

Abstract
Gliomas are the largest category of primary malignant brain tumors in adults, and glioblastomas account for nearly half of malignant gliomas. Glioblastomas are notoriously aggressive and drug-resistant, with a very poor 5 year survival rate of about 5%. New approaches to treatment are thus urgently needed. We previously identified an enzyme of fatty acid metabolism, very long-chain acyl-CoA synthetase 3 (ACSVL3), as a potential therapeutic target in glioblastoma. Using the glioblastoma cell line U87MG, we created a cell line with genomic deletion of ACSVL3 (U87-KO) and investigated potential mechanisms to explain how this enzyme supports the malignant properties of glioblastoma cells. Compared to U87MG cells, U87-KO cells grew slower and assumed a more normal morphology. They produced fewer, and far smaller, subcutaneous xenografts in nude mice. Acyl-CoA synthetases, including ACSVL3, convert fatty acids to their acyl-CoA derivatives, allowing participation in diverse downstream lipid pathways. We examined the effect of ACSVL3 depletion on several such pathways. Fatty acid degradation for energy production was not affected in U87-KO cells. Fatty acid synthesis, and incorporation of de novo synthesized fatty acids into membrane phospholipids needed for rapid tumor cell growth, was not significantly affected by lack of ACSVL3. In contrast, U87-KO cells exhibited evidence of altered sphingolipid metabolism. Levels of ceramides containing 18-22 carbon fatty acids were significantly lower in U87-KO cells. This paralleled the fatty acid substrate specificity profile of ACSVL3. The rate of incorporation of stearate, an 18-carbon saturated fatty acid, into ceramides was reduced in U87-KO cells, and proteomics revealed lower abundance of ceramide synthesis pathway enzymes. Sphingolipids, including gangliosides, are functional constituents of lipid rafts, membrane microdomains thought to be organizing centers for receptor-mediated signaling. Both raft morphology and ganglioside composition were altered by deficiency of ACSVL3. Finally, levels of sphingosine-1-phosphate, a sphingolipid signaling molecule, were reduced in U87-KO cells. We conclude that ACSVL3 supports the malignant behavior of U87MG cells, at least in part, by altering cellular sphingolipid metabolism.
Keywords: Glioma, GBM, very long-chain acyl-CoA synthetase 3, sphingolipid metabolism, U87MG cells
1. Introduction
Gliomas are the largest category of primary central nervous system (CNS) tumors in adults, accounting for ~80% of all primary intrinsic malignant CNS tumors.1 The World Health Organization (WHO) grades gliomas (Grades I-IV) by a combination of histopathologic and molecular criteria that predict clinical behavior. Low grade infiltrative gliomas (WHO grade II) are well-differentiated with typically indolent growth patterns and better patient prognoses.
Glioblastoma (GBM; WHO grade IV), is the most common and fatal type, accounting for nearly half of primary malignant brain tumors.1 Prognosis for patients with GBM remains very poor (median survival 15-18 moths) despite aggressive standard-of-care surgery, chemotherapy and radiation therapy and there is no therapy proven to substantially prolong survival after tumor recurrence.2,3 Therefore, new treatment strategies and new therapeutic targets are needed.
We previously found high levels of the enzyme very long-chain acyl-CoA synthetase 3 (ACSVL3; SLC27A3) in all grades of glioma histopathological specimens, including GBM.4 In support of our report, a query of The Cancer Genome Atlas TCGA_GBM (HG-UG133A) dataset using the GlioVis portal5 revealed significantly higher ACSVL3 mRNA expression in GBM vs. non-tumor brain tissue (Fig. 1A), and lower ACSVL3 expression was correlated with longer survival (Fig. 1C). In lower grade gliomas, other than anaplastic oligodendrogliomas, ACSVL3 expression levels were also lower and paralleled WHO grading (Fig. 1B).
Figure 1. ACSVL3/SLC27A3 mRNA expression in gliomas.

Using the GlioVis portal, we queried The Cancer Genome Atlas (TCGA) for expression of ACSVL3 in gliomas. In all cases, diagnosis was based on histology. (A) ACSVL3 mRNA expression in GBM (n=528) vs. non-tumor brain tissue (n=10). The TCGA dataset on the HG-U133A platform was used. (B) ACSVL3 mRNA expression in low-grade gliomas vs. GBM. O, oligodendroglioma (n=93); A, astrocytoma (n=131); AO, anaplastic oligodendroglioma (n=58); AA, anaplastic astrocytoma (n=120); GBM (n=225). The Chinese Glioma Genome Atlas dataset was used. (C) Kaplan-Meier survival plot for GBM patients with low (blue line, n=131) vs. high (red line, n=394) ACSVL3 mRNA expression. The cutoff was the lower quartile. Lower ACSVL3 expression was correlated with longer survival (log-rank p value = 8e-04; Wilcoxon p value = 0.0237).
Acyl-CoA synthetases (ACS) activate fatty acids for downstream metabolism by thioesterification to coenzyme A (CoA). The human genome encodes 26 ACSs.6 ACS subfamilies include enzymes with a preference for short-, medium-, long-, and very long-chain fatty acids. ACSVL3 is one of six members of the very long-chain family. U87MG cells, originally isolated from a 44 year old Caucasian male with GBM, and their tumor xenografts were used to investigate the mechanistic role of ACSVL3 in GBM.
We previously demonstrated that knockdown of ACSVL3 levels by RNA interference decreased the malignant behavior of this GBM model.4 Growth rates of monolayer cultures were significantly reduced, and fewer colonies were formed in suspension cultures. ACSVL3 knockdown U87MG cells produced fewer and slower-growing xenografts when implanted subcutaneously in nude mice, and growth of orthotopic intracranial xenografts was reduced by more than 85%. However, the precise role of ACSVL3 in fatty acid and lipid metabolism has not been elucidated. In this paper, we demonstrate that genomic knock-out of ACSVL3 in U87MG cells affects sphingolipid metabolism, and provides mechanistic insight into how this enzyme of fatty acid metabolism may be affecting GBM malignancy.
2. Materials and Methods
2.1. Reagents and general methods.
The following [1-14C]fatty acids were purchased from Moravek, Inc.: acetic (MC125), palmitic (MC121), stearic (MC172), behenic (MC2202), and lignoceric (MC1315). Western blotting was carried out as described previously,7 using polyclonal antibody to ACSVL3 produced in rabbits and purified as described previously4; the secondary antibody was goat anti-rabbit IgG horseradish peroxidase conjugate from Santa Cruz Biotechnology (sc2054). Lipid rafts were visualized using the Vybrant Lipid Raft Labeling kit (Molecular Probes) according to the manufacturer’s instructions. Unless otherwise noted, protein was measured by the method of Lowry et al.8 Results are presented as mean ± standard deviation. Except where noted, statistical significance was determined using Student’s t-test.
2.2. Production of ACSVL3 knockout cell line and culture conditions.
The U87 MG wild type (U87MG) cell line was obtained from American Type Culture Collection (Manassas, VA). U87MG cells were authenticated within a year of study by the Johns Hopkins Genetics Resources Core Facility by short tandem repeat analysis using the PowerPlex 16 HS kit (Promega). Unless otherwise indicated, all cell culture components were from Corning Cellgro. Cells were cultured in MEM (minimum essential medium, Eagle) containing 10% fetal bovine serum (FBS; Gemini Bioproducts) and supplemented with 10 mM Hepes, 1 mM pyruvate, and nonessential amino acids. Cells were incubated at 37°C in a humidified incubator with 5% CO2. The U87MG ACSVL3 knock out (U87-KO) cell line was produced using the zinc finger nuclease (ZFN) method, using reagents designed and supplied by Sigma-Aldrich (CompoZr™ Knockout Zinc Finger Nucleases). Two plasmids were obtained; pZFN1 (5’-TGTGCTTTCCCACCCTTCTA-3’) targets the forward genomic DNA strand just upstream of exon 2, and pZFN2 (5’- AGGTGAGGAGACTGGGGAGT-3’) targets the reverse strand just downstream of exon 2. Plasmids were co-transfected into U87MG cells using FuGENE (Promega). After 24 hours, cells were switched to culture medium containing Hygromycin (Roche; 0.2 mg/ml) to select for candidate knockout clones. After 6 days, living cells were harvested by gentle trypsinization and counted using a hemocytometer. Cells were plated into 96-well plates at an average density of 0.9 cells/well to produce clones generated from single cells. Clones were expanded and tested for ACSVL3 knockout by Western blot using affinity-purified anti-rabbit polyclonal antibody as previously described.9 Both genomic DNA and cDNA were prepared from clones and used for polymerase chain reaction- (PCR-) mediated amplification of the ZFN-targeted region. PCR was performed in a T100 Thermal Cycler (Bio-Rad). Samples amplified over 35 cycles using Phusion High Fidelity Polymerase (New England Biosciences). For each cycle the following temperatures and times were used: initial melting occurred at 98°C for 30 sec; melting 98°C for 8 sec; primers annealed at 58°C for 20 sec; extension at 72°C for 18 sec. Final extension was at 72°C for 8 min. ACSVL3 ZFN deletion forward primer sequence; 5’-CCTGCTGGAATTAGCGATT-3’. ACSVL3 ZFN deletion reverse primer sequence; 5’ –TGCCAGAGGTGAAGATGTA-3’. Cell growth rates in monolayer culture were assessed as previously described.4
2.3. Mouse xenografts.
All animal protocols were approved by the Johns Hopkins University Animal Care and Use Committee (ACUC). In vivo tumorigenesis of U87MG and U87-KO cells was assessed in 4- to 6-week old female mice as previously described.10 NIH III Xid/Beige/Nude mice (National Cancer Institute, Frederick, MD) were injected with 4 x 106 cells in 0.1 ml of PBS in the dorsal areas; both left and right flanks were injected. Tumor size was measured using calipers, and volume was estimated by the following formula: volume = (length x width2) / 2. Mice that were injected with U87MG cells were sacrificed 15 days after injection for humane considerations, and those that were injected with U87-KO cells were sacrificed on day 19. The xenografts were excised and weighed.
