

Abstract
We report a direct and modular method to access non-conjugated alkenyl amides containing the 8-aminoquinoline (AQ) directing auxiliary and related groups via cross-metathesis. In this way, readily available, AQ-containing, terminal β,γ-unsaturated amides can be coupled with various terminal alkenes to furnish internal alkene products that are otherwise difficult to prepare. The value of this family of products stems from their ability to participate in a number of directed alkene functionalization reactions.
Keywords: Alkenes, Alkene Functionalization, Directing group, Olefin metathesis, Grubbs catalyst
Graphical Abstract

1. Introduction
Metal-catalyzed directed alkene functionalization offers a strategy for converting readily accessible starting materials into value-added products with control of regio-, stereo-, and chemoselectivity.[1a–1f] This rapidly developing family of transformations has attracted considerable attention in recent years. Substrates that contain bidentate auxiliaries, as exemplified by alkenyl amides derived from 8-aminoquinoline (AQ), [1e–h] are privileged substrates that are compatible with a wide array of metals, redox manifolds, and coupling partners.[2–7] Typical substrates for these reactions include β,γ-unsaturated AQ amides with one or more substituents at the α, β, or γ positions. During the course of our work in this area, we recognized that substrates with some substitution patterns such as internal alkenes bearing an α-substituent [2a, 8] or a heteroatom substituent along the alkyl chain (i.e., -(CH2)nX at the γ position) (see below), are challenging to access using established methods.[2o, 3d, 9] This knowledge gap motivated the present study in which we describe the development of olefin cross-metathesis as a high-yielding method to access this family of compounds.
Previous studies pointed to the viability of the envisioned approach, while also illustrating potential complications. Our laboratory previously found that a cyclic alkenyl amide substrate could be prepared from an AQ-containing diene substrate via ring-closing metathesis upon treatment Hoveyda–Grubbs II catalyst.[4a] Loh and Wu prepared a series of AQ-containing alkenyl amides via cross-metathesis between vinyl acetic acid and various conjugated alkenes (e.g. ethyl acrylate), followed by amide coupling to install the AQ group.[2n] Based on these precedents and given the potential for the AQ-amide to sequester the Ru metathesis catalyst off-cycle, a key question that we wished to answer was whether intermolecular cross-metathesis (as opposed to intramolecular ring-closing metathesis) between an alkenyl AQ amide and a non-conjugated alkene partner would be feasible. [10,11].
2. Results and Discussions
To begin the study, we selected AQ amide 1a (1 equiv) and N-homoallyl phthalimide (2a) (3.0 equiv) as model substrates. A preliminary experiment revealed that commercially available Grubbs II catalyst effectively mediated the desired coupling (Table 1, entry 1). We tested catalyst loadings ranging from 1–10 mol% (entries 1–4) and found that increasing the amount of catalyst was beneficial up to 5 mol%, beyond which there was no meaningful increase in yield. Performing the reaction at higher concentration improved the yield, with 2.0 M (in DCM) identified as optimal (entries 5–7). We found that increasing the loading of coupling partner 2a to 5 equiv improved the yield of the desired cross-metathesis product (entry 8), as is commonly observed in cross-metathesis reactions.[10] The method of ethylene removal did not have a major effect on yield (entries 9–10), presumably because of the ample headspace of the vials used in these experiments. Across these experiments, the E/Z ratio varied only modestly (71:29–82:18), consistent with the reaction being under thermodynamic control.
Table 1.
Reaction optimization
|
| |||||
|---|---|---|---|---|---|
| Entry | Alkene | Grubbs II | DCM | Method | Yield (E:Z)a |
| 1 | 3.0 equiv | 1 mol% | 0.25 M | sealed vial | 36% (71:29) |
| 2 | 3.0 equiv | 2 mol% | 0.25 M | sealed vial | 51% (78:22) |
| 3 | 3.0 equiv | 5 mol% | 0.25 M | sealed vial | 57% (81:19) |
| 4 | 3.0 equiv | 10 mol% | 0.25 M | sealed vial | 58% (81:19) |
| 5 | 3.0 equiv | 5 mol% | 0.5 M | sealed vial | 60% (81:19) |
| 6 | 3.0 equiv | 5 mol% | 1.0 M | sealed vial | 62% (81:19) |
| 7 | 3.0 equiv | 5 mol% | 2.0 M | sealed vial | 66% (80:20) |
| 8 | 5.0 equiv | 5 mol% | 2.0 M | sealed vial | 72% (82:18) |
| 9 | 5.0 equiv | 5 mol% | 2.0 M | N2 (ballon) | 72% (82:18) |
| 10 | 5.0 equiv | 5 mol% | 2.0 M | N2 (purging) | 70% (76:24) |

1a (0.1 mmol), 1H NMR yield of crude reaction mixture with CI2CHCHCI2 as internal standard.
