

Abstract
Ischemic postconditioning (IPostC) protects against brain injury induced by stroke and is a potential strategy for ischemic stroke treatment. Understanding its underlying mechanisms and potential hurdles is essential for clinical translation. In this review article, we will summarize the current advances in IPostC for stroke treatment and the underlying protective mechanisms. Strong evidence suggests that IPostC reduces brain infarct size, attenuates blood-brain barrier (BBB) damage and brain edema, and improves neurological outcomes. IPostC also promotes neurogenesis and angiogenesis at the recovery phase of ischemic stroke. The protective mechanisms involve its effects on anti-oxidative stress, anti-inflammation, and anti-apoptosis. In addition, it regulates neurotransmitter receptors, ion channels, heat shock proteins (HSP) 40/70, as well as growth factors such as BDNF and VEGF. Furthermore, IPostC modulates several cell signaling pathways, including the PI3K/Akt, MAPK, NF-κB, and the Gluk2/PSD95/MLK3/MKK7/JNK3 pathways. We also discuss the potential hurdles for IPostC’s clinical translation, including insufficient IPostC algorithm studies, such as therapeutic time windows and ischemia-reperfusion periods and cycles, as well as its long-term protection. In addition, future studies should address confounding factors such as age, sex, and pre-existing conditions such as hypertension and hyperglycemia before stroke onset. At last, the combination of IPostC with other treatments, such as tissue plasminogen activator (t-PA), merits further exploration.
Keywords: stroke, cerebral ischemia, ischemic postconditioning, oxidative stress, neuroinflammation
1. Introduction
Stroke remains a leading cause of morbidity and mortality worldwide (Kuklina et al., 2012; Campbell et al., 2019), with ischemic stroke accounting for more than 85 percent of all stroke cases (Benjamin et al., 2018). Currently, the tissue plasminogen activator (t-PA) is the only FDA approved drug treatment used to dissolve blood clots due to ischemic stroke (Chapman et al., 2014). However, t-PA has a limited therapeutic time window of 4.5 hours, and treatment beyond this point increases the risk of hemorrhagic transformation 10-fold, a severe complication that could worsen stroke outcomes (Sussman and Connolly, 2013; Chen et al., 2017; Zhang et al., 2019). Recent advances showed that delayed endovascular thrombectomy improves functional outcomes from 6 to 16 and even 24h after ischemia, but this only applies to a small, select group of patients (Albers et al., 2018; Nogueira et al., 2018; Ma et al., 2019). Therefore, it is crucial to develop new therapeutic strategies for the treatment of ischemic stroke. As a result, scientists have been searching for drug candidates to target various ischemic cascades for stroke treatment. Although many drug candidates have been proven effective in attenuating brain injuries in animal stroke models, none have been successfully translated into clinical use (Hoyte et al., 2004). In facing these setbacks, which mainly target a single ischemic cascade or cell signaling pathway, scientists have explored whether endogenous protective mechanisms, such as pre- and postconditioning, could be employed for ischemic stroke treatment (Zhao, 2007).
Our group is among the first to demonstrate that ischemic postconditioning (IPostC) protects against ischemic brain injury (Zhao et al., 2006; Zhao, 2007). IPostC is defined in contrast to ischemic preconditioning (IPreC). While IPreC, which is induced by a sub-lethal ischemia, is conducted before stroke onset, IPostC is performed after stroke onset by a single or a series of mechanical interruptions of reperfusion. Classic postconditioning has evolved into a term that represents a broad range of stimuli or triggers, including pharmacological agents or brief episodes of ischemia conducted in a remote organ (Zhao, 2009; Xie et al., 2018). Remote IPostC shows neuroprotective effects in multiple cerebral vascular insult models, including the ischemic stroke model (Kitagawa et al., 2018; Liu et al., 2020) and the subarachnoid hemorrhage (SAH) model (Hu et al., 2018), It also accelerates hematoma resolution and improves neurological outcomes in the intracerebral hemorrhage (ICH) model via AMPK-dependent immune regulation (Vaibhav et al., 2018). In this brief article, we will exclusively review the protective effects of IPostC in ischemic stroke and its underlying protective mechanisms, and then discuss the potential problems and future directions in IPostC research.
2. IPostC neuroprotection effects
The protective effects of IPostC on ischemic injury and recovery are characterized by changes in infarction, neurological deficit, blood-brain barrier (BBB) leakage, brain edema, angiogenesis, and neurogenesis. Therefore, we will first summarize the protective effects of IPostC on these pathological events (Table 1).
Table 1.
Summary of representative studies on postconditioning for ischemic stroke treatment.
| Ischemia models/time period | Species | Sex | Age or body weight | Treatment time point after reperfusion | Treatment Periods | Cycle | Molecular mechanisms | Outcomes | Ref |
|---|---|---|---|---|---|---|---|---|---|
| MCAO 60 min | C57BL6 mice | Male | 23–26g | 10 min | 10 min I | 1 | Inhibited Leucocyte infiltration, microglial activation, NF-κB, MMP-9, proteasome activity; Increased HSP70.BDNF, VEGF | Reduced Infarct size, BBB damage, brain edema; Improved motor function, neurogenesis, angiogenesis | (Doeppner et al., 2017) |
| MCAO 45 min | C57BL6 mice | Male | 4–4.5 months | 2 min | 15s I/30s R | 3 | Inhibited miR-1/−12, let-7f; Increased miR-19a | Improved motor and cognitive function | (Miao et al., 2016) |
| MCAO 120 min | Wistar rats | Male | 7–8 weeks | Immediately | 30s R/30s I | 3 | Reduced protein carbonyl derivatives, advanced oxidation protein products, hydrogen peroxide; Increased SOD, CAT, proteasome activity | Reduced infarct size | (Li et al., 2012) |
| MCAO | Wistar rats | Male | 200–250g | 30 min | 10s I/30s R | 5 | Decreased ROS | Reduced infarct size, brain edema | (Rezazadeh et al., 2013) |
| 30s I/30s R | 5 | Reduced infarct size | |||||||
| 60s I/60s R | 5 | Reduced infarct size | |||||||
| MCAO 2 hours | Wistar rats | Male | 7–8 weeks | Immediately | 30s R/30s I | 3 | Increased IκBα, decreased p-IκBα, NF-κB, p- NF-κB, Caspase-3, Noxa, Bim and Bax | Reduced infarct size, apoptosis, improved neurological outcome | (Liang et al.; 2014) |
| MCAO 100min | 5D rats | Male | 10 min | 10 min I | 1 | Not studied | Reduced infarct size, improved neurological outcomes, neurogenesis, and angiogenesis | (Esposito et al., 2015) | |
| MCAO 100min | 5D rats | Male | 320–340g | 10 min | 10 min I | 1 | Increased VEGF | Reduced Infarct size; Improved neurological outcome; microglial polarization | (Esposito et al., 2018) |
| MCAO 60 min | SD rats | Male | 250–280g | Immediately | 30s R/30s I | 6 | Increased HSP70, bcl-2, SOD activity; Decreased caspase-3/9, MDA, inhibited cytochrome C and bax translocation | Reduced infarct size, and cell apoptosis; improved neurological outcomes | (Xing et al., 2008a) |
| MCAO 2,3, 4, 4.5, 6 hours | SD rats | Male | 7–8 weeks | Immediately | 10s I/10s R | 5 | Inhibited IL-1β, TLR2/4, IRAK4 | Reduced infarct size and cell apoptosis | (Wang et al., 2014) |
| MCAO 90 min | SD rats | Male | 7–8 weeks 270–300g | Immediately or 3 h | 10s I/10s R | 10 | Increased BDNF, p-CREB, | Reduced infarct size, brain edema, cell Hpoptosis, and stabilized cerebral blood flow | (Wu et al., 2015) |
| MCAO 120 min | SD rats | Male | 250–280g | Immediately | 30s R/30s I | 3 | Preserved claudin-5, occludin | Reduced infarct size, protected neurovascular unit, improved neurological outcome | (Han et al., 2014) |
| MCAO 120 min | SD rats | Female | 2–3 months 210–230g | Immediately | 10s R/10s I | 6 | Increased acid-sensing ion channel 2a | Protected neurons of hippocampal CA1 region | (Duanmu et al,, 2015) |
| MCAO 120 min | SD rats | Male | 250–280g | Immediately | 30s R/30s I | 3 | Inhibited AQP4 | Reduced infarct size and brain edema | (Han et al., 2015) |
| MCAO 120 min | SD Rat | Male | 250–300g | Immediately | 30s R/30s I | 3 | Not studied | Reduced infarct size | (Taskapilioglu et al., 2009) |
| MCAO | Rat | Male | Inhibited positive cell cycle regulators and ERK/CREB and GSK- 3β/CREB | (Zhao et al., 2013) | |||||
| MCAO 60 min | SD Rats | Male | 250–230g | Immediately | 30s R/30s I | 6 | Inhibited MPO, MDA, IL-1β, TNF-α, ICAM-1 | Reduced infarct size, cell necrosis and apoptosis, leucocyte infiltration and oxidative stress | (Xing et al., 2008b) |
| MCAO 100 min | SD rats | Male | 250–300g | 2 min | 2 min I/2 min R | 5 | Increased p-AKT, p-ERK, p-p38 | No protection on infarct volume | (Pignataro et al., 2008) |
| 5 min | 5 min I/5 min R | 3 | Reduced infarct size | ||||||
| 10 min | 10 min I | 1 | Reduced infarct size | ||||||
| 30 min | 10 min I | 1 | No protection | ||||||
| Thrombotic ischemia | Tree shrews | Male | 120–150g | 4 h after ischemia | 5 min I/5 min R | 3 | Inhibited MIPO, TLR4; Increased TLR4 at 72 hours | Reduced neuron death | (Feng et al., 2011) |
| Distal MCAO plus BCCA 30 min | SD rats | Male | 350–400g | Immediately or 3 h | 30s R/10s I | 3 | Decreased CHOP, caspase-12; Increased GRP7E, | Reduced infarct size and neuronal apoptosis | (Yuan et al., 2011) |
