

Abstract
Allostery is considered important in regulating protein’s activity. Drug development depends on the understanding of allosteric mechanisms, especially the identification of allosteric sites, which is a prerequisite in drug discovery and design. Many computational methods have been developed for allosteric site prediction using pocket features and protein dynamics. Here, we present an ensemble learning method, consisting of eXtreme gradient boosting (XGBoost) and graph convolutional neural network (GCNN), to predict allosteric sites. Our model can learn physical properties and topology without any prior information, and shows good performance under multiple indicators. Prediction results showed that 84.9% of allosteric pockets in the test set appeared in the top 3 positions. The PASSer: Protein Allosteric Sites Server (https://passer.smu.edu), along with a command line interface (CLI, https://github.com/smutaogroup/passerCLI) provide insights for further analysis in drug discovery.
1. Introduction
Allostery is the process in which proteins transmit the perturbation caused by the effect of binding at one site to a distal functional site.1 The allosteric process is fundamental in the regulation of activity. Compared with non-allosteric drugs, allosteric drugs have many advantages: they are conserved and highly specific;2 they can either activate or inhibit proteins; they can be used in conjunction with orthosteric (non-allosteric) drugs. Although allosteric drugs are important in the pharmaceutical industry,3 they are still poorly understood. 4 Most allosteric mechanisms remain elusive because of the difficulty of identifying potential allosteric sites.5
Many allosteric site prediction methods have been developed based on molecular dynamics (MD) simulations,6 normal mode analysis,7 two-state Gō models8 and machine learning (ML) models.9-11 Among the existing methods, AllositePro,12 AlloPred,13 SPACER14 and PARS15 are available as web servers or open-source packages. These previous studies have shown that it is promising to identify allosteric sites by combining static pocket features with protein dynamics. In these studies, static features are calculated by site descriptors describing physical properties of protein pockets, while the protein dynamics are extracted by MD simulation or perturbation.
Machine learning methods have been shown to be superior in the classification of protein pockets. For example, Allosite11 and AlloPred13 used support vector machine (SVM)16 with optimized features. Chen et al.17 used random forest (RF)18 to construct a three-way predictive model. With the development of ML, more advanced models have been developed and can contribute to the allosteric site classification. eXtreme gradient boosting (XGBoost)19 is one of the most powerful machine learning techniques in classification. It is an implementation of the gradient boosting algorithm with regularized terms to reduce overfitting. Compared with SVM and RF, XGBoost achieved superior predictive performance in the protein-protein interactions20 and hot spots.21
Though physical properties are largely contained in many methods, topological information is largely ignored and is considered important in classifying pockets. In order to explore the geometry features, an atomic graph is constructed for each pocket. Atoms are treated as nodes and the pairwise bond distances are calculated as edges.9 Graph convolutional neural networks (GCNNs),22 a popular concept in deep learning, have been applied in biological-related predictions, ranging from chemical reactions,23 molecular properties,24 to drug-target interactions.25
In this study, protein pockets are predicted using an ensemble learning method, which combines the results of XGBoost and GCNN. This model can learn both physical properties and topology information of allosteric pockets and has been proven to be superior to the single XGBoost and GCNN models. Various performance indicators validated the success of this ensemble learning method compared with previous methods.
2. Methods
2.1. Protein Database
The data used in the current work was collected from the Allosteric Database (ASD).26 There are a total of 1946 entries information of allosteric sites with different proteins and modulators. To ensure data quality, 90 proteins were selected using previous rules:11 protein structures with either resolution below 3Å or missing residues in the allosteric sites were removed; redundant proteins in the rest of the data that have more than 30% sequence identity were filtered out. The names and IDs of the 90 proteins extracted from the PDB Bank27,28 are listed in Table S1.
2.2. Site Descriptors
FPocket algorithm29 is used to detect pockets from the surface of the selected proteins. A pocket is labeled as either 1 (positive) if it contains at least one residue identified as binding to allosteric modulators or 0 (negative) if it does not contain such residues. Therefore, a single protein structure may have more than one positive label. A total of 2246 pockets were detected with 119 pockets being labeled as allosteric sites. There are 19 features calculated by the FPocket as shown in Table S2.