We next examined the substrate scope of the reaction. Whereas decreasing the linker length between the phthalimido group and the alkene to one methylene spacer led to a reduction in yield (3b, 38%), potentially due to inhibitory formation of a six-membered chelated ruthenium–alkylidene intermediate in this case, a longer tether length was well tolerated (3c, 80%). Using N-Boc pent-4-en-1-amine as the coupling partner, we examined the effect of substitution at the α-position of the alkenyl AQ amide and observed diminished yield as a function of increasing steric bulk (3d–3f). We next investigated alkenes that contain functional groups beyond cyclic imides and carbamates. We found that a free primary alcohol (3g), esters (3h and 3j), and a carbonate (3i) were all compatible. Lastly, N-protected anilines bearing alkenes connected at the ortho position reacted to furnish products 3k and 3l in synthetically useful yields. We also tested cross-metathesis between a γ,δ-unsaturated AQ amide (containing one additional methylene spacer compared to 1a) with representative terminal alkenes. However, these reactions only provided approximately 20% 1H NMR yield of the desired products, and purification proved to be difficult due to the presence of uncharacterized byproducts.
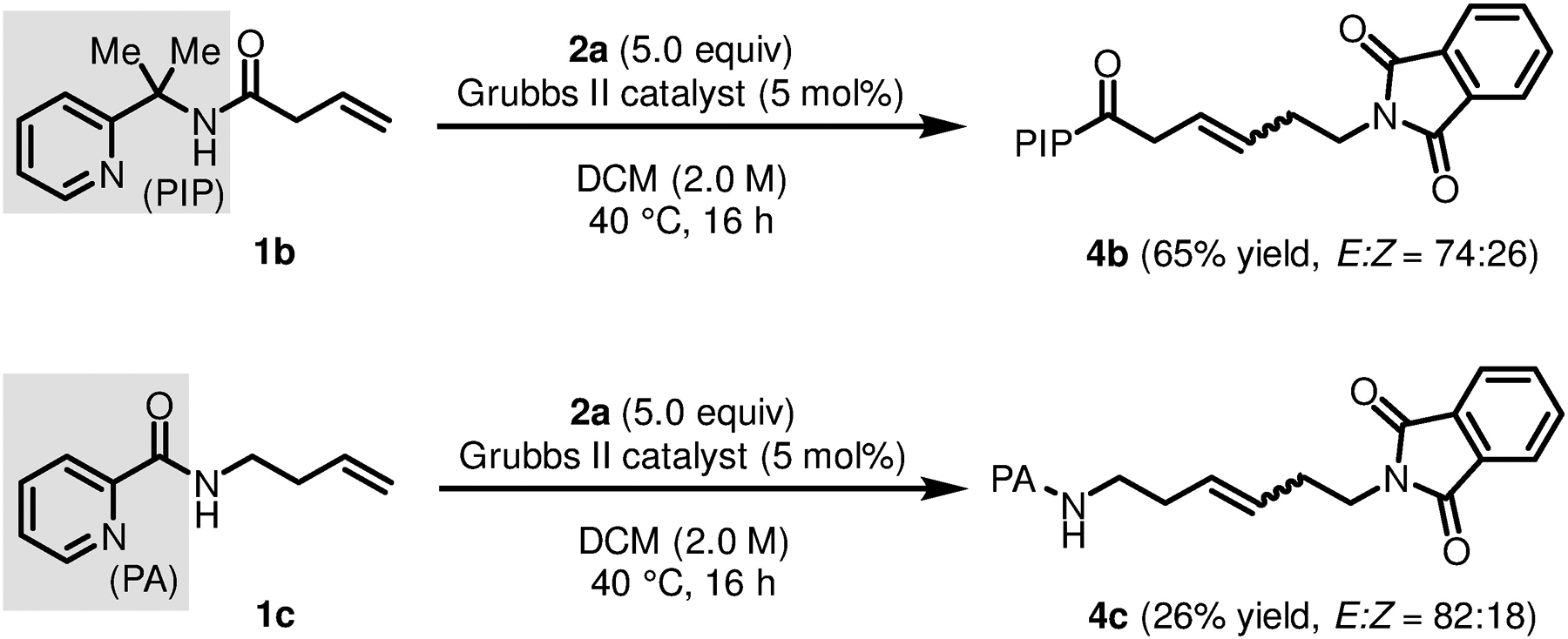
We also examined alkene coupling partners bearing different directing auxiliaries with established utility in synthesis. (Scheme 1) To our delight, an alkenyl amide derived from 2-(pyridin-2-yl)isopropyl amine (PIP) (1b) [2b, 3g, 12] underwent cross-metathesis to furnish 4b in good yield and with an E/Z ratio comparable to earlier examples. Moreover, this method was also effective for a homoallyl amine substrate bearing a picolinamide (PA) bidentate directing auxilary (1c) [2q, 5e, 6a, 6c, 13], though in this case the yield of 4c was only 26%.
Scheme 1:
Results with other directing auxiliaries
We illustrate the practical utility of this method by comparing the cross-metathesis route with the previous route from the literature involving decarboxylative aldol condensation for the preparation of 3d, an established substrate for Pd(II)-catalyzed cycloisomerization (Scheme 2).[2o] Whereas classical methods require de novo syntheses for any structural modifications, using cross-metathesis allows access to a wide variety of products from a small number of common alkene-containing building blocks. Although the lack of E/Z-stereochemical purity could be a disadvantage in some potential applications, many AQ-directed Pd(II)-catalyzed alkene functionalizations undergo in situ isomerization[14] and can even be performed in a stereoconvergent manner.[2d, 5b, 7a].
Scheme 2:

Comparison of different methods.
