mL) | 0.4 (0.3‐0.6) | 0.2 (0.1‐0.4) | .009 | 0.3 (0.1‐0.6) | 0.2 (0.1‐0.4) | .328 |
| Glucagon (pg/mL) | 113 (55‐163) | 148 (73‐320) | .429 | 113 (86‐132) | 236 (137‐582) | <.001 |
| Insulin/glucagon ratio | 3.5 (2.6‐5.8) | 1.1 (0.5‐3.2) | .017 | 3.0 (1.1‐4.7) | 0.9 (0.3‐1.8) | .003 |
| Lactate (mmol/L) | 3.2 (2.5‐4.0) | 4.4 (3.7‐4.7) | .052 | 1.8 (1.2‐3.3) | 2.1 (0.9‐3.9) | .328 |
| Triglycerides (mmol/L) | 0.6 (0.4‐0.7) | 1.7 (0.3‐2.7) | .082 | 0.5 (0.4‐0.6) | 3.5 (2.3‐8.3) | <.001 |
| Cholesterol (mmol/L) | 2.1 (1.6‐2.8) | 3.7 (1.7‐4.3) | .030 | 2.3 (2.0‐3.8) | 2.9 (1.6‐3.8) | .195 |
| Liver | ||||||
| G6P (nmol/g) | 406 (339‐505) | 1675 (819‐2306) | .004 | 422 (265‐483) | 2586 (1981‐3457) | <.001 |
| Glycogen (mg/g) | 51 (44‐57) | 66 (53‐77) | .017 | 18 (12‐30) | 54 (46‐62) | <.001 |
Note: Bold values indicate that p‐values <.05.
2.2. Differential contribution of de novo lipogenesis in normo‐ and hypoglycemic L‐G6pc −/− mice
It is generally assumed that increased glycolysis and de novo lipogenesis have major contributions to hepatic steatosis in GSD Ia. The enzymatic activities of the glycolytic enzymes GPI, ALD, GAPDH, L‐PK, and LDH were significantly increased in the liver of L‐G6pc −/− mice, irrespective of feeding state (Figure 2A, Table S2). To relate the activity of de novo lipogenesis to hepatic steatosis in normo‐ and hypoglycemic L‐G6pc −/− mice, we quantified the hepatic mRNA levels of transcription factors Srebp1c and Chrebpβ as well as of their glycolytic and lipogenic target genes (Figure 2B). Hepatic Srebp1c mRNA expression was lower in the hypoglycemic state but not affected by genotype, while Chrebpβ expression was 3‐fold induced in normoglycemic L‐G6pc −/− mice and 7‐fold in hypoglycemic L‐G6pc −/− mice compared to fed wild‐type controls. Similarly, the hepatic expression levels of L‐pk, Acaca, Fas, Elovl5, Elovl6, and Scd1 were increased under both conditions in L‐G6pc −/− mice as compared to wild‐type controls, with largest increments in hypoglycemic L‐G6pc −/− mice (Figure 2B).
FIGURE 2.
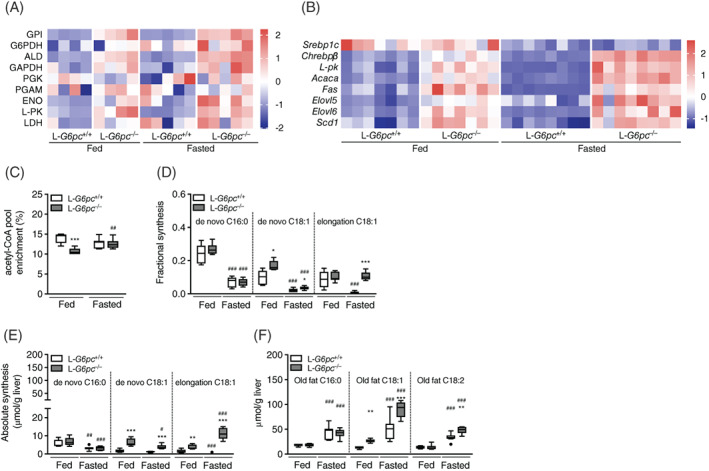
Differential contribution of de novo lipogenesis in fed and fasted L‐G6pc −/− mice. Heatmaps presenting Z‐score normalized (A) hepatic enzymatic activities and (B) hepatic gene expression levels in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 4‐8). C, Acetyl‐CoA pool enrichment and (D) fractional and (E) absolute de novo synthesis and chain elongation of palmitate (C16:0), oleate (C18:1) in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 5‐7). F, Hepatic palmitate (C16:0), oleate (C18:1), and linoleate (C18:2) derived from old fat in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 5‐8). Table S3 contains raw values and statistics for data presented in heatmaps
To establish the contribution of lipogenesis to hepatic steatosis in L‐G6pc −/− mice, we quantified hepatic de novo lipogenic fluxes using 13C‐acetate administered via drinking water during 24 hours prior to sacrifice. A significant reduction in acetyl‐CoA precursor pool enrichment was observed in normoglycemic L‐G6pc −/− mice, indicative of increased acetyl‐CoA turnover which is compatible with enhanced glycolysis (Figure 2C).22, 23 As expected, hepatic fractional de novo fatty acid synthesis of palmitate (C16:0) and oleate (C18:1), two major nonessential TG‐associated fatty acids, was significantly higher in fed compared to fasted wild‐type mice (Figure 2D). The accumulation of hepatic lipids in normo‐ and hypoglycemic L‐G6pc −/− mice was accompanied by an increase in fractional synthesis of oleate, while fractional synthesis of palmitate remained unchanged (Figure 2D). Absolute hepatic oleate synthesis via elongation of pre‐existing palmitate was increased in both normo‐ and hypoglycemic L‐G6pc −/− mice (Figure 2E), from which the latter was more pronounced. Similarly, fractional and absolute de novo synthesis of stearate (C18:0) were significantly increased in normoglycemic L‐G6pc −/− mice compared to wild‐type controls, while chain elongation was significantly increased in hypoglycemic L‐G6pc −/− mice only (Table S4). In terms of absolute hepatic fatty acid synthesis, lipid accumulation in normoglycemic L‐G6pc −/− mice mainly resulted from de novo oleate synthesis (Figure 2E). In contrast, elongation of pre‐existing palmitate to oleate was a major contributor of lipid accumulation in hypoglycemic L‐G6pc −/− mice (Figure 2E). The contribution of de novo lipogenesis to hepatic lipid accumulation in normoglycemic L‐G6pc −/− mice accounted for up to 20% (Figure 2D), while in hypoglycemic L‐G6pc −/− mice about 10% of the excess oleate synthesis was derived from elongation of pre‐existing palmitate (Figure 2D). Yet, the majority of excess hepatic fatty acids in both normo‐ and hypoglycemic L‐G6pc −/− mice was derived from “old fat” (Figure 2F, Table S2).