| Permanent distal (MCAO) plus CCAO 30 min | 5D rats | Male | 350–390g | Immediately | 30s R/10s I | 3 | Mot mentioned | Reduced brain infarct volume | (Gao et al,, 2008a) |
| 30s R/10s I | 10 | No protection | |||||||
| 10s R/10s I | 3 | No protection | |||||||
| 10s R/10s I | 10 | Reduced infarct size | |||||||
| 3 min | 10s I/10s R | 10 | No protection | ||||||
| Permanent distal (MCAO) plus CCAO 30 min | SD rats | Male | 350–450g | Immediately | 30s R/10s I | 3 | Inhibited superoxide | Reduced infarct size and cell apoptosis | (Zhao et al., 2006) |
| Permanent distal MCAO plus bilateral CCAO 30 min | SD rats; T-cell-deficient rats | Male | 230–250g | Immediately | 30s R/10s I | 3 | Increased p-Akt, Akt1/2/3, p-mTOR, mTOR, pS6K, S6K, p4EPB1, 4EPB1, GAP43 | Reduced infarct size and improved neurological outcomes | (Xie et al., 2013) |
| SD rats | Male | 350–390g | Immediately | 30s R/10s I | 3 | Increased p-Akt, p-GSK3β, p-β-catenin, p-JNK, p-ERK1/2, δPKC, εPKC | Long-term reduction of brain damage and improved neurological outcomes | (Gao et al., 2008b) | |
| SD rats | Male | 270–330g | 30 min | 10s I/10s R | 10 | Reduced infarct size | (Ren et al., 2008) | ||
| 30 min | 30s I/30s R | 3 | Reduced infarct size | ||||||
| 3h | 10s I/10s R | 10 | No protection | ||||||
| 3h | 30s I/30s R | 3 | Reduced infarct size | ||||||
| BCAO 10 min | Swiss mice | Male | 25 ± 2g | Immediately | 10s R/10s I | 3 | PI3K | Improved memory and motor coordination | (Rehni and Singh, 2007) |
| BCAO 12 min | Swiss mice | Male | 20 ± 5g | Immediately | 10s R/10s I | 3 | Increased nitrite/nitrate, GSH, eNOS; Inhibited AChE activity, TBRS | Reduced infarct size, improved memory and motor coordination | (Gulati et al., 2014) |
| BCAO 12 min | Swiss mice | Male | 22 ± 5g | Immediately | 10s R/10s I | 3 | Increased nitrite/nitrate, GSH; Inhibited AChE activity, TBARS | Reduced infarct size, improved memory and motor coordination | (Gulati and Singh, 2014) |
| BCAO 17 min | Swiss mice | Either sex | 25 ± 2g | Immediately | 10s R/10s I | 3 | Involved CCR2 | Reduced infarct size, improved memory and motor corrdiantion | (Rehni and Singh, 2012) |
| 4-VO 10 min | SD rats | Male | 200–220g | Immediately | 10s R/10s I | e | Inhibited caspase-3/6/9, bax; Increase bcl-2 | Protected hippocampal neurons and inhibited apoptosis | (Ding et al., 2012) |
| 4-VO 10 min | SD rats | Male | 200–220 g | Immediately | 10s R/10s I | 6 | Inhibited caspase-3/6/9, bax; Increase bcl-2 | Protected hippocampal neurons and inhibited apoptosis | (Ding et al., 2012) |
| 4-VO 10 min | SD rats | Male | 260–320g | Immediately | 15s R/15s I | 3 | Inhibited Cytochrome C translocation | Promoted neuron survival, spatial learning and memory; improved the disturbance of CBF | (Wang et al., 2008) |
| 30s R/30s I | 3 | Inhibited Cytochrome C translocation | Promoted neuron survival, spatial learning and memory; improved the disturbance of CBF | ||||||
| 60s R/15s I | 3 | None | None | ||||||
| 45 s | 15s I/15s R | 3 | Inhibited Cytochrome C translocation | Promoted neuron survival, spatial learning and memory; improved the disturbance of CBF | |||||
| 4-VO 10 min | SD rats | Male | 220–250g | Immediately | 10s R/10s I | 6 | Inhibited caspase-3; increased Bcl-2 | Reduced hippocampal neuron apoptosis | (Zhang et al., 2012) |
| 4-VO 15 min | SD rats | Male | 220–300g | 10 min | 3 min I | 1 | Stabilized VDAC1/2/3 expression and calcium homeostasis | Protected hippocampal neurons | (Yao et al., 2018) |
| 4-VO 15 min | SD rats | Male | 250–300g | 10 min | 3 mini | 1 | Inhibited GluK2-PSD-95-MLK3-MKK7-JNK3 | Protected hippocampal CA1 pyramidal neurons | (Liu et al., 2013) |
| 4-VO 15 min | Wistar rats | Male | 250–300g | Immediately | 30s R/15s I | 3 | Not studied | Reduced neuron apoptosis and neurodegeneration | (Xiang et al., 2018) |
| 4-VO 10 min | Wistar rats | Male | 250–300g | 2 days | 5 min I | 1 | Increased SOD and CAT activity | Reduced neuronal degeneration | (Danielisova et al., 2006) |
| 4-VO 10 min | Wistar Rats | Male | 250–350g | 2 days | 4 min or 6 min I | 1 | Increased MnSOD | Not studied | (Nemethova et al., 2008) |
| 4-VO 8, 10, 12 min | Wistar rats | Not mentioned | 250–350g | 2 days | 5, 6 min I | 1 | Not mentioned | Promoted survival of CA1 neurons, cortex and striatum neurons | (Burda et al., 2006) |
I, Ischemia; R, Reperfusion; 4-VO, four vessel occlusion; AQP4, Aquaporin 4; BCAO, bilateral carotid artery occlusion; BDNF, Brain-derived neurotrophic factor; CAT, Catalase; CCR2, C-C chemokine receptor type 2; CHOP, C/EBP-homologous protein; GAP43, Growth Associated Protein 43; Gluk2, Glutamatergic kainate receptor subunit 2; GSH, Glutathione; HSP70, heat shock protein 70; IRAK4, Interleukin-1 receptor-associated kinase 4; JNK3, c-Jun N-terminal kinase 3; MCAO, middle cerebral artery occlusion; MDA, Malondialdehyde; MLK3, Mixed-lineage protein kinase 3; MMP-9, Matrix metallopeptidase 9; MPO, Myeloperoxidase; mTOR, mammalian target of rapamycin; PSD-95, postsynaptic density protein 95; ROS, Reactive oxygen species; SOD, Superoxide dismutase; TBARS, Tihobarbituric acid reactive species; TLR4, Toll-like receptor 4; VDAC, Voltage-dependent anion channel; VEGF, Vascular endothelial growth factor
2.1. Brain infarction
We and others have reported that IPostC reduces brain infarction in focal and global cerebral ischemia in mice and rats (Xing et al., 2008b; Rehni and Singh, 2012; Gulati and Singh, 2014; Gulati et al., 2014; Han et al., 2014; Wang et al., 2014; Esposito et al., 2015; Wu et al., 2015; Doeppner et al., 2017). Most studies evaluated brain infarct sizes by using triphenyltetrazolium chloride (TTC) staining at 24h (Xing et al., 2008b, a; Taskapilioglu et al., 2009; Yuan et al., 2011; Rehni and Singh, 2012; Gulati and Singh, 2014; Gulati et al., 2014; Han et al., 2014; Han et al., 2015; Doeppner et al., 2017; Sun et al., 2018), 48h (Zhao et al., 2006; Ren et al., 2008; Xie et al., 2013; Wang et al., 2014), and 72h after ischemic onset (Xing et al., 2008b; Wu et al., 2015). Although one study suggests that early neuroprotection did not translate into long-term neuronal survival (Doeppner et al., 2017), we and others have shown that rapid IPostC, which was conducted within a few minutes after reperfusion, reduced infarct volume measured at 2 weeks or 1 month after ischemia (Gao et al., 2008; Esposito et al., 2015), suggesting long-term benefits of rapid IPostC.
We have found that ischemic severity determines the extent of protection that IPostC may afford, i.e. the longer the ischemic period, the less protection IPostC can achieve (Zhao et al., 2006). This finding is supported by another study, which shows that rapid IPostC did not reduce infarct sizes and brain edema when the ischemic period was extended to 4.5h (Wang et al., 2014).
We and others have evaluated the IPostC therapeutic time windows. We defined rapid IPostC when it is induced immediately or a few minutes after reperfusion onset, while delayed IPostC occurs when it is initiated a few hours after reperfusion onset. We found that delayed IPostC performed at 3h and 6h after ischemia/reperfusion robustly reduced infarction size in the rat stroke model (Ren et al., 2008). Compared with focal cerebral ischemia, the therapeutic time window in transient forebrain ischemia is longer, as delayed IPostC initiated as late as 2d after cerebral ischemia could still exert anti-oxidative stress effects and protect neurons (Burda et al., 2006; Danielisova et al., 2006; Nemethova et al., 2008).
Since preconditioning and postconditioning share some similar protective mechanisms, it is tempting to test whether preconditioning and postconditioning have synergistic effects. The combination of preconditioning with postconditioning showed greater neuroprotection (Wu et al., 2015). However, Taskapilioglu et al. (2009) showed contradictory results in that the combination of preconditioning and postconditioning did not yield further protection. Such inconsistent results could be due to the different experimental protocols employed in different studies.
In short, IPostC can reduce brain infarct size, and larger neuroprotection is achieved when IPostC is applied at an earlier time point of reperfusion. The exact therapeutic time window of IPostC varies in different stroke models, and whether preconditioning plus postconditioning has synergistic effects needs further investigation.
2.2. Neurological functions
Reductions in brain infarct sizes are often associated with neurological functional recovery. IPostC improves motor function, motor coordination, and short-term memory at the acute phase of ischemic stroke (Rehni and Singh, 2007; Xing et al., 2008b; Rehni and Singh, 2012; Gulati and Singh, 2014; Gulati et al., 2014; Han et al., 2014; Wu et al., 2015; Miao et al., 2016; Doeppner et al., 2017), attenuates neurologic deficit, and improves cognitive and memory function at the late phase of ischemic stroke (Rehni and Singh, 2007; Esposito et al., 2015; Miao et al., 2016). Notably, the improvement of neurological function at the acute phase by IPostC was abolished when the transient focal ischemia period was extended to 4.5h (Wang et al., 2014). We reported that delayed postconditioning at 6h after permanent focal ischemia provided long-term protection in attenuating motor asymmetry (Ren et al., 2008).