3. Conclusion
In conclusion, we have established a cross-metathesis approach to the synthesis of N-(quinoline-8-yl) alkenyl amides that grants access to a structurally diverse library of highly substituted alkenyl AQ amides from readily available alkene starting materials. Improved availability of this family of compounds, in turn, broadens the reach of AQ-directed alkene functionalization as a versatile synthetic strategy.
4. Experimental section
4.1. General Information
Unless otherwise noted, all materials were used as received from commercial sources without further purification. All terminal alkenes are synthesized according to literature procedures, with characterization data matching the reported data. Thin layer chromatography (TLC) was conducted with EMD gel 60 F254 pre-coated plates (0.25 mm) and visualized by UV light. Preparative thin layer chromatography (PTLC) was conducted with Analtech silica gel GF UV254 pre-coated plates (1.0 mm) and visualized using UV light. NMR spectra were recorded on JOEL-400 and AV-600 instruments. Spectra were internally referenced to SiMe4 or solvent signals. The following abbreviations (or combinations thereof) were used to explain multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet. High-resolution mass spectra (HRMS) for new compounds were recorded on an Agilent LC/MSD TOF mass spectrometer.
4.2. Experimental Procedure
To an 8-mL scintillation vial equipped with a Teflon-coated magnetic stir bar were added the AQ-containing alkenyl amide substrate 1 (0.1 mmol) and the appropriate alkene coupling partner 2 (5.0 equiv). The vial was transferred to a glovebox, and the Grubbs II catalyst (5 mol%) and DCM (0.05 mL) were added. The vial was sealed with a screw-top septum cap, removed from the glovebox, and placed in a heating block in oil bath that was pre-heated to 40 °C. The reaction was allowed to run for 16 h and then was cooled to room temperature. The reaction mixture was loaded directly onto a PTLC plate, which was developed to afford the pure product using hexanes: EtOAc as eluents.
4.2.1. (E/Z)-6-(1,3-dioxoisoindolin-2-yl)-N-(quinolin-8-yl)hex-3-enamide (3a)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), 2-(but-3-en-1-yl)isoindoline-1,3-dione (100 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3a as a white solid (30.0 mg, 78% yield, E/Z = 80:20). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 9.93 (s, 1H), 8.81 (dd, J = 4.3, 1.7 Hz, 1H), 8.69 (dd, J = 6.7, 2.3 Hz, 1H), 8.14 (dd, J = 8.2, 1.7 Hz, 1H), 7.76 (dd, J = 5.5, 3.1 Hz, 2H), 7.62 (dd, J = 5.5, 3.1 Hz, 2H), 7.52–7.40 (m, 3H), 6.06–5.51 (m, 2H), 3.90–3.80 (m, 2H), 3.25 (d, J = 5.9 Hz, 2H), 2.56 (q, J = 6.8 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 169.72, 168.66, 148.64, 138.80, 136.58, 134.65, 134.14, 132.43, 132.34, 128.21, 127.67, 125.94, 123.48, 121.95, 121.85, 116.66, 42.33, 37.74, 32.18. HRMS calcd. for C23H20N3O3+ [M+H]+: 386.1505, Found: 386.1503.
4.2.2. (E/Z)-5-(1,3-dioxoisoindolin-2-yl)-N-(quinolin-8-yl)pent-3-enamide (3b)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), 2-allylisoindoline-1,3-dione (94 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3b as a colorless oil (14.1 mg, 38%, E/Z = 67:33). The following NMR data corresponds to the E (major) isomer. 1H NMR (600 MHz, CDCl3) δ 10.59–9.79 (m, 1H), 8.90–8.52 (m, 2H), 8.13 (ddd, J = 8.2, 4.9, 1.6 Hz, 1H), 7.85 (dd, J = 5.4, 3.1 Hz, 2H), 7.71 (dd, J = 5.5, 3.0 Hz, 2H), 7.59–7.45 (m, 2H), 7.42 (dd, J = 8.2, 4.2 Hz, 1H), 6.30–5.78 (m, 2H), 4.39 (dd, J = 6.1, 1.3 Hz, 2H), 3.32 (dd, J = 7.1, 1.4 Hz, 2H). 13C NMR (151 MHz, CDCl3) δ 167.41, 147.80, 137.99, 135.80, 133.81, 133.53, 131.71, 128.44, 127.42, 126.91, 126.81, 126.34, 122.76, 121.16, 121.09, 115.98, 38.78, 34.15. HRMS calcd. for C22H18N3O3+ [M+H]+: 372.1348, Found: 372.1343.
4.2.3. (E/Z)-8-(1,3-dioxoisoindolin-2-yl)-N-(quinolin-8-yl)oct-3-enamide (3c)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), 2-(hex-5-en-1-yl)isoindoline-1,3-dione (115 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3c as a white solid (33.0 mg, 80%, E/Z = 82:18). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 10.02 (s, 1H), 8.76 (d, J = 2.3 Hz, 2H), 8.10 (dt, J = 8.4, 2.0 Hz, 1H), 7.95–7.73 (m, 2H), 7.72–7.60 (m, 2H), 7.56–7.45 (m, 2H), 7.39 (dd, J = 8.3, 4.0 Hz, 1H), 5.86–5.70 (m, 2H), 3.75–3.64 (m, 2H), 3.26 (d, J = 5.4 Hz, 2H), 2.31–2.17 (m, 2H), 1.78 (p, J = 7.3 Hz, 2H), 1.56 (p, J = 7.7 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 170.26, 168.75, 148.49, 138.81, 136.64, 136.54, 134.74, 134.16, 132.40, 128.19, 127.68, 123.46, 123.27, 121.87, 121.79, 116.64, 42.44, 38.10, 32.48, 28.38, 26.67. HRMS calcd. for C25H24N3O3+ [M+H]+: 414.1818, Found: 414.1813.