2.3. Adipose tissue lipolysis is enhanced in hypoglycemic L‐G6pc −/− mice
To investigate the origin of old fat accumulation in the liver of hypoglycemic L‐G6pc −/− mice, we analyzed alternative sources of hepatic TG input and output, that is, adipose tissue lipolysis, hepatic mitochondrial β‐oxidation and VLDL‐TG secretion and VLDL‐remnant uptake. Circulating nonesterified fatty acids (NEFAs), including oleate, were increased in both normo‐ and hypoglycemic L‐G6pc −/− mice compared to wild‐type controls, with highest concentrations in hypoglycemic L‐G6pc −/− mice (Figure 3A, Table S5). In normoglycemic GSD Ia patients, the concentrations of circulating NEFAs were also increased (Figure 3B, Table S5). Ex vivo glycerol release from adipose tissue was enhanced in hypoglycemic L‐G6pc −/− mice only (Figures 3C and S2A,B) and the phosphorylation of hormone sensitive lipase (HSL) on serine 563 was also increased in hypoglycemic L‐G6pc −/− mice compared to wild‐type littermates (Figure 3D‐F). In fasted L‐G6pc −/− mice, ex vivo glycerol release from adipose tissue positively correlated to pHSL S563 protein expression levels (Figure S2C). Adipose triglyceride lipase (ATGL) and perilipin (PLIN) protein levels in adipose tissue, essential for lipid mobilization, were not altered in L‐G6pc −/− mice (Figure 3F). Plasma levels of fibroblast growth factor 21 (FGF21) were increased in normo‐ and hypoglycemic L‐G6pc −/− mice as compared to wild‐type controls (Figure 3G). Increased adipose tissue lipolysis in fasted L‐G6pc −/− mice did not result in significant reductions in adipose tissue mass or whole‐body energy expenditure as compared to wild‐type controls (data not shown). Plasma ketone body concentrations were increased in both normo‐ and hypoglycemic L‐G6pc −/− mice as compared to wild‐type littermates, while total ketone body levels were significantly higher in hypo‐ vs normoglycemic L‐G6pc −/− mice (Figure 3H). Ex vivo hepatic mitochondrial β‐oxidation capacity was significantly increased in normo‐ and hypoglycemic L‐G6pc −/− mice (Figures 3I and S2D). Total hepatic acylcarnitine levels were not different between genotypes in fed or fasted state; however, C2 levels, the end product of β‐oxidation, were increased in hypoglycemic L‐G6pc −/− mice (Figure 3J, Table S6). Amino acid‐derived acylcarnitines C4 and C5 were increased in both normo‐ and hypoglycemic L‐G6pc −/− mice, while medium‐ and long‐chain acylcarnitines were not different (Table S6).
FIGURE 3.
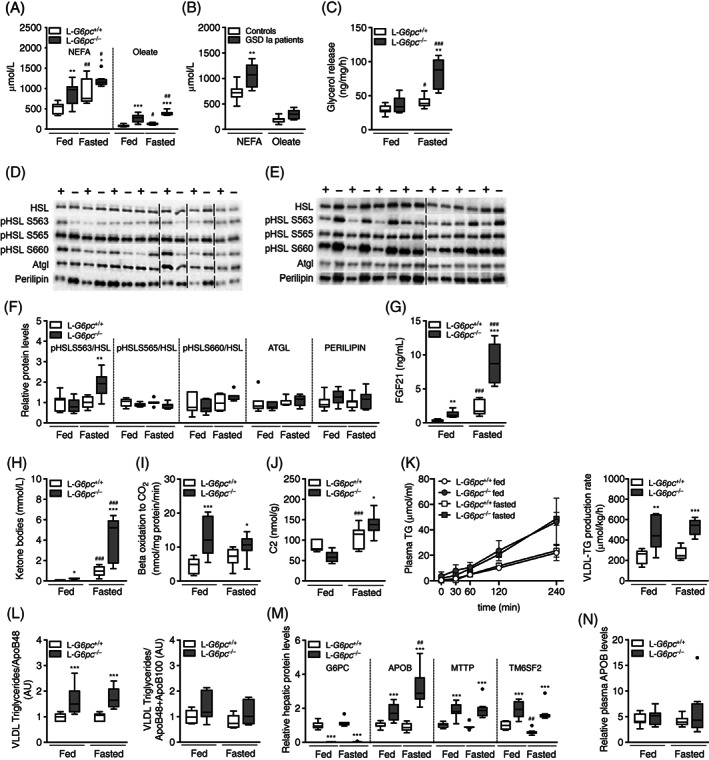
Adipose tissue lipolysis is induced in fasted L‐G6pc −/− mice. Plasma levels of nonesterified fatty acids (NEFAs) and oleate in (A) L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 5‐8) and in (B) normoglycemic GSD Ia patients and age‐ and sex‐matched controls (n = 7). C, Ex vivo glycerol release from adipose tissue isolated from L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 6‐7). Representative Western blots from adipose tissue of (D) fed and (E) fasted L‐G6pc −/− mice (indicated by −) and wild‐type littermates (indicated by +) for indicated proteins (n = 7), with boundaries between different sodium dodecyl sulfate poly acrylamide gel electrophoresis (SDS‐PAGE) gels indicated by vertical dashed lines. F, Quantification of Western blots for phosphorylated HSL at different sites normalized to total HSL, and ATGL and PLIN (n = 7). Plasma levels of (G) FGF21 and (H) total ketone bodies in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 5‐8). I, Ex vivo β‐oxidation capacity and (J) hepatic acylcarnitine C2 levels in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 5‐8). K, very‐low‐density lipoprotein‐triglyceride (VLDL‐TG) production curves (left) and production rates (right); L, Ratios of TG/ApoB48 (left) and TG/ApoB48+ApoB100 (right) in isolated VLDL as assessed by Western blot analysis (for full blots, see Figure S2E). M, Hepatic and (N) plasma protein levels assessed by targeted proteomics in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 6‐8), with APOB protein levels representing the sum of APOB48 and APOB100. ATGL, adipose triglyceride lipase; FGF21, fibroblast growth factor 21; GSD Ia, Glycogen Storage Disease type Ia; HSL, hormone sensitive lipase; PLIN, perilipin
To investigate whether altered hepatic VLDL‐TG secretion contributed to exacerbated liver steatosis or to hypertriglyceridemia in L‐G6pc −/− mice, we quantified hepatic VLDL‐TG secretion rates. VLDL‐TG secretion was doubled in L‐G6pc −/− mice as compared to controls, independent of feeding status (Figure 3K). The enhanced VLDL‐TG secretion rate in L‐G6pc −/− mice was associated with similar increases in the TG‐to‐Apolipoprotein B (ApoB) ratios, with a most marked effect on the TG‐to‐Apolipoprotein B48 ratio (Figures 3L and S2E). Hepatic ApoB protein and mRNA levels were elevated in normo‐ and hypoglycemic L‐G6pc −/− mice as compared to wild‐type controls (Figures 3M and S2F). Hepatic ApoB protein levels were further increased in hypoglycemic compared to normoglycemic L‐G6pc −/− mice (Figure 3M), while plasma ApoB levels were similar in L‐G6pc +/+ and L‐G6pc −/− mice under both conditions (Figure 3N). mRNA and protein levels of MTTP and TM6SF2, both involved in VLDL assembly, were increased in L‐G6pc −/− vs L‐G6pc +/+ mice, but unaffected by fasting (Figures 3M and S2F). Altogether, these data indicate that in acutely fasted L‐G6pc −/− mice, exacerbated liver steatosis was paralleled by enhanced adipose tissue lipolysis, but not by impaired β‐oxidation or reduced VLDL‐secretion.
2.4. VLDL catabolism is impaired in hypoglycemic L‐G6pc −/− mice
Because VLDL‐TG levels were increased in hypo‐ vs normoglycemic L‐G6pc −/− mice in the absence of an additional increase in VLDL‐TG secretion, we quantified VLDL catabolism in fed and fasted L‐G6pc −/− and L‐G6pc +/+ mice. In the fed state, the plasma decay of glycerol tri[3H]oleate‐labeled VLDL‐like particles was not different between L‐G6pc −/− mice and wild‐type littermates (Figure 4A,B). However, in the fasted state, elimination rate of labeled VLDL‐like particles was decreased by about 50% in L‐G6pc −/− mice compared to controls (Figure 4A). In the fasted state, uptake of TG‐derived fatty acids by muscle, epididymal white adipose tissue, and brown adipose tissue depots was significantly reduced (Figure 4B). VLDL‐TG catabolism is largely dependent on the activity of lipoprotein lipase (LPL), which is in turn controlled by apolipoproteins and angiopoietin‐like proteins.24, 25 Lpl mRNA levels in white adipose tissue were decreased in the fasted state yet were comparable in L‐G6pc −/− mice and wild‐type littermates (Figure 4C), while Angptl4 mRNA levels were unchanged (Figure 4C). Hepatic protein levels of APOC1 were unaffected in hypoglycemic L‐G6pc −/− mice while hepatic mRNA and protein levels of APOC2 were increased in both normoglycemic and hypoglycemic L‐G6pc −/− mice compared to controls (Figure 4D,E). Plasma protein levels of APOC3, ANGPTL3 and ANGPTL4, and APOC2/APOC3 ratios were not affected by genotype or feeding state (Figure 4F,G).