2.4. Acyl-CoA synthetase and fatty acid β-oxidation assays.
Activation of [1-14C]fatty acids to their CoA derivatives was assayed as previously described.7 Briefly, 5 nmol labeled fatty acid (~20,000 dpm/nmol) was solubilized using α-cyclodextrin and was incubated in a total volume of 0.25 ml with 40mM Tris, pH 7.5, 10mM ATP, 10mM MgCl2, 0.2 mM CoA, 0.2 mM dithiothreitol, and cell protein (15-50 μg) for 20 min at 37 °C. Reactants (fatty acids) and products (acyl-CoA) were separated by the method of Dole,11 and the latter quantitated by liquid scintillation counting.
To measure β-oxidation of [1-14C]palmitic acid to water-soluble products, cells were grown to confluence in triplicate wells of a 6-well plate. A labeling mix was prepared so that 0.75 ml contained 7.5 nmol radiolabeled palmitic acid (solubilized using α-cyclodextrin; ~20,000 dpm/nmol) and 2 mM carnitine in serum-free MEM. Growth medium was removed and the cells were washed with serum-free MEM. For each cell line, 0.75 ml labeling mix was added to duplicate wells; the third well (for protein assay) received 0.75 ml of MEM only. Cells were incubated in a CO2 incubator for 2 hr at 37°C, at which time 0.15 ml of 18% HClO4 was added to the label-containing duplicate wells. Plates were placed on ice for 1 hr. An aliquot (0.45 ml) of radiolabeled cell medium was extracted by the method of Folch et al.12 to separate the water-soluble products of β-oxidation from the 14C fatty acid substrate. Water-soluble radioactivity was quantitated by liquid scintillation counting. The wells that contained MEM only were washed, and cells were solubilized in 0.5 ml 1N NaOH for protein determination using the method of Lowry et al.8
2.5. Neutral lipid and phospholipid synthesis.
Cells were allowed to grow to confluence in 10 cm dishes. [1-14C]acetate (2 μCi) was added to each plate and allowed to incubate at 37°C in a 10% CO2 environment for 30 min. After incubation, the cells were harvested using trypsin and were washed twice with PBS. Portions of cell suspension containing 400 μg protein were extracted with chloroform:methanol (2:1) and subjected to thin-layer chromatography and visualization as described previously.7
2.6. Analysis of ceramides by LC-MS/MS.
Trypsinized cells were pelleted, washed, resuspended in 200 μl of dH2O, and disrupted using a three 5 second pulses with a probe sonicator (Missonix). 600 μl of methanol containing internal standard (C12:0 ceramide, Avanti Polar Lipids) was added, mixed, and then 800 μl of chloroform was then added. Samples were thoroughly mixed and then centrifuged at 14,000 rpm for 5 minutes to separate organic and aqueous phases cleanly. The separated lower organic phase was dried, resuspended, transferred to a separate glass vial and analyzed via LC-MS/MS.13-15
2.7. Ceramide Synthesis Assay.
For assay of fatty acid incorporation into ceramides, 5 nmol labeled fatty acid (~20,000 dpm/nmol) was solubilized using α-cyclodextrin and was incubated in a total volume of 0.25 ml with 40mM Tris, pH 8.0, 10mM ATP, 10mM MgCl2, 0.2 mM CoA, 0.2 mM dithiothreitol, 1 mM Serine, 0.2 mM NADPH, and cell protein (250 μg) for 2 hr at 37°C with gentle shaking. The reaction was stopped with 3.75 ml of 1:1 chloroform:methanol. Samples were extracted by the method of Folch et al.,12 and the lower (organic) phase was dried under N2. The resulting lipid pellet was resuspended in 50 μl 1:1 hexane:ethanol; 20 μl was spotted onto a silica gel G thin-layer chromatography plate that was developed with chloroform:MeOH:glacial acetic acid (94:1:5). Labeled ceramides were detected using a Fuji-Bas phosphorimager .
2.8. Sphingosine and sphingosine-1-phosphate (S1P) analysis.
Cells were grown to near confluence, harvested by scraping, and extracted using a modification of the method of Shaner et al.16 400 μg cell protein was extracted with 0.5 ml MeOH, 0.25 ml chloroform, and the internal standard cocktail for a total of 1 ng of each standard per sample (C17:0 sphingosine 1-phosphate, C17:0 sphinganine 1-phosphate, and C17:0 sphingosine; Avanti Polar Lipids). Samples were dispersed using a water bath sonicator and incubated at 48°C overnight. After cooling to room temperature, 75 μl of 1M KOH in MeOH was added to each tube and, after brief sonication, tubes were incubated for 2 hr at 37°C to cleave potentially interfering glycerolipids. After cooling to room temperature and neutralization with glacial acetic acid, tubes were centrifuged. Supernatants were further clarified by centrifugation through a Spin-X filter (Corning) and analyzed by LC-MS/MS as described.17,18
2.9. Ganglioside extraction and visualization.
U87MG and U87-KO cells were grown to near confluence, washed with phosphate-buffered saline, and harvested using a cell scraper. Gangliosides were extracted and separated by thin-layer chromatography as described by Schnaar and Needham.19 Gangliosides on thin-layer plates were visualized with resorcinol (0.3% resorcinol, 0.0031% CuSO4·5H2O, 30% HCl) spray. Reagents and ganglioside standards were the kind gift of Ronald Schnaar, Johns Hopkins Department of Pharmacology and Experimental Therapeutics.
2.10. Proteomic analysis.
U87MG and U87-KO cells were grown to near confluence. Biological replicates were harvested by gentle trypsinization and processed for analysis by LC-MS/MS as previously described.20,21 Briefly, cells were solubilized, reduced and alkylated, and digested with trypsin. Digested peptides were purified, lyophilized, and subjected to tandem mass tag (TMT) labeling, after which samples were pooled, vacuum dried, and fractionated by high pH reversed-phase liquid chromatography. Twenty-four fractions were collected and were subjected to LC-MS/MS analysis using an Orbitrap Elite mass spectrometer (Thermo Electron, Bremen, Germany) interfaced with Easy-nLC II nanoflow liquid chromatography system (Thermo Scientific, Odense, Denmark). Raw data was processed through the Proteome Discoverer 2.1.0.81 software suite to generate peak list files for the database searches. Data was searched against the human Refseq 73 protein database. Combined Sequest and Mascot search algorithms were used for the peptide identification; peptide quantification was carried out using reporter ion quantifier.
3. Results
3.1. Knockout of ACSVL3 decreases the malignant behavior of GBM cells.
We previously reported that siRNA-mediated knockdown of ACSVL3 in U87MG cells reduced growth in both monolayer and suspension culture, decreased tumor propagating potential and reduced tumor xenograft growth rates.4 To investigate mechanisms underlying these observations, we produced U87MG cells with homozygous genomic KO of ACSVL3 using zinc-finger nuclease technology. The resulting U87-KO cells were found to have an in-frame 210 bp deletion that included most of exon 2 (Fig. 2A). This deletion included part of Motif I, a catalytically critical region that forms part of the AMP-binding domain found in all ACSs.6,22,23 The deletion was confirmed by PCR (Fig. 2B) and Western blot (Fig. 2C).
Figure 2.