4.2.4. Tert-butyl (E/Z)-(8-oxo-8-(quinolin-8-ylamino)oct-5-en-1-yl)carbamate (3d)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), tert-butyl hex-5-en-1-ylcarbamate (100 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3d as a colorless oil (28.7 mg, 75%, E/Z = 82:18). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 10.02 (s, 1H), 8.82–8.73 (m, 2H), 8.15 (dd, J = 8.3, 2.2 Hz, 1H), 7.57–7.40 (m, 3H), 5.77 (dt, J = 11.0, 6.4 Hz, 2H), 4.54 (s, 1H), 3.27 (d, J = 6.0 Hz, 2H), 3.13 (p, J = 7.6, 6.9 Hz, 2H), 2.18 (q, J = 6.7 Hz, 2H), 1.58–1.47 (m, 4H), 1.42 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 170.31, 156.31, 148.51, 138.83, 136.82, 136.64, 134.74, 128.25, 127.71, 123.07, 121.94, 121.85, 116.66, 79.38, 42.43, 40.78, 32.62, 29.90, 28.74, 26.71. HRMS calcd. for C22H30N3O3+ [M+H]+: 384.2287, Found: 384.2281.
4.2.5. Tert-butyl (E/Z)-(7-methyl-8-oxo-8-(quinolin-8-ylamino)oct-5-en-1-yl)carbamate (3e)
The reaction was carried out according to the general procedure using alkene 1b (22.6 mg, 0.1 mmol), tert-butyl hex-5-en-1-ylcarbamate (100 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3e as a colorless oil (25.0 mg, 63%, E/Z = 89:11). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 10.08 (s, 1H), 8.82–8.74 (m, 2H), 8.14 (d, J = 8.3 Hz, 1H), 7.57–7.40 (m, 3H), 5.90–5.57 (m, 2H), 4.50 (s, 1H), 3.36–3.24 (m, 1H), 3.13 (q, J = 6.3 Hz, 2H), 2.15 (d, J = 6.7 Hz, 2H), 1.52 (dt, J = 7.5, 4.0 Hz, 4H), 1.42 (s, 9H), 1.40 (s, 3H). 13C NMR (100 MHz, CDCl3) δ 173.50, 156.30, 148.49, 138.93, 136.64, 134.91, 133.87, 130.42, 128.27, 127.75, 121.92, 121.72, 116.56, 79.39, 46.31, 40.79, 32.58, 29.91, 28.74, 26.76, 17.62. HRMS calcd. for C23H32N3O3+ [M+H]+: 398.2444, Found: 398.2444.
4.2.6. Tert-butyl (E/Z)-(7-benzyl-8-oxo-8-(quinolin-8-ylamino)oct-5-en-1-yl)carbamate (3f)
The reaction was carried out according to the general procedure using alkene 1c (30.2 mg, 0.1 mmol), tert-butyl hex-5-en-1-ylcarbamate (100 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3f as a colorless oil (18.9 mg, 40%, E/Z = 82:18). The following NMR data corresponds to the E (major) isomer. 1H NMR (600 MHz, CDCl3) δ 10.03 (d, J = 10.4 Hz, 1H), 8.83–8.76 (m, 2H), 8.16 (dd, J = 8.2, 1.7 Hz, 1H), 7.56 (dd, J = 9.6, 6.2 Hz, 1H), 7.51 (dd, J = 8.3, 1.4 Hz, 1H), 7.46 (dd, J = 8.2, 4.2 Hz, 1H), 7.31–7.23 (m, 4H), 7.23–7.16 (m, 1H), 5.71–5.60 (m, 2H), 4.43 (s, 1H), 3.53–3.31 (m, 2H), 3.17–2.99 (m, 2H), 2.93 (ddd, J = 32.5, 13.6, 8.6 Hz, 1H), 2.16–2.05 (m, 2H), 1.45 (d, J = 3.0 Hz, 9H), 1.40 (q, J = 6.3 Hz, 4H). 13C NMR (151 MHz, CDCl3) δ 171.41, 155.48, 147.68, 139.03, 138.08, 135.82, 134.65, 134.03, 128.85, 127.81, 127.77, 127.47, 126.93, 125.67, 121.12, 121.02, 115.95, 78.56, 53.50, 39.92, 37.71, 31.67, 28.81, 27.98, 25.75. HRMS calcd. for C29H36N3O3+ [M+H]+: 474.2757, Found: 474.2761.
4.2.7. (E/Z)-6-hydroxy-N-(quinolin-8-yl)hex-3-enamide (3g)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), but-3-en-1-ol (36 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3g as a colorless oil (20.0 mg, 78%, E/Z = 83:17). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 10.00 (s, 1H), 8.81 (t, J = 6.9 Hz, 2H), 8.17 (d, J = 8.2 Hz, 1H), 7.72–7.38 (m, 3H), 5.89 (dt, J = 11.4, 6.7 Hz, 2H), 3.83 (t, J = 5.9 Hz, 2H), 3.32 (d, J = 6.2 Hz, 2H), 2.86 (s, 1H), 2.45 (q, J = 6.1 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 169.58, 148.70, 138.48, 136.89, 134.17, 128.21, 127.56, 126.30, 124.77, 121.93, 121.74, 116.73, 61.67, 42.19, 36.43. HRMS calcd. for C15H17N2O2+ [M+H]+: 257.1290, Found: 257.1293.