2.3. Pearson Correlation Coefficient
Pearson correlation coefficient (PCC) measures the linear correlation of two variables. Given a pair of variables X and Y as {(x1, y1), …, (xn, yn)}, PCC (rX,Y) is calculated as
| (1) |
where n is the sample size and , are sample means. PCC has a value between −1 and +1. Positive value represents a positive correlation, and negative value represents a negative correlation. The absolute value indicates the degree of correlation. The larger the absolute value, the stronger the correlation.
2.4. eXtreme Gradient Boosting
Extreme gradient boosting is an ensemble learning method that combines several decision trees in sequence.
Let D = {(xi, yi)(∣D∣ = n, xi ϵ Rm, yi ϵ Rn)} represents a dataset with m features and n labels. The j-th decision tree in XGBoost predicts a sample (xi, yi) by:
| (2) |
where wq is the leaf weights of this decision tree. The final prediction of XGBoost is given by the summation of predictions from each decision tree:
| (3) |
where M is the total number of decision trees. To overcome overfitting introduced by decision trees, the objective function in XGBoost is composed of a loss function l and a regularization term Ω:
| (4) |
where with T represents the number of leaves and γ, λ are regularization parameters.
During training, XGBoost iteratively adds new decision trees. The prediction of the t-th iteration is expressed as:
| (5) |
Correspondingly, the objective function of the t-th iteration is:
| (6) |
XGBoost introduces both first derivative and second derivative of the loss function. By applying Taylor expansion on the objective function at second order, the objective function of the t-th iteration can be expressed as:
| (7) |
XGBoost can predict the labels of sample data with corresponding probabilities. For one pocket, XGBoost outputs the probability of this pocket being an allosteric pocket. This pocket is labeled as positive (allosteric) if the predicted probability is over 50% or negative otherwise.
There are only 5.3% of positive labels in this binary classification job, which means that the input data is highly imbalanced. To focus more on the limited positive labels, XGBoost uses “scale_pos_weight”, a parameter for controlling the balance of positive and negative weights. A typical value of this weight equals to the number of negative samples versus the number of positive samples.
In the current study, the maximum tree depth for base learners and the weights for positive labels were fine-tuned while keeping other parameters as default values. The XGBoost algorithm is implemented using Scikit-learn package30 version 0.23.2.
2.5. Graph Convolutional Neural Network
Graph convolutional neural network in this work follows this formula:22
| (8) |
where H(l) and H(l+1) represents the lth and l + 1th layer, respectively. , where N is the number of nodes and D is the number of features. Rectified linear unit (ReLU(x) = max(0,x)) is used as the activation function. W(l) denotes the weight matrix in the lth layer. D and A represent degree matrix and adjacent matrix, respectively, with . Renormalization (indicated by ~ symbol) is applied for the undirected graph G where each node is added with a self-connection. Therefore, Ã = A + IN where IN is the identity matrix.
A graph readout is calculated through the average of node features for each graph.
| (9) |
where hg is the readout result of graph g and hv is the node feature in node v. represents the nodes in graph g.
An example of 1-layer GCNN model is shown in Figure 1. A graph is first fed into a convolution layer. The in-degree and out-degree of a node refer to the number of edges coming into and going out from that node, respectively. In-degree of each node was calculated as the node feature. Graph feature is calculated as the average of node features in the readout layer with ReLU activation function. The output was further fed into a linear classification layer g, which predicts the probability of being an allosteric pocket. Previous research31 has shown the limitations of 1-layer GCNN. In the current study, atomic graphs of each protein pocket are constructed and fed into a 2-layer GCNN model. This model consists of two graph convolution layers, each with 256 dimensions of a hidden node feature vector, followed by a readout layer and a linear classification layer. The node degree is used as the initial node feature. The in-degree is the same as the out-degree in an undirected atomic graph. Graph representation is calculated as the average of node representations. The linear classification layer outputs the probabilities of pockets being allosteric sites.
Figure 1.

Architecture of 1-layer GCNN model. 1-layer GCNN is composed of a graph convolution layer, a readout layer and a linear classification layer. Rectified linear unit is used as the activation function. An atomic graph is constructed for a given pocket and GCNN predicts the probability of this pocket being an allosteric site.
To overcome the potential limitation in training GCNN with an imbalanced dataset, the ratio between negative labels and positive labels was fine-tuned. Specifically, in each protein, allosteric pockets (positive samples) were fully used while non-allosteric pockets (negative samples) were partially used. Each negative sample was randomly selected, and the total number of non-allosteric pockets equals the number of allosteric pockets times the ratio value.