FIGURE 4.
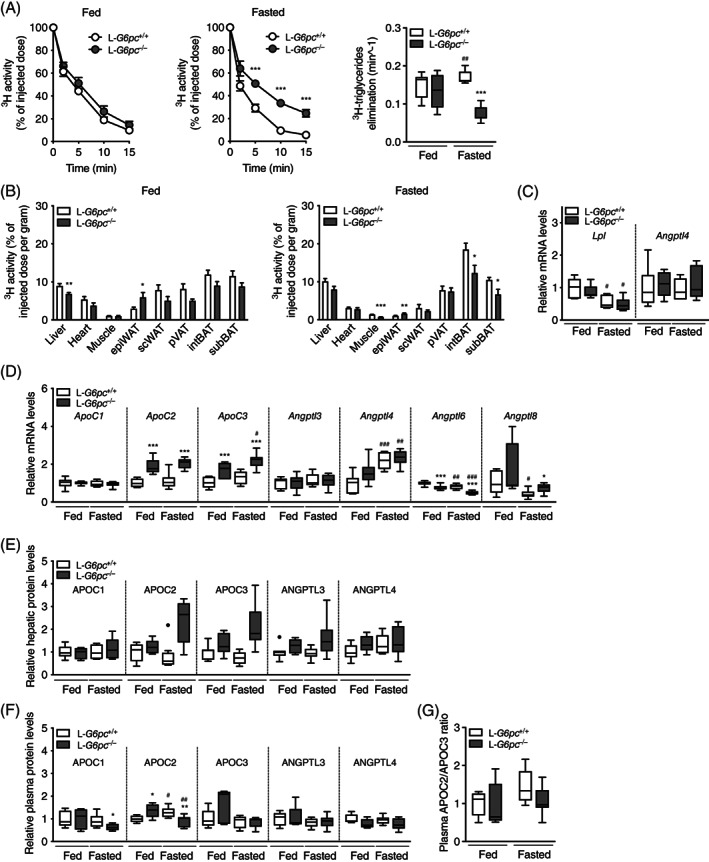
VLDL catabolism is impaired in fasted L‐G6pc −/− mice. A, Plasma clearance and plasma elimination rate of glycerol tri‐[3H]oleate‐labeled VLDL‐like particles in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 6‐8). B, 3H activity in various organs and expressed as percentage of the injected dose of glycerol tri[3H]oleate‐labeled very‐low‐density lipoprotein (VLDL)‐like particles per gram wet tissue weight (n = 7‐8). C, White adipose tissue mRNA levels of L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 5‐6). D, Hepatic gene expression and (E) hepatic protein levels assessed by targeted proteomics in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 6‐8). F, Plasma protein levels assessed by targeted proteomics and (G) Plasma APOC2/APOC3 protein ratio in L‐G6pc −/− mice and wild‐type littermates under fed and fasted conditions (n = 5‐8)
3. DISCUSSION
In the current study, we investigated the physiological mechanisms that link glycemia to hyperlipidemia in GSD Ia, by comparing fed (normoglycemic) to acutely fasted (hypoglycemic) L‐G6pc −/− mice. We found that excess TGs and cholesterol in GSD Ia patient serum were specifically associated with VLDL/chylomicrons, and that more severe liver steatosis and hypertriglyceridemia in hypoglycemic L‐G6pc −/− mice were associated with enhanced adipose tissue lipolysis and impaired VLDL‐TG catabolism, respectively (Figure S3). Altogether, our data suggest that increased VLDL‐TG secretion by the liver contributes to elevated serum TG concentrations in GSD Ia patients,17 and that impaired catabolism of TG‐rich lipoproteins explains the severe hyperlipidemia in patients with poor glycemic control.23, 26, 27
It is commonly assumed that hepatic steatosis and hypertriglyceridemia in GSD Ia result from an increase in hepatic de novo lipogenesis and VLDL secretion driven by a high glycolytic flux.22, 23, 26, 28 Our findings indicate that hepatic TG accumulation in normoglycemic L‐G6pc −/− mice mainly, but not exclusively, results from increased de novo lipogenesis. It should be noted that the contribution of lipogenesis was quantified over a 24 hour period and reached up to 20%, a value comparable to that observed in humans in the postprandial state.29 In fasted, hypoglycemic L‐G6pc −/− mice, enhanced adipose tissue lipolysis and subsequent uptake and elongation of fatty acids released by the adipose tissue were found to be the most predominant causes of hepatic TG accumulation. The increased contribution of adipose tissue‐derived circulating NEFAs to liver steatosis was further supported by the accumulation of linoleate (an essential fatty acid) which exclusively occurred in livers of hypoglycemic L‐G6pc −/− mice.
Adipose tissue lipolysis is, among other factors, controlled by HSL.30 Our data show increased phosphorylation of HSL at Ser‐563 and a tendency for increased phosphorylation at Ser‐660 (marking HSL activation) in hypoglycemic L‐G6pc −/− mice, while the inhibitory phosphorylation at Ser‐565 remained unaffected. These findings are in agreement with the increase in ex vivo adipose tissue lipolysis in these animals. Glucagon, catecholamines, and glucocorticoids, which are elevated in response to hypoglycemia, are well‐established enhancers of HSL‐mediated adipose tissue lipolysis via cAMP and β‐adrenergic signaling.30, 31, 32, 33, 34 In hypoglycemic L‐G6pc −/− mice, plasma glucagon levels were significantly increased, and the insulin‐to‐glucagon ratio was reduced (Table 1). We hypothesize that a lower insulin‐to‐glucagon ratio and/or increased catecholamine/glucocorticoid levels in fasted hypoglycemic L‐G6pc −/− mice35, 36, 37 increases adipose tissue lipolysis via enhanced HSL phosphorylation.31, 34, 38 We found that enhanced adipose tissue lipolysis in fasted L‐G6pc −/− mice was not associated with reduced adipose tissue mass (data not shown). Yet, it has been reported that L‐G6pc −/− mice display failure to thrive starting from 9 months after gene deletion,39 comparable to what is observed in patients. It is conceivable that prolonged (hepatic) G6pc deficiency imposes recurrent catabolic states that on the long run are reflected in reduced body weight or adiposity, while the catabolic response induced by a single fast within 2 weeks after hepatic G6pc deletion in the current study was relatively modest and did not (yet) translate into lower fat mass or body weight.
Our data highly indicate that enhanced VLDL‐TG secretion in L‐G6pc −/− mice mainly resulted from enhanced VLDL lipidation, rather than an increase in VLDL particle number. Consistent with these findings,40, 41 we observed that hepatic MTTP and TM6SF2 protein levels were increased and that circulating ApoB levels remained unchanged in L‐G6pc −/− mice. Importantly, our data also show that the increase in VLDL‐TG secretion was paralleled by impaired VLDL‐TG clearance in hypoglycemic L‐G6pc −/− mice only. This suggests that the activity of lipolytic enzymes such as LPL or hepatic lipase (HL) was reduced by a circulating factor that is specifically altered under fasted, hypoglycemic conditions, as was proposed previously.42 Among others, LPL and HL activities are regulated by apolipoproteins.25, 43 It has been reported that the apoC2/C3 ratio is reduced in GSD Ia VLDL,44 which may contribute to reduced LPL activity.45, 46 However, the data presented in the current study do not confirm a solid relationship between feeding state, apoC2/C3 ratios and VLDL levels or VLDL catabolism. Yet, this does not exclude the possibility that reduced availability of apoC2 to LPL contributed to impaired TG clearance and more severe hypertriglyceridemia in fasted L‐G6pc −/− mice. Interestingly, it has been proposed that circulating NEFAs inhibit LPL and TG hydrolysis, hence exerting product inhibition on its activity.42, 47, 48, 49, 50 Oleate, one of the most prevalent NEFAs, dose‐dependently inhibits LPL activity,47 probably by preventing ApoC2‐LPL interaction.51, 52 The observed increases in plasma NEFA and oleate concentrations in hypoglycemic L‐G6pc −/− mice may hence contribute to impaired VLDL catabolism independent of changes in ApoC2/C3 levels. This mechanism also likely explains previous findings from our laboratory in which plasma TG levels were increased in hypoglycemic rats treated with a glucose‐6‐phosphate transporter inhibitor, thereby acutely inducing GSD type 1b.22 In that study, VLDL‐TG secretion was not enhanced, yet NEFA concentrations were increased and presumably resulted in rapid inhibition of VLDL catabolism and hypertriglyceridemia.