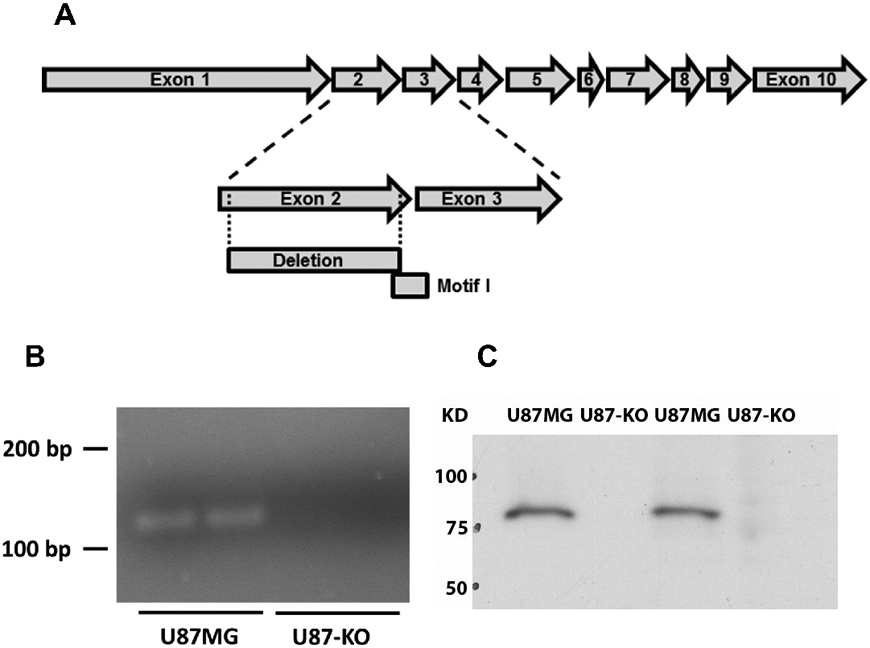
Creation of a U87MG cell line with genomic deletion of ACSVL3 (U87-KO). (A) Zinc-finger nuclease was used to delete a portion of ACSVL3 genomic DNA in U87MG cells. Sequencing of the U87-KO cell line revealed an in-frame 210 bp deletion in both genomic DNA and mRNA. The deletion contained most of Exon 2, and included a region encoding several amino acids of Motif I, a catalytically critical domain. (B) PCR amplification using primers specific for the deletion region show that DNA is amplified in the U87MG cells, and not the U87-KO line. (C) Western blotting shows a depletion of ACSVL3 in the U87-KO cell line.
When grown in monolayer culture, morphologic differences between U87MG and U87-KO cells were apparent. U87-KO cells (Fig. 3A, right) displayed a larger, flatter, and more contact-inhibited appearance than the spindly U87MG-WT cells (Fig. 3A, left) lacking contact-inhibition. The growth rate of U87-KO cells was significantly slower than that of U87MG cells (Fig. 3B) as evidenced by 88% fewer U87-KO cells than U87MG cells on post-inoculation day 11, a statistically significant difference (p<0.005).
Figure 3.
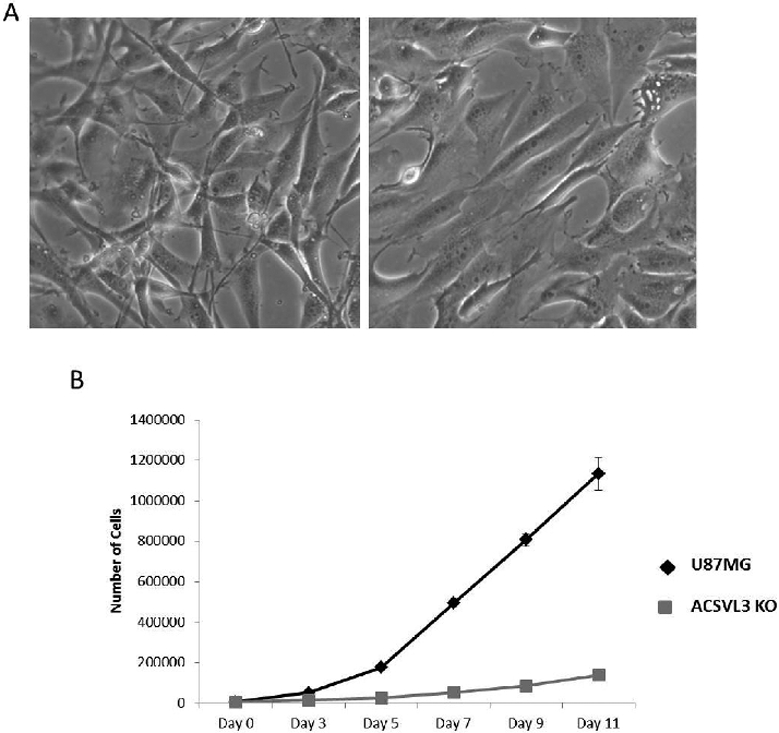
ACSVL3 KO cells have an altered morphology and grow significantly slower than the U87MG cells. (A) Morphology. Light microscopy reveals differences in morphology between the U87MG cells (left) and the U87-KO cells (right). The ACSVL3-deficient cells appear larger, flatter, take up more surface area, and exhibit less overgrowth. (B) Growth rate in monolayer culture was assessed as described in Methods. U87-KO cells (gray squares) grew significantly slower than U87MG cells (black diamonds).
To verify that in vivo tumorigenesis was reduced by ACSVL3 KO, nude mice were subcutaneously implanted with U87MG or U87-KO cells, and formation of xenografts was monitored. For each cell type, 5 mice were injected in both flanks. After one week of growth, the tumors were measured with calipers every other day for width and length (Fig. 4C). In mice bearing U87MG xenografts, tumors became quite large (Fig. 4A, B), necessitating sacrifice on day 15 for humane reasons; mice injected with U87-KO cells were sacrificed on day 19.
Figure 4. ACSVL3 depletion in U87MG cells produces fewer, smaller subcutaneous tumors in nude mice.

Either U87MG or U87-KO cells were injected into both flanks of nude mice (5 mice per group; 4 x 106 cells/injection site). (A) By day 15 post-injection, large xenografts were apparent at all injection sites in mice injected with U87MG cells (bottom row), and mice were humanely euthanized. Xenografts grown from U87-KO cells (top row) were allowed to grow to day 19 and were observed to be smaller, and in some cases, not readily detected. (B) After sacrifice on day 15 (U87MG0 or day 19 (U87-KO), xenografts were resected and weighed. Representative tumors produced from U87MG (left) and U87-KO cells (right) are shown. (C) Tumor size was measured throughout the study using calipers. U87MG xenografts grew significantly larger than U87-KO xenografts. Growth rates were significantly different (p<0.001) at all time points. (D) After harvesting, the tumors were weighed. Xenografts failed to initiate at 3 out of 10 injection sites for U87-KO cells, whereas tumors grew at all injection sites for U87MG cells. The U87MG tumors (n=10) weighed about 6-fold more than the U87-KO tumors (n=7). The difference was statistically significant (p<0.001).
Xenografts formed at all 10 sites in mice injected with U87MG cells. In contrast, tumors failed to form at 3 out of 10 sites injected with U87-KO cells. After sacrifice, U87MG xenografts were found to weigh significantly more than the U87-KO xenografts (Fig. 4D). The results of both in vitro and in vivo studies corroborate the results that were obtained previously with ACSVL3 knockdown cell lines.
3.2. Fatty acid β-oxidation and de novo membrane phospholipid synthesis are not affected in U87MG cells lacking ACSVL3.
Several possible mechanisms including effects on fatty acid β-oxidation and complex lipid synthesis were considered to explain why depleting U87MG cells of ACSVL3 inhibited cell growth and tumorigenesis. Cancer cells rely on glycolysis rather than fatty acid catabolism for energy production, while fatty acid synthesis is accelerated to provide substrates for the synthesis of membrane phospholipids needed by rapidly dividing cancer cells.24
To determine whether lack of ACSVL3 affected mitochondrial β-oxidation, we measured the ability of intact U87MG and U87-KO cells to degrade the 16-carbon saturated fatty acid, palmitate. Cells with a less malignant phenotype, such as U87-KO cells, might be expected to have increased rates of β-oxidation. However, U87-KO cells degraded palmitate at a rate of 0.53±0.08 nmol/hr/mg protein, which was slightly lower than that measured in U87MG cells (0.65±0.10 nmol/hr/mg protein); this difference was not statistically significant.
To sustain rapid proliferation, cancer cells rely on a continuous supply of phospholipids, particularly the major membrane lipids phosphatidylcholine (PC) and phosphatidylethanolamine (PE). To determine whether ACSVL3 depletion affected de novo phospholipid synthesis, U87MG and U87-KO cells were incubated with [1-14C]acetate. Lipids were extracted and phospholipid species separated by thin-layer chromatography.
As shown in Fig. 5, right, lack of ACSVL3 did not decrease the synthesis of PC or PE, as would be expected if this were the mechanism of decreased cell growth. We did note a slight decrease in synthesis of phosphatidylinositol, and an increase in phosphatidylserine synthesis. We also assessed [1-14C]acetate incorporation into neutral lipids (Fig 5, left). Although lack of ACSVL3 in KO cells had little effect on synthesis of cholesterol, free fatty acids, and triacylglycerol, there was significantly decreased synthesis of cholesterol esters.
Figure 5. Lipid synthesis is not significantly affected by depletion of ACSVL3 in U87MG cells.