2.3. Blood-brain barrier damage and brain edema
The neuroprotective effects of IPostC are usually linked to the preservation of the BBB integrity and reduction in brain edema in stroke models (Han et al., 2014; Doeppner et al., 2017; Xiang et al., 2018). During the cerebral ischemia-reperfusion injury, activation of the oxidative stress-mediated matrix metalloproteinases (MMPs) disrupts the tight junction proteins and extracellular matrix components of BBB (Haorah et al., 2007; Gu et al., 2012; Chen et al., 2018). IPostC significantly inhibits the oxidative stress and MMP-9 activity in the ischemic brain (Liu et al., 2012; Doeppner et al., 2017), preserves the ultrastructure of the neurovascular unit, protects the tight junction protein, claudin-5 and occludin, the extracellular matrix laminin, and fibronectin (Liu et al., 2012; Han et al., 2014). In addition, the protection of the BBB integrity is associated with the reduction of leukocyte infiltration and the attenuation of microglial activation (Doeppner et al., 2017). Therefore, IPostC protects the BBB possibly by inhibiting neuroinflammation and attenuating the oxidative stress-mediated activation of MMP-9, and preserving tight junction proteins and extracellular matrix.
The formation of brain edema during ischemic stroke could be due to both cytotoxic and vasogenic changes (Heo et al., 2005). Therefore, BBB protection could contribute to the attenuation of brain edema. Evidence showed that IPostC reduced brain edema in focal ischemia models, as calculated by brain swelling or wet-dry weight methods (Rezazadeh et al., 2013; Han et al., 2015). In addition, the reduction of brain edema is associated with the inhibition of membrane water channel aquaporin-4 (AQP4), which regulates water flux in the ischemic brain (Han et al., 2015).
2.4. Neurogenesis and angiogenesis
Neurogenesis and angiogenesis play important roles in brain functional recovery after stroke, which are promoted by IPostC. In a middle cerebral artery occlusion (MCAO) model, IPostC significantly enhanced neurogenesis and angiogenesis 2 weeks after ischemia, as indicated by the double staining of doublecortin/BrdU and collagen-IV/Ki67 in the ischemic brain (Anrather and Hallenbeck, 2013). In contrast, in another study using the mouse MCAO model, IPostC itself did not promote endogenous neurogenesis or angiogenesis when measured 28d after cerebral ischemia (Doeppner et al., 2017), but IPostC could facilitate the intracerebral transplant of neural progenitor cells and promoted cell survival in the post-ischemic brain (Doeppner et al., 2017). Therefore, IPostC may provide a favorable microenvironment for the ischemic brain repair process.
3. Molecular mechanisms involved in IPostC’s protection
IPostC triggers multiple molecular mechanisms, including its anti-oxidative, anti-inflammatory effects, anti-apoptotic effects, and its regulation of the neurotransmitter and ion channels that maintain calcium balance, as well as the induction of growth factors and heat shock proteins (Table 1, Figure 1). These cascades link to multiple kinase signaling pathways, including the phosphoinositide 3-kinase/protein kinase B (PI3K/Akt), mitogen-activated protein kinase (MAPK), and the nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB) pathways (Table 1, Figure 2), which are detailed below.
Figure 1.
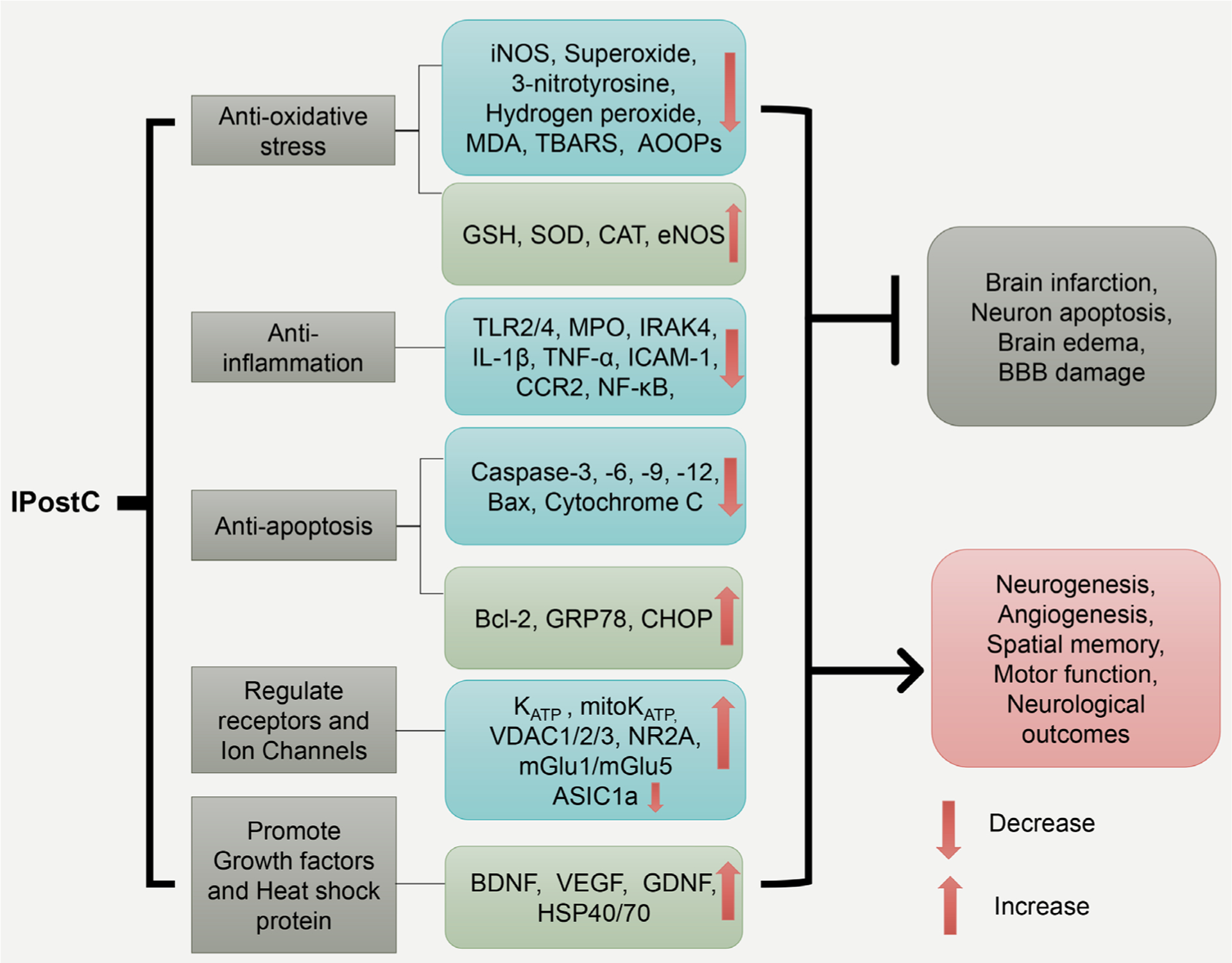
The schematic represents IPostC’s major protective mechanisms. The five major protective mechanisms mediated by IPostC are listed in the first row, and their corresponding molecules are shown in the second row. Together, these molecular events lead to the inhibition of brain infarction, neuronal apoptosis, edema, and BBB leakage, while promoting neurogenesis and angiogenesis related to functional recovery. Inducible nitric oxide synthase (iNOS); malondialdehyde (MDA); thiobarbituric acid reactive species (TBARS); advanced oxidation protein products (AOOPs); Glutathione (GSH); Superoxide dismutase (SOD); Catalase (CAT); endothelial nitric oxide synthase (eNOS); toll-like receptor 2/4 (TLR2/4); Myeloperoxidase (MPO); interleukin-1 receptor-associated kinase 4 (IRAK4); Interleukin 1 beta (IL-1β); Tumor necrosis factor α (TNF-α); Intercellular Adhesion Molecule 1 (ICAM-1); C-C chemokine receptor type 2 (CCR-2); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); glucose-regulated protein 78 (GRP78); C/EBP-homologous protein (CHOP); mitochondrial potassium ATP-dependent channel (mitoKATP); voltage-dependent anion channel protein (VDAC); glutamate receptor ε1 (NR2A); metabotropic glutamate receptor 1/5 (mGlu1/mGlu5); Acid-sensing ion channel 1a (ASIC1a); brain-derived neurotrophic factor (BDNF); vascular endothelial growth factor (VEGF); glial cell-derived neurotrophic factor (GDNF); heat shock protein 40/70 (HSP40/70); blood-brain barrier (BBB).
Figure 2.