Experimental data in GSD Ia patients suggest that altered VLDL metabolism may contribute to hypertriglyceridemia in GSD Ia.23, 26, 42, 44, 53, 54 It has been reported that ApoB turnover VLDL secretion is unchanged in GSD Ia patients in normoglycemic state.44, 54 Bandsma et al reported reduced ApoB100 turnover rates in two GSD Ia patients, and a higher turnover in another patient as compared to healthy controls.23 Our current data suggest that hepatic VLDL particle secretion remains (largely) unaffected in L‐G6pc −/− mice. Assessment of ApoB turnover rates therefore likely does not reveal increased VLDL‐TG secretion in GSD Ia. Our finding that excess TGs and cholesterol in patients are almost exclusively associated with TG‐rich lipoproteins is in agreement with our finding in mice that impaired VLDL catabolism is a major determinant of hyperlipidemia in GSD Ia. Although impaired VLDL catabolism has been proposed to contribute to hyperlipidemia in normoglycemic GSD Ia patients,23, 42, 44, 53, 55, 56 we show that VLDL catabolism was selectively inhibited in fasted L‐G6pc −/− mice. Besides the fact that the observed difference in lipid composition (Figure 3L) and basal plasma TG levels is not accounted for when using these model particles, it is unclear whether apoB, which is elevated in plasma of GSD Ia patients44, 55, 57 and livers of GSD Ia mice (Figure 3M), is exchanged with VLDL‐like particles. One methodological limitation of our study is therefore that we may not have accounted for altered lipid and apolipoprotein levels on VLDL catabolism in L‐G6pc −/− mice.
It should be noted that some differences exist between our preclinical findings and observations in GSD Ia patients. These may be related to multiple factors, such as (a) the use of hepatocyte‐specific instead of whole body G6pc knockout mice, (b) a lack of extra carbohydrate intake during the inactive phase in mice while GSD Ia patients regularly ingest complex carbohydrates during both day and night, (c) species differences between mice and humans, and (d) the experimental setup. In our study, hepatic G6pc deletion was induced during adulthood and analysis of TG metabolism was performed within 2 weeks after gene deletion, which provides insights into an early disease stage and does not account for pathophysiological adaptations occurring over a longer timeframe. In hypoglycemic L‐G6pc −/− mice, we observed enhanced β‐oxidation capacity and increased levels of ketone bodies as well as hepatic C2 acylcarnitines, suggesting that fatty acid oxidation was increased under hypoglycemic conditions. Based on these findings, we hypothesize that enhanced hepatic NEFA influx in hypoglycemic L‐G6pc −/− mice promoted hepatic fatty acid elongation, β‐oxidation, and ketogenesis. In contrast, hepatic C2 acylcarnitine levels were modestly reduced and plasma ketone body levels were only slightly increased in normoglycemic L‐G6pc −/− mice, despite an increase in hepatic β‐oxidation capacity ex vivo. These data suggest that in vivo β‐oxidation was not increased under fed conditions, consistent with findings in normoglycemic GSD Ia patients.23 Unlike fasted L‐G6pc −/− mice, GSD Ia is traditionally considered to cause nonketotic or hypoketotic hypoglycemia58, 59 despite elevated NEFA levels,60, 61 although GSD Ia patients may display elevated ketone concentrations.62, 63 This seemingly inconsistency may be related to the physiological state during which ketone levels were analyzed. With respect to plasma lactate levels, these tended to increase in fed L‐G6pc −/− mice as compared to wild‐type controls but were only slightly elevated in fasted L‐G6pc −/− mice, while circulating lactate levels are elevated in GSD Ia patients10, 62, 64 and in whole‐body G6pc −/− mice.65 We speculate that these discrepancies in plasma ketone body and lactate levels are related to an impairment of renal lactate excretion in GSD Ia patients and whole‐body G6pc knockouts,64, 65 that does not occur in hepatocyte‐specific G6pc knockout mice.21 Elevated lactate levels in GSD Ia patients and whole‐body G6pc knockout mice, in turn, likely inhibit ketogenesis.66 L‐G6pc −/− mice, on the other hand, retain functional renal lactate excretion, hence permitting ketogenesis, especially in hypoglycemic states. Our current data show a selective increase in adipose tissue lipolysis and inhibition of VLDL catabolism in fasted hypoglycemic L‐G6pc −/− mice. Previous studies have shown enhanced adipose tissue lipolysis in both normo‐ and hypoglycemic GSD Ia patients,60, 61 while TG hydrolysis activities were reduced under normoglycemia.42, 44, 53, 56, 67 The fact that adipocyte lipolysis and VLDL catabolism are also altered in well‐controlled patients is most likely explained by the presence of insulin resistance associated with dietary overtreatment,68 which also enhances adipose tissue lipolysis and circulating NEFA levels that may, in turn, inhibit VLDL catabolism.42, 47, 48, 49, 69, 70
According to the European Study on Glycogen Storage Disease type I, chronic serum TG concentrations <6.0 mmol/L are among the biomedical targets.4 The more recent American College of Medical Genetics and Genomics GSD I guidelines state that “elevated triglycerides and cholesterol above the normal ranges may persist in some patients with GSD I, despite appropriate dietary treatment”.13 In GSD Ia patients, serum TG levels can go up to 100 mmol/L,17 posing major health risk given that TG concentrations >11.3 mmol/L are associated with acute pancreatitis.71 Adenoma progression is significantly increased in GSD Ia subjects with a 5‐year mean TG concentration >5.6 mmol/L.14 How to translate these preclinical insights on the link between hypoglycemia and hypetriglyceridemia into management and monitoring of GSD Ia patients in clinical practice and future trial design? Our finding that hypoglycemia and hypertriglyceridemia are physiologically linked emphasizes the importance of diagnosing (asymptomatic) hypoglycemia. In this respect, better monitoring of glycemic control, for example by means of continuous glucose monitoring, will likely result in better control of hypertriglyceridemia. In addition, dedicated research on the role of factors that control VLDL‐TG catabolism, such as NEFA, hormones, apolipoproteins, and angiopoietin‐like proteins, will potentially result in identification of novel biomarkers that will aid better clinical management and monitoring of GSD Ia. With respect to monitoring, our results in L‐G6pc −/− mice illustrate that the timing of sampling vs meals is crucial to adequately interpret blood biomarkers. Many preclinical studies in GSD models are performed under fasting conditions, and time to hypoglycemia is considered an important outcome during clinical trials aiming at therapeutic interventions. On the other hand, for GSD Ia patients, self‐management and self‐monitoring of a strict dietary management aims to maintain glucose concentrations in a relatively narrow normal range, reflecting a small therapeutic window between undertreatment and overtreatment. The use of biomarkers in different stages of GSD Ia should be more clearly defined to (a) identify the risk of developing an illness, (b) screen for subclinical disease, (c) diagnose disease, (d) categorize disease severity, and (e) predict prognosis or complications. The thoughtful design of the current study with regard to physiological status, and the use of CRISPR‐cas9 mediated gene editing to generate a spectrum of GSD Ia disease phenotypes in mice (Rutten et al, in revision), will allow for identification of novel, clinically‐translatable biomarkers in GSD Ia.
In conclusion, our study identifies the physiological mechanism via which acute hypoglycemia is linked to hypertriglyceridemia in a liver‐specific mouse model for GSD Ia. Moreover, our work reveals a marked difference in the origin of hepatic steatosis in normoglycemic vs hypoglycemic GSD Ia mice. We propose that increased adipocyte lipolysis and the resulting elevated NEFA levels in GSD Ia impairs catabolism of TG‐rich lipoproteins, leading to hypertriglyceridemia, and that these changes are most marked in patients with poor metabolic control. Our findings highlight the contribution of hypoglycemia to GSD Ia pathophysiology. We therefore propose that preclinical studies in normo‐ vs hypoglycemic GSD Ia models will provide clinically relevant insights into the underlying mechanisms contributing to disease symptoms and complications in GSD Ia patients.