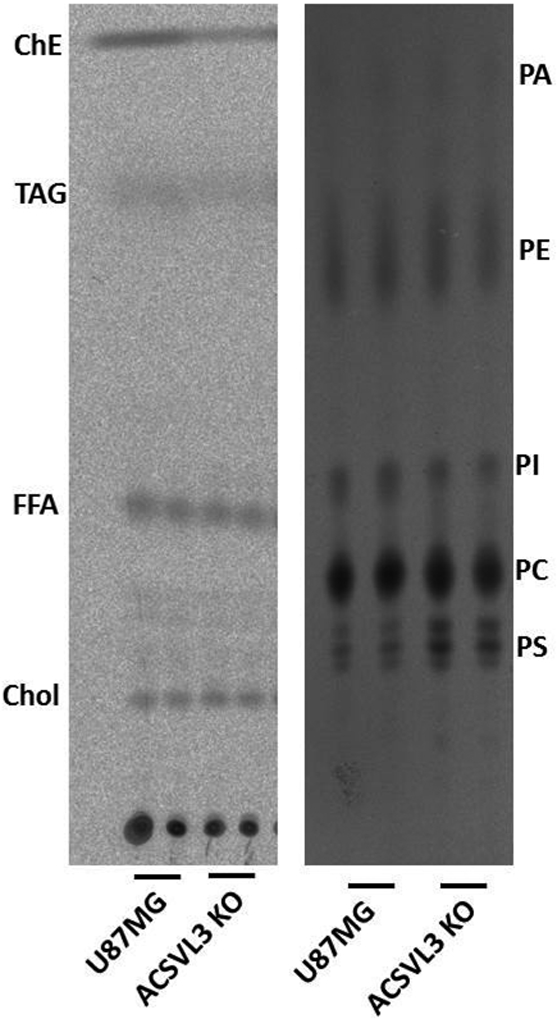
Lipids from cells incubated with [1-14C]acetate were separated using thin layer chromatography. Left, neutral lipids. Chol, cholesterol; FFA, free fatty acids; TAG, triacylglycerol; ChE, cholesterol esters. Diacylglycerols were observed as faint bands running between Chol and FFA. Right, phospholipids. PA, phosphatidic acid; PE, phosphatidylethanolamine; PI, phosphatidylinositol; PC, phosphatidylcholine; PS, phosphatidylserine. There were no significant differences in de novo lipid synthesis in ACSVL3-deficient KO cells when compared to U87MG cells.
3.3. U87MG cells lacking ACSVL3 have decreased levels of ceramides.
Our earlier investigations of U87MG cells with RNAi-mediated ACSVL3 knockdown revealed aberrant receptor tyrosine kinase-Akt pathway signaling.4 Plasma membrane microdomains, or lipid rafts, are thought to serve as platforms for signal transduction receptors, including receptor tyrosine kinases and G-protein coupled receptors.25,26 These microdomains are enriched in sphingolipids and cholesterol.27 Ceramide, itself a sphingolipid, is a central intermediate in the synthesis of more complex sphingolipids such as glycosphingolipids and sphingomyelin.
Therefore, we quantitated several ceramide species in U87MG and U87-KO cells. Ceramides contain two acyl chains. One, in the sphingoid base, is most often C18:1 (or C18:0 in dihydroceramides). The other, more variable, acyl group is amide-linked to the sphingoid base. As shown in Fig. 6 and Table 1, ceramides were generally lower in U87-KO cells, particularly those containing C18:0, C20:0, and C22:0 fatty acyl chains. Most of these changes were highly statistically significant. Interestingly, the levels of more abundant C16:0 and C24:0 ceramides were less affected by lack of ACSVL3.
Figure 6. Ceramide levels, particularly those with C18:0-C22:0 acyl chains , are decreased in U87-KO cells.
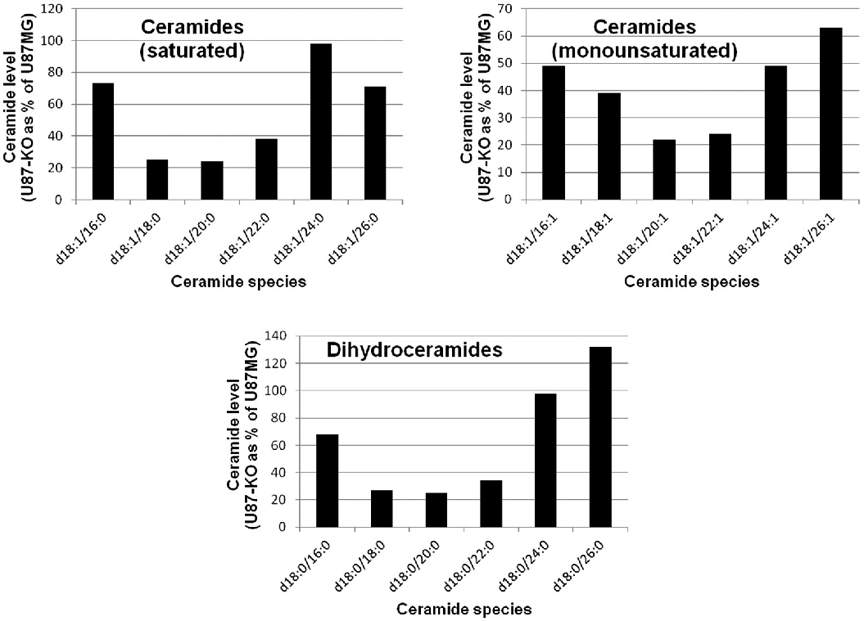
Ceramides were extracted from U87MG and U87-KO cells and quantitated by LC-MS/MS as described in Methods. Analyte levels were normalized by the amount of internal standard (IS) recovered. The amount of each ceramide species in U87-KO cells as a percentage of that in U87MG cells is shown. (Numerical data and statistics are found in Table 2.) Ceramides contain 2 acyl chains. The first listed (e.g. d18:1) is that in the sphingoid base, while the second listed (e.g. 16:0) is the amide-linked acyl group. Ceramides containing amide-linked C18-C22 acyl chains were, in general, more dramatically lowered by ACSVL3 KO than were C16, C24, and C26 acyl chains.
Table 1. Ceramide levels in U87MG and U87-KO cells.
Ceramides were extracted from quadruplicate plates of U87MG and U87-KO cells and quantitated by LC-MS/MS as described in Methods. Analyte levels were normalized by the amount of internal standard (IS) recovered. Mean ± SD is shown. Levels of ceramides were generally lower in the U87-KO cells, particularly in species containing 18-22 carbons in their amide-linked acyl chain. The significance of these changes were assessed using two-tailed Student’s t-test (*, p<0.05; ***, p<0.001). The percent changes as a result of ACSVL3 deficiency are shown in the last column, and are illustrated graphically in Figure 5.
| U87MG | U87-KO | Signifi- cance |
U87-KO as percent of U87MG |
|
|---|---|---|---|---|
| (analyte/IS) | ||||
| (mean ± SD) | ||||
| Ceramides (saturated) | ||||
| d18:1/16:0 | 1.90 ± 0.10 | 1.38 ± 0.08 | *** | 73% |
| d18:1/18:0 | 0.37 ± 0.03 | 0.094 ± 0.008 | *** | 25% |
| d18:1/20:0 | 0.093 ± 0.006 | 0.022 ± 0.002 | *** | 24% |
| d18:1/22:0 | 0.52 ± 0.04 | 0.20 ± 0.01 | *** | 38% |
| d18:1/24:0 | 3.06 ± 0.21 | 2.99 ± 0.08 | 98% | |
| d18:1/26:0 | 0.55 ± 0.10 | 0.39 ± 0.03 | * | 71% |
| Ceramides (monounsaturated) | ||||
| d18:1/16:1 | 0.008 ± 0.001 | 0.004 ± 0.001 | *** | 49% |
| d18:1/18:1 | 0.006 ± 0.000 | 0.002 ± 0.001 | *** | 39% |
| d18:1/20:1 | 0.009 ± 0.001 | 0.002 ± 0.000 | *** | 22% |
| d18:1/22:1 | 0.052 ± 0.005 | 0.012 ± 0.000 | *** | 24% |
| d18:1/24:1 | 0.64 ± 0.04 | 0.31 ± 0.02 | *** | 49% |
| d18:1/26:1 | 0.14 ± 0.01 | 0.087 ± 0.002 | *** | 63% |
| Dihydroceramides | ||||
| d18:0/16:0 | 0.025 ± 0.001 | 0.017 ± 0.001 | *** | 68% |
| d18:0/18:0 | 0.006 ± 0.000 | 0.002 ± 0.000 | *** | 27% |
| d18:0/20:0 | 0.007 ± 0.001 | 0.002 ± 0.000 | *** | 25% |
| d18:0/22:0 | 0.011 ± 0.001 | 0.004 ± 0.001 | *** | 34% |
| d18:0/24:0 | 0.067 ± 0.006 | 0.065 ± 0.003 | 98% | |
| d18:0/26:0 | 0.001 ± 0.000 | 0.001 ± 0.000 | 132% | |
3.4. Substrate specificity of ACSVL3.
The results of ceramide assays prompted us to investigate the ACS enzyme activity profile of U87MG and U87-KO cells. We assessed the ability of cell-derived proteins to activate saturated [1-14C]fatty acids containing 16, 18, 22, and 24 carbons. We found that lack of ACSVL3 significantly (p<0.001) reduced activation of 18- and 22-carbon fatty acids (Fig. 7). A potential decreased activation of the 16- and 24-carbon fatty acids was also observed, but the differences were not statistically significant. The dramatic reduction in specificity for the 18- to 22-carbon fatty acid substrates in U87-KO cells suggests that ACSVL3 may be activating these fatty acids for sphingolipid synthesis.