4.2.8. Benzyl (E/Z)-7-oxo-7-(quinolin-8-ylamino)hept-4-enoate (3h)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), benzyl pent-4-enoate (95 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3h as a colorless oil (22.5 mg, 60%, E/Z = 83:17). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 10.02 (s, 1H), 8.91–8.65 (m, 2H), 8.14 (dd, J = 8.3, 1.6 Hz, 1H), 7.61–7.46 (m, 2H), 7.41 (dd, J = 8.3, 4.3 Hz, 1H), 7.36–7.30 (m, 5H), 5.82 (m, 2H), 5.13 (s, 2H), 3.26 (d, J = 5.5 Hz, 2H), 2.67–2.49 (m, 4H). 13C NMR (100 MHz, CDCl3) δ 172.95, 169.72, 148.37, 138.61, 136.42, 136.07, 134.75, 134.47, 132.74, 128.68, 128.38, 128.03, 127.50, 124.07, 121.71, 121.68, 116.43, 66.42, 42.07, 34.09, 28.06. HRMS calcd. for C23H23N2O3+ [M+H]+: 375.1709, Found: 375.1709.
4.2.9. (E/Z)-benzyl (6-oxo-6-(quinolin-8-ylamino)hex-3-en-1-yl) carbonate (3i)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), benzyl but-3-en-1-yl carbonate (103 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3i as a colorless oil (25.3 mg, 65%, E/Z = 82:18). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 10.02 (s, 1H), 8.83–8.72 (m, 2H), 8.14 (dd, J = 8.3, 1.7 Hz, 1H), 7.59–7.47 (m, 2H), 7.43 (dd, J = 8.3, 4.2 Hz, 1H), 7.37–7.30 (m, 5H), 5.85 (qt, J = 15.5, 6.7 Hz, 2H), 5.14 (s, 2H), 4.31 (t, J = 6.9 Hz, 2H), 3.29 (d, J = 6.7 Hz, 2H), 2.55 (q, J = 6.7 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 169.72, 155.44, 148.66, 138.81, 136.63, 135.52, 134.65, 131.47, 128.92, 128.86, 128.68, 128.24, 127.69, 126.27, 121.95, 121.93, 116.66, 69.90, 67.67, 42.33, 32.39. HRMS calcd. for C23H23N2O4+ [M+H]+: 391.1658, Found: 391.1652.
4.2.10. (E/Z)-6-oxo-6-(quinolin-8-ylamino)hex-3-en-1-yl thiophene-2-carboxylate (3j)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), but-3-en-1-yl thiophene-2-carboxylate (91 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3j as a colorless oil (22.0 mg, 60%, E/Z = 79:11). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 10.04 (s, 1H), 8.84–8.71 (m, 2H), 8.14 (dd, J = 8.3, 1.7 Hz, 1H), 7.74 (dd, J = 3.8, 1.4 Hz, 1H), 7.60–7.35 (m, 4H), 7.00 (dd, J = 5.0, 3.7 Hz, 1H), 6.00–5.80 (m, 2H), 4.45 (t, J = 6.7 Hz, 2H), 3.32 (d, J = 6.2 Hz, 2H), 2.63 (q, J = 6.6 Hz, 2H). 13C NMR (100 MHz, CDCl3) δ 169.54, 162.32, 148.45, 138.57, 136.36, 134.42, 133.45, 132.39, 131.62, 129.93, 127.99, 127.75, 127.42, 126.01, 121.75, 121.68, 116.41, 64.40, 42.25, 32.24. HRMS calcd. for C20H19N2O3S+ [M+H]+: 367.1116, Found: 367.1115.
4.2.11. Ert-butyl (E/Z)-(2-(5-(naphthalen-1-ylamino)-5-oxopent-2-en-1-yl)phenyl)carbamate (3k)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), tert-butyl (2-allylphenyl)carbamate (117 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3k as a colorless oil (27.6 mg, 66%, E/Z = 76:24). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 9.94 (s, 1H), 9.03–8.60 (m, 2H), 8.16 (dd, J = 8.3, 1.7 Hz, 1H), 7.75 (d, J = 8.1 Hz, 1H), 7.56–7.41 (m, 3H), 7.32–7.18 (m, 2H), 7.14–6.93 (m, 1H), 6.59 (s, 1H), 6.07–5.63 (m, 2H), 3.71–3.16 (m, 4H), 1.42 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 169.49, 153.47, 148.37, 138.55, 136.51, 136.47, 134.43, 133.74, 132.20, 130.16, 128.06, 127.54, 127.48, 124.63, 124.50, 123.46, 121.75, 116.74, 116.58, 42.00, 35.41, 28.36. HRMS calcd. for C25H28N3O3+ [M+H]+: 418.2131, Found: 418.2130.