4. METHODS
4.1. Study approval
The study with human materials was performed in accordance with the Declaration of Helsinki and the institutional rules for studying biological rest materials and thereby approved for waived consent as it concerned retrospective, anonymous data. The Medical Ethical Committee of the University Medical Center Groningen stated that the Medical Research Involving Human Subjects Act was not applicable and that official study approval by the Medical Ethical Committee was not required (METc 2019/119).
All animal procedures were approved by the Dutch Central Committee Animal Experiments (Centrale Commissie Dierproeven) under permit number AVD105002015245 and DEC 6246 and adhered to guidelines set out in the 2010/63/EU directive.
4.2. Human subjects
For FPLC profiles, plasma samples from three GSD Ia patients (two female, one male; age range 32‐79 years) and one control subject were collected in fed state. For plasma NEFA profiles, plasma from seven GSD Ia patients (three female, four male; age range 22‐51 years) and age‐ and sex‐matched controls were randomly collected during the day in fed state. For patient demographics, see Table S1. Plasma samples were stored at −20°C until further analysis.
4.3. Animals
Male B6.G6pc lox/lox and B6.G6pc lox/lox.SAcreERT2/w mice39 (10‐16 weeks old) were housed in a light (lights on: 7:00 am‐7:00 pm) and temperature (21°C)‐controlled facility and fed a standard laboratory chow diet (RMH‐B, ABdiets, Woerden, The Netherlands). Animals received i.p. injections of tamoxifen for five consecutive days to generate hepatocyte‐specific G6pc‐deficient mice (L‐G6pc −/−) and wild‐type littermates (L‐G6pc +/+) by deletion of exon 3 of the G6pc gene, as described previously.39 Ten days after the last tamoxifen injection, mice were sacrificed by heart puncture (8:00 am) in either a fed or a 9 hour fasted (11:00 pm‐8:00 am) state. Tissues were quickly excised and stored at −80°C. Blood was centrifuged (4.000 rpm for 10 minutes at 4°C), and plasma was stored at −20°C.
Additional experimental and analytical procedures are described in the Supporting Information.
4.4. Statistics
Statistical analysis was performed using BrightStat software. Differences between two or multiple groups were tested by Mann‐Whitney U test or Kruskal‐Wallis H test followed by post hoc Conover pairwise comparisons, respectively. Data was represented by Tukey boxplots or means ± SEM. ***P < .001, **P < .01, *P < .05 indicates significance compared to wild‐type littermates. ###P < .001, ##P < .01, #P < .05 indicates significance compared to fed condition. Correlations were analyzed by Spearman's correlations coefficient using SPSS24.0 for Windows software (SPSS, Chicago, Illinois).
CONFLICT OF INTEREST
The authors declare no potential conflict of interest.
AUTHOR CONTRIBUTIONS
Designing research studies: Joanne A. Hoogerland, Brenda S. Hijmans, Sander Kooijman, Folkert Kuipers, Fabienne Rajas, Gilles Mithieux, Dirk‐Jan Reijngoud, Terry G. J. Derks, and Maaike H. Oosterveer; Conducting experiments: Joanne A. Hoogerland, Brenda S. Hijmans, Fabian Peeks, Justina C. Wolters, Sander Kooijman, Trijnie Bos, Aycha Bleeker, Henk Wolters, Albert Gerding, Karen van Eunen, Rick Havinga, and Amanda C. M. Pronk; Analyzing data: Joanne A. Hoogerland, Brenda S. Hijmans, Fabian Peeks, Justina C. Wolters, Theo H. van Dijk, Folkert Kuipers, Terry G. J. Derks, and Maaike H. Oosterveer; Writing the first draft of the manuscript: Joanne A. Hoogerland, Fabian Peeks, Folkert Kuipers, and Maaike H. Oosterveer; Critical revisions of the manuscript: Fabian Peeks, Sander Kooijman, Patrick C. N. Rensen, Theo H. van Dijk, Henk Wolters, Karen van Eunen, Fabienne Rajas, Gilles Mithieux, Folkert Kuipers, Terry G. J. Derks, and Maaike H. Oosterveer; Material support: Fabienne Rajas and Gilles Mithieux.
ETHICS STATEMENT
The Medical Ethical Committee of the University Medical Center Groningen stated that the Medical Research Involving Human Subjects Act was not applicable and that official study approval by the Medical Ethical Committee was not required (METc 2019/119).
PATIENT CONSENT STATEMENT
The study with human materials was performed in accordance with the Declaration of Helsinki and the institutional rules for studying biological rest materials and thereby approved for waived consent as it concerned retrospective, anonymous data.
DOCUMENTATION OF APPROVAL OF ANIMAL EXPERIMENTATION
All animal procedures were approved by the Dutch Central Committee Animal Experiments (Centrale Commissie Dierproeven) under permit number AVD105002015245 and DEC 6246 and adhered to guidelines set out in the 2010/63/EU directive.
Supporting information
Supplementary Figure 1. (A) Hepatic free cholesterol and (B) cholesteryl esters in L‐G6pc ‐/‐ mice and wildtype littermates under fed and fasted conditions (n=6‐8).
Supplementary Figure 2.Ex vivo glycerol release by adipose tissue expressed as a function of time in L‐G6pc ‐/‐ mice and wildtype littermates in (A) fed and (B) fasted conditions (n=6‐8). (C) Correlation between pHSL Ser563 and ex vivo adipose glycerol release under fasted conditions (n=6‐7). (D) Ex vivo partial β‐oxidation capacity in L‐G6pc ‐/‐ mice and wildtype littermates under fed and fasted conditions (n=5‐8). (E) Western blot for ApoB100 and ApoB48 in nascent VLDL isolated from fed and fasted L‐G6pc ‐/‐ mice (indicated by ‐) and wildtype littermates (indicated by +) (n=3) with 125 nmol of TG loaded in each lane. (F) Hepatic gene expression in L‐G6pc ‐/‐ mice and wildtype littermates under fed and fasted conditions (n=7‐8).
Supplementary Figure 3. Schematic overview of physiological adaptations in normo‐ and hypoglycemic L‐G6pc ‐/‐ mice.
In normoglycemic L‐G6pc ‐/‐ mice, hepatic de novo fatty acid synthesis contributes to hepatic lipid accumulation (left picture). In hypoglycemic L‐G6pc ‐/‐ mice, enhanced adipose tissue lipolysis and hepatic fatty acid elongation contribute to hepatic TG accumulation (right picture). VLDL‐TG levels and –secretion rates are increased in L‐G6pc ‐/‐ mice under both normo‐ and hypoglycemic conditions. VLDL catabolism is selectively inhibited in hypoglycemic L‐G6pc ‐/‐ mice, and associated with higher NEFA levels compared to normoglycemic L‐G6pc ‐/‐ mice.
Abbreviations: G6P, glucose‐6‐phosphate; G6PC, glucose‐6‐phosphatase; IDL, intermediate density lipoproteins; LDL, low density lipoprotein; LPL, lipoprotein lipase; NEFA, non‐esterified fatty acids; TG, triglycerides; VLDL, very low density lipoprotein.
Table S1. Demographics of GSD Ia patients
Table S2. Hepatic fatty acids (μmol/g liver) in liver of L‐G6pc ‐/‐ mice and wildtype littermates in fed state or after an overnight fast
Table S3. Individual enzyme activities and gene expression levels, normalized to values in fed L‐G6pc +/+ mice.