In constructing atomic graphs, the threshold of bond distance was also fine-tuned. Each atom was considered as a node in the atomic graph and the pairwise distances between atoms were calculated. If the distance is below a specified threshold, an edge is constructed connecting the two related nodes. Thus, the distance threshold controls the degree of local connectivity.
The GCNN model is implemented using Deep graph library (DGL) package32 version 0.4.3.
2.6. Performance Indicators
For binary classification, the results can be classified as Table 1. Various indicators were used to quantify model performance: precision (TP / (TP + FP)) measures how well the model can predict real positive labels; accuracy ((TP + TN) / (TP + FP + FN + TN)) measures the overall classification accuracy; recall (or sensitivity, TP / (TP + FN)) and specificity (TN / (TN + FP)) together measure the ability to classify TP and TN; F1 score (2 * precision * recall / (precision + recall)) is the weighted average of precision and recall. The higher the values of these indicators, the better the model’s performance.
Table 1.
Binary classification results. Confusion matrix can visualize and evaluate the performance of classification models. Rows represent the instances in predicted classes while columns represent the instances in real classes. True positive and true negative refer to the results where the model correctly predicts the positive and negative classes, respectively. Similarly, false positive and false negative refer to the results where the model incorrectly predicts the positive and negative classes, respectively.
| Real Positive | Real Negative | |
|---|---|---|
| Predicted Positive | True Positive (TP) | False Negative (FN) |
| Predicted Negative | False Positive (FP) | True Negative (TN) |
A receiver operating characteristic (ROC) curve was applied as another indicator to test model performance in binary classification. ROC curve is plotted as TP rate against FP rate with different threshold settings. The area under curve (AUC) is calculated for quantification. The upper limit for AUC value is 1.0. A dummy model should have an AUC value of 0.5.
The ranking information based on the predicted probabilities of protein pockets was also considered as an important indicator. Specifically, for each allosteric pocket, the ranking (predicted probability of being an allosteric site in descending order) was recorded and categorized as the first, second, third or other positions. The ranking result indicates how likely the allosteric pockets can appear in the top positions among all detected pockets in the same protein. A model with good performance should rank an allosteric pocket in the top positions.
3. Results
3.1. Feature Exploration
The distribution of 19 features is shown in Figure S1. While some exhibited long-tail distributions such as features 1 (Score) and 2 (Druggability score), data normalization is unnecessary since XGBoost does not require normal distribution. Instead, XGBoost, like other tree-based models, only focuses on the order, and whether features are normalized or not does not affect the prediction results. Violin plots are shown in Figure S2 to better distinguish the feature distribution of allosteric sites and non-allosteric sites.
The correlation matrix between these features is shown in Figure S3. Several features exhibited high correlations. For example, features 3 (Number of alpha spheres), 4 (Total SASA), 5 (Polar SASA), 6 (Apolar SASA), and 7 (Volume) are highly correlated with each other. Features 17 (Alpha sphere density) and 18 (Center of mass) are also strongly correlated with these five features.
For each feature value in a pocket, the inner ranking refers to the ranking position of this feature among values of other pockets in the same protein. Similar to the definition of inner ranking, the overall ranking refers to the ranking position of this feature value among values of other pockets in the overall dataset. Both sets of ranking features were normalized. The correlations between the original features and these two ranking feature sets were calculated and shown in Figure S3. The high negative values indicate strongly negative correlations between the original features and the ranking features. While feature rankings were calculated and applied as additional features in a previous study,13 in the current dataset, the high correlation indicated that the ranking features provided little additional information and thus were discarded.
3.2. Prediction performance of XGBoost
XGBoost model can overcome the limitation of data imbalance by controlling the weight difference between negative labels and positive labels. This parameter was fine-tuned along with the maximum depth of trees. The results are plotted in Figure 2. Two sets of parameters reached high F1 scores, and both were selected in the final model. The final XGBoost model is composed of two models, each with one set of parameters. The results in any given pocket are the averaged results predicted by these two models.
Figure 2.

Fine-tuned results of weight and depth parameters in XGBoost model. Two sets of parameters, depth 5 weight 40 and depth 8 weight 40, exhibited exceptional high F1 score and were selected for further prediction.
The results of the fine-tuned XGBoost model are listed in Table 2. Compared with the reference results, XGBoost model exhibited higher accuracy, precision, specificity, and ROC AUC values with comparable results in recall and F1 scores. Therefore, XGBoost model performs well in allosteric site prediction.