Figure 7. Total cellular activation of long- and very long-chain fatty acids is lower in ACSVL3 KO cells.
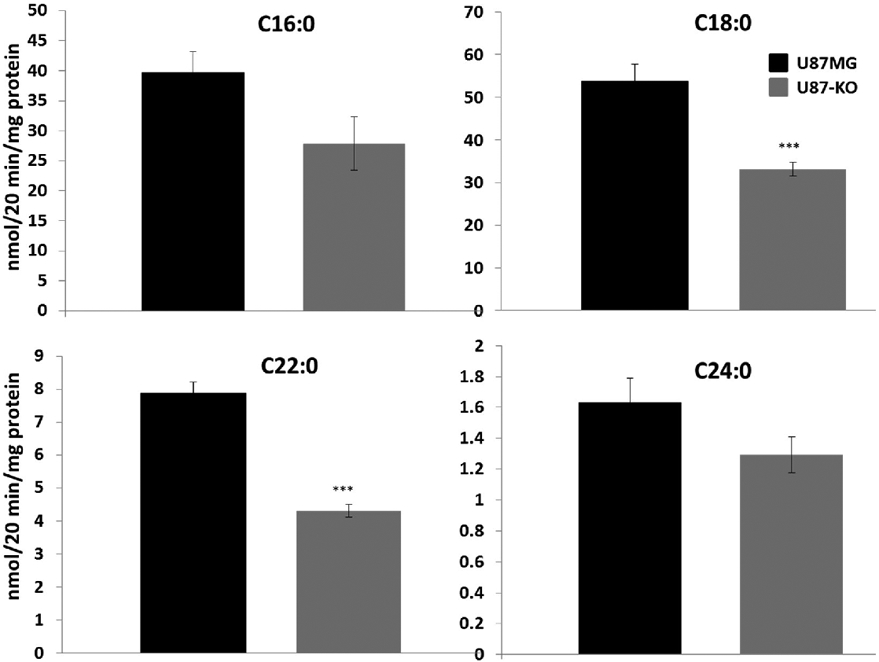
Cells were harvested and assayed for their ability to activate [1-14C]-labeled fatty acids. C16:0, palmitic acid (A) and C18:0, stearic acid (B) are considered long-chain fatty acids, while C22:0, behenic acid (C) and C24:0, lignoceric acid (D) are very long-chain fatty acids. Lack of ACSVL3 significantly (p<0.001) lowered the ability of U87MG cells to activate the 18- and 22-carbon fatty acid substrates. While activation of the 16- and 24-carbon fatty acid substrates trended lower in U87-KO cells, the decrease did not reach statistical significance.
3.5. Ceramide synthesis in U87MG cells lacking ACSVL3.
To explore further the possibility that ACSVL3 may partially control the acyl-chain profile of ceramides, we measured ceramide synthesis. U87MG and U87-KO cell-derived proteins were incubated with saturated [1-14C]fatty acids containing 16, 18, 22, and 24 carbons, and ceramides were resolved by thin-layer chromatography. Incorporation of the 18-carbon fatty acid into ceramides was slightly decreased in U87-KO cells; however, ceramide synthesis from 16-, 22-, and 24-carbon fatty acids was not noticeably reduced (Fig. 8).
Figure 8. Incorporation of radiolabeled C18:0 into ceramide is reduced in U87MG cells lacking ACSVL3.
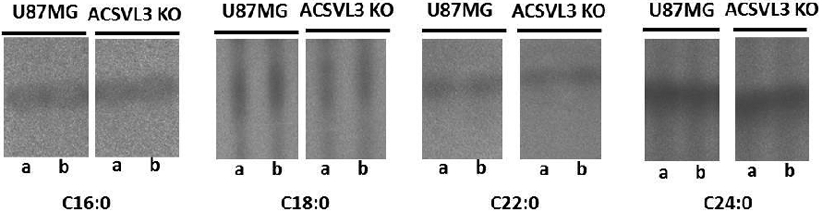
To assess ceramide synthesis, U87MG and U87-KO cells were incubated with [1-14C]-labeled fatty acids. Ceramides were extracted from the cells, and duplicate samples (a, b) were resolved by thin-layer chromatography. Decreased ceramide synthesis was observed with the 18-carbon fatty acid substrate, but not with the 16-, 22-, or 24-carbon fatty acid substrates.
3.6. Reduced levels of ceramide synthesis enzymes in U87-KO cells.
Synthesis of ceramide requires four enzymatic steps, catalyzed by serine palmitoyltransferase (SPTLC), 3-ketosphingosine reductase (KDSR), ceramide synthase (CERS), and dihydroceramide desaturase (DEGS). Proteomic analysis was carried out to determine whether lack of ACSLV3 affected expression of these enzymes. All three isoforms of SPTLC (SPTLC1, SPTLC2, and SPTLC3) were present in U87MG cells, and expression levels of all three were reduced in U87-KO cells (Table 2). SPTLC catalyzes the first, and rate-limiting, step of ceramide synthesis. Three of the 6 known CERS isoforms, CERS2, CERS5, and CERS6, were detected in U87MG cells and found at reduced levels in U87-KO cells. Levels of KDSR and DEGS were unchanged in U87-KO cells. These findings support our conclusion that ceramide synthesis is reduced in GBM cells lacking ACSVL3.
Table 2. Enzymes involved in ceramide synthesis are decreased in U87-KO cells.
Proteomic analysis revealed the presence of several enzymes involved in ceramide synthesis in U87MG and U87-KO cells. Three serine palmitoyltransferases (SPTLC1-3), catalyzing the first and regulated step of the pathway were detected, and the levels of all three were all decreased in cells lacking ACSVL3. Three of six known ceramide synthase isoforms (CERS2, 5, and 6), catalyzing the third step of ceramide synthesis, were present in the data set and all were decreased in U87-KO cells. Levels of the second (3-ketodihydrosphingosine reductase; KDSR) and fourth (dihydroceramide-Δ4-desaturase 1; DEGS1) enzymes of the pathway were unchanged.
| Protein | U87-KO/U87MG |
|---|---|
| SPTLC1 | 0.8 |
| SPTLC2 | 0.7 |
| SPTLC3 | 0.6 |
| KDSR | 1 |
| CERS2 | 0.6 |
| CERS5 | 0.9 |
| CERS6 | 0.5 |
| DEGS1 | 1 |
3.7. Lipid rafts and gangliosides are altered by ACSVL3 depletion.
Because our biochemical data and prior results suggested that lipid raft composition may be changed by ACSVL3 KO, we asked if lipid raft structural alterations were evident. Ganglioside GM1 has been proposed as a molecular marker for rafts.28 Fluorescently labeled cholera toxin B-subunit binds GM1 avidly, and was used to visualize rafts in U87MG and U87-KO cells. We predicted that KO cells would have less raft fluorescence than U87MG cells, reflective of the changes in RTK signaling. In contrast, we found the opposite (Fig. 9A and 9B), as more raft-associated fluorescence was detected in KO cells.
Figure 9. Lipid raft staining and ganglioside composition of U87MG and U87-KO cells.

(A,B) Raft visualization. Rafts in U87MG cells (A) and U87-KO cells (B) were detected by virtue of staining for the raft marker, ganglioside GM1. GM1 avidly binds cholera toxin B-subunit containing the fluorescent dye, alexa fluor 555. Increased raft staining was observed in U87-KO cells (B) relative to U87MG cells (A) suggesting that lack of ACSVL3 altered the cells’ ganglioside composition. C. Gangliosides were extracted and separated by thin-layer chromatography. The overall ganglioside level was decreased by ACSVL3 KO. In agreement with fluorescent labeling, levels of ganglioside GM1 were higher in KO cells, whereas levels of GM3 were higher in U87MG cells. Levels of GM2 (heavier band between GM1 and GM3) were relatively unchanged.
To investigate this further, we extracted gangliosides from U87MG and U87-KO cells and separated ganglioside species by thin-layer chromatography. We found a striking change in ganglioside composition when ACSVL3 was depleted (Fig. 9C). Overall ganglioside levels were lower in KO cells. GM3, a less-complex ganglioside was the predominant species in U87MG cells, and levels were lower in KO cells.