Table S4. Fractional and absolute synthesis of C18:0 in L‐G6pc ‐/‐ mice and wildtype littermates in fed state or after an overnight fast
Table S5. Non‐esterified fatty acids (μmol/L) in serum of normoglycemic GSD Ia patients (n = 7) and age and sex‐matched controls and in plasma of L‐G6pc ‐/‐ mice and wildtype littermates (n = 7‐8) in fed state or after an overnight fast
Table S6. Acylcarnitines (μmol/g) in liver of L‐G6pc ‐/‐ mice and wildtype littermates in fed state or after an overnight fast
Table S7. Taqman and SYBR Green qPCR primer and probe sequences
Table S8. Sequence of peptide standards
Appendix S1: Supporting information
ACKNOWLEDGMENTS
We thank Y. Lei, M. H. Koster, I. A. Martini, A. Jurdinski, K. Tholen, Y. van der Veen, T. Boer, M. Koehorst, R. Thomas, K. Alagere Krishnamurthy, and F. G. Perton for excellent technical assistance. The authors confirm independence from the sponsors; the content of the article has not been influenced by the sponsors. This research was supported by an unrestricted research grant from DSM Nutritional Products (Kaiseraugst, Switzerland) and cofinanced by the Ministry of Economic Affairs and Climate Policy by means of the PPP‐allowance made available by the Top Sector Life Sciences & Health to stimulate public private partnerships. F. P. is appointed as MD‐PhD by the UMCG, rewarded by the UMCG Junior Scientific Masterclass to F. P. and T. G. J. D. (MD‐PhD 16‐24). M. H. O is the recipient of a VIDI grant from the Dutch Scientific Organization (#91717373), and holds a Rosalind Franklin Fellowship from the University of Groningen. P. C. N. R. is supported by the Netherlands Cardiovascular Research Initiative: an initiative with support of the Dutch Heart Foundation (CVON2017 GENIUS‐II).
Hoogerland JA, Peeks F, Hijmans BS, et al. Impaired Very‐Low‐Density Lipoprotein catabolism links hypoglycemia to hypertriglyceridemia in Glycogen Storage Disease type Ia. J Inherit Metab Dis. 2021;44:879–892. 10.1002/jimd.12380
Communicating Editor: Gerard T. Berry
Funding information DSM Nutritional Products; Hartstichting, Grant/Award Number: CVON2017 GENIUS‐II; Ministry of Economic Affairs and Climate Policy; NWO‐VIDI, Grant/Award Number: #91717373; University Medical Center Groningen; University of Groningen
DATA AVAILABILITY STATEMENT
Raw data will be available when requested.
REFERENCES
- 1.Lei KJ, Chen YT, Chen H, et al. Genetic basis of glycogen storage disease type 1a: prevalent mutations at the glucose‐6‐phosphatase locus. Am J Hum Genet. 1995;57:766‐771. [PMC free article] [PubMed] [Google Scholar]
- 2.Bali DS, Chen Y‐T, Austin S, Goldstein JL. Glycogen Storage Disease Type I. Seattle, WA: University of Washington; 1993. [PubMed] [Google Scholar]
- 3.Froissart R, Piraud M, Boudjemline A, et al. Glucose‐6‐phosphatase deficiency. Orphanet J Rare Dis. 2011;6:27. 10.1186/1750-1172-6-27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Rake J, Visser G, Labrune P, et al. Guidelines for management of glycogen storage disease type I ‐ European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002a;161:S112‐S119. 10.1007/s00431-002-1016-7. [DOI] [PubMed] [Google Scholar]
- 5.Weinstein D, Wolfsdorf J. Effect of continuous glucose therapy with uncooked cornstarch on the long‐term clinical course of type 1a glycogen storage disease. Eur J Pediatr. 2002;161:S35‐S39. 10.1007/s00431-002-1000-2. [DOI] [PubMed] [Google Scholar]
- 6.Chen Y‐T, Bazzarre CH, Lee MM, et al. Type I glycogen storage disease: nine years of management with cornstarch. Eur J Pediatr. 1993;152:56‐59. 10.1007/BF02072090. [DOI] [PubMed] [Google Scholar]
- 7.Dambska M, Labrador E, Kuo C, Weinstein D. Prevention of complications in glycogen storage disease type Ia with optimization of metabolic control. Pediatr Diabetes. 2017;18:327‐331. 10.1111/pedi.12540. [DOI] [PubMed] [Google Scholar]
- 8.Okechuku GO, Shoemaker LR, Dambska M, et al. Tight metabolic control plus ACE inhibitor therapy improves GSD I nephropathy. J Inherit Metab Dis. 2017;40:703‐708. 10.1007/s10545-017-0054-2. [DOI] [PubMed] [Google Scholar]
- 9.Franco LM, Krishnamurthy V, Bali D, et al. Hepatocellular carcinoma in glycogen storage disease type Ia: a case series. J Inherit Metab Dis. 2005;28:153‐162. 10.1007/s10545-005-7500-2. [DOI] [PubMed] [Google Scholar]
- 10.Rake JP, Visser G, Labrune P, et al. Glycogen storage disease type I: diagnosis, management, clinical course and outcome. Results of the European Study on Glycogen Storage Disease Type I (ESGSD I). Eur J Pediatr. 2002b;161:S20‐S34. 10.1007/BF02679990. [DOI] [PubMed] [Google Scholar]
- 11.Chen Y‐T, Kishnani PS, Koeberl D. Glycogen storage diseases. In: Beaudet AL, Vogelstein B, Kinzler KW, et al., eds. The Online Metabolic and Molecular Bases of Inherited Disease. New York, NY: The McGraw‐Hill Companies, Inc.; 2014. [Google Scholar]
- 12.Howell RR, Stevenson RE, Ben‐Menachem Y, et al. Hepatic adenomata with type 1 glycogen storage disease. JAMA J Am Med Assoc. 1976;236:1481. 10.1001/jama.1976.03270140033019. [DOI] [PubMed] [Google Scholar]
- 13.Kishnani PS, Austin SL, Abdenur JE, et al. Diagnosis and management of glycogen storage disease type I: a practice guideline of the American College of Medical Genetics and Genomics. Genet Med. 2014;16:e1. 10.1038/gim.2014.128. [DOI] [PubMed] [Google Scholar]
- 14.Wang DQ, Fiske LM, Carreras CT, Weinstein DA. Natural history of hepatocellular adenoma formation in glycogen storage disease type I. J Pediatr. 2011;159:442‐446. 10.1016/j.jpeds.2011.02.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Peeks F, Boonstra WF, Baere L, et al. Research priorities for liver glycogen storage disease: an international priority setting partnership with the James Lind Alliance. J Inherit Metab Dis. 2020;43:279‐289. 10.1002/jimd.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bandsma RHJ, Smit GPA, Reijngoud D‐J, Kuipers F. Adiponectin levels correlate with the severity of hypertriglyceridaemia in glycogen storage disease Ia. J Inherit Metab Dis. 2009;32(suppl 1):S27‐S31. 10.1007/s10545-009-0993-3. [DOI] [PubMed] [Google Scholar]
- 17.Peeks F, Steunenberg TAH, de Boer F, et al. Clinical and biochemical heterogeneity between patients with glycogen storage disease type IA: the added value of CUSUM for metabolic control. J Inherit Metab Dis. 2017;40:695‐702. 10.1007/s10545-017-0039-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Beegle RD, Brown LM, Weinstein DA. Regression of hepatocellular adenomas with strict dietary therapy in patients with glycogen storage disease type I. JIMD Rep. 2014;18:23‐32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Matern D, Starzl TE, Arnaout W, et al. Liver transplantation for glycogen storage disease types I, III, and IV. Eur J Pediatr. 1999;158(suppl 2):S43‐S48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Parker P, Burr I, Slonim A, et al. Regression of hepatic adenomas in type Ia glycogen storage disease with dietary therapy. Gastroenterology. 1981;81:534‐536. [PubMed] [Google Scholar]