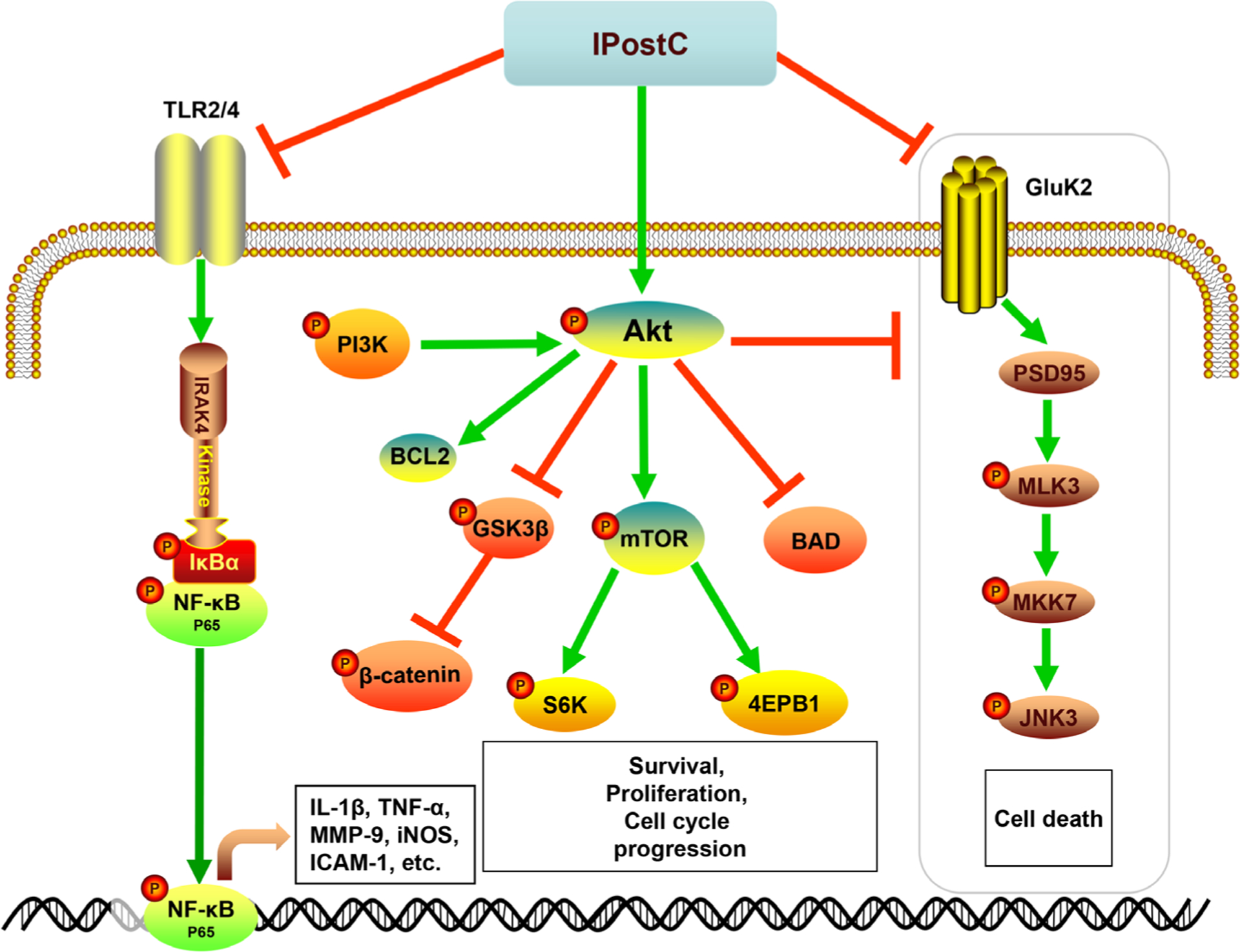
The diagram illustrates the major cellular and molecular signaling pathways activated by IPostC for neuroprotection. IPostC inhibits both the TLR2/4/NF-κB pathway and the GluK2/PSD-95/MLK3/MKK7/JNK3 signal complex to inhibit cell death, while promoting the PI3K/Akt pathway for neuronal survival. Toll-like receptor 2/4 (TLR2/4); interleukin-1 receptor-associated kinase 4 (IRAK4); nuclear factor kappa-light-chain-enhancer of activated B cells (NF-κB); NF-κB inhibitor α (IκB-α); protein kinase B (AKT); B-cell lymphoma 2 (Bcl-2); Bcl-2-associated death promoter (BAD); mammalian target of rapamycin (mTOR); ribosomal protein S6 kinase (S6K); eukaryotic initiation factor 4E-binding protein (4E-PB1); glutamatergic kainate receptor subunit 2 (Gluk2); postsynaptic density protein 95 (PSD95); mixed-lineage kinase 3 (MLK3); mitogen-activated protein kinase kinase 7 (MKK7); c-Jun N-terminal kinase 3 (JNK3).
3.1. Anti-oxidative actions
IPostC protects against oxidative stress. In both global and focal cerebral ischemia/reperfusion models, IPostC increases the activities of the endogenous antioxidant enzymes, superoxide dismutase (SOD) and catalase (CAT) (Danielisova et al., 2006; Nemethova et al., 2008; Xing et al., 2008b; Li et al., 2012), and inhibits inducible nitric oxide synthase (iNOS) expression (Wei et al., 2015). As a result, IPostC reduces oxidative products, including superoxide (Zhao et al., 2006), 3-nitrotyrosine (a footprint of peroxynitrite) (Zhao et al., 2014a), and hydrogen peroxide levels. IPostC also prevents lipid peroxidation and protein oxidation as indicated by the reduction of malondialdehyde (MDA), protein carbonyl derivatives, and advanced oxidation protein products (AOPPs) (Xing et al., 2008a, b; Li et al., 2012; Wei et al., 2015). In addition, IPostC reduces thiobarbituric acid reactive species (TBARS) levels, a lipid peroxidation marker, and improves glutathione levels (Gulati and Singh, 2014; Gulati et al., 2014; Doeppner et al., 2017; Xiang et al., 2018). Therefore, IPostC’s anti-oxidative effects may contribute to its neuroprotective effects against cerebral ischemia/reperfusion injury.
3.2. Regulation of neurotransmitter receptors and ion channels
The regulation of ion channels and neurotransmitter receptors by IPostC also contributes to its neuroprotection by maintaining ion stasis (Fan et al., 2017). For IPostC’s effects on the ion channels, Pateliya et al. (2008) showed that IPostC reduces brain infarct size by promoting the opioid receptor and subsequently opening KATP channels, and that the opioid receptor antagonist naloxone or the KATP channel blocker, glibenclamide, significantly abolishes IPostC’s protection. IPostC also protects against ischemic brain damage by modulating the mitochondrial potassium ATP-dependent channel (mitoKATP), and by inhibiting the mitochondrial permeability transition pore (MPTP) opening, subsequently preventing hyperpolarization (Robin et al., 2011). In addition, IPostC inhibits the transcription and expression of the acid-sensing ion channel, ASIC1a, a proton-gated cation channel activated during cerebral ischemia, leading to an excessive calcium entry in the temporoparietal cortex after transient focal ischemia (Pignataro et al., 2011). The down-regulation of ASIC1a by IPostC was dependent on the Akt signaling, as the Akt inhibitor, LY-294002, reversed ASIC1a modulation (Pignataro et al., 2011). Moreover, IPostC stabilizes the expressions of functional mitochondrial voltage-dependent anion channel proteins (VDAC1, VDAC2, and VDAC3), which are essential for calcium homeostasis (Yao et al., 2018).
The N-methyl-D-aspartate (NMDA) receptors are also important neuroprotection mediators of IPostC against cerebral ischemic injury (Celso Constantino et al., 2014). An in vivo study showed that the activation of the NMDA receptor subunit, NR2A, mediates the neuroprotective effects of IPostC by promoting activation of pro-survival signaling such as the extracellular signal regulated kinase (ERK) and cAMP response element binding proteins (CREB), and inducing brain-derived neurotrophic factor (BDNF) expression, which is blocked by the NR2A antagonist NVP-A (Zhang et al., 2015). An ex vivo study using rat organotypic hippocampal slices showed that metabotropic glutamate receptor 1/5 (mGlu1/mGlu5) contributes to IPostC’s protection, as the antagonist of mGlu1/mGlu5 abolished the protection (Scartabelli et al., 2008). This is strengthened by the fact that the mGlu1 and mGlu5 agonist 3,5-dihydroxyphenylglycine (DHPG) protects organotypic hippocampal slices from oxygen and glucose deprivation (OGD)-induced cell damage, which is dependent on Akt activation. Therefore, neurotransmitters and ion channel regulations, as well as the maintenance of ion homeostasis are important factors for IPostC-induced protection against ischemic brain injury.
3.3. Anti-apoptotic effects and the potential role of autophagic effects
IPostC’s anti-apoptotic effects have been reported by several ischemic stroke studies (Zhao et al., 2006; Xing et al., 2008a; Yuan et al., 2011; Ding et al., 2012; Zhang et al., 2012; Liang et al., 2014; Wang et al., 2014). IPostC significantly inhibits hippocampal neuronal apoptosis by inhibiting pro-apoptotic caspase-3 activation and promoting anti-apoptotic Bcl-2 expression (Zhang et al., 2012). In addition, IPostC down-regulates caspase-3/6/9 and pro-apoptotic Bax expression, and increases Bcl-2 in the hippocampal CA1 region (Ding et al., 2012). Moreover, IPostC also inhibits Bax translocation into the mitochondria and prevents the release of cytochrome C into the cytosol in a focal cerebral ischemia model (Xing et al., 2008b). Similarly, in a global ischemia model, IPostC inhibits cytosolic cytochrome C release, thus preventing subsequent neuronal cell death, and improves the learning and memory deficit (Wang et al., 2008). Furthermore, IPostC attenuates endoplasmic reticulum (ER)-stress-mediated apoptosis as indicated by the inhibition of C/EBP-homologous protein (CHOP), caspase-12, and increased levels of glucose-regulated protein 78 (GRP78) in the ischemic brain (Yuan et al., 2011). It is suggested that the PI3K/Akt pathway is necessary for such protection, as its inhibitor, LY294002, significantly abolishes IPostC’s effects on ER-stress related molecules and apoptosis (Yuan et al., 2011).
Autophagy also plays an important role in IPostC-mediated neuroprotection. One study reported that IPostC induces autophagy, which subsequently inhibits apoptosis (Sun et al., 2018). The neuroprotective effects of IPostC are reversed by the autophagy inhibitor, 3-methyladenine (3-MA) (Sun et al., 2018). Controversially, another study showed that IPostC inhibited autophagy, which contributed to neuroprotection (Gao et al., 2012). 3-MA attenuated the ischemic insult (Gao et al., 2012). Such contradictory results indicate the complex roles of autophagy in ischemic stroke injury probably depend on the severity of the ischemic event. Indeed, mild autophagy may protect the ischemic brain while excessive autophagy could cause damage (Feng et al., 2017). Therefore, IPostC may play an important role in balancing autophagy in the ischemic brain in different stroke models, and such roles need further verification.