In contrast, GM1 (which contains additional galactose and N-acetyl galactosamine residues compared to GM3) was nearly undetectable in U87MG cells, but was found at higher levels in KO cells. This is in agreement with the fluorescent raft staining pattern (Fig. 8A and 8B). Levels of GM2 (Fig. 8C, band between GM1 and GM3) were relatively unchanged by ACSVL3 depletion. These observations support our contention that ACSVL3 significantly affects sphingolipid metabolism and plasma membrane gangliosides in U87MG cells.
3.8. Levels of sphingosine and sphingosine-1-phosphate (S1P) are reduced in U87-KO cells.
In addition to their structural roles in lipid rafts, sphingolipids can themselves be signaling molecules. The ceramide metabolite, sphingosine, and its phosphorylated derivative, S1P, fall into this category. Ceramidase releases the nitrogen-bound fatty acid from ceramide, producing sphingosine; sphingosine kinases catalyze the phosphorylation of sphingosine to form S1P. We therefore measured these metabolites in U87MG and U87-KO cells. As shown in Fig. 10, there was a strong trend toward lower sphingosine levels in cells lacking ACSVL3 (p=0.06). S1P levels were significantly lower in U87-KO cells (p<0.05).
Figure 10. Sphingosine and S1P levels are decreased in ACSVL3-depleted U87MG cells.
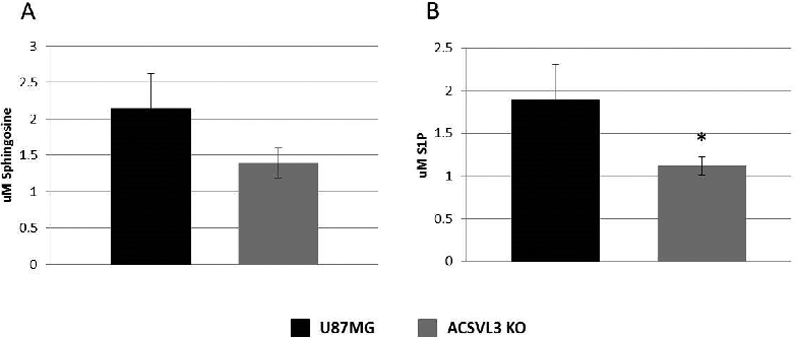
Cells grown to near confluence were harvested, extracted, and sphingosine and S1P levels were quantified using LC-MS/MS as described in Methods. (A) U87-KO cells had lower levels of sphingosine than U87MG cells, but the decrease did not reach statistical significance (p = 0.06). (B) S1P levels in U87-KO cells were significantly lower than in U87 MG cells (p<0.05).
Taken together, results presented here suggest that ACSVL3 maintains the malignant phenotype of U87MG cells, at least in part, by modulating sphingolipid metabolism.
4. Discussion
GBM remains the most deadly CNS malignancy, and continues to present significant therapeutic challenges.1 New drug targets representing novel mechanistic approaches could eventually improve this dire situation. There is a longstanding interest in the aberrant metabolism associated with malignant brain tumors exemplified by studies involving glycolytic metabolism and the Warburg effect29 and the more recent identification of IDH mutations causing excessive production of the onco-metabolite 4-hydroxybutyrate in low-intermediate grade glioma.30,31
We previously used an RNAi-mediated knockdown approach to show that reducing expression of ACSVL3 reduced both the in vitro and in vivo malignant behavior of U87MG cells.4 We have now produced a U87MG cell line with genomic knockout of ACSVL3 and confirmed the previous results suggesting that this fatty acid metabolism enzyme may be a novel therapeutic target in GBM.
Knockout of ACSVL3, a member of the very long-chain acyl-CoA synthetase family, significantly reduced growth rates and tumorigenicity of U87MG cells. Elucidating the mechanisms underlying these beneficial effects is needed both for understanding the pathophysiologic consequences of high levels of ACSVL3 expression in GBM, and for uncovering additional druggable metabolic pathways.
The functional role of ACSVL3 in overall cellular fatty acid metabolism is relatively unknown. Dissecting the role of an individual ACS in a given cell type is complicated by many factors. One such factor is that multiple ACSs are usually present in any given cell type. Our proteomic analysis revealed that U87MG cells express at least seven distinct long- (ACSL1, ACSL3, ACSL4, and ACSL5) and very-long chain (ACSVL3/SLC27A3, ACSVL4/SLC27A4, and ACSVL5/SLC27A1) ACSs.
When we probed The Cancer Genome Atlas (TCGA_GBM, HG-UG133A) dataset, no significant elevation of mRNA encoding ACSL1, ACSL3, ACSL4, and ACSL5 was found. While mean ACSVL3 mRNA expression was 27% higher in GBM vs. non-tumor tissue, levels of three ACSL enzymes were only marginally higher (ACSL1, 2%; ACSL4, 1%; ACSL5, 2%), and ACSL3 expression was 8% lower. SLC27A1 and SLC27A3 were not available in the HG-UG133A dataset.
All ACSs catalyze the ATP-dependent thioesterification of fatty acids to coenzyme A,6,32 and all can utilize a range of fatty acid chain lengths, and not a single fatty acid, as substrate. We previously tested only two fatty acid substrates – the 16-carbon saturated fatty acid (palmitic, a representative long-chain fatty acid) and the 24-carbon saturated fatty acid (lignoceric, a representative very long-chain fatty acid) – and found both to be activated by ACSVL3.9 The present study confirms this, and also finds that the 18- and 22-carbon saturated fatty acids (stearic and behenic, respectively) may be more preferred ACSVL3 substrates.
Although ACSs are categorized structurally by fatty acid substrate chain length preference (e.g. long- or very long-chain families), there is significant inter- as well as intra-family variability regarding substrate preference.6 Further complicating the picture are differing, and sometimes uncertain, subcellular locations of the various ACSs. For ACSVL3, previous studies indicated that the endogenous enzyme is found in small punctuate vesicles that partially colocalize with mitochondria.9,33
Once “activated” by an ACS, fatty acids can participate in numerous downstream pathways, such as degradation for energy production, incorporation into various species of complex lipids (triacylglycerol, phospholipids, sphingolipids, cholesteryl esters, plasmalogens), and covalent modification of proteins. In this work, several of these pathways were examined to determine if any were affected by lack of ACSVL3. Effects of ACSVL3 depletion on energy homeostasis, such as a fuel utilization switch from glucose to fatty acid catabolism, was one possibility. This would result from an increased rate of degradation of fatty acids. However, mitochondrial fatty acid β-oxidation rates were found to be slightly lower, rather than elevated, in U87-KO cells.
Fatty acid synthesis, on the other hand, is accelerated in most cancers, including GBM.24,34,35 This is consistent with a continuous need for membrane biosynthesis by rapidly dividing tumor cells. Fatty acid synthesis, as well as the downstream synthesis of both phospholipids and neutral complex lipids, can be assessed using radiolabeled acetate. Acetate is readily taken up by cells and converted to acetyl-CoA (via a short-chain ACS), the fundamental building block used for fatty acid synthesis.
We expected that knockout of ACSVL3, which dramatically slows the growth rate of U87MG cells, would substantially decrease the rate of fatty acid synthesis. Labeled fatty acids would then be incorporated into bulk membrane phospholipids (phosphatidylcholine and phosphatidylethanolamine) at lower levels. Neither of these predictions proved to be true, suggesting that ACSVL3 has a more subtle effect on lipid metabolism.
Signaling through receptor tyrosine kinases such as hepatocyte growth factor and epidermal growth factor receptor are known to drive the malignant behaviors of U87MG cells, at least in part, via downstream Akt signaling.4,36,37 We previously showed that RNAi-mediated knockdown of ACSVL3 disrupted RTK-Akt signaling.4 Lipid rafts – plasma membrane microdomains that are enriched in sphingolipids and cholesterol – are thought to be organizing centers or platforms for signaling receptors.25,26
Ceramide, itself a sphingolipid, is a requisite common intermediate in the synthesis of complex sphingolipids such as sphingomyelins and glycosphingolipids. Several lines of evidence presented here support the notion that lack of ACSVL3 disrupts ceramide synthesis in U87MG cells. Ceramides containing 18-22 carbon fatty acids were significantly less abundant in U87-KO cells. This fatty acid range parallels the substrate fatty acid chain length we observed for ACSVL3. When the 18-carbon fatty acid was used as substrate, ceramide synthesis was decreased. Proteomic analysis revealed reduced levels of key enzymes in ceramide synthesis in U87-KO cells. Overall, these results suggest that there is a decrease in ceramide synthesis machinery, resulting in lower levels of specific ceramide species, when ACSVL3 is depleted in U87MG cells.