- 21.Rajas F, Clar J, Gautier‐Stein A, Mithieux G. Lessons from new mouse models of glycogen storage disease type 1a in relation to the time course and organ specificity of the disease. J Inherit Metab Dis. 2015;38:521‐527. 10.1007/s10545-014-9761-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bandsma RH, Wiegman CH, Herling AW, et al. Acute inhibition of glucose‐6‐phosphate translocator activity leads to increased de novo lipogenesis and development of hepatic steatosis without affecting VLDL production in rats. Diabetes. 2001;50:2591‐2597. [DOI] [PubMed] [Google Scholar]
- 23.Bandsma RHJ, Prinsen BH, de Sain‐van der Velden M, et al. Increased de novo lipogenesis and delayed conversion of large VLDL into intermediate density lipoprotein particles contribute to hyperlipidemia in glycogen storage disease type 1a. Pediatr Res. 2008;63:702‐707. 10.1203/PDR.0b013e31816c9013. [DOI] [PubMed] [Google Scholar]
- 24.Connelly PW. The role of hepatic lipase in lipoprotein metabolism. Clin Chim Acta. 1999;286:243‐255. 10.1016/S0009-8981(99)00105-9. [DOI] [PubMed] [Google Scholar]
- 25.Kersten S. Physiological regulation of lipoprotein lipase. Biochim Biophys Acta. 2014;1841:919‐933. 10.1016/j.bbalip.2014.03.013. [DOI] [PubMed] [Google Scholar]
- 26.Bandsma RHJ, Rake J‐P, Visser G, et al. Increased lipogenesis and resistance of lipoproteins to oxidative modification in two patients with glycogen storage disease type 1a. J Pediatr. 2002;140:256‐260. 10.1067/mpd.2002.121382. [DOI] [PubMed] [Google Scholar]
- 27.Derks TGJ, van Rijn M. Lipids in hepatic glycogen storage diseases: pathophysiology, monitoring of dietary management and future directions. J Inherit Metab Dis. 2015;38:537‐543. 10.1007/s10545-015-9811-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Rajas F, Labrune P, Mithieux G. Glycogen storage disease type 1 and diabetes: learning by comparing and contrasting the two disorders. Diabetes Metab. 2013;39:377‐387. 10.1016/j.diabet.2013.03.002. [DOI] [PubMed] [Google Scholar]
- 29.Timlin MT, Parks EJ. Temporal pattern of de novo lipogenesis in the postprandial state in healthy men. Am J Clin Nutr. 2005;81:35‐42. 10.1093/ajcn/81.1.35. [DOI] [PubMed] [Google Scholar]
- 30.Duncan RE, Ahmadian M, Jaworski K, et al. Regulation of lipolysis in adipocytes. Annu Rev Nutr. 2007;27:79‐101. 10.1146/annurev.nutr.27.061406.093734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Du S, Joyner MJ, Curry TB, et al. Effect of β2‐adrenergic receptor polymorphisms on epinephrine and exercise‐stimulated lipolysis in humans. Physiol Rep. 2014;2:e12017. 10.14814/phy2.12017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Liljenquist JE, Bomboy JD, Lewis SB, et al. Effects of glucagon on lipolysis and ketogenesis in normal and diabetic men. J Clin Invest. 1974;53:190‐197. 10.1172/JCI107537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Perea A, Clemente F, Martinell J, et al. Physiological effect of glucagon in human isolated adipocytes. Horm Metab Res. 1995;27:372‐375. 10.1055/s-2007-979981. [DOI] [PubMed] [Google Scholar]
- 34.Yang L‐K, Tao Y‐X. Physiology and pathophysiology of the β3‐adrenergic receptor. Prog Mol Biol Transl Sci. 2019;161:91‐112. [DOI] [PubMed] [Google Scholar]
- 35.Mundy HR, Hindmarsh PC, Matthews DR, et al. The regulation of growth in glycogen storage disease type 1. Clin Endocrinol. 2003;58:332‐339. [DOI] [PubMed] [Google Scholar]
- 36.Rossi A, Simeoli C, Salerno M, et al. Imbalanced cortisol concentrations in glycogen storage disease type I: evidence for a possible link between endocrine regulation and metabolic derangement. Orphanet J Rare Dis. 2020;15(1):99. 10.1186/S13023-020-01377-W. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Walker EA, Ahmed A, Lavery GG, et al. 11β‐Hydroxysteroid dehydrogenase type 1 regulation by intracellular glucose 6‐phosphate provides evidence for a novel link between glucose metabolism and hypothalamo‐pituitary‐adrenal axis function. J Biol Chem. 2007;282:27030‐27036. 10.1074/jbc.M704144200. [DOI] [PubMed] [Google Scholar]
- 38.Frühbeck G, Méndez‐Giménez L, Fernández‐Formoso J‐A, et al. Regulation of adipocyte lipolysis. Nutr Res Rev. 2019;27:63‐93. 10.1017/S095442241400002X. [DOI] [PubMed] [Google Scholar]
- 39.Mutel E, Abdul‐Wahed A, Ramamonjisoa N, et al. Targeted deletion of liver glucose‐6 phosphatase mimics glycogen storage disease type 1a including development of multiple adenomas. J Hepatol. 2011;54:529‐537. 10.1016/j.jhep.2010.08.014. [DOI] [PubMed] [Google Scholar]
- 40.Atzel A, Wetterau JR. Mechanism of microsomal triglyceride transfer protein catalyzed lipid transport. Biochemistry. 1993;32:10444‐10450. [DOI] [PubMed] [Google Scholar]
- 41.Smagris E, Gilyard S, BasuRay S, et al. Inactivation of Tm6sf2, a gene defective in fatty liver disease, impairs lipidation but not secretion of very low density lipoproteins. J Biol Chem. 2016;291:10659‐10676. 10.1074/jbc.M116.719955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Muller DPR, Gamlen TR. The activity of hepatic lipase and lipoprotein lipase in glycogen storage disease: evidence for a circulating inhibitor of postheparin lipolytic activity. Pediat Res. 1984;18:881‐885. [DOI] [PubMed] [Google Scholar]
- 43.Larsson M, Vorrsjö E, Talmud P, et al. Apolipoproteins C‐I and C‐III inhibit lipoprotein lipase activity by displacement of the enzyme from lipid droplets. J Biol Chem. 2013;288:33997‐34008. 10.1074/jbc.M113.495366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Levy E, Thibault LA, Roy CC, et al. Circulating lipids and lipoproteins in glycogen storage disease type I with nocturnal intragastric feeding. J Lipid Res. 1988;29:215‐226. [PubMed] [Google Scholar]
- 45.Carlson LA, Ballantyne D. Changing relative proportions of apolipoproteins CII and CIII of very low density lipoproteins in hypertriglyceridaemia. Atherosclerosis. 1976;23:563‐568. [DOI] [PubMed] [Google Scholar]
- 46.Jong MC, Hofker MH, Havekes LM. Role of ApoCs in lipoprotein metabolism functional differences between ApoC1, ApoC2, and ApoC3. Arterioscler Thromb Vasc Biol. 1999;19:472‐484. [DOI] [PubMed] [Google Scholar]
- 47.Bengtsson G, Olivecrona T. Apolipoprotein CII enhances hydrolysis of monoglycerides by lipoprotein lipase, but the effect is abolished by fatty acids. FEBS Lett. 1979;106:345‐348. [DOI] [PubMed] [Google Scholar]
- 48.Bengtsson G, Olivecrona T. Lipoprotein lipase. Mechanism of product inhibition. Eur J Biochem. 1980;106:557‐562. [DOI] [PubMed] [Google Scholar]
- 49.Saxena U, Goldberg IJ. Interaction of lipoprotein lipase with glycosaminoglycans and apolipoprotein C‐II: effects of free‐fatty‐acids. Biochim Biophys Acta. 1990;1043:161‐168. [DOI] [PubMed] [Google Scholar]
- 50.Scow RO, Olivercrona T. Effect of albumin on products formed from chylomicron triacylclycerol by lipoprotein lipase in vitro. Biochim Biophys Acta. 1977;487:472‐486. [DOI] [PubMed] [Google Scholar]
- 51.Jackson RL, Shirai K, Harmony JAK, Quinn D. Mechanism of action of lipoprotein lipase: role of apolipoprotein C‐II. Prog Lipid Res. 1983;22:35‐78. 10.1007/978-3-642-81817-2_111. [DOI] [PubMed] [Google Scholar]