Table 2.
Evaluation and performance comparison of different models. The average values and standard errors of 6 indicators are calculated in 10 independent runs. The ensemble learning method can achieve better performance compared to single XGBoost and GCNN models.
| Accuracy | Recall | Precision | Specificity | F1 score | ROC AUC | |
|---|---|---|---|---|---|---|
| XGBoost | 0.969 ± 0.002a | 0.799 ± 0.023 | 0.732 ± 0.030 | 0.982 ± 0.003 | 0.764 ± 0.016 | 0.897 ± 0.016 |
| GCNN | 0.923 ± 0.006 | 0.604 ± 0.023 | 0.427 ± 0.046 | 0.943 ± 0.007 | 0.500 ± 0.031 | 0.832 ± 0.015 |
| Model ensembling | 0.974 ± 0.010 | 0.847 ± 0.095 | 0.726 ± 0.085 | 0.980 ± 0.013 | 0.782 ± 0.072 | 0.914 ± 0.018 |
| Allosite11 | 0.962 | 0.852 | 0.688 | 0.970 | 0.761 | 0.911 |
Standard error (SE) = Standard deviation (SD) / .
3.3. Prediction performance of GCNN
Unlike XGBoost, GCNN models suffer from imbalanced dataset. To address this problem, the ratio between negative labels and positive labels was evaluated first. The results are plotted in Figure 3A. A ratio of 2 (number of negative labels : number of positive labels =2 : 1) was selected. The distance threshold was further fine-tuned, and the results are plotted in Figure 3B. 10Å was selected as the distance cutoff when constructing atomic graphs.
Figure 3.

Fine-tuned results of ratio and distance threshold parameters in GCNN model. (A) The ratio between number of negative labels and number of positive labels was fine-tuned. Ratio of 2 is considered reaching a balance between recall and precision. (B) The atomic distance threshold was fine-tuned from 7 to 11 Å. There is no significant increase in F1 score after 10Å, which is selected as the distance cutoff. For each parameter value, GCNN was run 10 times independently.
The results of the fine-tuned GCNN model with 10 independent runs are listed in Table 2. Compared with XGBoost, GCNNs are less effective in classifying allosteric sites. However, it is expected that combining XGBoost and GCNN will result in better performance than either model.
3.4. Prediction performance of model ensembling
The ensemble learning model is composed of both XGBoost model and GCNN model. For a given pocket, physical properties are calculated and fed into the XGBoost model; a representative atomic graph is fed into the GCNN model. The final result is calculated as the averaged probability of these two models. This final model contains both the physical properties and topological features of protein pockets. The combined results are listed in Table 2. Compared with the XGBoost model, model ensembling leads to a 6.00% increase in recall, a 0.82% decrease in precision, and a 2.89% increase in F1 score. The AUC ROC value also had a 1.89% increase.
For each protein, the identified pockets are ranked based on the predicted probabilities. Overall, 60.7% of allosteric pockets are predicted as the first position, while 81.6% among the top 2 and 84.9% among the top 3. In other words, if a pocket is an allosteric pocket, there is a probability of 84.9% that it can be predicted in the top 3 among all detected pockets in the same protein. The prediction results, together with values reported in other studies, are listed in Table 3. It should be noted that the types and amounts of proteins in the test set are different from each other.
Table 3.
Probabilities of predicting allosteric sites in the top 3 positions. Ensemble learning method can rank an allosteric site in the top 3 positions with a probability of 84.9%, which is higher than previous results.
Not available in the reported results.
3.5. Novel Allosteric Sites Prediction
To test this ensemble learning method, two proteins not in the dataset (Table S1) were used. The predicted allosteric pockets of these two proteins are illustrated in Figure 4. These two proteins represent two different types of allosteric proteins. The second PDZ domain (PDZ2) is a dynamics-driven protein in human PTP1E protein which undergoes allosteric process upon binding with peptides.33 The light-oxygen-voltage (LOV) domain of Phaeodactylum tricornutum Aureochrome 1a (AuLOV) is a conformational-driven allosteric protein.34 AuLOV is a monomer in the dark state and undergoes dimerization upon blue light perturbation.35 In both cases, our prediction model ranks the allosteric sites as the top 1 with probabilities of 45.14% and 89.46%, respectively. This indicates that this model is capable of predicting both dynamics-driven and conformational-driven allosteric proteins. Apparently, the probability for the dynamics-driven allosteric protein is much smaller than the one of the conformational-driven allosteric protein, which is not unexpected.