4.2.12. Tert-butyl (E/Z)-(2-((6-(naphthalen-1-ylamino)-6-oxohex-3-en-1-yl)oxy)phenyl)carbamate (3l)
The reaction was carried out according to the general procedure using alkene 1a (21.2 mg, 0.1 mmol), tert-butyl (2-(but-3-en-1-yloxy)phenyl)carbamate (132 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 3l as a colorless oil (25.9 mg, 58%, E/Z = 75:25). The following NMR data corresponds to the E (major) isomer. 1H NMR (400 MHz, CDCl3) δ 10.03 (s, 1H), 8.87–8.66 (m, 2H), 8.15 (dd, J = 8.3, 1.7 Hz, 1H), 8.05 (s, 1H), 7.64–7.47 (m, 2H), 7.41 (dd, J = 8.3, 4.4 Hz, 1H), 7.10 (s, 1H), 7.01–6.75 (m, 3H), 6.11–5.86 (m, 2H), 4.36–4.05 (m, 2H), 3.35 (d, J = 6.0 Hz, 2H), 2.70 (q, J = 6.5 Hz, 2H), 1.49 (s, 9H). 13C NMR (100 MHz, CDCl3) δ 169.56, 152.86, 148.35, 146.71, 138.59, 136.46, 134.45, 131.84, 128.55, 128.05, 127.50, 125.90, 124.67, 122.36, 121.75, 121.40, 118.27, 116.51, 111.37, 68.02, 42.19, 32.76, 28.46. HRMS calcd. for C26H30N3O4+ [M+H]+: 448.2236, Found: 448.2236.
4.2.13. (E/Z)-6-(1,3-dioxoisoindolin-2-yl)-N-(2-(pyridin-2-yl)propan-2-yl)hex-3-enamide (4b)
The reaction was carried out according to the general procedure using alkene 1b (20.4 mg, 0.1 mmol), 2-(but-3-en-1-yl)isoindoline-1,3-dione (100 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 4b as a colorless oil (24.5 mg, 65%, E/Z = 74:26). The following NMR data corresponds to the E (major) isomer. 1H NMR (600 MHz, CDCl3) δ 8.51 (ddd, J = 4.9, 1.9, 1.0 Hz, 1H), 7.89 (s, 1H), 7.84 – 7.78 (m, 2H), 7.73 – 7.65 (m, 3H), 7.36 (dt, J = 8.1, 1.0 Hz, 1H), 7.18 (ddd, J = 7.5, 4.9, 1.0 Hz, 1H), 5.72 (dtt, J = 14.9, 6.8, 1.2 Hz, 1H), 5.68 – 5.60 (m, 1H), 3.79 (dd, J = 7.9, 6.7 Hz, 2H), 2.97 (dd, J = 7.0, 1.1 Hz, 2H), 2.57 – 2.45 (m, 2H), 1.70 (s, 6H). 13C NMR (151 MHz, CDCl3) δ 170.38, 168.70, 164.90, 147.97, 137.36, 134.22, 132.43, 131.18, 126.76, 123.55, 122.17, 119.75, 56.72, 42.12, 37.90, 32.09, 27.78. HRMS calcd. for C22H24N3O3+ [M+H]+: 378.1818, Found: 378.1808.
4.2.14. (E/Z)-N-(6-(1,3-dioxoisoindolin-2-yl)hex-3-en-1-yl)picolinamide (4c)
The reaction was carried out according to the general procedure using alkene 1c (17.6 mg, 0.1 mmol), 2-(but-3-en-1-yl)isoindoline-1,3-dione (100 mg, 0.5 mmol), Grubbs II (4.2 mg, 0.005 mmol) and DCM (0.05 mL). The reaction was allowed to run for 16 h, and the product was purified by PTLC to afford 4c as a white solid (9.2 mg, 26%, E/Z = 82:18). The following NMR data corresponds to the E (major) isomer. 1H NMR (600 MHz, CDCl3) δ 8.52 (dt, J = 4.8, 1.2 Hz, 1H), 8.19–8.14 (m, 1H), 8.04 (s, 1H), 7.85–7.79 (m, 3H), 7.73–7.66 (m, 2H), 7.40 (ddt, J = 7.6, 4.8, 1.4 Hz, 1H), 5.53 (dd, J = 6.9, 4.0 Hz, 2H), 3.73 (t, J = 7.2 Hz, 2H), 3.41 (t, J = 6.6 Hz, 2H), 2.44–2.37 (m, 2H), 2.30–2.26 (m, 2H). 13C NMR (151 MHz, CDCl3) δ 168.67, 164.51, 150.32, 148.38, 137.62, 134.24, 132.41, 130.12, 129.10, 126.37, 123.55, 122.47, 39.23, 38.00, 32.96, 31.95. HRMS calcd. for C20H20N3O3+ [M+H]+: 350.1505, Found: 350.1504.
Supplementary Material
Figure 1.

Background and project synopsis
Table 2.
Substrate scopea

|
1a (0.1 mmol). Percentages correspond to isolated yields, E/Z ratios determined by 1H NMR of the isolated product mixture.
Acknowledgments
This work was supported by the National Institutes of Health (R35GM125052). We gratefully acknowledge Umicore for their donation of Grubbs Catalyst® C848. We thank Camille Rubel for detailed proofreading of this manuscript.