3.4. Anti-neuroinflammatory effects
IPostC inhibits neuroinflammation in the post-ischemic brain. We reported that IPostC reduces leucocyte infiltration, including monocytes, CD4 T cells, CD8 T cells, and B cells, inhibits microglial activation in the ischemic brain, and attenuates systemic lymphopenia (Joo et al., 2013). Nevertheless, IPostC offered no protection against ischemic stroke in T cell-deficient nude rats, suggesting that IPostC protects the ischemic brain through T cell-mediated inflammation (Xie et al., 2013). We further found that IPostC inhibits T cell immunoglobulin and mucin domain containing-3 (Tim-3) expression and its ligand, galectin-9, which results in the elimination of type 1 helper T (Th1) cells (Wei et al., 2015). In addition to the lymphocytes, other studies report that IPostC also inhibits the innate immune response by down-regulating toll-like receptor-2/4 (TLR2/4) expression, thereby inhibiting myeloperoxidase (MPO) expression (Feng et al., 2011), and decreases the transcription and expression of interleukin-1 receptor-associated kinase 4 (IRAK4), subsequently down-regulating interleukin1 beta (IL-1β) in the ischemic brain (Wang et al., 2014). In addition, NF-κB is an essential downstream regulator of TLR’s activation in ischemic brain injury (Pradillo et al., 2009; Abdul et al., 2019). IPostC inhibits NF-κB expression and its translocation into the nucleus, and at the same time it promotes the expression of the NF-κB inhibitor, alpha (IκB-α) (Doeppner et al., 2017). In addition to inhibiting IL-1β, IPostC also decreases interleukin 6 (IL-6) (Kong et al., 2013), tumor necrosis factor alpha (TNF-α), and the intercellular adhesion molecule-1 (ICAM-1), the last being vital for leucocyte adhesion and infiltration (Xing et al., 2008a). Furthermore, C-C chemokine receptor type 2 (CCR-2) is involved in the neuroprotective effects of IPostC, as a selective CCR-2 antagonist abolished its neuroprotective effects (Rehni and Singh, 2012). Since CCR-2 is a pro-inflammatory monocyte marker, the role of monocytes in the neuroprotection of IPostC merits further investigation.
3.5. Increases in growth factors and heat shock proteins
IPostC promotes the expression of several growth factors in the ischemic brain, including BDNF and vascular endothelial growth factor (VEGF), which may contribute to neuronal repair and angiogenesis. For instance, IPostC enhances the concentrations of BDNF, glial cell-derived neurotrophic factor (GDNF), and VEGF in the ischemic brain with post-stroke neural progenitor cells (NPCs) transplantation, which is associated with enhanced neurogenesis and angiogenesis (Doeppner et al., 2017). Notably, the combination of early and delayed postconditioning further enhances BDNF expression in neurons and astrocytes in the ischemic brain, coupled with the induction of ERK and CREB phosphorylation, and further reduced infarct size (Wu et al., 2015). Similarly, IPostC induces neuronal VEGF expression in the ischemic penumbra and in cultured primary neurons, which binds to the VEGF receptor expressed on microglia, mediating microglial polarization into the anti-inflammatory M2 types (Esposito et al., 2018). Therefore, the release of VEGF may serve as a “help-me” signal for neurons to promote microglia and macrophages into beneficial functional types (Esposito et al., 2018).
Heat shock protein 40 and 70 (HSP40/HSP70) are important chaperones that assist in protein folding under physiological and stress conditions. IPostC enhances HSP70 expression and prevents HSP40 loss in ischemic CA1 tissue (Liang et al., 2012). The induction of HSP70 is vital for neuroprotection, and HSP70 knockdown reverses IPostC- induced reduction of infarction size and proteasome activation (Doeppner et al., 2017). Moreover, HSP70 induction by IPostC is associated with the inhibition of cytochrome c release, preventing cell apoptosis in a focal cerebral ischemia model (Xing et al., 2008b). Similarly, an in vitro study showed that postconditioning increases HSP70 expression and inhibits apoptosis of cultured primary cortical neurons exposed to OGD treatment (Zhao et al., 2014b). Therefore, the induction of HSPs is involved in IPostC neuroprotection possibly by attenuating neuronal cell apoptosis.
3.6. Regulation of kinase signaling pathways
Several protein kinase cell signaling pathways are involved in mediating the neuroprotective effects of IPostC in stroke models, which will be detailed in the following sections.
3.6.1. The PI3K/Akt pathway
Our group has studied the protective efficacy of IPostC and its associated mechanisms. We found that IPostC increases Akt activity and phosphorylation, which contributes to the protective effects of IPostC. The PI3K inhibitor, LY294002, reduces Akt activity, which partially suppresses the protective effects on infarct size reduction (Gao et al., 2008). Other research groups confirmed our results showing that PI3K/Akt inhibitors significantly reduced neuroprotective effects of IPostC (Rehni and Singh, 2007; Pignataro et al., 2008; Liu et al., 2013). The increase in Akt activity is associated with glycogen synthase kinase 3 beta (GSK3β) phosphorylation changes shortly after cerebral ischemia, and changes in the β-catenin phosphorylation pattern in the ischemic core and penumbra (Gao et al., 2008).
Akt directly and indirectly activates mammalian target of rapamycin (mTOR) signaling by enhancing its downstream molecules, including eukaryotic initiation factor 4E-binding protein (4E-BP1) and ribosomal protein S6 kinase (S6K), and promoting cell differentiation, growth, and survival (Martelli et al., 2010; Sabbah et al., 2011). We reported that IPostC increases phosphorylated Akt (p-Akt), phosphorylated mTOR (p-mTOR), phosphorylated S6K (p-S6K), and phosphorylated 4EBP1 (p-4EBP1) in the mTOR pathway and increases the presynaptic growth associated protein 43 (GAP43) protein levels (Xie et al., 2013). On the other hand, the administration of the mTOR inhibitor, rapamycin, abolishes the long-term protective effects of IPostC on reducing brain infarct size, and reduces GAP43 protein levels. Thus, the Akt-mediated mTOR pathway is important for the neuroprotection induced by IPostC (Xie et al., 2013).
Other groups have further explored the crosstalk between Akt and the GluK2–PSD-95–MLK3-JNK3 pathway. Postsynaptic density protein-95 (PSD-95) interacts with the glutamatergic kainate receptor subunit 2 (GluK2), and their downstream signaling molecules, such as mixed lineage kinase 3 (MLK3) and c-Jun N-terminal kinase 3 (JNK3) are important in mediating neuronal loss during cerebral ischemia (Pei et al., 2006; Hu et al., 2009). A recent study shows that IPostC suppresses the formation of the GluK2–PSD-95–MLK3-JNK3 signaling complex in the ischemic brain. These effects are abolished by Akt inhibition (Liu et al., 2013), suggesting that Akt signaling crosstalks with the GluK2–PSD-95–MLK3-JNK3 signaling pathway in ischemic brain injury (Figure 2).
3.6.2. The MAPKs signaling pathway
The MAPKs, including ERK1/2 and p38 MAPK, play important roles in regulating cell survival and cell death after cerebral ischemia (Lai et al., 2014). Yet their roles in IPostC are not fully understood. While one study showed that IPostC increased the phosphorylation of ERK1/2 and p38 MAPK in the ischemic cortex (Pignataro et al., 2008), another study showed contradictory results that IPostC reduced the phosphorylation of ERK1/2 and CREB in the ischemic brain (Zhao et al., 2013). Nevertheless, the ERK1/2 inhibitor, U0126, or p38 MAPK inhibitor, SB203580, did not abolish the neuroprotective effects of IPostC but did block the protective effects of IPreC (Pignataro et al., 2008). These results suggest that activation of ERK1/2 and p38 MAPK may not be necessary for IPostC’s neuroprotection. However, given these uncertain research results, dedicated studies are warranted to further verify the roles of ERK1/2 and p38 MAPK in the neuroprotection of IPostC.
3.6.3. The NF-κB pathway and PKC pathway
The NF-κB pathway is essential for mediating neuroinflammation after stroke. IPostC reduces NF-κB phosphorylation and inhibits its translocation from the cytosol to the nucleus (Liang et al., 2014; Doeppner et al., 2017). In addition, IPostC enhances the expression of the NF-κB inhibitor, IκBα, but inhibits its phosphorylation, increasing its ability to inhibit NF-κB (Liang et al., 2014). In the same study, the authors showed that the inhibition of NF-κB was associated with the reduction of pro-apoptotic proteins, including Noxa, Bim, Bax, and caspase-3, subsequently attenuated brain cell apoptosis, reduced infarct size, and improved neurological outcomes (Liang et al., 2014). Recent studies suggest that the activated TLR2/4 receptor is one of the upstream signaling pathways that activate NF-κB in ischemic injury (Liu et al., 2017; Wu et al., 2018). IPostC inhibited TLR2/4 and IRAK4 (Wang et al., 2014), which may contribute to the inhibition of NF-κB signaling for neuroprotection (Figure 2).
Another important kinase is protein kinase C (PKC). Delta protein kinase C (δPKC) cleavage or membrane translocation increases its activity and promotes neuronal death (Raval et al., 2005; Shimohata et al., 2007b), while an increase in εPKC levels promotes neuronal survival (Shimohata et al., 2007a). Our study showed that IPostC blocks δPKC cleavage and promotes the phosphorylation of εPKC, suggesting that the PKCs pathway is involved in IPostC’s neuroprotection (Gao et al., 2008).
4. Problems and prospects
Despite extensive IPostC studies in the past decade, many problems remain. We will now discuss the major problems and hurdles that may restrict IPostC’s clinical translation and future research directions.
4.1. IPostC algorithms
IPostC algorithms have great impacts on preventive outcomes (Wang et al., 2008; Lee et al., 2018). As summarized in Table 1, in most studies, when IPostC was induced immediately or shortly after reperfusion, it offered significant neuroprotection. Nevertheless, we have reported that the neuroprotection still existed when the IPostC was initiated 3h post-stroke (Ren et al., 2008). Notably, in a global cerebral ischemia model that produced delayed hippocampal neuron death, IPostC initiated as late as 2 days after ischemia still exerted neuroprotective effects, suggesting a wide therapeutic time window of IPostC in a global ischemia model (Burda et al., 2006). In addition to the therapeutic time window, other IPostC parameters are also important, including the duration and cycle numbers of occlusion and reperfusion, but the most optimal algorithms remain unknown. Future studies may further address these issues in different stroke models.
4.2. Long-term protection
It is important to know how long IPostC’s protection may last (Zhao, 2011). One report suggests that IPostC provided only temporary neuroprotection, but not when measured 28d after transient focal ischemia (Doeppner et al., 2017). Nevertheless, we and others have shown that IPostC offered long-term protection against infarction and neurological dysfunctions (Ren et al., 2008), and attenuated delayed neuron cell death in the hippocampus (Xiang et al., 2018). Such contradictory results may be explained by the different ischemia models (focal and global ischemia), animal strains (mouse and rat), or the protocols involved in different studies, which may be compared in future studies by using different models and different animal species and strains.