Figure 4.

Prediction results of two examples not included in the training set: (A) Dynamics-driven PDZ2 protein in bound states (PDB ID 3LNY); (B) The LOV domain of conformational-driven Phaeodactylum tricornutum Aureochrome 1a in dark state (PDB ID: 5DKK). Red regions are the most probable pockets in the predicted results with probabilities of (A) 45.14% and (B) 89.46% and are also the true allosteric sites.
3.6. Web and CLI Usage
A web server based on the allosteric prediction method developed in this study is implemented using a Python web framework, Django. JSmol36 is a JavaScript implementation of the Jmol package and is embedded in the web page for protein and pocket visualization. Web pages are rendered using Bootstrap. This server is named as Protein Allosteric Sites Server (PASSer). A workflow of PASSer is outlined in Figure 5.
Figure 5.
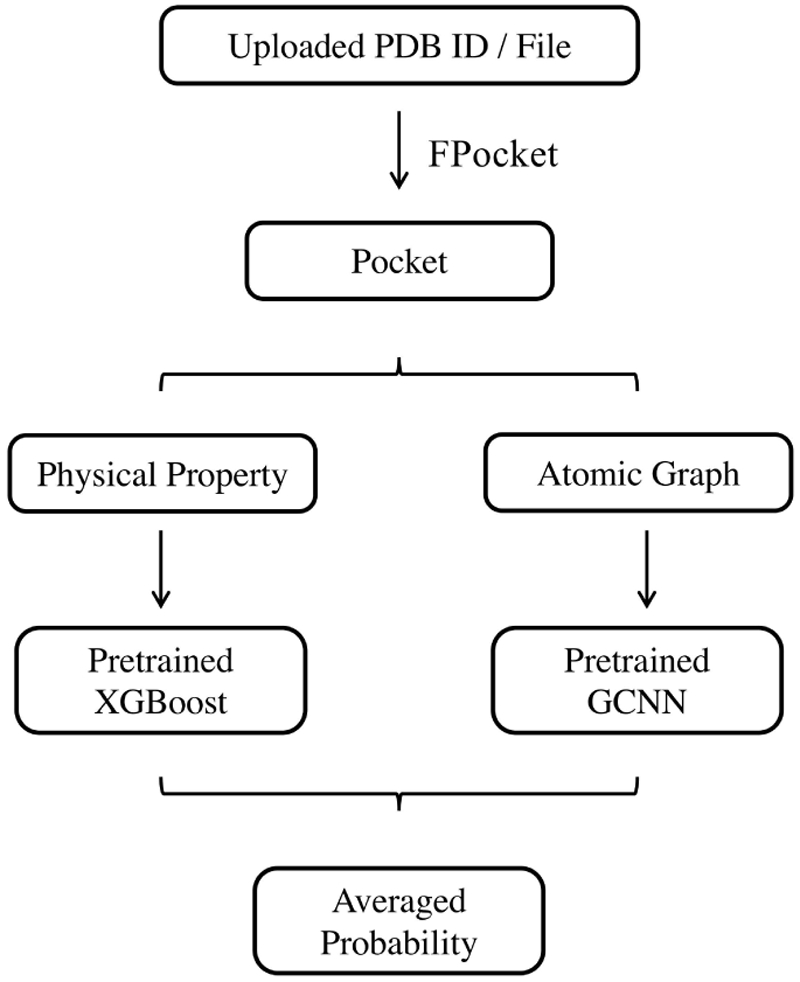
Web server workflow. User can upload either a PDB ID or a PDB file in the web server. FPocket is used to detect pockets. For each pocket, physical properties are calculated and predicted using a pretrained XGBoost model; while an atomic graph is constructed and fed into a pretrained GCNN model. The final probability is given by averaging results from both models.
An example of input and output of PASSer is displayed in Figure 6. Users can submit a PDB ID if available or upload a custom PDB file as shown in Figure 6A. By default, all chains in the protein are analyzed. Prediction results are displayed as two parts: top 3 pockets with the highest probability rendered with the protein structure (Figure 6B) and their probabilities (Figure 6C). For each pocket, the corresponding residues can be retrieved by clicking the “Show Residues” texts. Protein structure is visualized using JSmol. Each pocket is either displayed upon clicking its “Load pocket” icon or hidden by clicking its “Hide pocket” or overall “Reset” icons.