Levels of the ceramide-derived signaling molecule, S1P, was also lower when ACSVL3 was depleted. S1P has been reported to mediate growth, survival, invasion, migration, and angiogenesis in glioblastoma, and is associated with poor survival.38-40 Both the enzyme that synthesizes S1P (sphingosine kinase) and S1P receptors have been proposed as therapeutic targets for glioblastoma.41,42 Interestingly, S1P receptors are G protein-coupled receptors that have been found in lipid rafts.43 S1P was found to be a potent mitogenic factor for several glioblastoma cell lines, stimulating proliferation in U373MG cells through a G-coupled receptor and activating several different signaling pathways.44
Evidence suggests that S1P and its receptors are central players that regulate glioblastoma growth, migration, and invasion, and exogenously added S1P promotes glioma growth and enhances invasiveness.38,44,45 S1P was found to promote signaling through the growth factor receptor tyrosine kinase/phosphoinositide-3-kinase/Akt pathway, and inhibition of sphingosine kinase inhibited growth factor-stimulated Akt phosphorylation.41 This is consistent with our observations that ACSVL3 depletion reduces both S1P levels (this work) and Akt phosphorylation4 in U87MG cells. We were unable to show any changes in expression of enzymes involved in the synthesis of sphingosine (alkaline ceramidase) and S1P (sphingosine kinase), as they were below the level of detection in our proteomic analysis.
Because of the putative role of lipid rafts in oncogenic signal transduction, we also asked whether lack of ACSVL3 affected raft structure. A sphingolipid – ganglioside GM1 – is a widely-used marker for lipid rafts, and we expected that lack of ACSVL3 in U87-KO cells might reduce the number of rafts. Instead, there was significantly more raft staining in U87-KO cells, suggesting an increase rather than a decrease of GM1. Subsequent analysis of gangliosides in U87MG and U87-KO cells confirmed that GM1 was very low in U87MG cells, while GM3 levels were high.
GM3 and GM2 are intermediates in the synthesis of GM1, which is a more complex ganglioside. Addition of N-acetylglucosamine to GM3 yields GM2; further addition of a galactose moiety then produces GM1. The switch from GM3 in U87MG cells to GM1 in U87-KO cells is consistent with the more differentiated morphology of the latter cells. As was the situation for sphingosine and S1P, levels of enzymes of ganglioside synthesis were below the level of detection by proteomic analysis.
Our previous and present studies indicate that ACSVL3 is important to glioma cell proliferation, signaling, and the ability for the cells to form subcutaneous and orthotopic xenografts in nude mice.4 The malignancy-promoting effects of ACSVL3 are not limited to glioblastoma, as we also demonstrated high ACSVL3 levels in multiple types of lung tumor33; furthermore, knockdown of ACSVL3 reduced growth rates in both monolayer and suspension cultures of multiple lung cancer cell lines.33
We ask this fundamental question: Why is this specific ACS – an enzyme whose only known function is to synthesize acyl-CoAs – critical to maintaining the malignant properties of U87MG and other cancer cells? Here, we report that decreased availability of specific fatty acyl-CoA substrates, synthesized by ACSLV3, for sphingolipid synthesis affords one possible explanation. Figure 11 depicts our working model, which incorporates the main findings of this work, and provides a framework for guiding future research. Additional studies will be needed to thoroughly understand the role of ACSVL3 in sphingolipid metabolism and oncogenesis.
Figure 11. Lack of ACSVL3 abrogates malignant properties of U87MG cells via disruption of sphingolipid metabolism.

In this model, decreased activation of fatty acids containing 18-22 carbons (e.g. C18:0, lower left) in U87-KO cells leads to a decrease in synthesis of ceramides containing these fatty acids. This leads to decreased sphingolipid levels and altered ganglioside composition of lipid rafts, which are platforms for growth factor (GF) signaling through receptor tyrosine kinases (RTK). Decreased ceramide synthesis also reduces levels of the signaling sphingolipid, S1P. The combination of low S1P and aberrant GF/RTK signaling diminishes Akt-mediated malignant properties of U87MG cells, such as growth, lipid synthesis, and proliferation.
5. Conclusions
Knockout of the fatty acid metabolism enzyme, ACSVL3, in U87MG cells abrogated malignant properties of this GBM cell line, both in vitro and in mouse xenografts. Lack of ACSVL3 in U87-KO cells did not appreciably affect fatty acid degradation or synthesis, but rather had significant effects on ceramide and sphingolipid metabolism. Altered oncogenic signaling through membrane receptors in U87-KO cells likely results from changes in the sphingolipid and ganglioside composition of lipid rafts. Although additional research is needed to understand more fully the role of ACSVL3 in sphingolipid metabolism, this work reiterates our previous conclusion that ACSVL3 is a promising therapeutic target in GBM, and perhaps other cancers.
8. Acknowledgements
The authors thank Yanqiu Liu for expert technical assistance. We also thank Dr. Ronald Schnaar, Dept. of Pharmacology and Experimental Therapeutics, Johns Hopkins University School of Medicine, for assistance with ganglioside studies, and ganglioside standards. This work was supported by NIH grants R01 NS062043 (PAW) and F31 NS074713 (EAK).
9 References
- 1.Ostrom QT, Patil N, Cioffi G, Waite K, Kruchko C, Barnholtz-Sloan JS. CBTRUS statistical report: Primary brain and other central nervous system tumors diagnosed in the United States in 2013-2017. Neuro Oncol. 2020October30;22(12 Suppl 2):iv1–iv96. doi: 10.1093/neuonc/noaa200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weller M, van den Bent M, Hopkins K, et al. EANO guideline for the diagnosis and treatment of anaplastic gliomas and glioblastoma. Lancet Oncol. 2014;15(9):e395–403. doi: 10.1016/S1470-2045(14)70011-7 [DOI] [PubMed] [Google Scholar]
- 3.Oronsky B, Reid TR, Oronsky A, Sandhu N, Knox SJ. A Review of Newly Diagnosed Glioblastoma. Front Oncol. Published online 2021. February5;10:574012 doi: 10.3389/fonc.2020.574012 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Pei Z, Sun P, Huang P, Lal B, Laterra J, Watkins PA. Acyl-CoA synthetase VL3 knockdown inhibits human glioma cell proliferation and tumorigenicity. Cancer Res. 2009;69(24). doi: 10.1158/0008-5472.CAN-08-4689 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bowman RL, Wang Q, Carro A, Verhaak RGW, Squatrito M. GlioVis data portal for visualization and analysis of brain tumor expression datasets. Neuro Oncol. 2017. January;19(1):139–141. doi: 10.1093/neuonc/now247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Watkins PA, Maiguel D, Jia Z, Pevsner J. Evidence for 26 distinct acyl-coenzyme A synthetase genes in the human genome. J Lipid Res. 2007;48(12). doi: 10.1194/jlr.M700378-JLR200 [DOI] [PubMed] [Google Scholar]
- 7.Jia Z, Moulson CL, Pei Z, Miner JH, Watkins PA. Fatty acid transport protein 4 is the principal very long chain fatty acyl-CoA synthetase in skin fibroblasts. J Biol Chem. 2007;282(28). doi: 10.1074/jbc.M700568200 [DOI] [PubMed] [Google Scholar]
- 8.Lowry OH, Rosebrough NJ, Farr AL, Randall RJ. Protein measurement with the Folin phenol reagent. J Biol Chem. 1951;193:265–275. [PubMed] [Google Scholar]
- 9.Pei Z, Fraisl P, Berger J, Jia Z, Forss-Petter S, Watkins PA. Mouse very long-chain Acyl-CoA synthetase 3/fatty acid transport protein 3 catalyzes fatty acid activation but not fatty acid transport in MA-10 cells. J Biol Chem. 2004;279(52). doi: 10.1074/jbc.M410091200 [DOI] [PubMed] [Google Scholar]
- 10.Laterra J, Rosen E, Nam M, Ranganathan S, Fielding K, Johnston P. Scatter factor/hepatocyte growth factor expression enhances human glioblastoma tumorigenicity and growth. Biochem Biophys Res Commun. 1997;235(3):743–747. doi: 10.1006/bbrc.1997.6853. [DOI] [PubMed] [Google Scholar]