4.3. Sex
Stroke outcomes differ between males and females (Zhang et al., 2019). Sexual dimorphism exists in inflammatory responses, including microglial activation and polarization, and T cell response after stroke (Ahnstedt and McCullough, 2019; Kerr et al., 2019). Nevertheless, most studies were conducted in male rodents, and few studies have validated the protective effects of IPostC in female rodents. Although two studies have reported the protective effects of IPostC in female stroke models (Rehni and Singh, 2012; Duanmu et al., 2016), they only focused on neurons and neurological functions, and the underlying mechanisms are not known. Taken together, it is necessary to investigate the effects of IPostC in female stroke models, and compare them with males.
4.4. Age
Age is another important factor that affects stroke outcomes (Chen et al., 2010; Zhang et al., 2019). Aging could promote inflammatory responses, which exacerbate ischemic injury (Ritzel et al., 2018), and be a predictor of poor neurological outcomes and mortality (Kammersgaard et al., 2004; Saposnik et al., 2008). Unfortunately, all of the studies listed in Table 1 used young rodents in their experiments. We must keep in mind that the successful application of IPostC in young rodents is not equivalent to studies conducted in the aged group. Indeed, in the myocardial infarction model, postconditioning did not provide benefits to the elderly group while it protected the young age group (Somers et al., 2011). Therefore, it is important to verify whether IPostC protects against stroke and whether the known IPostC algorithms are still applicable in the aged group.
4.5. Confounding factors
Stroke patients usually have preexisting conditions such as hypertension, hyperlipidemia, and hyperglycemia, which greatly affect stroke outcomes (Luitse et al., 2013; Cipolla et al., 2017). Nevertheless, most studies were conducted in healthy animals without these confounding factors, and it remains unclear whether IPostC could exert beneficial effects against ischemic stroke in animals with one or more of these factors. These issues need to be addressed in future studies to promote IPostC’s clinical translation.
4.6. Combination therapy
Given that t-PA is the only FDA approved drug for ischemic stroke, it is important to test whether IPostC can prolong t-PA’s therapeutic time window and reduce t-PA-induced complications of hemorrhagic transformation. We have shown that delayed postconditioning could reverse infarction exacerbated by delayed t-PA treatment (Ren et al., 2008), suggesting that delayed IPostC could be a potential adjuvant therapy with t-PA. Further studies are needed to address whether delayed IPostC could prevent hemorrhagic transformation and improve neurological outcomes. Moreover, the combination of IPostC with other strategies are also important. For example, a study showed that IPostC could provide a favorable microenvironment for facilitating the homing of the transplanted neural stem cells and promoted NPC-mediated neurogenesis and angiogenesis (Doeppner et al., 2017). Therefore, future studies may test whether IPostC can be combined with other treatments to create novel strategies for stroke therapies.
5. Summary
In summary, IPostC is conducted by brief cycles of occlusion-reperfusion after ischemia, altering the hydrodynamics of reperfusion. The therapeutic effects of IPostC have been validated in various cerebral ischemia-reperfusion models with different animal strains, and different treatment protocols. IPostC reduces brain infarct, attenuates BBB damage, and brain edema in the acute phase, promotes neurogenesis and angiogenesis in the recovery phase, and subsequently improves neurological outcomes. Additionally, IPostC’s protection is linked to cellular and molecular mechanisms that inhibit oxidative stresse, inflammatory responses, apoptotic pathways, as well as modulating ion channels and heat shock proteins, and promoting growth factors such as VEGF and BDNF. Furthermore, IPostC modulates multiple kinase signaling pathways, including the PI3K/Akt/mTOR, the NF-κB, and the GluK2–PSD-95–MLK3-JNK3 pathways. Therefore, IPostC seems to have network regulators that modulate signaling pathways for attenuating ischemic brain damage (Anrather and Hallenbeck, 2013). Despite these studies, many critical issues have not been clarified. Future studies may further optimize IPostC algorithms, evaluate long-term outcomes, assess whether potential confounders such as age and sex affect IPostC, and investigate the potential of combining IPostC with other therapies to maximize its neuroprotection.
Acknowledgments
The authors wish to thank Ms. Felicia F. Beppu at the Department of Neurosurgery, Stanford University School of Medicine for language editing.
Funding
This work was supported by a National Institutes of Health Grant (R01NS064136C) (H.Z.).
References
- Abdul Y, Abdelsaid M, Li W, Webb RC, Sullivan JC, Dong G, Ergul A (2019) Inhibition of Toll-Like Receptor-4 (TLR-4) Improves Neurobehavioral Outcomes After Acute Ischemic Stroke in Diabetic Rats: Possible Role of Vascular Endothelial TLR-4. Mol Neurobiol 56:1607–1617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahnstedt H, McCullough LD (2019) The impact of sex and age on T cell immunity and ischemic stroke outcomes. Cell Immunol 345:103960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albers GW, Marks MP, Kemp S, Christensen S, Tsai JP, Ortega-Gutierrez S, McTaggart RA, Torbey MT, Kim-Tenser M, Leslie-Mazwi T (2018) Thrombectomy for stroke at 6 to 16 hours with selection by perfusion imaging. New England Journal of Medicine 378:708–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anrather J, Hallenbeck JM (2013) Biological networks in ischemic tolerance - rethinking the approach to clinical conditioning. Transl Stroke Res 4:114–129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benjamin EJ et al. (2018) Heart Disease and Stroke Statistics-2018 Update: A Report From the American Heart Association. Circulation 137:e67–e492. [DOI] [PubMed] [Google Scholar]
- Burda J, Danielisova V, Nemethova M, Gottlieb M, Matiasova M, Domorakova I, Mechirova E, Ferikova M, Salinas M, Burda R (2006) Delayed postconditionig initiates additive mechanism necessary for survival of selectively vulnerable neurons after transient ischemia in rat brain. Cell Mol Neurobiol 26:1141–1151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campbell BC, De Silva DA, Macleod MR, Coutts SB, Schwamm LH, Davis SM, Donnan GA (2019) Ischaemic stroke. Nature Reviews Disease Primers 5:1–22. [DOI] [PubMed] [Google Scholar]
- Celso Constantino L, Tasca CI, Boeck CR (2014) The Role of NMDA Receptors in the Development of Brain Resistance through Pre- and Postconditioning. Aging Dis 5:430–441. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chapman SN, Mehndiratta P, Johansen MC, McMurry TL, Johnston KC, Southerland AM (2014) Current perspectives on the use of intravenous recombinant tissue plasminogen activator (tPA) for treatment of acute ischemic stroke. Vasc Health Risk Manag 10:75–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Qi SH, Shen JG (2017) One-Compound-Multi-Target: Combination Prospect of Natural Compounds with Thrombolytic Therapy in Acute Ischemic Stroke. Curr Neuropharmacol 15:134–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen HS, Chen X, Li WT, Shen JG (2018) Targeting RNS/caveolin-1/MMP signaling cascades to protect against cerebral ischemia-reperfusion injuries: potential application for drug discovery. Acta Pharmacol Sin 39:669–682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen RL, Balami JS, Esiri MM, Chen LK, Buchan AM (2010) Ischemic stroke in the elderly: an overview of evidence. Nat Rev Neurol 6:256–265. [DOI] [PubMed] [Google Scholar]
- Cipolla MJ, Sweet JG, Chan SL (2017) Effect of hypertension and peroxynitrite decomposition with FeTMPyP on CBF and stroke outcome. J Cereb Blood Flow Metab 37:1276–1285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Danielisova V, Nemethova M, Gottlieb M, Burda J (2006) The changes in endogenous antioxidant enzyme activity after postconditioning. Cell Mol Neurobiol 26:1181–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ding ZM, Wu B, Zhang WQ, Lu XJ, Lin YC, Geng YJ, Miao YF (2012) Neuroprotective effects of ischemic preconditioning and postconditioning on global brain ischemia in rats through the same effect on inhibition of apoptosis. Int J Mol Sci 13:6089–6101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doeppner TR, Doehring M, Kaltwasser B, Majid A, Lin F, Bahr M, Kilic E, Hermann DM (2017) Ischemic Post-Conditioning Induces Post-Stroke Neuroprotection via Hsp70-Mediated Proteasome Inhibition and Facilitates Neural Progenitor Cell Transplantation. Mol Neurobiol 54:6061–6073. [DOI] [PubMed] [Google Scholar]
- Duanmu WS, Cao L, Chen JY, Ge HF, Hu R, Feng H (2016) Ischemic postconditioning protects against ischemic brain injury by up-regulation of acid-sensing ion channel 2a. Neural Regen Res 11:641–645. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito E, Hayakawa K, Maki T, Arai K, Lo EH (2015) Effects of Postconditioning on Neurogenesis and Angiogenesis During the Recovery Phase After Focal Cerebral Ischemia. Stroke 46:2691–2694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Esposito E, Hayakawa K, Ahn BJ, Chan SJ, Xing C, Liang AC, Kim KW, Arai K, Lo EH (2018) Effects of ischemic postconditioning on neuronal VEGF regulation and microglial polarization in a rat model of focal cerebral ischemia. J Neurochem 146:160–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan YY, Hu WW, Nan F, Chen Z (2017) Postconditioning-induced neuroprotection, mechanisms and applications in cerebral ischemia. Neurochem Int 107:43–56. [DOI] [PubMed] [Google Scholar]