Figure 6.

PASSer web server pages. (A) Users can either submit a PDB ID or a PDB file in the home page. (B) Predicted top 3 pockets are summarized in a table with corresponding probabilities and pocket residues. (C) Protein structures and pocket sites are displayed in an interactive window.
A command line interface (CLI) is provided to facilitate potential developments. Similar with the web usage, this CLI can take either a PDB ID or a local PDB file for predictions.
4. Discussion
The quality of the dataset used for training is critical. Classification models often fail in prediction performance, and lack the generalization with poorly-collected datasets, such as insufficient training data or high similarity between structures. ASD is an online database that provides allosteric proteins and sites with high resolution, bringing opportunities for allosteric site prediction. There are other databases, such as ASBench37 and sc-PDB,38 which can also be used to improve data quality and model performance.
In order to predict allosteric sites, proper pockets need to be identified on the surface of proteins. Several open-source pocket detection software has been developed. Previous results29 have shown that, the geometry-based algorithm FPocket is superior compared with other methods, such as PASS39 and LIGSITEcsc,40 and can cover known allosteric sites. In addition, FPocket is under active development and can be integrated with other methods to build a complete pipeline for site prediction.
Several computational methods have been developed for allosteric site prediction over the past few years. Due to the fast development of machine learning methods, many models integrate ML methods, such as support vector machine and random forest, for accurate predictions.11,17 One critical issue is that, many ML models fail when dealing with imbalanced datasets:41 in the allosteric site database, negative samples account for a majority of the dataset with a limited proportion of positive samples. Undersampling is one way to rebalance the dataset. For example, Allosite discarded some negative labels and used a ratio of 1:4 between positive and negative labels. However, undersampling leads to insufficient usage of the overall dataset. XGBoost as a gradient boosting method overcomes this data imbalance by controlling the relative weights between classes so that the dataset can be fully used. Various performance indicators, as listed in Table 2, validated the effectiveness of XGBoost for the identification of allosteric sites.
It is worth noting that some features are highly correlated, as shown in Figure S3. This collinearity should be addressed in regression models, which reduces the model precision and thus weakens prediction results. In contrast, XGBoost is free from this problem. When several features are found to be highly correlated to each other, XGBoost will choose one of these features. Therefore, collinearity does not affect the prediction results. Nevertheless, collinearity influences model interpretation, such as feature importance, which should be considered with caution.
Physical properties have been widely used to describe the characteristics of pockets. These features are normally calculated using static protein structures. To probe the dynamical behavior of pockets, normal mode analysis and MD simulations are normally conducted. 7,42 Results from these methods have shown that models can achieve satisfactory performance through the combination of both static features and protein dynamics. However, pocket geometry is often ignored, which could play an important role in prediction. Therefore, a graph convolutional neural network is applied to retain the topological information. Specifically, pockets are represented as undirected graphs at atomic level, and GCNN is designed to learn the local connectivity among atoms. While a previous study9 included energy-weighted covalent and weak bonds in the prediction of allosteric sites, it should be noted here that: (1) it is assumed that the physical properties are implicitly retained in the site descriptors and GCNN only studies the node degree; (2) GCNN does not require any a prior information about the location of active sites. The ensemble learning method, consisting of GCNN and XGBoost, exhibited higher performance compared with single models.
5. Conclusion
The proposed ensemble learning method involves XGBoost and GCNN, which can learn both the physical properties and topology of protein pockets. The results are comparable with previous studies and have a higher percentage of ranking allosteric sites at top positions. The web server provides a user-friendly interface. Protein structures and top pockets are visualized in an interactive window on the result page. This ensemble learning method, embedded in the PASSer and CLI, can help exploration on protein allostery and drug development.
Supplementary Material
Acknowledgment
Computational time was generously provided by Southern Methodist University’s Center for Research Computing. Research reported in this paper was supported by the National Institute of General Medical Sciences of the National Institutes of Health under Award No. R15GM122013.
Footnotes
Code availability
The PASSer server is available at https://passer.smu.edu. The command line interface is available on GitHub at https://github.com/smutaogroup/passerCLI.
Competing interests
The authors declare no competing interests
Data availability
The authors declare that all data supporting the findings of this study are available within the paper and its Supplementary Information files.