- 52.Quinn D, Shirai K, Jackson RL. Lipoprotein lipase: mechanism of action and role in lipoprotein metabolism. Prog Lipid Res. 1982;22:35‐78. [DOI] [PubMed] [Google Scholar]
- 53.Forget PP, Fernandes J, Begemann PH. Triglyceride clearing in glycogen storage disease. Pediatr Res. 1974;8:114‐119. 10.1203/00006450-197402000-00008. [DOI] [PubMed] [Google Scholar]
- 54.Wierzbicki AS, Watt GF, Lynas J, et al. Very low‐density lipoprotein apolipoprotein B‐100 turnover in glycogen storage disease type Ia (von Gierke disease). J Inherit Metab Dis. 2001;24:527‐534. [DOI] [PubMed] [Google Scholar]
- 55.Alaupovic P, Fernandes J. The serum apolipoprotein profile of patients with glucose‐6‐phosphatase deficiency. Pediatr Res. 1985;19:380‐384. [DOI] [PubMed] [Google Scholar]
- 56.Asayama K, Kato K, Anemiya S, Nozaki Y. Postheparin plasma lipases in patients with hepatic glycogenosis. Horm Metab Res. 1982;14:555‐555. 10.1055/s-2007-1019077. [DOI] [PubMed] [Google Scholar]
- 57.Fernandes J, Alaupovic P, Wit JM. Gastric drip feeding in patients with glycogen storage disease type I: its effects on growth and plasma lipids and apolipoproteins. Pediatr Res. 1989;25:327‐331. 10.1203/00006450-198904000-00002. [DOI] [PubMed] [Google Scholar]
- 58.Binkiewicz A, Senior B. Decreased ketogenesis in von Gierke's disease (type I glycogenosis). J Pediatr. 1973;83:973‐978. [DOI] [PubMed] [Google Scholar]
- 59.Fernandes J, Pikaar NA. Ketosis in hepatic glycogenosis. Arch Dis Child. 1972;47(251):41‐46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Havel RJ, Balasse EO, Williams HE, et al. Splanchnic metabolism in von Gierke's disease (glycogenosis type I). Trans Assoc Am Physicians. 1969;82:305‐323. [PubMed] [Google Scholar]
- 61.Ockerman PA. Glucose, glycerol and free fatty acids in glycogen storage disease type 1. Blood levels in the fasting and non‐fasting state. Effect of glucose and adrenalin administration. Clin Chim Acta. 1965;12:370‐382. [DOI] [PubMed] [Google Scholar]
- 62.Das AM, Lücke T, Meyer U, et al. Glycogen storage disease type 1: impact of medium‐chain triglycerides on metabolic control and growth. Ann Nutr Metab. 2010;56:225‐232. 10.1159/000283242. [DOI] [PubMed] [Google Scholar]
- 63.Duarte IF, Goodfellow BJ, Barros A, et al. Metabolic characterisation of plasma in juveniles with glycogen storage disease type 1a (GSD1a) by high‐resolution 1H NMR spectroscopy. NMR Biomed. 2007;20:401‐412. 10.1002/nbm.1073. [DOI] [PubMed] [Google Scholar]
- 64.Oster Y, Wexler ID, Heyman SN, Fried E. Recoverable, record‐high lactic acidosis in a patient with glycogen storage disease type 1: a mixed type A and type B lactate disorder. Case Rep Med. 2016;2016:4362743. 10.1155/2016/4362743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Peng W‐T, Pan C‐J, Lee EJ, et al. Generation of mice with a conditional allele for G6pc. Genesis. 2009;47:590. 10.1002/DVG.20538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.McGarry JD, Foster DW. The regulation of ketogenesis from oleic acid and the influence of antiketogenic agents. J Biol Chem. 1971;246:6247‐6253. [PubMed] [Google Scholar]
- 67.Fernandes J, Jansen H, Jansen TC. Nocturnal gastric drip feeding in glucose‐6‐phosphatase deficient children. Pediatr Res. 1979;13:225‐229. [DOI] [PubMed] [Google Scholar]
- 68.Bhattacharya K. Dietary dilemmas in the management of glycogen storage disease type I. J Inherit Metab Dis. 2011;34:621‐629. 10.1007/s10545-011-9322-8. [DOI] [PubMed] [Google Scholar]
- 69.Goodman DS. The interaction of human serum albumin with long‐chain fatty acid anions. Am Chem Soc. 1958;80:3892. [Google Scholar]
- 70.Kirkland JL, Hollenberg CH, Kindler S, Roncari DAK. Long‐chain fatty acids decrease lipoprotein lipase activity of cultured rat adipocyte precursors. Metabolism. 1994;43:144‐151. 10.1016/0026-0495(94)90236-4. [DOI] [PubMed] [Google Scholar]
- 71.Berglund L, Brunzell JD, Goldberg AC, et al. Evaluation and treatment of hypertriglyceridemia: an Endocrine Society clinical practice guideline. J Clin Endocrinol Metab. 2012;97:2969‐2989. 10.1210/jc.2011-3213. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1. (A) Hepatic free cholesterol and (B) cholesteryl esters in L‐G6pc ‐/‐ mice and wildtype littermates under fed and fasted conditions (n=6‐8).
Supplementary Figure 2.Ex vivo glycerol release by adipose tissue expressed as a function of time in L‐G6pc ‐/‐ mice and wildtype littermates in (A) fed and (B) fasted conditions (n=6‐8). (C) Correlation between pHSL Ser563 and ex vivo adipose glycerol release under fasted conditions (n=6‐7). (D) Ex vivo partial β‐oxidation capacity in L‐G6pc ‐/‐ mice and wildtype littermates under fed and fasted conditions (n=5‐8). (E) Western blot for ApoB100 and ApoB48 in nascent VLDL isolated from fed and fasted L‐G6pc ‐/‐ mice (indicated by ‐) and wildtype littermates (indicated by +) (n=3) with 125 nmol of TG loaded in each lane. (F) Hepatic gene expression in L‐G6pc ‐/‐ mice and wildtype littermates under fed and fasted conditions (n=7‐8).
Supplementary Figure 3. Schematic overview of physiological adaptations in normo‐ and hypoglycemic L‐G6pc ‐/‐ mice.
In normoglycemic L‐G6pc ‐/‐ mice, hepatic de novo fatty acid synthesis contributes to hepatic lipid accumulation (left picture). In hypoglycemic L‐G6pc ‐/‐ mice, enhanced adipose tissue lipolysis and hepatic fatty acid elongation contribute to hepatic TG accumulation (right picture). VLDL‐TG levels and –secretion rates are increased in L‐G6pc ‐/‐ mice under both normo‐ and hypoglycemic conditions. VLDL catabolism is selectively inhibited in hypoglycemic L‐G6pc ‐/‐ mice, and associated with higher NEFA levels compared to normoglycemic L‐G6pc ‐/‐ mice.
Abbreviations: G6P, glucose‐6‐phosphate; G6PC, glucose‐6‐phosphatase; IDL, intermediate density lipoproteins; LDL, low density lipoprotein; LPL, lipoprotein lipase; NEFA, non‐esterified fatty acids; TG, triglycerides; VLDL, very low density lipoprotein.
Table S1. Demographics of GSD Ia patients
Table S2. Hepatic fatty acids (μmol/g liver) in liver of L‐G6pc ‐/‐ mice and wildtype littermates in fed state or after an overnight fast
Table S3. Individual enzyme activities and gene expression levels, normalized to values in fed L‐G6pc +/+ mice.
Table S4. Fractional and absolute synthesis of C18:0 in L‐G6pc ‐/‐ mice and wildtype littermates in fed state or after an overnight fast
Table S5. Non‐esterified fatty acids (μmol/L) in serum of normoglycemic GSD Ia patients (n = 7) and age and sex‐matched controls and in plasma of L‐G6pc ‐/‐ mice and wildtype littermates (n = 7‐8) in fed state or after an overnight fast
Table S6. Acylcarnitines (μmol/g) in liver of L‐G6pc ‐/‐ mice and wildtype littermates in fed state or after an overnight fast
Table S7. Taqman and SYBR Green qPCR primer and probe sequences
Table S8. Sequence of peptide standards
Appendix S1: Supporting information
Data Availability Statement
Raw data will be available when requested.