- Feng J, Chen X, Shen J (2017) Reactive nitrogen species as therapeutic targets for autophagy: implication for ischemic stroke. Expert Opinion on Therapeutic Targets 21:305–317. [DOI] [PubMed] [Google Scholar]
- Feng R, Li S, Li F (2011) Toll-like receptor 4 is involved in ischemic tolerance of postconditioning in hippocampus of tree shrews to thrombotic cerebral ischemia. Brain Res 1384:118–127. [DOI] [PubMed] [Google Scholar]
- Gao L, Jiang T, Guo J, Liu Y, Cui G, Gu L, Su L, Zhang Y (2012) Inhibition of autophagy contributes to ischemic postconditioning-induced neuroprotection against focal cerebral ischemia in rats. PLoS One 7:e46092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Zhang H, Takahashi T, Hsieh J, Liao J, Steinberg GK, Zhao H (2008) The Akt signaling pathway contributes to postconditioning’s protection against stroke; the protection is associated with the MAPK and PKC pathways. J Neurochem 105:943–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu Y, Zheng G, Xu M, Li Y, Chen X, Zhu W, Tong Y, Chung SK, Liu KJ, Shen J (2012) Caveolin-1 regulates nitric oxide-mediated matrix metalloproteinases activity and blood-brain barrier permeability in focal cerebral ischemia and reperfusion injury. J Neurochem 120:147–156. [DOI] [PubMed] [Google Scholar]
- Gulati P, Singh N (2014) Evolving possible link between PI3K and NO pathways in neuroprotective mechanism of ischemic postconditioning in mice. Mol Cell Biochem 397:255–265. [DOI] [PubMed] [Google Scholar]
- Gulati P, Singh N, Muthuraman A (2014) Pharmacologic evidence for role of endothelial nitric oxide synthase in neuroprotective mechanism of ischemic postconditioning in mice. J Surg Res 188:349–360. [DOI] [PubMed] [Google Scholar]
- Han D, Sun M, He PP, Wen LL, Zhang H, Feng J (2015) Ischemic Postconditioning Alleviates Brain Edema After Focal Cerebral Ischemia Reperfusion in Rats Through Down-Regulation of Aquaporin-4. J Mol Neurosci 56:722–729. [DOI] [PubMed] [Google Scholar]
- Han D, Zhang S, Fan B, Wen LL, Sun M, Zhang H, Feng J (2014) Ischemic postconditioning protects the neurovascular unit after focal cerebral ischemia/reperfusion injury. J Mol Neurosci 53:50–58. [DOI] [PubMed] [Google Scholar]
- Haorah J, Ramirez SH, Schall K, Smith D, Pandya R, Persidsky Y (2007) Oxidative stress activates protein tyrosine kinase and matrix metalloproteinases leading to blood-brain barrier dysfunction. J Neurochem 101:566–576. [DOI] [PubMed] [Google Scholar]
- Heo JH, Han SW, Lee SK (2005) Free radicals as triggers of brain edema formation after stroke. Free Radic Biol Med 39:51–70. [DOI] [PubMed] [Google Scholar]
- Hoyte L, Kaur J, Buchan AM (2004) Lost in translation: taking neuroprotection from animal models to clinical trials. Exp Neurol 188:200–204. [DOI] [PubMed] [Google Scholar]
- Hu SQ, Zhu J, Pei DS, Zong YY, Yan JZ, Hou XY, Zhang GY (2009) Overexpression of the PDZ1 domain of PSD-95 diminishes ischemic brain injury via inhibition of the GluR6.PSD-95.MLK3 pathway. J Neurosci Res 87:3626–3638. [DOI] [PubMed] [Google Scholar]
- Hu X, Lv T, Yang SF, Zhang XH, Miao YF (2018) Limb remote ischemic postconditioning reduces injury and improves longterm behavioral recovery in rats following subarachnoid hemorrhage: Possible involvement of the autophagic process. Mol Med Rep 17:21–30. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- Joo SP, Xie W, Xiong X, Xu B, Zhao H (2013) Ischemic postconditioning protects against focal cerebral ischemia by inhibiting brain inflammation while attenuating peripheral lymphopenia in mice. Neuroscience 243:149–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kammersgaard LP, Jorgensen HS, Reith J, Nakayama H, Pedersen PM, Olsen TS (2004) Short- and long-term prognosis for very old stroke patients. The Copenhagen Stroke Study. Age Ageing 33:149–154. [DOI] [PubMed] [Google Scholar]
- Kerr N, Dietrich DW, Bramlett HM, Raval AP (2019) Sexually dimorphic microglia and ischemic stroke. CNS Neurosci Ther 25:1308–1317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitagawa K, Saitoh M, Ishizuka K, Shimizu S (2018) Remote Limb Ischemic Conditioning during Cerebral Ischemia Reduces Infarct Size through Enhanced Collateral Circulation in Murine Focal Cerebral Ischemia. J Stroke Cerebrovasc Dis 27:831–838. [DOI] [PubMed] [Google Scholar]
- Kong Y, Rogers MR, Qin X (2013) Effective Neuroprotection by Ischemic Postconditioning is Associated with a Decreased Expression of RGMa and Inflammation Mediators in Ischemic Rats. Neurochemical Research 38:815–825. [DOI] [PubMed] [Google Scholar]
- Kuklina EV, Tong X, George MG, Bansil P (2012) Epidemiology and prevention of stroke: a worldwide perspective. Expert Rev Neurother 12:199–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai TW, Zhang S, Wang YT (2014) Excitotoxicity and stroke: identifying novel targets for neuroprotection. Prog Neurobiol 115:157–188. [DOI] [PubMed] [Google Scholar]
- Lee JS, Song DJ, Hong JH, Kim TS, Joo SP (2018) Diverse Ischemic Postconditioning Protocols Affect the Infarction Size in Focal Ischemic Stroke. J Cerebrovasc Endovasc Neurosurg 20:159–167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li ZY, Liu B, Yu J, Yang FW, Luo YN, Ge PF (2012) Ischaemic postconditioning rescues brain injury caused by focal ischaemia/reperfusion via attenuation of protein oxidization. J Int Med Res 40:954–966. [DOI] [PubMed] [Google Scholar]
- Liang J, Yao J, Wang G, Wang Y, Wang B, Ge P (2012) Ischemic postconditioning protects neuronal death caused by cerebral ischemia and reperfusion via attenuating protein aggregation. Int J Med Sci 9:923–932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang J, Luan Y, Lu B, Zhang H, Luo YN, Ge P (2014) Protection of ischemic postconditioning against neuronal apoptosis induced by transient focal ischemia is associated with attenuation of NF-kappaB/p65 activation. PLoS One 9:e96734. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Yang J, Zhang C, Geng X, Zhao H (2020) Remote ischemic conditioning reduced cerebral ischemic injury by modulating inflammatory responses and ERK activity in type 2 diabetic mice. Neurochem Int 135:104690. [DOI] [PubMed] [Google Scholar]
- Liu J, Xu Q, Wang H, Wang R, Hou XY (2013) Neuroprotection of ischemic postconditioning by downregulating the postsynaptic signaling mediated by kainate receptors. Stroke 44:2031–2035. [DOI] [PubMed] [Google Scholar]
- Liu X, Zhang X, Wang F, Liang X, Zeng Z, Zhao J, Zheng H, Jiang X, Zhang Y (2017) Improvement in cerebral ischemia-reperfusion injury through the TLR4/NF-kappaB pathway after Kudiezi injection in rats. Life Sci 191:132–140. [DOI] [PubMed] [Google Scholar]
- Liu XR, Luo M, Yan F, Zhang CC, Li SJ, Zhao HP, Ji XM, Luo YM (2012) Ischemic postconditioning diminishes matrix metalloproteinase 9 expression and attenuates loss of the extracellular matrix proteins in rats following middle cerebral artery occlusion and reperfusion. CNS Neurosci Ther 18:855–863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luitse MJ, van Seeters T, Horsch AD, Kool HA, Velthuis BK, Kappelle LJ, Biessels GJ (2013) Admission hyperglycaemia and cerebral perfusion deficits in acute ischaemic stroke. Cerebrovasc Dis 35:163–167. [DOI] [PubMed] [Google Scholar]
- Ma H et al. (2019) Thrombolysis Guided by Perfusion Imaging up to 9 Hours after Onset of Stroke. N Engl J Med 380:1795–1803. [DOI] [PubMed] [Google Scholar]
- Martelli AM, Chiarini F, Evangelisti C, Grimaldi C, Ognibene A, Manzoli L, Billi AM, McCubrey JA (2010) The phosphatidylinositol 3-kinase/AKT/mammalian target of rapamycin signaling network and the control of normal myelopoiesis. Histol Histopathol 25:669–680. [DOI] [PubMed] [Google Scholar]
- Miao W, Bao TH, Han JH, Yin M, Zhang J, Yan Y, Zhu YH (2016) Neuroprotection induced by post-conditioning following ischemia/reperfusion in mice is associated with altered microRNA expression. Mol Med Rep 14:2582–2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nemethova M, Danielisova V, Gottlieb M, Burda J (2008) Post-conditioning exacerbates the MnSOD immune-reactivity after experimental cerebral global ischemia and reperfusion in the rat brain hippocampus. Cell Biol Int 32:128–135. [DOI] [PubMed] [Google Scholar]
- Nogueira RG, Jadhav AP, Haussen DC, Bonafe A, Budzik RF, Bhuva P, Yavagal DR, Ribo M, Cognard C, Hanel RA (2018) Thrombectomy 6 to 24 hours after stroke with a mismatch between deficit and infarct. New England Journal of Medicine 378:11–21. [DOI] [PubMed] [Google Scholar]
- Pateliya BB, Singh N, Jaggi AS (2008) Possible role of opioids and KATP channels in neuroprotective effect of postconditioning in mice. Biol Pharm Bull 31:1755–1760. [DOI] [PubMed] [Google Scholar]
- Pei DS, Wang XT, Liu Y, Sun YF, Guan QH, Wang W, Yan JZ, Zong YY, Xu TL, Zhang GY (2006) Neuroprotection against ischaemic brain injury by a GluR6–9c peptide containing the TAT protein transduction sequence. Brain 129:465–479. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Cuomo O, Esposito E, Sirabella R, Di Renzo G, Annunziato L (2011) ASIC1a contributes to neuroprotection elicited by ischemic preconditioning and postconditioning. Int J Physiol Pathophysiol Pharmacol 3:1–8. [PMC free article] [PubMed] [Google Scholar]
- Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, Gala R, Simon RP (2008) In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J Cereb Blood Flow Metab 28:232–241. [DOI] [PubMed] [Google Scholar]
- Pradillo JM, Fernandez-Lopez D, Garcia-Yebenes I, Sobrado M, Hurtado O, Moro MA, Lizasoain I (2009) Toll-like receptor 4 is involved in neuroprotection afforded by ischemic preconditioning. J Neurochem 109:287–294. [DOI] [PubMed] [Google Scholar]