References
- (1).Hilser VJ An ensemble view of allostery. Science 2010, 327, 653–654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Nussinov R; Tsai C-J; Csermely P Allo-network drugs: harnessing allostery in cellular networks. Trends in pharmacological sciences 2011, 32, 686–693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (3).Pei J; Yin N; Ma X; Lai L Systems biology brings new dimensions for structure-based drug design. Journal of the American Chemical Society 2014, 136, 11556–11565. [DOI] [PubMed] [Google Scholar]
- (4).Wenthur CJ; Gentry PR; Mathews TP; Lindsley CW Drugs for allosteric sites on receptors. Annual review of pharmacology and toxicology 2014, 54, 165–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).Motlagh HN; Wrabl JO; Li J; Hilser VJ The ensemble nature of allostery. Nature 2014, 508, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Laine E; Goncalves C; Karst JC; Lesnard A; Rault S; Tang W-J; Malliavin TE; Ladant D; Blondel A Use of allostery to identify inhibitors of calmodulin-induced activation of Bacillus anthracis edema factor. Proceedings of the National Academy of Sciences 2010, 107, 11277–11282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Panjkovich A; Daura X Exploiting protein flexibility to predict the location of allosteric sites. BMC bioinformatics 2012, 13, 273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (8).Qi Y; Wang Q; Tang B; Lai L Identifying allosteric binding sites in proteins with a two-state Go model for novel allosteric effector discovery. Journal of chemical theory and computation 2012, 8, 2962–2971. [DOI] [PubMed] [Google Scholar]
- (9).Amor BR; Schaub MT; Yaliraki SN; Barahona M Prediction of allosteric sites and mediating interactions through bond-to-bond propensities. Nature communications 2016, 7, 1–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Bian Y; Jing Y; Wang L; Ma S; Jun JJ; Xie X-Q Prediction of orthosteric and allosteric regulations on cannabinoid receptors using supervised machine learning classifiers. Molecular pharmaceutics 2019, 16, 2605–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Huang W; Lu S; Huang Z; Liu X; Mou L; Luo Y; Zhao Y; Liu Y; Chen Z; Hou T, et al. Allosite: a method for predicting allosteric sites. Bioinformatics 2013, 29, 2357–2359. [DOI] [PubMed] [Google Scholar]
- (12).Song K; Liu X; Huang W; Lu S; Shen Q; Zhang L; Zhang J Improved method for the identification and validation of allosteric sites. Journal of Chemical Information and Modeling 2017, 57, 2358–2363. [DOI] [PubMed] [Google Scholar]
- (13).Greener JG; Sternberg MJ AlloPred: prediction of allosteric pockets on proteins using normal mode perturbation analysis. BMC bioinformatics 2015, 16, 1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Goncearenco A; Mitternacht S; Yong T; Eisenhaber B; Eisenhaber F; Berezovsky IN SPACER: server for predicting allosteric communication and effects of regulation. Nucleic acids research 2013, 41, W266–W272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Panjkovich A; Daura X PARS: a web server for the prediction of protein allosteric and regulatory sites. Bioinformatics 2014, 30, 1314–1315. [DOI] [PubMed] [Google Scholar]
- (16).Suykens JA; Vandewalle J Least squares support vector machine classifiers. Neural processing letters 1999, 9, 293–300. [Google Scholar]
- (17).Chen AS-Y; Westwood NJ; Brear P; Rogers GW; Mavridis L; Mitchell JB A random forest model for predicting allosteric and functional sites on proteins. Molecular informatics 2016, 35, 125–135. [DOI] [PubMed] [Google Scholar]
- (18).Liaw A; Wiener M, et al. Classification and regression by randomForest. R news 2002, 2, 18–22. [Google Scholar]
- (19).Chen T; Guestrin C Xgboost: A scalable tree boosting system. Proceedings of the 22nd acm sigkdd international conference on knowledge discovery and data mining. 2016; pp 785–794. [Google Scholar]
- (20).Basit AH; Abbasi WA; Asif A; Gull S; Minhas FUAA Training host-pathogen protein–protein interaction predictors. Journal of bioinformatics and computational biology 2018, 16, 1850014. [DOI] [PubMed] [Google Scholar]
- (21).Li K; Zhang S; Yan D; Bin Y; Xia J Prediction of hot spots in protein–DNA binding interfaces based on supervised isometric feature mapping and extreme gradient boosting. BMC bioinformatics 2020, 21, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Kipf TN; Welling M Semi-supervised classification with graph convolutional networks. arXiv preprint arXiv:1609.02907 2016, [Google Scholar]