References and notes
- 1.a) For representative reviews, see:Hoveyda AH, Evans DA, Fu GC, Chem. Rev 93 (1993) 1307–1370. [Google Scholar]; b) Oestreich M, Eur. J. Org. Chem 5 (2005) 783–792. [Google Scholar]; c) Rousseau G, Breit B, Angew. Chem. Int. Ed 50 (2011) 2450–2494. [DOI] [PubMed] [Google Scholar]; d) Gurak JA Jr, Engle KM, Synlett 28 (2017) 2057–2065. [Google Scholar]; e) Lin C, Shen L, ChemCatChem 11 (2019) 961–968. [Google Scholar]; f) Wang Z-X, Bai X-Y, Li B-J, Chin. J. Chem 37 (2019) 1174–1180. [Google Scholar]; g) Daugulis O, Do H-Q, Shabashov D, Acc. Chem. Res 42 (2009) 1074–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Rouquet G, Chatani N, Angew. Chem. Int. Ed 52 (2013) 11726–11743. [DOI] [PubMed] [Google Scholar]
- 2.a) For AQ-directed hydrofunctionalization, see:Gurak JA Jr., Yang KS, Liu Z, Engle KM, J. Am. Chem. Soc 138 (2016) 5805–5808. [DOI] [PubMed] [Google Scholar]; b) Yang KS, Gurak JA Jr., Liu Z, Engle KM, J. Am. Chem. Soc 138 (2016) 14705–14712. [DOI] [PubMed] [Google Scholar]; c) Gurak JA Jr., Tran VT, Sroda MM, Engle KM, Tetrahedron 73 (2017) 3636–3642. [Google Scholar]; d) Wang H, Bai Z, Jiao T, Deng Z, Tong H, He G, Peng Q, Chen G, J. Am. Chem. Soc 140 (2018) 3542–3546. [DOI] [PubMed] [Google Scholar]; e) Lv H, Xiao L-J, Zhao D, Zhou Q-L, Chem. Sci 9 (2018) 6839–6843. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Wang C, Xiao G, Guo T, Ding Y, Wu X, Loh T-P, J. Am. Chem. Soc 140 (2018) 9332–9336. [DOI] [PubMed] [Google Scholar]; g) Matsuura R, Jankins TC, Hill DE, Yang KS, Gallego GM, Yang S, He M, Wang F, Marsters RP, McAlpine I, Engle KM, Chem. Sci 9 (2018) 8363–8368. [DOI] [PMC free article] [PubMed] [Google Scholar]; h) Shen H-C, Zhang L, Chen S-S, Feng J, Zhang B-W, Zhang Y, Zhang X, Wu Y-D, Gong L-Z, ACS Catal. 9 (2019) 791–797. [Google Scholar]; i) Nimmagadda SK, Liu M, Karunananda MK, Gao D-W, Apolinar O, Chen JS, Liu P, Engle KM, Angew. Chem. Int. Ed 58 (2019) 3923–3927. [DOI] [PMC free article] [PubMed] [Google Scholar]; j) Wei C, Ye X, Xing Q, Hu Y, Xie Y, Shi X, Org. Biomol. Chem 17 (2019) 6607–6611. [DOI] [PMC free article] [PubMed] [Google Scholar]; k) Zheng K, Xiao G, Guo T, Ding Y, Wang C, Loh T-P, Wu X, Org. Lett 22 (2020) 694–699. [DOI] [PubMed] [Google Scholar]; l) Bai Z, Bai Z, Song F, Wang H, Chen G, He G, ACS Catal. 10 (2020) 933–940. [Google Scholar]; m) Yang D, Huang H, Li M-H, Si X-J, Zhang H, Niu J-L, Song M-P, Org. Lett 22 (2020) 4333–4338. [DOI] [PubMed] [Google Scholar]; n) Guo T, Ding Y, Zhou L. Xu H, Loh T-P, Wu X, ACS Catal. 10 (2020) 7262–7268. [Google Scholar]; o) Wang X, Li Z-Q, Mai BK, Gurak JA, Xu JE, Tran VT, Ni H-Q, Liu Z, Liu Z, Yang KS, Xiang R, Liu P, Engle KM, Chem. Sci 11 (2020) 11307–11314. [DOI] [PMC free article] [PubMed] [Google Scholar]; p) Lu M-Z, Luo H, Hu Z, Shao C, Kan Y, Loh T-P, Org. Lett 22 (2020) 9022–9028. [DOI] [PubMed] [Google Scholar]; q) Jeon J, Lee C, Seo H, Hong S, J. Am. Chem. Soc 142 (2020) 20470–20480. [DOI] [PubMed] [Google Scholar]; r) Zhu C-F, Gao C-H, Hao W-J, Zhu Y-L, Tu S-J, Wang D-C, Jiang B, Org. Chem. Front 8 (2021) 127–132. [Google Scholar]
- 3.a) Dicarbofunctionalization:Liu Z, Zeng T, Yang KS, Engle KM, J. Am. Chem. Soc 138 (2016) 15122–15125. [DOI] [PubMed] [Google Scholar]; b) Derosa J, Tran VT, Boulous MN, Chen JS, Engle KM, J. Am. Chem. Soc 139 (2017) 10657–10660. [DOI] [PubMed] [Google Scholar]; c) Derosa J, van der Puyl VA, Tran VT, Liu M, Engle KM, Chem. Sci 9 (2018) 5278–5283. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Jeon J, Ryu H, Lee C Cho D, Baik M-H, Hong S, J. Am. Chem. Soc 141 (2019) 10048–10059. [DOI] [PubMed] [Google Scholar]; e) Zhang Y, Chen G, Zhao D, Chem. Sci 10 (2019) 7952–7957. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Pan R, Shi C, Zhang D, Tian Y, Guo S, Yao, A. Lin Org. Lett 21 (2019) 8915–8920. [DOI] [PubMed] [Google Scholar]; g) Yang T, Chen X, Rao W, Koh MJ, Chem, 6 (2020) 738–751. [Google Scholar]