- 11.Dole VP. A relation between non-esterified fatty acids in plasma and the metabolism of glucose. J Clin Invest. 1956;35:150–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Folch J, Lees M, Sloane-Stanley GH. A simple method for the isolation and purification of total lipids from animal tissues. J Biol Chem. 1957;226:457–509. [PubMed] [Google Scholar]
- 13.Bandaru VVR, Patel N, Ewaleifoh O, Haughey NJ. A failure to normalize biochemical and metabolic insults during morphine withdrawal disrupts synaptic repair in mice transgenic for HIV-gp120. J Neuroimmune Pharmacol. 2011. December;6(4):640–9. doi: 10.1007/s11481-011-9289-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.McFadden JW, Aja S, Li Q, et al. Increasing fatty acid oxidation remodels the hypothalamic neurometabolome to mitigate stress and inflammation. PLoS One. 2014. December26;9(12):e115642. doi: 10.1371/journal.pone.0115642 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Bandaru VVR, Troncoso J, Wheeler D, et al. ApoE4 disrupts sterol and sphingolipid metabolism in Alzheimer’s but not normal brain. Neurobiol Aging. 2009. April;30(4):591–9. doi: 10.1016/j.neurobiolaging.2007.07.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaner RL, Allegood JC, Park H, et al. Quantitative analysis of sphingolipids for lipidomics using triple quadrupole and quadrupole linear ion trap mass spectrometers. J Lipid Res. 2009. August;50(8):1692–707. doi: 10.1194/jlr.D800051-JLR200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Sullards MC, Merrill AH. Analysis of sphingosine 1-phosphate, ceramides, and other bioactive sphingolipids by high-performance liquid chromatography-tandem mass spectrometry. Sci STKE. 2001. January30;2001(67):pl1. doi: 10.1126/stke.2001.67.pl1 [DOI] [PubMed] [Google Scholar]
- 18.Petrache I, Natarajan V, Zhen L, et al. Ceramide upregulation causes pulmonary cell apoptosis and emphysema-like disease in mice. Nat Med. 2005. May;11(5):491–8. doi: 10.1038/nm1238 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Schnaar RL, Needham LK. Thin-layer chromatography of glycosphingolipids. Methods Enzym. 1994;230:371–389. doi: 10.1016/0076-6879(94)30025-9. [DOI] [PubMed] [Google Scholar]
- 20.Nirujogi RS, Wright JD, Manda SS, et al. Phosphoproteomic analysis reveals compensatory effects in the piriform cortex of VX nerve agent exposed rats. Proteomics. 2015. January;15(2-3):487–99. doi: 10.1002/pmic.201400371 [DOI] [PubMed] [Google Scholar]
- 21.Kim MS, Pinto SM, Getnet D, et al. A draft map of the human proteome. Nature. 2014. May29;509(7502):575–81. doi: 10.1038/nature13302 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Weimer PJ. End product yields from the extraruminal fermentation of various polysaccharide, protein and nucleic acid components of biofuels feedstocks. Bioresour Technol. 2010;102(3):3254–3259. doi:S0960-8524(10)01852-3 [pii] 10.1016/j.biortech.2010.11.050 [DOI] [PubMed] [Google Scholar]
- 23.Zou Z, DiRusso CC, Ctrnacta V, Black PN. Fatty acid transport in Saccharomyces cerevisiae. Directed mutagenesis of FAT1 distinguishes the biochemical activities associated with Fat1p. J Biol Chem. 2002;277(34):31062–31071. doi: 10.1074/jbc.M205034200. Epub 2002 Jun 6. PMID: 12052836. [DOI] [PubMed] [Google Scholar]
- 24.Röhrig F, Schulze A. The multifaceted roles of fatty acid synthesis in cancer. Nat Rev Cancer. 2016. November;16(11):732–749. doi: 10.1038/nrc.2016.89 [DOI] [PubMed] [Google Scholar]
- 25.Mollinedo F, Gajate C. Lipid rafts as major platforms for signaling regulation in cancer. Adv Biol Regul. 2015. January;57:130–46. doi: 10.1016/j.jbior.2014.10.003 [DOI] [PubMed] [Google Scholar]
- 26.Villar VAM, Cuevas S, Zheng X, Jose PA. Localization and signaling of GPCRs in lipid rafts. Methods Cell Biol. 2016;132:3–23. doi: 10.1016/bs.mcb.2015.11.008 [DOI] [PubMed] [Google Scholar]
- 27.Lingwood D, Simons K. Lipid rafts as a membrane-organizing principle. Science (80- ). 2010. January1;327(5961):46–50. doi: 10.1126/science.1174621 [DOI] [PubMed] [Google Scholar]
- 28.Simons K, Ikonen E. Functional rafts in cell membranes. Nature. 1997. June5;387(6633):569–72. doi: 10.1038/42408 [DOI] [PubMed] [Google Scholar]
- 29.Strickland M, Stoll EA. Metabolic reprogramming in glioma. Front Cell Dev Biol. 2017. April26;5:43. doi: 10.3389/fcell.2017.00043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Yan H, Parsons DW, Jin G, et al. IDH1 and IDH2 Mutations in Gliomas . N Engl J Med. 2009. February 19;360(8):765–73. doi: 10.1056/nejmoa0808710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Clark O, Yen K, Mellinghoff IK. Molecular pathways: Isocitrate dehydrogenase mutations in cancer. Clin Cancer Res. Published online 2016. doi: 10.1158/1078-0432.CCR-13-1333 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Watkins PA. Fatty acid activation. Prog Lipid Res. 1997;36(1). doi: 10.1016/S0163-7827(97)00004-0 [DOI] [PubMed] [Google Scholar]
- 33.Pei Z, Fraisl P, Shi X, et al. Very Long-Chain Acyl-CoA Synthetase 3: Overexpression and Growth Dependence in Lung Cancer. PLoS One. 2013;8(7). doi: 10.1371/journal.pone.0069392 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liu Q, Luo Q, Halim A, Song G. Targeting lipid metabolism of cancer cells: A promising therapeutic strategy for cancer. Cancer Lett. 2017. August10;401:39–45. doi: 10.1016/j.canlet.2017.05.002 [DOI] [PubMed] [Google Scholar]
- 35.Wakamiya T, Suzuki SO, Hamasaki H, et al. Elevated expression of fatty acid synthase and nuclear localization of carnitine palmitoyltransferase 1C are common among human gliomas. Neuropathology. 2014. October;34(5):465–74. doi: 10.1111/neup.12132 [DOI] [PubMed] [Google Scholar]
- 36.Zhang A, Long W, Guo Z, Guo Z, Cao BB. Downregulation of hepatoma-derived growth factor suppresses the malignant phenotype of U87 human glioma cells. Oncol Rep. 2012. July;28(1):62–8. doi: 10.3892/or.2012.1768 [DOI] [PubMed] [Google Scholar]
- 37.Huang W, Ding X, Ye H, Wang J, Shao J, Huang T. Hypoxia enhances the migration and invasion of human glioblastoma U87 cells through PI3K/Akt/mTOR/HIF-1α pathway. Neuroreport. 2018. December12;29(18):1578–1585. doi: 10.1097/WNR.0000000000001156 [DOI] [PubMed] [Google Scholar]
- 38.Van Brocklyn JR, Letterle CA, Snyder PJ, Prior TW. Sphingosine-1-phosphate stimulates human glioma cell proliferation through Gi-coupled receptors: Role of ERK MAP kinase and phosphatidylinositol 3-kinase β. Cancer Lett. 2002. July26;181(2):195–204. doi: 10.1016/S0304-3835(02)00050-2 [DOI] [PubMed] [Google Scholar]
- 39.Young N, Van Brocklyn JR. Roles of sphingosine-1-phosphate (S1P) receptors in malignant behavior of glioma cells. Differential effects of S1P2 on cell migration and invasiveness. Exp Cell Res. 2007. May1;313(8):1615–27. doi: 10.1016/j.yexcr.2007.02.009 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lepley D, Paik JH, Hla T, Ferrer F. The G protein-coupled receptor S1P2 regulates Rho/Rho kinase pathway to inhibit tumor cell migration. Cancer Res. 2005. May1;65(9):3788–95. doi: 10.1158/0008-5472.CAN-04-2311 [DOI] [PubMed] [Google Scholar]
- 41.Kapitonov D, Allegood JC, Mitchell C, et al. Targeting sphingosine kinase 1 inhibits Akt signaling, induces apoptosis, and suppresses growth of human glioblastoma cells and xenografts. Cancer Res. 2009. September1;69(17):6915–23. doi: 10.1158/0008-5472.CAN-09-0664 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Rostami N, Nikkhoo A, Ajjoolabady A, et al. S1PR1 as a Novel Promising Therapeutic Target in Cancer Therapy. Mol Diagnosis Ther. 2019. August;23(4):467–487. doi: 10.1007/s40291-019-00401-5 [DOI] [PubMed] [Google Scholar]
- 43.D’Aprile C, Prioni S, Mauri L, Prinetti A, Grassi S. Lipid rafts as platforms for sphingosine 1-phosphate metabolism and signalling. Cell Signal. 2021. April;80:109929. doi: 10.1016/j.cellsig.2021.109929 [DOI] [PubMed] [Google Scholar]
- 44.Van Brocklyn JR, Young N, Roof R. Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett. 2003. September10;199(1):53–60. doi: 10.1016/S0304-3835(03)00334-3 [DOI] [PubMed] [Google Scholar]
- 45.Young N, Pearl DK, Van Brocklyn JR. Sphingosine-1-phosphate regulates glioblastoma cell invasiveness through the urokinase plasminogen activator system and CCN1/Cyr61. Mol Cancer Res. 2009. January;7(1):23–32. doi: 10.1158/1541-7786.MCR-08-0061 [DOI] [PMC free article] [PubMed] [Google Scholar]