- (23).Coley CW; Jin W; Rogers L; Jamison TF; Jaakkola TS; Green WH; Barzilay R; Jensen KF A graph-convolutional neural network model for the prediction of chemical reactivity. Chemical science 2019, 10, 370–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ryu S; Kwon Y; Kim WY A Bayesian graph convolutional network for reliable prediction of molecular properties with uncertainty quantification. Chemical Science 2019, 10, 8438–8446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Torng W; Altman RB Graph convolutional neural networks for predicting drug-target interactions. Journal of Chemical Information and Modeling 2019, 59, 4131–4149. [DOI] [PubMed] [Google Scholar]
- (26).Huang Z; Zhu L; Cao Y; Wu G; Liu X; Chen Y; Wang Q; Shi T; Zhao Y; Wang Y, et al. ASD: a comprehensive database of allosteric proteins and modulators. Nucleic acids research 2011, 39, D663–D669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (27).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The protein data bank. Nucleic acids research 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Burley SK; Berman HM; Bhikadiya C; Bi C; Chen L; Di Costanzo L; Christie C; Dalenberg K; Duarte JM; Dutta S, et al. RCSB Protein Data Bank: biological macromolecular structures enabling research and education in fundamental biology, biomedicine, biotechnology and energy. Nucleic acids research 2019, 47, D464–D474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Le Guilloux V; Schmidtke P; Tuffery P Fpocket: an open source platform for ligand pocket detection. BMC bioinformatics 2009, 10, 1–11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V, et al. Scikit-learn: Machine learning in Python. the Journal of machine Learning research 2011, 12, 2825–2830. [Google Scholar]
- (31).Xu K; Hu W; Leskovec J; Jegelka S How powerful are graph neural networks? arXiv preprint arXiv:1810.00826 2018, [Google Scholar]
- (32).Wang M; Yu L; Zheng D; Gan Q; Gai Y; Ye Z; Li M; Zhou J; Huang Q; Ma C, et al. Deep graph library: Towards efficient and scalable deep learning on graphs. arXiv preprint arXiv:1909.01315 2019, [Google Scholar]
- (33).Zhou H; Dong Z; Tao P Recognition of protein allosteric states and residues: Machine learning approaches. Journal of Computational Chemistry 2018, 39, 1481–1490. [DOI] [PubMed] [Google Scholar]
- (34).Heintz U; Schlichting I Blue light-induced LOV domain dimerization enhances the affinity of Aureochrome 1a for its target DNA sequence. Elife 2016, 5, e11860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (35).Tian H; Trozzi F; Zoltowski BD; Tao P Deciphering the Allosteric Process of Phaeodactylum tricornutum Aureochrome 1a LOV Domain. The Journal of Physical Chemistry B 2020, [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Hanson RM; Prilusky J; Renjian Z; Nakane T; Sussman JL JSmol and the next-generation web-based representation of 3D molecular structure as applied to proteopedia. Israel Journal of Chemistry 2013, 53, 207–216. [Google Scholar]
- (37).Huang W; Wang G; Shen Q; Liu X; Lu S; Geng L; Huang Z; Zhang J ASBench: benchmarking sets for allosteric discovery. Bioinformatics 2015, 31, 2598–2600. [DOI] [PubMed] [Google Scholar]
- (38).Desaphy J; Bret G; Rognan D; Kellenberger E sc-PDB: a 3D-database of ligandable binding sites—10 years on. Nucleic acids research 2015, 43, D399–D404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Brady GP; Stouten PF Fast prediction and visualization of protein binding pockets with PASS. Journal of computer-aided molecular design 2000, 14, 383–401. [DOI] [PubMed] [Google Scholar]
- (40).Huang B; Schroeder M LIGSITE csc: predicting ligand binding sites using the Connolly surface and degree of conservation. BMC structural biology 2006, 6, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (41).Zhao X-M; Li X; Chen L; Aihara K Protein classification with imbalanced data. Proteins: Structure, function, and bioinformatics 2008, 70, 1125–1132. [DOI] [PubMed] [Google Scholar]
- (42).Penkler D; Sensoy O; Atilgan C; Tastan Bishop O Perturbation–response scanning reveals key residues for allosteric control in Hsp70. Journal of Chemical Information and Modeling 2017, 57, 1359–1374. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The authors declare that all data supporting the findings of this study are available within the paper and its Supplementary Information files.