- 4.a) Carboamination:Liu Z, Wang Y, Wang Z, Zeng T, Liu P, Engle KM, J. Am. Chem. Soc 139 (2017) 11261–11270. [DOI] [PubMed] [Google Scholar]; b) van der Puyl VA, Derosa J, Engle KM, ACS Catal. 9 (2019) 224–229. [Google Scholar]
- 5.a) Carbo- and aminoboration, see:Liu Z, Ni H-Q, Zeng T, Engle KM, J. Am. Chem. Soc 140 (2018) 3223–3227. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Liu Z, Li X, Zeng T, Engle KM, ACS Catal. 9 (2019) 3260–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]; c) Bai Z, Zheng S, Bai Z, Song F, Wang H, Peng Q, Chen G, He G, ACS Catal. 9 (2019) 6502–6509. [Google Scholar]; d) Shi P, Wang J, Gan Z, Zhang J, Zeng R, Zhao Y, Chem. Commun 55 (2019) 10523–10526. [DOI] [PubMed] [Google Scholar]; e) Liu Z, Chen J, Lu H-X, Li X, Gao Y, Coombs JR, Goldfogel M, Engle KM, Angew. Chem. Int. Ed 58 (2019) 17068–17073. [DOI] [PMC free article] [PubMed] [Google Scholar]; f) Bai Z, Zhang H, Wang H, Yu H, Chen G, He G, J. Am. Chem. Soc 143 (2021) 1195–1202. [DOI] [PubMed] [Google Scholar]; g) Zhang H, Lv X, Yu H, Bai Z, Chen G, He G, Org. Lett (2021) 10.1021/acs.orglett.1c01007. [DOI] [PubMed] [Google Scholar]
- 6.a) Other difunctionalization reactions:Zeng T, Liu Z, Schmidt MA, Eastgate MD, Engle KM, Org. Lett 20 (2018) 3853–3857. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Li Y, Liang Y, Dong J, Deng Y, Zhao C, Su Z, Guan W, Bi X, Liu Q, Fu J, J. Am. Chem. Soc 141 (2019) 18475–18485. [DOI] [PubMed] [Google Scholar]; c) Ni H-Q, Kevlishvili I, Bedekar PG, Barber JS, Yang S, Tran-Dubé M, Romine AM, Lu H-X, McAlpine IJ, Liu P, Engle KM, Nat. Commun 11 (2020) 6432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.a) Heck/C−H activation type functionalization:Liu M, Yang P, Karunananda MK, Wang Y, Liu P, Engle KM, J. Am. Chem. Soc, 140 (2018) 5805–5813. [DOI] [PMC free article] [PubMed] [Google Scholar]; b) Tang C, Zhang R, Zhu B, Fu J, Deng Y, Tian L, Guan W, Bi X. J. Am. Chem. Soc 140 (2018) 16929–16935. [DOI] [PubMed] [Google Scholar]; c) Lu H, Kang H, Zhou B, Xue X, Engle KM, Zhao D, Nat. Commun 10 (2019) 5025. [DOI] [PMC free article] [PubMed] [Google Scholar]; d) Deng Y, Zhao C, Zhou Y, Wang H, Li X, Cheng G-J, J. Fu. Org. Lett 22 (2020) 3524–3530. [DOI] [PubMed] [Google Scholar]
- 8.Rajendra G, Miller MJ, J. Org. Chem 52 (1987) 4471–4477. [Google Scholar]
- 9.Wang J, Chen J, Kee CW, Tan C-H, Angew. Chem. Int. Ed 51 (2012) 2382–2386. [DOI] [PubMed] [Google Scholar]
- 10.Chatterjee AK, Choi T-L, Sanders DP, Grubbs RH, J. Am. Chen. Soc, 125 (2003) 11360–11370. [DOI] [PubMed] [Google Scholar]
- 11.While this work was in progress, an unoptimized version of the present method was applied to access one substrate in Ref. 2o.
- 12.Zhang Q, Shi B-F, 10.1021/acs.accounts.1c00168. [DOI]
- 13.a) Zang Z−, Sheng Zhao S Karnakanti C-L Liu P-L Shao Y. He, Org. Lett 18 (2016) 5014–5017. [DOI] [PubMed] [Google Scholar]; b) Schreib BS, Carreira EM, J. Am. Chem. Soc 141 (2019) 8758–8763. [DOI] [PubMed] [Google Scholar]; c) Mao C-L, Zhao S, Zang Z-L, Xiao L, Zhou C-H, He Y, Cai G-X, J. Org. Chem 85 (2020) 774–787. [DOI] [PubMed] [Google Scholar]
- 14.Matsuura R, Karunananda MK, Liu M, Nguyen N, Blackmond DG, Engle KM, ACS Catal. 11 (2021) 4239–4246. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.