- Raval AP, Dave KR, Prado R, Katz LM, Busto R, Sick TJ, Ginsberg MD, Mochly-Rosen D, Perez-Pinzon MA (2005) Protein kinase C delta cleavage initiates an aberrant signal transduction pathway after cardiac arrest and oxygen glucose deprivation. J Cereb Blood Flow Metab 25:730–741. [DOI] [PubMed] [Google Scholar]
- Rehni AK, Singh N (2007) Role of phosphoinositide 3-kinase in ischemic postconditioning-induced attenuation of cerebral ischemia-evoked behavioral deficits in mice. Pharmacol Rep 59:192–198. [PubMed] [Google Scholar]
- Rehni AK, Singh TG (2012) Involvement of CCR-2 chemokine receptor activation in ischemic preconditioning and postconditioning of brain in mice. Cytokine 60:83–89. [DOI] [PubMed] [Google Scholar]
- Ren C, Gao X, Niu G, Yan Z, Chen X, Zhao H (2008) Delayed postconditioning protects against focal ischemic brain injury in rats. PLoS One 3:e3851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rezazadeh H, Hoseini Kahnuee M, Roohbakhsh A, Shamsizadeh A, Rahmani MR, Bidaki R, Amin F, Kamali B, Bakhshi H, Allahtavakoli M (2013) Neuroprotective consequences of postconditioning on embolic model of cerebral ischemia in rat. Iran J Basic Med Sci 16:144–149. [PMC free article] [PubMed] [Google Scholar]
- Ritzel RM, Lai YJ, Crapser JD, Patel AR, Schrecengost A, Grenier JM, Mancini NS, Patrizz A, Jellison ER, Morales-Scheihing D, Venna VR, Kofler JK, Liu F, Verma R, McCullough LD (2018) Aging alters the immunological response to ischemic stroke. Acta Neuropathol 136:89–110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Robin E, Simerabet M, Hassoun SM, Adamczyk S, Tavernier B, Vallet B, Bordet R, Lebuffe G (2011) Postconditioning in focal cerebral ischemia: role of the mitochondrial ATP-dependent potassium channel. Brain Res 1375:137–146. [DOI] [PubMed] [Google Scholar]
- Sabbah DA, Brattain MG, Zhong H (2011) Dual inhibitors of PI3K/mTOR or mTOR-selective inhibitors: which way shall we go? Curr Med Chem 18:5528–5544. [DOI] [PubMed] [Google Scholar]
- Saposnik G, Cote R, Phillips S, Gubitz G, Bayer N, Minuk J, Black S (2008) Stroke outcome in those over 80: a multicenter cohort study across Canada. Stroke 39:2310–2317. [DOI] [PubMed] [Google Scholar]
- Scartabelli T, Gerace E, Landucci E, Moroni F, Pellegrini-Giampietro DE (2008) Neuroprotection by group I mGlu receptors in a rat hippocampal slice model of cerebral ischemia is associated with the PI3K-Akt signaling pathway: a novel postconditioning strategy? Neuropharmacology 55:509–516. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Zhao H, Steinberg GK (2007a) εPKC may contribute to the protective effect of hypothermia in a rat focal cerebral ischemia model. Stroke 38:375–380. [DOI] [PubMed] [Google Scholar]
- Shimohata T, Zhao H, Sung JH, Sun G, Mochly-Rosen D, Steinberg GK (2007b) Suppression of δ PKC Activation after Focal Cerebral Ischemia Contributes to the Protective Effect of Hypothermia. Journal of Cerebral Blood Flow & Metabolism 27:1463–1475. [DOI] [PubMed] [Google Scholar]
- Somers SJ, Lacerda L, Opie L, Lecour S (2011) Age, genetic characteristics and number of cycles are critical factors to consider for successful protection of the murine heart with postconditioning. Physiol Res 60:971–974. [DOI] [PubMed] [Google Scholar]
- Sun Y, Zhang T, Zhang Y, Li J, Jin L, Sun Y, Shi N, Liu K, Sun X (2018) Ischemic Postconditioning Alleviates Cerebral Ischemia-Reperfusion Injury Through Activating Autophagy During Early Reperfusion in Rats. Neurochem Res 43:1826–1840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sussman ES, Connolly ES Jr. (2013) Hemorrhagic transformation: a review of the rate of hemorrhage in the major clinical trials of acute ischemic stroke. Front Neurol 4:69–69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taskapilioglu MO, Alkan T, Goren B, Tureyen K, Sahin S, Taskapilioglu O, Korfali E (2009) Neuronal protective effects of focal ischemic pre- and/or postconditioning on the model of transient focal cerebral ischemia in rats. J Clin Neurosci 16:693–697. [DOI] [PubMed] [Google Scholar]
- Vaibhav K, Braun M, Khan MB, Fatima S, Saad N, Shankar A, Khan ZT, Harris RBS, Yang Q, Huo Y, Arbab AS, Giri S, Alleyne CH Jr., Vender JR, Hess DC, Baban B, Hoda MN, Dhandapani KM (2018) Remote ischemic postconditioning promotes hematoma resolution via AMPK-dependent immune regulation. J Exp Med 215:2636–2654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang JY, Shen J, Gao Q, Ye ZG, Yang SY, Liang HW, Bruce IC, Luo BY, Xia Q (2008) Ischemic postconditioning protects against global cerebral ischemia/reperfusion-induced injury in rats. Stroke 39:983–990. [DOI] [PubMed] [Google Scholar]
- Wang Y, Ge P, Yang L, Wu C, Zha H, Luo T, Zhu Y (2014) Protection of ischemic post conditioning against transient focal ischemia-induced brain damage is associated with inhibition of neuroinflammation via modulation of TLR2 and TLR4 pathways. J Neuroinflammation 11:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei D, Xiong X, Zhao H (2015) Tim-3 cell signaling and iNOS are involved in the protective effects of ischemic postconditioning against focal ischemia in rats. Metab Brain Dis 30:483–490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu G, McBride DW, Zhang JH (2018) Axl activation attenuates neuroinflammation by inhibiting the TLR/TRAF/NF-kappaB pathway after MCAO in rats. Neurobiol Dis 110:59–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu H, Yang SF, Dai J, Qiu YM, Miao YF, Zhang XH (2015) Combination of early and delayed ischemic postconditioning enhances brain-derived neurotrophic factor production by upregulating the ERK-CREB pathway in rats with focal ischemia. Mol Med Rep 12:6427–6434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiang J, Andjelkovic AV, Zhou N, Hua Y, Xi G, Wang MM, Keep RF (2018) Is there a central role for the cerebral endothelium and the vasculature in the brain response to conditioning stimuli? Cond Med 1:220–232. [PMC free article] [PubMed] [Google Scholar]
- Xie R, Li J, Zhao H (2018) The underlying mechanisms involved in the protective effects of ischemic postconditioning. Conditioning medicine 1:73. [PMC free article] [PubMed] [Google Scholar]
- Xie R, Wang P, Ji X, Zhao H (2013) Ischemic post-conditioning facilitates brain recovery after stroke by promoting Akt/mTOR activity in nude rats. J Neurochem 127:723–732. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xing B, Chen H, Zhang M, Zhao D, Jiang R, Liu X, Zhang S (2008a) Ischemic post-conditioning protects brain and reduces inflammation in a rat model of focal cerebral ischemia/reperfusion. J Neurochem 105:1737–1745. [DOI] [PubMed] [Google Scholar]
- Xing B, Chen H, Zhang M, Zhao D, Jiang R, Liu X, Zhang S (2008b) Ischemic postconditioning inhibits apoptosis after focal cerebral ischemia/reperfusion injury in the rat. Stroke 39:2362–2369. [DOI] [PubMed] [Google Scholar]
- Yao GY, Zhu Q, Xia J, Chen FJ, Huang M, Liu J, Zhou TT, Wei JF, Cui GY, Zheng KY, Hou XY (2018) Ischemic postconditioning confers cerebroprotection by stabilizing VDACs after brain ischemia. Cell Death Dis 9:1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan Y, Guo Q, Ye Z, Pingping X, Wang N, Song Z (2011) Ischemic postconditioning protects brain from ischemia/reperfusion injury by attenuating endoplasmic reticulum stress-induced apoptosis through PI3K-Akt pathway. Brain Res 1367:85–93. [DOI] [PubMed] [Google Scholar]
- Zhang H, Lin S, Chen X, Gu L, Zhu X, Zhang Y, Reyes K, Wang B, Jin K (2019) The effect of age, sex and strains on the performance and outcome in animal models of stroke. Neurochem Int 127:2–11. [DOI] [PubMed] [Google Scholar]
- Zhang W, Wang B, Zhou S, Qiu Y (2012) The effect of ischemic post-conditioning on hippocampal cell apoptosis following global brain ischemia in rats. J Clin Neurosci 19:570–573. [DOI] [PubMed] [Google Scholar]
- Zhang X, Zhang Q, Tu J, Zhu Y, Yang F, Liu B, Brann D, Wang R (2015) Prosurvival NMDA 2A receptor signaling mediates postconditioning neuroprotection in the hippocampus. Hippocampus 25:286–296. [DOI] [PubMed] [Google Scholar]
- Zhao H (2007) The protective effect of ischemic postconditioning against ischemic injury: from the heart to the brain. J Neuroimmune Pharmacol 2:313–318. [DOI] [PubMed] [Google Scholar]
- Zhao H (2009) Ischemic postconditioning as a novel avenue to protect against brain injury after stroke. J Cereb Blood Flow Metab 29:873–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H (2011) The Protective Effects of Ischemic Postconditioning against Stroke: From Rapid to Delayed and Remote Postconditioning. Open Drug Discov J 5:138–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao H, Sapolsky RM, Steinberg GK (2006) Interrupting reperfusion as a stroke therapy: ischemic postconditioning reduces infarct size after focal ischemia in rats. J Cereb Blood Flow Metab 26:1114–1121. [DOI] [PubMed] [Google Scholar]
- Zhao H, Luo Y, Liu X, Wang R, Yan F, Liu X, Li S, Leak RK, Ji X (2013) Ischemic post-conditioning partially reverses cell cycle reactivity following ischemia/reperfusion injury: a genome-wide survey. CNS Neurol Disord Drug Targets 12:350–359. [DOI] [PubMed] [Google Scholar]
- Zhao H, Wang R, Tao Z, Gao L, Yan F, Gao Z, Liu X, Ji X, Luo Y (2014a) Ischemic postconditioning relieves cerebral ischemia and reperfusion injury through activating T-LAK cell-originated protein kinase/protein kinase B pathway in rats. Stroke 45:2417–2424. [DOI] [PubMed] [Google Scholar]
- Zhao JH, Meng XL, Zhang J, Li YL, Li YJ, Fan ZM (2014b) Oxygen glucose deprivation post-conditioning protects cortical neurons against oxygen-glucose deprivation injury: role of HSP70 and inhibition of apoptosis. J Huazhong Univ Sci Technolog Med Sci 34:18–22. [DOI] [PubMed] [Google Scholar]